

ABSTRACT
Magnitude and diversity of gut microbiota and metabolic systems are critical in shaping human health and diseases, but it remains largely unclear how complex metabolites may selectively regulate gut microbiota and determine health and diseases. Here, we show that failures or compromised effects of anti-TNF-α therapy in inflammatory bowel diseases (IBD) patients were correlated with intestinal dysbacteriosis with more pro-inflammatory bacteria, extensive unresolved inflammation, failed mucosal repairment, and aberrant lipid metabolism, particularly lower levels of palmitoleic acid (POA). Dietary POA repaired gut mucosal barriers, reduced inflammatory cell infiltrations and expressions of TNF-α and IL-6, and improved efficacy of anti-TNF-α therapy in both acute and chronic IBD mouse models. Ex vivo treatment with POA in cultured inflamed colon tissues derived from Crohn’s disease (CD) patients reduced pro-inflammatory signaling/cytokines and conferred appreciable tissue repairment. Mechanistically, POA significantly upregulated the transcriptional signatures of cell division and biosynthetic process of Akkermansia muciniphila, selectively increased the growth and abundance of Akkermansia muciniphila in gut microbiota, and further reprogrammed the composition and structures of gut microbiota. Oral transfer of such POA-reprogrammed, but not control, gut microbiota induced better protection against colitis in anti-TNF-α mAb-treated recipient mice, and co-administration of POA with Akkermansia muciniphila showed significant synergistic protections against colitis in mice. Collectively, this work not only reveals the critical importance of POA as a polyfunctional molecular force to shape the magnitude and diversity of gut microbiota and therefore promote the intestinal homeostasis, but also implicates a new potential therapeutic strategy against intestinal or abenteric inflammatory diseases.
KEYWORDS: Gut microbiota, Biological therapy, Inflammatory bowel diseases, Akkermansia muciniphila, TNF-α
Introduction
Physiological functions of gut microbiota and metabolic systems (including host and microbial metabolism) play a critical role in modulation of human health and diseases1,2. Unraveling the complexity and diversity of multiplex molecular and chemical interaction networks in gut microbiota and metabolic systems have extremely important implications for understanding pathogenesis mechanisms and development of better therapies for diseases2–4. However, it is full of great challenges to achieve in-depth and precise regulation of microbial and metabolic networks. While previous studies have shown that metabolite profiles may selectively shape the structure, function, or strain-fitness of gut bacteria5,6, it remains largely unknown how more diverse metabolism pathways producing highly diverse molecular structures may selectively regulate growth and physiology of certain bacterial species or strains and ultimately affect the entire gut microbiota structure and metabolic activity.
Inflammatory bowel diseases (IBD), including ulcerative colitis (UC) and Crohn’s disease (CD), are chronic gastrointestinal inflammation with disorders of highly-dynamic gut microbiota and metabolites4,7. It is thought that the development, progression, and poor prognostic outcomes of IBD result from multiple pathogenesis factors involving genetic predisposition8, intestinal microbiome disturbance9, abnormal immune balance10, and mucosal barrier destruction leading to aberrant host-microbial interactions11. Particularly, IBD are associated with reductions of gut microbial diversity, blooms of pro-inflammatory bacteria and fungi, and perturbations of intestinal metabolites4,7,12–14. However, the exact effects and mechanisms of molecular and chemical networks between gut microbiota and metabolic systems on the development, progression, and prognosis of IBD remain largely unknown.
Mucosal integrity provides an intact barrier for colonization and growth of gut microbiota, limiting translocations of pathogenic bacteria or bacterial components and products, and sustaining immune tolerance15, which may serve as a fundamental anatomic and physiological basis for maintaining the delicate balance or homeostasis of chemical networks between gut microbiota and metabolic systems. Thus, promoting intestinal mucosal healing is thought to be one of the most promising therapeutic strategies in IBD16. However, it is unclear how to restore the homeostasis of such networks between gut microbiota and metabolic systems, which may promote the healing of intestinal mucosal integrity and therefore further develop a new therapeutic strategy to control IBD or other intestinal diseases.
In this study, we found that failure of anti-TNF-α treatment in IBD patients attributed to unresolved intestinal inflammation, mucosal non-healing, intestinal dysbacteriosis and lipid abnormalities. Metabolomics analyses revealed that lipid metabolism abnormalities, particularly decreased levels of an anti-inflammatory metabolite, palmitoleic acid (POA), were related to nonresponse to anti-TNF-α therapy, and oral supplement with palmitoleic acid (POA) could significantly repair mucosal barriers and reduce inflammation in both murine models of colitis and highly inflamed colon tissues derived from Crohn’s disease (CD) patients. Mechanistically, palmitoleic acid (POA) rewired disrupted intestinal barrier and reprogrammed gut microbiota with selectively increasing the abundance of anti-inflammatory gut bacteria such as Akkermansia muciniphila. Transcriptomic analysis demonstrated increased expression of cell division, ATP synthesis coupled electron transport and biosynthetic process in Akkermansia muciniphila in response to palmitoleic acid (POA). Moreover, palmitoleic acid (POA) dramatically enhanced the efficacy of anti-TNF-α therapy against colitis in mice.
Therefore, this study not only uncovers previously unrecognized pathogenesis mechanisms of IBD but also provides a potential new therapeutic strategy of IBD and other intestinal or abenteric inflammatory diseases via promoting the homeostasis of molecular and chemical networks between gut microbiota and metabolic systems.
Results
Non-responsiveness to anti-TNF-α therapy, extensive inflammation and mucosal injury are associated with enrichment for pro-inflammatory gut bacteria in IBD patients
Anti-TNF-α therapy is still the most conventionally and widely used biological therapies of IBD, but there are still huge unmet clinical needs for improvement of the efficacy and safety of anti-TNF-α therapy14,17,18. Disruptions of molecular communications and homeostasis in gut microbiota structure and metabolic activity is implicated in the onset and progression of IBD7,19, but it is still unclear whether and how such disruptions are correlated with clinically unmet efficacy of anti-TNF-α therapy. Thus, we hypothesized that disrupted homeostasis in gut microbiota and metabolic systems would have a profound impact on the efficacy of anti-TNF-α therapy.
To address this, we initially assessed the progression and clinical characteristics of IBD patients receiving British Society of Gastroenterology consensus guidelines-based anti-TNF-α (Infliximab, IFX) therapy20. Efficacy or responses to therapy of anti-TNF-α were assessed by the severity of symptoms21,22, classifying patients as either responders [(R) complete or partial response or stable disease≥12 weeks; n = 19] or non-responders [(NR) clinical remission<12 weeks or progressive disease; n = 12] (see Methods for details). Endoscopic analysis serves as a golden standard in examining and assessing the disease severity in the intestinal mucosa as well as evaluating the efficacy of treatments23. Endoscopic analyses suggested that, compared with the healthy controls, anti-TNF-α therapy responders exhibited clinical remission and mucosal healing while non-responders still presented moderate or severe disease with ulcers, stenosis, and fistulas (Figure 1a, b). Compared with responders, non-responders showed higher levels of C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) in the serum (Supplementary Figure S1, A to C), more severe pathology with more extensive mucosal damages and ulcerations in mucosal biopsies (Figure 1c, d). Moreover, mucosal biopsies of non-responders exhibited more infiltrations of inflammatory cells (e.g., CD8+ and CD11b+ cells) and glandular destructions characterized by lower expressions of intestinal mucosal integrity markers, ZO-1 and Occludin (Figure 1e, f and Supplementary Figure S1, D and E). Thus, these data suggest that the compromised effects of anti-TNF-α therapy were associated with unresolved intestinal inflammation and failed functional repairment of colon.
Figure 1.
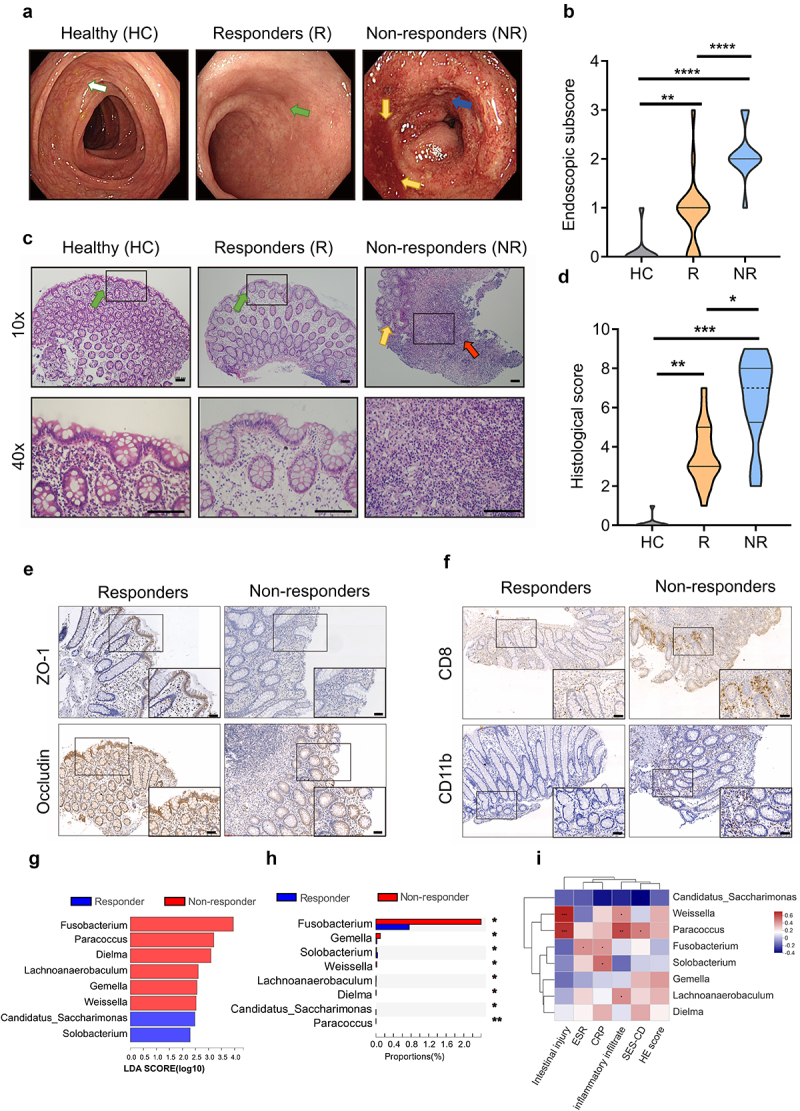
Gut dysbacteriosis is correlated with the compromised effects of anti-TNF-α therapy in IBD patients.
Note: (a) Representative endoscopic images of healthy controls (HC), anti-TNF-α therapy (Infliximab, IFX) responders (R) and non-responders (NR) with IBD. White arrow shows intact intestinal mucosa with clear blood vessels. Green arrow shows the repairment of damaged intestinal mucosa while yellow arrows show extensive mucosal erosion with bleeding. Blue arrow indicates endoscopic ulcers.
(b) Endoscopic subscore for evaluating mucosal appearance of healthy controls (HC), anti-TNF-α therapy responders (R) and non-responders (NR) with IBD.
(c) Representative images of the Hematoxylin and eosin (H&E)-stained colon biopsy sections of healthy controls (HC), anti-TNF-α therapy responders (R) and non-responders (NR) with IBD. Top: 10×, scale bar = 100 μm; Bottom: 40×, scale bar = 100 μm. The black-boxed areas at the top are enlarged in bottom right corner. Green arrow indicates dense and tightly connected intestinal epithelial cells, and yellow arrow shows extensive damage and loss of mucosal epithelial layer. Red arrow marks lesions and infiltration of inflammatory cells.
(d) Histological score for loss of epithelium, crypt damage, depletion of goblet cells and infiltration of inflammatory cells in healthy controls (HC), anti-TNF-α therapy responders (R) and non-responders (NR) with IBD.
(e) Immunohistochemical images of colon sections stained for epithelial marker including ZO-1 and Occludin in anti-TNF-α therapy responders (R) and non-responders (NR) with IBD. A high-magnification of the region is shown (bottom right). Scale bar = 50 μm.
(f) Immunohistochemical images of colon sections stained for CD8+ and CD11b+ cells in colonic lamina propria in anti-TNF-α therapy responders (R) and non-responders (NR) with IBD. A high-magnification of the region is shown (bottom right). Scale bar = 50 μm
(g) Differences in microbial taxa at genus level between anti-TNF-α therapy responders (R) and non-responders (NR) with IBD were calculated by LDA effect size (LEfSe). (P < 0.01 and LDA score>2.0). Higher abundant genera in NR are shaded with red and higher abundant genera in R are shaded with blue.
(h) Proportions of different microbial genus in feces between anti-TNF-α therapy responders (R) and non-responders (NR) with IBD. Significance was tested using Wilcoxon rank-sum test with FDR multiple test correction.
(i) Pearson correlation analysis shows the association between differential microbial abundance and clinical features.
Data represent means ± SEM (HC = 10, R = 19, NR = 12); *P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001; NS, no statistical significance. P values were calculated by Kruskal-Wallis test [(b) and (d)], Wilcoxon rank-sum test (h) and Pearson correlation analysis (i).
Although the alterations of gut microbiota are associated with the effects of anti-TNF-α therapy against IBD14,18,24,25, the precise molecular forces for modulating gut microbiota to improve anti-TNF-α therapy for IBD remain largely unknown. To elucidate such molecular forces, we first addressed the roles of compositional differences and functional diversity of gut microbiome in the efficacy of anti-TNF-α therapy. Fecal samples of anti-TNF-α therapy responders and non-responders with IBD were collected and sequenced by 16S rRNA. α-diversity analyses such as Shannon and Simpson index showed unobservable significant difference in microbial community richness between responders and non-responders (Supplementary Figure S2A). However, LEfSe analysis suggested that non-responders showed higher abundance of Fusobacterium, Paracoccus, Dielma, Lachnoanaerobaculum, Gemella and Weissella, while responders exhibited higher abundance of Candidatus Saccharimonas, Solobacterium at genus level (Figure 1g, h). Remarkably, compared with those in responders, there was a higher abundance of pro-inflammatory bacteria that have been implicated with exacerbation of IBD, such as Escherichia-Shigella, Fusobacterium and Gemella26–28, in non-responders (Supplementary Figure S2B). Thus, there is a significant correlation between shift of gut microbial compositions toward higher abundance of pro-inflammatory bacteria and the compromised efficacy of anti-TNF-α therapy.
We further investigated whether the disease burden and clinical characteristics of IBD patients were associated with alterations of the gut microbiota compositions. We found that most bacterium enriched in non-responders were positively correlated with characteristic of the inflammatory cell infiltration and mucosal non-healing in anti-TNF-α therapy non-responders with IBD (Figure 1i). Enriched pathogenic Fusobacterium was positively associated with inflammatory markers CRP and ESR, while high abundance of Paracoccus and Weissella were positively correlated with inflammatory infiltrations and intestinal injuries (Figure 1i). Collectively, these data suggested that compromised responsiveness to anti-TNF-α therapy was highly associated with increased abundance of pro-inflammatory bacteria and that such gut dysbacteriosis was positively correlated with less favorable clinical outcomes of IBD patients.
Metabolomics analyses reveal that lipid metabolism abnormalities characterized with declined abundance of palmitoleic acid (POA) are significantly related to failure of anti-TNF-α therapy
Since compromised responsiveness to anti-TNF-α therapy was associated with gut dysbacteriosis, we then further analyzed whether such alterations of gut microbial ecosystem may also correlate with alternations of profiles of gut metabolites, which may further molecularly contribute to disrupted homeostasis in microbial ecosystems. To address this, LC-MS was carried out to analyze the metabolism profiles of fecal and serum samples of anti-TNF-α responders and non-responders with IBD. We noticed that approximately 34% of fecal metabolites with differential abundance were lipid and lipid-like molecules, implicating that anti-TNF-α non-responders showed aberrant compositions and abundance of lipid metabolites in intestine (Figure 2, a and b). KEGG pathway classification also demonstrated that metabolism of lipid such as cis-12-octadecenoic acid methyl ester and 7-dehydrodesmosterol correlated to the responsiveness of anti-TNF-α therapy (Figure 2, b and c).
Figure 2.
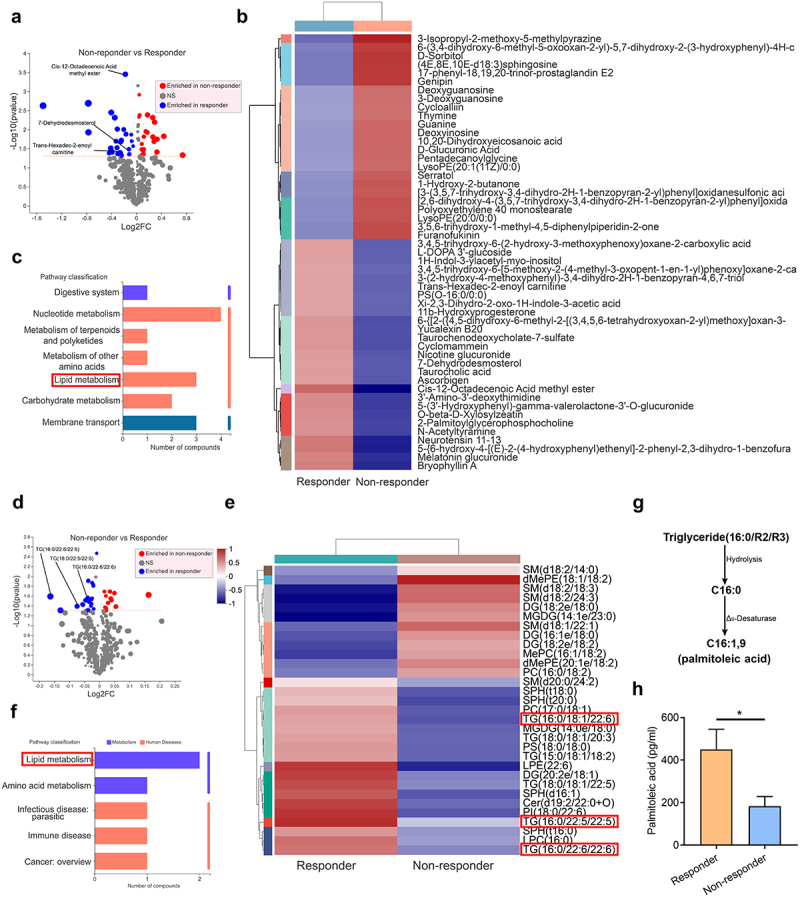
Differential metabolic profiles between anti-TNF-α therapy responders (R) and non-responders (NR) highlight that aberrant lipid metabolism with decreased palmitoleic acid (POA) are associated with compromised efficacy of anti-TNF-α therapy.
Note: (a) Volcano plot showing the differential fecal metabolites between anti-TNF-α therapy responders (R) and non-responders (NR) with IBD. The red points are 24 metabolites enriched in anti-TNF-α non-responders (NR) while the blue points are 27 metabolites enriched in anti-TNF-α responders (R) (R = 19, NR = 12).
(b) Heatmap of differential fecal metabolite profile in anti-TNF-α therapy responders (R) and non-responders (NR) with IBD. The metabolites with red box were enriched in responders and classified into lipid metabolism according to KEGG pathway.
(c) KEGG pathway classification of differential fecal metabolites between anti-TNF-α therapy responders (R) and non-responders (NR) with IBD.
(d) Volcano plot showing the differential serum metabolites between anti-TNF-α therapy responders (R) and non-responders (NR) with IBD. The red points are 12 metabolites enriched in anti-TNF-α non-responders (NR) while the blue points are 19 metabolites enriched in anti-TNF-α responders (R) (R = 6, NR = 6).
(e) Heatmap of differential serum metabolite profiles in anti-TNF-α therapy responders (R) and non-responders (NR) with IBD. The metabolites with red box were enriched in responders and classified into lipid metabolism according to KEGG pathway.
(f) KEGG pathway classification of differential serum metabolites between anti-TNF-α therapy responders (R) and non-responders (NR) with IBD.
(g) Black arrow indicates the metabolic pathway of triglycerides (16:0/R2/R3) to palmitoleic acid (C16:1).
(h) Palmitoleic acid is much higher in plasma samples in anti-TNF-α therapy responders (R) than non-responders (NR) with IBD (R = 26, NR = 24).
Data represent mean ± SEM; *P < 0.05. P values were calculated by two-tailed unpaired Student’s t test (H). [The details for screening statistically differential metabolites in (a) and (d) are shown in Materials and Methods]
To systematically evaluate the lipid metabolic profiles, we comparatively investigated serum lipid profiles between anti-TNF-α responders and non-responders with IBD. We discovered abnormal lipid metabolism in non-responders with down-regulation of triglycerides (TG), diacylglycerol (DG), lysophospholipid (LPC) and other compounds containing long-chain fatty acyl-CoA chain (Figure 2, d and e), which could further be hydrolyzed into long-chain fatty acids (LCFAs). In line with these findings, enhanced lipid metabolism was noted in serum of responders (Figure 2f). Triglycerides are the most common bioactive lipid, and triglycerides (16:0/R2/R3) enriched in responders could be hydrolyzed into C16:0 (palmitic acid, PA), which could be further desaturated to C16:1 (palmitoleic acid, POA) (Figure 2, e and g). Indeed, the relative concentrations of palmitoleic acid (POA) were significantly lower in plasma from non-responders of anti-TNF-α therapy than those from responders (Figure 2h). In agreement with our findings, long-chain fatty acids (LCFAs) were found to be the most numerous depleted classes of metabolites in stools of IBD patients29 and inflammatory intestinal tissues contained much lower concentrations of palmitoleic acid (POA) than adjacent non-inflammatory tissues did in colitis patients30.
To validate that these reduced concentrations of palmitoleic acid (POA) in patients with compromised responsiveness of anti-TNF-α therapy is at least partially ascribed to gut dysbacteriosis, mice were treated with a cocktail of broad-spectrum antibiotics (Abx) before and during dextran sulfate sodium salt (DSS) treatment. Mice receiving anti-TNF-α mAb injection underwent rapid loss of diversity of gut microbiota upon the oral treatment of broad-spectrum antibiotics (ampicillin, neomycin, vancomycin, and metronidazole), exacerbated colitis with rapid body weight loss, increased disease activity index (DAI) score and bloody diarrhea, compared with mice treated with IgG antibody control (Supplementary Figure S4, A to C). Although anti-TNF-α mAb treatment conferred some benefits, characterized by reduced colonic inflammation, preserved colon length, and muted histopathological changes (Supplementary Figure S4, C to F), the antibiotics treatment remarkably impaired the benefits of anti-TNF-α mAb, characterized by more disruptions of epithelial cells, loss of goblet cells, more infiltrations of inflammatory cells and higher histological scores, as well as decreased palmitoleic acid (POA) concentrations in serum of mice with colitis (Supplementary Figure S4, E to G). Thus, these data suggested that gut dysbacteriosis led to reduced abundance levels of palmitoleic acid (POA) and inefficacy of anti-TNF-α therapy.
Oral administration of palmitoleic acid (POA) but not palmitic acid (PA) promotes mucosal healing and attenuates inflammation in DSS-induced acute and chronic colitis in mice with no observable toxicity
Next, we explored whether palmitoleic acid (POA) really played a causal but not correlative role in regulating the disease outcomes of IBD. To test the potential anti-inflammatory effects of palmitoleic acid (POA), PBMCs derived from IBD patients were stimulated with palmitoleic acid (POA), and palmitic acid (PA) was served as a control. We observed significant ex vivo effects reducing the productions of TNF-α and IL-6, two important cytokines had been implicated to be involved in promoting IBD pathogenesis10,11, by palmitoleic acid (POA) but not palmitic acid (PA) in PBMCs from IBD patients (Supplementary Figure S5). We then employed routine DSS-induced acute and chronic colitis models, respectively, to evaluate the exact role of palmitoleic acid (POA) on colitis development and progression. DSS-treated mice with palmitoleic acid (POA) treatment showed less colitis development, as shown by improved body weight regain and reduced DAI score (Figure 3a, b), slighter bloody diarrhea and rectal bleeding (Figure 3c), and longer colons (Figure 3d, f), less infiltrations of inflammatory cells (Figure 3g-i), lower expression of CD11b in the mucosa and minimal reductions of goblet cells and crypts, and higher expression of epithelial markers ZO-1 and Occludin (Figure 3j, and Supplementary Figure S7A). We also compared the anti-colitis effects of palmitoleic acid (POA) and palmitic acid (PA) using DSS-induced model (Supplementary Figure S6). Palmitoleic acid (POA) showed much better anti-colitis effects than palmitic acid (PA) (Supplementary Figure S6). Furthermore, murine colon contained only very low background levels of palmitoleic acid (POA), and the concentrations of palmitoleic acid (POA) in murine colon after palmitoleic acid (POA) treatments increased significantly, with an average increase of 50% than mice without treatments, suggesting that a considerable proportion of dietary palmitoleic acid (POA) could successfully reach the colon and exert anti-colitis effects (Supplementary Figure S7D).
Figure 3.
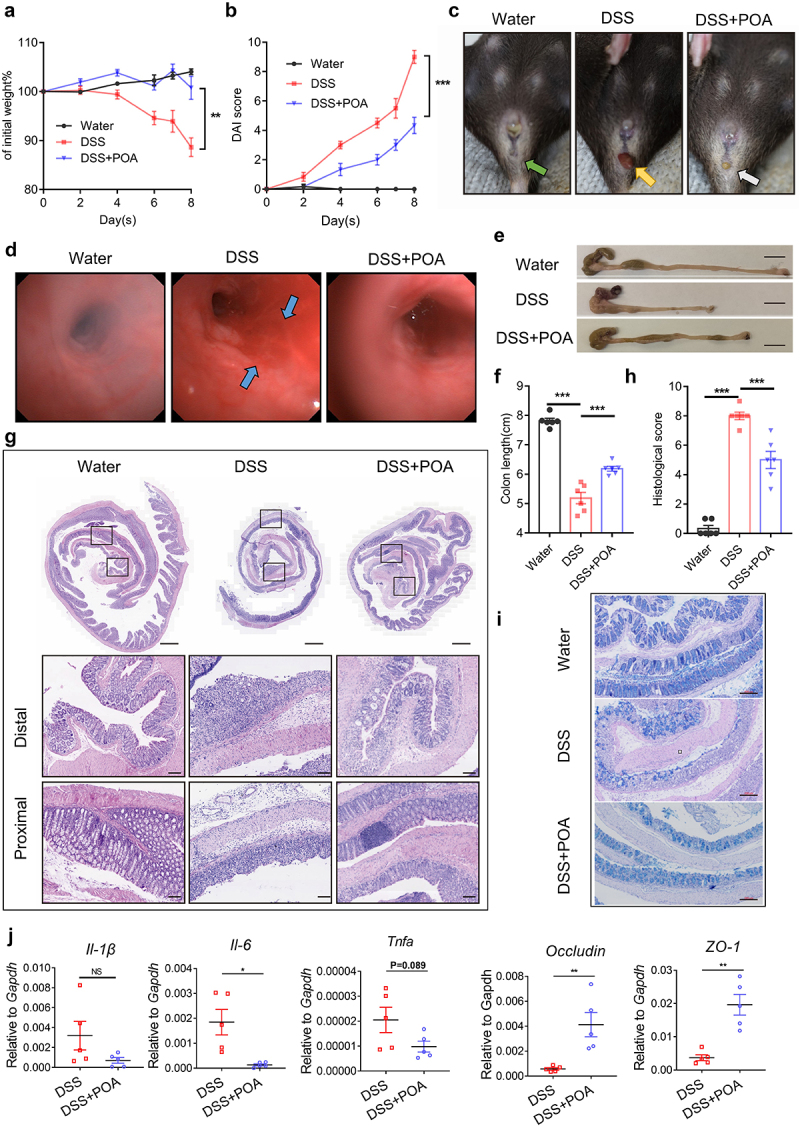
Oral supplement with palmitoleic acid (POA) ameliorates colon inflammation in DSS-induced acute colitis in mice.
Note: (a) Changes in the body weight (of initial weight) of mice treated with water, DSS, or DSS plus palmitoleic acid (POA).
(b) Disease activity index (DAI) score for colitis models including body weight loss, stool consistency, and fecal bleeding.
(c) Images showing the development of rectal bleeding and fecal blood of mice treated with water, DSS, or DSS plus palmitoleic acid (POA). Green arrow indicates normal anus while orange arrow indicates bloody stool. White arrow indicated mild diarrhea with no obvious bloody stools.
(d) Representative endoscopic images of mice treated with water, DSS, or DSS plus palmitoleic acid (POA) at the end of DSS treatment. Blue arrow showed extensive mucosa damage with bleeding.
(e) Gross anatomy of colons of mice treated with water, DSS, or DSS plus palmitoleic acid (POA). Scale bar = 1.0 cm.
(f) Colon lengths of mice treated with water, DSS, or DSS plus palmitoleic acid (POA).
(g) Hematoxylin and eosin (H&E) staining of “Swiss roll” colon sections from mice treated with water, DSS, or DSS plus palmitoleic acid (POA). Top: original magnification, scale bar = 500 μm. Middle: distal colon sections, scale bar = 100 μm. Bottom: proximal colon sections, scale bar = 100 μm.
(h) Histological scores for inflammation, crypt damage, depletion of goblet cells and infiltration of inflammatory cells.
(i) Alcian Blue Periodic acid Schiff (AB-PAS) staining was performed to visualize the presence of goblet cells in the gut sections (scale bar = 200 μm).
(j) The expression of Il-1β, Il6, Tnfα, Occludin and ZO-1 was measured by real-time qPCR.
Data represent mean ± SEM (n = 5-6 mice per group); *P < 0.05; **P < 0.01; ***P < 0.001; NS, no statistical significance. P values were calculated by unpaired Student’s t test [(a), (b), (f), (h) and (j)]. At least two biological repeats were performed.
It is worth to note that mice treated with palmitoleic acid (POA) and water control showed a similar food and water consumption during DSS administration, suggesting that the attenuation of colitis was not attributed to food and DSS consumption (Supplementary Figure 8). Also, although butyrate, a short-chain fatty acid (SCFA) metabolite, showed some protection effects against colitis31, palmitoleic acid (POA) treatment exhibited a much better curative effect with an increased colon length and decreased inflammatory infiltrates than butyrate did (Supplementary Figure S9). Moreover, DSS-treated mice receiving palmitoleic acid (POA) showed significantly lower expressions of pro-inflammatory IL-6 and TNF-α in serum and colons (Figure 3j, and Supplementary Figure S7, B and C). Also, palmitoleic acid (POA) selectively suppressed TNF-α and IL-6 production but not IFN-γ/IL-2/IL-4/IL-10/IL-17A in LPS-stimulated RAW 264.7 cells (Supplementary Figure 10, A to C). Further immunoblot analysis also showed that palmitoleic acid (POA) resulted in the reduced expressions of COX2, pSTAT3, and/or pNF-κB in either RAW 264.7 cells or peripheral blood mononuclear cells (PBMCs) derived from IBD patients (Supplementary Figure S10, D and E). Thus, palmitoleic acid (POA) might exert anti-inflammatory activity against colon inflammation via targeting the IL-6/STAT3 and TNF-α/NF-κB pathways.
Additionally, we investigated whether palmitoleic acid (POA) provided protections in chronic colitis. Mice were treated with DSS to induce chronic colitis for three consecutive 7-day cycles. Oral treatments of palmitoleic acid (POA) showed decreased DAI and longer colons (Supplementary Figure 11, A to D), exhibited less epithelial loss, ulceration, and inflammatory cell infiltration (Supplementary Figure 11E). Thus, palmitoleic acid (POA) induced similar protections against chronic colitis as like in acute colitis. Together, these data collectively suggest that palmitoleic acid (POA) could effectively inhibit inflammation, repair intestinal mucosal barrier, and attenuate both acute and chronic colitis in mouse models.
Furthermore, the safety profile of palmitoleic acid (POA) was evaluated in mice by oral gavage of different doses (0.1 g/kg, 0.5 g/kg, 2.5 g/kg). It showed that mice orally gavaged with several doses of palmitoleic acid (POA) (0.1 g/kg, 0.5 g/kg, 2.5 g/kg) and water control showed no significant differences in weight changes, colon length and spleen weight (Supplementary Figure 12, A to C). Compared with the control group, palmitoleic acid (POA) significantly increased superoxide dismutase (SOD) (Supplementary Figure 12D) but not triglycerides (TG), low-density lipoprotein (LDL), high-density lipoprotein (HDL) and cholesterol (CHO) expression (Supplementary Figure 12E). Activities of alkaline phosphatase (ALKP), alanine aminotransferase (ALT), and aspartate aminotransferase (AST) maintain in the normal ranges, indicating no observable liver damages of palmitoleic acid (POA) (Supplementary Figure 12F). Parameters of renal function including urea nitrogen (UREA) and creatinine (CREA) were also within the normal levels as well (Supplementary Figure 12 G). Also, no apparent liver histological changes were identified upon palmitoleic acid (POA) treatments (Supplementary Figure 12 H). Thus, palmitoleic acid (POA) had well tolerable safety profiles for next-step in-depth mechanism and potential translational research.
Palmitoleic acid (POA) exerts anti-colitis effects in 2,4,6-trinitrobenzenesulfonic acid (TNBS) -induced colitis model in mice
To explore the breadth of potential therapeutic applications of palmitoleic acid (POA) against colitis, we utilized TNBS-induced colitis, which is recognized as one of the closest models mimicking with Crohn’s disease (CD)32. BALB/c mice were subjected to intrarectal administration of TNBS in 50% ethanol to establish TNBS-induced colitis model. TNBS-treated mice rapidly developed bloody diarrhea, extensive wasting syndrome and a weight loss with a mortality rate of 60% (Supplementary Figure 13A). However, compared with water control treatment, TNBS-treated mice with oral palmitoleic acid (POA) treatment showed higher survival rate of 83% and recovered body weight, decreased colonic weight/length ratio (a macroscopic indicator of inflammation), and lower Wallace (indications of extensive inflammatory lesions) (Supplementary Figure 13, A to C).
Also, compared with TNBS-treated mice receiving water, TNBS-treated mice receiving palmitoleic acid (POA) showed a significant trend of increased colon length (Supplementary Figure 13D) and displayed less ulceration, milder colonic tissue damage, much fewer infiltrations of inflammatory cells in intestinal barrier and less goblet cell loss (Supplementary Figure 13, E to G), particularly higher expression of epithelial markers including ZO-1 and Occludin (Supplementary Figure 13 H), and lower frequency of CD8+, ly6G+ and CD11b+ cells in colonic lamina propria (Supplementary Figure 13, I and J). Collectively, these results suggest that oral administration with palmitoleic acid (POA) protected against intestinal inflammation and attenuated TNBS-induced colitis.
Palmitoleic acid (POA) ex vivo exerts appreciable tissue repairment and anti-IL-6/TNF-α effects on highly inflamed colon derived from Crohn’s disease (CD) patients
Then, we analyzed whether palmitoleic acid (POA) could confer some therapeutic effects against human IBD. To address this, we collected highly inflamed colonic samples from Crohn’s disease (CD) patients, and these tissues were ex vivo co-cultured with vehicle control (Ctrl) and palmitoleic acid (POA), respectively. Histology analysis revealed reduced pathological impairments in palmitoleic acid (POA)-treated inflamed colon tissues with less epithelial injury, immune cell infiltration and edema (Figure 4, b and c). Supplementation with palmitoleic acid (POA) selectively decreased colonic production of TNF-α and IL-6, but not IL-2/IL-4/IL-10/IFN-γ/IL-17A (Figure 4, d to g). Moreover, Alcian Blue Periodic acid Schiff (AB-PAS) staining showed that numbers of goblet cells in palmitoleic acid (POA)-treated group was significantly larger than that in the control group (Figure 4i). Palmitoleic acid (POA) treatment could support better intestinal integrity and suppress neutrophil infiltration as palmitoleic acid (POA)-treated colon tissue showed increased ZO-1 expressions and diminished infiltrations of CD11b+ cells in colonic tissues (Figure 4, j and k). Taken together, palmitoleic acid (POA) ex vivo exerted anti-inflammatory effects and mucosal repairment in highly inflamed tissues from Crohn’s disease (CD) patients.
Figure 4.
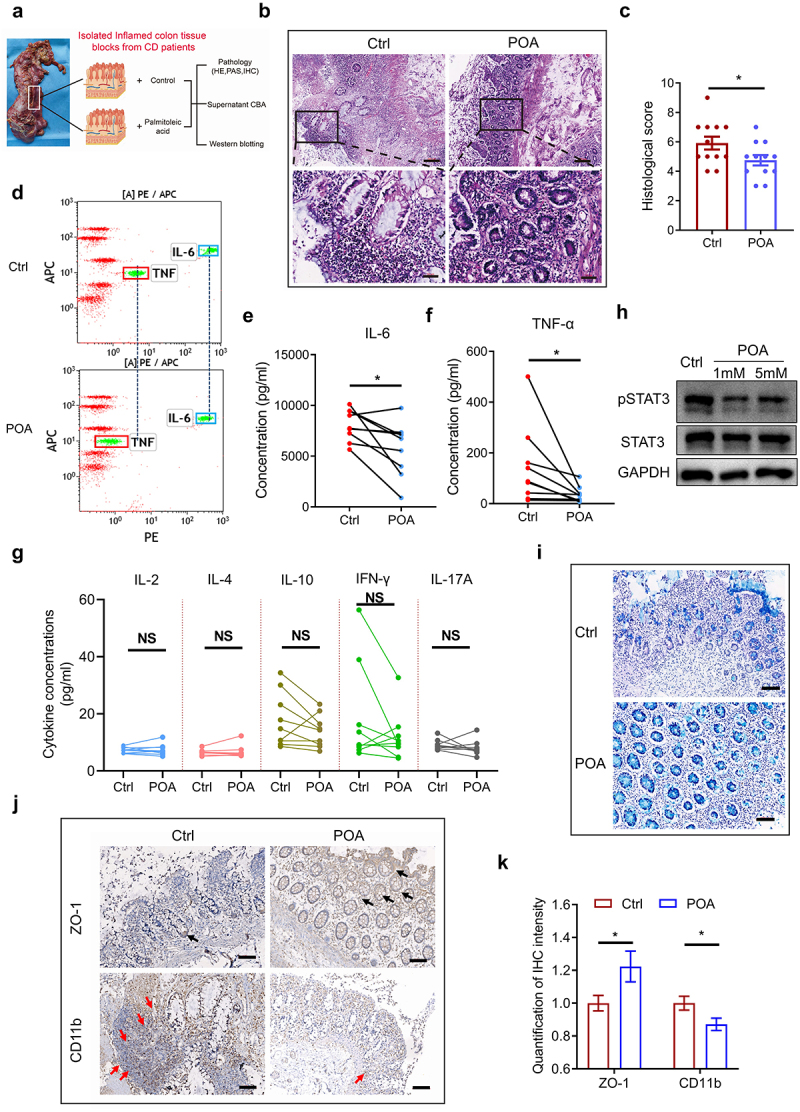
Palmitoleic acid (POA) ex vivo exerts tissue repairment and anti-inflammatory effects on highly inflamed colon tissues derived from Crohn’s disease (CD) patients.
Note: (a) Workflow showing the processing of freshly collected surgical highly inflamed colon resections from Crohn’s disease (CD) patients for pathology, Cytometric Bead Array (CBA) assay and western blotting.
(b-c) HE staining was performed to reveal the pathology of highly inflamed colon tissues co-cultured with vehicle control (Ctrl) or palmitoleic acid (POA) ex vivo. Top: scale bar = 200 μm; Bottom: scale bar = 50 μm. The black-boxed areas at the top are enlarged below (n = 12).
(d) Representative CBA analysis of culture supernatants of highly inflamed colon tissues co-cultured with vehicle control (Ctrl) or palmitoleic acid (POA) using human Th1/Th2/Th17 CBA kit.
(e-g) The expressions of IL-6, TNF-α, IL-2, IL-4, IL-10, IFN-γ and IL-17A in culture supernatants of highly inflamed colon tissues co-cultured with vehicle control (Ctrl) or palmitoleic acid (POA). Boxed areas mark the fluorescent clusters of IL-6 (blue) and TNF-α (red), respectively, and dashed lines mark the shift of fluorescent clusters of IL-6 and TNF-α, respectively. No significant effect on expressions of IL-2, IL-4, IL-10, IFN-γ and IL-17A in inflamed colon tissues between treatments of palmitoleic acid (POA) and Ctrl.
(h) STAT3 and pSTAT3 expressions in highly inflamed colon tissues co-cultured with vehicle control (Ctrl) or palmitoleic acid (POA).
(i) Alcian Blue Periodic acid Schiff (AB-PAS) staining was performed to visualize the presence of goblet cells in the gut sections (scale bar = 100 μm).
(j-k) IHC staining and relative intensity of ZO-1 and CD11b in highly inflamed colon tissues co-cultured with vehicle control (Ctrl) or palmitoleic acid (POA) ex vivo (scale bar = 100 μm).
Data represents mean± SEM; *P < 0.05; NS, no statistical significance. P values were calculated by two-tailed unpaired (c) and paired Student’s t test [(e), (f), (g) and (k)].
Palmitoleic acid (POA) shapes colonic gut microbiota composition with selectively increased abundance of anti-inflammatory Akkermansia muciniphila
Since intestinal metabolites play a significant role in shaping the gut microbiota5,33, we then hypothesize that such differentially abundant metabolites between anti-TNF-α responders and non-responders with IBD could serve as a molecular force to regulate the growth and physiology of gut bacteria. We therefore further investigated how palmitoleic acid (POA) altered the functional and compositional diversity of gut microbiota. To address this, we performed 16S rRNA sequencing to analyze the colon contents from DSS-treated mice receiving treatments of palmitoleic acid (POA) and water, respectively (Figure 5, a to d). Analysis of β-diversity showed significantly distinct species diversity between DSS-treated mice receiving treatments of palmitoleic acid (POA) and water (Figure 5a), although the α-Diversity using Shannon and Simpson index showed no significant species richness between DSS-treated mice receiving treatments of palmitoleic acid (POA) and water (Supplementary Figure 14, A and B). Compared with mice treated with DSS, DSS-treated mice receiving palmitoleic acid (POA) showed higher phylum abundance of Firmictues and Verrucomicrobia, and higher genus abundance of beneficial genera including Akkermansia, Lactobacillus and Bifidobacterium (Figure 5b, and Supplementary Figure 14 C). These data suggest that oral administrations of palmitoleic acid (POA) could effectively change the composition and diversity of gut microbiome.
Figure 5.
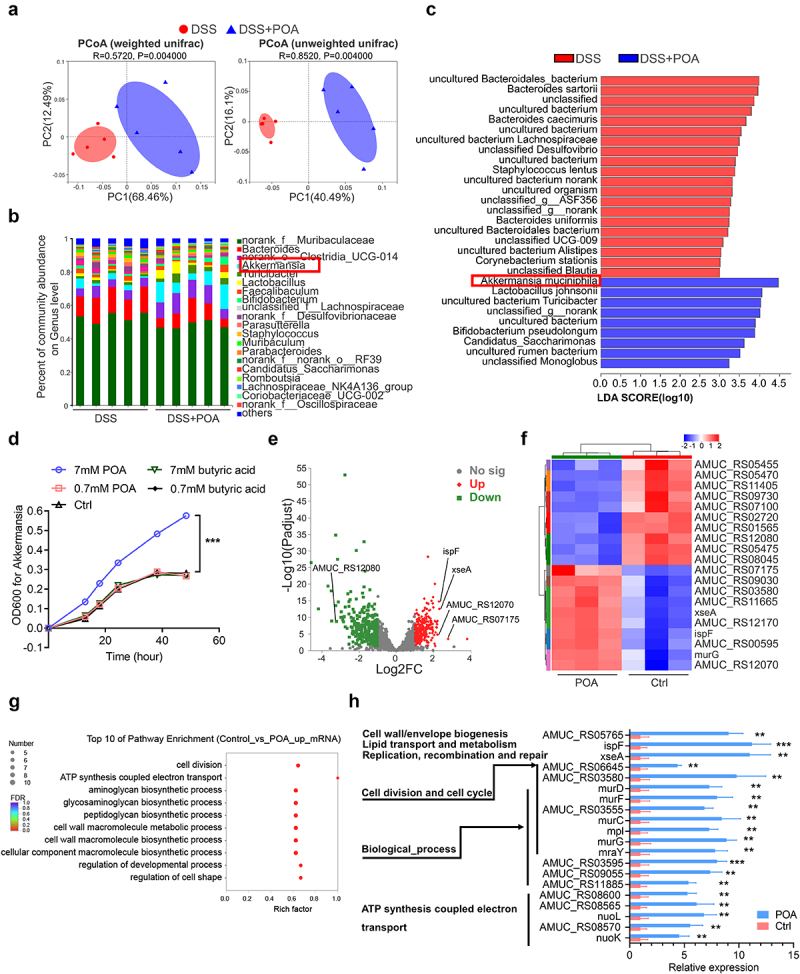
Palmitoleic acid (POA) reprograms gut microbiota compositions, enhances the growth of Akkermansia muciniphila via upregulating transcription signatures of cell division and biosynthetic process, increases the intestinal abundance of Akkermansia muciniphila.
Note: (a) Principal coordinate analysis (PCoA) plots on OUT level were based on weighted and unweighted unifrac. Red points indicated the microbiota enriched in DSS-treated mice and blue points indicated the microbiota enriched in DSS-treated mice receiving palmitoleic acid (POA) [analysis of similarities (ANOSIM): R = 0.5720, P = 0.004 and R = 0.8520, P = 0.004 with 999 permutations].
(b) The relative abundance of bacteria on genus level of DSS-treated mice receiving water or palmitoleic acid (POA).
(c) Linear discriminant analysis (LDA) coupled with effect size measurements (LEfSe) identified the species with different abundance of DSS-treated mice receiving water or palmitoleic acid (POA). Higher abundant species in DSS-treated mice are shaded with red and higher abundant species in DSS-treated mice receiving palmitoleic acid (POA) are shaded with blue (P < 0.05 and LDA score>3.0). qPCR validation of the relative abundance of different microbial taxa was shown in Supplementary Figure 14.
(d) The growth curve of Akkermansia muciniphila cultured with 7 mM palmitoleic acid (POA), 0.7 mM palmitoleic acid (POA), vehicle control (Ctrl), 7 mM butyric acid or 0.7 mM butyric acid, in an anaerobic cabinet in BHI broth supplemented with 0.25% mucin. The growth curve of other bacteria was shown in Supplementary Figure 15.
(e) Volcano plot of differential expressed genes (DEGs) in Akkermansia muciniphila in response to 7 mM palmitoleic acid (POA). Red spots represent upregulated genes, and green spots represent downregulated genes (FC ≥ 2 and p-adjust≤0.05). Each group contains data for three independent samples.
(f)Fold changes of the expression of top 10 upregulated or downregulated genes response to 7 mM palmitoleic acid (POA) compared with control in Akkermansia muciniphila.
(g) GO enrichment analysis of upregulated DEGs in Akkermansia muciniphila in response to 7 mM palmitoleic acid (POA). The plot showed top 10 functional classifications of GO categories including cellular component, biological process, and molecular function. GO enrichment analysis of downregulated DEGs in Akkermansia muciniphila in response to 7 mM palmitoleic acid (POA) was shown in Supplementary Figure 16.
(h) The expression levels of upregulated DEGs related to functional classifications of GO categories in Akkermansia muciniphila in response to 7 mM palmitoleic acid (POA).
Data represent mean± SEM (n = 5 mice per group); **P < 0.01; ***P < 0.001; NS, no statistical significance. P values were calculated by two-tailed unpaired Student’s t test [(d) and (h)].
Importantly, LefSe analysis indicated that nine bacterial species including Akkermansia muciniphila, Lactobacillus johnsonii and Bifidobacterium pseudolongum were enriched in DSS-treated mice receiving oral treatments of palmitoleic acid (POA), while other twenty taxa were enriched in DSS-treated mice receiving water control (Figure 5c). Remarkably, Akkermansia muciniphila was the bacterial species with the most significantly enriched and highest abundance in DSS-treated mice receiving oral treatments of palmitoleic acid (POA) (Figure 5c and Supplementary Figure 14, D and F).
To verify whether palmitoleic acid (POA) directly enhances the growth of these bacterial species observed in colons of DSS-treated mice receiving oral treatments of palmitoleic acid (POA), we then performed in vitro co-culture analyses of Akkermansia muciniphila and Fusobacterium nucleatum with palmitoleic acid (POA). Palmitoleic acid (POA) with two different concentrations (0.7 mM and 7 mM, respectively) were included in culture medium of these bacterial species to observe the effects of palmitoleic acid (POA) on the growth of these bacterial species. Notably, in consistent with those in vivo analyses in mice (Figure 5c and Supplementary Figure 14, D and F), palmitoleic acid (POA) with 7 mM concentration significantly facilitated the growth of Akkermansia muciniphila with accelerated exponential growth and prolonged stationary phase growth but inhibited the growth of Fusobacterium nucleaum in vitro (Figure 5d, and Supplementary Figure 15), suggesting the species-specific effects of palmitoleic acid (POA) on promoting the growth of anti-inflammatory Akkermansia muciniphila.
Transcriptional changes emphasize responses of cell division, energy production and biosynthetic process to palmitoleic acid (POA) in Akkermansia muciniphila
To elucidate the molecular mechanisms governing the bacterial responses to palmitoleic acid (POA), we analyzed the transcriptomes of Akkermansia muciniphila after co-cultured with palmitoleic acid (POA). Transcriptomics analyses of Akkermansia muciniphila revealed that the addition of palmitoleic acid (POA) led to up-regulation of 286 genes and down-regulation of 305 genes (Figure 5e). Significantly altered transcriptional signatures including cell division, lipid transport and metabolism, coenzyme transport and metabolism, cell motility, energy production and conversion, post translational modification, and other uncertain general function (Supplementary Table 3). In the presence of palmitoleic acid (POA), genes related to replication (xseA), cell division (AMUC_RS03580), and nucleotide transport (AMUC_RS00595) were upregulated while genes related to coenzyme transport and metabolism (AMUC_RS05455), cell motility (AMUC_RS12080), energy production and conversion (AMUC_RS11405) and post translational modification (AMUC_RS07100) were downregulated (Figure 5, f and g, and Supplementary Table 3). Notably, concomitant with augmented nucleotide transport metabolism was upregulation of the genes related to energy production (AMUC_RS12070) following treatment with palmitoleic acid (POA) (Supplementary Table 3). These results suggested that palmitoleic acid (POA) could boost the growth, energy production and biosynthetic pathway of Akkermansia muciniphila.
Further GO enrichment analysis showed that up-regulated differential expressed genes (DEGs) in Akkermansia muciniphila in response to palmitoleic acid (POA) were mainly clustered in cell division, ATP synthesis coupled electron transport and biosynthetic process (Figure 5, g and h). Additionally, down-regulated transcriptional activities were mainly clustered in calcium ion binding, RNA helicase activity, anion transport, negative regulation of cellular process and other biological process (Supplementary Figure 16). Together, these data highlighted that palmitoleic acid (POA) could transcriptionally upregulate genes involving in pathways of cell division and biosynthetic process to promote the proliferation of Akkermansia muciniphila.
Co-administration of palmitoleic acid (POA) and Akkermansia muciniphila synergistically maintains intestinal barrier and controls chronic colitis in mice
Given that palmitoleic acid (POA) shapes colonic gut microbiota composition with increased abundance of Akkermansia muciniphila, and we and other colleagues have recently shown that Akkermansia muciniphila could confer significant anti-inflammatory protection effects34,35, we thus postulated that there should be synergistic effects between palmitoleic acid (POA) and Akkermansia muciniphila in control of colitis. Such postulation is well justified because: (i) palmitoleic acid (POA) shows a remarkable ability to repair gut mucosal tissue, which is necessary for bacterial colonization and growth niches such as Akkermansia muciniphila; (ii) palmitoleic acid (POA) may reprogram the gut microbial structures via favoring the growth of beneficial gut bacteria such as Akkermansia muciniphila.
Indeed, compared with DSS-induced chronic colitis mice treated with only palmitoleic acid (POA) or Akkermansia muciniphila, mice receiving co-administrations of palmitoleic acid (POA) and Akkermansia muciniphila exhibited higher intestinal abundance of Akkermansia muciniphila, much less ulceration, reduced inflammatory cell infiltration and more advanced healing of the eroded mucosal barrier (Supplementary Figure 17, and Supplementary Figure 18). Thus, co-administrations of palmitoleic acid (POA) and Akkermansia muciniphila not only favored the intestinal growth and abundance of Akkermansia muciniphila but also synergistically conferred therapeutic effects against colitis with Akkermansia muciniphila. Thus, these data collectively suggest that palmitoleic acid (POA) reprogrammed the gut microbiota with increased abundance of Akkermansia muciniphila, and co-administration of palmitoleic acid (POA) and Akkermansia muciniphila synergisticallyfacilitated the control of colitis.
Gut microbiota reprogrammed by palmitoleic acid (POA) contributes to improved effects of anti-TNF-α therapy against colitis
We then investigated whether palmitoleic acid (POA)-reprogrammed gut microbiome characterized with higher abundance of Akkermansia muciniphila and other beneficial bacteria could really confer protective effect against colitis. To address this, littermate mice were orally treated with palmitoleic acid (POA) or water control, respectively, for 7 days in co-transfer experiments (Figure 6a), and then the feces from donor mice fed with palmitoleic acid (POA) or water group were transferred to antibiotic-pretreated recipient mice. After one-week fecal transplantation, recipient and donor mice were treated with 2.5% DSS and Infliximab (IFX, an anti-TNF-α mAb widely used for biological therapy) injection (Figure 6a). Recipient mice orally treated with feces derived from palmitoleic acid (POA)-fed donor mice gained less weight loss and longer colon length compared with recipient mice that received feces from chow-fed donor mice in response to therapy of anti-TNF-α (Infliximab, IFX) during DSS-induced colitis (Figure 6, b to d). In addition, mice transplanted with palmitoleic acid (POA)- treated gained less weight loss fecal samples developed lighter colitis features and exhibited alleviated pathological features with less inflammatory cell infiltration and more intact colonic architecture with few observable ulcerations (Figure 6, e and f). Together, these data suggested that improved therapeutic benefits of anti-TNF-α against colitis might be ascribed to reprogrammed gut microbial community in mice after oral treatments of palmitoleic acid (POA).
Figure 6.

Palmitoleic acid (POA)-educated microbiota improves the efficacy of anti-TNF-α therapy against colitis.
Note: (a) Experimental diagram for determining whether the increased anti-TNF-α responsiveness to colitis achieved by palmitoleic acid (POA) treatment is transferable. Conventionally raised mice were fed with water or palmitoleic acid (POA) for 7 days. Fecal homogenates from water or palmitoleic acid (POA)-treated mice were orally transmitted into antibiotics-pretreated recipient mice. After one-week transplantation, all mice were treated with 2.5% DSS and anti-TNF-α mAb (Infliximab, IFX) for 7 days.
(b) Changes in the body weight (of initial weight) of DSS-treated mice, donor mice treated with water or palmitoleic acid (POA), and recipient mice orally transmitted with feces of donor mice after DSS administration.
(c) Gross anatomy of colons of DSS-treated mice, donor mice treated with water or palmitoleic acid (POA), and recipient mice orally transmitted with feces of donor mice. Scale bar = 1.0 cm.
(d) Colon lengths of DSS-treated mice, donor mice treated with water or palmitoleic acid (POA), and recipient mice orally transmitted with feces of donor mice.
(e) Representative images of pathologic colon sections. Top: original magnification, scale bar = 500 μm. Middle: distal colon sections, scale bar = 100 μm. Bottom: proximal colon sections, scale bar = 100 μm.
(f) Colon histological scores for inflammation, crypt damage, depletion of goblet cells and infiltration of inflammatory cells.
Data represent mean ± SEM (n = 5 mice per group); *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; NS, no statistical significance. P values were calculated by two-tailed unpaired Student’s t test [(b), (d) and (f)]. At least two biological repeats were performed.
Palmitoleic acid (POA) promotes mucosal barrier healing and improves the efficacy of anti-TNF-α therapy against colitis in mice
Since palmitoleic acid (POA) showed the therapeutic effects of maintaining intestinal epithelial integrity and alleviating inflammation in IBD mouse models, we next explored whether palmitoleic acid (POA) increased the efficacy of anti-TNF-α therapy against colitis. Mice were administrated with 2.5% DSS and treated with palmitoleic acid (POA) or water, respectively, followed by intraperitoneally (i.p.) injected with anti-TNF-α mAb after DSS administration. Mice receiving co-administrations of palmitoleic acid (POA) plus anti-TNF-α mAb exhibited less weight loss and lower DAI compared with mice receiving only anti-TNF-α mAb or palmitoleic acid (POA) (Figure 7, a and b). Remarkably, most mice receiving co-administration of palmitoleic acid (POA) plus anti-TNF-α mAb showed more significant attenuation of diarrhea and improved colon length (Figure 7, c to e). Histopathologically, mice receiving combination treatments of palmitoleic acid (POA) with anti-TNF-α mAb showed much better preservation of mucosal integrity with more observable goblet cells and crypts, less inflammation and ulceration (Figure 7, f to i). Taken together, these data suggested that palmitoleic acid (POA) contributed to better responsiveness of anti-TNF-α biological therapy against colitis.
Figure 7.
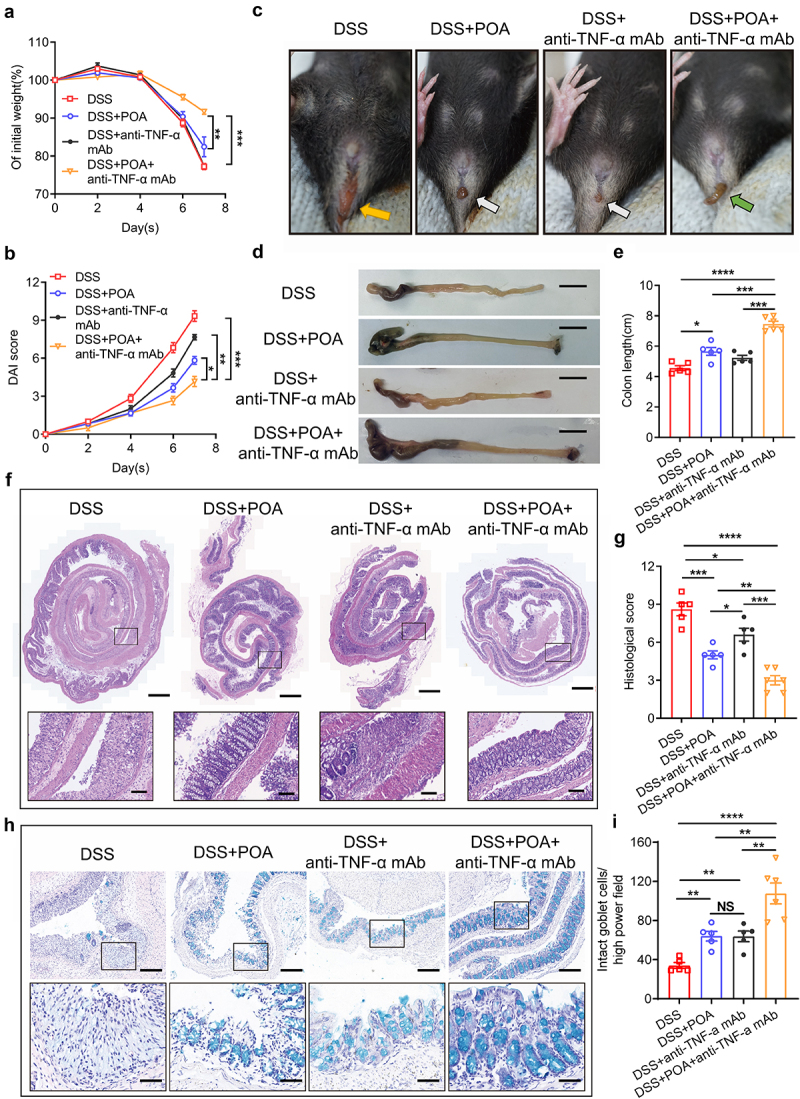
Oral administration of palmitoleic acid (POA) enhances therapeutic effects of anti-TNF-α mAb against colitis in mice.
Note: (a) Changes in the body weight (of initial weight) of DSS-induced mouse colitis models treated with water, palmitoleic acid (POA), anti-TNF-α mAb (Infliximab, IFX), and palmitoleic acid (POA) plus anti-TNF-α mAb (Infliximab, IFX) after DSS administration.
(b) Disease activity index (DAI) score for colitis models including body stool consistency and fecal bleeding.
(c) Images showing the development of severe diarrhea and rectal bleeding of DSS-induced mouse colitis models treated with water, palmitoleic acid (POA), anti-TNF-α mAb (Infliximab, IFX), and palmitoleic acid (POA) plus anti-TNF-α mAb (Infliximab, IFX) at the end of DSS treatment. Orange arrow indicates severe diarrhea and bloody stools while white arrow indicates mild diarrhea with no obvious bloody stools. Green arrow shows no obvious diarrhea with no obvious bloody stools.
(d) Gross anatomy of colons of DSS-induced mouse colitis models treated with water, palmitoleic acid (POA), anti-TNF-α mAb (Infliximab, IFX), and palmitoleic acid (POA) plus anti-TNF-α mAb (Infliximab, IFX). Scale bar = 1.0 cm.
(e) Colon lengths of DSS-induced mouse colitis models treated with water, palmitoleic acid (POA), anti-TNF-α mAb (Infliximab, IFX), and palmitoleic acid (POA) plus anti-TNF-α mAb (Infliximab, IFX).
(f) Representative images of pathologic colon sections of DSS-induced mouse colitis models treated with water, palmitoleic acid (POA), anti-TNF-α mAb (Infliximab, IFX), and palmitoleic acid (POA) plus anti-TNF-α mAb (Infliximab, IFX). Top: original magnification, scale bar = 500 μm. Bottom: proximal colon sections, scale bar = 100 μm. The black-boxed areas at the top are enlarged below.
(g) Colon histological scores for inflammation, crypt damage, depletion of goblet cells and infiltration of inflammatory cells.
(h) Alcian Blue Periodic acid Schiff (AB-PAS) staining of the colon tissue to visualize goblet cell within the mucosa. Top: low power field, scale bar = 200 μm. Bottom: high power field, scale bar = 50 μm. The black-boxed areas at the top are enlarged below.
(i) Quantification of AB-PAS-positive cell in high power field of the colon tissue.
Data represent mean ± SEM (n = 5-6 mice per group); *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; NS, no statistical significance. P values were calculated by two-tailed unpaired Student’s t test [(a), (b), (e), (g)and (i)]. At least two biological repeats were performed.
Discussion
Due to the challenges of dramatic fluctuations of diversity and magnitude of microbiota and metabolite network in host9, precise orchestration and underlying mechanisms of chemically and molecularly complex regulation networks in host physiology and immune homeostasis remain largely unraveled36. This study has revealed the critical role of molecular regulation in gut microbiome by modulating homeostasis among gut microbiota, metabolic systems, intestinal mucosal barrier, and immune network. Particularly, this study suggested the previous unrecognized polyfunctionality and mechanisms of palmitoleic acid (POA) in both effectively promoting mucosal healing and selectively modulating abundance, composition, structure, and function of gut microbiota to alleviate diseases and enhance biological therapy in the context of this complex host-microbiota interplay networks, providing potential novel prevention and therapeutic strategies against intestinal and abenteric diseases.
Molecularly complex regulation of abundance, composition, structure and function of gut microbiota requires exact understanding of the role of a designated molecule, but most of our previous understandings are limited in short-chain fatty acids (SCFAs)37, glycans6, sugar38 and other molecules39,40. Increasing evidence has revealed the important roles of dietary long-chain unsaturated fatty acids in regulating inflammatory responses13. Palmitic acid (PA) is the most common saturated fatty acid and metabolized to oleic acid, stearic acid, sphingolipids as well as palmitoleic acid (POA)41. Palmitic acid (PA) might trigger TLR responses and endoplasmic reticulum (ER) stress in macrophages, damage gut barriers with decreased abundance of Akkermansia muciniphila and increase inflammatory cytokine productions42–44. Excessive intake of long-chain polyunsaturated unsaturated fatty acids (PUFAs) such as n-3 and n-6 fatty acids might be associated with inflammatory diseases including IBD because of the increased TLR2-mediated ER stress and chemokine productions45. Different from palmitic acid (PA) and other LCFAs, palmitoleic acid (POA) is an omega-7 monounsaturated fatty acid with a double bond and one of the most abundant monounsaturated fatty acids in plants and marine sources46,47, which might have anti-inflammatory capabilities48. Our data demonstrated that palmitoleic acid (POA) but not palmitic acid (PA) displayed anti-inflammatory and anti-colitis properties via regulating structure and function of gut microbiota. However, it remains largely unknown whether and how other LCFAs regulate gut microbiota and further mediate IBD pathogenesis.
This work demonstrated in vivo and in vitro effects of palmitoleic acid (POA) on selectively increasing the abundance of beneficial gut bacteria such as Akkermansia muciniphila and significantly extend the diversity of molecule arsenal and metabolic pathways capable of reprogramming such highly dynamic and complex bacterial communities. The concentrations of palmitoleic acid (POA) in the host should be highly-dynamic and variable because of the alteration of gut microbiota, dietary intake, and other physiological or pathological conditions that may influence metabolic processes. It was estimated that approximately 10 ~ 17% of palmitic acid (PA), which can later be metabolized into palmitoleic acid (POA), would be absorbed in mesenteric lymphatic vessels and that over 80% of palmitic acid (PA) might reach colon in adult rats49. Given that palmitoleic acid (POA) is also a microbial metabolite preferentially produced by some gut bacteria such as Akkermansia muciniphila34,50, this study reveals previously unknown positive feedback loop from higher abundance of beneficial gut bacteria, higher levels of selected bacterial metabolites to preferred gut microbiota structure for better health and disease therapies. Also, in other words, the development and progression of inflammatory diseases (e.g., IBD in this study) and limited effects of therapy (e.g., anti-TNF-α mAb) of these inflammatory diseases may attribute to destructions or failed reconstructions of this positive feedback loop.
Anti-TNF-α antibody is the earliest, most widely used, and most well-evaluated biological therapy for IBD, but the irresponsiveness or therapeutic resistance is thought to represent the main cause for the limited success of IBD therapy51. Although treatments for IBD act by inhibiting inflammatory cytokines or infiltration of inflammatory cell subsets have been made significant progresses51, mucosal healing and regeneration are a most promising strategy and a most critical clinical indication during IBD therapy16. This study shows that, palmitoleic acid (POA), a long-chain fatty acid (LCFA) with higher abundance in responders receiving anti-TNF-α-based biological therapy, is a protective metabolite with multi-potency critical for reprogramming gut microbiota structure and inducing tissue repair and homeostasis after intestinal damage in IBD. Indeed, this study presents a head-to-head comparison in a highly relevant mouse IBD model that a long-chain fatty acid (LCFA) displays a superior capability than butyrate in intestinal mucosal healing and regeneration. Such long-chain fatty acid (LCFA)-conferred tissue repair and homeostasis recovery in intestinal damage in IBD may be ascribed to: (i) the direct regulation of expression of molecule components of the mucosal tight junction (e.g., Occludin, ZO-1); (ii) suppression of systemic and mucosal inflammation, which further facilitates the reduction or inhibition of intestinal damages in IBD; (iii) reprogramming of gut microbiota toward a more favorable structures and functions against IBD. Notably, palmitoleic acid (POA) significantly upregulated the growth fitness of Akkermansia muciniphila and inhibited some detrimental pro-inflammatory bacteria such as Fusobacterium, and this antibacterial effect of palmitoleic acid (POA) might have contributed to the gut microbiota-reprogramming capability of palmitoleic acid (POA) against colitis. Thus, palmitoleic acid (POA) and palmitoleic acid (POA)-reprogrammed gut microbiota may show broader spectrum roles in enhancing efficacy of other biological therapies against IBD and other inflammatory disorders. However, more in-depth mechanisms underlying the palmitoleic acid (POA)-regulated mucosal healing and regeneration need to be uncovered.
Accumulating evidence has revealed that IBD patients and health controls may display significantly differential signatures of blood and intestinal metabolites12,29,52, and remissions to biological therapy of IBD patients were associated with changes of blood and intestinal metabolome14,17,18,24,53,54. For example, higher fecal butyrate and substrates were significantly correlated with clinical benefits of anti-TNF therapy14, and serum secondary bile acids may be a valuable metabolic signature for prediction of clinical remission upon receiving anti-TNF or anti-IL12/23 biological therapeutics24. These metabolic signatures may serve as not only critical biomarkers for deciphering the pathogenesis mechanisms of IBD and predicting disease prognosis and therapeutics outcomes of IBD but also useful intervention targets for IBD therapeutics. However, few, if there any, evidence has been presented that any of these metabolic markers or signatures might successfully improve biological therapy of IBD. Thus, this study presents the first evidence demonstrating that a designated metabolite signature (i.e., palmitoleic acid (POA)) may facilitate the biological therapy of IBD.
Overall, this study reveals the critical role of palmitoleic acid (POA) in modulating molecular and chemical network of gut microbiota and gut metabolites, which determines the magnitude and diversity of a beneficial gut microbial ecosystem. Such palmitoleic acid (POA)-modulated magnitude and diversity of gut microbial ecosystem may help us to uncover the pathogenesis mechanisms and develop new therapeutics of IBD and other intestinal and abenteric diseases.
Materials and methods
Ethics statement
IBD patients and healthy controls were recruited at the First Affiliated Hospital of Sun Yat-sen University [Ethics Number: (2019)348], confirmed by radiological, endoscopic, and histological findings. The scoring criteria of mucosal appearance at endoscopy refers to Chinese Grading System of Crohn’s Disease (CGSCD) or Mayo Score for ulcerative colitis. Prior to enrollment, all participants provided written informed consent. Response was defined as clinical remission in diarrhea, abdominal cramping, endoscopy, and mucosal biopsy for at least 12 weeks after initiation. Non-response was defined as outcomes not meeting above definitions or adverse effects (for example, infusion reactions due to immunogenicity) at 12 weeks after initial therapy. Clinical specimens including stool samples, blood, and colon tissues were obtained from IBD patients as well as healthy controls. Feces and serum were frozen at −80°C. The characteristics of the patients are listed in Supplementary Table 1. Study procedures were conducted according to the Declaration of Helsinki.
Fecal genomic DNA extraction, 16S rRNA gene sequencing and taxonomic annotation
Fecal DNA was extracted using the EZNA ® soil DNA Kit (Omega Bio-tek, Norcross, GA, U.S.) according to manufacturer’s instructions. The hypervariable region V3-V4 of the bacterial 16S rRNA gene were amplified with primer pairs 338F (5’-ACTCCTACGGGAGGCAGCAG-3’) and 806 R(5’-GGACTACHVGGGTWTCTAAT-3’) by an ABI GeneAmp® 9700 PCR thermocycler (ABI, CA, USA). PCR products were purified with an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) and quantified using QuantiFluor™-ST (Promega, USA). Purified and pooled amplicon libraries were paired-end sequenced (2 × 300) on the Illumina MiSeq platform (Illumina, San Diego, USA) according to the standard protocols by Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China). The raw reads were demultiplexed, quality-filtered, merged and clustered into OTUs with a 97% similarity cutoff using UPARSE (version 7.1, http://drive5.com/uparse/), and chimeric sequences were identified and removed using UCHIME. The taxonomy of each OTU representative sequence was analyzed by RDP Classifier version 2.255 against the 16S rRNA database (e.g. Silva v137) using confidence threshold of 0.7.
Fecal UHPLC-MS/MS untargeted metabolism
50 mg solid samples were weighed for the metabolomics study, and the metabolites were extracted using a 400 µL methanol: water (4:1, v/v) solution. The mixture was allowed to settle at −20°C and treated by high throughput tissue crusher Wonbio-96c (Shanghai wanbo biotechnology co., LTD) at 50 Hz for 6 min, then followed by vortex for 30 s and ultrasound at 40 kHz for 30 min at 4°C. The samples were placed at −20°C for 30 min to precipitate proteins. After centrifugation at 13,000 × g at 4°C for 15 min, the supernatant was carefully transferred to sample vials for UHPLC-MS/MS analysis.
Chromatographic separation of the metabolites was performed on a Thermo UHPLC system equipped with an ACQUITY BEH C18 column (100 mm × 2.1 mm i.d., 1.7 µm; Waters, Milford, USA). The mass spectrometric data was collected using a Thermo UHPLC-Q Exactive Mass Spectrometer equipped with an electrospray ionization (ESI) source operating in either positive or negative ion mode. After UPLC-TOF/MS analyses, the raw data were imported into the Progenesis QI 2.3 (Nonlinear Dynamics, Waters, USA) for peak detection and alignment. Statistically differential metabolites between responders’ and non-responders’ stools were selected with VIP value>1 and p value<0.05. A total of 51 differential peaks were selected including 24 peaks in ESI+ and 27 peaks in ESI-. Differential metabolites between two groups were summarized, and mapped into their biochemical pathways through metabolic enrichment and pathway analysis based on database search (HMDB, http://www.hmdb.ca/; KEGG, https://www.kegg.jp/kegg/pathway.html). These metabolites can be classified according to the pathways they involved or the functions they performed. Enrichment analysis was usually to analyze a group of metabolites in a function node whether appears or not.
Serum UHPLC-MS/MS lipid metabolism
200 µL liquid sample was extracted using 80 µL methanol and 400 µL MTBe. After centrifugation (13000 × g) for 15 min at 4°C and concentration, 80 µL supernatant was transferred to sample vials for LC-MS/MS analysis. Chromatographic separation of the lipids was performed on a Thermo UHPLC Vanquish Horizon system equipped with an ACQUITY BEH C18 column (100 mm × 2.1 mm i.d., 1.7 µm; Waters, Milford, USA). The mass spectrometric data was collected using a Thermo Q-Exactive Mass Spectrometer equipped with an electrospray ionization (ESI) source operating in either positive or negative ion mode.
After UPLC-MS analyses, the raw data were imported into the Lipid Search (Thermo, CA) for peak detection, alignment, and identification. The preprocessing results generated a data matrix that consisted of the lipid class, retention time (RT), mass-to-charge ratio (m/z) values, and peak intensity. Features which the relative standard deviation (RSD) of QC > 30% were discarded. Statistically differential metabolites between responders’ and non-responders’ serum were selected with VIP value>1 and p value<0.05. A total of 31 differential lipids including 24 peaks in ESI+ and 7 peaks in ESI- were identified and mapped into biochemical pathways through metabolic enrichment and pathway analysis based on database search (KEGG, https://www.kegg.jp/kegg/pathway.html).
Induction of colitis
The animal study was approved by the Animal Ethics Committee of Sun Yat-sen University (SYSU-IACUC-2021–000828). To establish an acute colitis model, oral administration of 2.5% dextran sodium sulfate (DSS) (Molecular weight 36,000–50000, Yeasen) in drinking water was conducted for 7 days in C57BL/6 female mice. For the chronic colitis model, mice were treated with 2.0% DSS to induce chronic colitis for three consecutive 7-day cycles. Each DSS cycle is separated from normal drinking water. For anti-TNF-α antibody treatment, mice were injected with 10 mg/kg Infliximab (170277-31-3) or isotype control IgG (BE0091, Bio X Cell) for three times a week. For dietary treatment of metabolites, 0.5 g/kg palmitoleic acid (Aladdin) or butyrate (Aladdin) was orally gavaged to mice every two days. For oral bacteria transfer, mice were orally gavaged with 200 µl suspension containing 2 × 108 bacteria for 3 times per week. Animals were daily monitored for the appearance of diarrhea, body weight loss, and survival. The signs of bloody diarrhea were measured using BASO fecal OB-II (BASO diagnostic Inc., Zhuhai, China). Disease activity index (DAI) consisted of the following parameters: body weight loss (0, < 5% weight loss; 1, 5–10% weight loss; 2, 10–15% weight loss; 3, 15–20% weight loss; and 4, > 20% weight loss), stool consistency (0, formed pellets; 2, pasty/semi-formed stool; and 4, liquid stool) and fecal bleeding (0, no rectal bleeding; 2, hemoccult-positive; and 4, visible gross bleeding) as described previously56.
For TNBS-induced colitis, anesthetized BALB/c mice (17–22 g) were sensitized with 2.5% TNBS (Sigma-Aldrich) together with acetone and olive oil57. After 1 week, the mice were given 100 μl of 2.5% TNBS in 50% ethanol via a flexible feeding tube that maintained their heads in a vertical position for 10 min after an overnight fast. Colon inflammation was evaluated by the ratio of weight to length (anus to cecum). The colons were opened and examined to evaluate macroscopic lesions according to the Wallace criteria58. Colon sections were stained with H&E and scored according to the Ameho criteria59.
Depletion of commensal bacteria and measurement of antibiotics effect
Commensal bacteria depletion was achieved by administering an antibiotic cocktail containing metronidazole (1 g/L), ampicillin (1 g/L), neomycin (1 g/L) and vancomycin (0.5 g/L) prepared in autoclaved water (pH 7.2) and filter sterilized, as we previously performed60. To confirm the effect of antibiotics in mice, fecal samples were collected and homogenized in PBS. After serial dilutions, bacterial suspension was plated on brain-heart infusion agar (BHI) and cultivated under aerobic and anaerobic conditions.
Safety assessment
C57BL/6 mice were randomly divided into groups and maintained in a pathogen-free facility under a day-night cycle. Mice were orally treated with POA at different doses of 0.1 g/kg, 0.5 g/kg or 2.5 g/kg, and mice in the control group were treated with an equal volume of vehicle control by gavage. After 14-day treatment, anesthetized mice were sacrificed their colons, spleens and livers were collected. Colon length and spleen weight were calculated while SOD levels and lipid profile in serum including TG, CHO, HDL, LDL were analyzed. Serums were also collected to test hepatic enzymes including AST, ALP, and ALT, and kidney function UREA and CREA.
Fecal microbiome transplantation (FMT)
Mice were orally administrated with palmitoleic acid (POA) or water for 1 week. For the fecal transplants, fresh fecal pellets were collected from donor groups in PBS with a final concentration of 50 mg feces/ml. Fecal homogenates were centrifuged at 1000 rpm to pellet large particles and the supernatant used for FMT treatment. 200 µL of fecal supernatant was orally administered into microbiota-depleted recipient mice. To deplete microbiota, mice were treated with a cocktail of antibiotics for 1 week as described above. Microbial depletion was confirmed by bacterial colony assays. After two-week fecal transplantation, donor and recipient mice were treated with 2.5% DSS to induce colitis and injected with anti-TNF-α mAb (Infliximab, IFX).
Culture of inflamed colon explants
Patient colon tissues were weighted, washed three times with cold PBS with vortex and cultured in 1 ml DMEM (per 100 mg tissue) containing 10% FBS and 5 × penicillin/streptomycin for 12 hours. Palmitoleic acid (POA) or vehicle control was added to the culture medium of colon explants. The supernatants were centrifuged to remove floating tissue debris, then were analyzed for cytokine concentrations by Cytometric Bead Array (CBA) assay and normalized to explant weight.
Histopathological assessment and immunohistochemistry imaging
Mouse colon tissues and patients’ colon tissues were harvested. The colon tissues using the Swiss-roll technique were fixed in 4% paraformaldehyde, embedded in paraffin. Tissue sections of the whole colon were stained with hematoxylin & eosin (H&E) or Alcian Blue Periodic acid Schiff (AB-PAS). The histopathology was scored by two independent pathologists. Three independent parameters were measured: the extent of inflammation (0, none; 1, slight; 2, moderate; 3, severe; 4, massive), the extent of crypt damage (0, none; 1, the basal one-third portion damaged; 2, the basal two-thirds portion damaged; 3, the entire crypt damaged but the surface epithelium intact; 4, the entire crypt and epithelium lost) and percentage of involvement (0, none; 1, 0–25% involved; 2, 25–50% involved; 3, 50–75% involved; 4, 75–100% involved).
For immunohistochemistry (IHC), 4% paraformaldehyde-fixed and paraffin-embedded tissue sections were de-paraffinized using xylene and hydrated in an alcohol gradient. The sections were treated with 10 μM citrate buffer for antigen retrieval and washed with 1 × PBS (pH 7.2). The endogenous peroxidase activity was quenched with methanol and 3% H2O2, and then the sections were blocked with 4% goat serum for 30 min. The tissues were stained with anti‐ZO-1 antibody (AF4144, Affinity), anti-Occludin antibody (DF7504, Affinity), anti-CD8 antibody (AF5126, Affinity), anti-Ly6G antibody (#87048, CST) or anti-CD11b antibody (ab133347, Abcam), overnight at 4°C followed by a biotinylated secondary IgG and then incubated with streptavidin-peroxidase for 1 hour. DAB was used for detection. The sections were counterstained with Mayer’s hematoxylin. All histological assessments were performed by two independent blinded observers. The images were collected using a Zeiss microscope (AxioScan.Z1).
Western blotting
Cells and tissues were lysed with Protein Extraction Kit containing protease and phosphatase inhibitors (CW0791). The protein samples with 1 × SDS loading buffer were boiled at 100°C for 10 min. and run on 10% SDS-page gels (PG112). The gels were transferred onto a PVDF membrane (Millipore) by wet transfer. The membranes were incubated in a blocking solution with 4% nonfat milk in TBS-T. The membranes were incubated with primary antibodies in 4% nonfat milk in TBS-T at 4°C overnight and with the secondary antibodies in TBS‐T for 1 hour at room temperature. The blots were visualized by ECL kit.
Cytometric Bead Array (CBA) assay
The levels of cytokines in mice serum or culture supernatants, including IL-2, IL-4, IL-6, IFN-γ, TNF, IL-17A, and IL-10, were examined using the CBA assay (BD Biosciences, San Diego, CA) as we previously performed34. The data were collected on the Beckman Coulter Gallios (Beckman) using flowjo software (BD Biosciences). The concentrations of each cytokine were revealed by the fluorescence intensity. Cytokine concentrations were calculated relative to the standard dilution curve.
RNA extraction and real-time qPCR
Total RNA was prepared from tissues using AG RNAex Pro Reagent (AG21102). The concentration of RNA was determined by Nanodrop 2000 spectrometer. First-strand cDNA was synthesized using the Evo M-MLV RT Premix for qPCR (AG11706) following the manufacturer’s instructions; qPCR was performed using the SYBR Green Premix Pro Taq HS qPCR Kit (AG11701) to determine the expression of target cDNA and bacterial DNA. GAPDH and 16S rDNA was used as an endogenous control to normalize gene expression, respectively. Relative mRNA expression levels were presented as means ± SEM. Statistical differences were analyzed by the student’s t-test. For primer sequences, see Supplementary Table 2.
Liquid culture of Akkermansia muciniphila and other bacteria
Cultures of Akkermansia muciniphila (ATCC BAA-835) were grown in brain heart infusion (BHI) medium with 0.25% mucin for at least 72 hours at 37°C in anaerobic conditions (atmosphere 5% H2, 20% CO2, 75% N2). Cultures of Fusobacterium nucleatum (ATCC 23,726) were grown in BHI medium for at least 48 hours at 37°C in anaerobic conditions. The identity of each strain was confirmed by sequencing the 16S ribosomal RNA using primers 27F (5’- AGAGTTTGATCMTGGCTCAG) and 1492 R (5’- GGTTACCTTGTTACGACTT). Strains were preserved in cryotubes at −80°C in growth medium containing 20% glycerol. The liquid was shaken to ensure homogeneity and bacterial growth was monitored in Spectrophotometer (absorbance at 600 nm).
RNA-seq transcriptomics
Akkermansia muciniphila (ATCC BAA-835) were grown in brain heart infusion (BHI) medium with 0.25% mucin with or without palmitoleic acid (POA) at 37°C in anaerobic conditions (atmosphere 5% H2, 20% CO2, 75% N2). The cells were pelleted by centrifugation at 10,000 × g for 10 min at 4°C. Total RNA was extracted from the tissue using TRIzol® Reagent according the manufacturer’s instructions (Invitrogen) and genomic DNA was removed using DNase I (Takara). Three independently RNA samples from each group were used for RNA-Seq. RNA-seq transcriptome library was prepared following TruSeqTM RNA sample preparation Kit from Illumina (San Diego, CA) using 2 μg of total RNA. Illumina sequencing was performed using the Illumina HiSeq×TEN. Data analyses were performed using DESeq2. Genes exhibiting 2-fold changes in expression were statistically significant as determined by Student’s t-test (p < 0.05). DEGs GO enrichment analysis is used to identify statistically significantly enriched GO term using Fisher’s exact test. The purpose of performing FDR correction is to reduce the Type-1 error by Bonferroni, Holm, BY, BH (multiple hypothesis test method). After multiple testing correction, GO terms with adjusted p-value≤0.05 are significantly enriched in DEGs.
Statistical analysis
Statistical analysis was performed using GraphPad Prism (version 8.0). The statistical significance of differences between two groups was analyzed with the unpaired Student’s t-test or Mann-Whitney test. For multiple group comparisons and repeated measures, analysis of variance (ANOVA) or Kruskal-Wallis test. All P values were two-sided. P values less than 0.05 were considered statistically significant. *P < 0.05; ** P < 0.01; *** P < 0.001; NS, no statistical significance. In figure legends, n represents the number of samples.
Supplementary Material
Acknowledgments
This work was partially supported by grants from the Natural Science Foundation of China (82072250 to G.C.Z.).
Funding Statement
The work was supported by the Natural Science Foundation of China [82072250].
Author Contributions
Y.W.C., Q.D.M., Z.X.C., T.L. Y.J.C. L.J.Y., Y.S, L.X., and L.N.L. performed the experiments. Y.W.C., Q.D.M., Z.X.C., T.L., Y.J.C., S.M.T., Z.Y.W., T.M.C., B.Y.O., L.M.C., Z.Y.Z., Y.Y., L.J.Y., Y.S, S.E.Z, L.X., L.N.L., J.L., H.B.S., S.H.Z., L.C.Z., and G.C.Z. analyzed the data. Y.W.C., J.H., Y.W. and M.D.Z. contributed materials/analysis tools. Y.W.C., Q.D.M., L.J.Y., Y.S, L.X., L.N.L., G.C.Z., S.H.Z., and L.C.Z. drafted, discussed, and revised the manuscript. S.H.Z., L.C.Z., and G.C.Z. conceived this study.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials. The datasets generated for this study can be found in the http://www.ncbi.nlm.nih.gov/bioproject/916399 (BioProject ID: PRJNA916399) and http://www.ncbi.nlm.nih.gov/bioproject/917464 (BioProject ID: PRJNA917464)
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2023.2211501.
References
- 1.Fischbach MA, Segre JA.. Signaling in host-associated microbial communities. Cell. 2016;164:1288–26. doi: 10.1016/j.cell.2016.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cani PD. Microbiota and metabolites in metabolic diseases. Nat Rev Endocrinol. 2019;15(2):69–70. doi: 10.1038/s41574-018-0143-9. [DOI] [PubMed] [Google Scholar]
- 3.Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol. 2021;19:55–71. doi: 10.1038/s41579-020-0433-9. [DOI] [PubMed] [Google Scholar]
- 4.de Souza HSP, Fiocchi C, Iliopoulos D, de Souza HSP. The IBD interactome: an integrated view of aetiology, pathogenesis and therapy. Nat Rev Gastroenterol Hepatol. 2017;14(12):739–749. doi: 10.1038/nrgastro.2017.110. [DOI] [PubMed] [Google Scholar]
- 5.Makki K, Deehan EC, Walter J, Bäckhed F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe. 2018;23(6):705–715. doi: 10.1016/j.chom.2018.05.012. [DOI] [PubMed] [Google Scholar]
- 6.Park S-Y, Rao C, Coyte KZ, Kuziel GA, Zhang Y, Huang W, Franzosa EA, Weng J-K, Huttenhower C, Rakoff-Nahoum S. Strain-level fitness in the gut microbiome is an emergent property of glycans and a single metabolite. Cell. 2022;185(3):513–29 e21. doi: 10.1016/j.cell.2022.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schirmer M, Garner A, Vlamakis H, Xavier RJ. Microbial genes and pathways in inflammatory bowel disease. Nat Rev Microbiol. 2019;17:497–511. doi: 10.1038/s41579-019-0213-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nayar S, Morrison JK, Giri M, Gettler K, Chuang LS, Walker LA, Ko HM, Kenigsberg E, Kugathasan S, Merad M, et al. A myeloid–stromal niche and gp130 rescue in NOD2-driven Crohn’s disease. Nature. 2021;593(7858):275–281. doi: 10.1038/s41586-021-03484-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halfvarson J, Brislawn CJ, Lamendella R, Vazquez-Baeza Y, Walters WA, Bramer LM, D’Amato M, Bonfiglio F, McDonald D, Gonzalez A, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol. 2017;2(5):17004. doi: 10.1038/nmicrobiol.2017.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14(5):329–342. doi: 10.1038/nri3661. [DOI] [PubMed] [Google Scholar]
- 11.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Bhosle A, Bae S, McIver LJ, Pishchany G, Accorsi EK, Thompson KN, Arze C, Wang Y, Subramanian A, et al. Discovery of bioactive microbial gene products in inflammatory bowel disease. Nature. 2022;606(7915):754–760. doi: 10.1038/s41586-022-04648-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adolph TE, Meyer M, Schwärzler J, Mayr L, Grabherr F, Tilg H. The metabolic nature of inflammatory bowel diseases. Nat Rev Gastroenterol Hepatol. 2022;19(12):753–767. doi: 10.1038/s41575-022-00658-y. [DOI] [PubMed] [Google Scholar]
- 14.Aden K, Rehman A, Waschina S, Pan W-H, Walker A, Lucio M, Nunez AM, Bharti R, Zimmerman J, Bethge J, et al. Metabolic functions of gut microbes associate with efficacy of tumor necrosis factor antagonists in patients with inflammatory bowel diseases. Gastroenterol. 2019;157(5):1279. doi: 10.1053/j.gastro.2019.07.025. [DOI] [PubMed] [Google Scholar]
- 15.Caruso R, Lo BC, Nunez G. Host–microbiota interactions in inflammatory bowel disease. Nat Rev Immunol. 2020;20(7):411–426. doi: 10.1038/s41577-019-0268-7. [DOI] [PubMed] [Google Scholar]
- 16.Villablanca EJ, Selin K, Hedin CRH. Mechanisms of mucosal healing: treating inflammatory bowel disease without immunosuppression? Nat Rev Gastroenterol Hepatol. 2022;19(8):493–507. doi: 10.1038/s41575-022-00604-y. [DOI] [PubMed] [Google Scholar]
- 17.Yang Y, Gharaibeh RZ, Newsome RC, Jobin C. Amending microbiota by targeting intestinal inflammation with TNF blockade attenuates development of colorectal cancer. Nat Cancer. 2020;1(7):723–734. doi: 10.1038/s43018-020-0078-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhuang X, Tian Z, Feng R, Li M, Li T, Zhou G, Qiu Y, Chen B, He Y, Chen M, et al. Fecal microbiota alterations associated with clinical and endoscopic response to infliximab therapy in Crohn’s disease. Inflamm Bowel Dis. 2020;26(11):1636–1647. doi: 10.1093/ibd/izaa253. [DOI] [PubMed] [Google Scholar]
- 19.Mills RH, Dulai PS, Vázquez-Baeza Y, Sauceda C, Daniel N, Gerner RR, Batachari LE, Malfavon M, Zhu Q, Weldon K, et al. Multi-omics analyses of the ulcerative colitis gut microbiome link Bacteroides vulgatus proteases with disease severity. Nature Microbiol. 2022;7(2):262–276. doi: 10.1038/s41564-021-01050-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamb CA, Kennedy NA, Raine T, Hendy PA, Smith PJ, Limdi JK, Hayee B, Lomer MCE, Parkes GC, Selinger C, et al. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut. 2019;68(Suppl 3):s1–106. doi: 10.1136/gutjnl-2019-318484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allez M, Karmiris K, Louis E, Van Assche G, Ben-Horin S, Klein A, Van der Woude J, Baert F, Eliakim R, Katsanos K, et al. Report of the ECCO pathogenesis workshop on anti-TNF therapy failures in inflammatory bowel diseases: definitions, frequency and pharmacological aspects. J Crohns Colitis. 2010;4(4):355–366. doi: 10.1016/j.crohns.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 22.Sands BE, Anderson FH, Bernstein CN, Chey WY, Feagan BG, Fedorak RN, Kamm MA, Korzenik JR, Lashner BA, Onken JE, et al. Infliximab maintenance therapy for fistulizing Crohn’s disease. N Engl J Med. 2004;350(9):876–885. doi: 10.1056/NEJMoa030815. [DOI] [PubMed] [Google Scholar]
- 23.Battat R, Sandborn WJ . Advances in the comprehensive management of postoperative Crohn’s disease. Clin Gastroenterol Hepatol. 2022:20(7):1436–1449. doi: 10.1016/j.cgh.2021.03.048. [DOI] [PubMed] [Google Scholar]
- 24.Lee JWJ, Plichta D, Hogstrom L, Borren NZ, Lau H, Gregory SM, Tan W, Khalili H, Clish C, Vlamakis H, et al. Multi-omics reveal microbial determinants impacting responses to biologic therapies in inflammatory bowel disease. Cell Host Microbe. 2021;29(8):1294–304.e4. doi: 10.1016/j.chom.2021.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ventin-Holmberg R, Eberl A, Saqib S, Korpela K, Virtanen S, Sipponen T, Salonen A, Saavalainen P, Nissilä E. Bacterial and fungal profiles as markers of infliximab drug response in inflammatory bowel disease. J Crohns Colitis. 2021;15(6):1019–1031. doi: 10.1093/ecco-jcc/jjaa252. [DOI] [PubMed] [Google Scholar]
- 26.Boudeau J, Glasser AL, Masseret E, Joly B, Darfeuille-Michaud A, Orndorff PE. Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn’s disease. Infect Immun. 1999;67(9):4499–4509. doi: 10.1128/IAI.67.9.4499-4509.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Engevik MA, Danhof HA, Ruan W, Engevik AC, Chang-Graham AL, Engevik KA, Shi Z, Zhao Y, Brand CK, Krystofiak ES, et al. Fusobacterium nucleatum secretes outer membrane vesicles and promotes intestinal inflammation. mBio. 2021;12(2):12. doi: 10.1128/mBio.02706-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yao Y, Ni H, Wang X, Xu Q, Zhang J, Jiang L, Wang B, Song S, Zhu X. A new biomarker of fecal bacteria for non-invasive diagnosis of colorectal cancer. Front Cell Infect Microbiol. 2021;11:744049. doi: 10.3389/fcimb.2021.744049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser H, Reinker S, Vatanen T, Hall AB, Mallick H, McIver LJ, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nature Microbiol. 2019;4(2):293–305. doi: 10.1038/s41564-018-0306-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishida T, Miwa H, Shigematsu A, Yamamoto M, Iida M, Fujishima M. Increased arachidonic acid composition of phospholipids in colonic mucosa from patients with active ulcerative colitis. Gut. 1987;28:1002–1007. doi: 10.1136/gut.28.8.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504(7480):446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 32.Morris GP, Beck PL, Herridge MS, Depew WT, Szewczuk MR, Wallace JL. Hapten-induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology. 1989;96:795–803. doi: 10.1016/S0016-5085(89)80079-4. [DOI] [PubMed] [Google Scholar]
- 33.Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1(6):6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen L, Zhang G, Li G, Wang W, Ge Z, Yang Y, He X, Liu Z, Zhang Z, Mai Q, et al. Ifnar gene variants influence gut microbial production of palmitoleic acid and host immune responses to tuberculosis. Nat Metab. 2022;4(3):359–373. doi: 10.1038/s42255-022-00547-3. [DOI] [PubMed] [Google Scholar]
- 35.Bae M, Cassilly CD, Liu X, Park S-M, Tusi BK, Chen X, Kwon J, Filipčík P, Bolze AS, Liu Z, et al. Akkermansia muciniphila phospholipid induces homeostatic immune responses. Nature. 2022;608(7921):168–173. doi: 10.1038/s41586-022-04985-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kolodziejczyk AA, Zheng D, Elinav E. Diet–microbiota interactions and personalized nutrition. Nat Rev Microbiol. 2019;17(12):742–753. doi: 10.1038/s41579-019-0256-8. [DOI] [PubMed] [Google Scholar]
- 37.Dalile B, Van Oudenhove L, Vervliet B, Verbeke K. The role of short-chain fatty acids in microbiota–gut–brain communication. Nat Rev Gastroenterol Hepatol. 2019;16(8):461–478. doi: 10.1038/s41575-019-0157-3. [DOI] [PubMed] [Google Scholar]
- 38.Khan S, Waliullah S, Godfrey V, Khan MAW, Ramachandran RA, Cantarel BL, Behrendt C, Peng L, Hooper LV, Zaki H. Dietary simple sugars alter microbial ecology in the gut and promote colitis in mice. Sci Transl Med. 2020;12(567):12. doi: 10.1126/scitranslmed.aay6218. [DOI] [PubMed] [Google Scholar]
- 39.Fornelos N, Franzosa EA, Bishai J, Annand JW, Oka A, Lloyd-Price J, Arthur TD, Garner A, Avila-Pacheco J, Haiser HJ, et al. Growth effects of N-acylethanolamines on gut bacteria reflect altered bacterial abundances in inflammatory bowel disease. Nat Microbiol. 2020;5(3):486–497. doi: 10.1038/s41564-019-0655-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strandwitz P, Kim KH, Terekhova D, Liu JK, Sharma A, Levering J, McDonald D, Dietrich D, Ramadhar TR, Lekbua A, et al. GABA-modulating bacteria of the human gut microbiota. Nat Microbiol. 2019;4(3):396–403. doi: 10.1038/s41564-018-0307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hosomi K, Kiyono H, Kunisawa J. Fatty acid metabolism in the host and commensal bacteria for the control of intestinal immune responses and diseases. Gut Microbes. 2020;11(3):276–284. doi: 10.1080/19490976.2019.1612662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Korbecki J, Bajdak-Rusinek K. The effect of palmitic acid on inflammatory response in macrophages: an overview of molecular mechanisms. Inflamm Res. 2019;68(11):915–932. doi: 10.1007/s00011-019-01273-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghezzal S, Postal BG, Quevrain E, Brot L, Seksik P, Leturque A, Thenet S, Carrière V. Palmitic acid damages gut epithelium integrity and initiates inflammatory cytokine production. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865(2):158530. doi: 10.1016/j.bbalip.2019.158530. [DOI] [PubMed] [Google Scholar]
- 44.Kunisawa J, Hashimoto E, Inoue A, Nagasawa R, Suzuki Y, Ishikawa I, Shikata S, Arita M, Aoki J, Kiyono H. Regulation of intestinal IgA responses by dietary palmitic acid and its metabolism. J Immunol. 2014;193(4):1666–1671. doi: 10.4049/jimmunol.1302944. [DOI] [PubMed] [Google Scholar]
- 45.Schwärzler J, Mayr L, Vich Vila A, Grabherr F, Niederreiter L, Philipp M, Grander C, Meyer M, Jukic A, Tröger S, et al. PUFA-Induced metabolic enteritis as a fuel for Crohn’s disease. Gastroenterology. 2022;162(6):1690–1704. doi: 10.1053/j.gastro.2022.01.004. [DOI] [PubMed] [Google Scholar]
- 46.Ozogul Y, Duysak O, Ozogul F, Özkütük AS, Türeli C. Seasonal effects in the nutritional quality of the body structural tissue of cephalopods. Food Chem. 2008;108(3):847–852. doi: 10.1016/j.foodchem.2007.11.048. [DOI] [PubMed] [Google Scholar]
- 47.Yang B, Kallio HP. Fatty acid composition of lipids in sea buckthorn (Hippophaë rhamnoides L.) berries of different origins. J Agric Food Chem. 2001;49(4):1939–1947. doi: 10.1021/jf001059s. [DOI] [PubMed] [Google Scholar]
- 48.Mao C, Xiao P, Tao XN, Qin J, He QT, Zhang C, Guo S-C, Du Y-Q, Chen L-N, Shen D-D, et al. Unsaturated bond recognition leads to biased signal in a fatty acid receptor. Science. 2023;380(6640):eadd6220. doi: 10.1126/science.add6220. [DOI] [PubMed] [Google Scholar]
- 49.Bernard A, Echinard B, Carlier H. Differential intestinal absorption of two fatty acid isomers: elaidic and oleic acids. Am J Physiol. 1987;253(6):G751–9. doi: 10.1152/ajpgi.1987.253.6.G751. [DOI] [PubMed] [Google Scholar]
- 50.Kolouchová I, Sigler K, Schreiberová O, Masák J, Řezanka T. New yeast-based approaches in production of palmitoleic acid. Bioresour Technol. 2015;192:726–734. doi: 10.1016/j.biortech.2015.06.048. [DOI] [PubMed] [Google Scholar]
- 51.D’Haens GR, van Deventer S. 25 years of anti-TNF treatment for inflammatory bowel disease: lessons from the past and a look to the future. Gut. 2021;70:1396–1405. doi: 10.1136/gutjnl-2019-320022. [DOI] [PubMed] [Google Scholar]
- 52.Lavelle A, Sokol H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat Rev Gastro Hepat. 2020;17(4):223–237. doi: 10.1038/s41575-019-0258-z. [DOI] [PubMed] [Google Scholar]
- 53.Friedrich M, Pohin M, Jackson MA, Korsunsky I, Bullers SJ, Rue-Albrecht K, Christoforidou Z, Sathananthan D, Thomas T, Ravindran R, et al. IL-1-driven stromal–neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat Med. 2021;27(11):1970–1981. doi: 10.1038/s41591-021-01520-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ding NS, McDonald JAK, Perdones-Montero A, Rees DN, Adegbola SO, Misra R, Hendy P, Penez L, Marchesi JR, Holmes E, et al. Metabonomics and the gut microbiome associated with primary response to anti-TNF therapy in Crohn’s disease. J Crohns Colitis. 2020;14(8):1090–1102. doi: 10.1093/ecco-jcc/jjaa039. [DOI] [PubMed] [Google Scholar]
- 55.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microb. 2007;73(16):5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- 57.Wirtz S, Popp V, Kindermann M, Gerlach K, Weigmann B, Fichtner-Feigl S, Neurath MF. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat Protoc. 2017;12(7):1295–1309. doi: 10.1038/nprot.2017.044. [DOI] [PubMed] [Google Scholar]
- 58.Wallace JL, MacNaughton WK, Morris GP, Beck PL. Inhibition of leukotriene synthesis markedly accelerates healing in a rat model of inflammatory bowel disease. Gastroenterology. 1989;96:29–36. doi: 10.1016/0016-5085(89)90760-9. [DOI] [PubMed] [Google Scholar]
- 59.Ameho CK, Adjei AA, Harrison EK, Takeshita K, Morioka T, Arakaki Y, Ito E, Suzuki I, Kulkarni AD, Kawajiri A, et al. Prophylactic effect of dietary glutamine supplementation on interleukin 8 and tumour necrosis factor α production in trinitrobenzene sulphonic acid induced colitis. Gut. 1997;41(4):487–493. doi: 10.1136/gut.41.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang F, Yang Y, Chen L, Zhang Z, Liu L, Zhang C, Mai Q, Chen Y, Chen Z, Lin T, et al. The gut microbiota mediates protective immunity against tuberculosis via modulation of lncRNA. Gut Microbes. 2022;14(1):2029997. doi: 10.1080/19490976.2022.2029997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials. The datasets generated for this study can be found in the http://www.ncbi.nlm.nih.gov/bioproject/916399 (BioProject ID: PRJNA916399) and http://www.ncbi.nlm.nih.gov/bioproject/917464 (BioProject ID: PRJNA917464)