


Abstract
Understanding the pathological impact of non-coding genetic variation is a major challenge in medical genetics. Accumulating evidences indicate that a significant fraction of genetic alterations, including structural variants (SVs), can cause human disease by altering the function of non-coding regulatory elements, such as enhancers. In the case of SVs, described pathomechanisms include changes in enhancer dosage and long-range enhancer-gene communication. However, there is still a clear gap between the need to predict and interpret the medical impact of non-coding variants, and the existence of tools to properly perform these tasks. To reduce this gap, we have developed POSTRE (Prediction Of STRuctural variant Effects), a computational tool to predict the pathogenicity of SVs implicated in a broad range of human congenital disorders. By considering disease-relevant cellular contexts, POSTRE identifies SVs with either coding or long-range pathological consequences with high specificity and sensitivity. Furthermore, POSTRE not only identifies pathogenic SVs, but also predicts the disease-causative genes and the underlying pathological mechanism (e.g, gene deletion, enhancer disconnection, enhancer adoption, etc.). POSTRE is available at https://github.com/vicsanga/Postre.
INTRODUCTION
Structural Variants (SVs) represent one of the greatest sources of genetic variation in the human genome (1,2). Recent estimates based on the use of short and long read sequencing indicate that a typical human genome contains more than 10.000 SVs, which most often (90%) reside within non-coding sequences (3,4). This group of genetic alterations includes: deletions, duplications, inversions, insertions, and translocations. Their size ranges from a few base pairs (<50) to several megabases (Mb) (5). SVs can cause phenotypic diversity and a broad set of human disorders, including congenital abnormalities and various cancer types (6). The pathogenicity of SVs can be triggered by two main mechanisms: (i) direct effects on genes (e.g. ‘coding’ pathomechanisms: changes in gene dosage, gene truncations, formation of fusion transcripts) and (ii) changes in the non-coding regulatory landscape of disease relevant genes that alter their expression levels (i.e. ‘long-range’ pathomechanisms) (6–8). However, it is currently challenging to predict or interpret the functional consequences of SVs. This is particularly true for SVs that affect gene expression through long-range regulatory mechanisms (3,6,8) due to the limited understanding of the non-coding regulatory genome and the cell-type specific functions of distal regulatory elements, such as enhancers.
Genome-wide association studies (GWAS) for human complex diseases highlight the functional and medical relevance of non-coding sequences, as the vast majority (>90%) of complex-disease associated variants are located within the non-coding fraction of the human genome (9–12). Although genetic studies of mendelian disorders have preferentially focused on protein-coding sequences (13,14), accumulating evidences indicate that non-coding alterations can often contribute to this type of diseases (15–23). Non-coding variants associated with either complex or mendelian disorders are mainly located within putative enhancers (10,24). Enhancers are distal cis-regulatory elements (25,26) that positively control the expression of their target genes in space and time, and are major determinants of cell-type specific gene expression programs (27–30). Enhancers have been globally identified in hundreds of human cell types and tissues using universal epigenomic approaches (31–33). Enhancers can control gene expression over large genomic distances (>1 Mb) (34–36), often skipping proximal genes while controlling the expression of more distally located ones (37). Consequently, linking enhancers with their target genes is not an obvious task. In this regard, 3D genome organization studies based on Hi-C technology (38) revealed that genomes are organized in large (Mb-scale) self-interacting domains often referred to as topologically associating domains (TADs) (39). TADs represent fundamental regulatory domains as (i) they facilitate enhancer–gene interactions within them (6,39–41) and (ii) insulate genes from contacting ectopic enhancers located in other TADs (42). SVs can disrupt TAD organization, which in turn can rewire enhancer-gene communication and lead to pathological changes in gene expression (6,7,42). This can occur through two alternative long-range regulatory mechanisms: (i) SVs can lead to gene silencing (loss of function (LOF)) by disconnecting genes from their cognate enhancer/s (i.e. enhancer disconnection) (43), or through the deletion of enhancers (i.e. enhancer deletion) (44); (ii) SVs can lead to gains in gene expression (gain of function (GOF)) by enabling enhancers to interact with non-target genes (i.e. enhancer adoption/hijacking) (45), or by duplicating enhancers (46). Nevertheless, predicting the pathological consequences of TAD disruption is complicated by the fact that, besides TAD organization, other genetic and epigenetic features also contribute to productive gene-enhancer communication (7,47–53).
Since gene expression programs change in space and time, it is fundamental to assess the pathological impact of SVs in the relevant cellular contexts. For instance, a deletion in chromosome (Chr) 17 causes a pathogenic downregulation of SOX9 in the neural crest but not in other cell types, such as embryonic stem cells (ESCs) or chondrocytes (36). Importantly, this deletion eliminates enhancers that are specifically active in neural crest cells (NCC) and that, consequently, control SOX9 expression in this cell type but not in others. Additionally, the same SVs might differentially affect the expression of the same gene depending on the cellular context. For example, a deletion could eliminate enhancers in one cell type in which the gene is active and lead to gene silencing (pathogenic LOF), whereas in another cell type in which the same gene is inactive, the deletion could eliminate a TAD boundary and lead to gene overexpression through enhancer adoption mechanisms (pathogenic GOF) (6). Therefore, although the recurrency of non-coding SVs within specific genomic loci could help identifying the genes involved in certain disorders, the exact pathomechanisms can only be predicted and experimentally validated if cell type specific regulatory landscapes (i.e. enhancers and TADs) are considered. During the last few years, and partly due to the efforts of large international consortia, different types of genomic data (e.g. epigenomic, transcriptomic and Hi-C data) have been generated in hundreds of different human cell types and tissues (54,55). Furthermore, most of this data is available through public databases such as GEO (56). In principle, these genomic datasets could be integrated in order to predict the pathological consequences of SVs in a cell type-specific manner (7). Nonetheless, finding the appropriate datasets for the disease of interest, as well as their subsequent processing, analysis and integration can be time-consuming and may require advanced bioinformatic skills. Consequently, the diagnosis and interpretation of how SVs might cause human disease remains complicated and the pathogenicity of SVs identified in hundreds of patients is currently unknown (often referred to as Variants of Uncertain Significance (VUS)) (57). To overcome these limitations, it is essential to implement user-friendly computational tools that can be used by a broad scientific community (7,8).
Taking the previous challenges into account, we developed POSTRE (Prediction Of STRuctural variant Effects), a computational tool that, thanks to its user-friendly graphical interface, facilitates the cell type-specific analysis of SVs implicated in congenital disorders. Compared to other SV analysis tools, POSTRE is able to predict both coding and long-range pathomechanisms in a cell type/tissue-specific manner. The current version of POSTRE can handle SVs potentially implicated in limb, craniofacial, cardiac and neurodevelopmental congenital abnormalities, thus covering a broad set of congenital diseases (58–63). Here we extensively describe how POSTRE works and illustrate how it can predict the pathological consequences of SVs, particularly those implying long-range regulatory mechanisms, with high sensitivity and specificity.
MATERIALS AND METHODS
Key definitions
The terms LOF, GOF, candidate gene, causative gene and phenotype appear recurrently throughout the manuscript. To avoid misunderstandings, it is important to clearly define these terms:
LOF (Loss of Function): pathogenic loss of gene function or expression through either coding or non-coding mechanisms.
GOF (Gain of Function): pathogenic gain of gene function or expression through either coding or non-coding mechanisms.
Candidate gene: gene whose regulatory domain (TAD) or sequence (e.g gene deletion) is altered by a SV. A candidate gene is not necessarily involved in the disease etiology (i.e. candidate genes include both causative and non-causative genes).
Causative gene: gene predicted to be involved in the disease etiology. All causative genes are candidate genes.
Phenotype: the phenotypes of the investigated patients and associated SVs are described according to the Human Phenotype Ontology (HPO) (64), which provides a standarized vocabulary of phenotypic abnormalities. Check the genePhenoScore Methods section for more details.
POSTRE pathogenicity score
POSTRE is a software developed to predict the pathogenicity and underlying pathomechanisms (‘coding’ or ‘long-range’) of SVs implicated in a broad set of congenital disorders. The tool requires as input: (i) the type of SV (i.e. deletion, duplication, inversion or translocation), (ii) the genomic coordinates of the SV breakpoints and (iii) the patient phenotype/s (i.e. type of congenital abnormality) (Figure 1).
Figure 1.

POSTRE Overview. (A) User Input: For each SV identified in a patient, POSTRE requires (i) the type of SV (Deletion, Duplication, Inversion or Translocation), (ii) the genomic coordinates of the SV breakpoints at base-pair (bp) resolution and (iii) the patient's phenotype (i.e. type of congenital abnormality) according to HPO terms. (B) Software internal proceeding: cell types/tissues considered relevant for the patient's phenotype are selected. For each of the selected cell types/tissues, specific genomic data (e.g. gene expression profiles, enhancer maps) are loaded to predict the SV pathogenicity. Each of the cell types/tissues considered as relevant for the patient's phenotype is independently assessed. TADs are used as a proxy to determine the regulatory domains and genes potentially affected by the SV. To evaluate the pathogenicity of the candidate genes several features, such as dosage sensitivity (DS) or previous associations with the patient phenotype, are considered. In addition, for those candidate genes that are not directly disrupted by by the SV, long-range regulatory mechanisms resulting in either a gain (GOF) or loss (LOF) of gene expression are considered. (C) Output: heatmap providing an overview of the predictions for each candidate gene and relevant cell type/tissue. Genes considered as pathogenic are highlighted in red (Loss of Function, LOF) or green (Gain of Function, GOF). For genes predicted as pathogenic, a detailed report describing the genetic and molecular basis of the prediction is provided. The report includes information such as a simplified graphical abstract of how the SV affects the candidate gene, details about enhancer changes in the regulatory domain of the candidate gene or a link to visualize relevant genomic data in a genome browser.
When evaluating the pathogenicity of a SV, POSTRE first considers the phenotype/s associated with the SV in order to select cell types/tissues relevant for such phenotype/s (Supplementary Data 1) (e.g, two different embryonic brain prefrontal cortex datasets for neurodevelopmental defects). Next, for each of the selected cell types/tissues, an independent pathogenic prediction is performed using cell-type/tissue specific genomic data (i.e. gene expression profiles, enhancer maps and TAD maps; Supplementary Data 1). More specifically, the SV breakpoints are mapped in the context of TADs identified in the selected cell-type/tissue. Once the affected TADs (i.e. regulatory domains) are determined, all the genes located within them are selected as potential candidates associated with the patient disease (i.e. candidate genes). Subsequently, POSTRE integrates a set of genetic and genomic features in order to assign a pathogenicity score (PS) to each candidate gene considering in parallel both LOF and GOF scenarios. These gene PS are independently calculated in each of the cell types/tissues considered as relevant for a given phenotype. In addition, POSTRE offers the possibility to compute the PS using two alternative running modes (i.e. Standard and High-Specificity; see details below). Finally, all candidate genes are ranked according to their PS in each of the considered cell types/tissues.
The features used to calculate the gene PS can be broadly divided into the following categories or sub-scores:
Gene-phenotype relationships (genePhenoScore)
Gene expression (geneExpressionScore)
Enhancer activity (geneEnhancerScore)
-
Gene features (geneFeatureScore):
Dosage sensitivity (dosageSensitivityScore)
Breadth of polycomb domains in promoter regions (polycombScore)
Gene-phenotype relationships: computing the genePhenoScore
The genePhenoScore metric is used to quantify the relationship between the candidate genes and the SV associated phenotype. Before describing how the genePhenoScore was calculated, we briefly explain how the associations between genes and phenotypes were established.
Firstly, we obtained a collection of Human Phenotype Ontology (HPO) terms (64) for each of the considered patient phenotypic categories (i.e. Cardiovascular, Craniofacial/Head-Neck, Limbs and Neurodevelopmental). HPO terms represent standardized phenotypic categories that facilitate the annotation of clinical abnormalities. HPO terms are organized following a hierarchical and nested structure. For example, the HPO term Absent speech (HP:0001344) belongs, following a hierarchical order, to the Delayed speech and language (HP:0000750), Neurodevelopmental abnormality (HP:0012759), Abnormality of the nervous system (HP:0000707) and Phenotypic abnormality (HP:0000118) categories. In POSTRE, a set of reference HPOs, together with all their nested and more specific terms, were selected for each of the considered phenotypes. Following the previous example, when considering neurodevelopmental phenotypes we used Neurodevelopmental abnormality (HP:0012759) as a reference HPO, which, among others includes the Delayed speech language (HP:0000750) and Absent speech (HP:0001344) HPO terms. In this regard, the reference HPOs selected for the patient phenotypes considered in POSTRE were:
Cardiovascular: Abnormality of the cardiovascular system (HP:0001626)
Head-Neck: Abnormality of head or neck (HP:0000152)
Limbs: Abnormality of limbs (HP:0040064)
Neurodevelopmental: Neurodevelopmental abnormality (HP:0012759)
To retrieve all the HPO terms associated with each of the reference HPOs, we used the hp.obo file from the Downloads/Ontology section in the HPO website (64).
On the other hand, human genes were also annotated according to HPO terms. For each gene present in OMIM (65), all its associated HPO terms were retrieved using the genes_to_phenotype.txt file from the Downloads/Annotation section in the HPO website (64). Next, genes associated with at least one of the HPO terms present within the reference HPO phenotypic groups described above were linked with the main patient phenotypes considered by POSTRE (i.e. Cardiovascular, Head-Neck, Limbs or Neurodevelopmental). For example, if a gene is only associated with the Delayed speech language (HP:0 000 750) term, it will still be linked with the Neurodevelopmental phenotype because it is contained within the more general Neurodevelopmental abnormality (HP:0012759) HPO category.
A similar approach was used to associate human genes with mouse phenotypes as defined in the Mammalian Phenotype Ontology (MPO) (66). MPO terms, which are equivalent to HPOs, are also organized in a hierarchical and nested manner. As described for HPOs, a set of reference MPOs and all their nested and more specific terms were selected for each of the considered phenotypes:
Cardiovascular: Cardiovascular system phenotype (MP:0005385)
Head-Neck: Craniofacial phenotype (MP:0005382) and abnormal neck morphology (MP:0012719)
Limbs: Limbs/digits/tail phenotype (MP:0005371)
Neurodevelopmental: Behavior/neurological phenotype (MP:0005386)
To retrieve all the MPO terms associated with each of the reference MPOs, we used the mp.obo file from the OBO Foundry website (67). Next, to link human genes with MPO terms, we used the HMD_HumanPhenotype.rpt file from MGD (68). Lastly, human genes associated with at least one of the MPO terms present within the reference MPO phenotypic groups described above were linked with the main patient phenotypes considered by POSTRE (i.e. Cardiovascular, Head-Neck, Limbs or Neurodevelopmental).
The previous relationships between human genes and human/mouse phenotypes were used to compute the genePhenoscore. When considering the phenotype associated with a SV (POSTRE input), a candidate gene receives a genePhenoscore = 1 if it is associated with the same phenotype in humans, a genePhenoscore = 0.5 if it is associated with the same phenotype in mice but not in humans (this is only applied in the Standard Running Mode) and a genePhenoScore = 0 otherwise. These genePhenoscore criteria are applied by default, but they can be modified by the user with the Advanced Parameters options: if the Gene-PatientPheno option is set to ‘No’, a candidate gene will get a genePhenoscore = 1 as far as it is associated with at least one phenotype in humans (regardless of whether it matches the phenotype associated with the SV), and a genePhenoscore = 0.5 if it is associated with at least one phenotype in mice but not in humans (this is only applied in the Standard Running Mode) and a genePhenoScore = 0 otherwise.
Dosage sensitivity: computing the dosageSensitivityScore
The dosageSensitivityScore is used to quantify the dosage sensitivity of the candidate genes. It is based on Haploinsufficiency (HI) and Triplosensitivity (TS) scores, depending on whether LOF or GOF scenarios are considered, respectively.
HI gene scores were retrieved from two different sources: (i) HI scores ranging between 0 (low HI) and 1 (High HI) were obtained for each gene from (69–71); (ii) ClinGen (ClinGen_haploinsufficiency_gene_GRCh37.bed) (72): genes with strong evidence for dosage sensitivity (ClinGen score = 3) were assigned a HI score = 1. Then, between these two alternative HI scores, the highest one was selected for each gene. Next, genes with HI scores ≥ 0.85 were assigned such value as their final_HI_score, while genes with HI scores < 0.85 were assigned a final_HI_score = 0.
TS scores ranging between 0 (low TS) and 1 (High TS) were obtained for each autosomal gene from (71). Next, genes with TS scores ≥ 0.85 were assigned such value as their final_TS_score, while genes with TS scores < 0.85 were assigned a final_TS_score = 0.
Genes that are not located in autosomal chromosomes (e.g ChrX) lack TS scores. Therefore, since HI genes are also usually TS (71), for genes in sexual chromosomes the final_HI_scores were used as proxy for TS (i.e. final_TS_score = final_HI_score).
Note: For the HI and TS scores, the thresholds were set to 0.85 based on the analyses of the causative genes reported for positive control patients (Tables 1 and 2), since among the HI and TS scores for these causative genes, the lowest HI/TS score was 0.85.
Table 1.
Overview of POSTRE analysis for SVs causing congenital abnormalities through experimentally validated long-range mechanisms

|
Table 2.
Overview of POSTRE analysis for SVs previously predicted to cause congenital abnormalities through long-range mechanisms

|
Finally, the dosageSensitivityScore is computed as follows:
dosageSensitivityScoreLOF = final_HI_score
dosageSensitivityScoreGOF = final_TS_score
Breadth of polycomb domains in promoter regions: computing the polycombScore
The polycombScore was used to quantify the breadth of polycomb domains in gene promoters. Previous work showed that genes whose promoters are covered by broad polycomb/H3K27me3 domains when inactive largely correspond with major developmental genes frequently implicated in congenital disorders (73,74). Moreover, these developmental genes display high enhancer responsiveness (49,75,76). The polycombScore ranges from 0 (no or narrow polycomb domain) to 1 (broad polycomb domain).
To compute the polycombScore two different approaches were considered.
Approach#1: H3K27me3 ChIP-seq data generated in ESC (GSE24447; H3K27me3: SRR067372, input: SRR067371), NCC (GSE108518; H3K27me3: SRR6418921, input: SRR6418919) and cardiomyocytes (GSE85628; H3K27me3: SRR4032228, input: SRR4032231) were used independently to call H3K27me3 peaks using MACS2 (77) with the broad peak calling mode. Peaks with a fold-enrichment >3 and Q-value <0.1 were considered. Subsequently, peaks within 1 kb of each other were merged using bedtools (78), and associated with a protein-coding gene when overlapping a transcription start site (TSS). Next, as previously described in (49), the size distribution of H3K27me3 peaks associated with TSS was analyzed with findiplist() (inflection R package; https://cran.r-project.org/web/packages/inflection/vignettes/inflection.html). The findiplist() function provides the coordinates of the inflection point, as well as the upper and lower limit values containing it. The H3K27me3 peak size corresponding to the upper limit value was selected as a cutoff for each sample: 6689 bp for ESC, 6719 bp for NCC, and 8020 bp for cardiomyocytes (Supplementary Figure S1). Lastly, genes with H3K27me3 peaks ≥ than the cutoff calculated for each cell type (e.g. 6689 bp for ESC) in at least one of the corresponding cell types (ESC, NCC or cardiomyocytes) were assigned a polycombScore = 1. All other genes were assigned a polycombScore = 0.
Approach#2: It is based on the repressive tendency score (RTS) described in (74), a metric that quantifies the association between genes and broad H3K27me3 domains. This metric is computed considering data generated in hundreds of different cell types. RTS ranges from 0 (no or narrow polycomb domain) to 1 (broad polycomb domain). Based on RTS, three groups of genes were defined in (74): (i) ‘regulatory’ genes (high RTS), (ii) ‘structural’ genes (medium RTS) and (iii) ‘housekeeping’ genes (low RTS). ‘Regulatory’ genes were assigned a polycombScore = 1 and the rest were assigned a polycombScore = 0.
Finally, each gene was assigned the highest polycombScore obtained using any of the two approaches described above.
Computing the geneFeatureScore
This metric results from the aggregation of the polycombScore and the dosageSensitivityScore.
For this metric to be >0 either the polycombScore or the dosageSensitiveScore must be ≥0.85 in the Standard running mode, while both scores must be ≥0.85 in the High-Specificity running mode. The geneFeatureScore is computed as follows:
 |
 |
Gene expression: computing the geneExpressionScore
The geneExpressionScore is used to incorporate gene expression levels into POSTRE’s PS. A detailed list of the gene expression datasets considered for the different patient phenotypes, together with the methodologies used to quantify gene expression in each dataset, is provided in Supplementary Data 1.
Considering the expression status of the candidate genes is particularly relevant for loss of function (LOF) situations, as the disease-causative genes must be expressed in the relevant cell types/tissues (for both ‘coding’ and ‘long-range’ cases). We defined a minimum (FPKM = 1) and a maximum (FPKM = 10) expression threshold: genes with FPKM ≤ 1 are considered as not expressed and are assigned a geneExpressionScore = 0; genes with FPKM ≥ 10 are considered to be expressed and are assigned a geneExpressionScore = 1; genes with 1 < FPKM < 10 are subject to a 0–1 min-max normalization to determine its geneExpressionScore, considering as min-max values the expression thresholds.
Changes in enhancer activity: computing the geneEnhancerScore
The geneEnhancerScore quantifies changes in enhancer activity between Control and Patient alleles. Firstly, enhancers are annotated in the different cell types/tissues considered by POSTRE. To annotate enhancers, we considered H3K27ac peaks located at least (minimum required distance) 10kb away from any protein-coding gene TSS. Depending on the cell type/tissue, H3K27ac peaks were either already available or called using MACS2 (77). For some cell types/tissues, chromatin accessibility (e.g. DNAse-seq, ATAC-seq) data and/or ChIP-seq data for other regulatory proteins (e.g. p300) were available in addition to H3K27ac. In those cases, enhancers were identified as regions showing overlapping peaks for H3K27ac and other available proteins/chromatin accessibility. The ChIP-Seq datasets used to annotate enhancers in the different cell types/tissues together with the methodologies applied in each case are more extensively described in Supplementary Data 1. In addition, H3K27ac ChIP-seq bigwig files were also obtained for each of the relevant cell types/tissues. These H3K27ac bigwig files were either already available in public repositories or generated with deepTools (79) using bamCoverage (reads per genome coverage (RPGC) normalization). Next, for each cell type/tissue, the enhancer lists and H3K27ac bigwig files were combined to quantify enhancer activity as the maximum H3K27ac levels for each enhancer using the bigWigAverageOverBed UCSC binary tool. As a result, each enhancer was assigned an enhancerIndividualActivity score according to the following formula:
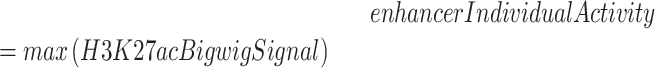 |
Then, for each candidate gene an enhancerActivityControl metric is calculated as the sum of the enhancerIndividualActivity scores assigned to the enhancers located within the control (i.e. without SV) regulatory domain of the candidate gene (Supplementary Figures S2 and S3). Likewise, an enhancerActivityPatient metric is calculated as the sum of enhancerIndividualActivity scores assigned to the enhancers located within the rearranged regulatory domain of the candidate gene (see ‘Rearranged regulatory domains’ section below for more details) (Supplementary Figures S2 and S3). We assume that individual enhancer activities are not affected by the SVs (e.g. H3K27ac levels of an enhancer are the same after relocation in the genome by an inversion). For LOF, the enhancerActivityPatient value is computed only considering the cognate enhancers that remain within the rearranged regulatory domain of the candidate gene (enhancerActivityPatientLOF). In contrast, for GOF, the enhancerActivityPatient value is computed considering both the remaining cognate enhancers as well as the ectopic enhancers located within the rearranged regulatory domain of the candidate gene (enhancerActivityPatientGOF). These distinct criteria for either LOF or GOF were defined taking into consideration our current understanding of enhancer-gene communication (80,81). Briefly, although compensatory mechanisms (i.e. no changes in gene expression) whereby cognate enhancers are substituted by ectopic ones due to SVs are theoretically possible, to the best of our knowledge, there are not reported evidences to support them. In contrast, enhancer adoption mechanisms leading to GOF effects have been previously described, particularly for genes displaying broad polycomb domains and characterized by their high enhancer responsiveness (45,49,75,76).
Next, the final geneEnhancerScores are computed as follows:
geneEnhancerScoreLOF: If enhancerActivityPatientLOF < enhancerActivityControl (i.e. the candidate gene shows higher enhancer activity in the Control/wild-type regulatory domain), then geneEnhancerScoreLOF = 1. Otherwise, the geneEnhancerScoreLOF = 0.
geneEnhancerScoreGOF: If enhancerActivityPatientGOF > enhancerActivityControl (i.e. the candidate gene shows higher enhancer activity in the Patient/rearranged regulatory domain), then geneEnhancerScoreGOF = 1. Otherwise, the geneEnhancerScoreGOF = 0.
Rearranged regulatory domains
For each candidate gene, its control regulatory domain (containing its cognate enhancers) is delimited by the coordinates of the TAD where the gene is located (Supplementary Figures S2a and S3a). To predict long-range (enhancer mediated) pathogenic effects, it is essential to properly reconstruct the patient regulatory domains (i.e. rearranged regulatory domains). Depending on the SV type, and whether the SV is Intra-TAD (not crossing a TAD boundary and, thus, only affecting one TAD) or Inter-TAD (crossing a TAD boundary and, thus, affecting multiple TADs), the rearranged regulatory domains are computed differently:
Deletions
Intra-TAD: The rearranged regulatory domain includes the enhancers located in the control regulatory domain, minus those located in the deleted area (Supplementary Figure S2b)
Inter-TAD: The rearranged regulatory domain includes the enhancers located in the non-deleted parts of the two TADs that are merged due to the loss of a TAD boundary (Supplementary Figure S2c).
Inversions
Intra-TAD: inversions within one TAD are considered to simply relocate enhancers inside of the same regulatory domain. As currently implemented in POSTRE, these rearrangements are not considered to alter the regulatory domain of the candidate gene (Supplementary Figure S2d).
Inter-TAD: inversions crossing TAD boundaries will lead to the shuffling of two TADs at each of the inversion breakpoints. At each breakpoint, the regulatory domain of a candidate gene will include the enhancers located in the non-inverted part of its original TAD plus the enhancers located in the inverted part of the other TAD (Supplementary Figure S2e).
Translocations
Balanced translocations are modeled by considering that, for the two affected chromosomes (e.g. ChrA and ChrB), the fragment of ChrA that is associated with the ChrA centromere merges with the fragment from ChrB that is not associated with ChrB centromere, and vice versa (Supplementary Figure S3b). Translocations will lead to the shuffling of two TADs at each of the translocation breakpoints. At each breakpoint, the regulatory domain of a candidate gene will include the enhancers located in the non-translocated part of its original TAD plus the enhancers located in the translocated part of the other TAD (Supplementary Figure S3b).
Duplications
Intra-TAD: The rearranged regulatory domain includes the enhancers located in the control regulatory domain plus those that are duplicated (Supplementary Figure S3c).
Inter-TAD: If the duplication includes a TAD boundary, then two different scenarios are considered: (i) if the duplication does not include the candidate gene (i.e. the gene is not duplicated), then the candidate gene regulatory domain is not considered to be affected (Supplementary Figure S3d); (ii) if the duplication includes the candidate gene (i.e. the gene is duplicated), a new regulatory domain including the candidate gene will be created (i.e. novel or neo-TAD). In this context, the enhancers potentially regulating the candidate gene will be all those present in the control regulatory domain plus those located in the newly created regulatory domain (Supplementary Figure S3e).
Calculating the pathogenic score (PS)
To calculate the PS, the sub-scores described in previous sections are integrated. For each candidate gene and cell type/tissue analyzed, PS are computed assuming both LOF and GOF scenarios. Moreover, PS are computed differently depending on whether or not the SVs directly affect gene sequences.
I. Long-range pathomechanisms: candidate genes sequence not directly affected by the SV
PS for LOF
 |
where enhScLOF corresponds with geneEnhancerScoreLOF, expSc with geneExpressionScore, featScLOF with geneFeatureScoreLOF and phenoSc with genePhenoScore.
PS for GOF
Long-range GOF pathomechanisms can occur through (i) upregulation of already active genes due to duplications directly affecting some of its cognate enhancers; (ii) upregulation of either active or inactive genes due to interactions with ectopic enhancers (i.e. enhancer adoption). The PSlongRangeGOF is computed differently for these two GOF scenarios:
If the SV is predicted to cause ectopic interactions between the candidate gene and non-cognate enhancers (e.g. through TAD fusion, neo-TAD formation or TAD shuffling), the geneExpressionScore is not considered relevant:
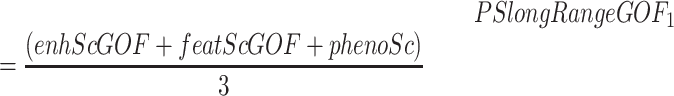 |
where enhScGOF corresponds with geneEnhancerScoreGOF, featScGOF with geneFeatureScoreGOF and phenoSC with genePhenoScore.
If the SV is not predicted to cause ectopic interactions between the candidate gene and non-cognate enhancers (e.g. intra-TAD duplication), the geneExpressionScore is considered important:
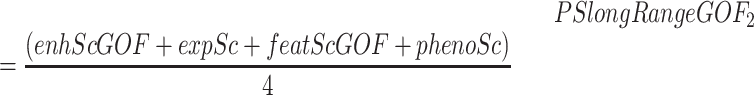 |
where enhScGOF corresponds with geneEnhancerScoreGOF, expSc with geneExpressionScore, featScGOF with geneFeatureScoreGOF and phenoSc with genePhenoScore.
II. Coding pathomechanisms: candidate genes sequence directly affected (deleted, truncated or duplicated) by the SV
If the candidate gene is deleted or truncated, only LOF will be evaluated and the PS for GOF will be set to 0. If the gene is duplicated, only GOF will be assessed and the PS for LOF will be set to 0.
PS for LOF
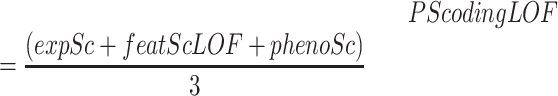 |
where expSc corresponds with geneExpressionScore, featScLOF with geneFeatureScoreLOF and phenoSc with genePhenoScore.
PS for GOF
 |
where expSc corresponds with geneExpressionScore, featScGOF with geneFeatureScoreGOF and phenoSc with genePhenoScore.
If the SVs cause a gene deletion or truncation, then long-range pathomechanisms are not considered for those candidate genes. However, if the SVs cause gene duplication/s, long-range pathomechanisms involving enhancer adoption and the formation of new TADs (neo-TAD) can also occur (6,82). Hence, for SVs resulting in gene duplications, coding and long-range GOF PS are computed and the highest one is selected as the most likely pathomechanism.
Given all the previous PS models, POSTRE applies them as follows:
POSTRE evaluates whether the SV directly affects the candidate gene sequence (i.e. ‘coding’) or not (i.e.‘long-range’).
Based on the impact of the SV over the candidate gene, the corresponding GOF and LOF PS models are applied.
Each candidate gene gets two PS scores: PS_GOF and PS_LOF. If any of these two PS scores is higher than a pathogenic threshold (0.8), then a detailed report describing the predicted pathomechanism is provided.
POSTRE software installation
POSTRE is built with the Shiny framework (83) and, thus, most of its code is written in R. Regarding the user interface, it has been developed using Shiny libraries and custom html, css and javascript code.
The full set of instructions (including videos and tutorials) explaining how to download and run POSTRE is provided in GitHub https://github.com/vicsanga/Postre. Briefly, the user needs to install R (version > = 3.5.0) and then simply run the following command in the R
console: source(‘https://raw.githubusercontent.com/vicsanga/Postre/main/Postre_wrapper.R’).
POSTRE can also be easily uploaded to a server (e.g. shinyapps.io) and accessed online as a normal web app. More details on POSTRE can be found in the dedicated GitHub webpage (https://github.com/vicsanga/Postre).
POSTRE main functionalities
Single SV Submission: Allows the user to submit one SV and analyze it in the context of one or multiple phenotypes. If pathogenic events are found, POSTRE provides a detailed report explaining the molecular basis of the predictions. This report contains a set of graphics and text that facilitate the interpretation of the SV pathogenicity. It also provides links to different external resources (e.g. Online Mendelian Inheritance in Man (OMIM) (65), MedGen (84) and Mouse Genome Database (MGD) (68)) containing information about the gene/s affected by the SV. Lastly, the user can also visualize disease-relevant genomic data within the affected locus through the UCSC genome browser (85).
Multiple SVs Submission: Allows the user to submit multiple SVs simultaneously and to assign multiple phenotypes to each of them. All the downstream predictions are summarized in a set of tables: (i) a table presenting a pathogenic score and labelling each analyzed SV as pathogenic or not-pathogenic; (ii) a table summarizing the detected pathogenic events by phenotype, gene, and type of pathological mechanism (i.e. coding or long-range). In addition, a detailed report for each of the submitted SVs can be easily obtained by executing a Single Submission job for the SV of interest.
Explore previous SVs: Allows the user to navigate POSTRE predictions for patient SVs reported in public databases. The predicted pathogenic events are presented through different tables, as the ones obtained upon Multiple SVs Submission. The detailed report for each of the analyzed SVs can be easily obtained by executing a Single Submission job for the SV of interest.
Advanced features: When running a single SV or a multiple SV submission job, POSTRE allows the user to modify the default configuration with different parameters:
The running mode can be either: (i) the Standard Running Mode or the (ii) High-Specificity Running Mode (differences between the running modes are described in previous ‘Methods’ sections). To predict a candidate gene as disease causative, POSTRE requires by default that such gene is linked in humans or mice with the same phenotype associated with the investigated SV. If this requirement is inactivated, then the only requirement for a disease causative gene is to be associated with any kind of disease phenotype and not necessarily with the one associated with the SV. Users can also upload their own TAD map. In this case, the uploaded TAD map will be used as a reference for any of the predictions performed, regardless of the selected phenotype/s.
TAD maps annotation
POSTRE uses TADs as a proxy of gene regulatory domains in order to predict the long-range pathomechanisms. POSTRE also allows users to upload their own TAD maps to be used as reference when performing the predictions. All genes found within the TAD/s affected by a SV are initially considered as potentially involved in the patient phenotype (i.e. candidate genes). Depending on the patient phenotype, POSTRE uses TAD maps generated in specific cell types/tissues (Supplementary Data 1). When TAD maps are not available for a cell type/tissue of interest, ESC TAD maps are used instead (based on the general stability of TADs among different cell types (39)). TAD maps were obtained from publically available repositories through different approaches (Supplementary Data 1):
TAD coordinates already available (no processing needed).
TAD coordinates inferred from available TAD boundary maps. In this case, TADs are defined as the regions located between neighboring TAD boundaries.
TAD coordinates obtained with DomainCaller (https://github.com/XiaoTaoWang/domaincaller/) and the 50kb contact matrices provided in .hiC files.
Additional tools and resources
The genomic data displayed in the UCSC genome browser and accessible through POSTRE reports is hosted either at the CyVerse Discovery Environment (https://de.cyverse.org), or at the GEO database (56).
Reference genome
The reference genome currently handled by POSTRE is hg19. Accordingly, all genomic coordinates provided in the manuscript and the supplementary material correspond with this reference genome.
Randomizing SVs location in the genome
To randomize the location of ‘real’ SVs (genetic alterations, either pathogenic or not pathogenic, found in existent individuals), each SV was randomly relocated in the genome while maintaining its size in a 1000 iteration process. In addition, for deletions, duplications and inversions, the randomly selected chromosomes were large enough to accommodate the randomized rearrangements, since some of the SVs might be larger than some chromosomes. When randomizing the location of a SV, all its originally assigned phenotypes were maintained. Overall, a new set of randomized SVs with the same properties as those of the ‘real’ SVs (size, phenotypes), but with different locations in the genome, were obtained during each iteration.
Matching SVs by number of candidate genes
To match SVs based on the number of associated candidate genes, two different groups of SVs were considered: (i) the set of ‘real’ SVs (either pathogenic or not pathogenic) and (ii) all the randomized SVs generated during the iterations described above. Then, in a 1000 iteration process, 25 000 SVs were randomly selected from the randomized SV dataset. Next, each ‘real SV’ was matched with one of the randomized SVs based on the number of candidate genes by applying the nearest neighbor matching method (without replacement and ratio = 1) using MatchIt (https://cran.r-project.org/web/packages/MatchIt/MatchIt.pdf). In addition, to minimize possible confounding factors, only matches between SVs of the same type and associated with the same phenotype were allowed. In the end, 1000 subsets of random SVs matched by the number of associated candidate genes with the ‘real’ SVs were obtained.
Benchmarking of SV analysis tools
Pathogenic scores (PS) were computed for different SV datasets using CADD-SV, STRVCTVRE, TADA and SVScore (86–89). Similarly to POSTRE, TADA and STRVCTVRE provide pathogenic scores for the analyzed SVs in a 0–1 range (0 = not pathogenic; 1 = pathogenic) and, thus, no further processing was required. For CADD-SV, the maximum of span and flank raw scores were used as an indicator of SV pathogenicity, followed by a min-max-normalization. For SVScore, pathogenic scores were computed with default parameters, followed by a min-max normalization. For CADD-SV and SVScore, the min and max values for the normalization were determined considering all the pathogenic scores computed by each tool after analyzing all the SVs associated with the patient and healthy control cohorts used in this work.
POSTRE computes PS for each of the phenotypes associated with a SV, whereas the rest of the tools compute one PS per SV. To eliminate this difference, during the benchmarking, for each SV we only considered the maximum PS computed by POSTRE among all the phenotypes associated with a SV. For the tools that can only analyze copy number variants (CNVs) (i.e. CADD-SV, TADA and STRVCTVRE), the PS for inversions and translocations were set to 0. Similarly, since STRVCTVRE only predicts pathogenicity for SVs that affect exons, the PS scores for SVs not affecting exons were set to 0. Moreover, since TADA can only handle SVs in autosomal chromosomes, the PS for SVs located in sexual chromosomes were set to 0.
TADEUS2 (90) prioritizes candidate genes using (i) haploinsufficiency scores, (ii) number of interrupted enhancer-promoter interactions, (iii) the distance from the SV breakpoints and (iv) the phenotypic data associated to the candidate genes in different databases. Overall, these four features are combined into a gene PS that ranges from 0 to 4. Based on the FOXG1 case study available in TADEUS2 webpage (https://tadeus2.mimuw.edu.pl), we considered that a candidate gene was predicted as disease causative by TADEUS2 if its PS ≥ 3.
POSTRE test files
The coordinates of the SVs in positive control patients (Table 1 and Table 2), the 270 patients analysed in Figure 4 and the 500 largest SVs found in healthy individuals in gnomAD (analysed in Table 3) can be downloaded in a POSTRE friendly format from the testFiles directory available in POSTRE’s GitHub page (https://github.com/vicsanga/Postre). These files can be directly uploaded and analyzed with POSTRE with the Multiple SV submission form. For more details about how to create a file for a multiple SV submission with POSTRE, check POSTRE’s user guide.
Figure 4.

POSTRE analysis for a cohort of 270 patients involving 417 SV-phenotype associations. (A) Frequency of the different types of SVs in the patient cohort. (B) Percentage of SVs predicted as pathogenic by POSTRE for the indicated phenotypes using the Standard or High-Specificity modes. N = total number of SVs associated with each of the indicated phenotypes. NDD: neurodevelopmental defects. (C) Percentage of SV-phenotype associations predicted as pathogenic for the 270 patient cohort (Pathogenic, blue) and for all the SVs found in healthy individuals analyzed in Table 3 (Healthy, purple). (D) Pie chart showing the relative abundance of the main pathological mechanisms (i.e. coding GOF, coding LOF, long-range GOF, long-range LOF) among the SV-phenotype associations predicted as pathogenic for the 270 patient cohort. (E) Percentage of predicted coding and long-range pathomechanisms when considering: (i) all SVs, (ii) only deletions (Del) and duplications (Dup) or (iii) only inversions (Inv) and translocations (Trans) in the SV-phenotype associations predicted as pathogenic for the 270 patient cohort. (F) Violin plots showing the number of candidate (purple, left) and causative (green, right) genes identified by POSTRE (Standard running mode) for each of the analyzed SV-phenotype associations predicted to be pathogenic. Each dot corresponds to a SV-phenotype association. RM= running mode.
Table 3.
Analysis of non-pathogenic (control) SVs with POSTRE

|
RESULTS
POSTRE overview
POSTRE is a software developed to predict the pathological impact of SVs implicated in a broad set of congenital abnormalities (i.e. limb, craniofacial/head&neck, cardiac or neurodevelopmental). In comparison with previous tools (87) (see POSTRE benchmarking section for more details), POSTRE can analyze SVs with direct effects on protein coding genes as well as SVs acting through long-range regulatory mechanisms that alter gene expression. Furthermore, POSTRE can analyze not only Copy Number Variants (CNVs) (i.e. deletions and duplications) (86–88), but also inversions and balanced translocations. In this section we provide an overview of the tool (Figure 1).
In order to analyze SVs identified in patients with congenital defects, POSTRE requires three main inputs (Figure 1a): (i) the type of SV (i.e. deletion, duplication, inversion or translocation) (ii) the genomic coordinates of the SV breakpoints and (iii) the patient phenotype/s (i.e. type of congenital abnormality: limb, craniofacial/head&neck, cardiac or neurodevelopmental). Once the input data is submitted (Figure 1b), the cell types/tissues considered most relevant for the patient phenotype are selected for downstream analysis (Supplementary Data 1). Moreover, for each cell type/tissue, multiple developmental stages and/or in vitro differentiation time-points are typically analyzed. For instance, NCC obtained at two different time-points through an in vitro differentiation protocol are used for the study of craniofacial abnormalities (43,91), while two different developmental stages of the embryonic brain prefrontal cortex are analyzed for neurodevelopmental disorders (92). POSTRE uses three main types of genomic information for each of the selected cell types/tissues: (i) gene expression profiles (based on RNA-Seq), (ii) active enhancer maps (based on ChIP-Seq) and (iii) TAD maps (based on Hi-C). Both gene expression profiles and active enhancer maps are specific for each cell type/tissue and developmental stage. However, due to the relative scarcity of Hi-C data, some of the TAD maps used by POSTRE were generated in cellular contexts different than the ones relevant for each phenotype (e.g. ESC TAD map for limb phenotypes; Supplementary Data 1). This is justified by the general stability of TADs among different cell types (39) and by the use of TAD maps generated in ESC to successfully annotate regulatory domains disrupted by SVs associated with limb or craniofacial abnormalities (43,45). Once the relevant genomic data is loaded, the impact of the SV is independently evaluated for all the considered cell types/tissues. Firstly, the SV breakpoints are mapped to the genome attending to the location of TADs, which are used as a proxy to determine the regulatory domains affected by the genetic rearrangement. Subsequently, all the genes located in the disrupted TADs are considered as potentially affected by the SV (i.e. candidate genes) through either coding or long-range mechanisms. Then, all the candidate genes are ranked according to multiple features in order to estimate their likelihood of being involved in the patient disease (i.e. causative genes). A summary of these features is presented below:
-
Gene features:
Dosage sensitivity (e.g. haploinsufficiency or triplosensitivity): deviations from the normal dosage (i.e. number of copies), or expression levels, can be detrimental for some but not all genes. Hence, it is important to know whether the affected genes are dosage sensitive or not.
Breadth of polycomb domains in promoter regions: genes whose promoters are covered by broad polycomb/H3K27me3 domains when inactive often correspond with major developmental genes implicated in congenital disorders (73,74). Moreover, this type of genes are characterized by their high enhancer responsiveness (49,75,76), which might be attributed to the presence of promoter CpG islands that prevent DNA methylation, facilitate enhancer-gene communication and, overall, provide a permissive chromatin environment.
Gene expression: the expression status of the candidate genes is particularly relevant for LOF situations, as in those cases, the disease-causative genes must be expressed in the relevant cellular context to be involved in the patient condition. For GOF situations gene expression is also considered, except when enhancer adoption is predicted, given that a pathogenic upregulation of a disease causative gene can occur either through the overexpression of an already active gene, or through the ectopic activation of an inactive gene (see Methods for more details).
Enhancer changes: for the candidate genes that are not directly disrupted by the SV, long-range regulatory mechanisms are considered instead. In addition, for duplicated genes, given the possibility of enhancer adoption through neo-TAD formation (82), long-range pathogenic events are also evaluated (see Methods for more details). Briefly, the enhancer activity associated to each candidate gene is estimated as the sum of the H3K27ac levels present at all the enhancers found within their TAD in the presence (patient) or absence (control) of the SV (93) (see Materials and Methods and Supplementary Figures S2 and S3 for more details). Then, differences in enhancer activity between the patient and control situations are computed for each candidate gene. For LOF, the candidate genes should lose enhancer activity (i.e. control enhancer activity > patient enhancer activity) through the deletion of cognate enhancers or enhancer disconnection pathomechanisms (43,44). For GOF, the candidate genes should gain enhancer activity (i.e. control enhancer activity < patient enhancer activity) through the duplication of cognate enhancers (46,94,95) or enhancer adoption/hijacking pathomechanisms (96).
Gene-phenotype annotation: genes previously associated with the patient phenotypic category (e.g. limb malformation) are considered particularly relevant. This information is obtained from OMIM (65) and MGD (68) databases.
Based on the previous criteria, each candidate gene will receive an overall pathogenic score (PS) between 0 and 1, with a PS = 1 meaning that the gene fulfills all the pathogenic criteria for a certain cell type/tissue (see Methods for more details). Then, these pathogenic scores are used to rank all the candidate genes across the considered cell types/tissues (Figure 1c). Moreover, genes with a PS ≥ 0.8 are highlighted as potentially pathogenic and a detailed report is provided for each of them. These reports contain a set of graphics, text and links to different external resources (e.g. UCSC genome browser with enhancer and TAD maps) to illustrate why a candidate gene is predicted as pathogenic in a specific cell type/tissue due to the SV (Supplementary Data 2).
In addition, POSTRE allows users to simultaneously analyze multiple SVs, each potentially associated with multiple phenotypes, in an automatic and sequential manner.
POSTRE performance with experimentally validated SVs causing disease through long-range mechanisms
To evaluate POSTRE performance, we initially focused on a set of six patients with SVs causing congenital abnormalities (limb or craniofacial) compatible with POSTRE’s analysis pipeline (Table 1a; Supplementary Data 3 and 4). Importantly, each of these SVs pathogenically affects the expression levels of only one gene through experimentally validated long-range pathomechanisms (36,43,45,46,82). A summary of POSTRE’s results for these six patients is shown in Table 1b-c and more details can be found in Supplementary Data 4.
After mapping the breakpoints of the six SVs with respect to TADs in the relevant cell types/tissues, a total of 70 candidate genes (genes whose TADs are disrupted) were identified (Table 1b). Remarkably, for five out of six patients, POSTRE successfully predicted the single causative gene whose expression is affected by each SV as well as the implicated long-range pathomechanism (Table 1c). Overall, POSTRE achieved a sensitivity of 83% and a specificity of 100% (i.e. no false positive genes predicted) at the gene level for this limited, albeit relevant, patient group (Table 1c). For example, for a patient with craniofacial abnormalities carrying a heterozygous inversion in Chr6 (i.e. patient Nr 1 in Table 1a and Supplementary Data 3), POSTRE predicted the physical disconnection between TFAP2A and some of its cognate enhancers in NCC (i.e. enhancer disconnection) (Figure 2). POSTRE’s report indicates that this enhancer disconnection can lead to the loss of TFAP2A expression in NCC and the emergence of craniofacial defects, in agreement with the experimentally validated pathomechanism (43). POSTRE assigned TFAP2A a high pathogenic score because (i) it is highly expressed in NCC, (ii) it loses enhancer activity in NCC due to the inversion, (iii) it is a dosage sensitive gene, (iv) its promoter displays a broad polycomb domain when it is inactive and (v) it has been previously associated with craniofacial (head&neck) abnormalities in OMIM and MGD (Figure 2; Supplementary Figure S4; Supplementary Table S1). Similarly, for a patient with limb abnormalities carrying a heterozygous deletion in Chr2 (i.e. patient Nr 4 in Table 1a and Supplementary Data 3), POSTRE successfully predicted a pathogenic gain of PAX3 expression in the limb due to an enhancer adoption mechanism (Figure 3). It is worth mentioning that, although the deletion eliminates one of the EPHA4 alleles, POSTRE did not predict this gene as pathogenic. This is in perfect agreement with the experimental data (45) and highlights the relevance of considering long-range mechanisms even when protein-coding genes are directly affected by SVs. POSTRE assigned PAX3 a high pathogenicity score because (i) it gains enhancer activity, (ii) it is a dosage sensitive gene, (iii) its promoter displays a broad polycomb domain when it is inactive and (iv) it has been previously associated with limb abnormalities in OMIM and MGD (Figure 3; Supplementary Figure S5; Supplementary Table S2).
Figure 2.

POSTRE results for the TFAP2A patient (Patient Nr1 in Table 1). (A) An inversion in Chr6 was previously shown to cause craniofacial abnormalities through the disconnection between TFAP2A and its Neural Crest Cell (NCC) cognate enhancers, resulting in the haploinsufficient expression of TFAP2A in NCC (43). The inversion was analyzed by POSTRE and, among the candidate genes, TFAP2A was the only one considered as pathogenic in two human NCC in vitro differentiation stages (NeuralCrestEarly and NeuralCrestLate) and the embryonic palate from Carnegie stage 20 (Palate CS20) (see Supplementary Data 1 for more details). The maximum pathogenic score (Max PS) computed for every candidate gene among the different cell types/tissues is shown to the right. (B) Enhancer activity (H3K27ac levels; see Materials and Methods) and number of enhancers associated with TFAPA2A in NCC (NeuralCrestLate) are shown in the absence (Control) or presence (Patient) of the inversion. (C) Genome browser view of the two TADs affected by the inversion. The inversion breakpoints are highlighted in red and the TFAP2A cognate enhancers in NeuralCrestLate in green. (D) Graphical abstract illustrating the changes in the regulatory landscape of TFAP2A due to the inversion in NeuralCrestLate. The green ovals represent enhancers (enh).
Figure 3.

POSTRE results for the PAX3 patient (Patient Nr4 in Table 1). (A) A deletion in Chr2 was previously shown to cause limb abnormalities through an enhancer adoption mechanism leading to PAX3 ectopic expression in the developing limb bud (45). The deletion was analyzed by POSTRE and, among the candidate genes, PAX3 was the only one considered as pathogenic in two human limb bud developmental stages (EmbryonicLimb1, EmbryonicLimb2) (See Supplementary Data 1 for more details). The maximum pathogenic score (Max PS) computed for every candidate gene among the different cell types/tissues is shown to the right. (B) Enhancer activity (H3K27ac levels) and number of enhancers associated with PAX3 in embryonic limb buds (EmbryonicLimb1) are shown in the absence (Control) or presence (Patient) of the deletion. (C) Genome browser view of the two TADs affected by the deletion. The deletion is highlighted in orange and the limb bud active enhancers in EmbryonicLimb1 in green. (D) Graphical abstract illustrating the changes in the regulatory landscape of PAX3 in EmbryonicLimb1 due to the deletion. The green ovals represent enhancers (enh).
For one of the six analyzed patients POSTRE did not predict the causative gene due to limitations in the available genomic data (Patient Nr 6 in Table 1a and Supplementary Data 3). Briefly, previous work showed that, in this patient, a duplication in Chr7 spanning the ZRS enhancer led to abnormally high SHH expression levels in the embryonic limb (46). Notably, the ZRS enhancer is active and controls SHH expression in a rather limited number (<4%) of cells within the developing limb bud (i.e. zone of polarizing activity (ZPA)) (97). In contrast, the genomic data used by POSTRE was generated from bulk human limb buds rather than isolated ZPA cells. As a result, it was not possible to detect the ZRS enhancer, which precluded the prediction of SHH as the relevant gene in this patient.
POSTRE performance with SVs previously predicted to cause human congenital abnormalities through long-range mechanisms
Next, we focused on a set of 41 patients with phenotypes compatible with POSTRE and carrying SVs previously predicted to cause congenital abnormalities through long-range mechanisms that, nevertheless, have not been fully experimentally validated (Table 2; additional information for these patients, including the SV genomic coordinates, can be found in Supplementary Data 3) (98). The experimental validation of the long-range pathomechanisms predicted in these patients was considered incomplete because (i) the impact of the SV in gene expression is either not evaluated, or not evaluated in the appropriate cell type/tissue for the investigated diseases (e.g. analysis in patient blood cells for a neurodevelopmental disorder) and/or (ii) the exact patient SV is not analysed (e.g. engineered deletion created to evaluate part of the effects of a translocation which relocates some enhancers away from its target gene) and, thus, the full impact of the SV is not known (e.g. total number of disease causative genes).
When analyzing these 41 patients with POSTRE using the Standard mode, the previously proposed causative genes and long-range pathomechanisms were successfully predicted in 95% of the cases (Table 2, Supplementary Data 4). The proposed pathological mechanisms included (i) LOF by enhancer deletion and enhancer disconnection, and (ii) GOF by enhancer duplication (Table 2). For the cases where the tool identifies the causative gene and where multiple SVs were available, predictions were still pathogenic regardless of whether the SV breakpoints were close or distally located with respect to the target genes, as depicted for SATB2 (Supplementary Figure S6). The sensitivity dropped to 66% when using the High specificity mode. The reason for this reduction is that some of the causative genes, MEF2C and SLC2A1, do not display broad polycomb domains when they are not active, one of the gene features required by the High-specificity mode. Among the analyzed patients, it is worth highlighting one of them (DECIPHER ID: 260 836; FOXG1 patient Nr 8 in Supplementary Data 3) carrying a deletion located close to FOXG1 and showing neurodevelopmental defects. There has been some discrepancy regarding the mechanism whereby this deletion might affect FOXG1 expression. It was originally proposed that the deletion could eliminate a TAD boundary and cause FOXG1 overexpression through an enhancer adoption mechanism (99). However, subsequent work indicated that this deletion could lead to FOXG1 downregulation by eliminating some of its cognate brain enhancers (100), which is in agreement with FOXG1 haploinsufficiency causing brain congenital abnormalities (e.g. Rett Syndrome) (101). Importantly, POSTRE also predicted that this deletion could cause a loss of FOXG1 expression in the brain prefrontal cortex through the deletion of relevant enhancers (Supplementary Figure S7).
In addition to the accurate identification of the previously proposed causative genes, POSTRE predicted a few additional genes as causative for some of the analyzed SVs (Table 2, Supplementary Figure S8). For example, for one of the deletions predicted to cause the loss of MEF2C expression in the embryonic brain prefrontal cortex through a long-range mechanism (MEF2C patient Nr 11 in Supplementary Data 3), POSTRE also identified NR2F1 as a potentially causative gene (Supplementary Figure S9). In this case, POSTRE predicted a coding LOF mechanism, as the deletion eliminates one of the NR2F1 alleles. Both MEF2C and NR2F1 have been previously associated with neurodevelopmental defects (102,103). Therefore, the deletion could cause the loss of both NR2F1 and MEF2C function through distinct mechanisms, which in turn might define the phenotypic spectrum of this patient. This illustrates how direct and long-range effects could be simultaneously elicited by a single SV (99,104).
These examples (FOXG1 patient Nr 8 and MEF2C patient Nr 11) highlight that it is important to consider all possible mechanisms (i.e. coding LOF, coding GOF, long-range LOF, long-range GOF) when evaluating the pathogenic impact of SVs, as this might provide a better understanding of the etiology and phenotypic variability of human disease.
It is worth mentioning that POSTRE always computes in parallel LOF and GOF scores for the candidate genes (see Methods). Consequently, POSTRE might predict that, due to the same SV, a particular candidate gene loses some cognate enhancers (i.e. LOF), while simultaneously gaining a larger amount of ectopic enhancers (i.e. GOF). When this situation is encountered POSTRE will predict both LOF and GOF for the same candidate gene, since it is not possible to determine which of the two pathomechanisms might prevail. This is well illustrated by one of the SATB2 cases presented in Table 2 (SATB2 patient Nr 6 in Supplementary Data 3), in which POSTRE predicts that an inversion causes SATB2 to lose 23 cognate enhancers (40% reduction in cognate enhancer activity; LOF) and to gain 34 ectopic enhancers (19.3% increase in total enhancer activity; GOF) in the embryonic prefrontal cortex (Supplementary Figure S10, Supplementary Table S3).
Lastly, we analyzed a set of previously reported patients with SVs (n = 21) acting through ‘coding’ pathomechanisms and affecting disease causative genes included in Tables 1 and 2 (Supplementary Table S4, Supplementary Data 3, Supplementary Data 4). POSTRE correctly identified the causative gene and the suspected ‘coding’ pathomechanism (e.g. deletion, truncation or duplication of a disease causative gene) for all the analyzed SVs (Supplementary Table S4).
Assessment of type-1 error rate (false positives)
Having shown that, when analyzing previously described pathogenic SVs, POSTRE can effectively discriminate between disease causative and non-causative genes (Table 1), we then wanted to assess POSTRE capacity to discriminate between non-pathogenic and pathogenic SVs. This is particularly important considering the abundance of structural variation in humans, with a typical genome carrying >10 000 SVs (3,4). For this purpose, SVs from healthy individuals available in gnomAD (105) were selected, as the majority of these variants are expected to be non-pathogenic. Firstly, we randomly selected 10 000 SVs (8136 Deletions, 1821 Duplications, 43 Inversions and 0 translocations) and analyzed them using the four different phenotypic categories currently handled by POSTRE (i.e. craniofacial/head&neck, neurodevelopmental, limb and cardiovascular). Each SV-phenotype association is independently analyzed, resulting in a total of 40 000 SV-phenotype predictions. Chiefly, POSTRE did not predict any pathogenic effect for most of the SV-phenotype associations (96.8% non-pathogenic for the Standard mode; 99.2% non-pathogenic for the High Specificity mode) (Table 3a). In general, pathogenic SVs tend to be larger and, consequently, affect a higher number of genes than non-pathogenic ones (99,106). This size difference is readily observed between previously reported pathogenic SVs analyzed with POSTRE (Tables 1 and 2) and those selected from healthy individuals (Table 3b). To evaluate whether these size differences could explain the low % of pathogenic predictions among SVs from healthy individuals, we also tested POSTRE performance after selecting the largest SVs present among healthy individuals. Briefly, considering that the smallest SV included in Table 1 was 75 kb, we selected 2980 SVs > 75 kb (11 920 SV-phenotype associations), as well as the top 500 largest SVs from gnomAD (2000 SV-phenotype associations). Importantly, POSTRE still reported most of these SV-phenotype associations as non-pathogenic, even for the top 500 largest SVs (98.3% for High-Specificity mode; 89.5% for Standard mode) (Table 3a). Furthermore, considering the 500 largest SVs and the Standard running mode, we found that the percentage of SVs predicted as pathogenic was significantly smaller (P-value < 1e-3) than upon randomization of their genomic locations (Supplementary Figure S11a). This holds also true after matching the original and randomized SVs by the number of candidate genes (Supplementary Figure S11b). These randomization tests suggest that non-pathogenic SVs are not randomly distributed across the human genome and support the use of SVs from healthy individuals to estimate false positives.
On the other hand, the fact that some SVs found in healthy individuals are predicted as pathogenic by POSTRE does not necessarily imply that these are false positive predictions. For example, some of these SVs might indeed be pathogenic but, due to the incomplete penetrance and variable expressivity that characterizes many congenital disorders, might not always result in a phenotypic abnormality (107). In agreement with this possibility, when analyzing with TADA (88) those CNVs (TADA can only handle deletions and duplications) from healthy individuals that POSTRE predicted as either pathogenic (for any of the SV associated phenotypes) or non-pathogenic, TADA pathogenic scores were significantly higher for the CNVs predicted as pathogenic by POSTRE (Supplementary Figure S11c). In addition, TADA also classified as pathogenic a large percentage of the CNVs that POSTRE predicted as pathogenic (Supplementary Figure S11d).
Altogether, these results show that POSTRE controls well the Type-1 errors (false positives).
Database of POSTRE predictions for patients with congenital abnormalities carrying SVs
POSTRE can be used to analyze large cohorts of patient SVs described in the literature and/or deposited in databases (104,108–111), many of which are currently considered as variants of uncertain significance (VUS) (57). The systematic analysis of large SV cohorts might (i) identify relevant disease loci based on recurrence, (ii) predict novel pathological mechanisms for already known disease-relevant genes and (iii) provide testable hypothesis regarding the pathogenicity of VUS. To illustrate this, we analyzed a set of 270 patients described in (104,110) (Supplementary Data 3) carrying balanced SVs and CNVs (Figure 4A) and displaying phenotypes compatible with POSTRE. Many of these SVs are associated with multiple phenotypes (e.g. craniofacial and neurodevelopmental alterations), and we independently analysed each SV with respect to each of its associated phenotypes with POSTRE (417 SV-phenotype associations) (Figure 4B). POSTRE predicted pathogenic events in 59% and 23% of the SV-phenotype associations analysed with the Standard and High-Specificity modes, respectively (Figure 4B, Supplementary Data 4), which is considerably higher than for the healthy controls described in Table 3 (Figure 4C). Furthermore, these percentages of pathogenic predictions were significantly higher than those obtained upon randomization of the SV locations in the human genome (P-value < 1e-3) (Supplementary Figure S12).
POSTRE does not simply identify SVs with pathogenic potential, but also predicts the cellular context in which the SVs might elicit their pathogenic effects as well as their potential mechanism of action. In this regard, when considering all the predicted pathological events with the Standard running mode, 57% corresponded to long-range regulatory effects (Figure 4D, E), highlighting the potential prevalence of this type of pathomechanism. Moreover, ‘long-range’ pathological mechanisms are particularly abundant (80%) among balanced SVs (i.e. inversions and translocations), while ‘coding’ pathomechanisms are more common (70%) among CNVs (i.e. deletions, duplications) (Figure 4D, E). On the other hand, when considering POSTRE pathogenic predictions (Standard running mode) for all 270 patient SVs, the average number of candidate genes was 31.3, while the average number of predicted causative genes was 1.8 (Figure 4F). The average number of candidate genes was clearly higher than for the patients described in Table 2 (8.3; Supplementary Figure S8), probably because the SVs in these 270 patients tend to be larger (Supplementary Figure S13). However, the number of predicted causative genes was rather similar between these two patient cohorts (1.8 versus 1.2; Figure 4F and Supplementary Figure S8). This suggests that POSTRE can facilitate the prioritization of disease-relevant genes even when many candidate genes are potentially affected by large SVs. Nevertheless, 41% of the 417 SV-phenotype associations were not predicted as pathogenic by POSTRE, which highlights the need to keep improving POSTRE scoring criteria, the functional characterization of the non-coding genome and to incorporate genomic data from additional human cell types in order to reveal the full pathogenic spectrum of structural variation.
Overall, for more than half of the analyzed SVs in this 270 patient cohort, POSTRE predicted a pathogenic event as well as an underlying pathomechanism (Figure 4, Supplementary Data 4), which can improve the current understanding of these disorders and facilitate the design of experimental strategies to uncover their molecular etiology (7). All these predictions are available through POSTRE’s Explore Previous SVs section, enabling users to browse them by patient, gene, phenotype or pathological mechanism. To illustrate the usefulness of these results, we now describe POSTRE predictions for a couple of the investigated patients (Figure 5):
Figure 5.

Long-Range LOF predictions for UTR8 and DGAP270 patients. (A) Patient UTR8 displays craniofacial abnormalities (Pierre Robin Sequence) and carries a translocation with breakpoints in Chr10 and Chr17, which is currently classified as a VUS in ClinVar. (B) The UTR8 translocation was analyzed by POSTRE and, among the candidate genes, SOX9 was the only one considered as pathogenic in two NCC in vitro differentiation stages (NeuralCrestEarly, NeuralCrestLate) (see Supplementary Data 1 for more details). (C) Genome browser view of one of the TADs affected by the UTR8 translocation, in which SOX9 is located. The translocation breakpoint is highlighted in red and the NCC active enhancers in NeuralCrestLate in green. (D) Enhancer activity (H3K27ac levels) and number of enhancers associated with SOX9 in NCC (NeuralCrestLate) are shown in the absence (Control) or presence (Patient) of the UTR8 translocation. (E) Patient DGAP270 displays neurodevelopmental defects and carries an inversion in ChrX, which is currently classified as a VUS in ClinVar. (F) The DGAP270 inversion was analyzed by POSTRE and, among the predicted disease causative genes, PCDH19 showed the highest pathogenic score in the embryonic prefrontal cortex gestation week 18 (PfcGw18) (see Supplementary Data 1 for more details). (G) Genome browser view of one of the TADs affected by the DGAP270 inversion, in which PCDH19 is located. The inversion breakpoint is highlighted in red and the prefrontal cortex active enhancers in PfcGw18 in green. (H) Enhancer activity (H3K27ac levels) and number of enhancers associated with PCDH19 in the prefrontal cortex (PfcGw18) are shown in the absence (Control) or presence (Patient) of the DGAP270 inversion.
Patient UTR8 (dbVar): patient with Pierre Robin sequence (PRS), a syndrome associated with craniofacial abnormalities. This patient carries a translocation (UTR8 in dbVar, 268030 in ClinVar) considered as a VUS in ClinVar (Figure 5A). POSTRE predicted that this translocation could cause a pathogenic loss of SOX9 expression in NCC through an enhancer disconnection mechanism (Figure 5B–D) (please note that for one of the considered NCC stages, a potential GOF through enhancer adoption is also predicted, although with a smaller PS). This LOF prediction is supported by previous work (36), where some of the NCC enhancers that are disconnected from SOX9 due to the translocation were deleted in hESC using CRISPR/Cas9 technology. Notably, the deletion of those enhancers significantly and specifically reduced SOX9 expression upon differentiation of hESC into NCC, supporting that SOX9 LOF through long-range mechanisms could lead to PRS.
Patients U152 and DGAP270 (dbVAR): patients with neurodevelopmental defects carrying a deletion (U152, dbVar) or an inversion (DGAP270, dbVar) in ChrX. In both cases, POSTRE predicted LOF for PCDH19 in the brain prefrontal cortex through either coding (U152) or long-range regulatory mechanisms (DGAP270). PCDH19 is associated with neurodevelopmental disorders, especially with epilepsy and intellectual disability (112–114). In the case of DGAP270, the associated inversion was listed as a VUS in ClinVar (ID: 267917) (Figure 5E). Notably, POSTRE predicted that the inversion could cause a loss of PCDH19 expression in the embryonic brain prefrontal cortex through an enhancer disconnection mechanism (Figure 5F–H). In addition, a potential GOF through enhancer adoption for SHROOM4 and FOXP3 was also predicted for the DGAP270 patient, although with a smaller PS than for PCDH19. Moreover, while PCDH19 is predicted as disease causative with both the Standard and High-Specificity Running Modes, SHROOM4 and FOXP3 are only predicted as disease causative with the Standard Running Mode.
POSTRE benchmarking
Several computational tools to analyze SVs have been developed during the last few years: TADA (88), CADD-SV (86), SVScore (89), STRVCTVRE (87), TADeus2 (90), ClinTAD (115), VEP (116). A detailed comparison between POSTRE and other available tools is presented below (see Methods, Figure 6 and Table 4 for more details):
Figure 6.

Benchmarking of SV analysis tools. (A–D) The SVs found in the different patient cohorts were analyzed with POSTRE, SVScore (89), CADD-SV (86), TADA (88) and STRVCTVRE (87). (A, B) Pathogenic scores for all the SVs found in (A) the 270 patient cohort described in Figure 4 or (B) the positive control patients with long-range pathomechanisms described in Tables 1 and 2. (C, D) Pathogenic scores for the CNVs found in (A) the 270 patient cohort described in Figure 4 or (B) the CNVs present in the positive control patients with long-range pathomechanisms described in Tables 1 and 2. (E) Percentage of SVs predicted as pathogenic by either POSTRE (blue) or TADA (green) when considering all the SVs or only CNVs found in the indicated patient cohorts (i.e. the positive control patients from Tables 1 and 2; 270 patient cohort described in Figure 4). In addition, the percentage of SVs (99% CNVs) predicted as pathogenic when considering the SVs (N = 12 718) found in the healthy individuals analyzed in Table 3. (F) Ratio between Sensitivity (fraction of SVs predicted as pathogenic from the 270 patient cohort described in Figure 4) and 1 - Specificity (fraction of SVs predicted as pathogenic from all the SVs found in the healthy individuals analyzed in Table 3), considering either all SVs or only CNVs. (G) Identifying and ranking disease causative genes, comparison between POSTRE and TADEUS2 (90). Percentage of SVs among selected positive control patients from Table 1 and Table 2 where: (i) the known causative gene is predicted, (ii) additional causative genes are predicted, and (iii) the known causative gene is ranked first among all the considered candidate genes.
Table 4.
POSTRE comparison with other SV analysis tools (86�90,115,116)

|
Type of SVs
Most of the currently available tools to interpret SVs (e.g. TADA, STRVCTVRE, VEP, CADD-SV, ClinTAD) can only analyze CNVs (i.e. deletions and duplications). In contrast, POSTRE, SVScore and TADeus2 can also analyze inversions and translocations.
Computing pathogenic scores
Some tools provide pathogenic scores (PS) for each of the analyzed SVs (e.g. POSTRE, TADA, CADD-SV, SVScore, STRVCTVRE). These PS can be very useful to rank and prioritize SVs among large SV cohorts. Several tools (POSTRE, TADA, STRVCTVRE) provide PS in a 0 (not pathogenic) – 1 (pathogenic) range. POSTRE PS for a given SV corresponds with the maximum PS computed for the candidate genes associated with the SV (POSTRE predicts one PS per SV-associated phenotype, but for this task only the maximum PS among all the computed SV-phenotype associations was considered for each SV). To compare POSTRE PS with the ones computed by other methods (STRVCTVRE, TADA, SVScore and CADD-SV), we computed PS for the SVs found in the patient cohorts described in previous sections (positive controls with long-range pathogenic events: Tables 1 and 2; 270 patient cohort: Figure 4). For SVScore and CADD-SV, PS were normalized to 0–1 (see Materials and Methods).
When considering all SVs, POSTRE outperformed the rest of the tools and its PS were higher on average (Figure 6 a-b). This is expected given that most of the available tools can only handle CNVs (STRVCTVRE, CADD-SV, TADA), and, thus, inversions and translocations were assigned a PS = 0 when analyzed with these tools (see Materials and Methods). When considering only CNVs from the 270 patients cohort, the PS were quite similar for all the tools (Figure 6C). Since coding pathomechanisms are particularly abundant among these SVs (Figure 4E), this suggests that all tools show a similar performance when analyzing this type of pathomechanism. In contrast, when analyzing CNVs from the positive control patients (Tables 1 and 2), which are enriched in long-range pathomechanisms, the highest PS were obtained with POSTRE, followed by CADD-SV and TADA (Figure 6d). Overall, STRVCTVRE was the tool providing the smallest PS (Figure 6d), which is expected as this tool only considers coding pathomechanisms.
Pathogenic labelling
Most of the tools that compute PS for SVs do not clearly classify those variants as pathogenic or non-pathogenic (pathogenic labelling). In contrast, both POSTRE and TADA explicitly label pathogenic SVs, thus enabling us to compare these two tools for this task (POSTRE predicts pathogenicity per SV-associated phenotype, but for this task SVs were considered pathogenic if pathogenicity was predicted for any phenotype). Firstly, when considering all positive controls in Tables 1 and 2, POSTRE clearly outperformed TADA, as 44/47 cases (94%) were predicted pathogenic by POSTRE vs 18/47 (38%) by TADA (Figure 6e). This is expected since TADA can only analyze CNVs. Based on this limitation, we then restricted our analyses to the CNVs from the positive control patients (22 of the 47 SVs in positive control patients are CNVs). POSTRE and TADA predicted pathogenicity for 20/22 (91%) and 18/22 (82%) CNVs, respectively (Figure 6E). Therefore, when considering only CNVs, although POSTRE identified more positive controls as pathogenic, there were very few SVs not correctly predicted as pathogenic by each tool: POSTRE failed to identify patients Nr6 in Table 1 (SHH) and NR2F2 Nr 1 in Supplementary Data 3, while TADA did not predict pathogenicity for patient Nr 2 in Table 1 (SOX9) in addition to patients MEF2C Nr 7, ARX Nr 1 and NR2F2 Nr 1 in Supplementary Data 3. Furthermore, we analyzed the SVs from the 270 patient cohort described in previous sections. When evaluating all these SVs, POSTRE predicted a larger percentage as pathogenic (64% versus 48%), while if we only considered CNVs (144 SVs), TADA predicted more of them as pathogenic (90% vs 58%) (Figure 6e). POSTRE might show lower sensitivity for this set of CNVs because it performs cell-type specific predictions; i.e. POSTRE will not predict a SV as pathogenic unless a meaningful pathological mechanism is detected for a relevant cell type/tissue (e.g. a deletion near an active disease relevant gene will not be predicted pathogenic unless some active enhancers are found to be deleted in the same cell type/tissue), while TADA predictions are not limited by the cellular context restriction. Lastly, when analyzing SVs from healthy individuals (99% are CNVs), POSTRE displayed higher specificity than TADA (POSTRE predicted 11% as pathogenic and TADA 24%) (Figure 6e). Overall, if the prediction results for both patients and healthy controls are considered together, we found that the sensitivity/(1-specificity) ratios are larger for POSTRE (Figure 6f) when considering either all SVs (POSTRE = 5.8, TADA = 2) or only CNVs (POSTRE = 5.3, TADA = 3.8).
Ranking genes based on pathogenicity
Among the benchmarked tools, only POSTRE and TADEUS2 predict and rank disease-causative genes. However, TADEUS2 can only analyze one SV at a time and the prediction results have to be screened manually, thus complicating a comparative analysis for large SV datasets. Therefore, in order to compare POSTRE and TADEUS2 for this task, we selected 21 SVs associated with positive control patients (Table 1 patients: all SVs (n = 6); Table 2 patients: for each gene of interest (SATB2, ARX, FOXG1, MEF2C, DLX5-6, BCL11B, SLC2A1, NR2F2, CTNNA2) we selected one SV of each type (i.e. one inversion, one translocation and one deletion) (n = 15)). Notably, POSTRE predicted the correct disease-causative gene in 18/21 cases (86%) and TADEUS2 in 14/21 cases (67%) (Figure 6g). POSTRE failed to identify SHH, NR2F2 and CTNNA2 as disease causative genes, while TADEUS did not predict SOX9, KCNJ2, MEF2C (in 1 out of 3 cases), DLX5-6 (in 1 out of 3 cases), BCL11B, NR2F2 and CTNNA2. Furthermore, we also compared the number of SVs for which additional disease-causative genes (besides the already known ones) were predicted. POSTRE predicted additional causative genes for 3/21 (14%) SVs and TADEUS2 for 15/21 (71%) (Figure 6g). Lastly, we also compared how often the known disease-causative genes were ranked as the most pathogenic according to the PS computed by each tool. In 19/21 cases (90%), POSTRE ranked the known disease-causative gene as the most pathogenic, whereas this number dropped to 10/21 (48%) for TADEUS2. Overall, these comparisons suggest that POSTRE shows a better performance when identifying and ranking disease causative genes. Furthermore, in contrast to POSTRE, TADEUS2 only considers the loss of enhancer-promoter interactions due to SVs, but not GOF long-range pathomechanisms.
Information about disease mechanisms
Pathogenic scores do not provide information regarding the mechanisms (e.g. causative gene/s, disease-relevant cellular context, ‘coding’ vs ‘long-range’ mechanisms, etc.) whereby a SV might actually cause a disease. In this regard, there are tools that provide different layers of descriptive information for the genomic loci affected by the SVs (e.g. VEP (116), ClinTAD (115), TADEUS2 (90), CADD-SV(86) or POSTRE), including the genes directly affected or located close to the SV breakpoints, the recurrence of SVs within the same locus in patients with similar phenotypes, the presence of cis-regulatory elements, etc. In this context, the tools with graphical user interfaces (GUIs) (e.g. POSTRE, TADEUS2, VEP) generally provide more and easier to understand information.
Overall, POSTRE is rather unique regarding its annotation features:
POSTRE performs cell-type specific predictions attending to the patient phenotype. Considering the cellular context in which a SV might manifest its pathogenicity is particularly relevant in the case of long-range pathomechanisms, as enhancers display high tissue specificity. However, with a few exceptions (99,104), most available tools do not consider the patient phenotype when estimating the pathogenicity of SVs. POSTRE takes into account the patient phenotype in order to select the genomic data and cellular context/s in which the impact of the SV should be modeled, as well as to prioritize the disease-causative genes. Although this strategy allows POSTRE to accurately predict both coding and long-range pathomechanisms, it also restricts its applicability to certain diseases/phenotypes. However, this limitation should be reduced as functional genomic data becomes available for an increasing number of human cell types/tissues and is incorporated into POSTRE’s pipeline.
POSTRE provides the users with cell-type specific pathogenic reports. These reports can inform about the developmental stages or cellular contexts where the SV might exert a pathogenic effect. Moreover, the reports can also facilitate the design of downstream experimental or in silico analyses to further characterize and validate the disease etiology and the SV pathomechanisms.
POSTRE is, to the best of our knowledge, the only tool that distinguishes between GOF and LOF mechanisms and that can detect all major types of long-range pathomechanisms (i.e. enhancer adoption, enhancer duplication, enhancer disconnection and enhancer deletion).
Multiple SVs as input
Considering the large number of SVs typically present in a human genome (around 10 000) (3,4) as well as the increasing sizes of the patient cohorts in which SVs are identified (104,110), SV analysis tools should be ideally able to accept multiple SVs as input. However, not all the evaluated tools (Table 4) offer this possibility and some of them can only analyze one SV at a time (e.g. TADeus2).
Required computational skills and user experience
Tools designed to predict the pathogenicity of SVs or any other genetic alteration should be ideally usable by a broad scientific community, including scientists with limited computational skills. This is particularly relevant considering that the in silico interpretation of SVs might often involve the usage of not one, but several, computational tools, each of them with its own strengths and weaknesses (117). One way to improve tool usability is through the development of GUIs. However, some of the current available SV prediction tools (e.g. TADA) work strictly through command line interfaces, thus complicating their usage. In contrast, POSTRE can be executed with a user-friendly GUI that provides the user with detailed graphical and written reports for those SVs predicted as pathogenic.
DISCUSSION
SVs can affect gene function through either coding (e.g. gene deletions, gene fusions) or long-range (e.g. enhancer adoption, enhancer disconnection) mechanisms, which in turn can significantly contribute to human disease, phenotypic diversity and evolution (1,2,6,118). SVs acting through coding mechanisms have been described for multiple human disorders (119–121). In addition, recent advances in whole-genome sequencing are revealing that SVs with long-range pathological consequences might be also highly prevalent (8,121,122). However, the prediction of these long-range pathomechanisms is challenging, partly due to the still incomplete characterization of enhancers across different human cell types and our limited understanding of the factors controlling enhancer-gene communication. Consequently, there is a lack of tools for predicting and annotating the pathological effects of SVs considering both coding and long-range mechanisms. In this work, we have presented POSTRE, a user-friendly software that can be used to analyze SVs implicated in a broad range of congenital abnormalities.
In comparison with previous computational tools dedicated to the pathological analysis of SVs (see POSTRE benchmarking section for more details), POSTRE offers several advantages:
POSTRE is not restricted to CNVs, as it can handle all major types of SVs (i.e. deletions, inversions, duplications and translocations).
POSTRE is capable of predicting both coding and long-range pathogenic effects and can distinguish between a wide variety of pathomechanisms (e.g. gene deletion, gene truncation, gene duplication, enhancer adoption, enhancer disconnection, enhancer deletion, enhancer duplication). Long-range mechanisms are predicted assuming that genes and enhancers located within the same TAD can effectively communicate with each other (42) and that developmental genes (i.e. broad polycomb domains in promoter regions) show high enhancer responsiveness (49,75,76). However, accumulating evidences indicate that additional factors (e.g. linear distance, CpG islands, promoter DNA methylation, type of core promoter elements) can also influence enhancer-gene compatibility within TADs (49,52,53,81,123). As these factors are uncovered and characterized, they could be incorporated into POSTRE’s scoring system in order to further improve its specificity. It is worth mentioning that SVs can be implicated in congenital defects with a recessive inheritance (e.g. a point mutation affecting one allele and a CNV deleting the other). However, as currently implemented, POSTRE is oriented towards the prediction of SVs causing dominant disorders and its capacity to identify SVs implicated in recessive phenotypes has not been specifically evaluated.
POSTRE considers the cellular context to predict the pathological effects of SVs. To achieve this, POSTRE uses functional genomics information (e.g. gene expression levels, enhancer maps) generated in cell types/tissues deemed important for the patient phenotype (e.g. different brain developmental stages for neurodevelopmental defects). As a result, POSTRE not only predicts whether a SV is likely to be pathogenic but also the cellular context where such pathogenicity might be manifested, which can facilitate downstream experimental and/or in silico analyses. Furthermore, the independent evaluation of a given SV in various cell types/tissues offers the possibility of identifying either different candidate genes or different pathological mechanisms involving the same candidate gene depending on the cellular context (6). However, considering the cellular context can also introduce certain limitations, as illustrated by the patient described in Table 1 in whom upregulation of SHH due to the duplication of its cognate ZRS enhancer causes limb abnormalities (46). The ZRS enhancer drives SHH expression specifically within the ZPA (124), which only represents a small fraction of all the limb cells during embryogenesis. Consequently, since POSTRE uses bulk genomic data generated in whole limb buds (125,126), the ZRS enhancer can not be identified and SHH appears as an inactive gene, thus precluding POSTRE from successfully predicting the effects of the patient duplication. In addition to the problem of using bulk data from heterogeneous tissues, the lack of genomic data for the appropriate developmental stages or differentiation time points may also affect the identification of pathologically relevant genes. Nevertheless, recent advances in single-cell genomics are expected to dramatically expand the catalogue of cell types and developmental stages in which gene expression profiles and enhancer maps can be explored (127–129). Therefore, as single-cell datasets are generated in human embryos, we will incorporate them into POSTRE in order to expand its applicability towards additional congenital abnormalities and improve its current sensitivity.
One major topic in the field of data sciences, particularly in the health care area, is model interpretability (130,131). Dealing with complex models complicates the user capacity to understand and learn from the predictions, which, in the biomedicine field, can limit the impact that such models can have on disease diagnosis or treatment (132,133). POSTRE’s scoring criteria are built on a set of simple and comprehensible rules based on the current knowledge about enhancers and the pathological relevance of genes, thus conceptually resembling the scoring criteria proposed in (90,104). Hence, since the scoring criteria are simple, it is also easier to explain the results to the user. However, there is an increasing tendency towards using artificial intelligence to create prediction models. Currently, the standard option to create such models consists on providing a group of variables (predictors) measured in a set of observations and with a certain outcome associated to each observation (response variable). Next, during a training phase, a machine learning method automatically assigns a weight to each of the predictors and builds a model to predict the response variable (134). During this phase, complex interactions among the predictors may be automatically established in the model, which complicate the capacity to interpret it afterwards. On top of that, if the training data does not faithfully recapitulate the general population, it can create biases in the algorithm criteria through overfitting (134). Since databases used to train SV classifiers are biased towards coding deleterious effects due to the scarcity of reported SV with long-range pathogenic effects, the classifiers can also suffer from the same bias and, thus, might not be appropriate for the prediction of non-coding (e.g. long-range) pathomechanisms, as reported for TADA (88). In the case of POSTRE, since coding and long-range effects are evaluated independently (see Methods), the problem of coding effects dominating with respect to long-range effects is avoided, and overfitting in this context is minimized.
In summary, POSTRE is a user-friendly software to predict the pathomechanisms whereby SVs can cause congenital disorders. POSTRE can handle all major types of SVs, considers both coding and long-range mechanisms and performs its predictions in a cellular context-dependent manner. Altogether, these features make POSTRE rather unique among SV prediction tools, particularly with respect to enhanceropathies, for which there is still a clear need for tools capable of linking non-coding variation with human disease (24).
DATA AVAILABILITY
POSTRE is publicly available at https://github.com/vicsanga/Postre and https://doi.org/10.5281/zenodo.7732896. Its source code and the full set of instructions (including videos and tutorials) explaining how to download and run it are provided there. POSTRE software is distributed under a GNU General Public License v3.0.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Maria Mariner Fauli for her advice on POSTRE’s graphical design and help with the elaboration of some figures. We would also like to thank Magdalena Laugsch, Julia Baptista, Ayat Essabi, Judith Zaugg and all the Rada-Iglesias lab members for insightful comments and suggestions.
Contributor Information
Víctor Sánchez-Gaya, Institute of Biomedicine and Biotechnology of Cantabria (IBBTEC), CSIC/Universidad de Cantabria, Albert Einstein 22, 39011 Santander, Spain.
Alvaro Rada-Iglesias, Institute of Biomedicine and Biotechnology of Cantabria (IBBTEC), CSIC/Universidad de Cantabria, Albert Einstein 22, 39011 Santander, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Víctor Sánchez-Gaya is supported by a doctoral fellowship from the University of Cantabria (Spain); Work in the Rada-Iglesias laboratory is supported by the EMBO Young Investigator Programme [PGC2018-095301-B-I00, PID2021-123030NB-I00] funded by MCIN/AEI/10.13039/501 100 011 033 and by ‘ERDF A way of making Europe’ [RED2018-102553-T (REDEVNEURAL 3.0)] funded by MCIN/AEI/10.13039/501 100 011 033; ERC CoG ‘PoisedLogic’ [862 022] funded by the European Research Council and grant ‘ENHPATHY’ H2020-MSCA-ITN-2019-860002 funded by the European Commission. Funding for open access charge: Grants.
Conflict of interest statement. None declared.
REFERENCES
- 1. Feuk L., Carson A.R., Scherer S.W.. Structural variation in the human genome. Nat. Rev. Genet. 2006; 7:85–97. [DOI] [PubMed] [Google Scholar]
- 2. Stankiewicz P., Lupski J.R.. Structural variation in the human genome and its role in disease. Annu. Rev. Med. 2010; 61:437–455. [DOI] [PubMed] [Google Scholar]
- 3. Lappalainen T., Scott A.J., Brandt M., Hall I.M.. Genomic analysis in the age of human genome sequencing. Cell. 2019; 177:70–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Collins R.L., Brand H., Karczewski K.J., Zhao X., Alföldi J., Francioli L.C., Khera A.V., Lowther C., Gauthier L.D., Wang H.et al.. A structural variation reference for medical and population genetics. Nature. 2020; 581:444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ho S.S., Urban A.E., Mills R.E.. Structural variation in the sequencing era. Nat. Rev. Genet. 2020; 21:171–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Spielmann M., Lupiáñez D.G., Mundlos S.. Structural variation in the 3D genome. Nat. Rev. Genet. 2018; 19:453–467. [DOI] [PubMed] [Google Scholar]
- 7. Sánchez-Gaya V., Mariner-Faulí M., Rada-Iglesias A.. Rare or overlooked? Structural disruption of regulatory domains in human neurocristopathies. Front. Genet. 2020; 11:688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krude H., Mundlos S., Øien N.C., Opitz R., Schuelke M.. What can go wrong in the non-coding genome and how to interpret whole genome sequencing data. Medizinische Genet. 2021; 33:121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J.et al.. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012; 337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krijger P.H.L., De Laat W.. Regulation of disease-associated gene expression in the 3D genome. Nat. Rev. Mol. Cell Biol. 2016; 17:771–782. [DOI] [PubMed] [Google Scholar]
- 11. Zhu Y., Tazearslan C., Suh Y.. Challenges and progress in interpretation of non-coding genetic variants associated with human disease. Exp. Biol. Med. 2017; 242:1325–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Elgar G., Vavouri T.. Tuning in to the signals: noncoding sequence conservation in vertebrate genomes. Trends Genet. 2008; 24:344–352. [DOI] [PubMed] [Google Scholar]
- 13. French J.D., Edwards S.L.. The role of noncoding variants in heritable disease. Trends Genet. 2020; 36:880–891. [DOI] [PubMed] [Google Scholar]
- 14. Smedley D., Schubach M., Jacobsen J.O.O.B., Köhler S., Zemojtel T., Spielmann M., Jäger M., Hochheiser H., Washington N.L.L., McMurry J.A.A.et al.. A whole-genome analysis framework for effective identification of pathogenic regulatory variants in Mendelian disease. Am. J. Hum. Genet. 2016; 99:595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chahal G., Tyagi S., Ramialison M.. Navigating the non-coding genome in heart development and Congenital Heart Disease. Differentiation. 2019; 107:11–23. [DOI] [PubMed] [Google Scholar]
- 16. Villar D., Frost S., Deloukas P., Tinker A.. The contribution of non-coding regulatory elements to cardiovascular disease. Open Biol. 2020; 10:200088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Flöttmann R., Kragesteen B.K., Geuer S., Socha M., Allou L., Sowińska-Seidler A., Bosquillon De Jarcy L., Wagner J., Jamsheer A., Oehl-Jaschkowitz B.et al.. Noncoding copy-number variations are associated with congenital limb malformation. Genet. Med. 2018; 20:599–607. [DOI] [PubMed] [Google Scholar]
- 18. Valente E.M., Bhatia K.P.. Solving Mendelian mysteries: the non-coding genome may hold the key. Cell. 2018; 172:889–891. [DOI] [PubMed] [Google Scholar]
- 19. Medico-Salsench E., Karkala F., Lanko K., Barakat T.S.. The non-coding genome in genetic brain disorders: new targets for therapy?. Essays Biochem. 2021; 65:671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang Y., Muzny D.M., Reid J.G., Bainbridge M.N., Willis A., Ward P.A., Braxton A., Beuten J., Xia F., Niu Z.et al.. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013; 369:1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weedon M.N., Cebola I., Patch A.M., Flanagan S.E., De Franco E., Caswell R., Rodríguez-Seguí S.A., Shaw-Smith C., Cho C.H.H., Allen H.L.et al.. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat. Genet. 2014; 46:61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lettice L.A., Horikoshi T., Heaney S.J.H., Van Baren M.J., Van Der Linde H.C., Breedveld G.J., Joosse M., Akarsu N., Oostra B.A., Endo N.et al.. Disruption of a long-range cis-acting regulator for Shh causes preaxial polydactyly. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:7548–7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haro E., Petit F., Pira C.U., Spady C.D., Lucas-Toca S., Yorozuya L.I., Gray A.L., Escande F., Jourdain A.S., Nguyen A.et al.. Identification of limb-specific Lmx1b auto-regulatory modules with Nail-patella syndrome pathogenicity. Nat. Commun. 2021; 12:5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Claringbould A., Zaugg J.B.. Enhancers in disease: molecular basis and emerging treatment strategies. Trends Mol. Med. 2021; 27:1060–1073. [DOI] [PubMed] [Google Scholar]
- 25. Ong C.-T., Corces V.G.. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 2011; 12:283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wittkopp P.J., Kalay G.. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 2012; 13:59–69. [DOI] [PubMed] [Google Scholar]
- 27. Wray G.A. The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 2007; 8:206–216. [DOI] [PubMed] [Google Scholar]
- 28. Bulger M., Groudine M.. Enhancers: the abundance and function of regulatory sequences beyond promoters. Dev. Biol. 2010; 339:250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bulger M., Groudine M.. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011; 144:327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Buecker C., Wysocka J.. Enhancers as information integration hubs in development: lessons from genomics. Trends Genet. 2012; 28:276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heintzman N.D., Hon G.C., Hawkins R.D., Kheradpour P., Stark A., Harp L.F., Ye Z., Lee L.K., Stuart R.K., Ching C.W.et al.. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009; 459:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A.et al.. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rada-Iglesias A., Bajpai R., Swigut T., Brugmann S.A., Flynn R.A., Wysocka J.. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011; 470:279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lettice L.A. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 2003; 12:1725–1735. [DOI] [PubMed] [Google Scholar]
- 35. Sagai T., Hosoya M., Mizushina Y., Tamura M., Shiroishi T.. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 2005; 132:797–803. [DOI] [PubMed] [Google Scholar]
- 36. Long H.K., Osterwalder M., Welsh I.C., Hansen K., Davies J.O.J., Liu Y.E., Koska M., Adams A.T., Aho R., Arora N.et al.. Loss of extreme long-range enhancers in human neural crest drives a craniofacial disorder. Cell Stem Cell. 2020; 27:765–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sanyal A., Lajoie B.R., Jain G., Dekker J.. The long-range interaction landscape of gene promoters. Nature. 2012; 489:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O.et al.. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nora E.P., Dekker J., Heard E.. Segmental folding of chromosomes: a basis for structural and regulatory chromosomal neighborhoods?. Bioessays. 2013; 35:818–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rao S.S.P., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S.et al.. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lupiáñez D.G., Spielmann M., Mundlos S.. Breaking TADs: how alterations of chromatin domains result in disease. Trends Genet. 2016; 32:225–237. [DOI] [PubMed] [Google Scholar]
- 43. Laugsch M., Bartusel M., Rehimi R., Alirzayeva H., Karaolidou A., Crispatzu G., Zentis P., Nikolic M., Bleckwehl T., Kolovos P.et al.. Modeling the pathological long-range regulatory effects of human structural variation with patient-specific hiPSCs. Cell Stem Cell. 2019; 24:736–752. [DOI] [PubMed] [Google Scholar]
- 44. Benko S., Fantes J.A., Amiel J., Kleinjan D.J., Thomas S., Ramsay J., Jamshidi N., Essafi A., Heaney S., Gordon C.T.et al.. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat. Genet. 2009; 41:359–364. [DOI] [PubMed] [Google Scholar]
- 45. Lupiáñez D.G., Kraft K., Heinrich V., Krawitz P., Brancati F., Klopocki E., Horn D., Kayserili H., Opitz J.M., Laxova R.et al.. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015; 161:1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lohan S., Spielmann M., Doelken S.C., Flöttmann R., Muhammad F., Baig S.M., Wajid M., Hülsemann W., Habenicht R., Kjaer K.W.et al.. Microduplications encompassing the sonic hedgehog limb enhancer ZRS are associated with haas-type polysyndactyly and Laurin-Sandrow syndrome. Clin. Genet. 2014; 86:318–325. [DOI] [PubMed] [Google Scholar]
- 47. Ghavi-Helm Y. Functional consequences of chromosomal rearrangements on gene expression: not so deleterious after all?. J. Mol. Biol. 2019; 432:665–675. [DOI] [PubMed] [Google Scholar]
- 48. Ghavi-Helm Y., Jankowski A., Meiers S., Viales R.R., Korbel J.O., Furlong E.E.M.. Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression. Nat. Genet. 2019; 51:1272–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pachano T., Sánchez-Gaya V., Ealo T., Mariner-Faulí M., Bleckwehl T., Asenjo H.G., Respuela P., Cruz-Molina S., Muñoz-San Martín M., Haro E.et al.. Orphan CpG islands amplify poised enhancer regulatory activity and determine target gene responsiveness. Nat. Genet. 2021; 53:1036–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Batut P.J., Bing X.Y., Sisco Z., Raimundo J., Levo M., Levine M.S.. Genome organization controls transcriptional dynamics during development. Science. 2022; 375:566–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bergman D.T., Jones T.R., Liu V., Ray J., Jagoda E., Siraj L., Kang H.Y., Nasser J., Kane M., Rios A.et al.. Compatibility rules of human enhancer and promoter sequences. Nature. 2022; 607:176–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zuin J., Roth G., Zhan Y., Cramard J., Redolfi J., Piskadlo E., Mach P., Kryzhanovska M., Tihanyi G., Kohler H.et al.. Nonlinear control of transcription through enhancer-promoter interactions. Nature. 2022; 604:571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ringel A.R., Szabo Q., Chiariello A.M., Chudzik K., Schöpflin R., Rothe P., Mattei A.L., Zehnder T., Harnett D., Laupert V.et al.. Repression and 3D-restructuring resolves regulatory conflicts in evolutionarily rearranged genomes. Cell. 2022; 185:3689–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R.et al.. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Satterlee J.S., Chadwick L.H., Tyson F.L., McAllister K., Beaver J., Birnbaum L., Volkow N.D., Wilder E.L., Anderson J.M., Roy A.L.. The NIH common fund/roadmap epigenomics program: successes of a comprehensive consortium. Sci. Adv. 2019; 5:eaaw6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M.et al.. NCBI GEO: archive for functional genomics data sets - Update. Nucleic Acids Res. 2013; 41:D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Federici G., Soddu S.. Variants of uncertain significance in the era of high-throughput genome sequencing: a lesson from breast and ovary cancers. J. Exp. Clin. Cancer Res. 2020; 39:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Trainor P.A. Craniofacial birth defects: the role of neural crest cells in the etiology and pathogenesis of Treacher Collins syndrome and the potential for prevention. Am. J. Med. Genet. Part A. 2010; 152A:2984–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jeste S.S. Neurodevelopmental behavioral and cognitive disorders. Contin. Lifelong Learn. Neurol. 2015; 21:690–714. [DOI] [PubMed] [Google Scholar]
- 60. Kirby R.S. The prevalence of selected major birth defects in the United States. Semin. Perinatol. 2017; 41:338–344. [DOI] [PubMed] [Google Scholar]
- 61. Hansen B.H., Oerbeck B., Skirbekk B., Petrovski B.É., Kristensen H.. Neurodevelopmental disorders: prevalence and comorbidity in children referred to mental health services. Nord. J. Psychiatry. 2018; 72:285–291. [DOI] [PubMed] [Google Scholar]
- 62. Wu W., He J., Shao X.. Incidence and mortality trend of congenital heart disease at the global, regional, and national level, 1990-2017. Medicine (Baltimore). 2020; 99:e20593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Barik S., Pandita N., Paul S., Kumari O., Singh V.. Prevalence of congenital limb defects in Uttarakhand state in India – A hospital-based retrospective cross-sectional study. Clin. Epidemiol. Glob. Heal. 2021; 9:99–103. [Google Scholar]
- 64. Köhler S., Gargano M., Matentzoglu N., Carmody L.C., Lewis-Smith D., Vasilevsky N.A., Danis D., Balagura G., Baynam G., Brower A.M.et al.. The human phenotype ontology in 2021. Nucleic Acids Res. 2021; 49:D1207–D1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Amberger J., Bocchini C.A., Scott A.F., Hamosh A.. McKusick's Online Mendelian Inheritance in Man (OMIM®). Nucleic Acids Res. 2009; 37:D793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Smith C.L., Goldsmith C.A.W., Eppig J.T.. The Mammalian Phenotype Ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol. 2005; 6:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jackson R., Matentzoglu N., Overton J.A., Vita R., Balhoff J.P., Buttigieg P.L., Carbon S., Courtot M., Diehl A.D., Dooley D.M.et al.. OBO Foundry in 2021: operationalizing open data principles to evaluate ontologies. Database (Oxford). 2021; 2021:baab069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bult C.J., Blake J.A., Smith C.L., Kadin J.A., Richardson J.E., Anagnostopoulos A., Asabor R., Baldarelli R.M., Beal J.S., Bello S.M.et al.. Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2019; 47:D801–D806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Huang N., Lee I., Marcotte E.M., Hurles M.E.. Characterising and Predicting Haploinsufficiency in the Human Genome. PLos Genet. 2010; 6:e1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B.et al.. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Collins R.L., Glessner J.T., Porcu E., Lepamets M., Brandon R., Lauricella C., Han L., Morley T., Niestroj L.M., Ulirsch J.et al.. A cross-disorder dosage sensitivity map of the human genome. Cell. 2022; 185:3041–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rehm H.L., Berg J.S., Brooks L.D., Bustamante C.D., Evans J.P., Landrum M.J., Ledbetter D.H., Maglott D.R., Martin C.L., Nussbaum R.L.et al.. ClinGen — The Clinical Genome Resource. N. Engl. J. Med. 2015; 372:2235–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rehimi R., Nikolic M., Cruz-Molina S., Tebartz C., Frommolt P., Mahabir E., Clément-Ziza M., Rada-Iglesias A.. Epigenomics-based identification of major cell identity regulators within heterogeneous cell populations. Cell Rep. 2016; 17:3062–3076. [DOI] [PubMed] [Google Scholar]
- 74. Shim W.J., Sinniah E., Xu J., Vitrinel B., Alexanian M., Andreoletti G., Shen S., Sun Y., Balderson B., Boix C.et al.. Conserved epigenetic regulatory logic infers genes governing cell identity. Cell Syst. 2020; 11:625–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kraft K., Magg A., Heinrich V., Riemenschneider C., Schöpflin R., Markowski J., Ibrahim D.M., Acuna-Hidalgo R., Despang A., Andrey G.et al.. Serial genomic inversions induce tissue-specific architectural stripes, gene misexpression and congenital malformations. Nat. Cell Biol. 2019; 21:305–310. [DOI] [PubMed] [Google Scholar]
- 76. Xu Z., Lee D.S., Chandran S., Le V.T., Bump R., Yasis J., Dallarda S., Marcotte S., Clock B., Haghani N.et al.. Structural variants drive context-dependent oncogene activation in cancer. Nature. 2022; 612:564–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Feng J., Liu T., Qin B., Zhang Y., Liu X.S.. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012; 7:1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pachano T., Haro E., Rada-Iglesias A.. Enhancer-gene specificity in development and disease. Development. 2022; 149:dev186536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Galouzis C.C., Furlong E.E.M.. Regulating specificity in enhancer-promoter communication. Curr. Opin. Cell Biol. 2022; 75: [DOI] [PubMed] [Google Scholar]
- 82. Franke M., Ibrahim D.M., Andrey G., Schwarzer W., Heinrich V., Schöpflin R., Kraft K., Kempfer R., Jerković I., Chan W.L.et al.. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 2016; 538:265–269. [DOI] [PubMed] [Google Scholar]
- 83. Chang W., Cheng J., Allaire J., Sievert C., Schloerke B., Xie Y., Allen J., McPherson J., Dipert A., Borges B.. shiny: Web Application Framework for R. 2023; R package version 1.7.4.9002.
- 84. Louden D.N. MedGen: NCBI’s portal to information on medical conditions with a genetic component. Med Ref Serv Q. 2020; 39:183–191. [DOI] [PubMed] [Google Scholar]
- 85. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kleinert P., Kircher M.. A framework to score the effects of structural variants in health and disease. Genome Res. 2022; 32:gr.275995.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sharo A.G., Hu Z., Sunyaev S.R., Brenner S.E.. StrVCTVRE: a supervised learning method to predict the pathogenicity of human genome structural variants. Am. J. Hum. Genet. 2022; 109:195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hertzberg J., Mundlos S., Vingron M., Gallone G.. TADA—a machine learning tool for functional annotation-based prioritisation of pathogenic CNVs. Genome Biol. 2022; 23:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ganel L., Abel H.J., Hall I.M.. SVScore: an impact prediction tool for structural variation. Bioinformatics. 2017; 33:1083–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Poszewiecka B., Pienkowski V.M., Nowosad K., Erôme J.′, Robin D., Gogolewski K., Gambin A.. TADeus2: a web server facilitating the clinical diagnosis by pathogenicity assessment of structural variations disarranging 3D chromatin structure. Nucleic Acids Res. 2022; 1:13–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Prescott S.L., Srinivasan R., Marchetto M.C., Grishina I., Narvaiza I., Selleri L., Gage F.H., Swigut T., Wysocka J.. Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest. Cell. 2015; 163:68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Markenscoff-Papadimitriou E., Whalen S., Przytycki P., Thomas R., Binyameen F., Nowakowski T.J., Kriegstein A.R., Sanders S.J., State M.W., Pollard K.S.et al.. A chromatin accessibility atlas of the developing human telencephalon. Cell. 2020; 182:754–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Fulco C.P., Nasser J., Jones T.R., Munson G., Bergman D.T., Subramanian V., Grossman S.R., Anyoha R., Doughty B.R., Patwardhan T.A.et al.. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 2019; 51:1664–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Klopocki E., Ott C.E., Benatar N., Ullmann R., Mundlos S., Lehmann K.. A microduplication of the long range SHH limb regulator (ZRS) is associated with triphalangeal thumb-polysyndactyly syndrome. J. Med. Genet. 2008; 45:370–375. [DOI] [PubMed] [Google Scholar]
- 95. Cox J.J., Willatt L., Homfray T., Woods C.G.. A SOX9 duplication and familial 46,XX developmental testicular disorder. N. Engl. J. Med. 2011; 364:91–93. [DOI] [PubMed] [Google Scholar]
- 96. Lettice L.A., Daniels S., Sweeney E., Venkataraman S., Devenney P.S., Gautier P., Morrison H., Fantes J., Hill R.E., Fitzpatrick D.R.. Enhancer-adoption as a mechanism of human developmental disease. Hum. Mutat. 2011; 32:1492–1499. [DOI] [PubMed] [Google Scholar]
- 97. Vandermeer J.E., Smith R.P., Jones S.L., Ahituv N.. Genome-wide identification of signaling center enhancers in the developing limb. Dev. 2014; 141:4194–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. D’haene E., Vergult S.. Interpreting the impact of noncoding structural variation in neurodevelopmental disorders. Genet. Med. 2021; 23:34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ibn-Salem J., Köhler S., Love M.I., Chung H.-R., Huang N., Hurles M.E., Haendel M., Washington N.L., Smedley D., Mungall C.J.et al.. Deletions of chromosomal regulatory boundaries are associated with congenital disease. Genome Biol. 2014; 15:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Mehrjouy M.M., Fonseca A.C.S., Ehmke N., Paskulin G., Novelli A., Benedicenti F., Mencarelli M.A., Renieri A., Busa T., Missirian C.et al.. Regulatory variants of FOXG1 in the context of its topological domain organisation /631/208/200 /631/208/1516 article. Eur. J. Hum. Genet. 2018; 26:186–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kumakura A., Takahashi S., Okajima K., Hata D. A haploinsufficiency of FOXG1 identified in a boy with congenital variant of Rett syndrome. Brain Dev. 2014; 36:725–729. [DOI] [PubMed] [Google Scholar]
- 102. Tocco C., Bertacchi M., Studer M.. Structural and functional aspects of the neurodevelopmental gene NR2F1: from animal models to human pathology. Front. Mol. Neurosci. 2021; 14:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zhang Z., Zhao Y.. Progress on the roles of MEF2C in neuropsychiatric diseases. Mol. Brain. 2022; 15:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Middelkamp S., Vlaar J.M., Giltay J., Korzelius J., Besselink N., Boymans S., Janssen R., de la Fonteijne L., van Binsbergen E., van Roosmalen M.J.et al.. Prioritization of genes driving congenital phenotypes of patients with de novo genomic structural variants. Genome Med. 2019; 11:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P.et al.. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020; 581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Rodriguez-Revenga L., Mila M., Rosenberg C., Lamb A., Lee C.. Structural variation in the human genome: the impact of copy number variants on clinical diagnosis. Genet. Med. 2007; 9:600–606. [DOI] [PubMed] [Google Scholar]
- 107. Kingdom R., Wright C.F.. Incomplete penetrance and variable expressivity: from clinical studies to population cohorts. Front. Genet. 2022; 13:920390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Firth H.V., Richards S.M., Bevan A.P., Clayton S., Corpas M., Rajan D., Van Vooren S., Moreau Y., Pettett R.M., Carter N.P.. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am. J. Hum. Genet. 2009; 84:524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Lappalainen I., Lopez J., Skipper L., Hefferon T., Spalding J.D., Garner J., Chen C., Maguire M., Corbett M., Zhou G.et al.. DbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res. 2013; 41:D936–D941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Redin C., Brand H., Collins R.L., Kammin T., Mitchell E., Hodge J.C., Hanscom C., Pillalamarri V., Seabra C.M., Abbott M.-A.et al.. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat. Genet. 2017; 49:36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W.et al.. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018; 46:D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Smith L., Singhal N., El Achkar C.M., Truglio G., Rosen Sheidley B., Sullivan J., Poduri A.. PCDH19-related epilepsy is associated with a broad neurodevelopmental spectrum. Epilepsia. 2018; 59:679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Symonds J.D., Zuberi S.M., Stewart K., McLellan A., O’Regan M., MacLeod S., Jollands A., Joss S., Kirkpatrick M., Brunklaus A.et al.. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain. 2019; 142:2303–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Samanta D. PCDH19-related epilepsy syndrome: a comprehensive clinical review. Pediatr. Neurol. 2020; 105:3–9. [DOI] [PubMed] [Google Scholar]
- 115. Spector J.D., Wiita A.P.. ClinTAD: a tool for copy number variant interpretation in the context of topologically associated domains. J. Hum. Genet. 2019; 64:437–443. [DOI] [PubMed] [Google Scholar]
- 116. McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F.. The ensembl variant effect predictor. Genome Biol. 2016; 17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E.et al.. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015; 17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Real F.M., Haas S.A., Franchini P., Xiong P., Simakov O., Kuhl H., Schöpflin R., Heller D., Moeinzadeh M.H., Heinrich V.et al.. The mole genome reveals regulatory rearrangements associated with adaptive intersexuality. Science. 2020; 370:208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Nanni L., Ming J.E., Bocian M., Steinhaus K., Bianchi D.W., De Die-Smulders C., Giannotti A., Imaizumi K., Jones K.L., Del Campo M.et al.. The mutational spectrum of the sonic hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Hum. Mol. Genet. 1999; 8:2479–2488. [DOI] [PubMed] [Google Scholar]
- 120. Milunsky J.M., Maher T.A., Zhao G., Roberts A.E., Stalker H.J., Zori R.T., Burch M.N., Clemens M., Mulliken J.B., Smith R.et al.. TFAP2A mutations result in branchio-oculo-facial syndrome. Am. J. Hum. Genet. 2008; 82:1171–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Spielmann M., Mundlos S.. Looking beyond the genes: the role of non-coding variants in human disease. Hum. Mol. Genet. 2016; 25:R157–R165. [DOI] [PubMed] [Google Scholar]
- 122. Zhang F., Lupski J.R.. Non-coding genetic variants in human disease. Hum Mol Genet. 2015; 24:R102–R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Arnold C.D., Zabidi M.A., Pagani M., Rath M., Schernhuber K., Kazmar T., Stark A.. Genome-wide assessment of sequence-intrinsic enhancer responsiveness at single-base-pair resolution. Nat. Biotechnol. 2017; 35:136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hill R.E., Lettice L.A.. Alterations to the remote control of Shh gene expression cause congenital abnormalities. Philos. Trans. R. Soc. B Biol. Sci. 2013; 368:20120357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Gerrard D.T., Berry A.A., Jennings R.E., Piper Hanley K., Bobola N., Hanley N.A.. An integrative transcriptomic atlas of organogenesis in human embryos. Elife. 2016; 5:e15657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Gerrard D.T., Berry A.A., Jennings R.E., Birket M.J., Zarrineh P., Garstang M.G., Withey S.L., Short P., Jiménez-Gancedo S., Firbas P.N.et al.. Dynamic changes in the epigenomic landscape regulate human organogenesis and link to developmental disorders. Nat. Commun. 2020; 11:3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Abe Y., Sakata-Yanagimoto M., Fujisawa M., Miyoshi H., Suehara Y., Hattori K., Kusakabe M., Sakamoto T., Nishikii H., Nguyen T.B.et al.. A single-cell atlas of non-haematopoietic cells in human lymph nodes and lymphoma reveals a landscape of stromal remodelling. Nat. Cell Biol. 2022; 24:565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Eraslan G., Drokhlyansky E., Anand S., Fiskin E., Subramanian A., Slyper M., Wang J., Wittenberghe N.V., Rouhana J.M., Waldman J.et al.. Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function. Science. 2022; 376:eabl4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Tabula Sapiens Consortium* Jones R.C., Karkanias J., Krasnow M.A., Pisco A.O., Quake S.R., Salzman J., Yosef N., Bulthaup B., Brown P.et al.. The Tabula Sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science. 2022; 376:eabl4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Lipton Z.C. The Mythos of Model Interpretability. Commun. ACM. 2016; 61:35–43. [Google Scholar]
- 131. Reddy S. Explainability and artificial intelligence in medicine. Lancet Digit Health. 2022; 4:e214–e215. [DOI] [PubMed] [Google Scholar]
- 132. Vellido A. The importance of interpretability and visualization in machine learning for applications in medicine and health care. Neural Comput. Appl. 2020; 32:18069–18083. [Google Scholar]
- 133. Quinn T.P., Jacobs S., Senadeera M., Le V., Coghlan S.. The three ghosts of medical AI: can the black-box present deliver?. Artif. Intell. Med. 2022; 124:102158. [DOI] [PubMed] [Google Scholar]
- 134. Nichols J.A., Herbert Chan H.W., Baker M.A.B.. Machine learning: applications of artificial intelligence to imaging and diagnosis. Biophys. Rev. 2019; 11:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
POSTRE is publicly available at https://github.com/vicsanga/Postre and https://doi.org/10.5281/zenodo.7732896. Its source code and the full set of instructions (including videos and tutorials) explaining how to download and run it are provided there. POSTRE software is distributed under a GNU General Public License v3.0.