


Abstract
In response to different stimuli many transcription factors (TFs) display different activation dynamics that trigger the expression of specific sets of target genes, suggesting that promoters have a way to decode dynamics. Here, we use optogenetics to directly manipulate the nuclear localization of a synthetic TF in mammalian cells without affecting other processes. We generate pulsatile or sustained TF dynamics and employ live cell microscopy and mathematical modelling to analyse the behaviour of a library of reporter constructs. We find decoding of TF dynamics occurs only when the coupling between TF binding and transcription pre-initiation complex formation is inefficient and that the ability of a promoter to decode TF dynamics gets amplified by inefficient translation initiation. Using the knowledge acquired, we build a synthetic circuit that allows obtaining two gene expression programs depending solely on TF dynamics. Finally, we show that some of the promoter features identified in our study can be used to distinguish natural promoters that have previously been experimentally characterized as responsive to either sustained or pulsatile p53 and NF-κB signals. These results help elucidate how gene expression is regulated in mammalian cells and open up the possibility to build complex synthetic circuits steered by TF dynamics.
Graphical Abstract
Graphical Abstract.
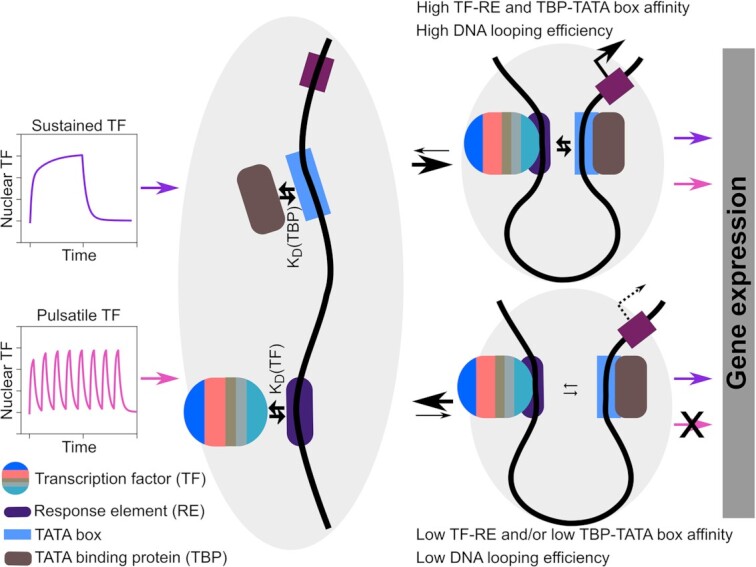
Mammalian promoters decode transcription factor dynamics. DifferentTF dynamics are distinguished by promoters characterized by inefficientcoupling between TF binding and PIC formation and stabilization.
INTRODUCTION
Gene expression is a tightly regulated, complex biological process that turns a specific DNA sequence, the gene, into RNA or protein. It comprises several steps, among which are transcription, mRNA translation and protein folding and degradation (1). In eukaryotes, transcription of protein-coding genes is carried out by RNA polymerase II (Pol II) and typically initiates at the transcription start site (TSS) found at the 5′ end of a gene within the core promoter. This is the location at which the general transcription machinery –constituted by Pol II and its associated general transcription factors (GTFs) (2)– assemble, forming the pre-initiation complex (PIC) (3). The core promoter has specific sequence motifs, which are known to recruit GTFs to mediate PIC assembly. The two Pol II core promoter motifs capable of nucleating the PIC are the TATA box and the Initiator element (Inr) (4). Core promoters usually have low basal activity and are regulated by distal DNA elements –the enhancers– as well as chromatin state (5,6). Enhancers are the genomic regions at which transcription factors (TFs) and co-factors are recruited (7,8). TFs bind specific DNA sequences called response elements (REs) (9). Individual TFs often control a multitude of genes (10), and they act either as repressors or activators. DNA loop formation brings the TF-bound RE(s) and the core promoter into close proximity, allowing the TF to recruit the GTFs (11,12).
Specialization of TFs for certain targets is one of the mechanisms used by cells to start different gene expression programs under specific conditions. The human genome codes for ∼1600 TFs (8). Likely the necessity to maintain a manageable genome size led to the evolution of other strategies consenting cells to re-use the same TF in multiple ways. One strategy consists in post-translationally modifying TFs to modulate their stability, localization as well as affinity for the DNA, co-activators and/or the GTFs (13,14). In the past decade, another cellular strategy emerged as an effective way of achieving multiplexing: controlling TF dynamics, that is, the time-resolved activity of the TF (10,15,16). In particular, several studies have shown that TFs accumulate in the nucleus –where they are active– either in a single, sustained pulse, or in repeated pulses of distinct frequencies and amplitudes depending on the stimulus sensed by the cells (10,17). p53, for instance, in response to UV irradiation, was shown to accumulate in the nucleus in a single pulse, with amplitude proportional to the UV dose; in response to γ-radiation, instead, p53 displays pulses of fixed amplitude (18). Artificially turning the natural p53 pulsatile dynamics in response to γ-radiation into a single sustained pulse increased the frequency of cells going into senescence instead of recovering from the DNA damage, suggesting that TF dynamics directly influence cell fate decisions (19).
If TF dynamics dictate which genes are activated, then promoters have a way to decode them. TF dynamics decoding at the level of the promoter has been so far studied in the yeast Saccharomyces cerevisiae (20) and the filamentous fungus Neurospora crassa (21). A systematic analysis of the regulatory elements necessary to render a mammalian promoter sensitive to TF dynamics is missing. Moreover, the role played by translation initiation efficiency in the decoding process remains unclear.
Here, we apply optogenetic perturbations to control the nuclear localization of a synthetic TF, and generate defined TF dynamics. We then study the ability of these TF dynamics to activate a library of synthetic promoters built of well-defined and characterized parts. Furthermore, we investigate the effect of translation initiation rates in transmitting TF dynamics to downstream gene expression programs. Combining experiments and mathematical modelling, we show that different TF dynamics are distinguished by promoters characterized by inefficient coupling between TF binding and PIC formation and stabilization, and that inefficient translation of the mRNA amplifies the decoding process achieved at the promoter level. We use promoters that respond differently to synTF dynamics to build a synthetic circuit that allows generating two distinct gene expression programs. Finally, we analyse a set of p53- and NF-κB-responsive promoters known to be activated by either sustained or pulsatile TF dynamics and find that we can recapitulate their behaviour based on our understanding of how promoters decode TF dynamics.
MATERIALS AND METHODS
Bacterial strains for molecular cloning
Chemically competent E. coli TOP10 or DH5α cells were used for the transformation of circular plasmid DNA. For plasmid amplification, kanamycin or ampicillin was used as a selection agent at a final concentration of 50 or 100 μg ml−1, respectively. All bacterial cells were incubated in lysogeny broth medium (LB) and on LB agar plates containing the appropriate antibiotic.
Cell lines and maintenance
HEK293 and HeLa cells were maintained at 37°C and 5% CO2 in phenol red-free Dulbecco's modified Eagle's medium (Gibco, Thermo Fisher Scientific) (DMEM) supplemented with 10% fetal calf serum (Sigma-Aldrich (MilliporSigma)), 2 mM l-glutamine (Gibco, Thermo Fisher Scientific), 100 U ml−1 penicillin and 10 mg ml−1 streptomycin (Gibco, Thermo Fisher Scientific).
Molecular cloning
PCR for molecular cloning
Single-stranded primer deoxyribonucleotides with a final concentration of 100 μM were ordered from Sigma Aldrich or Eurofins Genomics. PCR reactions with plasmid and genomic DNA templates were performed using the Phusion High-Fidelity 2x Master Mix or Q5 High-Fidelity 2x Master Mix (New England Biolabs) according to the manufacturer's protocol. Samples were purified by DNA agarose gel electrophoresis followed by gel extraction using the QIAquick Gel Extraction Kit (Qiagen).
Molecular cloning using gibson assembly
Plasmid pDN98 (22) was used as parental plasmid for the construction of the synthetic TF (synTF). The LexA dimerization domain was amplified from genomic DNA extracted from E. coli (TOP10 strain) and inserted between the LexA DNA-binding domain and the VP48 transactivation domain in pDN98 to generate plasmid pEA00.
The iRFP670 coding sequence was PCR-amplified from plasmid pNLS-iRFP670 (23) (gift from Vladislav Verkhusha; Addgene plasmid #45466) with a primer containing the sequence encoding the CAAX motif and cloned in place of the firefly luciferase gene into plasmid pDN100 (22). The DNA sequence encompassing the synthetic promoter, the reporter gene and the bovine growth hormone (BGH) terminator was then PCR-amplified from this modified pDN100 plasmid and cloned into pEA00 upstream of the CMV promoter (synTF promoter) in a tandem orientation, giving rise to plasmid pEA01 (synPlasmid1). Insertion of the reporter construct in a convergent orientation downstream of the SV40 terminator led to the construction of pEA02 (synPlasmid2). Reversing the orientation of the synTF construct in pEA01 gave rise to pEA03 (synPlasmid3). All other reporter library constructs were generated by modifying an element in the promoter of the synthetic reporter in pEA01. The sequences of the promoter elements used in this study are given in Supplementary Table S1. All reporter constructs designed in this study and their constituent elements are listed in Supplementary Table S2.
The gene encoding the MS2 coat protein (MCP) was amplified from plasmid ubc-nls-ha-MCP-VenusN-nls-ha-PCP-VenusC (24) (gift from Robert Singer; Addgene plasmid #52985). The IRES-SV40/NLS-MCP gene sequence together with the full-length mVenus gene amplified from pTriEx-NTOM20-mVenus-Zdk2 (25) (gift from Klaus Hahn; Addgene plasmid #81011) was inserted after the stop codon of synTF in pEA01. The 12xMBS-PBS sequence was PCR-amplified from plasmid Pcr4-12xMBS-PBS (24) (gift from Robert Singer; Addgene plasmid #52984) and cloned after the stop codon of the reporter gene sequence. For better foci visualization, the BGH promoter was removed to allow for a longer 3′UTR, which permits the nascent RNA to be bound long enough for it to be visualized. The complete list of all plasmids is given in Supplementary Table S3. The list of all primers used in this study is given in Supplementary Table S4.
All plasmids were constructed using Gibson assembly (26). Gibson assemblies were performed using 50 ng backbone DNA in a 10 μl reaction volume and a molar 1:1–3 backbone:insert ratio, using the NEBuilder HiFi DNA Assembly Master Mix (2x) (NEB) for 20–40 min at 50 °C. Agarose gel-purified DNA fragment concentrations were determined using a spectrophotometer (NanoDrop One, Thermo Fisher Scientific).
DNA agarose gel electrophoresis
Gels were prepared with 1% agarose (Agarose Standard, Carl Roth) in 0.5x TAE-buffer and 1:50 000 Ethidium Bromide (Roth), running for 20–30 min at 130 V. For analysis, 1 kb Plus DNA Ladder (NEB) was used. The samples were mixed with gel loading dye (purple, 6x) (NEB).
Bacterial transformation with plasmid DNA
Chemical transformation was performed by mixing 10 μl of Gibson reaction with 50 μl of chemically competent cells and incubating the mixture on ice for 30 min. Cells were then heat-shocked at 42 °C for 90 s, further incubated on ice for 5 min and finally mixed with 450 μl LB medium. Transformed cells were incubated at 37°C on a shaker for 30–45 min before plating on agar plates containing the antibiotic. Plates were incubated overnight at 37 °C or for 48 h at room temperature.
Plasmid DNA purification and sanger sequencing
Individual clones were picked from the agar plate and inoculated in 2–3 ml LB medium with kanamycin or ampicillin and incubated for about 6–8 h. Plasmid DNA was purified with the QIAprep Plasmid MiniSpin kit (QIAGEN) according to the manufacturer's protocol. Plasmids were sent for Sanger sequencing (GATC-Biotech/Eurofins) and analysed using APE (https://jorgensen.biology.utah.edu/wayned/ape/).
Plasmid transfection
Cells were transfected with the calcium phosphate transfection protocol. DNA amounts were kept constant in all of the experiments to yield reproducible complex formation and comparable results. A total amount of 500 ng of DNA was used to transfect cells seeded in Ibidi μ-dish 4-well dishes (Ibidi GmbH, Germany). pBlue-ScriptIIS/K was used as stuffer plasmid (1:200 DNA of insert:stuffer DNA ratio). Cells were plated 1 day before transfection (75 000 cells in 250 μl of culture medium per well of Ibidi μ-dish 4-well dishes).
Cellular imaging and optogenetic stimulation
Microscopy was performed always 24 h posttransfection. Cells were maintained at 37°C and 5% CO2 in a dark incubation chamber for the duration of the microscopy session. Images in the mCherry and iRFP670 channels were acquired in confocal modality on a Zeiss LSM 800 confocal microscope equipped with a motorized stage, a Plan-Apochromat 40×/1.4 numerical aperture oil immersion objective (Zeiss), a laser module containing 405, 488, 561 and 640 nm lasers and an electronically switchable illumination and detection module. Images in the mCherry channel were acquired with the following settings: 0.15% of 561 nm excitation laser using 37 μm pinhole aperture and 700 V gain. Images in the iRFP670 channel were acquired with 0.30% of 640 nm excitation laser using 41 μm pinhole aperture and 650 V gain. The confocal microscope was also equipped with a colibri light source consisting of 385, 425, 469, 511, 555 and 631 nm LEDs for widefield epifluorescence microscopy. Blue light activation was performed by exciting cells with 0.5% of 469 nm LED light in widefield microscopy mode using the 38 HE GFP filter set from Zeiss. The 0.5% light intensity, which corresponded to 6.44 W m−2 of light as measured with the LI-250A light sensor (LI-COR Biosciences), was achieved by filtering out 75% of the 2% LED intensity. Automated cell focusing was done using mCherry as the reference channel. The sustained dynamics were generated by illuminating cells with blue light (GFP channel) for 125 ms every 45 s for 2 h. The pulsatile dynamics were generated by illuminating the cells with the same illumination scheme used for sustained dynamics for 15 min, followed by a dark phase of either 15 (high-frequency pulses) or 30 (low-frequency pulses) min, and repeating this cycle 8x. For time-lapse tracking of synTF-mCherry localization, confocal mCherry images were taken every 5 min during activation and dark recovery phases. All image acquisitions were done using the ZenBlue software. mCherry and iRFP670 confocal images were taken prior to starting the optogenetic stimulation and post activation, every 30 min for 5h. Nascent RNA transcripts were visualized on the same microscope with an Axiocam503 camera and an 63x/1.4 Plan-Apochromat oil-immersion objective (Zeiss). Blue light activation of cells was performed with the same setup as above except that 0.95% 469 nm LED light intensity was used to account for the change of objective. This LED intensity corresponded to 6.79 W m−2 of light. Images in the YFP channel (to image mVenus) were acquired using 5% of 511 nm LED light in widefield microscopy mode using the 46 HE YFP filter set from Zeiss in a Z-stack of 16–18 sections with 0.75 μm step size. Images were acquired every 5 min during the blue light activation phases. Since Z-stack imaging in the YFP channel was phototoxic to the cells when performed for a prolonged time, the pulsatile dynamics in the experiments for nascent RNA transcripts visualization was performed repeating the 15 min blue light activation six instead of eight times.
Predicting DNA curvature and flexibility
Seq1 and seq2 were analysed using the cgDNAweb + webserver (27) (https://cgdnaweb.epfl.ch/) with parameter set 4. The .pdb models were then aligned based on their coordinates using PyMol 2.4.1 (28).
Modelling
Model simulations were performed in python v3.8.3.final.0 using the Anaconda v2020.07 distribution. Numerical simulations were performed using the odeint function in SciPy v1.5.0 scipy.integrate module, which is used as a wrapper for the LSODA ordinary differential equation solver for stiff or non-stiff systems from the FORTRAN library odepack. Initial conditions were set according to experimental data at time t = 0 or were fitted from the sustained dynamics time-course data. The variables of interest were plotted using the matplotlib library. The list of all model parameters is given in Supplementary Table S5.
Quantification and statistical analysis
Automated quantification of mCherry and iRFP670 signals
The algorithm for automatic segmentation of the nucleus and cytosol in the absence of nuclear or plasma membrane markers has been described in detail in (29). Briefly, to segment the nucleus we applied the same base network as in (29) and used the off-the-shelf Mask R-CNN (30) trained with the mCherry channel only. To augment the data and create nearly realistic input images, we also used the elastic deformations of U-Net (31) to help improve the generalization capabilities of the network. To detect segmentation errors, we used both data uncertainty (aleatoric) and model uncertainty (epistemic). We modelled the former by learning the noise scale during training and computing the entropy of the class pseudo-probabilities for each pixel at test time as in (32). For the later, we used the Winner-Takes-All (33) method, which trains a single network with multiple heads and only updates the head with the best prediction every iteration. We chose this combination since it performed best in (29). To improve the output of Mask R-CNN, we computed the tracks as described in (29). We applied the suggested hyperparamters α = 0.7 and β = 0.85 to decide which frames needed to be updated. We considered a simple yet effective warping strategy by estimating the shift and scaling parameters computed between the not yet updated and neighbouring nuclei predictions. Likewise, we implicitly assumed that the shape of the nuclei does not change over short time windows and only allowed slight deformations to occur. Although flow-based methods tend to perform better according to (29), we did not use them to reduce the computational burden. To mitigate this slight drop in performance, we applied a sampling strategy before measuring the fluorescence. We reported the average fluorescence of nucleus, cytosol and membrane per cell and frame. Instead of using the full prediction mask to compute the average, we sampled a subset of pixels that had higher chances to belong to the corresponding structure. For nucleus and cytosol, segmentation errors occur mainly on the border. Therefore, we gradually eroded the segmentation mask as long as it contained >2000 pixels. We then superposed the binary mask with mCherry channel and computed the average signal. Measuring the fluorescence for the membrane is very challenging since it is a very thin structure. Moreover, touching cells cause interference that amplify the signal. Thus, we used the iRFP670 channel and computed a skeleton. Notice that this skeleton might miss cells because of very low signal and might add artefacts in case of very high signal over surfaces. To avoid these errors, we relied on the cytosol masks and computed candidate pixels for the membrane by dilating the cytosol once. Then we removed intersections between candidate membranes of touching cells and pixels that were very close to the border of the image. Finally, we superposed the skeleton and the candidate membranes and computed the average based on the intersection. If there was no signal in the skeleton, we completely relied on the candidate membrane inferred from the cytosol.
Quantification of mVenus signal
The maximum projection of the Z-stacks was computed in ImageJ (34) to bring all detected foci onto a single plane. To quantify nascent RNA, individual cells were first cropped and passed through a nascent RNA quantification pipeline as described in (35). The first step in the pipeline was removal of the fluorescent background signal using a Gaussian filter. The filtered image was then subtracted from the original image to obtain an image which has the RNA foci features preserved without fluorescence background. A 2D Gaussian function was then fitted to the pixel intensity surface of each cell. The volume of the fitted function was used to represent the mean nascent RNA levels. The parameters of the 2D Gaussian function were limited to exclude fitting to background intensity fluctuations and large aggregates of the fluorescent proteins.
Reporter mRNA abundance quantification
1.5 × 105 HeLa cells maintained at 37°C and 5% CO2 in phenol red-free DMEM supplemented with 10% fetal calf serum (Sigma-Aldrich (MilliporSigma)), 2 mM L-glutamine (Gibco, Thermo Fisher Scientific), 100 U ml−1 penicillin and 10 mg ml−1 streptomycin (Gibco, Thermo Fisher Scientific) were seeded in a six-well plate (Thermofisher Scientific (Nunclon Delta Surface)) 24 h before transfection. Cells were transfected with the plasmid of interest using TransIT-X2 (Mirus Bio) according to the manufacturer's protocol. 24 h post-transfection, cells were activated with blue light in a custom light box equipped with blue LEDs. Cells were activated by illuminating the bottom of the plate with 28.31 W m−2 blue light for 2 sec every 30 sec for 2 h and then kept 30 min in the dark to allow processing of most mRNAs before harvesting the cells. RNA isolation and purification were done using the RNeasy kit from Qiagen according to the manufacturer's protocol. Reverse transcription of the mRNA was done using the RevertAid First Strand cDNA-synthesis kit (Thermofisher Scientific) according to the manufacturer's protocol. qPCR was performed using the PowerUp™ SYBR™ Green Master Mix (Thermofisher Scientific) according to the manufacturer's protocol on a CFX Opus 384 qPCR machine (BioRad). The aslov gene was used as the reference gene to normalize the results to the transfection efficiency (reporter and synTF fused to the modified AsLOV domain (LINuS) are encoded by the same plasmid).
Reporter mRNA degradation rate estimation
7.5 × 105 HeLa cells maintained in DMEM containing 10% FCS, 2 mM l-glutamine, 100 U ml−1 penicillin and 10 mg ml−1 streptomycin were seeded in a 10 cm culture dish and cultured in 5% CO2 at 37°C for 24 h. Cells were then transfected with the plasmid of interest using TransIT-X2 (Mirus Bio) according to the manufacturer's protocol. 24 h post-transfection, cells were collected and distributed into four 15-ml falcon tubes and actinomycin D (1:1000 dilution from 5 mg/ml stock in DMSO) was immediately added. Cells in the 15-ml tubes were harvested at 0, 1, 2, 3 h, respectively. RNA isolation, reverse transcription and qPCR were performed as described above.
Analysis of NF-κB- and p53-responsive promoters
The list of NF-κB target genes was extracted from (36), while the list of p53 target genes was extracted from (19). For the subsequent analysis, we considered only genes driven by promoters harbouring at least one experimentally validated response element (RE; Supplementary Tables S6 and S7). The RE sequences were then extended on both sides to obtain 20 bp-long oligonucleotides that could be directly used in the Fabian-variant web server (37) to estimate RE strength relative to the reference sequences ‘gatcgggggatttcccatcg’ for NF-κB and ‘ggacatgcccgggcatgtcc’ for p53. The transcriptional start sites were manually extracted from the NCBI databank (https://www.ncbi.nlm.nih.gov/) and were used to locate the TATA box and the Initiator element (Inr) (38). The ‘TATAWAW’ (where W is either A or T) consensus sequence with two allowable mismatches was used to extract the TATA box coordinates. Promoters with a TATA box identified within –35/–20 of the TSS were considered as TATA box-containing promoters. The ‘YYANWYY’ (where Y is C or T, N is any base, and W is A or T) consensus sequence with three allowable mismatches but conserved A at position 3 was used to extract the coordinates of the Inr. The distance λ was calculated as the number of base pairs between the RE and the TATA box or the Inr.
Statistics and reproducibility
Statistics were calculated using the scipy.stats python module. No statistical method was used to predetermine sample size. The experiments were not randomized. Dividing cells, cells that detached during the experiments and cells without well-defined nucleus at the start of the data acquisition were excluded from the analysis. Additionally, cells were stratified based on initial nuclear synTF levels and amplitude during blue light activation to allow for fair comparison of data from different dynamics and promoters. All experimental findings were reproduced in at least three biologically independent experiments.
RESULTS
Different TF dynamics can be imposed using an engineered light-responsive synthetic TF
We constructed a synthetic TF (synTF) fusing to the well-characterized E. coli repressor protein LexA (39) three copies of residues 436–447 of the herpes simplex virus type 1 transcription factor VP16 (VP48) (40), the fluorescent protein mCherry and LINuS, an optogenetic tool consisting of a light-inducible nuclear localization signal (NLS) that allows accumulating synTF in the nucleus or the cytosol upon blue light illumination and incubation in the dark, respectively (22) (Figure 1A and Supplementary Figure S1A). By selecting the appropriate light regime, we can generate different synTF dynamics; here we opted for a single sustained pulse (we refer to this as sustained activation), pulses with ∼30-min period (designated 15–15 pulses) and pulses with ∼45-min period (designated 15–30 pulses) (Supplementary Figure S1B). synTF transcriptional activity is quantified at the reporter nascent RNA (that is, RNA in the process of being synthesized via transcription and, therefore, still attached to the DNA template; Supplementary Figure S1C) and protein (Supplementary Figure S1D) levels. To capture differences due solely to TF dynamics, and not cumulative TF levels, the experiments are assigned different durations, but they all comprise the same five-hour waiting time at the end of the last activation phase to allow for the maturation of the iRFP670 reporter protein (23) (Figure 1B). The workflow starts with a transient transfection step, explicitly chosen to obtain information about the sensitivity of the promoters to synTF amplitude, followed by the application of the dynamics, and the automated quantification of the time-lapse microscopy images (Figure 1C). The image processing pipeline is based on a neuronal network developed by us for the specific task of segmentation in the absence of dedicated nuclear and plasma membrane fluorescent markers (29). The data are finally clustered into bins based on nuclear synTF level (mCherry signal) at t = 0 for fair comparison of different dynamics and promoters.
Figure 1.

Design of molecular components and experimental setup used to study how transcription factor dynamics are decoded by mammalian gene regulatory elements. (A) Schematic representation of the expected behavior of the synthetic transcription factor (synTF) in darkness (left) and during blue light illumination (right). Blue light leads to the exposure of the nuclear localization signal (NLS), which, in turn, leads to the nuclear import of synTF. LexA, full-length bacterial repressor used as DNA binding domain (DBD) within synTF. VP48, 3x residues 436–47 of the VP16 transactivation domain (TAD). NES, nuclear export signal. (B) Schematic representation of the illumination schemes used in this study to achieve similar cumulative TF levels (area under the curve) for the different TF dynamics. (C) Schematic overview of the experimental setup. (D) Schematic representation of the reporter libraries used in this study.
To understand the contribution of specific regulatory elements to the TF dynamics decoding process, we built a library of reporter constructs made of well-characterized DNA elements. As the gene regulatory elements, we focused on TF binding site (RE), core promoter, and 5′UTR. We created two types of libraries: one in which the promoter varies, and one in which the 5′UTR does (Figure 1D and Supplementary Table S2). The TF and the reporter gene are encoded on the same plasmid to ensure that lack of iRFP670 signal in individual cells be not due to the absence of the reporter gene (Figure 1C).
Mammalian promoters can decode TF dynamics
We started analysing the first promoter in the library, promoter p1, which is characterized by four repeats of a strong RE, and a strong TATA box (Supplementary Table S2). We imposed to the cells the three different light regimes for sustained and pulsatile dynamics, and measured synTF nuclear concentration (Figure 2A), and mean reporter nascent RNA (Figure 2B) and protein (Figure 2C) levels over time. Nascent RNA visualization was performed only during the blue light illumination phases not to activate the LOV domain within LINuS, which would result in synTF nuclear localization and promoter binding. The RNA transcription status during the dark phases can be deduced from the first image acquired in each activation phase.
Figure 2.

A promoter with strong RE and TATA box does not distinguish TF dynamics. (A-C) Quantification of synTF nuclear translocation (A), mean reporter nascent RNA (B) and protein (C) levels over time for the indicated TF dynamics in combination with promoter p1. Light blue shadowing, blue light illumination phase. Together with the experimental data (black dots), fitted (violet line; for sustained dynamics) and simulated (pink line; for both pulsatile dynamics) values are shown. The mathematical model used is shown in Figure 4 and the equations are described in the Supplememtary Text. (D, G, J) Quantification of mean nascent RNA per cell per minute (calculated averaging over the entire blue light illumination phase) for the indicated synTF dynamics in combination with promoters p1 (D), p2 (G) and p3 (J). (E, H, K) Quantification of reporter protein expression levels at the end of the experiment for the indicated synTF dynamics in combination with promoters p1 (E), p2 (H) and p3 (K). (F, I, L) Quantification of reporter protein expression levels at the end of the experiment for high (dark colors) or low (light colors) synTF amplitudes for the indicated synTF dynamics in combination with promoters p1 (F), p2 (I) and p3 (L). (E, F, H, I, K, L) P-values were calculated with the Welch's t-test. ns, non signficant (P > 0.05). (E) *P-value = 0.0244; **P-value = 0.003. (L) *P-value (from left to right) = 0.02552, 0.01285, 0.03517. Data represent mean ± s.e.m. of at least n = 20 individual cells, imaged on at least n = 3 biologically independent experiments.
All three dynamics elicited a robust activation of promoter p1. The RNA data showed that there is no transcriptional shutdown during the dark phases for the pulses (Figure 2B, middle and right panels). When comparing the mean reporter nascent RNA levels per cell per minute, we did not find any difference between conditions (Figure 2D), corroborating the conclusion that promoter p1 does not distinguish dynamics. Interestingly, at the protein level, both pulsatile dynamics triggered a higher response than sustained activation (Figure 2E). Promoter p1 does not respond significantly to synTF amplitude (Figure 2F).
When we tested a version of promoter p1 with the Inr in place of the TATA box as core promoter motif, we found only background levels of the reporter protein for sustained synTF dynamics. Hence all promoters we discuss have the TATA box as core promoter motif.
Next, we analysed promoters p2 and p3 (Supplementary Table S2), which are characterized by either the same strong RE as promoter p1, but a weak TATA box (p2) or the same strong TATA box, but a weak RE (p3). We again imposed the three light regimes and measured synTF nuclear concentration (Supplementary Figure S2A), and mean reporter nascent RNA (Supplementary Figure S2B, D) and protein (Supplementary Figure S2C, E) levels over time. Interestingly, we observed a refractory response for promoter p2 under sustained activation, reflected in the decrease of the mean nascent RNA levels during the activation phase (Supplementary Figure S2B, left panel). We define as refractory a promoter for which the PIC cannot be assembled despite the TF being bound at the RE(s). Differently than for promoter p1, we found lower mean reporter nascent RNA levels per cell per minute for the pulsatile than for the sustained dynamics for both promoters (Figure 2G, J). Thus, these promoters are able to distinguish TF dynamics. At the protein level, however, we found no significant difference at the end of the experiment (Figure 2H, K). While promoter p2 is insensitive to synTF amplitude (Figure 2I), promoter p3 is (Figure 2L). This is in line with the fact that promoter p3 features a weak RE, which requires a higher synTF amplitude to achieve full promoter activation.
Finally, we analysed promoter p4, which combines a weak RE (four repeats thereof) with a weak TATA box (Supplementary Table S2, and Figure 3A–C). For promoter p4, refractoriness was more prominent than for promoter p2 (compare Supplementary Figure S2B and Figure 3B, left panels). Despite being overall a weaker promoter than p1-p3, p4 is the best at distinguishing synTF dynamics. Indeed, the two pulsatile dynamics elicited only a mild activation of promoter p4, and, in this case, this is seen both, at the reporter RNA (Figure 3D) and protein (Figure 3E) levels. Interestingly, the difference between the responses of promoter p4 to two different synTF amplitudes appears not statistically significant for sustained and 15–30 pulses (Figure 3F). Nonetheless, since promoter p4 is characterized by a weak RE, we consider this promoter sensitive to amplitude. Lack of statistical significance is justified by the noisy nature of this promoter (Supplementary Figure S3). The more stochastic nature of promoter p4 compared to promoters p1-p3 is likely due to its strong refractory behaviour, because exiting the refractory state is a random process (41).
Figure 3.

A promoter with weak RE and TATA box distinguishes TF dynamics. (A-C) Quantification of synTF nuclear translocation (A), mean reporter nascent RNA (B) and protein (C) levels over time for the indicated TF dynamics. Light blue shadowing, blue light illumination phase. Together with the experimental data (black dots), fitted (violet line; for sustained dynamics) and simulated (pink line; for both pulsatile dynamics) values are shown. The mathematical model used is shown in Figure 4 and the equations are described in the Supplememtary Text. (D) Quantification of mean nascent RNA per cell per minute for the indicated synTF dynamics. (E) Quantification of mean reporter protein expression levels at the end of the experiment for the indicated synTF dynamics. (F) Quantification of mean reporter protein expression levels at the end of the experiment for high (dark colors) or low (light colors) synTF amplitudes for the indicated synTF dynamics. (E, F) P-values were calculated with the Welch's t-test. ns, non signficant (P > 0.05). (E) *, P-value (from left to right) = 0.0217; 0.0212. (F) *P-value = 0.03292. (A–F) Data represent mean ± s.e.m. of at least n = 20 individual cells, imaged on at least n = 3 biologically independent experiments. The promoter used in these experiments was p4.
Versions of promoters p1 and p2 with an even stronger RE (promoters p10 and p11, respectively; Supplementary Table S2) were fully activated by the low synTF levels present in the nucleus in the dark. Therefore, these constructs are not light-sensitive and have not been further analysed.
Weak coupling between TF binding and PIC assembly is the key feature that allows a promoter to distinguish dynamics
To gain a mechanistic understanding of the reasons why promoter p4 is the best at distinguishing synTF dynamics, while p1 cannot do it at all, we developed a compartmental mathematical model of gene expression based on ordinary differential equations (Figure 4A). We opted for a three-state promoter model, whereby the promoter can be unbound (synTF is unbound), active (synTF is bound to a certain fraction of the REs, the TATA binding protein (TBP) is bound to the TATA box and, consequently, the PIC can be assembled) and refractory (synTF is bound to the REs, but the PIC cannot be assembled). synTF can bind to all or a fraction of the REs present in the promoter. We model the DNA looping –needed to bring the TF in close proximity to the TATA box for recruitment of the GTFs (11,12)– by dividing the rate of PIC assembly by a factor, jm, that accounts for the distance between the last RE and the TATA box, given that the efficiency of DNA looping between two points decreases with increasing distance between them (42,43). We finally assume that the maturation of the RNA and the fluorescent reporter protein occur at constant rates, while protein translation depends on mRNA concentration and ability of the ribosome to find the start codon (see Supplementary Text for the equations and a detailed description of the model).
Figure 4.

A mathematical model of gene expression mechanistically explains how a mammalian promoter can distinguish TF dynamics. (A) Schematic showing the reactions included in the mathematical model. The equations are explained in the Supplementary Text. (B,C,E) Model predictions (left panel; fitted (violet bar; for sustained dynamics) and simulated (pink bar; for both pulsatile dynamics)) and experimental data (right panel) of the mean reporter protein levels at the end of the experiment for the indicated synTF dynamics in combination with promoters p5 (B), p6 (C) and p7 (E). (D) Curvature of the indicated DNA sequences modeled using the cgDNAweb + web server (27). (B, C, E) Data represent mean ± s.e.m. of at least n = 20 individual cells, imaged on at least n = 3 biologically independent experiments. P-values were calculated with the Welch's t-test. ns, non signficant (P > 0.05). **, P-value = 0.00369.
We assigned physiologically reasonable values to some parameters based on the literature (44,45) (Supplementary Table S5), measured mRNA half-life using qPCR after actinomycin D treatment (Supplementary Figure S4) and further used the data obtained with the sustained dynamics with promoter p2 (due to better resolution of reporter expression at lower synTF levels) to fit the remaining parameters (Supplementary Figure S2A–C left panels and Supplementary Figure S5). Promoter p1-specific parameters were calculated fitting the model to the data obtained for this promoter under sustained synTF dynamics (Figure 2A–C, left panels). We then used the model to simulate synTF dynamics, and mean reporter nascent RNA and protein levels over time for the two pulsatile dynamics with promoter p1. We found a good agreement between the simulations and the experimental data (Figure 2A–C, middle and right panels).
Having now a mathematical model able to describe the behaviour of promoter p1, we used it to understand why this promoter generates higher reporter protein levels at the end of the experiment for pulsatile synTF (Figure 2C, E), despite being insensitive to TF dynamics, as seen by the RNA data (Figure 2B, D). We hypothesized that the reason for this behaviour lied in the potential higher effective cumulative synTF levels obtained with the pulsatile experiments, which run for a longer period of time than the experiment for sustained dynamics. Using the model, we determined the threshold for synTF concentration above which the reporter protein levels are above the half-maximal value (Supplementary Figure S2F) and used this value to quantify the effective cumulative levels for synTF for sustained and 15–30 pulses (Supplementary Figure S2G). We found that, indeed, they are higher for the pulsatile dynamics. After normalizing the experimental mean reporter protein levels at the end of the experiments against the effective cumulative synTF levels found in silico, the difference among the dynamics disappears (Supplementary Figure S2H).
The model successfully predicts the behaviour of promoters p2–p4, when the promoter-specific parameters are updated using the sustained dynamics data (Supplementary Figure S2A–E, and Figure 3A–C, left panels). Importantly, the models describing the four promoters are identical in the set of reactions, the only difference being the values of the promoter-specific parameters, which require a fitting step for each promoter (done using the data for sustained synTF).
Because the model of gene expression we developed captures all experimental data, we conclude that the processes we included are sufficient to explain how a mammalian promoter distinguishes TF dynamics. Our model offers the following mechanistic explanation: if the coupling between TF binding and PIC assembly is very efficient, any TF binding event, even a brief one, will be sufficient to recruit the GTFs, assemble the PIC and initiate transcription. The coupling is efficient, for instance, if the TF binds with high affinity to the RE, and/or the TBP binds with high affinity to the TATA box. A promoter, such as p4, with weak RE and TATA box, has inefficient coupling and, therefore, is less activated by pulsatile dynamics, characterized by a shorter residence time of the TF in the nucleus, and, consequently, at the promoter.
To demonstrate that the efficiency of coupling is the key feature that renders a mammalian promoter sensitive to TF dynamics, we used our mathematical model to predict whether we could turn promoter p1 from insensitive to sensitive to TF dynamics by creating a new version of it (which we call promoter p5), with longer distance between the last RE and the TATA box (Supplementary Table S2 and Figure 1D). Indeed, as previously mentioned, the longer the distance, the less efficient the DNA looping, and, consequently, the coupling between TF binding and GTFs recruitment. After fitting the promoter-specific parameters using the data obtained with the sustained dynamics, we used the model to predict the behaviour of promoter p5 under synTF pulses. The simulations indicate that promoter p5 is activated twice as much by sustained dynamics than low-frequency pulses (Figure 4B, left panel). We constructed promoter p5 by inserting a random DNA sequence of 147 bp (seq1) between the last RE and the TATA box in promoter p1 (λ = 196 bp; Supplementary Table S2 and Figure 1D). Despite producing overall much less reporter protein than promoter p1 under sustained dynamics (compare Figures 2E and 4B, right panel), promoter p5 barely responded to the 15–30 pulses (Figure 4B, right panel), implying that decreasing the coupling efficiency was sufficient to render promoter p1 sensitive to dynamics. When in silico introducing the same distance between the RE and the TATA box into promoter p2 (creating promoter p6), we found a similar trend, with sustained activation leading to higher protein production than pulses (Figure 4C, left panel). To validate the model predictions, we constructed promoter p6 by inserting the same random sequence seq1 into promoter p2 (Supplementary Table S2). However, promoter p6 was too weak to be experimentally analysed (Figure 4C, right panel).
Sequence length is, however, not the only important parameter for DNA looping. Reasoning that DNA looping efficiency could be improved by changing the DNA sequence keeping the same distance between the RE and the TATA box, we cloned promoter p7 using a sequence predicted to be prone to looping (46) (seq2; Figure 4D). The model predicted that promoter p7 produces more protein than promoter p6 and that it is insensitive to dynamics (Figure 4E, left panel), which we experimentally confirmed (Figure 4E, right panel). This suggests that, as far as the coupling between TF binding and GTFs recruitment is efficient, even promoters with overall low activity can respond well to low-frequency pulses, becoming unable to filter them out.
Inefficient translation initiation strengthens the ability of a promoter to decode TF dynamics
As mentioned above, promoters p2 and p3––that contain four REs––distinguish synTF dynamics, but this is visible only at the level of the RNA (Figure 2G, H, J and K). One explanation for this behaviour is that, when the RNA is efficiently translated into protein, the difference at the RNA level gets lost at the protein level, when the RNA levels are high. This compensation does not occur anymore when the RNA levels are much lower: promoter p8, which is a version of promoter p2 with two instead of four REs is able to sense dynamics also at the protein level (Supplementary Figure S6). Bulk mRNA quantification using qPCR showed that promoter p4, which also senses dynamics, is characterised by the lowest mRNA abundance among the four promoter constructs p1-p4 (Supplementary Figure S7).
To further test the hypothesis that translation efficiency might play a role in transmitting the information encoded in TF dynamics, we built a small library of reporter constructs, whereby the promoter is fixed, but the 5′UTR varies (Figure 1D and Supplementary Table S2). In particular, we tested the role played by mRNA scanning by the ribosome to locate the start codon, not other processes involved in mRNA translation. We took promoter p2 and, as the first step, decreased the distance between the TATA box and the start codon ATG, creating construct 5UTR1 (Figure 5A). This should eliminate any secondary RNA structures that may affect mRNA translation (47) and should increase the efficiency of the translation initiation complex to locate the start codon (48–52). The second construct, 5UTR2, is similar to 5UTR1, with the only difference that we exchanged the G at position –6 relative to the start codon to A, which should reduce translation efficiency (53,54) (Figure 5A). Finally, we created a construct, 5UTR3, lacking the Kozak sequence (Figure 5A). We transfected these constructs in HEK293 cells, imposed the two light regimes for sustained and low-frequency pulsatile dynamics, and measured synTF nuclear concentration and mean reporter protein levels over time (Supplementary Figure S8, A and B). The first observation we made is that, indeed, decreasing the length of the 5′UTR has a positive effect on reporter gene expression, since construct 5UTR1 leads to twice as high mean reporter protein levels than the original construct with promoter p2 with the longer 5′UTR under both, sustained dynamics and 15–30 pulses (compare Figures 2H and 5C, left panel). Construct 5UTR2 gave rise to ∼1.2 times lower mean reporter protein levels under both conditions (Figure 5C). Neither construct showed sensitivity to synTF dynamics. Construct 5UTR3, which is characterized by the lowest translation initiation efficiency due to complete lack of the Kozak consensus sequence, interestingly distinguishes TF dynamics (Figure 5D). Notably, in this case, it is not the mRNA level that makes the difference, but rather the efficiency of its translation. As a matter of fact, constructs 5UTR1 and 3 lead to similar mRNA abundances (Supplementary Figure S7).
Figure 5.

Decreasing translation initiation efficiency amplifies the ability of the promoter to sense TF dynamics. (A) Schematic showing the features of the constructs in the 5′UTR library. The Kozak sequence is shown in gold, the mutated Kozak sequence in orange, and the random sequence in black. The start codon is shown in bold, while the TATA box in blue. (B) Quantification of mean reporter protein levels over time for the indicated TF dynamics. Together with the experimental data (black dots), fitted (violet line; for sustained dynamics) and simulated (pink line; for both pulsatile dynamics) values are shown. The mathematical model used is shown in Figure 4 and the equations are described in the Supplememtary Text. (C, D) Quantification of reporter protein expression levels at the end of the experiment for the indicated synTF dynamics in combination with the indicated constructs. P-values were calculated with the Welch's t-test. ns, non signficant (P > 0.05). *, P-value = 0.0419;.***, P-value = 0.0002. (B, C) Data represent mean ± s.e.m. of at least n = 20 individual cells, imaged on at least n = 3 biologically independent experiments.
Our mathematical model could correctly predict the behaviour of the constructs in this library under the two pulsatile dynamics, after updating the translation parameters using the data from the sustained dynamics (Figure 5B and Supplementary Figure S8).
Two distinct gene expression programs can be achieved with synTF using promoters p1 and p4
We sought to exploit the fact that different promoters respond differently to synTF dynamics to build a synthetic circuit that allows generating distinct expression programs using a single TF. We cloned two fluorescent reporter proteins under the control of promoters p1 and p4, because p1 does not distinguish synTF dynamics, while p4 does (Figure 6A). As a consequence, only for sustained synTF dynamics, both promoters are activated and both reporter proteins are expected to be expressed at high levels (Figure 6B). For pulsatile synTF dynamics, only the reporter under the p1 promoter is expected to be expressed at high levels (Figure 6B). We transiently transfected HeLa cells with this circuit and imposed the two different dynamics on synTF using light. We found that, indeed, we could obtain the two distinct gene expression programs (both proteins highly expressed versus only one protein highly expressed) by simply regulating synTF dynamics (Figure 6C).
Figure 6.

Obtaining two gene expression programs with a single TF is possible using promoters that are differently activated by TF dynamics. (A) Schematic representation of the construct. (B) Truth table showing the output of the circuit depending on synTF dynamics. 0 signifies low expression of the reporter protein; 1 signifies high expression of the reporter protein. (C) Bar plot showing the fold change in reporter expression for the indicated dynamics. The data represent the mean ± s.e.m of three biologically independent experiments.
NF-κB- and p53-responsive promoters can be classified as responsive to sustained or pulsatile signal based on some of the promoter features identified in this study
We asked whether the knowledge obtained using the simplified synthetic setup could be generalized also to natural mammalian promoters and TFs. Unfortunately, for such natural systems many parameters (e.g. RE or TATA box strength) have not been experimentally determined and can only be estimated using algorithms (37). The great majority of mammalian promoters lack a TATA box (38), which is the core promoter element we focused on in our study. Moreover, the relationship between the dynamics and the corresponding gene expression programs is established only for a few mammalian TFs (19,36,55). Finally, other mechanisms can be in place to decode TF dynamics, such as negative feedback and regulation at the protein level (56), which confound the interpretation of the results (e.g. certain genes might be activated better by pulses than sustained signal, but the promoter itself is not responsible for this). Despite these challenges, we went on to analyse two TFs for which the link between dynamics and target genes is known: nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (36,57–60) and p53 (18,19).
NF-κB regulates innate and adaptive immune responses as well as inflammation (61). Transient NF-κB pulses resulting from TNFα treatment have been shown to induce the expression of inflammatory response genes, while a sustained signal induced by lipopolysaccharides (LPS) leads to the expression of adaptive immune response genes (Figure 7A) (36,57–60,62–64). p53 is a stress sensor which initiates various antiproliferative programs to allow cells to repair the damage caused by the stress or, if unable to do so, to undergo apoptosis or enter into senescence, and for this reason it is a potent tumour suppressor (65). While γ-radiation induces pulses of p53, UV radiation leads to sustained p53; in turn, these different dynamics have been shown to trigger the expression of distinct set of genes: either those mediating cell cycle arrest and p53 regulation, or those causing apoptosis and senescence, respectively (Figure 7B) (19).
Figure 7.

Promoter features allow the classification of NF-κB- and p53-responsive promoters into sensitive and insensitive to dynamics. (A, B) Schematic representation of how different NF-κB (A) and p53 (B) dynamics lead to the expression of different target genes. (C, D) Plot showing NF-κB-responsive promoters according to the mean distance between the REs and the core promoter (TATA box for all promoters except TRAF1, for which the Inr was considered) normalized to the longest distance (x-axis) and the mean RE strength normalized to the strongest RE (y-axis) (C) and p53-responsive promoters according to the type of core promoter (x-axis) and the mean RE strength normalized to the strongest RE (y-axis) (D). Each dot represents a promoter, color-coded to indicate activation by pulsaltile (magenta) or sustained (violet) TF signal, as in (A). The size of the dots increases with the number of REs.
We wanted to see if we could distinguish promoters responding to pulses from promoters responding to sustained signal using some of the promoter features we found to be involved in TF dynamics decoding. We thought that the following features could be extracted from available databanks and previous studies: sequence, position and number of experimentally validated REs; position and sequence of the TATA box; distance between RE and TATA box. As for the strength of each RE, this is mostly unknown, but it could be estimated with algorithms (37). Additionally, for promoters lacking a TATA box, we could extract the position and sequence of the Inr, and calculate its distance from the RE. The Inr can be considered as a weak core promoter (66,67), which is in line with our observations that promoters built with this core promoter element show only background reporter levels.
Of the selected 11 NF-κB-responsive promoters, 10 have the TATA box as core promoter (Supplementary Table S6). For promoters with two or more of them, REs have different strengths. We calculated the mean RE strength and the mean distance between the REs and the TATA box (or the Inr, for the TRAF1 promoter). We then plotted the promoters based on these two features (Figure 7C). According to our understanding of how promoters decode TF dynamics, we would predict that promoters falling into the upper left quadrant would be activated also by pulses (that is, they would not distinguish dynamics), given that the coupling between TF-RE binding events and PIC assembly would be strong (short distance between RE and TATA box/Inr and strong REs). Promoters falling into the bottom right quadrant would, instead, be able to filter pulses out and would need a sustained TF signal, given that the coupling would be inefficient (longer distance between RE and TATA box/Inr, and weak REs). We found that the promoters clustered mostly in the two expected quadrants, with those known to respond to TNFα being in the upper left quadrant and those responding to LPS being in the bottom right one (Figure 7C). The same is seen when considering the distance between the last RE and the TATA box/Inr, with the exception of the TNF promoter (Supplementary Figure S9). For this promoter, however, it has been reported that additional regulation by the CREB protein is needed for the effective transcriptional induction by the last RE alone (68). This could explain why this promoter would still be able to filter pulses out.
Among the selected 11 p53-responsive promoters, only three possess a TATA box (Supplementary Table S7). Interestingly, we noticed that these promoters drive the expression of genes involved in cell cycle arrest or p53 regulation, which are known to respond to p53 pulses (19). Thus, it appears that, for p53, the type of core promoter (weak versus strong) correlates well with the ability of promoters to filter p53 pulses out or not. When using the mean RE strength as additional feature, we found that the two remaining promoters driving the expression of genes involved in cell cycle arrest or p53 regulation (XPC and PPM1D) clustered in the upper left quadrant, having very strong mean RE (Figure 7D). We cannot exclude that the coupling between p53 binding and PIC assembly be strong in these promoters despite their harbouring the Inr as core promoter, making them sensitive to pulses. Nonetheless, Purvis and colleagues showed that these two genes are actually more transcribed by sustained than pulsatile p53 (19). Our predictions are in line with these results. The fact that p53-responsive promoters responding to pulses do not have strong REs corroborates our conclusions that it is the interplay between TF binding and PIC assembly that is important for TF decoding rather than the mere affinity of the TF for its cognate DNA motif.
DISCUSSION
In this study, we combined experimental and computational methods to decipher how mammalian promoters decode TF dynamics. We opted for a synthetic biology approach: we studied a synthetic TF (synTF) and synthetic promoters, all made of well-known and characterized parts. Being synTF orthogonal to the mammalian cells, we expected it not to be subject to endogenous regulation, which isolates our system from all other cellular processes and makes the data interpretation reliable. Nonetheless, the TAD within synTF comes from VP16, a transcription factor of the Herpes simplex virus that infects the peripheral and central nervous systems in humans and that interacts with a multitude of human proteins involved in transcriptional regulations, such as acetyltransferases and general TFs (69). Therefore, a TF based on the VP16 TAD is suitable to study gene expression regulation in mammalian cells.
Working with synTF additionally consented us to be quantitative, knowing the affinities of the DBD and the TBP for the RE and the TATA box, respectively.
It was interesting for us to see that a two-fold decrease in binding affinity between the TBP and the TATA box had a measurable effect. In our study, we call the TATA box bound by the TBP with KD = 4 nM and the RE bound by synTF with KD = 5.64 nM weak, despite these values being per se still in the nanomolar range.
To impose different dynamics to synTF we used an optogenetic tool, LINuS (22), that allowed us to directly manipulate, with blue light, the nuclear concentration of synTF. We designed our experiments in a way that cells in all conditions would be exposed to the same total amount of light, permitting comparisons among the three investigated TF dynamics without differential interference of light.
To focus on TF dynamics, we decided to fix the cumulative TF levels, as previously done for p53 (19). Some promoters, like promoter p1 in our library, are, indeed, sensitive to cumulative TF levels, rather than dynamics, and having different values for this parameter would confound the data interpretation. Although an alternative way to obtain similar cumulative levels across conditions would have been to fix the experimental time and change the amplitude of the signal in the three conditions, we opted against this choice, because some promoters are sensitive to TF amplitude, which would have precluded the possibility to isolate only effects due to dynamics. Since promoters p1 and p2 are insensitive to synTF amplitude (Figure 2F, I), we performed in this case also the experiment fixing the time and lowering the amplitude for the sustained dynamics to obtain similar cumulative synTF levels as for the pulses (Supplementary Figure S2I). We found consistent results with those obtained with variable experimental time (Supplementary Figure S2J, K), thus we concluded that our choice to fix cumulative levels by changing experimental time is valid.
In parallel to detecting the reporter protein, we established an RNA visualization method compatible with living cells to more directly monitor promoter activity. Nascent RNA measurements allowed us to realize that promoters p2 and p3 distinguish dynamics (Figure 2G, J), although this is not seen at the protein level when the mRNA is efficiently translated (Figure 2H, K). The limitation of our setup––that relies on mVenus to visualize nascent RNA––is that we cannot take images in this channel during the dark phases of the pulsatile dynamics, because the illumination would lead to activation of LINuS within synTF and, consequently, to unwanted nuclear import and reporter gene expression. Despite this limitation, we expected the first visualization at the beginning of each blue light activation phase to be indicative of whether transcriptional shut down had occurred during the dark phase. Because nascent RNA levels after each dark incubation phase were similar to those obtained during blue light activation (Figure 2B, Supplementary Figure S2B, D, middle and right panels), we conclude that, at the population level, transcriptional shutdown during the phases in which synTF is in the cytoplasm does not occur. Looking at individual cells, we observed that some cells shut down, while others either don’t or do, but re-initiate RNA transcription during the dark phases (Supplementary Movie S1). The appearance of RNA foci during the phases in which the synTF is not nuclear, and therefore unbound from the REs, could be explained by transcription re-initiation events that do not involve the TF (70,71).
Interestingly, for some of the promoters, namely p2 and p4, we observed refractoriness (Supplementary Figure S2B and Figure 3B, left panels). The molecular mechanism behind this effect is not very well understood. It could be due to lack of GTFs locally available at the promoter to initiate PIC assembly. Refractoriness has been observed, for instance, in Neurospora crassa (72), in yeast for the GAL promoters (73) and in the Notch signaling pathway (74); thus, this phenomenon is observed in natural systems. It is noteworthy that we observed it also in our synthetic system.
Nucleosome positioning has been previously suggested to play a critical role in the ability of a promoter to decode TF dynamics (20). Clearly, the presence of a nucleosome at the promoter renders the coupling between TF binding events and PIC assembly inefficient, thus it is in line with our explanation of what makes a promoter sensitive to TF dynamics. Nonetheless, our data indicate that there are other ways to achieve this, independent of the chromatin status.
We found particularly interesting the observation that lowering the translation initiation efficiency amplifies the ability of promoter p2 to sense dynamics. This is explained considering that mRNA translation and degradation compete with one another, with high ribosome occupancy protecting mRNA from degradation (75). If the mRNA is inefficiently translated, higher mRNA concentrations will be required to improve the chances of successfully initiating translation on high enough a fraction of mRNA molecules before they are degraded.
While it was pleasing to see that we could use some of the features identified in this study using a synthetic biology setup to understand the response of natural promoters to TF dynamics (Figure 7), we are reluctant to conclude that it is possible to predict for every promoter in the human genome whether it will be responsive to TF dynamics or not, because natural systems are very complex and often have regulatory elements which we did/could not investigate. For instance, many promoters harbour response elements of several TFs, which could play a synergistic or antagonist role; there are multiple feedback loops that can influence how dynamics are read. For the Ras/ERK pathway, for example, it was shown that immediate-early genes (IEGs) respond better to ERK pulses of intermediate frequency than to sustained ERK (56). The mechanism that allows IEGs to filter out a sustained ERK signal involves negative feedback enacted by ERK negative regulators. Thus, while the promoter per se could be classified as one not sensitive to dynamics, the downstream gene could be activated better by pulsatile TF signal. Moreover, ∼45% of human promoters do not contain either the TATA box or the Inr (38,76). Since we did not study promoters with other core promoters than these two, we could not conclude anything for these promoters.
For synthetic biology applications, it is often desirable to activate different gene expression programs with a minimal set of biological parts. While it is possible to control two genes using distinct promoters responsive to different TFs, we show here we can use a single TF, as far as promoters differentially responding to its dynamics are employed (Figure 6C). At this stage, the maximum number of programs achievable is two –program A (proteins X and Y both highly expressed) and program B (protein X highly expressed, protein Y expressed at low levels)– because all the promoters in our library that respond to pulsatile synTF signals respond to the sustained signal, too. It would be advantageous to have promoters that respond much better to pulses than to sustained TF dynamics. In this case, a third program would be possible, namely protein Y highly expressed. While we demonstrated this principle here only with fluorescent proteins, if the proteins being controlled have a biological function, this circuit would allow achieving specific biological outputs employing a single TF. Natural signalling pathways exist for which certain target genes are more efficiently activated by pulsatile than sustained signal. We mentioned above the case of the Ras/ERK pathway, for which, due to the action of negative regulators, a stronger response to ERK pulses of intermediate frequency than to sustained ERK can be achieved (56). Our synthetic gene network does not comprise negative feedback; therefore, in our setup, promoters that are activated by pulses are also necessarily activated by sustained synTF signal. Our mathematical model, though, shows that it is possible to accumulate higher reporter protein levels for pulsatile than sustained synTF dynamics if synTF residing in the nucleus for long enough would repress, instead of activate the promoter (Supplementary Text andSupplementary Figure S10). Recently, it has been shown that a TF that activates gene expression when localized into small clusters becomes a repressor when localized into larger droplets (77). This work indicates that, in principle, it would be possible to turn synTF into a repressor when at high levels (that is, under sustained light illumination), if a droplet-forming mechanism is included.
Finally, it can be reasoned that the regulation of the interplay between specific TFs and GTFs may represent a strategy used by cells to not only distinguish TF dynamics, but also improve the specificity of the transcriptional response (78).
In summary, here we provided a mechanistic understanding of the features that render mammalian promoters sensitive to TF dynamics. Our results will help interpret how natural signalling pathways are activated by different dynamic inputs as well as pave the way to control complex biological functions using minimal circuits based on a single TF.
DATA AVAILABILITY
This study includes no data deposited in external repositories. Requests for materials should be directed to Barbara Di Ventura (barbara.diventura@uni-freiburg.de). All plasmids can be obtained under a Material Transfer Agreement.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Robert Grosse, Peter Walentek, Wolfgang Schamel and Johan Elf for critical reading of the manuscript, Jens Timmer and Clemens Kreutz for valuable input on the mathematical model, Mehmet A. Öztürk and all members of the Di Ventura lab for useful discussions. We thank Vladislav Verkhusha for donating plasmid pNLS-iRFP670 (Addgene plasmid #45466), Robert Singer for donating plasmids Pcr4-12xMBS-PBS and Ubc-NLS-HA-MCP-mVenusN-NLS-HA-PCP-mVenusC (Addgene plasmids #52984 and #52985), and Klaus Hahn for donating plasmid pTriEx-NTOM20-mVenus-Zdk2 (25) (Addgene plasmid #81011).
Contributor Information
Enoch B Antwi, Heidelberg Biosciences International Graduate School (HBIGS), University of Heidelberg, Heidelberg, Germany; Signalling Research Centres BIOSS and CIBSS, University of Freiburg, Freiburg, Germany; Faculty of Biology, Institute of Biology II, University of Freiburg, Freiburg, Germany.
Yassine Marrakchi, Signalling Research Centres BIOSS and CIBSS, University of Freiburg, Freiburg, Germany; Department of Computer Science, University of Freiburg, Freiburg, Germany.
Özgün Çiçek, Department of Computer Science, University of Freiburg, Freiburg, Germany.
Thomas Brox, Signalling Research Centres BIOSS and CIBSS, University of Freiburg, Freiburg, Germany; Department of Computer Science, University of Freiburg, Freiburg, Germany; BrainLinks-BrainTools, University of Freiburg, Freiburg, Germany.
Barbara Di Ventura, Signalling Research Centres BIOSS and CIBSS, University of Freiburg, Freiburg, Germany; Faculty of Biology, Institute of Biology II, University of Freiburg, Freiburg, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Federal Ministry of Education and Research (BMBF) [031L0079 to B.D.V.]; Excellence Initiative of the German Federal and State Governments BIOSS (Centre for Biological Signalling Studies; EXC-294); CIBSS (Center for Integrative Signalling Studies; EXC-2189). Funding for open access charge: house funds.
Conflict of interest statement. None declared.
REFERENCES
- 1. Buccitelli C., Selbach M.. mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 2020; 21:630–644. [DOI] [PubMed] [Google Scholar]
- 2. Hampsey M. Molecular genetics of the RNA polymerase II general transcriptional machinery. Microbiol. Mol. Biol. Rev. 1998; 62:465–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haberle V., Stark A.. Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol. 2018; 19:621–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roy A.L., Singer D.S.. Core promoters in transcription: old problem, new insights. Trends Biochem. Sci. 2015; 40:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zabidi M.A., Stark A.. Regulatory enhancer-core-promoter communication via transcription factors and cofactors. Trends Genet. 2016; 32:801–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kornberg R.D., Lorch Y.. Primary role of the nucleosome. Mol. Cell. 2020; 79:371–375. [DOI] [PubMed] [Google Scholar]
- 7. Kadonaga J.T. Regulation of RNA polymerase II transcription by sequence-specific DNA binding factors. Cell. 2004; 116:247–257. [DOI] [PubMed] [Google Scholar]
- 8. Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T.. The human transcription factors. Cell. 2018; 172:650–665. [DOI] [PubMed] [Google Scholar]
- 9. Zhu L., Huq E.. Mapping functional domains of transcription factors. Methods Mol. Biol. 2011; 754:167–184. [DOI] [PubMed] [Google Scholar]
- 10. Purvis J.E., Lahav G.. Encoding and decoding cellular information through signaling dynamics. Cell. 2013; 152:945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bartman C.R., Hsu S.C., Hsiung C.C.S., Raj A., Blobel G.A.. Enhancer regulation of transcriptional bursting parameters revealed by forced chromatin looping. Mol. Cell. 2016; 62:237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Petrascheck M., Escher D., Mahmoudi T., Verrijzer C.P., Schaffner W., Barberis A.. DNA looping induced by a transcriptional enhancer in vivo. Nucleic. Acids. Res. 2005; 33:3743–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moore J.W., Loake G.J., Spoel S.H.. Transcription dynamics in plant immunity. Plant Cell. 2011; 23:2809–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tootle T.L., Rebay I.. Post-translational modifications influence transcription factor activity: a view from the ETS superfamily. Bioessays. 2005; 27:285–298. [DOI] [PubMed] [Google Scholar]
- 15. Behar M., Barken D., Werner S.L., Hoffmann A.. The dynamics of signaling as a pharmacological target. Cell. 2013; 155:448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Behar M., Hoffmann A.. Understanding the temporal codes of intra-cellular signals. Curr. Opin. Genet. Dev. 2010; 20:684–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Muta Y., Matsuda M., Imajo M.. Divergent dynamics and functions of ERK MAP kinase signaling in development, homeostasis and cancer: lessons from fluorescent bioimaging. Cancers (Basel). 2019; 11:513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Batchelor E., Loewer A., Mock C., Lahav G.. Stimulus-dependent dynamics of p53 in single cells. Mol. Syst. Biol. 2011; 7:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Purvis J.E., Karhohs K.W., Mock C., Batchelor E., Loewer A., Lahav G.. p53 Dynamics control cell fate. Science. 2012; 336:1440–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hansen A.S., O’Shea E.K. Promoter decoding of transcription factor dynamics involves a trade-off between noise and control of gene expression. Mol. Syst. Biol. 2013; 9:704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li C., Cesbron F., Oehler M., Brunner M., Höfer T.. Frequency modulation of transcriptional bursting enables sensitive and rapid gene regulation. Cell Syst. 2018; 6:409–423. [DOI] [PubMed] [Google Scholar]
- 22. Niopek D., Benzinger D., Roensch J., Draebing T., Wehler P., Eils R., Di Ventura B.. Engineering light-inducible nuclear localization signals for precise spatiotemporal control of protein dynamics in living cells. Nat. Commun. 2014; 5:4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shcherbakova D.M., Verkhusha V.V.. Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat. Methods. 2013; 10:751–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu B., Chen J., Singer R.H.. Background free imaging of single mRNAs in live cells using split fluorescent proteins. Sci. Rep. 2014; 4:3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang H., Vilela M., Winkler A., Tarnawski M., Schlichting I., Yumerefendi H., Kuhlman B., Liu R., Danuser G., Hahn K.M.. LOVTRAP: an optogenetic system for photoinduced protein dissociation. Nat. Methods. 2016; 13:755–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A., Smith H.O.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009; 6:343–345. [DOI] [PubMed] [Google Scholar]
- 27. De Bruin L., Maddocks J.H.. CgDNAweb: a web interface to the cgDNA sequence-dependent coarse-grain model of double-stranded DNA. Nucleic Acids Res. 2018; 46:W5–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Delano W.L. The PyMOL Molecular Graphics System. 2002; [Google Scholar]
- 29. Çiçek Ö., Marrakchi Y., Boasiako Antwi E., Di Ventura B., Brox T.. Recovering the imperfect: cell segmentation in the presence of dynamically localized proteins. Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics). 2020; 12446:Springer Science and Business Media Deutschland GmbH; 85–93. [Google Scholar]
- 30. He K., Gkioxari G., Dollár P., Girshick R.. Mask R-CNN. IEEE Transactions on Pattern Analysis and Machine Intelligence. 2017; 42:386–397. [DOI] [PubMed] [Google Scholar]
- 31. Ronneberger O., Fischer P., Brox T.. U-Net: convolutional networks for biomedical image segmentation. MICCAI. 2015; 9351:234–241. [Google Scholar]
- 32. Kendall A., Gal Y.. What uncertainties do we need in Bayesian deep learning for computer vision. 2017; arXiv doi:05 October 2017, preprint: not peer reviewedhttps://arxiv.org/abs/1703.04977.
- 33. Ilg E., Çiçek Ö., Galesso S., Klein A., Makansi O., Hutter F., Brox T.. Uncertainty estimates and multi-hypotheses networks for optical flow. 2018; arXiv doi:20 December 2018, preprint: not peer reviewedhttps://arxiv.org/abs/1802.07095.
- 34. Schneider C.A., Rasband W.S., Eliceiri K.W.. NIH image to ImageJ: 25 years of image analysis. Nat. Methods. 2012; 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rullan M., Benzinger D., Schmidt G.W., Milias-Argeitis A., Khammash M.. An optogenetic platform for real-time, single-cell interrogation of stochastic transcriptional regulation. Mol. Cell. 2018; 70:745–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Werner S.L., Barken D., Hoffmann A.. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 2005; 309:1857–1861. [DOI] [PubMed] [Google Scholar]
- 37. Steinhaus R., Robinson P.N., Seelow D.. FABIAN-variant: predicting the effects of DNA variants on transcription factor binding. Nucleic Acids Res. 2022; 50:W322–W329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang C., Bolotin E., Jiang T., Sladek F.M., Martinez E.. Prevalence of the initiator over the TATA box in human and yeast genes and identification of DNA motifs enriched in human TATA-less core promoters. Gene. 2007; 389:52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Howard-Flanders P., Boyce R.P., Theriot L.. Three loci in Escherichia coli K-12 that control the excision of pyrimidine dimers and certain other mutagen products from DNA. Genetics. 1966; 53:1119–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Triezenberg S.J., Kingsbury R.C., McKnight S.L.. Functional dissection of VP16, the trans-activator of herpes simplex virus immediate early gene expression. Genes Dev. 1988; 2:718–729. [DOI] [PubMed] [Google Scholar]
- 41. Harper C.V., Finkenstädt B., Woodcock D.J., Friedrichsen S., Semprini S., Ashall L., Spiller D.G., Mullins J.J., Rand D.A., Davis J.R.E.et al.. Dynamic analysis of stochastic transcription cycles. PLoS Biol. 2011; 9:e1000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nolis I.K., McKay D.J., Mantouvalou E., Lomvardas S., Merika M., Thanos D.. Transcription factors mediate long-range enhancer-promoter interactions. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:20222–20227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ringrose L., Chabanis S., Angrand P.O., Woodroofe C., Stewart A.F.. Quantitative comparison of DNA looping in vitro and in vivo: chromatin increases effective DNA flexibility at short distances. EMBO J. 1999; 18:6630–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hahn S., Buratowski S., Sharp P.A., Guarente L.. Yeast TATA-binding protein TFIID binds to TATA elements with both consensus and nonconsensus DNA sequences. Proc. Natl. Acad. Sci. U.S.A. 1989; 86:5718–5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang A.P.P., Pigli Y.Z., Rice P.A.. Structure of the LexA–DNA complex and implications for SOS box measurement. Nature. 2010; 466:883–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lowary P.T., Widom J.. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998; 276:19–42. [DOI] [PubMed] [Google Scholar]
- 47. Haimov O., Sinvani H., Dikstein R.. Cap-dependent, scanning-free translation initiation mechanisms. Biochim. Biophys. Acta - Gene Regul. Mech. 2015; 1849:1313–1318. [DOI] [PubMed] [Google Scholar]
- 48. Pestova T.V., Kolupaeva V.G.. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 2002; 16:2906–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hinnebusch A.G. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. 2011; 75:434–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hinnebusch A.G. The scanning mechanism of eukaryotic translation initiation. Annu. Rev. Biochem. 2014; 83:779–812. [DOI] [PubMed] [Google Scholar]
- 51. Araujo P.R., Yoon K., Ko D., Smith A.D., Qiao M., Suresh U., Burns S.C., Penalva L.O.F.. Before it gets started: regulating translation at the 5′ UTR. Comp. Funct. Genomics. 2012; 2012:475731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leppek K., Das R., Barna M.. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018; 19:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mohan R.A., van Engelen K., Stefanovic S., Barnett P., Ilgun A., Baars M.J.H., Bouma B.J., Mulder B.J.M., Christoffels V.M., Postma A.V.. A mutation in the Kozak sequence of GATA4 hampers translation in a family with atrial septal defects. Am. J. Med. Genet. Part A. 2014; 164:2732–2738. [DOI] [PubMed] [Google Scholar]
- 54. De Angioletti M., Lacerra G., Sabato V., Carestia C.. β+45 G → C: a novel silent β-thalassaemia mutation, the first in the Kozak sequence. Br. J. Haematol. 2004; 124:224–231. [DOI] [PubMed] [Google Scholar]
- 55. Marshall C.J. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995; 80:179–185. [DOI] [PubMed] [Google Scholar]
- 56. Wilson M.Z., Ravindran P.T., Lim W.A., Toettcher J.E.. Tracing information flow from erk to target gene induction reveals mechanisms of dynamic and combinatorial control. Mol. Cell. 2017; 67:757–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pulendran B., Kumar P., Cutler C.W., Mohamadzadeh M., Dyke T.V., Banchereau J.. Lipopolysaccharides from distinct pathogens induce different classes of immune responses In vivo. J. Immunol. 2001; 167:5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Covert M.W., Leung T.H., Gaston J.E., Baltimore D.. Achieving stability of lipopolysaccharide-induced NF-κb activation. Science. 2005; 309:1854–1857. [DOI] [PubMed] [Google Scholar]
- 59. Hoffmann A., Levchenko A., Scott M.L., Baltimore D.. The iκb-NF-κb signaling module: temporal control and selective gene activation. Science. 2002; 298:1241–1245. [DOI] [PubMed] [Google Scholar]
- 60. Nelson D.E., Ihekwaba A.E.C., Elliott M., Johnson J.R., Gibney C.A., Foreman B.E., Nelson C., See V., Horton C.A., Spiller D.G.et al.. Oscillations in NF-κb signaling control the dynamics of gene expression. Science. 2004; 306:704–708. [DOI] [PubMed] [Google Scholar]
- 61. Liu T., Zhang L., Joo D., Sun S.C.. NF-κb signaling in inflammation. Signal Transduct. Target. Ther. 2017; 2:17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lee T.K., Denny E.M., Sanghvi J.C., Gaston J.E., Maynard N.D., Hughey J.J., Covert M.W.. A noisy paracrine signal determines the cellular NF-κb response to lipopolysaccharide. Sci. Signal. 2009; 2:ra65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sung M.H., Salvatore L., De Lorenzi R., Indrawan A., Pasparakis M., Hager G.L., Bianchi M.E., Agresti A.. Sustained oscillations of NF-κb produce distinct genome scanning and gene expression profiles. PLoS One. 2009; 4:e7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tay S., Hughey J.J., Lee T.K., Lipniacki T., Quake S.R., Covert M.W.. Single-cell NF-B dynamics reveal digital activation and analogue information processing. Nature. 2010; 466:267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kennedy M.C., Lowe S.W.. Mutant p53: it's not all one and the same. Cell Death Differ. 2022 295. 2022; 29:983–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Emami K.H., Navarre W.W., Smale S.T.. Core promoter specificities of the Sp1 and VP16 transcriptional activation domains. Mol. Cell. Biol. 1995; 15:5906–5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Javahery R., Khachi A., Lo K., Zenzie-Gregory B., Smale S.T.. DNA sequence requirements for transcriptional initiator activity in mammalian cells. Mol. Cell. Biol. 1994; 14:116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yao J., Mackman N., Edgington T.S., Fan S.T.. Lipopolysaccharide induction of the tumor necrosis factor-α promoter in human monocytic cells: regulation by egr-1, c-jun, and NF-κb transcription factors. J. Biol. Chem. 1997; 272:17795–17801. [DOI] [PubMed] [Google Scholar]
- 69. Hirai H., Tani T., Kikyo N.. Structure and functions of powerful transactivators: VP16, MyoD and FoxA. Int. J. Dev. Biol. 2011; 54:1589–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yean D., Gralla J.D.. Transcription reinitiation rate: a potential role for TATA box stabilization of the TFIID:TFIIA:DNA complex. Nucleic. Acids. Res. 1999; 27:831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Joo Y.J., Ficarro S.B., Soares L.M., Chun Y.J., Marto J.A., Buratowski S.. Downstream promoter interactions of TFIID tafs facilitate transcription reinitiation. Genes Dev. 2017; 31:2162–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cesbron F., Oehler M., Ha N., Sancar G., Brunner M.. Transcriptional refractoriness is dependent on core promoter architecture. Nat. Commun. 2015; 6:6753. [DOI] [PubMed] [Google Scholar]
- 73. Hsu C., Scherrer S., Buetti-Dinh A., Ratna P., Pizzolato J., Jaquet V., Becskei A.. Stochastic signalling rewires the interaction map of a multiple feedback network during yeast evolution. Nat. Commun. 2012 31. 2012; 3:682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Viswanathan R., Hartmann J., Pallares Cartes C., De Renzis S.. Desensitisation of Notch signalling through dynamic adaptation in the nucleus. EMBO J. 2021; 40:e107245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jia L., Mao Y., Ji Q., Dersh D., Yewdell J.W., Qian S.B.. Decoding mRNA translatability and stability from the 5′ UTR. Nat. Struct. Mol. Biol. 2020; 27:814–821. [DOI] [PubMed] [Google Scholar]
- 76. Ngoc L.V., Cassidy C.J., Huang C.Y., Duttke S.H.C., Kadonaga J.T.. The human initiator is a distinct and abundant element that is precisely positioned in focused core promoters. Genes Dev. 2017; 31:6–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chong S., Graham T.G.W., Dugast-Darzacq C., Dailey G.M., Darzacq X., Tjian R.. Tuning levels of low-complexity domain interactions to modulate endogenous oncogenic transcription. Mol. Cell. 2022; 82:2084–2097. [DOI] [PubMed] [Google Scholar]
- 78. Wunderlich Z., Mirny L.A.. Different gene regulation strategies revealed by analysis of binding motifs. Trends Genet. 2009; 25:434–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study includes no data deposited in external repositories. Requests for materials should be directed to Barbara Di Ventura (barbara.diventura@uni-freiburg.de). All plasmids can be obtained under a Material Transfer Agreement.