

Abstract
Viruses are diverse biological entities that influence all life. Even with limited genome sizes, viruses can manipulate, drive, steal from, and kill their hosts. The field of virus genomics, using sequencing data to understand viral capabilities, has seen significant innovations in recent years. However, with advancements in metagenomic sequencing and related technologies, the bottleneck to discovering and employing the virosphere has become the analysis of genomes rather than generation. With metagenomics rapidly expanding available data, vital components of virus genomes and features are being overlooked, with the issue compounded by lagging databases and bioinformatics methods. Despite the field moving in a positive direction, there are noteworthy points to keep in mind, from how software-based virus genome predictions are interpreted to what information is overlooked by current standards. In this review, we discuss conventions and ideologies that likely need to be revised while continuing forward in the study of virus genomics.
Introduction
Genomics approaches for the study of viruses (infecting eukarya and archaea) and bacteriophages (phage; viruses infecting bacteria) has taken off in the last few years, much in part due to our ability to understand and interpret viral genomes from metagenomes. In fact, it is common to find a publication describing environmental virus genomics from the last few years that indicate viruses as the most abundant and diverse biological entities on the planet. As a scientific community, we are recognizing the extensive footprint viruses leave on all environments where life exists. For example, examining viral genomes has allowed us to discover metabolic genes encoded by viruses such as for photosynthesis and sulfur oxidation, and extrapolate the impacts of virus-directed metabolism on various biogeochemical processes [1–8]. Investigating viral genomes has also aided in the innovation of novel CRISPR-based genome editing technologies [9–11], further development of phage therapy applications [12,13], broader understanding of human gut dysbiosis [14–16], and more.
Unseen to our daily lives, viruses and phages are constantly modifying the planet around us through manipulation and/or lysis of their hosts [17]. Unfortunately, only a small fraction of all viruses that are estimated to exist have been cultivated in the laboratory. This has led to great interest in utilizing next-generation sequencing and metagenomics specifically, to catalog, explore, describe, and understand the diversity of viral genomes [18–21]. Through metagenomic methods and technologies, thousands of viral genomes can be acquired from a single mixed metagenome (mixed community) or virome (virus-specific) sample.
There are two general methods by which to obtain genomic information to study viruses using metagenomics: extraction and sequencing of viromes, and virus prediction from mixed microbial metagenomes (Figure 1). A virome differs from a conventional mixed microbial metagenome in that it is the physical separation, collection, and sequencing of virus-like particles (VLPs) from a sample. Methodologies of VLP collection vary considerably and require modification depending on the source environment (e.g., soil, aquatic, human gut). Each method comes with its own use-case utilities, biases, and ease-of-use, and no one method is globally accepted in the field. A virome can be described as an in situ method of virus discovery. On the other hand, virus prediction is the in silico discovery of virus sequences from a metagenome, or even a virome; a software tool or manual sequence inspection is used to separate viral from non-viral sequences within a mixed community. Notably, there are distinct differences between these two methods that impact the way in which the data is analyzed. For studies specifically focused on the viral fraction of an ecosystem, VLP sequencing of the virome can yield results best suited for studying viral communities [22]. Virome samples are often better at capturing low abundance viruses but may exclude viral genomes that are in an intracellular state (e.g., non-replicating proviruses and virocells) [17]. Conversely, predicting viral sequences from bulk metagenomes can provide context of the viruses and microbes together within the same sample, such as allowing for more accurate host predictions or identifying intracellular viral genomes [23,24].
Figure 1. Sample collection and metagenomic sequencing of viruses.
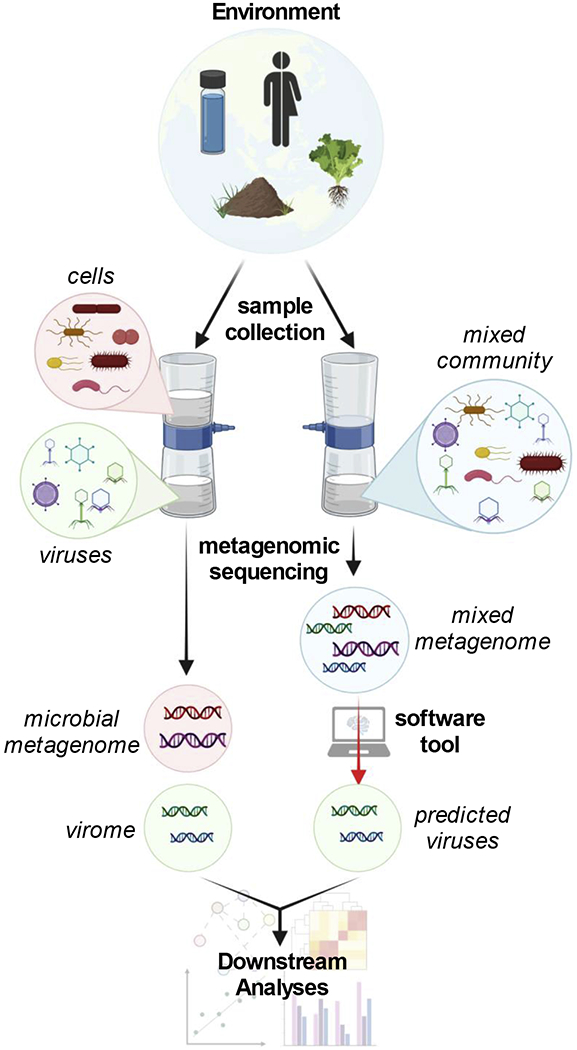
Virus genomes can be identified by physical separation from cells (left) or by software tool prediction (right) preceding downstream analyses.
In the last few years there has been a rapid expansion in the knowledge of viruses on a global genomics level by using metagenomes. Here, we slow down and take a step back to ask what is being overlooked? Considering the current state of virus genomics, where should conventions be broken, and innovations be made? To do this, we will explore some of the methods available to extract viral sequences from metagenomes and describe best practices of how those sequences can or should be analyzed. Here, we will focus on software-based virus prediction methods and their benefits, utilities, flaws, biases, and future directions.
Sweeping contamination under the rug: balancing recovery and false discovery
Virus prediction from mixed metagenomes is powerful in that it allows for an entire sample to have nucleotides extracted and sequenced while maintaining the integrity of the original microbial community comprised of organisms and viruses. A substantial number of software tools are currently available to predict viruses from nucleotides with varying methods, degrees of precision, and recovery capabilities [25–36]. In all cases, it is vital to consider the reality of these predictions in that all computational methods have drawbacks (Figure 2a, Table 1).
Figure 2. Conceptual summary diagram.
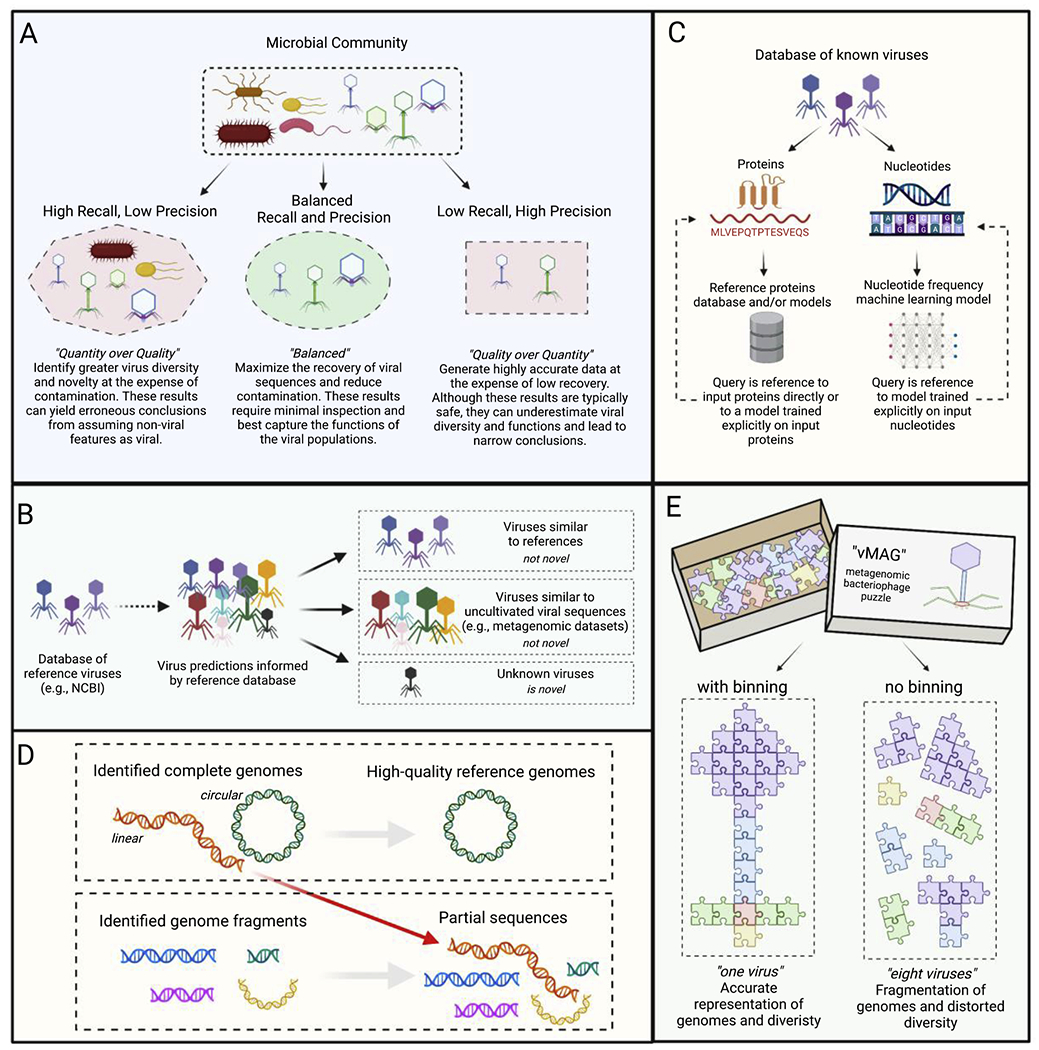
A: comparison of general virus prediction strategies utilized by software tools, from variable recall and precision capabilities to a balanced approach. B: categorization of virus predictions as “not novel” or “novel” according to similarity to reference databases and datasets of uncultivated viral sequences. C: the reference-free fallacy; visualization of how virus prediction software tools, whether protein annotation-based (left) or nucleotide feature-based (right), are all inherently referenced-based. D: the fate of complete linear versus circular viral genomes in interpreting metagenomic data. E: illustration of a viral genome either binned into a vMAG (left) or analyzed as individual fragments (right); each sequence fragment is represented by puzzle pieces.
Table 1.
Recommendations for the questions, biases, and pitfalls posed in each section.
| Sweeping contamination under the rug: balancing recovery and false discovery All software tools that predict viruses from metagenomes can make mistakes |
|---|
| 1. Using multiple virus prediction tools and combining results can strengthen predictions by mitigating the biases and pitfall of each individual tool 2. In published work, report all parameters and thresholds used for predicting viruses, including methods of manual curation 3. Selecting low thresholds when running software or retaining low probability predictions will often generate “more data” at the expense of that data being low quality (i.e., contaminated) 4. Read the tool’s publication (if available) in addition to the software documentation to best understand the tool’s utility, pitfalls, and performance benchmarks |
| Of reference and reality The reliance of most software tools on reference databases is a source of bias |
| 1. Consider homology search to additional curated databases in addition to NCBI databases when reporting novel sequences or gene features |
| The reference-free fallacy: no such thing as a reference-free virus prediction No current tool for predicting virus sequences is reference-free |
| 1. Repeated training tools on NCBI databases has led to overlap in training and testing datasets across tools, making benchmarks increasingly difficult to perform without bias. Including non-NCBI databases in training, testing, and curating databases can reduce bias 2. Avoid falsely assuming database-independent machine learning models, whether trained on protein annotations or nucleotide features, overcome the necessity for reference-based searches |
| Linear genomes can be complete: where did all the linear genomes go? Emphasis is placed on circular genomes as complete, excluding linear genomes |
| 1. Although complete, linear genomes may be identified as high quality or near complete, the lack of circularization signatures underemphasizes these genomes in databases or analyses 2. A metagenomics-scale approach to identify complete viral genomes without terminal repeats may reduce the bias towards circular genomes. Until such a tool is available, it is necessary to keep in mind the possibility of underrepresenting linear genomes |
| Metagenomes are puzzles: an unfinished puzzle is still just pieces Not all metagenomic viral scaffolds represent the whole genome |
| 1. The inclusion of binning in virus analysis pipelines and constructing viral metagenome-assembled genomes (vMAGs) will likely better represent true composition of viruses and viral diversity |
Virus prediction, for the vast majority of implementations, do not encompass all viruses in a sample due to loss in recovery, low sequencing depth of the viruses compared to microbes, or biases against certain viral families. Therefore, when using software to predict viral sequences, the recovered viruses will represent a subset of the true composition. These results can be influenced by the specific computational methods utilized by different tools or universal limitations in available methods [37]. For example, all currently available tools are limited by known virus diversity and struggle to predict viruses with entirely novel sequences. Many tools are also biased toward dsDNA viruses and phages due to dsDNA-centric databases and sequencing methods. Likewise, viral genome sequences comprised mostly of genes or features common to both viruses and organisms are difficult to identify accurately. These biases have the potential to leave behind viruses with novelty to reference databases or regions of recent recombination without close inspection [38,39]. In general, all software tools can only find viruses that appear similar to what we already know about due to reliance on reference-based prediction methods (see the reference-free fallacy below). This limitation has been addressed by incorporating non-reference (e.g., metagenomic) sequences into software training algorithms, but with the caveat that contamination of virus predictions or virome extractions is not uncommon [25,40].
Contamination, or false discovery, of non-viral sequences is a feature of all virus prediction software and should not be ignored. That is, not all recovered sequences predicted to be viruses should be included haphazardly into analyses [41]. In most cases, the time, expertise, and/or computational resources are not available to manually validate all recovered viruses. However, the reality behind the precision of predictions should be made clear, such as providing details of how the prediction results may have been validated including software-specific cutoffs and identification of viral hallmark genes [42]. This is especially relevant when considering the ratio of recovery to precision. For example, reporting numbers of high virus identifications (high recovery) at the expense of the validity of those identifications (low precision) yields seemingly valuable but fundamentally flawed data. Low precision can result from the poor performance of a software tool, incorrect usage of a software tool (e.g., wrong implementation or retaining low probability or scored predictions), inclusion of many short sequence fragments (e.g., less than 3 kb), and other factors.
The following sections stem from the original biases and limitations of the current state of virus prediction. By exploring these topics, we aim to shed light on the potential advancements in computational methods or inconsistencies in interpretations for viral metagenomic data.
Of reference and reality
Many of the gold standards (i.e., trusted reference sequences) for viral genomes are deposited in public repositories such as NCBI databases [43,44]. These sequences are utilized by various software tools beyond virus prediction, such as for prediction of hosts of viruses, prediction of virus taxonomy, functional annotation, genome quality assessment, and more [45–47]. However, this presents significant biases owing to the small and non-diverse composition of NCBI databases, relative to nature. The diversity of viruses by taxonomy and sequence composition within NCBI databases is estimated to be far less than what can be identified in nature and is primarily limited to viruses that have been cultivated on a limited number of hosts, mostly those of clinical significance or as a model research system [48]. Considering virus prediction software tools are reliant on these reference databases, it is clear that there are pitfalls associated with assuming that reference sequences fully mimic natural reality.
Similarly, the designation of viral genomes as “novel” according to a database search is not equivalent to true novelty. True novelty refers to if a given genome has yet to be identified by other sources and is not deposited in another database. For example, a search of NCBI databases excludes the majority of metagenome-derived viral sequences, many of which can be found throughout the literature and in curated databases [21,23,40,49]. Therefore, a virus may be novel with regard to reference database sequences, but not actually represent a truly novel sequence. Another source of novelty can be if the given sequence contains features yet to be discovered or broader implications that have yet to be identified. For example, the identification of crAssphages as highly abundant in the human gut came after representative sequences were deposited into databases [50] (Figure 2b, Table 1).
The reference-free fallacy: no such thing as a reference-free virus prediction
Many virus prediction software tools are based on bona fide genomes derived from NCBI RefSeq, which is mainly composed of isolated and cultivated viruses that serve as reference systems. There are two broad categories of tools according to the methods used: nucleotide sequence features (e.g., VirFinder) and protein similarity (e.g., VIBRANT), or a hybrid of both (e.g., VirSorter2) [25–27]. For either category, machine learning has become a powerful approach for identifying patterns to increase prediction reliability and specificity [51]. However, this has led to some misconceptions to believe that “reference-free” refers to complete independence from reference databases, whereas “reference-based” refers to the use of protein annotation methods based on the annotations of reference viruses. Conversely, we advocate there is no tool completely reference-free and rather all tools are inherently reference-based in some manner (Figure 2c, Table 1).
For a tool that utilizes protein annotation, the reliance on reference sequences is in the form of prediction models built from a protein database [52–54], which is a clear reference-dependent method. Namely, only reference proteins are able to be annotated, queried, and subsequently analyzed. On the other hand, a tool that strictly uses sequence features (e.g., tetra-nucleotide frequency) does not necessarily need to rely on a database, but can rather rely on a machine learning model. This machine learning model can be perceived as reference-free, but similar to a protein database, the model too is dependent on the reference sequences used to train it. Therefore, for both categories of tools there is a direct reliance on reference sequences, making them both inherently reference based. A more accurate distinction would be “database-dependent” or “database-free” methods. Even manual verification of virus predictions is not reference-free as this method typically involves searching through protein annotations (e.g., phage structural hallmark proteins) and other reference-informed signatures (e.g., gene density and gene strand switch frequency) [55].
Moreover, it is important to note that the reference sequences used to compare, train and test software tools and/or machine learning models typically all come from the same genetic pool (i.e., NCBI databases). This perpetuates biases: biases against rare virus groups and biases in accurate comparisons. First, it is estimated that the true diversity of viruses in nature has yet to be captured by the sequences available on NCBI databases [19,49,56]. This results in a lack of representation of more rare viruses or simply those that have yet to be isolated/cultivated [39,57–59]. Since virus prediction tools are inherently reference-based, this leads to perpetual biases towards identifying viruses we already know about, with rare occasions of identifying a truly novel species [57]. Second, the utilization of NCBI databases for assessing available software tools results in an inherent loss of fair comparisons. It is becoming increasingly difficult to generate a comparison dataset of gold standard viral sequences that does not, in some capacity, represent the sequences used to train existing tools. This is due to the limited size of NCBI databases. Especially for tools that utilize machine learning, evaluating a tool with a sequence that was used to train that tool results in inflated, positive performance. The common work around is to only include viral sequences submitted to NCBI databases after the dates of publication for tools to compare, but this also results in biases, such as the inclusion of viruses nearly identical to those submitted previously. This latter example can be addressed by removing any identical sequences via dereplication, though this is seldom employed. In attempts to solve this issue and generate comprehensive, fair datasets for future software tool development and comparison, more focus and better curation standards need to be placed on the construction of reference sequence datasets.
Linear genomes can be complete: where did all the linear genomes go?
Identifying complete viral genomes from sequencing data allows for more robust analyses compared to fragmented, partial genomes. Automated methods to predict complete viral genomes focus on circularization signatures, namely the identification of terminal nucleotide repeats (direct or inverted) of free viral sequences or insertion sites of viruses integrated into their host’s genome (proviruses) [25,26,29,30,34,47]. For free (lytic cycle) viruses, the identification of circularization can typically indicate with confidence that the given genome is complete. However, this method discounts complete linear genomes, such as those without identifiable terminal repeats [60].
Thus far, no high-throughput informatics method exists for the identification of complete linear genomes in the absence of circularization signatures [47,61]. This results in over-emphasizing circular genomes as the only gold standards in generating metagenomic-based reference genomes or the highest quality genomes in genomic datasets. Though these conclusions are not flawed on their own as correctly identified circular genomes are certainly of high quality, barring false positives [62], this overall bias against linear genomes has infiltrated the currently available literature (Figure 2d, Table 1). Speculatively, the ability to identify complete, linear virus genomes may allow for a more holistic view of a viral community or lead to novel discoveries of underappreciated viral groups.
Metagenomes are puzzles: an unfinished puzzle is still just pieces
Metagenomic assemblies reconstruct thousands to millions of sequence fragments (contigs) representing partial genomes, and rarely complete genomes. A common practice in the study of bacterial and archaeal genomes is to reconstruct metagenome-assembled genomes (MAGs) [63,64]. This is typically done through a method termed binning where anywhere from two to hundreds or even thousands of contigs may be grouped into a single, putative genome (bin). When using short read (e.g., 75-300 bp) sequencing technology and assembly, many resulting contigs are less than 5 kb in length, with relatively few exceeding 20 kb. Consequently, bacterial and archaeal genomes that generally exceed 1,000 kb must be computationally binned into MAGs. Though long-read (e.g., 1-20 kb) technologies are advancing these boundaries, the construction of MAGs is typically still required. For bacteria and archaea, several software tools are available for binning and constructing MAGs [65–70].
Viral genomes range from as small as 3 kb to greater than 2,000 kb. Many identified phages are members of the class Caudoviricetes (formerly Caudovirales) which range considerably in size, but most are approximately 30 kb to 200 kb [71]. Interestingly, the convention accepted in descriptions of viruses derived from viromes or predicted from metagenomes is that a single contig represents an uncultivated viral genome (UViG) or virus population [19]. To assume each sequence represents a separate genome likely far overestimates viral diversity within a sample given the expected fragmentation of viral genomes. This is especially true for viruses that are rarer and would likely result in high genome fragmentation after assembly. The construction of viral metagenome-assembled genomes (vMAGs) would better represent the true composition of viruses within a sample. Importantly, UViGs still have utility in that any viral sequence left unbinned may represent an entire viral population, contrary to what is accepted for bacteria and archaea where unbinned sequences are typically discarded (Figure 2e, Table 1). This can be achieved by binning vMAGs using short- or long-read sequencing [72]. Despite this, few studies bin vMAGs, and those that do bin typically focus on viruses with the largest genomes [5,73–75]. This conspicuous discrepancy of binning bacteria and archaea, but not viruses, is a convention that likely hinders advancement in the field of viral metagenomics. Development of virus binning tools, such as vRhyme [76], will fuel this advancement.
Conclusions
Virus genomics, specifically metagenomics, allows for the circumvention of conventional cultivation approaches to study viruses, their impacts on microbial communities, biogeochemistry, applications for biotechnology, human medicine, and more. After sequencing a sample, it has become just a few keystrokes and a click of a button to obtain a list of the viruses present. The outcome is that our knowledge of viral genomic diversity has increased at a near exponential rate over the last few years, opening new and exciting opportunities. However, this has been at the expense of biasing conclusions due to tools, methodologies, and conventions that lag data acquisition.
We are led to several overarching questions. Are virus predictions capturing the true nature of a community of viruses? Are heavily reference-guided predictions making it easy to miss any undiscovered novelty without studious inspection? Are conventions in identifying high-quality and complete viral genomes ignoring entire viral groups with unique genome architecture? Is the field as a whole moving too fast to fully consider the scope of the genomes presented?
There is no single set of answers to address all these questions easily. Rather, recognizing the limitations of the available methods will help to best work towards an optimized, efficient, and accurate approach to handle the rapid, near-constant flow of sequencing information. The goal is a fair, holistic representation of the global virosphere to best understand how viruses influence all life.
Acknowledgements
We thank Evelien Adriaenssens and Jelle Matthijnssens for the invitation to contribute to this special series. We thank members of the Anantharaman laboratory at the University of Wisconsin-Madison for helpful feedback and discussions. K.K. was supported by a Wisconsin Distinguished Graduate Fellowship Award from the University of Wisconsin-Madison, and a William H. Peterson Fellowship Award from the Department of Bacteriology, University of Wisconsin-Madison. This research was supported by National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM143024, and by the National Science Foundation under grant number DBI2047598. Figures were created with Biorender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors declare no conflicts of interest.
References
- [1].Bragg JG, Chisholm SW: Modeling the Fitness Consequences of a Cyanophage-Encoded Photosynthesis Gene. PLOS ONE 2008, 3:e3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mann NH, Cook A, Millard A, Bailey S, Clokie M: Bacterial photosynthesis genes in a virus. Nature 2003, 424:741. [DOI] [PubMed] [Google Scholar]
- [3].Roux S, Hawley AK, Beltran MT, Scofield M, Schwientek P, Stepanauskas R, Woyke T, Hallam SJ, Sullivan MB: Ecology and evolution of viruses infecting uncultivated SUP05 bacteria as revealed by single-cell- and meta-genomics. eLife Sciences 2014, 3:e03125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Trubl G, Jang HB, Roux S, Emerson JB, Solonenko N, Vik DR, Solden L, Ellenbogen J, Runyon AT, Bolduc B, et al. : Soil Viruses Are Underexplored Players in Ecosystem Carbon Processing. mSystems 2018, 3:e00076–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chen L-X, Méheust R, Crits-Christoph A, McMahon KD, Nelson TC, Slater GF, Warren LA, Banfield JF: Large freshwater phages with the potential to augment aerobic methane oxidation. Nat Microbiol 2020, 5:1504–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Roux S, Brum JR, Dutilh BE, Sunagawa S, Duhaime MB, Loy A, Poulos BT, Solonenko N, Lara E, Poulain J, et al. : Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537:689–693. [DOI] [PubMed] [Google Scholar]
- [7].Kieft K, Breister AM, Huss P, Linz AM, Zanetakos E, Zhou Z, Rahlff J, Esser SP, Probst AJ, Raman S, et al. : Virus-associated organosulfur metabolism in human and environmental systems. Cell Rep 2021, 36:109471. [DOI] [PubMed] [Google Scholar]
- [8].Kieft K, Zhou Z, Anderson RE, Buchan A, Campbell BJ, Hallam SJ, Hess M, Sullivan MB, Walsh DA, Roux S, et al. : Ecology of inorganic sulfur auxiliary metabolism in widespread bacteriophages. Nat Commun 2021, 12:3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mojica FJM, Díez-Villaseñor C, García-Martínez J, Soria E: Intervening Sequences of Regularly Spaced Prokaryotic Repeats Derive from Foreign Genetic Elements. J Mol Evol 2005, 60:174–182. [DOI] [PubMed] [Google Scholar]
- [10].Pourcel C, Salvignol G, Vergnaud GY 2005: CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology [date unknown], 151:653–663. [DOI] [PubMed] [Google Scholar]
- [11].Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. : Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fujimoto K, Kimura Y, Shimohigoshi M, Satoh T, Sato S, Tremmel G, Uematsu M, Kawaguchi Y, Usui Y, Nakano Y, et al. : Metagenome Data on Intestinal Phage-Bacteria Associations Aids the Development of Phage Therapy against Pathobionts. Cell Host & Microbe 2020, 28:380–389.e9. [DOI] [PubMed] [Google Scholar]
- [13].Chatterjee A, Willett JLE, Nguyen UT, Monogue B, Palmer KL, Dunny GM, Duerkop BA: Parallel Genomics Uncover Novel Enterococcal-Bacteriophage Interactions. mBio [date unknown], 11:e03120–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mangalea MR, Paez-Espino D, Kieft K, Chatterjee A, Chriswell ME, Seifert JA, Feser ML, Demoruelle MK, Sakatos A, Anantharaman K, et al. : Individuals at risk for rheumatoid arthritis harbor differential intestinal bacteriophage communities with distinct metabolic potential. Cell Host & Microbe 2021, 29:726–739.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shkoporov AN, Clooney AG, Sutton TDS, Ryan FJ, Daly KM, Nolan JA, McDonnell SA, Khokhlova EV, Draper LA, Forde A, et al. : The Human Gut Virome Is Highly Diverse, Stable, and Individual Specific. Cell Host & Microbe 2019, 26:527–541.e5. [DOI] [PubMed] [Google Scholar]
- [16].Clooney AG, Sutton TDS, Shkoporov AN, Holohan RK, Daly KM, O’Regan O, Ryan FJ, Draper LA, Plevy SE, Ross RP, et al. : Whole-Virome Analysis Sheds Light on Viral Dark Matter in Inflammatory Bowel Disease. Cell Host & Microbe 2019, 26:764–778.e5. [DOI] [PubMed] [Google Scholar]
- [17].Howard-Varona C, Lindback MM, Bastien GE, Solonenko N, Zayed AA, Jang H, Andreopoulos B, Brewer HM, Rio TG del, Adkins JN, et al. : Phage-specific metabolic reprogramming of virocells. ISME J 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Camarillo-Guerrero LF, Almeida A, Rangel-Pineros G, Finn RD, Lawley TD: Massive expansion of human gut bacteriophage diversity. Cell 2021, 184:1098–1109.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Roux S, Adriaenssens EM, Dutilh BE, Koonin EV, Kropinski AM, Krupovic M, Kuhn JH, Lavigne R, Brister JR, Varsani A, et al. : Minimum Information about an Uncultivated Virus Genome (MIUViG). Nature Biotechnology 2019, 37:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Paez-Espino D, Zhou J, Roux S, Nayfach S, Pavlopoulos GA, Schulz F, McMahon KD, Walsh D, Woyke T, Ivanova NN, et al. : Diversity, evolution, and classification of virophages uncovered through global metagenomics. Microbiome 2019, 7:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gregory AC, Zayed AA, Conceição-Neto N, Temperton B, Bolduc B, Alberti A, Ardyna M, Arkhipova K, Carmichael M, Cruaud C, et al. : Marine DNA Viral Macro- and Microdiversity from Pole to Pole. Cell 2019, 177:1109–1123.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Santos-Medellin C, Zinke LA, ter Horst AM, Gelardi DL, Parikh SJ, Emerson JB: Viromes outperform total metagenomes in revealing the spatiotemporal patterns of agricultural soil viral communities. ISME J 2021, 15:1956–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gregory AC, Zablocki O, Zayed AA, Howell A, Bolduc B, Sullivan MB: The Gut Virome Database Reveals Age-Dependent Patterns of Virome Diversity in the Human Gut. Cell Host & Microbe 2020, 28:724–740.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kieft K, Anantharaman K: Deciphering active prophages from metagenomes. bioRxiv 2021, doi: 10.1101/2021.01.29.428894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Guo J, Bolduc B, Zayed AA, Varsani A, Dominguez-Huerta G, Delmont TO, Pratama AA, Gazitúa MC, Vik D, Sullivan MB, et al. : VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 2021, 9:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kieft K, Zhou Z, Anantharaman K: VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020, 8:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ren J, Ahlgren NA, Lu YY, Fuhrman JA, Sun F: VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Saw AK, Raj G, Das M, Talukdar NC, Tripathy BC, Nandi S: Alignment-free method for DNA sequence clustering using Fuzzy integral similarity. Scientific Reports 2019, 9:3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS: PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 2016, 44:W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Song W, Sun H-X, Zhang C, Cheng L, Peng Y, Deng Z, Wang D, Wang Y, Hu M, Liu W, et al. : Prophage Hunter: an integrative hunting tool for active prophages. Nucleic Acids Res 2019, 47:W74–W80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Antipov D, Raiko M, Lapidus A, Pevzner PA: MetaviralSPAdes: assembly of viruses from metagenomic data. Bioinformatics 2020, 36:4126–4129. [DOI] [PubMed] [Google Scholar]
- [32].Deaton J, Yu FB, Quake SR: PhaMers identifies novel bacteriophage sequences from thermophilic hot springs. bioRxiv 2017, doi: 10.1101/169672. [DOI] [Google Scholar]
- [33].Zheng T, Li J, Ni Y, Kang K, Misiakou M-A, Imamovic L, Chow BKC, Rode AA, Bytzer P, Sommer M, et al. : Mining, analyzing, and integrating viral signals from metagenomic data. Microbiome 2019, 7:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Roux S, Enault F, Hurwitz BL, Sullivan MB: VirSorter: mining viral signal from microbial genomic data. PeerJ 2015, 3:e985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Amgarten D, Braga LPP, da Silva AM, Setubal JC: MARVEL, a Tool for Prediction of Bacteriophage Sequences in Metagenomic Bins. Frontiers in Genetics 2018, 9:304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Aylward FO, Moniruzzaman M: ViralRecall—A Flexible Command-Line Tool for the Detection of Giant Virus Signatures in ‘Omic Data. Viruses 2021, 13:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ponsero AJ, Hurwitz BL: The Promises and Pitfalls of Machine Learning for Detecting Viruses in Aquatic Metagenomes. Frontiers in Microbiology 2019, 10:806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Roux S, Krupovic M, Daly RA, Borges AL, Nayfach S, Schulz F, Sharrar A, Carnevali PBM, Cheng J-F, Ivanova NN, et al. : Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth’s biomes. Nature Microbiology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Devoto AE, Santini JM, Olm MR, Anantharaman K, Munk P, Tung J, Archie EA, Turnbaugh PJ, Seed KD, Blekhman R, et al. : Megaphages infect Prevotella and variants are widespread in gut microbiomes. Nature Microbiology 2019, 4:693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Paez-Espino D, Eloe-Fadrosh EA, Pavlopoulos GA, Thomas AD, Huntemann M, Mikhailova N, Rubin E, Ivanova NN, Kyrpides NC: Uncovering Earth’s virome. Nature 2016, 536:425–430. [DOI] [PubMed] [Google Scholar]
- [41].Roux S, Krupovic M, Debroas D, Forterre P, Enault F: Assessment of viral community functional potential from viral metagenomes may be hampered by contamination with cellular sequences. Open Biology [date unknown], 3:130160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pratama AA, Bolduc B, Zayed AA, Zhong Z-P, Guo J, Vik DR, Gazitúa MC, Wainaina JM, Roux S, Sullivan MB: Expanding standards in viromics: in silico evaluation of dsDNA viral genome identification, classification, and auxiliary metabolic gene curation. PeerJ 2021, 9:e11447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. : Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 2016, 44:D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW: GenBank. Nucleic Acids Res 2016, 44:D67–D72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jang HB, Bolduc B, Zablocki O, Kuhn JH, Roux S, Adriaenssens EM, Brister JR, Kropinski AM, Krupovic M, Lavigne R, et al. : Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nature Biotechnology 2019. [DOI] [PubMed] [Google Scholar]
- [46].Ecale Zhou CL, Malfatti S, Kimbrel J, Philipson C, McNair K, Hamilton T, Edwards R, Souza B: multiPhATE: bioinformatics pipeline for functional annotation of phage isolates. Bioinformatics 2019, 35:4402–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nayfach S, Camargo AP, Schulz F, Eloe-Fadrosh E, Roux S, Kyrpides NC: CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat Biotechnol 2021, 39:578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Dion MB, Oechslin F, Moineau S: Phage diversity, genomics and phylogeny. Nature Reviews Microbiology 2020. [DOI] [PubMed] [Google Scholar]
- [49].Roux S, Páez-Espino D, Chen I-MA, Palaniappan K, Ratner A, Chu K, Reddy TBK, Nayfach S, Schulz F, Call L, et al. : IMG/VR v3: an integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Research 2021, 49:D764–D775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dutilh BE, Cassman N, McNair K, Sanchez SE, Silva GGZ, Boling L, Barr JJ, Speth DR, Seguritan V, Aziz RK, et al. : A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nature Communications 2014, 5:4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Auslander N, Gussow AB, Koonin EV: Incorporating Machine Learning into Established Bioinformatics Frameworks. International Journal of Molecular Sciences 2021, 22:2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Grazziotin AL, Koonin EV, Kristensen DM: Prokaryotic Virus Orthologous Groups (pVOGs): a resource for comparative genomics and protein family annotation. Nucleic Acids Res 2017, 45:D491–D498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].UniProt Consortium T: UniProt: the universal protein knowledgebase. Nucleic Acids Res 2018, 46:2699–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zayed AA, Lücking D, Mohssen M, Cronin D, Bolduc B, Gregory AC, Hargreaves KR, Piehowski PD, White RA, Huang EL, et al. : efam: an expanded, metaproteome-supported HMM profile database of viral protein families. Bioinformatics 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Shaffer M, Borton MA, McGivern BB, Zayed AA, La Rosa SL, Solden LM, Liu P, Narrowe AB, Rodríguez-Ramos J, Bolduc B, et al. : DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Research 2020, 48:8883–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Brister JR, Ako-Adjei D, Bao Y, Blinkova O: NCBI viral genomes resource. Nucleic Acids Res 2015, 43:D571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kauffman KM, Hussain FA, Yang J, Arevalo P, Brown JM, Chang WK, VanInsberghe D, Elsherbini J, Sharma RS, Cutler MB, et al. : A major lineage of non-tailed dsDNA viruses as unrecognized killers of marine bacteria. Nature; London 2018, 554:118–122,122A-122T. [DOI] [PubMed] [Google Scholar]
- [58].Krishnamurthy SR, Janowski AB, Zhao G, Barouch D, Wang D: Hyperexpansion of RNA Bacteriophage Diversity. PLOS Biology 2016, 14:e1002409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Callanan J, Stockdale S, Shkoporov A, Draper L, Ross RP, Hill C: Expansion of known ssRNA phage genomes: From tens to over a thousand. Science Advances 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Casjens SR, Gilcrease EB: Determining DNA Packaging Strategy by Analysis of the Termini of the Chromosomes in Tailed-Bacteriophage Virions. Methods Mol Biol 2009, 502:91–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Beaulaurier J, Luo E, Eppley JM, Uyl PD, Dai X, Burger A, Turner DJ, Pendelton M, Juul S, Harrington E, et al. : Assembly-free single-molecule sequencing recovers complete virus genomes from natural microbial communities. Genome Res 2020, 30:437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Roux S, Emerson JB, Eloe-Fadrosh EA, Sullivan MB: Benchmarking viromics: an in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ 2017, 5:e3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, Schulz F, Jarett J, Rivers AR, Eloe-Fadrosh EA, et al. : Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nature Biotechnology 2017, 35:725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF: Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 2004, 428:37–43. [DOI] [PubMed] [Google Scholar]
- [65].Wu Y-W, Tang Y-H, Tringe SG, Simmons BA, Singer SW: MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2014, 2:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Nissen JN, Johansen J, Allesøe RL, Sønderby CK, Armenteros JJA, Grønbech CH, Jensen LJ, Nielsen HB, Petersen TN, Winther O, et al. : Improved metagenome binning and assembly using deep variational autoencoders. Nature Biotechnology 2021. [DOI] [PubMed] [Google Scholar]
- [67].Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, Lahti L, Loman NJ, Andersson AF, Quince C: Binning metagenomic contigs by coverage and composition. Nature Methods 2014, 11:1144–1146. [DOI] [PubMed] [Google Scholar]
- [68].Uritskiy GV, DiRuggiero J, Taylor J: MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sieber CMK, Probst AJ, Sharrar A, Thomas BC, Hess M, Tringe SG, Banfield JF: Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nature Microbiology 2018, 3:836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, Wang Z: MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7:e7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Turner D, Kropinski AM, Adriaenssens EM: A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13:506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Warwick-Dugdale J, Solonenko N, Moore K, Chittick L, Gregory AC, Allen MJ, Sullivan MB, Temperton B: Long-read viral metagenomics captures abundant and microdiverse viral populations and their niche-defining genomic islands. PeerJ 2019, 7:e6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Moniruzzaman M, Martinez-Gutierrez CA, Weinheimer AR, Aylward FO: Dynamic genome evolution and complex virocell metabolism of globally-distributed giant viruses. Nat Commun 2020, 11:1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Schulz F, Andreani J, Francis R, Boudjemaa H, Bou Khalil JY, Lee J, La Scola B, Woyke T: Advantages and Limits of Metagenomic Assembly and Binning of a Giant Virus. mSystems 2020, 5:e00048–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Anantharaman K, Duhaime MB, Breier JA, Wendt KA, Toner BM, Dick GJ: Sulfur Oxidation Genes in Diverse Deep-Sea Viruses. Science 2014, 344:757–760. [DOI] [PubMed] [Google Scholar]
- [76].Kieft K, Adams A, Salamzade R, Kalan L, Anantharaman K: vRhyme enables binning of viral genomes from metagenomes. bioRxiv 2021, doi: 10.1101/2021.12.16.473018. [DOI] [PMC free article] [PubMed] [Google Scholar]