

Abstract
The NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome is a multimeric, cytoplasmic, protein complex that regulates maturation and secretion of interleukin (IL)-1β, a potent pro-inflammatory cytokine. Critical to host defense against pathogens, IL-1β amplifies early innate immune responses by activating transcription of numerous other cytokines and chemokines. Excessive IL-1β is associated with poor outcomes in inflammatory illnesses, such as sepsis and the acute respiratory distress syndrome (ARDS). Tight regulation of this signaling axis is vital, but little is known about mechanisms to limit excessive inflammasome activity. Here we identify the deubiquitinase STAM-binding protein (STAMBP) as a negative regulator of the NLRP3 inflammasome. In monocytes, knockout of STAMBP by CRISPR/Cas9 gene editing increased expression of numerous cytokines and chemokines in response to Toll-like receptor (TLR) agonists or bacterial lipopolysaccharide (LPS). This exaggerated inflammatory response was dependent on IL-1β signaling, and STAMBP knockout directly increased release of IL-1β with TLR ligation. While STAMBP does not modulate NLRP3 protein abundance, cellular depletion of the deubiquitinase increased NLRP3 K63 chain polyubiquitination resulting in increased NLRP3 inflammasome activation. These findings describe a unique mechanism of non-degradative ubiquitination of NLRP3 by STAMBP to limit excessive inflammasome activation and to reduce injurious IL-1β signaling.
Keywords: Ubiquitin, Deubiquitinase, Inflammasome, Interleukin-1β, innate immunity
1. INTRODUCTION
Inflammatory cytokines are key effectors in host defense against invading pathogens. When excessive, circulating cytokines contribute to host morbidity and mortality in inflammatory diseases, including the acute respiratory distress syndrome (ARDS)(1, 2) and sepsis (3, 4). Interleukin (IL)-1β is one such pro-inflammatory cytokine. Produced in response to a variety of environmental insults, IL-1β coordinates host response by activating transcription of numerous other cytokines and chemokines including IL-6(5), IL-8(6), CXCL1, CXCL2, and CXCL3 (7). While crucial for survival against bacterial (8), viral (9), and fungal (10) infections, excessive IL-1β is associated with increased mortality in sepsis (3) and ARDS (11). Most cytokines are regulated transcriptionally, but secretion of IL-1β requires further processing by a multimeric, cytoplasmic, protein complex, the NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome. The NLRP3 inflammasome assembles as multiple heterotrimeric subunits comprised of: (i) NLRP3, functioning as a cytoplasmic pattern recognition receptor, (ii) an adaptor protein ASC (apoptosis-associated speck-like protein), and (iii) caspase-1, which enzymatically cleaves pro-IL-1β and pro-IL-18 into their mature forms (12). In mouse models, these inflammasome constituents are required to defend against infection (8). However, knockout of NLRP3 and caspase-1 protects mice from injury in models of sterile inflammation induced by bacterial endotoxin (13, 14). Effective host defense requires an adequate, yet tightly regulated cytokine response to limit tissue injury.
An appropriate innate immune response to noxious stimuli requires alteration in both the abundance and function of cellular proteins to restore homeostasis to the cells, tissues and organism. A major mechanism of protein regulation is the reversible, post-translational modification, ubiquitination. A small ~8kDa protein, ubiquitin molecules are covalently attached to lysine residues of substrate proteins. As ubiquitin itself contains seven lysines, ubiquitin can bind to a substrate protein as a monomer, linear chain, or branched chain. The characteristics of the bound ubiquitin direct the substrate protein for trafficking to another cellular compartment(15). For example, substrate proteins modified by ubiquitin chains linked at the lysine 48 (K48) residue often traffic to the proteasome for degradation. Similarly, ubiquitination with lysine 63 (K63) linked chains directs cargo proteins to the endo-lysosomal pathway. While ubiquitination is well known to regulate protein abundance, ubiquitination also modulates protein and cellular function by directing non-degradative trafficking. Recently, Maresin1, a lipid mediator, inhibited the NLRP3 inflammasome activation and IL-1β secretion in macrophages (16). This inhibitory effect was associated with K63-linked ubiquitination of NLRP3. Aside from this study, mechanisms to limit injurious or excessive IL-1β driven inflammation via feedback inhibition of NLRP3 remain unclear.
Proteins are ubiquitinated through the activity of ubiquitin ligases, and ubiquitin chains are removed by deubiquitinases (DUBs). This dynamic balance or “proteostasis” directs vital cellular processes including host defense(17). Substrate ubiquitination occurs through the coordinated activity of ubiquitin activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin (E3) ligases. Over 700 E3 ligases are known. The E3 ligases attach a ubiquitin chain of a specific configuration (K48, K63, etc.) and target specific motifs, often another post-translational modification, such as phosphorylation within substrates. As antagonists regulating ubiquitin-dependent processes, there are ~90 known human DUB proteins, divided into five families (18). Given the paucity of DUBs compared to ubiquitin ligases, the biologic function of many DUBs remains undescribed. DUBs cleave specific ubiquitin linkage types (i.e. some are specific to K63-linked chains) or can be promiscuous and cleave multiple ubiquitin chain types (19). As DUBs target ubiquitin chains as opposed to substrates, the mechanisms by which DUBs specifically direct cellular processes remains unclear. Proteins modified by K63-linked ubiquitin chains are often sorted to the endo-lysosomal pathway. Ubiquitinated proteins are chaperoned through the endosomes by the Endosomal Sorting Complexes Required for Transport (ESCRTs). Two endosome-resident DUBs, STAM-binding protein (STAMBP) and USP8, function to regulate ESCRT trafficking of ubiquitinated proteins. Our laboratory demonstrated that STAMBP regulates degradation of the inflammasome protein NLRP7 (20) and others show that it modulates sorting of the chemokine receptor CXCR4 (21). Other innate signaling proteins such as NOD1 and NOD2 are known to traffic to the endosome upon activation (22). The inflammasome protein NLRP3 (NACHT, LRR and PYD domains-containing protein 3) is known to be K63 polyubiquitinated and yet the role of endosome-associated DUBs in regulation of these proteins, including NLRP3 has not been investigated.
Here we describe the deubiquitinase STAMBP as a central modulator in innate immune signaling through regulation of the NLRP3 inflammasome activity. In a monocyte cell line, genetic knockout of STAMBP increases IL-1β secretion and increases subsequent IL-1β-driven cytokine and chemokine gene expression. STAMBP does not appear to regulate protein abudance of inflammasome components NLRP3, ASC, pro-caspase-1 or pro-IL-1β, either at basal levels or in response to endotoxin. Instead, STAMBP triggers NLRP3 K63-linked polyubiquitination in response to endotoxin, which seems to function as an activating and non-degradative signal for the NLRP3 inflammasome. These findings demonstrate a unique molecular mechanism of NLRP3 inflammasome regulation by STAMBP by non-degradative ubiquitin modification.
2. MATERIALS AND METHODS
2.1. Cell culture:
THP-1 and HEK-293 were purchased from the American Type Culture Collection (ATCC). THP-1 cells were cultured in RPMI supplemented with 10% FBS. For experiments, THP-1 cells were centrifuged and resuspended in RPMI supplemented with 1% FBS. HEK-293 cells were cultured in DMEM supplemented with 10% FBS. As indicated, THP-1 cells were treated with crude LPS (Sigma, L4391), 20 μM of MG-132 (Sigma), or 100 μM of leupeptin (Sigma). As indicated, cells were incubated with neutralizing mouse IgG monoclonal antibody against human IL-1β (InvivoGen, mabg-hil1b-3) or a β-galactosidase mouse IgG antibody (InvivoGen, mabg1-ctrlm).
2.2. CRISPR/Cas9 STAMBP knockout THP-1 cell line:
Control sgRNA and sgRNA targeted against STAMBP (see Supplementary Table 2) were inserted into the backbone plasmid lentCRISPRv2. HEK-293T cells were transfected with the 3rd generation lentiviral packaging plasmids pMD2.G, pRSV-Rev, pMDLg/pRRE and the lentCRISPRv2 backbone with targeting sgRNA. Single clones of THP-1 KO monocytes were generated via lentiviral transduction and selection with puromycin.
2.3. Quantitative PCR:
Total cellular RNA was collected from cells using the Monarch Total RNA Miniprep Kit (New England Biolabs), following the manufacturer’s protocol. The cellular RNA was then used to create cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to the manufacturer’s protocol. qPCR was performed using SYBR Select Master Mix (Applied Biosystems) according to the manufacturer’s protocol with 20 ng cDNA as a template and primer concentration of 200 nM. Each biological replicate was performed in at least technical duplicate; data was analysed using the ΔΔCq method. qPCR primer sequences are available in Supplementary Table 1.
2.4. Cytokine release assays:
THP-1 cells were counted, normalized and divided into 12-well plates prior to treatment. Cells were stimulated with LPS as indicated. Cytokine release was quantified from clarified culture supernatant using either human IL-1β (Invitrogen, 88–7261) or human TNFα (Invitrogen, 88–7346) ELISA kits according to the manufacturer’s protocol. Each experiment was replicated and performed in triplicate.
2.5. siRNA Transfection:
All siRNA was purchased from the predesigned DsiRNA library from IDT, specifically STAMBP (hs.Ri.STAMBP.13). A universal negative control (DS NC1) were purchased from IDT as negative controls. THP-1 cells (1 × 106 cells per mL) were transfected with 100 nM siRNA duplexes using GenMute siRNA Transfection Reagent (SignaGen Laboratories) for 24 h. Cell were washed with fresh media and incubated for an additional 24–48 h before assay. HEK293 cells (70% confluent) were transfected with 10 nM siRNA duplexes using GenMute for 72 h. Cells were washed and fresh media was added before assay.
2.6. Immunoblotting:
Cells were lysed in RIPA buffer, sonicated and clarified by centrifugation. Lysates were diluted in SDS protein sample buffer. Cell supernatants were collected as indicated. Prior to electrophoresis, supernatants were concentrated by Amicon Ultra 0.5mL centrifugal filters (Sigma, Z677094) then diluted in SDS protein sample buffer. Proteins were separated by electrophoresis and transferred to a nitrocellulose membrane. Blots were blocked in 5% milk, followed by probing overnight with antibodies: NLRP3 (rabbit, Abcam, ab210491), NALP7 (rabbit, Abcam, ab105405), STAMBP (rabbit, Abcam, ab108301), USP8 (rabbit, Cell Signaling, 1183S), ASC (rabbit, AdipoGen, AG-25B-006-C100), Ub P4D1 (mouse, Cell Signaling, 3936S), V5-tag (rabbit, Cell Signaling, 13202S), FLAG (mouse, Sigma Aaldrich, F1804-2000G), HA (mouse, Santa Cruz Biotechnology, sc-7392), TLR2 (rabbit, Cell Signaling, 12276S), TLR4 (mouse, Santa Cruz Biotechnology, sc-293072), MyD88 (rabbit, Cell Signaling 4283S), TRIF (rabbit, Cell Signaling, 4596S), Ub HRP-conj (Cell Signaling, 14049S). All antibody solutions were diluted 1:1,000, except gasdermin (1:600). Pro-caspase-1 and pro-IL-1β antibodies were a gift from Dr. Mark Wewers. Following addition of secondary antibodies (goat anti-mouse, Biorad, 170–6516, and goat anti-rabbit, Biorad, 170–6515; 1:2,000 dilution), membranes were developed using a Western Bright Sirius immunoblotting detection kit (Advansta) and imaged using Biorad imaging software. Single band intensity was quantified using ImageJ software.
2.7. Lactate Dehydrogenase (LDH) cytotoxicity assay:
THP-1 cells were counted, normalized and divided into 12-well plates prior to treatment. Cells were stimulated with LPS as indicated. LDH release was quantified from clarified culture supernatant using CyQUANT LDH Cytotoxicity Assay kits (Invitrogen #C20300) according to the manufacturer’s protocol. LDH activity was assayed in technical triplicate for each replicate.
2.8. ASC speck immunofluorescence:
THP-1 cells were counted and normalized prior to treatment with LPS for 4 h. Cells were washed in PBS, prior to fixation in 4% paraformaldehyde. The samples were loaded into an automated cytospin machine (Thermo Scientific Cytospin 4) following the manufacturer’s instructions and centrifuged (500 rpm for 5 m). Slides were fixed using 4% paraformaldehyde for 10 m, permeabilized with 0.1% Triton-X100 for 10 m, and then blocked with 1% BSA in PBS for 30 m. Cells were probed with primary antibodies ASC (rabbit, AdipoGen, AG-25B-006-C100, 1:50 in 1% BSA in PBS) for 2 h. Following washing, cells were incubated with Alexa Fluor conjugate secondary antibodies (Thermo Fisher, 1:100 dilution for 1 h) and briefly stained with DAPI. Cells were imaged using an Invitrogen EVOS M7000 Imaging System at 40x original magnification. For each biological replicate, three investigators independently counted ASC specks and nuclei in each high power field for at least 200 cells in each condition.
2.9. FLICA assay:
THP-1 cells (1 × 106 cells per mL) were exposed to FAM-FLICA reagent (Immunochemistry Technologies, 98) and LPS (100 ng/mL) or control for 4 h. FAM-FLICA reagent was prepared according to the manufacturer’s protocol. Cells were washed and resuspended in wash buffer, followed by flow cytometry analysis using the 488 nm excitation channel.
2.10. TUBEs (Tandem Ubiquitin Binding Entities) and immunoprecipitation:
Ubiquitin immunoprecipitation was performed under non-denaturing conditions. Lysates were harvested in PBS + 0.5% Triton X-100 buffer containing Tandem Ubiquitin Binding Entities (TUBEs), which bind to ubiquitin chains. We used biotin-tagged TUBEs that specifically bind to K48-linked chains (Life Sensors, UM307 ), K63-linked chains (Life Sensors, UM304), or all ubiquitin chains (Life Sensors, UM301). Biotin-tagged TUBEs are immunopurified from lysates using Streptavidin magnetic beads (Dynabeads, MyOne, Invitrogen #65001) using the manufacturer’s protocol. FLAG pulldown was performed using Anti-FLAG M2 Magnetic Beads (Sigma, M8823) under non-denaturing conditions. Lysates were harvested in PBS + 0.5% Triton X-100 buffer, prior to centrifugation and pulldown proceeded according to the protocol provided by the manufacturer.
2.11. Cloning and mutagenesis:
NLRP3, STAMBP, and USP8 cDNA with complete protein coding gene sequences, all in pLX304 vector, were purchased from DNASU. The coding sequences from NLRP3, STAMBP and USP8 plasmids were cloned using the Phusion High-Fidelity DNA Polymerase kit (New England BioLabs). Site-directed mutagenesis was performed using the QuikChange II XL Site-Directed Mutagenesis kit (Agilent). After cloning, cDNA was ligated into pcDNA3.1D/V5-His vector (Invitrogen) and constructs were verified by Sanger sequencing. Plasmids were overexpressed in HEK-293 cells using XtremeGene HP DNA Transfection Reagent (Roche) for 24–48 h before assay.
3. RESULTS
3.1. STAMBP knockout increases innate immune cytokine and chemokine expression.
Prior in vitro studies demonstrate a role for STAMBP in innate immune signaling (20, 21). To further characterize immune modulation by STAMBP, we silenced STAMBP gene expression in human-derived THP-1 monocytic cells using the CRISPR/Cas9 gene editing system, creating stable STAMBP-/- and Cas9 control THP-1 cell lines. Wild-type THP-1 monocytes upregulate transcription of many inflammatory cytokines (IL-1β, IL-6, IL-8 and TNF) and chemokines (CCL2, CCL3, CCL4, CCL5 and CCL9) in response to the TLR2/4 ligand bacterial endotoxin (23). We hypothesized that STAMBP is necessary for normal homeostatic control of these cytokines and chemokines in response to TLR agonism by endotoxin. As these cytokines and chemokines are exclusively regulated at the level of transcription, with the exception of IL-1β, we assessed expression of transcripts with quantitative PCR. As expected, transcript expression of all assayed cytokines and chemokines increased in both the STAMBP-/- and Cas9 cells with endotoxin exposure. However, STAMBP-/- cells displayed significantly higher fold increases of cytokine and chemokine mRNA with endotoxin exposure compared to the Cas9 controls (Fig. 1A), supporting a hyperinflammatory phenotype in the STAMBP-/- THP-1 monocytes. IL-1β is known to induce the many of the assayed cytokines and chemokines assayed here, as it is robustly secreted by monocytes in response to LPS (5–7).To investigate if secreted IL-1β was necessary for the observed exaggeration of innate immune signaling with STAMBP knockout, we pre-treated STAMBP-/- and Cas9 control THP-1 cells with IL-1β neutralizing antibody or an antibody targeting mouse β-galactosidase as a control prior to exposure to endotoxin. IL-1β neutralizing antibody partially reversed the hyper-inflammatory phenotype displayed by STAMBP-/- THP-1 cells (Fig. 1B), supporting the hypothesis that secreted IL-1β was required for the observed increases in inflammatory phenotytpe observed with STAMBP depletion.
Figure 1: STAMBP knockout with LPS exposure triggers increases in multiple cytokines and chemokines.
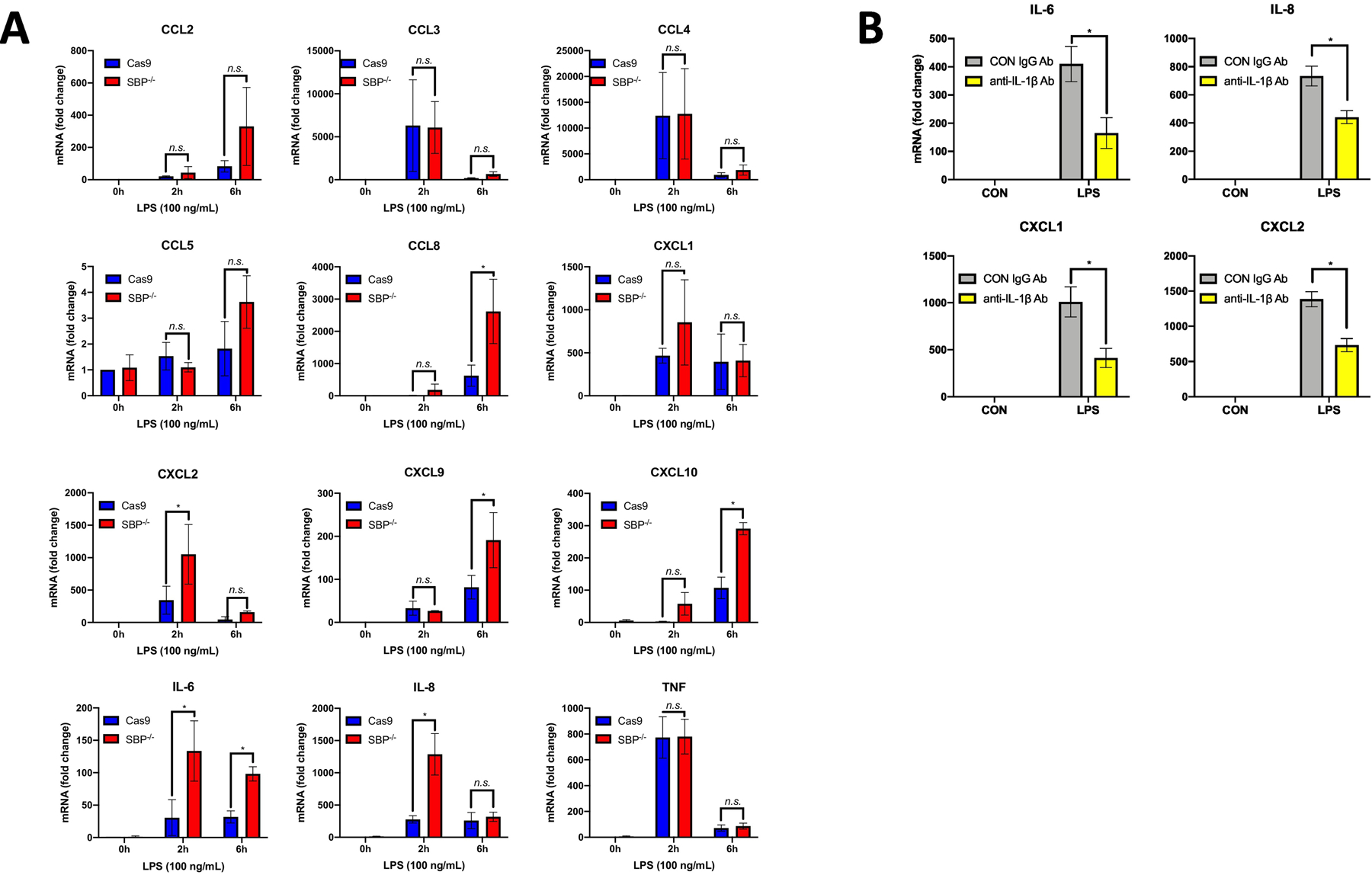
(A) Cas9 control and STAMBP-/- (SBP-/-) THP-1 monocytes (1 x 106 cells/mL) were exposed to LPS (100ng/mL) for 2h or 6h, prior to harvest of total cellular RNA. Twelve known LPS-inducible cytokines and chemokines were assayed by qPCR. STAMBP-/- THP-1 cells had higher transcription of numerous cytokines (IL-6 and IL-8) and chemokines (CCL8, CXCL2, CXCL9 and CXCL10) compared to controls. Data shown as mean of 3 separate experiments ± SEM. *P<0.05 compared to time matched Cas9 control, as determined by ANOVA with post hoc multiple comparisons test. (B) STAMBP-/- THP-1 cells (1 x 106 cells/mL) were pre-treated with anti-IL-1β neutralizing antibody or IgG isotype control (both at 100ng/mL) immediately preceding exposure to LPS (100ng/mL) for 3h. Pre-treatment with IL-1β neutralizing antibody decreased transcription of inflammatory cytokines and chemokines. Data shown as mean of 4 separate experiments ± SEM. *P<0.05 as determined by Mann-Whitney test comparing LPS-treated anti- IL-1β neutralizing antibody to the IgG control.
3.2. STAMBP knockout increases IL-1β secretion in THP-1 monocytes after endotoxin.
STAMBP knockout enhances mRNA expression of inflammatory cytokines and chemokines seemingly due to enhanced IL-1β signaling. Expressed as a pro-protein, pro-IL-1β requires additional processing at the inflammasome prior to secretion as mature IL-1β. Accordingly, measurement of secreted, mature IL-1β is more indicative of inflammatory signaling than assessing transcription. To assess whether STAMBP modulates LPS-mediated IL-1β release, STAMBP-/- and Cas9 control THP-1 cells were treated with LPS, and IL-1β release was evaluated by ELISA. STAMBP-/- cells displayed significantly increased IL-1β secretion within 2 hours of exposure to LPS, and IL-1β secretion remained increased up to 6 hours compared to controls (Fig. 2A). There were no significant differences in secreted TNF. We next assessed caspase-1 and IL-1β abundance in cell lysates and supernatants from STAMBP-/- and Cas9 control THP-1 cells with LPS exposure. We observed no significant differences in protein abundance of pro-caspase-1 or pro-IL-1β in the cell lysates. However, secretion of mature IL-1β and cleaved caspase-1 was increased in STAMBP-/- cells compared to controls (Fig. 2B). These findings recapitulate our ELISA data and suggest that increased IL-1β secretion may be due to increased inflammasome activation, as evidenced by increased cleaved caspase-1. We also assessed the dose response to bacterial endotoxin. STAMBP-/- THP-1 cells released substantially more IL-1β compared to controls in response to LPS at concentrations as low as 10 ng/mL (Fig. 2C). These findings support the hypothesis that STAMBP knockout enhances IL-1β signaling.
Figure 2: STAMBP knockout enhances LPS-induced IL-1β release.
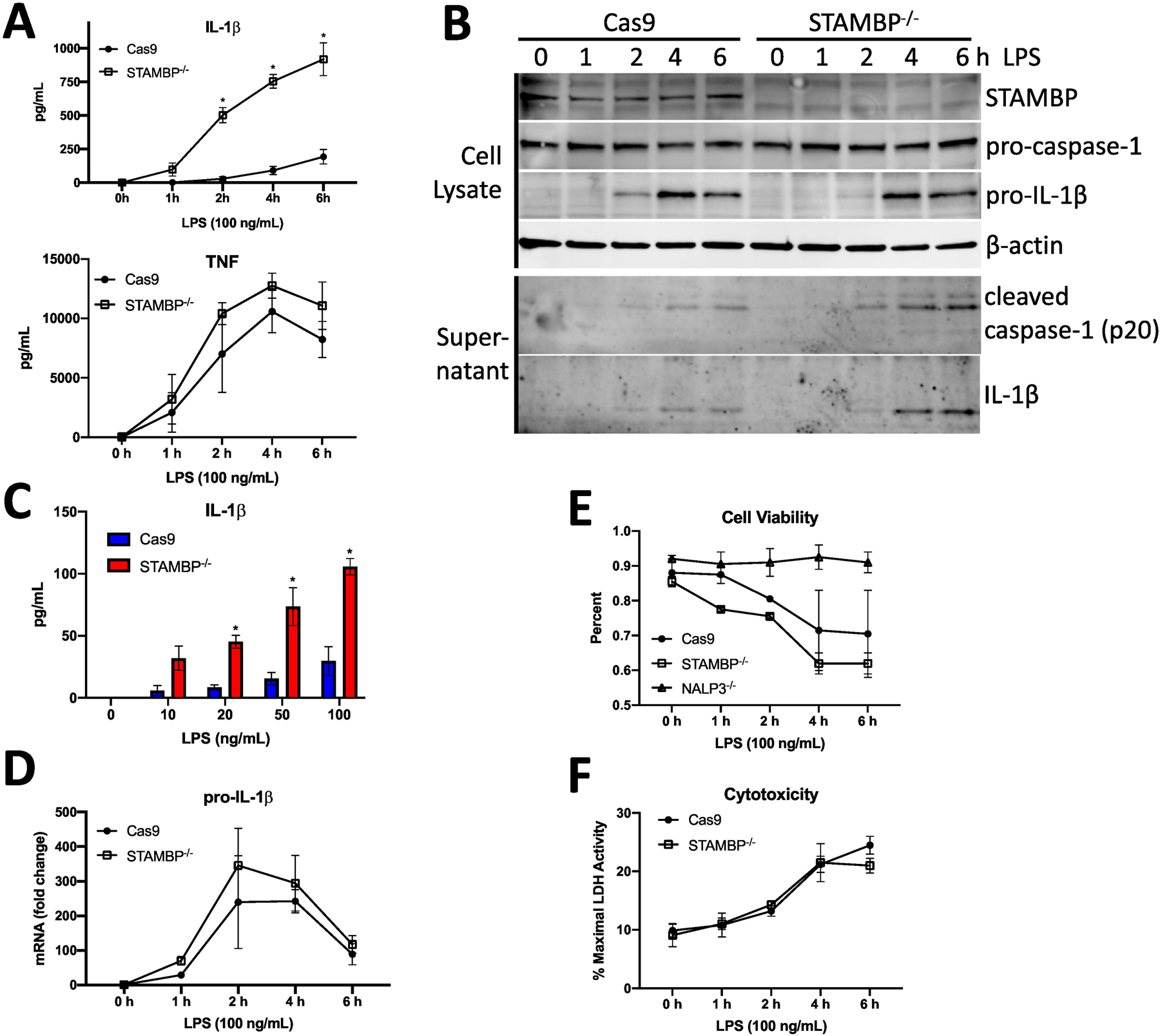
(A) Cas9 control and STAMBP-/- THP-1 monocytes (1 x 106 cells/mL) were exposed to LPS (100ng/mL) for the indicated times, prior to measurement of cytokines by ELISA. IL-1β but not TNF secretion was increased across time points in STAMBP-/- cells compared to controls. (B) Cas9 control and STAMBP-/- THP-1 monocytes (1 x 106 cells/mL) were exposed to LPS (100ng/mL) for the indicated times. Cell pellets and supernatants were separated, prior to analysis by immunoblotting. Mature IL-1β and cleaved caspase-1 were more abundant in the supernatants of STAMBP-/- cells compared to controls. (C) Cas9 control and STAMBP-/- THP-1 monocytes (1 x 106 cells/mL) were exposed to LPS at indicated concentrations for 4 hours, prior to measurement of IL-1β by ELISA. IL-1β secretion was increased in STAMBP-/- monocytes compared to Cas9 controls. (D-F) Cas9 control and STAMBP-/- THP-1 monocytes (1 x 106 cells/mL) were exposed to LPS (100ng/mL) for the indicated times, prior to measurement of (D) pro-IL-1β mRNA by qPCR, (E) cell viability by a trypan blue assay, and (F) cytotoxicity by LDH release assay. (A, C-F) Graphs represent mean ± SEM of three separate experiments. P values determined by ANOVA with post hoc multiple comparisons test. (A,C) *P<0.05 compared to time matched Cas9 control.
To begin to investigate the mechanism by which STAMBP knockout mediates enhanced IL-1β release, we first evaluated changes in transcription of pro-IL-1β. STAMBP knockout did not significantly alter transcription of pro-IL-1β in response to endotoxin (Fig. 2D). As IL-1β release is often coupled with inflammasome activation and pyroptosis, we assessed cell viability and observed no significant difference in cell death in STAMBP-/- cells compared to the Cas9 controls (Figs. 2E, 2F). As we observed increased cleaved caspase-1 secretion by Western blot in the STAMBP knockout cells, we next assayed NLRP3 inflammasome activation as a possible mechanism of increased IL-1β release with STAMBP knockout. ASC oligomerization is a requisite step for inflammasome assembly and function and can be visualized in cells as a relatively large ASC speck (24). We measured ASC speck formation by immunofluorescence microscopy and observed increased speck formation in STAMBP-/- cells compared to controls (Fig. 3A, arrows). Caspase-1 activation occurs following ASC oligomerization. We next measured active caspase-1 using a fluorescent inhibitor of caspase activation (FAM-FLICA); this inhibitor binds irreversibly to active caspase-1, trapping the fluorescent compound inside the cells. Cells containing active caspase-1 display higher fluorescent signal intensity. Compared to Cas9 controls, STAMBP-/- THP-1 cells displayed a higher percentage of cells with activated caspase-1 and higher mean fluorescence intensity in response to bacterial endotoxin, as measured by flow cytometry (Fig. 3B). These findings indicate that STAMBP knockout results in higher levels of active caspase-1. As caspase-1 is downstream of NLRP3, this supports the hypothesis that STAMBP knockout results in increased NLRP3 inflammasome activation.
Figure 3: STAMBP knockout enhances LPS-induced inflammasome activation.
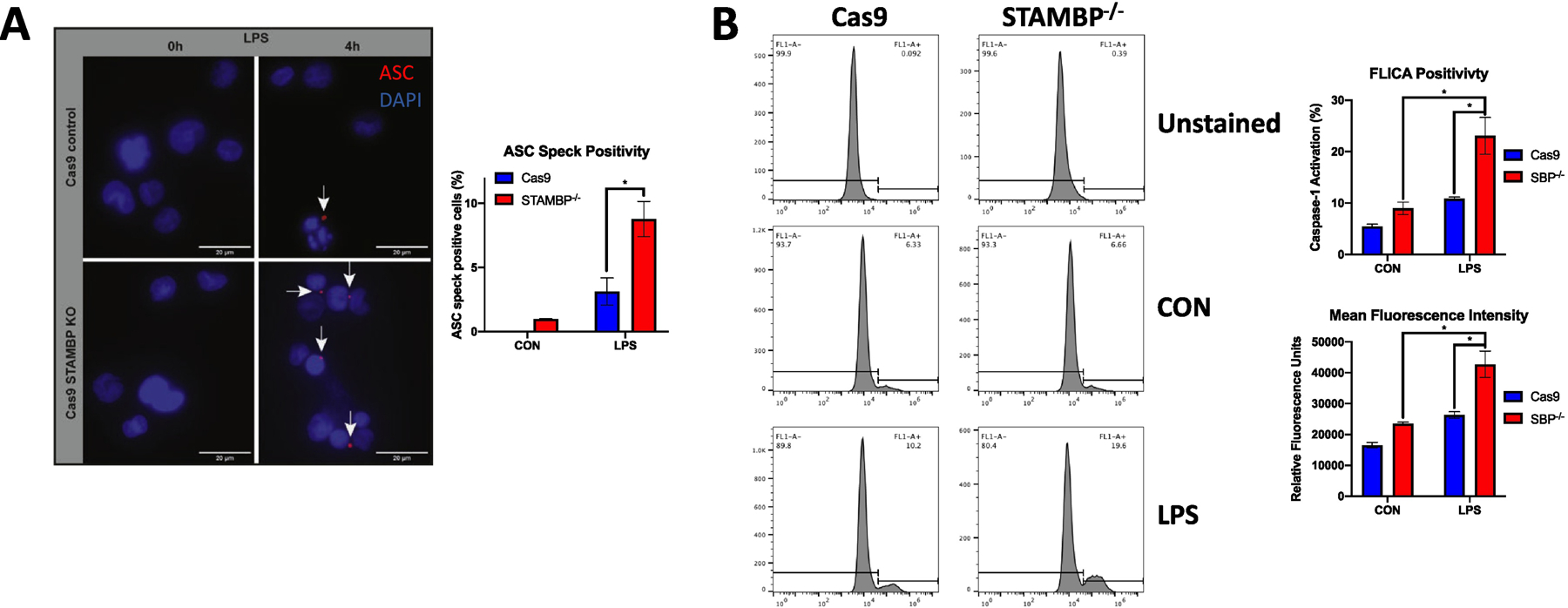
(A) Cas9 control and STAMBP-/- THP-1 monocytes (1 x 106 cells/mL) were exposed to LPS (100ng/mL) for 4h prior to immunofluorescence co-staining of ASC (red) and DAPI (blue). STAMBP knockout increased the percentage of THP-1 cells (visualized by nuclei) that demonstrated formation of ASC specks (indicated by white arrows). Data shown as representative images. Bar graph (right) show mean % ASC speck positive cells ± SEM of three separate experiments. (B) FAM- FLICA assay. Cas9 control and STAMBP-/- THP-1 monocytes (1 x 106 cells/mL) were exposed to LPS (100ng/mL) for 4h and FAM-FLICA reagent was added for the final 1h of incubation. Cells were washed and retained FAM-FLICA was analyzed by flow cytometry. STAMBP knockout increased the population of THP-1 cells that stained positive for the carboxyfluorescein derivative substrate (that binds to activated caspase-1 in cells), and mean fluorescence intensity was increased in STAMBP-/- cells compared to controls. Data shown as representative histograms, with brackets showing the threshold fluorescence for cells with inactive (left) versus active (right) caspase-1. Bars graphs (right) show % FLICA positivity (upper) and mean fluorescence intensity (lower). (A-B) *P<0.05 compared to the unstimulated STAMBP-/- cells or the LPS-treated Cas9 control cells, as indicated.
3.3. STAMBP deubiquitinates NLRP3 but does not regulate its degradation.
NLRP3 inflammasome activity as shown above is activated by STAMBP knockout in THP-1 cells. We hypothesized that STAMBP deubiquitinates NLRP3, which modulates NLRP3 activity. First, we confirmed poly-ubiquitination of NLRP3 in THP-1 monocytes and demonstrate both K48- and K63-linked NLRP3 polyubiquitination using Tandem Ubiquitin Binding Entities (TUBEs), which pull down all ubiquitinated proteins or proteins with a specific ubiquitin modification (K48 vs. K63) (Fig. 4A). This recapitulates findings of prior groups in our cell model system (25, 26). In co-immunoprecipitation studies, ectopically expressed STAMBP-V5 with FLAG-NLRP3 demonstrated that the two proteins interact in cells (Fig. 4B). Next, we assayed the impact of STAMBP on NLRP3 ubiquitination in a cell system utilizing both overexpression and knockdown. Overexpression of STAMBP using a mammalian expression vector resulted in a decrease in polyubiquitinated NLRP3. Further, overexpression of STAMBP D348A, which lacks DUB activity, did not impact ubiquitination of NLRP3 (Fig 4C). We silenced STAMBP expression with siRNA and overexpressed FLAG-tagged NLRP3 in HEK-293 cells. STAMBP knockdown resulted in increases in polyubiquitinated NLRP3 versus a control construct (Fig. 4D). Next, we assayed K63-linked ubiquitination of NLRP3 in the presence of bacterial endotoxin. In the STAMBP-/- THP-1 cells, we observed an increase in K63-ubiquitinated NLRP3 with exposure to endotoxin, and this trend differed from the Cas9 controls cells (Fig. 4E). This suggests that STAMBP is necessary to limit K63-linked ubiquitination of NLRP3 following endotoxin exposure. Collectively, these data support a functional relationship via physical association of NLRP3 with STAMBP, whereby STAMBP modulates NLRP3 polyubiquitination through its catalytic activity.
Figure 4: NLRP3 is deubiquitinated by STAMBP.
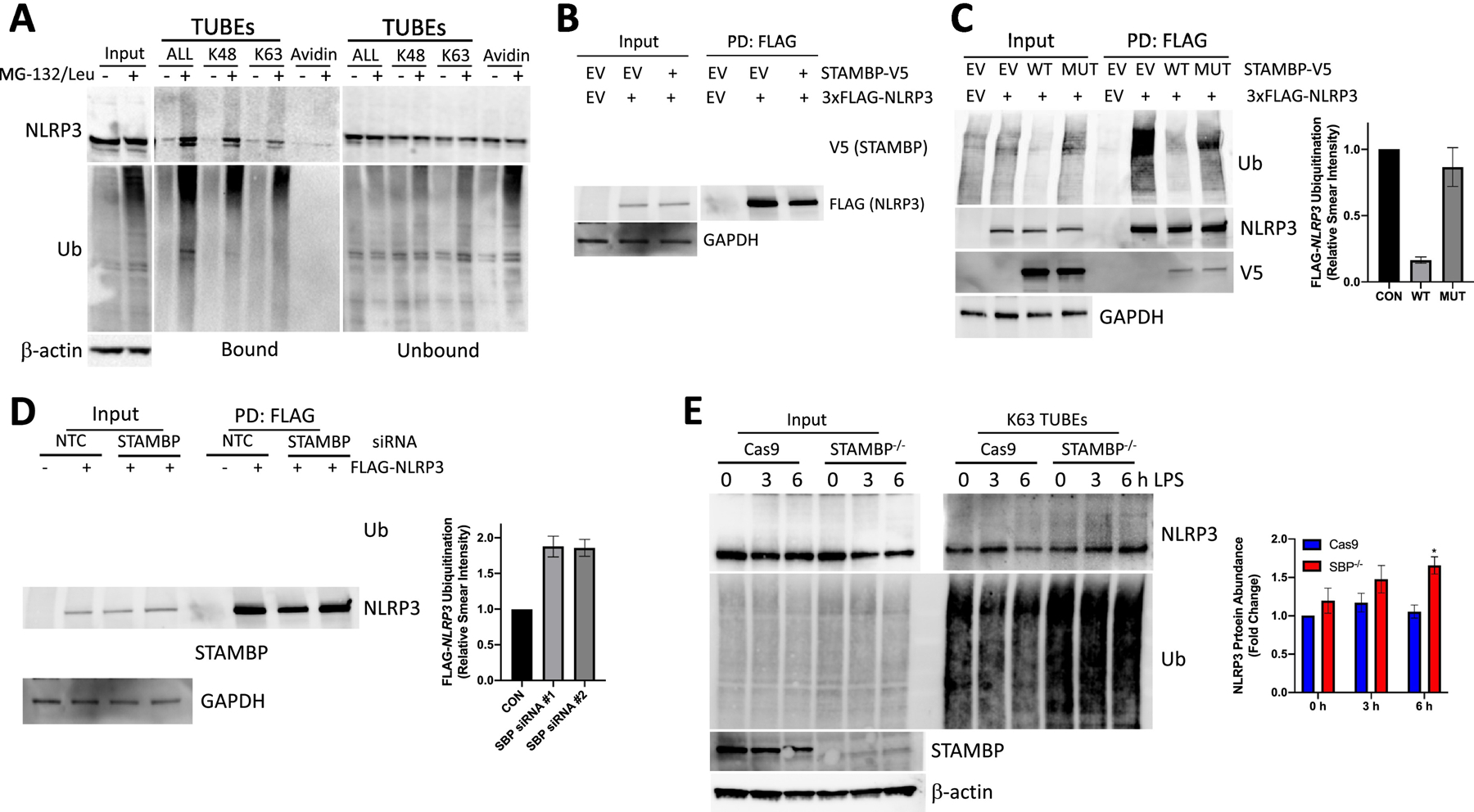
(A) Wild type THP-1 cells were treated with MG-132 and leupeptin for 4 hours, as indicated, prior to pulldown of ubiquitinated proteins using TUBEs (Tandem Ubiquitin Binding Entities), specific to K48-linked, K63-linked or all ubiquitin chains (ALL). Endogenous NLRP3 protein demonstrates both K48-linked and K63-linked ubiquitin modification. (B) HEK-293 cells were transfected with plasmids encoding STAMBP-V5, FLAG-NLRP3, or empty vector (EV). Cells were immunoprecripitated with FLAG affinity matrix and immunoprecipitate samples assayed for V5, FLAG, and GAPDH by immunoblotting. (C) HEK-293 cells were transfected with plasmids encoding FLAG-NLRP3, WT-STAMBP-V5, mutant STAMBP(D348A)-V5, or empty vector (EV), followed by harvest and FLAG pulldown. Overexpression of catalytically active STAMBP-V5 decreased levels of immunopurified Ub-NLRP3. Overexpression of STAMBP with a mutated DUB active site (STAMBP D348A) did not affect levels of immunopurified Ub-NLRP3. Right, densitometic analysis of the immunoreactive Ub signal, normalized to NLRP3 in the FLAG pulldown condition. (D) HEK-293 cells were co-transfected with siRNA targeting STAMBP or siRNA targeting a non-targeting control (NTC) and a plasmid encoding FLAG-NLRP3 prior to harvest and immunoprecipitation of the FLAG-NLRP3. STAMBP knockdown resulted in increased immunoreactive Ub-FLAG-NLRP3. Right, densitometic analysis of the immunoreactive Ub signal, normalized to NLRP3 in the FLAG pulldown condition. (E) THP-1 cells were treated with LPS (100ng/mL) along a time course and harvested in lysis buffer containing biotin-tagged K63-specific TUBEs, followed by streptavidin pulldown and immunopurification of K63-ubiquitinated substrates. Protein abundance was assayed by immunoblotting. Representative images show increased endogenous Ub-NLRP3 in the STAMBP-/- THP-1 cells with LPS exposure, compared to the Cas9 control cells. Right, densitometic analysis of the immunoreactive Ub-NLRP3 signal in the K63 TUBEs condition. (A-E) Data shown as representative images. (C-E) Graphs represent mean ± SEM of three separate experiments. (E) *P<0.05 as determined by ANOVA with post hoc multiple comparisons test compared to unstimulated Cas9 control.
3.4. STAMBP does not impact NLRP3 protein abundance with endotoxin exposure.
Activation of cytokine release through NLRP3 could occur by increasing protein concentrations of the inflammasome or via post-translational modification of preformed NLRP3. With regard to the former possibility, we did not observe differences in transcription of pro-IL-1β (Fig. 2D) or mRNA of NLRP3 (data not shown). We further evaluated if STAMBP regulates the NLRP3 inflammasome through modulation of protein abundance. Prior reports have demonstrated K48 polyubiquitination of NLRP3 triggers its degradation by the proteasome (27, 28). As STAMBP exclusively cleaves K63-linked ubiquitin chains, we hypothesized that STAMBP knockout would not impact NLRP3 protein abundance. Basal NLRP3 protein was modestly decreased in the stable knockout STAMBP THP-1 monocytes compared to Cas9 controls (Fig. 5A). We next exposed cells to endotoxin to assess how LPS may impact NLRP3 protein with STAMBP knockout. Prior in vitro studies demonstrate decreases in NLRP3 protein with exposure to LPS (26). We observed a modest dose- (Fig. 5B) and time-dependent (Fig. 5C) decrease in NLRP3 protein abundance in the Cas9 control cells. Contrary to trends observed in the Cas9 cells, the STAMBP-/- THP-1 cells displayed lower basal NLRP3 protein, but the endotoxin-induced decrease in NLRP3 abundance was not observed (Figs. 5B, 5C). There were no significant differences in expression of other inflammasome components with endotoxin exposure when comparing the Cas9 cells and STAMBP-/- monocytes, specifically ASC, pro-caspase-1 and pro-IL-1β (Fig. 5B, 5C). These findings suggest that observed differences in IL-1β secretion after endotoxin exposure are not due to STAMBP regulating abundance of the NLRP3 inflammasome. Finally, we assayed proteins upstream of the NLRP3 inflammasome, as differences in abundance of upstream activators may impact inflammasome activators. Toll-like receptors 2 and 4 (TLR2 and TLR4) activate the NLRP3 inflammasome in response to ligation with LPS. Signal transduction occurs through MyD88 to activate the canonical inflammasome or alternatively non-canonical inflammasome activation occurs through the activity of TRIF. CRISPR/Cas9 knockout of STAMBP in THP-1 cells did not impact protein abundance of TLR2, TLR4, MyD88 or TRIF (Fig. 5D). Taken together, these results suggest strongly that LPS exposure to THP-1 monocytes depleted in STAMBP triggers K63 polyubiquination of NLRP3 that results in its activation, releasing pro-inflammatory cytokines.
Figure 5: STAMBP knockout does not significantly impact cellular abundance of inflammasome proteins with endotoxin exposure.
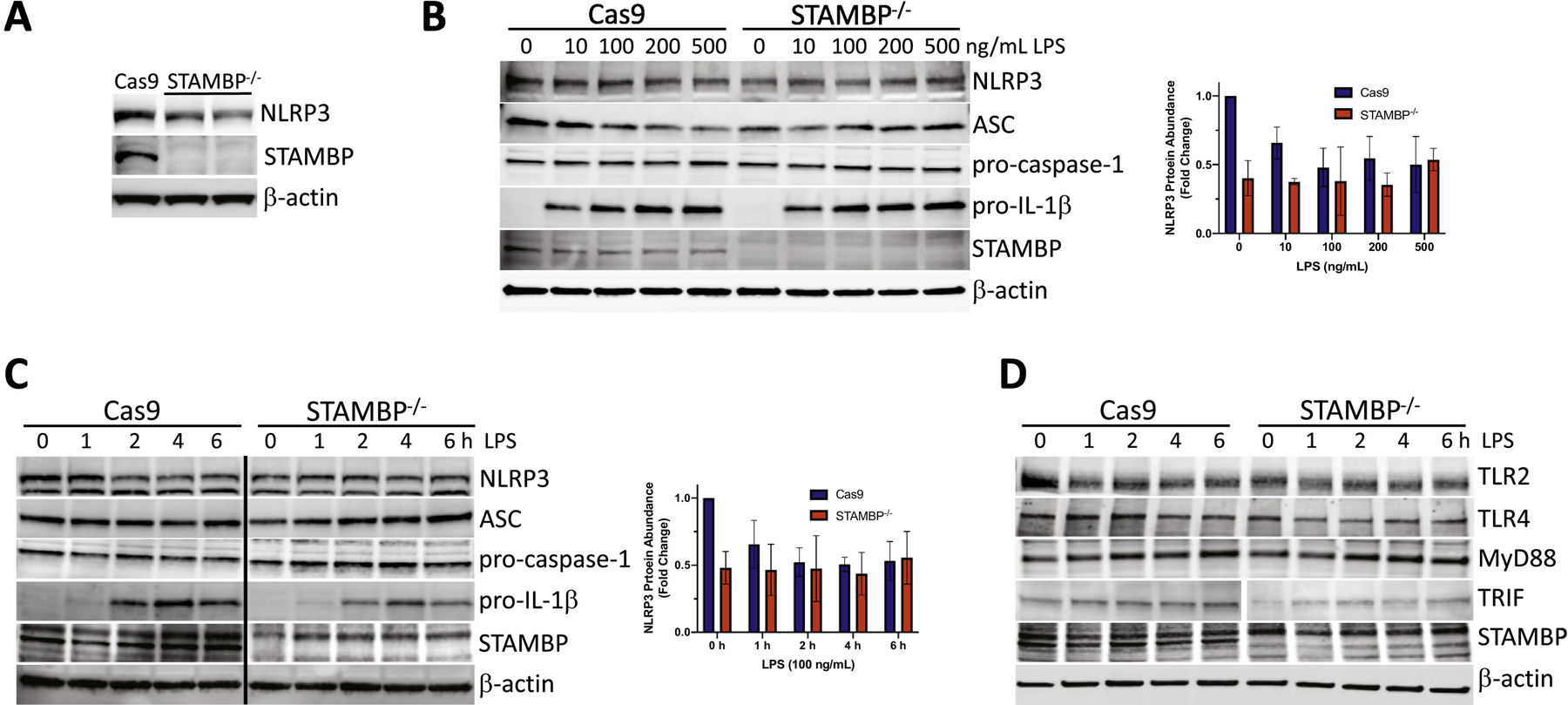
(A) Baseline levels of NLRP3 were assayed by immunoblotting in STAMBP-/- THP-1 cells compared to Cas9 controls and demonstrate decreased basal NLRP3 protein abundance. (B-D) Cas9 control and STAMBP-/- THP-1 monocytes (1 x 106 cells/mL) were exposed to LPS followed by immunoblotting. Inflammasome components NLRP3, ASC, pro-caspase-1 and pro-IL-1β were not significantly different in STAMBP-/- THP-1 monocytes compared to Cas9 THP-1 controls following exposure to (B) various concentrations of LPS for 16 hours or (C) following LPS exposure (100ng/mL) for the indicated time points. Graphs shown to right of immunoblots represent results of densitometic analysis of the NLRP3 signal, normalized to β-actin. Graphs represent mean ± SEM of three separate experiments. (D) Immunoreactive levels of upstream NLRP3 activating proteins TLR2, TLR4, MyD88 and TRIF were not significantly different in STAMBP-/- THP-1 cells compared to Cas9 controls following exposure to LPS (100ng/mL) for the indicated time points.
4. DISCUSSION
These studies demonstrate a unique ability of the endosome-associated DUB STAMBP to regulate inflammasome activity and IL-1β secretion through polyubiquitination of NLRP3. The new findings of the study are that (i) STAMBP modulates inflammasome activation and IL-1β secretion in monocytes, (ii) NLRP3, through its deubiquitination by STAMBP, represents a new client for the DUB, and (iii) NLRP3 K-63 linked polyubiquitination triggered by STAMPB cellular depletion appears to modulate inflammasome activity but not by increasing NLRP3 protein abundance.
Prior studies have demonstrated the importance of the NLRP3 inflammasome – IL-1β signaling axis as a central mediator of the innate immune response. Consistent with this, STAMBP knockout resulted in increased expression of many pro-inflammatory cytokines. This effect was dependent on intact IL-1β signaling (Fig. 1). As the key regulator of IL-1β secretion, ubiquitination of NLRP3 and the NLRP3 inflammasome has been extensively studied, but a consensus model for modulation of NLRP3 protein abundance or inflammasome activation has remained elusive. Ubiquitination of NLRP3 has been demonstrated to both negatively regulate inflammasome activity in some studies (25, 27, 29) and conversely, enhances inflammasome activation in others (30, 31). Our data suggest that NLRP3 ubiquitination, likely K63-linked, provides a signal for activation and inflammasome assembly. Consistent with prior studies (25, 26, 30), we demonstrate poly-ubiquitination of NLRP3 with both K48- and K63-linked ubiquitin chains, raising the possibility that different ubiquitination events regulate different cellular processes. Multiple reports agree that NLRP3 is degraded within the proteasome, depenedent on K48-linked ubiquitination. K63-linked ubiquitination does not seem to impact protein abundance and may alternatively be a required signal for inflammasome activation as suggested recently (16). The results here suggest that K63 NLRP3 deubiquitination resulting from STAMBP represents an novel feedback inhibitory control mechanism to temper inflammation dring endotoxin stress.
Many E3 ligases and DUBs have been reported to affect NLRP3 ubiquitination, but again, these studies are difficult to reconcile into a unified model. The E3 ligases FbxL2(27) and TRIM31(28) have both been shown to ubiquitinate NLRP3, directing proteasomal degradation. Knockdown of the deubiquitinase BRCC3 increases NLRP3 ubiquitination and inhibits inflammasome activity (25), opposite of our observed phenotype with STAMBP knockout. Like STAMBP, BRCC3 is a K63-specific DUB. Our studies demonstrated subtle differences in NLRP3 protein abundance with stable CRISPR/Cas9 knockout, questioning whether a stable BRCC3 knockout would recapitulate the published phenotype that utilized only transient knockdown in mouse macrophages. A recent study demonstrated sequential ubiquitination of NLRP3 by two E3 ligases, RNF125 and Cbl-b, whereby NLRP3 was K63-ubiquitinated by RNF125, followed by K48-linked modification by Cbl-b. K63-linked ubiquitination was increased with stimulation with LPS and ATP (26). Again, the K48 polyubiquitin modification seems to direct proteasomal degradation of NLRP3 to limit excessive inflammasome activity, but the role of the K63-linked polyubiqutination remains unclear. One hypothesis is that K63 ubiquitination can function as an activating signal that may direct subcellular trafficking. Here knockout of STAMBP preserves NLRP3 abundance with K63 modification after endotoxin exposure, allowing for faster and more robust inflammasome assembly and activity. Coupling activation with recruitment of an E3 ligase, Cbl-b, that enacts degradation is consistent with the theme of feedback inhibition exhibited by many cellular signaling pathways.
Our studies highlight a critical role for STAMBP in limiting injurious inflammation caused by excessive inflammasome acivity. Clinically, STAMBP has only been associated with microcephaly-capillary malformation syndrome, characterized by profound neurologic growth deficits, refractory seizures, and profound developmental delay (32). STAMBP functions in the endo-lysosomal pathway and interacts with the Endosomal Sorting Complexes Required for Transport (ESCRTs) (33). Most ESCRT proteins are essential to cellular function, and mutations are often lethal in humans. The role of STAMBP in innate immune signaling has not been extensively characterized. Our group previously demonstrated a functional relationship between STAMBP and another inflammasome protein NLRP7, whereby STAMBP knockdown enhanced NLRP7 degradation and decreased IL-1β release in differentiated THP-1 macrophages (20). In this study, we demonstrate STAMBP-mediated ubiquitination of NLRP3 to provide a non- degradative signal, and STAMBP knockout increased IL-1β secretion in undifferentiated THP-1 monocytes (Fig. 2). These differential effects observed with STAMBP knockout highlight the different immune profiles of monocytes and macrophages. Prior groups have demonstrated differences in NLR expression with monocyte differentiation and macrophage polarization (34). Further, monocytes and macrophages require different signals for IL-1β release, suggesting differential regulation of inflammasome activation in these models (35) and opportunity for further investigation. Another protein regulated by STAMBP is the chemokine receptor CXCR4. Ubiquitination of CXCR4 is not directly modulated by STAMBP. Instead, STAMBP regulates ubiquitination of ESCRT-0 components STAM1 and HRS, which facilitate endocytic trafficking and degradation of CXCR4(21). These studies highlight a number of mechanisms whereby a DUB, such as STAMBP, can direct cellular processes. Given the paucity of DUBs in the human genome, it is likely that each DUB, such as STAMBP, effects a number of protein substrates, directly or indirectly to provide a degradative or alternative trafficking signal.
5. CONCLUSIONS
This study demonstrates an important role for STAMBP as a central regulator of innate immunity through its direct modulation of the NLRP3 inflammasome and IL-1β secretion.
The NLRP3 inflammasome is polyubiquitinated.
STAMBP deubiquitinates NLRP3.
STAMBP DUB activity limits NLRP3 inflammasome activity and excessive IL-1β secretion to protect against injurious inflammation.
Supplementary Material
Highlights:
This study demonstrates an important role for STAMBP as a central regulator of innate immunity through its direct modulation of the NLRP3 inflammasome and IL-1β secretion.
The NLRP3 inflammasome is polyubiquitinated.
STAMBP deubiquitinates NLRP3.
STAMBP DUB activity limits NLRP3 inflammasome activity and excessive IL-1β secretion to protect against injurious inflammation.
Acknowledgments
This work was also supported by NIH grants UH3HL123502, R01HL096376, R01HL097376, R01HL098174, R01HL081784, and P01HL114453 to R.K.M. JB designed the study, performed experiments, analyzed results and wrote manuscript. FJ, TRS, and NP performed experiments and analyzed data. JDL and RKM reviewed the data and edited manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Credit Author Statement:
JB designed the study, performed experiments, analyzed results and wrote manuscript. FJ, TRS, and NP performed experiments and analyzed data. JDL and RKM reviewed the data and edited manuscript.
REFERENCES:
- 1.Bos LD, Schouten LR, van Vught LA, Wiewel MA, Ong DSY, Cremer O, Artigas A, Martin-Loeches I, Hoogendijk AJ, van der Poll T, Horn J, Juffermans N, Calfee CS, Schultz MJ, consortium M. Identification and validation of distinct biological phenotypes in patients with acute respiratory distress syndrome by cluster analysis. Thorax 2017; 72: 876–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA, Network NA. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med 2014; 2: 611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wiersinga WJ, Leopold SJ, Cranendonk DR, van der Poll T. Host innate immune responses to sepsis. Virulence 2014; 5: 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pruitt JH, Copeland EM, 3rd, Moldawer LL. Interleukin-1 and interleukin-1 antagonism in sepsis, systemic inflammatory response syndrome, and septic shock. Shock 1995; 3: 235–251. [DOI] [PubMed] [Google Scholar]
- 5.van Scheppingen J, Mills JD, Zimmer TS, Broekaart DWM, Iori V, Bongaarts A, Anink JJ, Iyer AM, Korotkov A, Jansen FE, van Hecke W, Spliet WG, van Rijen PC, Baayen JC, Vezzani A, van Vliet EA, Aronica E. miR147b: A novel key regulator of interleukin 1 beta- mediated inflammation in human astrocytes. Glia 2018; 66: 1082–1097. [DOI] [PubMed] [Google Scholar]
- 6.Song H, Chan J, Rovin BH. Induction of chemokine expression by adiponectin in vitro is isoform dependent. Transl Res 2009; 154: 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chevillard G, Derjuga A, Devost D, Zingg HH, Blank V. Identification of interleukin-1beta regulated genes in uterine smooth muscle cells. Reproduction 2007; 134: 811–822. [DOI] [PubMed] [Google Scholar]
- 8.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 2011; 29: 707–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas PG, Dash P, Aldridge JR Jr., Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, Doherty PC, Kanneganti TD. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 2009; 30: 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vonk AG, Netea MG, van Krieken JH, Iwakura Y, van der Meer JW, Kullberg BJ. Endogenous interleukin (IL)-1 alpha and IL-1 beta are crucial for host defense against disseminated candidiasis. J Infect Dis 2006; 193: 1419–1426. [DOI] [PubMed] [Google Scholar]
- 11.Meduri GU, Headley S, Kohler G, Stentz F, Tolley E, Umberger R, Leeper K. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest 1995; 107: 1062–1073. [DOI] [PubMed] [Google Scholar]
- 12.Schroder K, Tschopp J. The inflammasomes. Cell 2010; 140: 821–832. [DOI] [PubMed] [Google Scholar]
- 13.Sarkar A, Hall MW, Exline M, Hart J, Knatz N, Gatson NT, Wewers MD. Caspase-1 regulates Escherichia coli sepsis and splenic B cell apoptosis independently of interleukin-1beta and interleukin-18. Am J Respir Crit Care Med 2006; 174: 1003–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 1995; 80: 401–411. [DOI] [PubMed] [Google Scholar]
- 15.Ciechanover A Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol 2005; 6: 79–87. [DOI] [PubMed] [Google Scholar]
- 16.Zheng S, Ma M, Li Z, Hao Y, Li H, Fu P, Jin S. Posttreatment of Maresin1 Inhibits NLRP3 inflammasome activation via promotion of NLRP3 ubiquitination. FASEB J 2020. [DOI] [PubMed]
- 17.Weathington NM, Sznajder JI, Mallampalli RK. The emerging role of the ubiquitin proteasome in pulmonary biology and disease. Am J Respir Crit Care Med 2013; 188: 530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol 2009; 10: 550–563. [DOI] [PubMed] [Google Scholar]
- 19.Ritorto MS, Ewan R, Perez-Oliva AB, Knebel A, Buhrlage SJ, Wightman M, Kelly SM, Wood NT, Virdee S, Gray NS, Morrice NA, Alessi DR, Trost M. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat Commun 2014; 5: 4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bednash JS, Weathington N, Londino J, Rojas M, Gulick DL, Fort R, Han S, McKelvey AC, Chen BB, Mallampalli RK. Targeting the deubiquitinase STAMBP inhibits NALP7 inflammasome activity. Nat Commun 2017; 8: 15203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sierra MI, Wright MH, Nash PD. AMSH interacts with ESCRT-0 to regulate the stability and trafficking of CXCR4. J Biol Chem 2010; 285: 13990–14004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakamura N, Lill JR, Phung Q, Jiang Z, Bakalarski C, de Maziere A, Klumperman J, Schlatter M, Delamarre L, Mellman I. Endosomes are specialized platforms for bacterial sensing and NOD2 signalling. Nature 2014; 509: 240–244. [DOI] [PubMed] [Google Scholar]
- 23.Alasoo K, Martinez FO, Hale C, Gordon S, Powrie F, Dougan G, Mukhopadhyay S, Gaffney DJ. Transcriptional profiling of macrophages derived from monocytes and iPS cells identifies a conserved response to LPS and novel alternative transcription. Sci Rep 2015; 5: 12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bryan NB, Dorfleutner A, Rojanasakul Y, Stehlik C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol 2009; 182: 3173–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 2013; 49: 331–338. [DOI] [PubMed] [Google Scholar]
- 26.Tang J, Tu S, Lin G, Guo H, Yan C, Liu Q, Huang L, Tang N, Xiao Y, Pope RM, Rajaram MVS, Amer AO, Ahmer BM, Gunn JS, Wozniak DJ, Tao L, Coppola V, Zhang L, Langdon WY, Torrelles JB, Lipkowitz S, Zhang J. Sequential ubiquitination of NLRP3 by RNF125 and Cbl-b limits inflammasome activation and endotoxemia. J Exp Med 2020; 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han S, Lear TB, Jerome JA, Rajbhandari S, Snavely CA, Gulick DL, Gibson KF, Zou C, Chen BB, Mallampalli RK. Lipopolysaccharide Primes the NALP3 Inflammasome by Inhibiting Its Ubiquitination and Degradation Mediated by the SCFFBXL2 E3 Ligase. J Biol Chem 2015; 290: 18124–18133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song H, Liu B, Huai W, Yu Z, Wang W, Zhao J, Han L, Jiang G, Zhang L, Gao C, Zhao W. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat Commun 2016; 7: 13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 2012; 287: 36617–36622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duong BH, Onizawa M, Oses-Prieto JA, Advincula R, Burlingame A, Malynn BA, Ma A. A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity 2015; 42: 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzuki S, Mimuro H, Kim M, Ogawa M, Ashida H, Toyotome T, Franchi L, Suzuki M, Sanada T, Suzuki T, Tsutsui H, Nunez G, Sasakawa C. Shigella IpaH7.8 E3 ubiquitin ligase targets glomulin and activates inflammasomes to demolish macrophages. Proc Natl Acad Sci U S A 2014; 111: E4254–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDonell LM, Mirzaa GM, Alcantara D, Schwartzentruber J, Carter MT, Lee LJ, Clericuzio CL, Graham JM Jr., Morris-Rosendahl DJ, Polster T, Acsadi G, Townshend S, Williams S, Halbert A, Isidor B, David A, Smyser CD, Paciorkowski AR, Willing M, Woulfe J, Das S, Beaulieu CL, Marcadier J, Consortium FC, Geraghty MT, Frey BJ, Majewski J, Bulman DE, Dobyns WB, O’Driscoll M, Boycott KM. Mutations in STAMBP, encoding a deubiquitinating enzyme, cause microcephaly-capillary malformation syndrome. Nat Genet 2013; 45: 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCullough J, Row PE, Lorenzo O, Doherty M, Beynon R, Clague MJ, Urbe S. Activation of the endosome-associated ubiquitin isopeptidase AMSH by STAM, a component of the multivesicular body-sorting machinery. Curr Biol 2006; 16: 160–165. [DOI] [PubMed] [Google Scholar]
- 34.Awad F, Assrawi E, Jumeau C, Georgin-Lavialle S, Cobret L, Duquesnoy P, Piterboth W, Thomas L, Stankovic-Stojanovic K, Louvrier C, Giurgea I, Grateau G, Amselem S, Karabina SA. Impact of human monocyte and macrophage polarization on NLR expression and NLRP3 inflammasome activation. PLoS One 2017; 12: e0175336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, van de Veerdonk FL, Ferwerda G, Heinhuis B, Devesa I, Funk CJ, Mason RJ, Kullberg BJ, Rubartelli A, van der Meer JW, Dinarello CA. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 2009; 113: 2324–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.