

Abstract
Recently developed chemical and enzyme-based technologies to install serine ADP-ribosylation onto synthetic peptides have enabled new approaches to study poly(ADP-ribose) polymerase (PARP) biology. Here, we establish a generalizable strategy to prepare ADP-ribosylated peptides that are compatible with N-terminal, C-terminal, and sequential protein ligation reactions. Two unique protein-assembly routes are employed to generate full-length linker histone constructs that are homogeneously ADP-ribosylated at known DNA damage-dependent modification sites. We found that serine mono-ADP-ribosylation is sufficient to alleviate linker histone-dependent chromatin compaction and that this effect is amplified by ADP-ribose chain elongation. Our work will greatly expand the scope of ADP-ribose-modified proteins that can be constructed via semisynthesis, which is rapidly emerging as a robust approach to elucidate the direct effects that site-specific serine mono- and poly-ADP-ribosylation have on protein function.
Graphical Abstract.
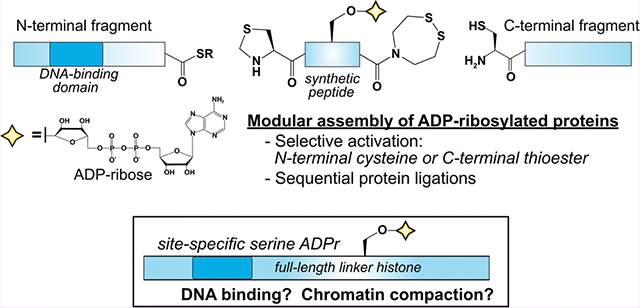
Protein ADP-ribosylation (ADPr) is a nicotinamide adenine dinucleotide (NAD+)-dependent post-translational modification that targets hundreds of proteins in mammalian cells to regulate diverse signaling processes.1 Among the most widespread forms of this modification is DNA damage-induced serine ADPr, which is catalyzed by the poly(ADP-ribose) polymerase (PARP)1/2–histone PARylation factor 1 (HPF1) complex.2–5 While intense efforts have been directed toward understanding PARP1/2–HPF1 regulatory mechanisms,6–8 substrate preferences,4,9,10 and factors that govern ADP-ribose chain elongation11–13 many questions remain surrounding how specific mono- and poly-ADPr events affect target protein function. A greater understanding of ADPr-mediated signaling mechanisms could expand the effective use of PARP1/2 inhibitors for the treatment of homology repair-deficient cancers and other diseases14 and has the potential to uncover alternative treatment strategies.
We and others recently developed synthetic and chemoenzymatic strategies to prepare peptides bearing site-specific serine ADPr.12,15,16 These approaches are compatible with peptide thioesters to enable native chemical ligation, which is a cornerstone of protein post-translational modification research that can now be applied to the study of serine ADPr. However, the core histones H2B and H3 remain the only two full-length serine ADP-ribosylated proteins that have been assembled via semisynthesis.12,17 Notably, both H2B and H3 were prepared via a single ligation reaction wherein the ADP-ribosylated N-terminal peptide thioester fragment was ligated to a recombinant C-terminal fragment. More modular functionalization of ADP-ribosylated peptides, including an N-terminal cysteine and a latent C-terminal thioester, is necessary to grant access to modification sites throughout an entire protein sequence. This design would vastly expand the number of semisynthetically modified PARP1/2–HPF1 substrates that can be functionally characterized in biochemical, biophysical, and cell-based assays.
The disordered, lysine-rich C-terminal domain of linker histone H1 contains several serine residues that act as acceptor sites for DNA damage-induced ADPr.4,8,9,18 It is clear that PARP1/2 activity is required to release linker histone H1 from chromatin at DNA damage sites, which contributes to local chromatin relaxation and DNA repair.18,19 Multiple mechanisms to explain how PARP1 activity impacts H1 function have been put forth, including the following: (i) PARP1 displaces H1 from chromatin by engaging an overlapping nucleosome binding site, (ii) a noncovalent H1–poly-ADPr interaction disrupts the H1/DNA interface, and (iii) polyanionic ADP-ribose chains reduce the affinity between the highly basic H1 C-terminus and the negatively charged chromatin polymer.19–22 Additionally, H1 ADPr may induce downstream protein-modification cascades or ADP-ribose-binding protein recruitment events that influence H1 function. With semisynthetic ADP-ribosylated H1 constructs, chromatin structure analysis can be performed in the absence of confounding variables that include the PARP1 enzyme, core histone ADPr, and other protein modifications or DNA repair factors. Thus, the impact that site-specific mono- and poly-ADPr have on H1 function can be directly interrogated.
We chose to study the linker histone H1.2 as it is an abundant variant and a known target of DNA damage-induced serine ADPr.8,18 There are four PARP1/2–HPF1 substrate motifs (Lys-Ser) in H1.2 (Figure 1A), of which several have been reported as ADPr acceptor sites in mammalian cell-based assays. To determine the primary H1.2 ADPr sites, we introduced wild-type or serine-to-alanine 6×His-tagged H1.2 transgenes into HEK293T cells for ADPr analysis. Following exposure to an oxidative DNA damage agent, cells were collected and lysed in a denaturing buffer that included 6 M urea to rapidly quench all ADPr and glycohydrolase activity. Lysates were then cleared and passed over a Ni–nitriloacetic acid (NTA) column to enrich H1.2 transgenes for Western blot analysis (Figure 1B). We found that all detectable H1.2 ADPr occurs at the S150 and S188 sites, as confirmed by the S150A/S188A double mutant. This modification site preference is also maintained in reconstituted ADPr assays comprising the PARP1/2–HPF1 complex and recombinant, full-length H1.2 constructs (Figure 1C). While the S86 site falls within the folded domain of H1.2 and is likely sterically protected from PARP1 activity, the S150, S173, and S188 sites are located within the C-terminal disordered region. Unlike S150 and S188, the S173 site is immediately followed by a proline residue, which we hypothesized may prevent modification by the PARP1–HPF1 complex. To test this concept, an reversed-phase high-performance liquid chromatography (RP-HPLC)/mass spectrometry (MS)-based assay was employed to analyze PARP1–HPF1 activity on H1.2 peptide fragments (Figure 1D, Figure S1). Indeed, while ADPr could not be detected on the H1.2166–181 fragment, the H1.2143–158, H1.2182–195, and an H1.2166–181 bearing a P174A mutation were quantitatively ADP-ribosylated after a 20 min incubation with PARP1–HPF1 (1 μM/25 μM) at 30 °C. Thus, a proline residue directly C-terminal to the Lys-Ser motif renders the neighboring serine a poor PARP1–HPF1 substrate.
Figure 1.
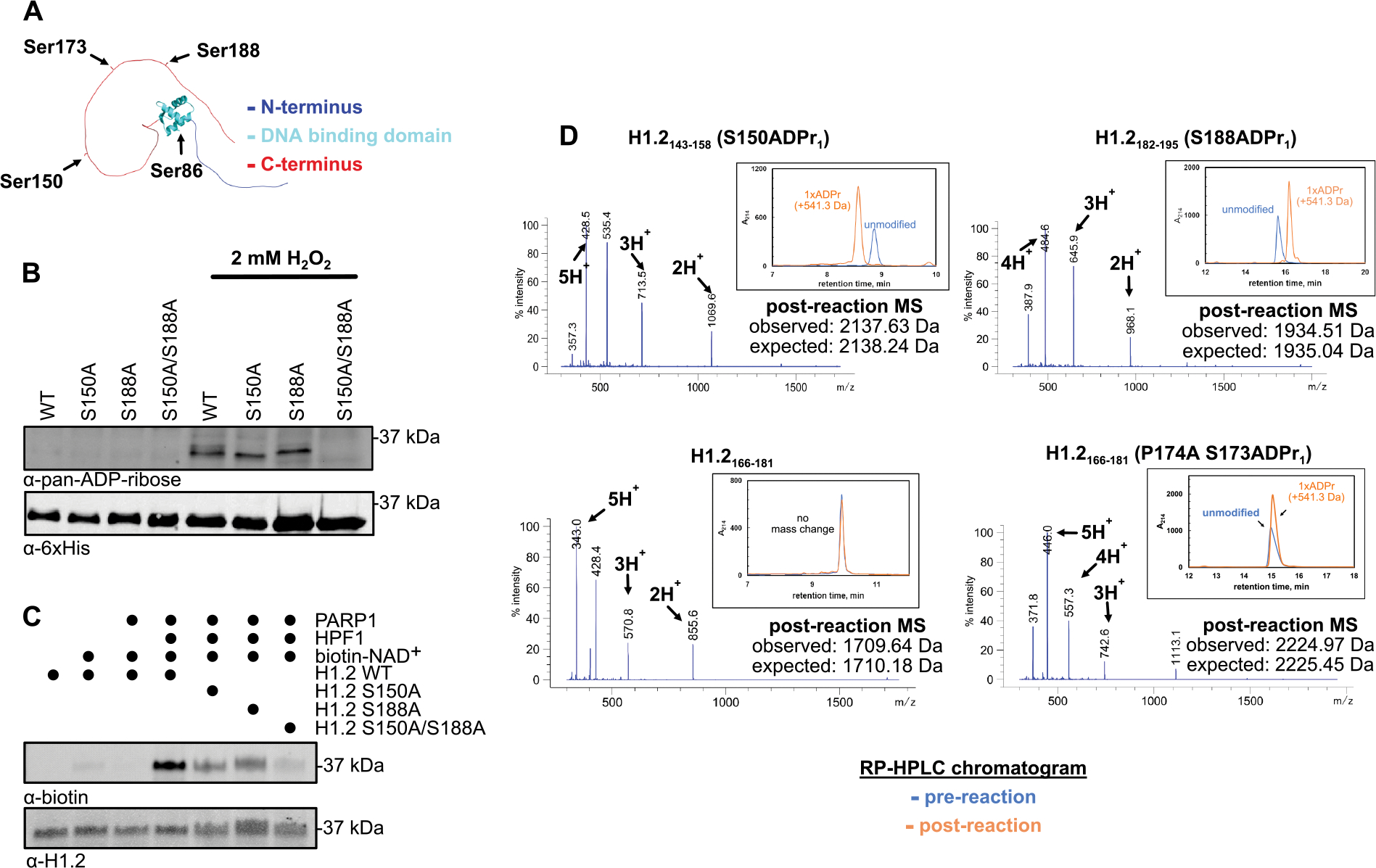
Histone H1.2 S150 and S188 are the main DNA-damage dependent ADP-ribosylation sites. (A) AlphaFold-predicted three-dimensional structure of full-length H1.2 with serine residues that occur within a Lys-Ser motif indicated. (B) Western blot analysis of the indicated 6×His-tagged H1.2 transgenes that were enriched from HEK293T cells via Ni-NTA affinity pre- and post-exposure to hydrogen peroxide (2 mM, 15 min). (C) Western blot analysis of reconstituted, biotin-NAD+-based PARP1–HPF1 ADPr assays consisting of the indicated recombinant enzymatic components and full-length H1.2 constructs. (D) RP-HPLC analysis of the indicated synthetic H1.2 peptide fragments pre- and post-incubation with reconstituted PARP1–HPF1 and NAD+. Intact ESI-MS analysis of each post-reaction H1.2 fragment is shown.
With key H1.2 serine ADPr target sites established, we set out to prepare semisynthetic H1.2 constructs modified with mono-ADP-ribose at the S150 or S188 site. We envisioned a three-piece native chemical ligation strategy to gain synthetic access to the S150 site (Figure 2A). A recombinant piece 1 thioester fragment (amino acids 2–141; H1.22–141) was prepared via an intein fusion-based approach, and a recombinant piece 3 fragment (amino acids 163–213, A163C; H1.2163–213) was prepared via standard procedures (Figure S2). The synthetic piece 2 fragment (amino acids 142–162, A142C; H1.2142–162) was initially functionalized with an N-terminal cysteine and a C-terminal acyl hydrazide, which can be converted to a thioester in a biorthogonal reaction23 to enable sequential protein ligations starting from the N-terminus. Following peptide synthesis, H1.2142–162 was incubated with the PARP1–HPF1 complex in the presence of poly(ADP-ribose) glycohydrolase (PARG) to install the mono-ADP-ribose modification at S150, the sole serine residue in the peptide. However, post-reaction analysis revealed that the H1.2142–162 fragment is modified with two ADP-ribose moieties, and a similar peptide substrate bearing a S150A mutation maintained a single PARP1–HPF1 modification site (Figure S3A). Considering that the highly similar H1.2143–158 S150A construct was not a PARP1–HPF1 substrate, we hypothesized that the H1.2142–162 secondary modification site is dependent upon the N-terminal cysteine residue. To unambiguously identify the modification site, we incubated the unmodified S150A mutant peptide bearing an N-terminal cysteine and the ADP-ribosylated variant of this peptide with iodoacetamide. Interestingly, while the unmodified S150A mutant could be labeled with iodoacetamide as confirmed by RP-HPLC/MS analysis, the ADP-ribosylated variant of this peptide was resistant to iodoacetamide treatment (Figure S3B). We note that free ADP-ribose, but not NAD+, was sufficient to ADP-ribosylate the N-terminal cysteine residue in the absence of PARP1–HPF1, albeit with reduced efficiency (Figure S3C). Therefore, both enzymatic and nonenzymatic activities contribute to modification of the thiol moiety of N-terminal cysteine residues.
Figure 2.
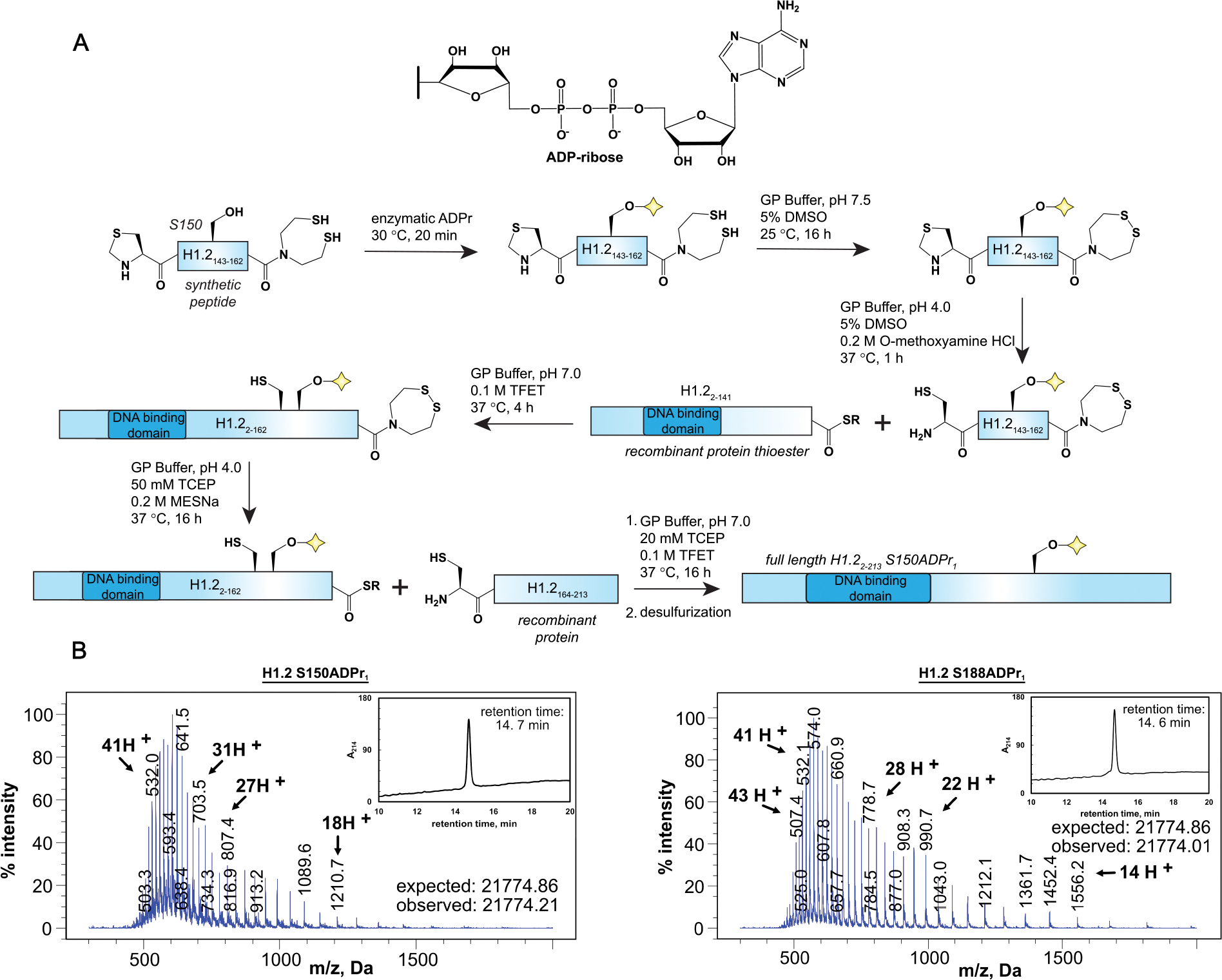
Semisynthetic preparation of full-length, mono-ADP-ribosylated H1.2 constructs. (A) Semisynthetic scheme to prepare full-length H1.2 S150ADPr1. GP buffer = 6 M guanidine-HCl, 0.1 M sodium phosphate; SR = MES thioester. Preparation of SEA resin, “Enzymatic ADPr” conditions and “desulfurization” conditions are described in the Supporting Information. For the semisynthetic scheme to prepare full-length H1.2 S150ADPr1, see Figure S4. (B) RP-HPLC and ESI-MS analysis of the full-length H1.2 S150ADPr1 and full-length H1.2 S188ADPr1 proteins.
Our discovery that N-terminal cysteine side chains are efficiently modified in the ADPr reaction is a critical point to consider when preparing ADP-ribosylated proteins via the chemoenzymatic strategy. All ADP-ribosylated fragments that require an N-terminal cysteine for downstream ligation reactions must maintain a thiol protecting group through the enzymatic ADPr step. Furthermore, the peptide–ADP-ribose linkage must remain stable under conditions required to liberate the free thiol after the peptide ADPr reaction. Considering that serine ADPr is an acid-stable modification, an N-terminal thiazolidine (Thz) protection strategy was pursued, as this moiety can be rapidly and quantitatively converted to cysteine in the presence of methoxyamine hydrochloride under acidic conditions.24 Initial N-terminal Thz H1.2142–162 synthesis efforts revealed that the Thz moiety is not stable during the acyl hydrazide to thioester conversion reaction, as previously reported.25 We therefore prepared H1.2142–162 as a C-terminal bis(2-sulfanylethyl)amido (SEA) peptide (Figure 2A and S2). While the oxidized SEA group is inert in native chemical ligation reactions, the reduced SEA moiety is highly reactive with small molecule thiols to generate a C-terminal thioester. Thus, the user can selectively deprotect the free N-terminal Thz or the oxidized SEA group (via incubation with 10 mM tris(2-carboxyethyl)phosphine (TCEP)) depending on the desired sequential ligation directionality. While alternative strategies such as Dawson’s 3,4-diaminobenzoic acid (Dbz) resin for thioester peptide preparation or disulfide-based cysteine protection approaches could be useful here, neither offer a straightforward workflow for N-terminus to C-terminus protein assembly with ADP-ribosylated peptides. More importantly, we found that both Thz and oxidized SEA deprotection reactions are compatible with the ADP-ribose moiety.
ADP-ribosylated H1.2142–162 was prepared by incubating the Thz/reduced SEA peptide with the PARP1–HPF1 complex, and the mono-ADP-ribosylated product was purified via RP-HPLC (Figure S2). As observed previously, near quantitative conversion of starting material to mono-ADP-ribosylated product was achieved with up to ~20 mg of peptide.12 Next, the peptide was resuspended in a mild oxidation buffer to fully oxidize the SEA moiety, and the Thz deprotection reaction was performed to liberate the N-terminal cysteine (Figure S2). A native chemical ligation reaction with H1.22–141 was carried out, and the ligated product (H1.22–162 S150ADPr1) was purified via RP-HPLC (Figure S2). An SEA to thioester conversion step was used to generate the H1.22–162 S150ADPr1 thioester fragment, which was directly employed in a second ligation reaction with H1.2163–213 (Figure S2). Finally, a desulfurization reaction was carried out to convert ligation junction cysteines to native alanine residues and obtain the full-length H1.2 S150ADPr1 construct (Figure 2B). A similar synthetic scheme was effective to produce the full-length H1.2 S188ADPr1 construct (Figure 2B and S4). In this case, the synthetic fragment (amino acids 177–213) was prepared with an N-terminal Thz and C-terminal acid moiety, as a two-piece ligation strategy was sufficient to access the S188 ADPr site (Figure S5). We therefore expect the Thz-based N-terminal cysteine protection strategy will be broadly compatible with our chemoenzymatic peptide ADPr technology and, when combined with the C-terminal SEA moiety, will grant synthetic access to many unexplored ADPr sites.
We have previously shown that PARP1 efficiently elongates ADP-ribose chains from mono-ADP-ribosylated serine residues in the absence of HPF1.12 Indeed, this catalytic property of PARP1 is maintained on the full-length, mono-ADP-ribosylated H1.2 constructs prepared herein. We found that PARP1 catalyzes ADP-ribose chain polymerization from both the S150ADPr1 and S188ADPr1 sites to a similar extent in unlabeled NAD+ and biotinylated NAD+-based ADPr assays (Figure 3A,B). Importantly, the ADPr activity observed in PARP1 elongation assays represents single ADP-ribose chains emanating from the pre-installed H1.2 mono-ADPr site, as only trace levels of activity could be detected on the unmodified H1.2 substrate.
Figure 3.
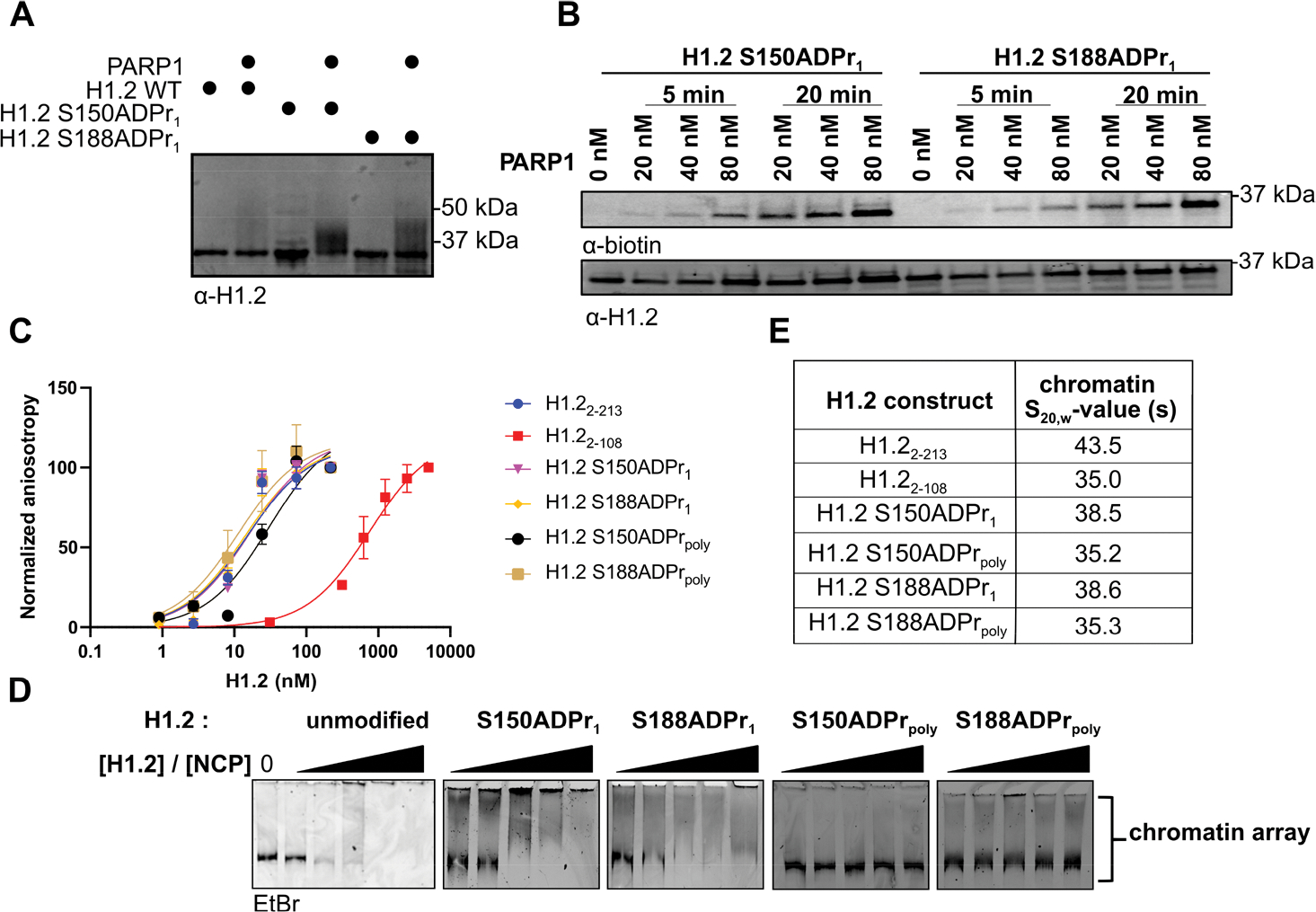
Site-specific linker histone serine ADP-ribosylation directly abrogates its ability to induce chromatin compaction. (A) Western blot analysis of PARP1 elongation activity on the indicated unmodified or mono-ADP-ribosylated full-length H1.2 substrate protein. (B) Western blot analysis of PARP1 elongation activity time course on the indicated mono-ADP-ribosylated full-length H1.2 substrate in the presence of biotinylated NAD+. (C) Fluorescence polarization assays to evaluate binding affinities of the indicated H1.2 constructs to a fluorescein-labeled 30 base pair DNA fragment. (D) Native 3% TBE gel electrophoresis analysis of chromatin arrays (5 nM array = 60 nM nucleosome) in the presence of increasing concentrations of the indicated H1.2 construct (120, 240, 480, 960, and 1200 nM). (E) Sedimentation coefficient values (S20,w) of chromatin arrays (15 nM array = 180 nM nucleosome) in the presence of the indicated H1.2 construct (180 nM) as determined via analytical ultracentrifugation.
The linker histone C-terminal domain is known to enhance DNA binding affinity.26 A fluorescence polarization-based 30-mer DNA binding assay was employed to determine if site-specific serine mono-ADPr impacts the H1/DNA interaction (Figure 3C and Table S1). While an H1.2 construct lacking the C-terminal domain (H1.22–108) exhibited a 50-fold reduction in DNA binding affinity, mono-ADPr did not significantly impact the H1.2/DNA interaction. We next sought to test the possibility that H1.2 poly-ADPr is required to disrupt the H1.2/DNA interface. To this end, the full-length H1.2 S150ADPr1 and H1.2 S188ADPr1 constructs were incubated with PARP1 and NAD+, and products bearing variable length ADP-ribose chains were isolated via RP-HPLC fractionation. Gel migration and mass analysis of the poly-ADP-ribosylated H1 products showed a chain length distribution ranging primarily from 4 to 18 ADP-ribose units (Figure S6), and the purified constructs (H1.2 S150ADPrpoly and H1.2 S188ADPrpoly) were employed in DNA interaction assays. Again, no significant impact on DNA affinity was observed. While these results suggest that H1.2 poly-ADPr does not directly impact DNA binding activity, it should be noted that longer ADP-ribose chain lengths or chromatin templates may be required to elicit such an effect.
We next explored the effects of site-specific mono- and poly-ADPr on H1-induced chromatin compaction. Chromatin array substrates (5 nM) comprising 12 evenly spaced nucleosomes on a single DNA template were incubated with increasing concentrations of the unmodified and mono- and poly-ADP-ribosylated H1 constructs (Figure 3D). Native gel shift-based compaction assays revealed that unmodified H1 constructs abrogated gel migration of all array species at 240 nM (4:1 molar ratio of H1:nucleosome), indicating a strong compaction effect. In stark contrast, no chromatin compaction was observed in the presence of the poly-ADP-ribosylated H1 constructs at concentrations as high as 600 nM (10:1 molar ratio of H1.2/nucleosome). A more modest effect was observed with mono-ADP-ribosylated H1 constructs, which maintained their ability to stimulate chromatin compaction albeit at higher concentrations relative to the unmodified protein. Thus, site-specific H1.2 serine ADPr is sufficient to abrogate its chromatin-compaction activity, and this effect is amplified by ADP-ribose chain elongation.
To further characterize H1.2 ADPr and its impact on chromatin structure, H1.2 constructs were incubated with chromatin arrays at a 12:1 molar ratio (an equimolar ratio of H1.2/nucleosome), and chromatin sedimentation velocities were analyzed via analytical ultracentrifugation (Figure 3E and Table S2). Consistent with gel mobility results, the chromatin arrays that were incubated with unmodified H1.2 displayed the highest sedimentation coefficient (S20,w = 43.5 S) and thus the greatest level of compaction. Again, we found that mono-ADPr at either the S150 or S188 site was sufficient to reduce chromatin compaction levels as evidenced by the reduced sedimentation coefficients (S20,w = 38.5 and 38.6 S, respectively) relative to the unmodified H1.2-treated arrays. These compaction deficiencies were even more pronounced in array samples treated with poly-ADP-ribosylated H1 constructs, wherein H1.2 S150ADPrpoly and H1.2 S188ADPrpoly S20,w values were 35.2 and 35.3 S, respectively. Interestingly, compaction levels measured in the presence of H1.2 S150ADPrpoly, H1.2 S188ADPrpoly, or the H1.22–108 construct lacking the C-terminal domain were nearly identical. Therefore, poly-ADPr at a single serine site is sufficient to prevent the H1.2 C-terminal disordered domain from contributing to chromatin compaction.
In closing, we have developed a strategy to prepare ADP-ribosylated peptides that can be assembled into full-length proteins using sequential native chemical ligation reactions. We found that site-specific serine mono-ADPr on the linker histone H1.2 is sufficient to induce chromatin decompaction and that this effect is amplified by ADP-ribose chain elongation. Importantly, our approach allows us to unambiguously attribute these chromatin compaction deficiencies to H1.2 ADPr. The results presented here are consistent with a model wherein H1.2 serine ADPr induces chromatin relaxation, but additional factors are necessary to displace H1.2 from DNA damage sites. Future studies employing ADP-ribosylated H1.2 molecules will enable such aspects of this pathway to be explored and may also shed light on molecular determinants of site-specific ADP-ribosylhydrolase activity. More broadly, our work demonstrates the utility of an expanded semisynthetic toolkit to study site-specific serine ADPr and its impact on protein function and genome structure.
Supplementary Material
ACKNOWLEDGMENTS
We thank D. Nijhawan, B. Tu, and members of the Liszczak laboratory for insightful discussions. We thank A. Lemoff and the UT Southwestern Proteomics Core for technical assistance. This work was supported by grants from the Welch Foundation (I-2039-20200401 to G.L.), the Cancer Prevention Research Institute of Texas (RR180051 to G.L.), and the American Cancer Society (UTSW-IRG-17-174-13). G.L. is the Virginia Murchison Linthicum Scholar in Medical Research.
Footnotes
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.2c00091
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.2c00091.
Detailed description of the materials and methods, MS and RP-HPLC characterizations of protein fragments and full-length proteins, N-terminal cysteine ADPr data, schematic of the two-piece assembly strategy used to access the H1.2 S188ADPr site, full data from fluorescence polarization experiments and analytical ultracentrifugation, and uncropped gels and blots presented in this study (PDF)
Contributor Information
Kyuto Tashiro, Department of Biochemistry, University of Texas Southwestern Medical Center, Dallas, Texas 75390–9038, United States.
Jugal Mohapatra, Department of Biochemistry, University of Texas Southwestern Medical Center, Dallas, Texas 75390–9038, United States.
Chad A. Brautigam, Departments of Biophysics and Microbiology, University of Texas Southwestern Medical Center, Dallas, Texas 75390–9038, United States
Glen Liszczak, Department of Biochemistry, University of Texas Southwestern Medical Center, Dallas, Texas 75390–9038, United States.
REFERENCES
- (1).Gupte R; Liu Z; Kraus WL PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 2017, 31, 101–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Gibbs-Seymour I; Fontana P; Rack JGM; Ahel I HPF1/C4orf27 Is a PARP-1-Interacting Protein that Regulates PARP-1 ADP-Ribosylation Activity. Mol. Cell 2016, 62, 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bonfiglio JJ; Fontana P; Zhang Q; Colby T; Gibbs-Seymour I; Atanassov I; Bartlett E; Zaja R; Ahel I; Matic I Serine ADP-Ribosylation Depends on HPF1. Mol. Cell 2017, 65, 932–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Larsen SC; Hendriks IA; Lyon D; Jensen LJ; Nielsen ML Systems-wide Analysis of Serine ADP-Ribosylation Reveals Widespread Occurrence and Site-Specific Overlap with Phosphorylation. Cell Rep 2018, 24, 2493–2505. [DOI] [PubMed] [Google Scholar]
- (5).Palazzo L; Leidecker O; Prokhorova E; Dauben H; Matic I; Ahel I Serine is the major residue for ADP-ribosylation upon DNA damage. Elife 2018, 7, e34334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rudolph J; Roberts G; Muthurajan UM; Luger K HPF1 and nucleosomes mediate a dramatic switch in activity of PARP1 from polymerase to hydrolase. Elife 2021, 10, e65773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Suskiewicz MJ; Zobel F; Ogden TEH; Fontana P; Ariza A; Yang JC; Zhu K; Bracken L; Hawthorne WJ; Ahel D; et al. HPF1 completes the PARP active site for DNA damage-induced ADP-ribosylation. Nature 2020, 579, 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hendriks IA; Buch-Larsen SC; Prokhorova E; Elsborg JD; Rebak A; Zhu K; Ahel D; Lukas C; Ahel I; Nielsen ML The regulatory landscape of the human HPF1- and ARH3-dependent ADP-ribosylome. Nat. Commun. 2021, 12, 5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Leidecker O; Bonfiglio JJ; Colby T; Zhang Q; Atanassov I; Zaja R; Palazzo L; Stockum A; Ahel I; Matic I Serine is a new target residue for endogenous ADP-ribosylation on histones. Nat. Chem. Biol. 2016, 12, 998–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Liszczak G; Diehl KL; Dann GP; Muir TW Acetylation blocks DNA damage-induced chromatin ADP-ribosylation. Nat. Chem. Biol. 2018, 14, 837–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Langelier MF; Billur R; Sverzhinsky A; Black BE; Pascal JM HPF1 dynamically controls the PARP1/2 balance between initiating and elongating ADP-ribose modifications. Nat. Commun. 2021, 12, 6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mohapatra J; Tashiro K; Beckner RL; Sierra J; Kilgore JA; Williams NS; Liszczak G Serine ADP-ribosylation marks nucleosomes for ALC1-dependent chromatin remodeling. Elife 2021, 10, e71502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Prokhorova E; Agnew T; Wondisford AR; Tellier M; Kaminski N; Beijer D; Holder J; Groslambert J; Suskiewicz MJ; Zhu K; et al. Unrestrained poly-ADP-ribosylation provides insights into chromatin regulation and human disease. Mol. Cell 2021, 81, 2640–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Curtin NJ; Szabo C Poly(ADP-ribose) polymerase inhibition: past, present and future. Nat. Rev. Drug Discov 2020, 19, 711–736. [DOI] [PubMed] [Google Scholar]
- (15).Bonfiglio JJ; Leidecker O; Dauben H; Longarini EJ; Colby T; San Segundo-Acosta P; Perez KA; Matic I An HPF1/PARP1-Based Chemical Biology Strategy for Exploring ADP-Ribosylation. Cell 2020, 183, 1086–1102. [DOI] [PubMed] [Google Scholar]
- (16).Voorneveld J; Rack JGM; Ahel I; Overkleeft HS; van der Marel GA; Filippov DV Synthetic alpha- and beta-Ser-ADP-ribosylated Peptides Reveal alpha-Ser-ADPr as the Native Epimer. Org. Lett. 2018, 20, 4140–4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hananya N; Daley SK; Bagert JD; Muir TW Synthesis of ADP-Ribosylated Histones Reveals Site-Specific Impacts on Chromatin Structure and Function. J. Am. Chem. Soc. 2021, 143, 10847–10852. [DOI] [PubMed] [Google Scholar]
- (18).Li Z; Li Y; Tang M; Peng B; Lu X; Yang Q; Zhu Q; Hou T; Li M; Liu C; et al. Destabilization of linker histone H1.2 is essential for ATM activation and DNA damage repair. Cell Res. 2018, 28, 756–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Strickfaden H; McDonald D; Kruhlak MJ; Haince JF; Th’ng JPH; Rouleau M; Ishibashi T; Corry GN; Ausio J; Underhill DA; et al. Poly(ADP-ribosyl)ation-dependent Transient Chromatin Decondensation and Histone Displacement following Laser Microirradiation. J. Biol. Chem. 2016, 291, 1789–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Althaus FR Poly ADP-ribosylation: a histone shuttle mechanism in DNA excision repair. J. Cell Sci. 1992, 102, 663–670. [DOI] [PubMed] [Google Scholar]
- (21).Azad GK; Ito K; Sailaja BS; Biran A; Nissim-Rafinia M; Yamada Y; Brown DT; Takizawa T; Meshorer E PARP1-dependent eviction of the linker histone H1 mediates immediate early gene expression during neuronal activation. J. Cell Biol. 2018, 217, 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Poirier GG; de Murcia G; Jongstra-Bilen J; Niedergang C; Mandel P Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. U. S. A. 1982, 79, 3423–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zheng JS; Tang S; Qi YK; Wang ZP; Liu L Chemical synthesis of proteins using peptide hydrazides as thioester surrogates. Nat. Protoc 2013, 8, 2483–2495. [DOI] [PubMed] [Google Scholar]
- (24).Bang D; Pentelute BL; Kent SB Kinetically controlled ligation for the convergent chemical synthesis of proteins. Angew. Chem., Int. Ed. Engl. 2006, 45, 3985–3988. [DOI] [PubMed] [Google Scholar]
- (25).Fang GM; Wang JX; Liu L Convergent chemical synthesis of proteins by ligation of peptide hydrazides. Angew. Chem., Int. Ed. Engl. 2012, 51, 10347–10350. [DOI] [PubMed] [Google Scholar]
- (26).White AE; Hieb AR; Luger K A quantitative investigation of linker histone interactions with nucleosomes and chromatin. Sci. Rep 2016, 6, 19122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.