

Abstract

Following a single photon VUV absorption, the N2 molecule dissociates into distinct channels leading to N atoms of different reactivities. The optically accessible singlets are bound, and dissociation occurs through spin–orbit induced transfer to the triplets. There is a forest of coupled electronic states, and we here aim to trace a path along the nonadiabatic couplings toward a particular exit channel. To achieve this, we apply a time-reversed quantum dynamical approach that corresponds to a dissociation running back. It begins with an atom–atom relative motion in a particular product channel. Starting with a Gaussian wave packet at the dissociation region of N2 and propagating it backward in time, one can see the population transferring among the triplets due to a strong nonadiabatic interaction between these states. Simultaneously, the optically active singlets get populated because of spin–orbit coupling to the triplets. Thus, backward propagation traces the nonradiative association of nitrogen atoms.
Even for the simpler diatomic molecules, there is a maze of higher energy excited electronic states.1 High resolution spectroscopy has made major steps in understanding the structure of the stationary states.2,3 But the dynamics of these states is still a subject of active research; see, for example, the literature4−8 and, of course, the very elegant attosecond studies of H2 and HD and their ions.9,10 There have been corresponding developments in theoretical and computational dynamics.11−14 Two dimensional and higher order spectroscopy has also made significant contributions.15,16 Laser methods17−19 have enabled a detailed analysis of what are the exit channels. Toward these developments, we discuss a complementary computational approach to photodissociation dynamics. It is here applied to molecular nitrogen, N2, in the VUV region,20−23 for which very detailed experimental studies of the branching into different electronic exit channels are available.24−26
The singlet 1Σu+ states of N2 that are optically accessible by one photon VUV spectroscopy are energetically not able to dissociate at these energies. Rather, the dissociating states are of higher multiplicity, primarily triplets and quintets. They are accessed by spin–orbit coupling from the singlet states.27−29 The states of given symmetry and multiplicity are mixed by strong nonadiabatic terms.30 The considerable spatial and state selectivity of the spin–orbit coupling; see, for example refs (2 and 31), is displayed for N2 in ref (32) and here in Scheme 1.
Scheme 1. Spin–Orbit Couplings between the Electronic States of N2 Included in the Hamiltonian.

Singlet 1Σu+ states are coupled to triplet 3Σu and 3Πu (1st order coupling), the triplets 3Σu– and 3Πu are coupled to one another and to quintet 5Πu (2nd order coupling). The magnetic quantum number for each electronic state, mS, is indicated in parentheses. The different columns are for different spin multiplicities.
The experiments24−26 that resolve the dissociation into final channels clearly show that the branching fraction into each channel depends on the energy and/or the mass in a distinctly nonmonotonic fashion. Furthermore, it makes a difference if the initially optically excited state has a Σ or a Π character and, for each symmetry, if it has primarily a valence excited or a Rydberg character. The stationary singlet states are often a nonadiabatic mixture of these two bonding characteristics,20,30 and typically one is more dominant.
To trace the exit dynamics we here perform a time-reversed propagation of the dissociating N2 molecule. Starting from a state localized on a particular exit potential and integrating it backward in time. This highlights the role of the doorways to the individual exit channels. The essential technical implementation is as follows. Let ψ(R, t) be a wave function that is propagated forward in time: iℏ∂ψ(R, t)/∂t = Hψ(R, t). Next, take the complex conjugate of the Schrödinger equation iℏ∂ψ*(R, t)/∂(−t) = Hψ*(R, t) where the Hamiltonian is Hermitian. Changing t to –t shows that ψ*(R, −t) is a wave function propagating backward in time,33
| 1 |
Explicitly, for a diatomic molecule already fairly well separated into atoms such that the potential energy is more or less flat, we represent the initial state for the propagation reversed in time as a Gaussian wave packet along the interatomic distance R of width σR. Following the notation of Heller,34,35 we have a parameter α that specifies the width, Im(α) = 1/4σR, of the coherent distribution of momenta about its mean. After taking the complex conjugate of the Gaussian wave function
| 2 |
We see that the sign of the mean momentum ⟨P⟩ has been reversed. This is closely analogous to what one would do to propagate backward in time in a computation using classical dynamics. The three time-independent parameters in the Gaussian wave function, ⟨P⟩, ⟨R⟩ and α, determine the mean energy of the initial state for the backward propagation and its coherent width as follows. ⟨P⟩2/2μ is the exit kinetic energy, the total energy minus the threshold energy of that exit channel, and μ is the reduced mass. The wider is the Gaussian in R, the narrower is its distribution in momentum about the mean. The specific experiments we aim to simulate resolve different vibrational singlet states so their coherent width in energy needs to be far less than a vibrational spacings in the singlets. The Gaussian begins its journey on the flat part of the exit potential, so we need that ⟨R⟩, the initial mean displacement of the Gaussian, is chosen to be significantly larger than the width σR. In the following we apply these ideas to the nonradiative recombination of a pair of N atoms.
The VUV photodissociation of N2 is indirect and proceeds through the triplet and quintet electronic states.3,24,27,36−38 We here consider three optically accessible bound singlet 1Σu+ and six dissociative states: one 3Σu, four 3Πu and one 5Πu electronic states (Figure 1A). This set of states was defined based on our recent computational simulation32 of the experimentally measured branching fractions into N(4S3/2) + N(2DJ) and N(4S3/2) + N(2PJ) product channels shown with the blue and orange arrows in Figure 1A, respectively. The triplet 3Πu states have been known to be the primary channels for the photodissociation of the N2 molecule.27,36,37 A special case is at an excitation energy of 110,144 cm–1 where the dissociation of the triplet 3Πu states is particularly slow and the contribution of the 13Σu– and 25Πu electronic states is found to be dominant.32
Figure 1.

(A) Potential energy curves of the optically accessible singlet 1Σu+ (in black), one 3Σu, four 3Πu, and one 5Πu adiabatic electronic states of N2 as functions of the internuclear distance R. Different electronic states are color coded as shown in the figure. The computed potentials are shifted in energy by 850 cm–1 to be in agreement with high resolution spectra as reported in.20 The arrows indicate two dissociation channels to N(4S3/2) + N(2DJ) (channel I, blue) and N(4S3/2) + N(2PJ) (channel II, orange). (B) Nonadiabatic coupling terms among singlet 1Σu+ states (top panel in B) and triplet 3Πu states (bottom panel in B). (C) Spin–orbit coupling terms between triplet 3Πu states and 11Σu states (top panel in C) and 21Σu+ states (bottom panel in C).
Each triplet 3Πu state is degenerate because of its spin multiplicity and its 2-fold spatial degeneracy due to its cylindrical symmetry about the molecular axis. Of the three degenerate spin states only two, mS = +1 and mS = −1, can be relevant here because the spin–orbit coupling from the singlet 1Σu+ states does not access the state of zero magnetic quantum number. Similar comments apply to the quintet 5Πu state. Scheme 1 shows the spin–orbit coupling pattern for the states included in our basis for the backward propagation. In terms of orders of perturbation theory, the singlet states that are optically connected to the ground state are shown as coupled to first order by the spin–orbit terms to triplet states. In second order, the triplet states are spin–orbit coupled among themselves and to the quintet state.
Technically,
the computations backward in time start from a Gaussian
wave packet localized in the exit channel region on a grid of N–N
internuclear distance R. We mainly examine time-reversed
propagation starting from two different electronic states, 13Πu or 23Πu. For each state of a given total energy,
we here take a mixed (meaning not coherent) state where the four degenerate
states have each the same amplitude of 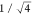 . We illustrate the time evolution in the
population distribution among the coupled triplet and singlet states
in Figure 2.
. We illustrate the time evolution in the
population distribution among the coupled triplet and singlet states
in Figure 2.
Figure 2.

Backward propagation all the way to the singlets. (A, B) Population of triplet 3Πu states along the grid at early time (t = 200 fs) after an initial 13Πu (A, top row) or, independently, a23Πu (B, top row) state of the same total energy (E = 113,551 cm–1) and the same width in energy (σE = 90 cm–1) start moving back to the region of interactions. Singlet 1Σu+ states (A and B, bottom rows) become populated due to slow spin–orbit induced transfer from the triplets. (C, D) Population redistribution of triplet 3Πu (top rows) and 1Σu singlet states (bottom rows) during time-reversed propagation at longer time (t = 1000 fs) evolve to a very similar triplet and singlet components. Panels E–H show a corresponding example for the total energy of 114,538 cm–1.
Parts A and B (left top panel) of Figure 2 show the early time dynamics when the backward moving initial triplet state reaches the region of interactions, i.e., when the internuclear distance is less than 5 au. At this time the tail of the initial Gaussian wave packet passes the potential energy barriers and transfers population to other 3Πu triplet electronic states which are coupled by strong nonadiabatic interaction (Figure 1B). The singlet and triplet states are coupled by weak spin–orbit terms (Figure 1C) and therefore the singlet 1Σu+ states are populated from the triplet 3Πu during the time-reversed propagation. This can be seen from the localization of the population in these states along R in Figure 2, parts A and B (computed for 200 fs), and in movies of the dynamics that are included in the Supporting Information.
Propagating an initial triplet state that is narrow in energy makes it possible to access singlet states that correspond to a particular vibrational level of the adiabatic singlet potential. The backward time evolution of the two initial wave packets is driven by sequence of nonadiabatic interactions between the triplets and so are not identical at the early times, compare parts A and B of Figure 2. However, at longer times both initial triplet electronic states, 13Πu and 23Πu of the same total energy lead back to the singlet and triplet states of a similar character as shown in parts C and D of Figure 2. The final distribution along R is similar to the eigenstate of similar energy; see Figure S1 of the Supporting Information. The time evolution of a Rydberg state is shown in Figure 2E–H: the initial redistribution of population is similar to the one shown in Figure 2, parts A and B. Also for this incoming state the distribution at longer time is similar to the eigenstate of the same energy; see Figure S2 of the Supporting Information.
By comparing the localization in the singlet state in Figure 2, parts C and D, one can notice that there is about 50 times more population in the singlet state for the 23Πu initial state then for the 13Πu one. The two exit channels correspond to N atoms in different electronic states. So, the different yield in N atom recombination is consistent with the expectation of different reactivity for different N atomic states. Therefore, the time-reversed dynamics performed at a particular total energy of the molecule shows the pathway of the electronic system to each channel and the most probable channel for dissociation. More on this in the discussion of Figure 3, below.
Figure 3.

Branching fraction into N(4S3/2) + N(2PJ) (channel II) computed with backward propagation (red sticks) versus experimental data of Song et al.25 (black open circles). The dynamics at the energies of 113,551 and 114,538 cm–1 that have a rather different branching ratio are shown in detail in Figure 2.
The experiments of Walter et al.24 and of Song et al.25 showed the nonmonotonic energy dependence of the photodissociation branching ratios of the N2 molecule. This experimental result is a laser-induced dynamics at a well-defined wavelength starting from the ground electronic state. Our backward dynamical computation was used to account for the branching as due to the relative reactivity of states of different channels as shown in Figure 3. To quantitatively determine the nonradiative recombination efficiency, we examined the time profile of the population in the singlet states as produced by the backward propagation from states in either channels I or II. The evolution backward in time shows an early induction period where population is exchanged followed by a longer time stable population of the singlet states. This can be seen in detail in the movies that are part of the Supporting Information. A particular example is shown in Figure 2, panels C, D, G, and H. We use this distribution to determine the magnitude of the singlet state produced by the recombination. Thereby we determine the branching ratio through the backward computation, where Figure 3 shows a comparison with experiment.
In the energy range from 108,000 to 116,000 cm–1 both dissociation channels I and II are open. The first peak into the channel II is at 110,144 cm–1 and it is related to a very long predissociation lifetime of the singlet state24,32 which allows the triplet 13Σu– and quintet 25Πu states to contribute to the photodissociation. According to selection rules,21Σu and 3Σu– are coupled by the LSZ spin–orbit term; see Scheme 1. However, the population transfer between 3Σu and 1Σu+ is less efficient than that between 3Πu and 1Σu. Only when the lifetime of 1Σu+ is rather long, a few hundred nanoseconds, the accumulation due to transfer from 3Σu to 1Σu+ can be noticeable.
A significant rise of dissociation into channel II occurs between 112,000 and 114,500 cm–1 which is between two avoiding crossing regions: the lower is between 23Πu and 33Πu at 4 au and the higher one is between 33Πu and 43Πu at 3.5 au (Figure 1A). Here the singlets become resonant with the repulsive triplet potential formed by 23Πu, 33Πu, and 43Πu states (Figure 1A). An example of population distribution can be seen in Figure 2A–D.
The shape of potential energy curves of 3Πu states guides the wave packet. A drastic change in the branching fractions at 114,500 cm–1 is induced by trapping of the triplets’ population in the shallow well at 3 au (see the potential of 33Πu in Figure 1A). Figure 2E–2H shows both singlets and triplets populated at this energy during backward dynamics. Higher in energy, after 115,000 cm–1, the singlets are in resonance with the outgoing states of the repulsive triplet potential.
To summarize, we presented a complementary computational approach that proceeds backward in time from the exit region of the products of the photodissociation into the interaction region. In the backward direction, we start from a high energy electronically excited state so that the energy acquisition in the forward direction by optical excitation from the ground state need not be simulated. The reverse motion at a sharp energy begins with a broad Gaussian wave packet, and its propagation requires only a relatively short time interval to populate all the way back to the singlets. The Supporting Information provides access to movies showing the time evolution of the density of the vibrational state in two different exit channels that are also shown for two time points in Figure 2. Experiments and also our simulations forward in time32 demonstrate that the branching fraction into the product channels varies nonmonotonically with the total energy. Figure 3 proves that this is equally the result when we integrate backward. Last, the relative efficiency of nonradiative recombination of N atoms in different electronic states at the same total energy is closely matched by the branching ratios for the photodissociation.
The delayed unimolecular dissociation of electronically excited N2 means a longer than simply expected duration for the recombination of two N atoms. It will be a “sticky” collision, and thereby, such mechanisms as a third body induced recombination can be more facile. Computationally our approach allows a more efficient route to exploring the dynamics in the exit region.
Quantum Dynamical Simulations
We use the time-dependent Schrödinger equation to compute the quantum dynamics for 9 excited electronic states of definite multiplicity. The triplet and quintet states are degenerate because of multiplicity or spatial symmetry. The equation of motion for the amplitudes Cn, j* = Ψn(Rj) at a given electronic state n and grid point j are the solutions of
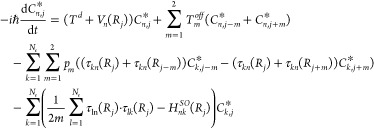 |
3 |
Here Td and Toff are diagonal and off-diagonal kinetic energy terms, respectively, and Vn(R) denotes the potential energy of an electronic state. The nonadiabatic couplings τkn(R) between electronic states n and k are scaled by the finite difference momentum terms, pm. We use the five-point finite difference method39 to approximate the momentum and kinetic energy terms for the wave function defined on the grid. The total length of the internuclear grid is 67 au with spacing between grid points is 5 × 10–3 au. The time step in the propagation is 10–4 fs.
The initial state is a Gaussian specified in eq 1, where ⟨R⟩ is 37 au with a width of 4.0 au, which is equivalent to σE of 90 cm–1.
Acknowledgments
The authors thank Dr. Ksenia Komarova for fruitful discussions on the methodological and technical aspects of the time-reversed propagation and quantum chemical calculations. N.G. and R.D.L. acknowledge financial support by the NSF–BSF Grant No. 2019722 Astronomy and Astrophysics. F.R. acknowledges the support of the Fonds National de la Recherche (F.R.S.-FNRS, Belgium), #T0205.20.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.3c00666.
Comparison between states obtained from diagonalization and dynamics for two examples, total energy of 113,551 and 114,538 cm–1. (PDF)
Movie for time-reversed propagation at total energy of 113,551 cm–1 using two initial states, 13Πu and 23Πu (MP4)
Movie for time-reversed propagation at total energy of 114,538 cm–1 using (MP4)
The authors declare no competing financial interest.
Supplementary Material
References
- Gilmore F. R. Potential Energy Curves for N2, NO, O2 and Corresponding Ions. J. Quant. Spectrosc. Radiat. Transfer 1965, 5, 369–389. 10.1016/0022-4073(65)90072-5. [DOI] [Google Scholar]
- Lefebvre-Brion H.; Field R. W.. The Spectra and Dynamics of Diatomic Molecules; Elsevier: Amsterdam, 2004. [Google Scholar]
- Li X.; Heays A. N.; Visser R.; Ubachs W.; Lewis B. R.; Gibson S. T.; Van Dishoeck E. F. Photodissociation of Interstellar N2. Astron. Astrophys. 2013, 555, A14. 10.1051/0004-6361/201220625. [DOI] [Google Scholar]
- Li W.; Jaroń-Becker A. A.; Hogle C. W.; Sharma V.; Zhou X.; Becker A.; Kapteyn H. C.; Murnane M. M. Visualizing Electron Rearrangement in Space and Time during the Transition from a Molecule to Atoms. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20219–20222. 10.1073/pnas.1014723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakitzis T. P.; Kandel S. A.; Zare R. N. Photolysis of ICl Causes Mass-Dependent Interference in the Cl(2P3/2) Photofragment Angular Momentum Distributions. J. Chem. Phys. 1998, 108, 8291–8294. 10.1063/1.476257. [DOI] [Google Scholar]
- Warrick E. R.; Cao W.; Neumark D. M.; Leone S. R. Probing the Dynamics of Rydberg and Valence States of Molecular Nitrogen with Attosecond Transient Absorption Spectroscopy. J. Phys. Chem. A 2016, 120, 3165–3174. 10.1021/acs.jpca.5b11570. [DOI] [PubMed] [Google Scholar]
- Trabattoni A.; Klinker M.; González-Vázquez J.; Liu C.; Sansone G.; Linguerri R.; Hochlaf M.; Klei J.; Vrakking M. J. J.; Martín F.; et al. Mapping the Dissociative Ionization Dynamics of Molecular Nitrogen with Attosecond Time Resolution. Phys. Rev. X 2015, 5, 041053 10.1103/PhysRevX.5.041053. [DOI] [Google Scholar]
- Eckstein M.; Yang C. H.; Kubin M.; Frassetto F.; Poletto L.; Ritze H. H.; Vrakking M. J. J.; Kornilov O. Dynamics of N2 Dissociation upon Inner-Valence Ionization by Wavelength-Selected XUV Pulses. J. Phys. Chem. Lett. 2015, 6, 419–425. 10.1021/jz5025542. [DOI] [PubMed] [Google Scholar]
- Sansone G.; Kelkensberg F.; Pérez-Torres J. F.; Morales F.; Kling M. F.; Siu W.; Ghafur O.; Johnsson P.; Swoboda M.; Benedetti E.; et al. Electron Localization Following Attosecond Molecular Photoionization. Nature 2010, 465, 763–766. 10.1038/nature09084. [DOI] [PubMed] [Google Scholar]
- Kling M. F.; Siedschlag C.; Verhoef A. J.; Khan J. I.; Schultze M.; Uphues T.; Ni Y.; Uiberacker M.; Drescher M.; Krausz F.; et al. Control of Electron Localization in Molecular Dissociation. Science 2006, 312, 246–248. 10.1126/science.1126259. [DOI] [PubMed] [Google Scholar]
- Nisoli M.; Decleva P.; Calegari F.; Palacios A.; Martín F. Attosecond Electron Dynamics in Molecules. Chem. Rev. 2017, 117, 10760–10825. 10.1021/acs.chemrev.6b00453. [DOI] [PubMed] [Google Scholar]
- Attosecond Molecular Dynamics, 1st ed.; Vrakking M. J. J., Lepine F., Eds.; Royal Society of Chemistry: London, 2018. [Google Scholar]
- Cistaro G.; Malakhov M.; Esteve-Paredes J. J.; Uría-Álvarez A. J.; Silva R. E. F.; Martín F.; Palacios J. J.; Picón A. Theoretical Approach for Electron Dynamics and Ultrafast Spectroscopy (EDUS). J. Chem. Theory Comput. 2023, 19, 333–348. 10.1021/acs.jctc.2c00674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann Y.; Hollas D.; Curchod B. F. E. Extending the Applicability of the Multiple-Spawning Framework for Nonadiabatic Molecular Dynamics. J. Phys. Chem. Lett. 2022, 13, 12011–12018. 10.1021/acs.jpclett.2c03295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrick E. R.; Fidler A. P.; Cao W.; Bloch E.; Neumark D. M.; Leone S. R. Multiple Pulse Coherent Dynamics and Wave Packet Control of the N2 A″ 1Σ+g Dark State by Attosecond Four-Wave Mixing. Faraday Discuss. 2018, 212, 157–174. 10.1039/C8FD00074C. [DOI] [PubMed] [Google Scholar]
- Ramasesha K.; Leone S. R.; Neumark D. M. Real-Time Probing of Electron Dynamics Using Attosecond Time-Resolved Spectroscopy. Annu. Rev. Phys. Chem. 2016, 67, 41–63. 10.1146/annurev-physchem-040215-112025. [DOI] [PubMed] [Google Scholar]
- Zare R. N.Angular Momentum: Understanding Spatial Aspects in Chemistry and Physics, 1st ed.; Wiley-Interscience: New York, 1991. [Google Scholar]
- Bernstein R. B.Chemical Dynamics via Molecular Beam and Laser Techniques; Oxford University Press: New York, 1982. [Google Scholar]
- Hu W.; Liu Y.; Luo S.; Li X.; Yu J.; Li X.; Sun Z.; Yuan K. J.; Bandrauk A. D.; Ding D. Coherent Interference of Molecular Electronic States in NO by Two-Color Femtosecond Laser Pulses. Phys. Rev. A 2019, 99, 011402 10.1103/PhysRevA.99.011402. [DOI] [Google Scholar]
- Spelsberg D.; Meyer W. Dipole-Allowed Excited States of N2: Potential Energy Curves, Vibrational Analysis, and Absorption Intensities. J. Chem. Phys. 2001, 115, 6438–6449. 10.1063/1.1400139. [DOI] [Google Scholar]
- Little D. A.; Tennyson J. An Ab Initio Study of Singlet and Triplet Rydberg States of N2. J. Phys. B At. Mol. Opt. Phys. 2013, 46, 145102. 10.1088/0953-4075/46/14/145102. [DOI] [Google Scholar]
- Adamson S. O.; Kuverova V. V.; Ozerov G. K.; Golubkov G. V.; Nabiev S. S.; Golubkov M. G. Ab Initio Calculation of the Lowest Singlet and Triplet Excited States of the N2 Molecule. Russ. J. Phys. Chem. B 2018, 12, 620–631. 10.1134/S1990793118040024. [DOI] [Google Scholar]
- Qin Z.; Zhao J.; Liu L. Radiative Transition Probabilities between Low-Lying Electronic States of N2. Mol. Phys. 2019, 117, 2418–2433. 10.1080/00268976.2018.1562579. [DOI] [Google Scholar]
- Walter C. W.; Cosby P. C.; Helm H. N(4S0), N(2D0), and N(2P0) Yields in Predissociation of Excited Singlet States of N2. J. Chem. Phys. 1993, 99, 3553–3561. 10.1063/1.466152. [DOI] [Google Scholar]
- Song Y.; Gao H.; Chang Y. C.; Hammouténe D.; Ndome H.; Hochlaf M.; Jackson W. M.; Ng C. Y. Quantum-State Dependence of Product Branching Ratios in Vacuum Ultraviolet Photodissociation of N2. Astrophys. J. 2016, 819, 23. 10.3847/0004-637X/819/1/23. [DOI] [Google Scholar]
- Liu M.; Jiang P.; Lu L.; Yin T.; Ma L.; Cheng M.; Yin Q.; Gao H. Strong Isotope-Dependent Photodissociation Branching Ratios of N 2 and Their Potential Implications for the 14N/15N Isotope Fractionation in Titan’s Atmosphere. Astrophys. J. 2021, 923, 196. 10.3847/1538-4357/ac2f97. [DOI] [Google Scholar]
- Lewis B. R.; Gibson S. T.; Zhang W.; Lefebvre-Brion H.; Robbe J. M. Predissociation Mechanism for the Lowest 1Πu States of N2. J. Chem. Phys. 2005, 122, 144302. 10.1063/1.1869986. [DOI] [PubMed] [Google Scholar]
- Hochlaf M.; Ndome H.; Hammoutène D.; Vervloet M. Valence-Rydberg Electronic States of N2: Spectroscopy and Spin-Orbit Couplings. J. Phys. B At. Mol. Opt. Phys. 2010, 43, 245101. 10.1088/0953-4075/43/24/245101. [DOI] [Google Scholar]
- Hochlaf M.; Ndome H.; Hammoutène D. Quintet Electronic States of N2. J. Chem. Phys. 2010, 132, 104310. 10.1063/1.3359000. [DOI] [PubMed] [Google Scholar]
- Stahel D.; Leoni M.; Dressler K. Nonadiabatic Representations of the 1Σu+ and 1Πu States of the N2 Molecule. J. Chem. Phys. 1983, 79, 2541–2558. 10.1063/1.446166. [DOI] [Google Scholar]
- McGlynn S. P.; Azumi T.; Kinoshita M.. Molecular Spectroscopy of the Triplet State; Prentice-Hall: New Jersey, 1969. [Google Scholar]
- Gelfand N.; Komarova K.; Remacle F.; Levine R. D. On the energy-specific photodissociation pathways of 14N2 and 14N15N isotopomers to N atoms of different reactivity: a quantum dynamical perspective. Astrophys. J. 2023, 948, 58. 10.3847/1538-4357/acbef8. [DOI] [Google Scholar]
- Levine R. D.Quantum Mechanics of Molecular Rate Processes; Dover Publications: Mineola, NY, 2011. [Google Scholar]
- Heller E. J. Time-Dependent Approach to Semiclassical Dynamics. J. Chem. Phys. 1975, 62 (4), 1544–1555. 10.1063/1.430620. [DOI] [Google Scholar]
- Tannor D.Introduction to Quantum Mechanics: A Time-Dependent Perspective; University Science Books: Sausalito, CA, 2006. [Google Scholar]
- Helm H.; Cosby P. C. Product Branching in Predissociation of the e 1Πu, E′1Σu+, and B′ 1Σu+ States of N2. J. Chem. Phys. 1989, 90, 4208–4215. 10.1063/1.455777. [DOI] [Google Scholar]
- Lefebvre-Brion H.; Lewis B. R. Comparison between Predissociation Mechanisms in Two Isoelectronic Molecules: CO and N2. Mol. Phys. 2007, 105, 1625–1630. 10.1080/00268970701390180. [DOI] [Google Scholar]
- Heays A. N.; Bosman A. D.; Van Dishoeck E. F. Photodissociation and Photoionisation of Atoms and Molecules of Astrophysical Interest. Astron. Astrophys. 2017, 602, A105. 10.1051/0004-6361/201628742. [DOI] [Google Scholar]
- Fornberg B. Generation of Finite Difference Formulas on Arbitrarily Spaced Grids. Math. Comput. 1988, 51, 699. 10.1090/S0025-5718-1988-0935077-0. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.