

Abstract
Disturbance of the cholinergic system plays a crucial role in the pathological progression of neurological diseases that cause dyskinesia-like behaviors. However, the molecular mechanisms underlying this disturbance remain elusive. Here, we showed that cyclin-dependent kinase 5 (Cdk5) was reduced in cholinergic neurons of midbrain according to the single-nucleus RNA sequencing analysis. Serum levels of CDK5 also decreased in patients with Parkinson’s disease accompanied by motor symptoms. Moreover, Cdk5 deficiency in cholinergic neurons triggered paw tremors, abnormal motor coordination, and motor balance deficits in mice. These symptoms occurred along with cholinergic neuron hyperexcitability and increases in the current density of large-conductance Ca2+-activated K+ channels (BK channels). Pharmacological inhibition of BK channels restrained the excessive intrinsic excitability of striatal cholinergic neurons in Cdk5-deficient mice. Furthermore, CDK5 interacted with BK channels and negatively regulated BK channel activity via phosphorylation of threonine-908. Restoration of CDK5 expression in striatal cholinergic neurons reduced dyskinesia-like behaviors in ChAT-Cre;Cdk5f/f mice. Together, these findings indicate that CDK5-induced phosphorylation of BK channels involves in cholinergic-neuron-mediated motor function, providing a potential new therapeutic target for treating dyskinesia-like behaviors arising from neurological diseases.
Introduction
Dyskinesia-like behaviors are common neurological disorders occurring in neurological diseases such as Parkinson’s disease (PD), ataxia, dystonia, and Huntington’s disease [1,2]. Abnormal corticothalamic projections to the striatum contribute to the pathophysiology of these movement syndromes [3,4]. The striatum is required for motor planning, sequencing, and execution [5]. Within the striatal circuit, which is associated with the control of motor coordination and cognitive function, cholinergic neurons play an important regulatory role [6,7].
Increased cholinergic activity and acetylcholine levels are characteristics of PD and influence the functional output of striatal circuits [8,9]. However, anticholinergics, including trihexyphenidyl and procyclidine, are not currently commonly used to treat dyskinesia-like behaviors in PD due to their adverse effects, such as confusion and hallucinations [10,11]. As these traditional anticholinergic drugs indirectly inhibit central hypercholinergic excitability by extensively blocking central acetylcholine receptor or reducing acetylcholine [11], a new motor-function-related target is needed to precisely regulate cholinergic activity.
Striatal cholinergic neurons have extensive axonal fields that project locally to affect the output of striatal projection neurons [12], which display specific firing properties of tonic firing with a transient pause [13]. Striatal cholinergic neurons also show 2 periodic patterns: rhythmic bursts and single spikes [14,15]. Aberrant K+ channels activity alters the excitability of striatal cholinergic neurons, which is associated with pathological process of dyskinesia and PD. For example, the decreased Kv1.3 currents induce hyperexcitability of cholinergic neurons in the striatum of PD mice [16]. Although previous findings have suggested that delayed rectifier, hyperpolarization-activated, and calcium-dependent K+ channels contribute to the spontaneous firing patterns of striatal cholinergic neurons [14,15,17], studies have yet to identify the precise biochemical substrate that modifies electrophysiological currents or explains these changes.
Cyclin-dependent kinase 5 (CDK5) is a serine/threonine protein kinase involved in a variety of critical functions related to neural signaling pathways and neuronal activity [18,19]. For example, CDK5 suppresses both presynaptic dopamine release and postsynaptic dopamine signaling in striatal neurons [20,21]. In addition, specific knockdown of Cdk5 in the dorsal striatum disrupted the excitatory/inhibitory synaptic balance [19]. Furthermore, medium spiny neurons of the nucleus accumbens in Cdk5-conditional knockout (cKO) mice exhibited elevated intrinsic neuronal activity [22]. Therefore, it is essential to establish the causality in the association between CDK5 signaling and cholinergic neuronal activity; hence, an approach will provide a viable rescue strategy to reverse the cellular adaptations and behavioral symptoms of neurological disorders.
Here, we report the potential roles of CDK5 in regulating striatal cholinergic neuron activity via Ca2+-activated K+ channels (BK channels). Loss of cholinergic CDK5 induces hyperactivity of striatal cholinergic neurons and abnormal motor function in mice. Combining gene technology with electrophysiology, we demonstrate that CDK5 binds to BK channels and limits BK-mediated currents by phosphorylating threonine-908 (T908). More importantly, restoration of CDK5 expression in striatal cholinergic neurons alleviated the motor dysfunction induced by cholinergic-neuron-specific Cdk5 deficiency. Together, our pharmacological and genetic findings suggest that CDK5 signaling may represent a viable target to ameliorate dyskinesia-like disorders.
Results
Decreased CDK5 mRNA and serum CDK5 in PD patients
Lesions of cholinergic systems contribute to motor and nonmotor syndromes in PD [9]. Here, we filtered 41,435 cells from a Gene Expression Omnibus dataset (GSE157783) through quality control; this dataset contains single-nucleus RNA sequencing (snRNA-seq) data from 6 patients with PD and 5 controls collected postmortem from the midbrain (Fig. S1A to C). We analyzed and counted the number of genes expressed in each cell, resulting in data from 41,427 cells and 19,832 gene features. Then, we mapped single-nucleus transcriptomes through uniform manifold approximation and projection (UMAP). According to the previously well-identified marker genes and machine learning cross-validation of cell type annotation, the cells were divided into the following 10 cell types: astrocytes, cholinergic neurons, dopaminergic neurons (DaNs), endothelial cells, ependymal cells, γ-aminobutyric acidergic (GABAergic) neurons, microglia, oligodendrocytes, oligodendrocyte precursor cells (OPCs), and pericytes (Fig. 1A). Next, to investigate the molecular differences between patients with PD and healthy controls (HCs), we validated the top 10 differentially expressed genes (DEGs; specifically, the top 10 downregulated DEGs) in patients with PD; CDK5 was identified as the most substantially downregulated DEG in patients with PD (Fig. 1B). Specifically, this gene was markedly decreased in midbrain cholinergic neurons in patients with PD (Fig. 1C), the neurons that were filtered by the markers acetylcholinesterase (ACHE) and choline acetyltransferase (CHAT) (Fig. S1D) through model-based analysis of single-cell transcriptomics. We also found a decreased protein level of CDK5 (Fig. 1D and E) in the serum of patients with PD (Table S1).
Fig. 1.

Decreased CDK5 mRNA and serum CDK5 levels in patients with PD. (A) Uniform manifold approximation and projection (UMAP) plot of all 41,427 midbrain cells from HCs and patients with PD. Colors indicate different cell types. (B) The top 10 downregulated genes in patients with PD according to model-based analysis of single-cell transcriptomics. (C) The mRNA expression of CDK5 in patients with PD decreased in all midbrain cell types and especially in cholinergic neurons compared with that of HCs. *P < 0.05 and **P < 0.01, unpaired 2-tailed Student’s t test. Data represent the means ± SEM. (D and E) Representative bands of WBs and quantification of CDK5 in the serum of patients with PD (n = 6) and HCs (n = 6), **P < 0.01, unpaired 2-tailed Student’s t test. Data represent the means ± SEM. DaNs, dopaminergic neurons; GABA, GABAergic neurons; OPCs, oligodendrocyte precursor cells; a.u., arbitrary units.
Mice with cholinergic-neuron-specific Cdk5 deficiency exhibit tremor and motor dysfunction
To examine the role of CDK5 in cholinergic neurons, we first confirmed CDK5 expression in cholinergic neurons through immunostaining. The data showed that CDK5 colocalized with choline acetyltransferase (ChAT)-positive cholinergic neurons (Fig. 2A), which suggested that CDK5 was enriched in cholinergic neurons. Then, we crossed Cdk5f/f mice, in which exon 1 to exon 5 of Cdk5 were flanked by loxP sites, with ChAT-Cre mice to generate cholinergic-neuron-specific Cdk5-cKO mice (Fig. 2B and C). The immunostaining results indicated that ChAT-positive neurons did not express CDK5 in ChAT-Cre;Cdk5f/f mice (Fig. 2D). In addition, cholinergic-neuron-specific Cdk5 deletion had no effect on body weight, brain weight, or the organ index (Fig. S2A to C).
Fig. 2.
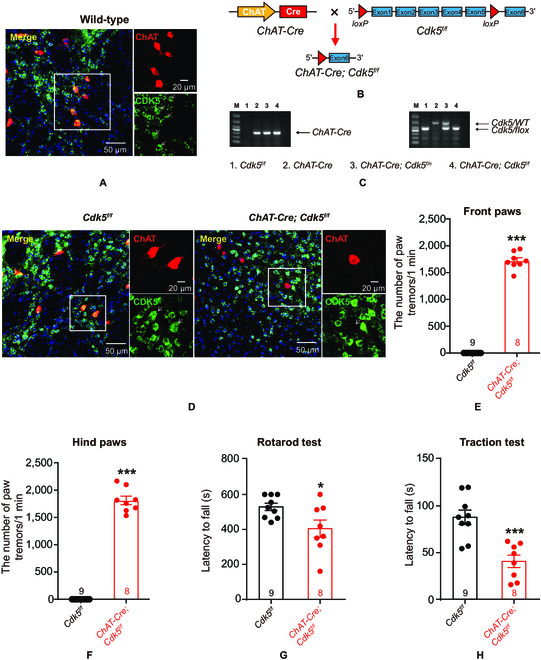
Mice with cholinergic-neuron-specific Cdk5 deficiency exhibit tremor and motor dysfunction. (A) Representative image of CDK5 and ChAT in wild-type mice. The slices were stained with an anti-CDK5 antibody (green), anti-ChAT antibody (red), and 4′,6-diamidino-2-phenylindole (blue; marker for nuclei). Scale bar, 50 μm; magnified image, 20 μm. (B) The strategy used to generate cholinergic-neuron-specific Cdk5-cKO mice (ChAT-Cre;Cdk5f/f mice). Floxed Cdk5 transgenic mice (Cdk5f/f) were crossed with ChAT-Cre mice. (C) Genotyping of ChAT-Cre;Cdk5f/f mice. ChAT-Cre primers generated a 300-bp product, and Cdk5 primers generated a 460-bp product of the loxp-flanked allele or a 660-bp product of the wild-type allele. (D) Representative images of CDK5 and ChAT in Cdk5f/f mice and ChAT-Cre;Cdk5f/f mice. Scale bars, 50 μm; magnified images, 20 μm. (E and F) The number of paw tremors in ChAT-Cre;Cdk5f/f mice (n = 8) and Cdk5f/f mice (n = 9). Paw tremors in ChAT-Cre;Cdk5f/f mice were recorded for 1 min. ***P < 0.001, unpaired 2-tailed Student’s t test. Data represent the means ± SEM. (G and H) Quantification of the time spent in the rotarod (G) and traction (H) tests. Cdk5f/f mice (n = 9) and ChAT-Cre;Cdk5f/f mice (n = 8). *P < 0.05 and ***P < 0.001, unpaired 2-tailed Student’s t test. Data represent the means ± SEM.
Cholinergic neurons are implicated in motor control and motor processes; thus, we first explored whether Cdk5 loss affected motor behavior in ChAT-Cre;Cdk5f/f mice. Video data (Movie S1) showed that all Cdk5-deficient mice exhibited paw tremors (Fig. 2E and F). In the rotarod test, ChAT-Cre;Cdk5f/f mice showed a shorter latency to fall from the rotating rod than Cdk5f/f mice (Fig. 2G). A similar result was also seen in the traction test (Fig. 2H). However, gait analysis revealed no difference between ChAT-Cre;Cdk5f/f mice and Cdk5f/f mice (Fig. S2D). To exclude the influence of alterations in the function of limb skeletal muscles on motor ability, we performed limb electromyography (EMG) in anesthetized mice [23]. The EMG results indicated no impairment of electrical activity in the limbs (Fig. S2G and H). The grip strength test showed that the Cdk5 deficiency in cholinergic neuron weakens grip strength (Fig. S2I and J). Moreover, our results revealed no difference in performance in the Y-maze test (Fig. S2K) or the open-field test (Fig. S2L) between the groups.
Together, these results confirm that CDK5 plays a role in basic cholinergic-neuron-mediated motor function.
Enhanced intrinsic excitability of striatal cholinergic neurons in mice with cholinergic-neuron-specific Cdk5 deficiency
Striatal cholinergic neurons are involved in motor function, and, according to the Human Protein Atlas, Cdk5 mRNA is enriched in the basal ganglia (Fig. S3A). Next, to validate the specific role of CDK5 in striatal cholinergic neurons in motor function, we administered a bilateral injection of adeno-associated virus (AAV) vectors carrying rAAV-ChAT-Cre-EGFP or rAAV-ChAT-EGFP (as the control) into the striatum of Cdk5f/f mice to conditionally knock out Cdk5 in cholinergic neurons (Fig. 3A). In the striatum, the immunostaining data showed that the ratio of enhanced green fluorescent protein (EGFP)+-ChAT+ cells/ChAT+ cells was 88.94% ± 3.19%, and the ratio of EGFP+-ChAT+ cells/EGFP+ cells was 82.75% ± 2.42% (Fig. S3B and C), which indicated high infection efficiency. Twenty-one days after virus injection, the immunohistochemistry results indicated reduced expression of CDK5 in striatal cholinergic neurons in mice injected with rAAV-ChAT-Cre-EGFP (Fig. 3B). Behavioral analyses were performed to evaluate the motor function of striatal cholinergic Cdk5-deficient mice. The striatal cholinergic Cdk5-deficient mice exhibited static paw tremors in all 4 limbs compared with the control mice (Fig. 3C and D). During the 10-min rotarod test, the Cdk5-cKO mice exhibited a shorter latency to fall from the rotating rod than the control mice (Fig. 3E). Similarly, the Cdk5-cKO mice showed a shorter latency to fall from the metallic wire in the traction test (Fig. 3F). These results showed that the loss of Cdk5 in striatal cholinergic neurons impaired the motor function of mice.
Fig. 3.

Elevation of intrinsic striatal cholinergic neuron excitability in mice with cholinergic-neuron-specific Cdk5 deficiency. (A) Schematic diagrams of the injection of rAAV-ChAT-Cre-EGFP vectors or control vectors (rAAV-ChAT-EGFP) into the striatum of Cdk5f/f mice. (B) Representative confocal images to identify Cdk5-knockout efficiency in striatal cholinergic neurons. Scale bars, 50 μm; magnified images, 20 μm. (C and D) The number of paw tremors in Cdk5f/f mice injected with rAAV-ChAT-Cre-EGFP (n = 5) or rAAV-ChAT-EGFP (n = 5) vectors. Paw tremors were recorded for 1 min. ***P < 0.001, unpaired 2-tailed Student’s t test. Data represent the means ± SEM. (E and F) Quantification of the time spent in the rotarod (E) and traction (F) tests. Cdk5f/f mice injected with rAAV-ChAT-EGFP (n = 11) and rAAV-ChAT-Cre-EGFP (n = 9). *P < 0.05 and **P<0.01, unpaired 2-tailed Student’s t test. Data represent the means ± SEM. (G) Representative images of EGFP-positive neurons in the striata of Cdk5f/f mice injected with AAV vectors. Scale bar, 20 μm. (H and I) Representative APs and quantification of the AP frequency in EGFP-positive neurons in the striata of Cdk5f/f mice injected with rAAV-ChAT-Cre-EGFP (n = 14 cells from 3 mice) or rAAV-ChAT-EGFP (n = 14 cells from 3 mice) following current injections at 20 pA. ***P < 0.001, 2-way ANOVA followed by Tukey’s multiple comparisons test. Data represent the means ± SEM. (J) The AP half-width in cholinergic neurons in the striata of Cdk5f/f mice injected with rAAV-ChAT-EGFP (n = 14 cells from 3 mice) and rAAV-ChAT-Cre-EGFP (n = 14 cells from 3 mice). **P < 0.01, unpaired 2-tailed Student’s t test. Data represent the means ± SEM. (K and L) Quantification of the AP amplitude and AP firing threshold in eGFP-positive neurons in the striata of Cdk5f/f mice injected with rAAV-ChAT-EGFP (n = 14 cells from 3 mice) and rAAV-ChAT-Cre-EGFP (n = 14 cells from 3 mice) under whole-cell recording. AP amplitude, P = 0.8376; AP firing threshold, P = 0.7283; unpaired 2-tailed Student’s t test. Data represent the means ± SEM. (M to P) Intrinsic membrane properties of striatal cholinergic neurons from Cdk5f/f mice injected with rAAV-ChAT-EGFP (n = 14 cells from 3 mice) and rAAV-ChAT-Cre-EGFP (n = 14 cells from 3 mice). Membrane potential, P = 0.7873; input resistance: P = 0.7818; Tau, P = 0.3343, Cm: P = 0.0976; unpaired 2-tailed Student’s t test. Data represent the means ± SEM. STR, striatum; RMP, resting membrane potential; ns, not significant.
To further address the effect of Cdk5 deficiency on the firing properties of striatal cholinergic neurons, we conducted electrophysiological recordings according to the whole-cell mode (Fig. 3G) [8]. Cdk5 deficiency increased the frequency (Fig. 3H and I) and reduced the half-width (Fig. 3J) of action potentials (APs) in striatal cholinergic neurons in rAAV-ChAT-Cre-EGFP-injected mice compared with rAAV-ChAT-EGFP-injected mice. Other AP properties, including amplitude and firing threshold, did not differ between the 2 groups of mice (Fig. 3K and L). Moreover, no significant changes were found in the resting membrane potential, membrane time constant (Tau), input resistance (Rin), or membrane capacitance (Cm) between the 2 groups of mice (Fig. 3M to P).
We also performed electrophysiological recordings in striatal cholinergic neurons of ChAT-Cre;Cdk5f/f mice. To accurately identify cholinergic neurons, we generated ChAT-Cre;Cdk5f/f;ChAT eGFP mice. Consistent with striatal cholinergic neurons in Cdk5-cKO mice, the frequency of APs in cholinergic neurons was increased (Fig. S3D), and the AP half-width was reduced (Fig. S3E) in ChAT-Cre;Cdk5f/f;ChAT eGFP mice. Other AP properties (amplitude and threshold) were unaltered (Fig. S3F and G). Furthermore, cell-attached recordings indicated no change in the spontaneous firing rate of these neurons in ChAT-Cre;Cdk5f/f;ChAT eGFP mice (Fig. S3H and I). Whole-cell recordings showed no significant changes in the resting membrane potential, Rin, Tau, or Cm (Fig. S3J to M). These data suggest that CDK5 mediates the electrophysiological activity of cholinergic neurons.
Elevation of BK currents is associated with hyperexcitability of striatal cholinergic neurons in ChAT-Cre;Cdk5f/f;ChAT eGFP mice
Voltage-gated Na+ channels and K+ channels are associated with the generation of APs and determine AP properties [24]. To test whether CDK5 alters Na+ channels and K+ channels, resulting in the hyperactivity of cholinergic neurons, we recorded Na+ and K+ currents under voltage-clamp conditions. We observed an increased density of K+ currents (Fig. 4A) and no change in the density of Na+ currents (Fig. S4A). There are numerous K+ currents including the following 3 main types: transient outward K+ currents (IA currents), delayed rectifier K+ currents (Ik currents), and calcium-activated K+ currents (BK currents) [25]. We observed no change in the density of Ik currents (Fig. 4B) or IA currents (Fig. 4C). However, BK channels were previously reported to regulate AP half-width; blockage of BK channels increased the AP half-width [26,27]. We used iberiotoxin (IBTX), a highly specific BK channel blocker, to isolate BK currents [28]. As shown in Fig. 4D, BK current density was increased in ChAT-Cre;Cdk5f/f;ChAT eGFP mice. More importantly, the hyperactivity of cholinergic neurons in ChAT-Cre;Cdk5f/f;ChAT eGFP mice was reversed by IBTX (Fig. 4E); thus, BK channels may induce the alteration in cholinergic neuron activity. In addition, CDK5 has been reported to regulate voltage-gated Ca2+ channels [29]. We therefore recorded Ca2+ currents in striatal cholinergic neurons under voltage-clamp conditions. However, we found no difference in the Ca2+ current density between groups in mice (Fig. S4B and C). These electrophysiology data demonstrate that increased BK currents due to Cdk5 deficiency led to hyperexcitability of striatal cholinergic neurons.
Fig. 4.

Increased BK currents are associated with hyperexcitability of striatal cholinergic neurons in ChAT-Cre;Cdk5f/f;ChAT eGFP mice. (A) Representative K+ currents and related summary data for striatal cholinergic neurons from ChAT-Cre;Cdk5f/f;ChAT eGFP mice (n = 20 cells from 4 mice) and ChAT eGFP;Cdk5f/f mice (n = 19 cells from 4 mice). **P < 0.01, 2-way ANOVA followed by Tukey’s multiple comparisons test. Data represent the means ± SEM. (B) Representative delayed rectifier K+ currents (IK currents) and related summary data for striatal cholinergic neurons from ChAT-Cre;Cdk5f/f;ChAT eGFP mice (n = 17 cells from 2 mice) and ChAT eGFP;Cdk5f/f mice (n = 26 cells from 3 mice). P = 0.9991, 2-way ANOVA followed by Tukey’s multiple comparisons test. Data represent the means ± SEM. (C) Representative transient outward K+ currents (IA currents) and related summary data for striatal cholinergic neurons from ChAT-Cre;Cdk5f/f;ChAT eGFP mice (n = 20 cells from 4 mice) and ChAT eGFP;Cdk5f/f mice (n = 19 cells from 4 mice). P = 0.9986, 2-way ANOVA followed by Tukey’s multiple comparisons test. Data represent the means ± SEM. (D) Representative BK currents (acquired by subtracting currents recorded after the application of IBTX from currents before the application of IBTX) and related summary data for striatal cholinergic neurons from ChAT-Cre;Cdk5f/f;ChAT eGFP mice (n = 16 cells from 4 mice) and ChAT eGFP;Cdk5f/f mice (n = 16 cells from 4 mice). ***P < 0.001, 2-way ANOVA followed by Tukey’s multiple comparisons test. Data represent the means ± SEM. (E) Representative APs and quantification of the AP frequency of eGFP-positive neurons in the striata of ChAT-Cre;Cdk5f/f;ChAT eGFP mice (n = 28 cells from 5 mice) and ChAT eGFP;Cdk5f/f mice (n = 28 cells from 5 mice) with or without the application of IBTX following current injections at 20 pA from 0 to 180 pA. ***P < 0.001, 2-way ANOVA followed by Tukey’s multiple comparisons test. Data represent the means ± SEM. DMSO, dimethyl sulfoxide.
CDK5 negatively regulates BK channel activity via phosphorylation of T908
To explore how Cdk5 deficiency contributes to the elevation of the BK-channel-mediated current density, we examined the association between CDK5 and BK channels. Coimmunoprecipitation and Western blotting data suggested that CDK5 binds directly to the BK channel in striatal cholinergic neurons (Fig. 5A). To further determine the role of interaction between CDK5 and BK channels, we constructed mslo (encoding BK)-eGFP (BK-eGFP) and Cdk5-Flag plasmids and transiently transfected them into Neuro 2A (N2A) cells (Fig. 5B). Confocal microscopy indicated that CDK5 and BK channels were colocalized in the N2A cell membrane (Fig. 5C). Similarly, coimmunoprecipitation data indicated that CDK5 binds directly to BK channels (Fig. 5D). Moreover, whole-cell patch-clamp recording suggested null N2A cells with a small current increase. These currents were up to 6-fold larger in BK-eGFP-transfected N2A cells than in null N2A cells and were strongly inhibited in BK-eGFP- and Cdk5-Flag-cotransfected N2A cells (Fig. 5E and F).
Fig. 5.

CDK5 negatively regulates BK channel activity via phosphorylation of T908. (A) Proteins pulled down with an anti-CDK5 antibody in the striatum were probed with an anti-BK channel antibody. (B) Schematic diagrams of the BK-channel-encoding mslo and Cdk5 plasmids. (C) Representative images of the localization of CDK5 (red) and BK channels (GFP; green) in N2A cells after transfection with BK and Cdk5 plasmids. Scale bars, 5 μm. (D) Proteins pulled down with an anti-eGFP antibody in N2A cells after 3×Flag-labeled Cdk5 and BK-eGFP cotransfection were probed with an anti-Flag antibody. WCL, whole-cell lysate. (E) Representative BK current traces from N2A cells under voltage-clamp recording (10-mV increment). N2A cells were cotransfected with BK-eGFP and Flag (BK-eGFP + Flag) or BK-eGFP and Cdk5-Flag (BK-eGFP + Cdk5-Flag). (F) The current density of BK currents in N2A cells transfected with or without Cdk5 plasmids. eGFP + Flag (n = 13), BK-eGFP + Flag (n = 12), and BK-eGFP + Cdk5-Flag (n = 7). ***P < 0.001, 2-way ANOVA followed by Tukey’s multiple comparisons test. Data represent the means ± SEM. (G) CDK5 phosphorylated sites on BK channel proteins were predicted with NetPhos 3.1. (H) Representative BK current traces under voltage-clamp recording (10-mV increment) from N2A cells cotransfected with BK-eGFP or BKT908A-eGFP and Cdk5-Flag or Flag. (I) Current–voltage relationships of BK currents for (H). BK-eGFP + Flag (n = 10), BK-eGFP + Cdk5-Flag (n = 13), BKT908A-eGFP + Flag (n = 7), and BKT908A-eGFP + Cdk5-Flag (n = 12). ***P<0.001, 2-way ANOVA followed by Tukey’s multiple comparisons test. Data represent the means ± SEM. IgG, immunoglobulin G; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
BK channels have multiple phosphorylation patterns [30]. Phosphorylation of serine-695 by protein kinase C blocks BK channel activity, and phosphorylation of serine-1151 by protein kinase C enhances the BK channel response to activation by guanosine 3′,5′-monophosphate-dependent protein kinase [31]. For BK channel activation, adenosine 3′,5′-monophosphate (cAMP)-dependent protein kinase must phosphorylate serine-899 in the α subunit of BK channels [32]. We used NetPhos 3.1 to predict the site of the BK channel protein targeted by CDK5 (scores greater than 0.5 were considered valid) [33] and identified 4 potential sites, i.e., serine-716 (S716), serine-720 (S720), threonine-908 (T908), and threonine-1112 (T1122) (Fig. 5G). To determine the site that CDK5 targets, we constructed BK channel mutants (BKS716A-eGFP, BKS720A-eGFP, BKT908A-eGFP, or BKT1112A-eGFP), cotransfected them with or without the Cdk5-Flag plasmid into N2A cells, and performed electrophysiology recordings. As expected, the increase in the BK current density induced by BK-eGFP transfection was blocked by cotransfection of BK-eGFP and Cdk5-Flag in N2A cells. More interestingly, there was no difference in the BK current density among N2A cells transfected with BK-eGFP and Flag, BKT908A-eGFP and Flag, or BKT908A-eGFP and Cdk5-Flag (Fig. 5H and I). However, the BK current density was decreased in N2A cells cotransfected with Cdk5-Flag, BKS716A-eGFP, BKS720A-eGFP, or BKT1112A-eGFP (Fig. S5A). Moreover, the BK current density did not differ among N2A cells transfected with BKS716A-eGFP, BKS720A-eGFP, or BKT1112A-eGFP compared to those transfected with BK-eGFP (Fig. S5B). These data indicate that CDK5 negatively regulates BK currents via phosphorylation of T908.
Restoration of CDK5 expression in striatal cholinergic neurons ameliorates motor dysfunction
To examine whether restoring CDK5 expression would alleviate motor dysfunction in ChAT-Cre;Cdk5f/f mice, we bilaterally injected an AAV-expressing Cdk5 or mCherry (pAAV-EF1a-DIO-Cdk5-mCherry or pAAV-EF1a-DIO-mCherry) into the striata of ChAT-Cre;Cdk5f/f mice and an AAV-expressing mCherry into the striata of ChAT-Cre mice (Fig. 6A and B). After injection, we observed a large population of mCherry-labeled ChAT-positive neurons (Fig. 6C), indicating that CDK5 was expressed specifically in cholinergic neurons. Subsequently, we performed motor-related behavioral tests. The front paw tremors of ChAT-Cre;Cdk5f/f mice were alleviated by injection of pAAV-EF1a-DIO-Cdk5-mCherry, but there was no change in hind paw tremors (Fig. 6D and E). Furthermore, the results of the rotarod and traction tests indicated that restoration of CDK5 expression improved motor performance (Fig. 6F and G). To explore the effect of restoring CDK5 expression on electrical activity in the striatum, we recorded local field potentials (LFPs) in the striata of AAV-injected Cdk5f/f and ChAT-Cre;Cdk5f/f mice. We recorded 0- to 100-Hz LFP oscillations in the striata of freely behaving mice. We found that theta (4 to 12 Hz), beta (13 to 35 Hz), low-gamma (36 to 65 Hz), and high-gamma (66 to 95 Hz) oscillations were decreased in the striata of ChAT-Cre;Cdk5f/f mice; these decreases were significantly reversed by Cdk5 overexpression (Fig. 6H to M).
Fig. 6.

Restoration of CDK5 expression in striatal cholinergic neurons improves the motor function of ChAT-Cre;Cdk5f/f mice. (A) Schematic diagrams of ChAT-Cre-dependent AAV vectors for the overexpression of Cdk5. (B) Schematic diagram of the injection of Cre-dependent AAV vectors into the bilateral striatum of mice. (C) Representative images of mCherry/ChAT-positive cells in the striata of ChAT-Cre;Cdk5f/f mice. ChAT (green), CDK5 (mCherry; red); Scale bar, 500 μm (left) and 100 μm (right). (D and E) The number of front paw tremors (D) and hind paw tremors (E). The number of paw tremors in ChAT-Cre;Cdk5f/f mice was recorded for 1 min. ChAT-Cre + AAV-mCherry mice (n = 4), ChAT-Cre;Cdk5f/f + AAV-mCherry mice (n = 4), and ChAT-Cre;Cdk5f/f + AAV-Cdk5-mCherry mice (n = 4). **P < 0.01 and ***P < 0.001, unpaired 2-tailed Student’s t test. Data represent the means ± SEM. (F and G) Quantification of the time spent on the accelerating rotarod and in the traction test. ChAT-Cre + AAV-mCherry mice (n = 9), ChAT-Cre;Cdk5f/f + AAV-mCherry mice (n = 8), and ChAT-Cre;Cdk5f/f + AAV-Cdk5-mCherry mice (n = 8). *P < 0.05, **P < 0.01, and ***P < 0.001, one-way ANOVA followed by Tukey’s multiple comparisons test. Data represent the means ± SEM. (H) Relative LFP power values in the striata of ChAT-Cre + AAV-mCherry mice (gray; n = 5), ChAT-Cre;Cdk5f/f + AAV-mCherry mice (red; n = 5) and ChAT-Cre;Cdk5f/f + AAV-Cdk5-mCherry mice (green; n = 5) following 32-channel recording electrode implantation (shaded bands indicate the SEM). (I) Representative LFP spectrograms from ChAT-Cre + AAV-mCherry mice, ChAT-Cre;Cdk5f/f + AAV-mCherry mice, and ChAT-Cre;Cdk5f/f + AAV-Cdk5-mCherry mice. (J to M) Relative LFP power values for different frequencies in the striata of mice. Theta (4 to 12 Hz) (J), beta (13 to 35 Hz) (K), low-gamma (36 to 65 Hz) (L), and high-gamma (66 to 95 Hz) (M) power in the striatum. ChAT-Cre + AAV-mCherry mice (n = 5), ChAT-Cre;Cdk5f/f + AAV-mCherry mice (n = 5), and ChAT-Cre;Cdk5f/f+ AAV-Cdk5-mCherry mice (n = 5), *P < 0.05, **P < 0.01, and ***P < 0.001, one-way ANOVA followed by Tukey’s multiple comparisons test. Data represent the means ± SEM.
Discussion
In the present study, we illustrated that the BK channel is a CDK5 target that plays essential roles in modulating the activity of striatal cholinergic neurons, which are implicated in motor control and motor processes. By elucidating the electrophysiological mechanisms underlying Cdk5 deficiency-mediated motor dysfunction, we found hyperexcitability of striatal cholinergic neurons, reduced AP half-width, and increased BK current density according to whole-cell recordings in ChAT-Cre;Cdk5f/f mice. At the cellular level, further mechanistic data suggested that CDK5 phosphorylates BK at T908 and negatively regulates BK currents. In addition, we demonstrated that restoration of CDK5 expression in striatal cholinergic neurons ameliorated cholinergic-neuron Cdk5-deficit-triggered dyskinesia-like behaviors.
The cardinal dyskinesia symptoms in PD include static tremor, rigidity, and postural instability [34]. Abnormal activity of striatal cholinergic neurons has been identified to be essential to dyskinesia-like behaviors and motor symptoms of PD. The upregulation of regulator of G protein signaling 4 (RGS4) and the activation D5-cAMP-ERK1/2 signaling pathway are reported to mediate hyperexcitability of striatal cholinergic neurons [35,36]. Neuronal CDK5 is required for spatial learning and recurrent excitatory circuits in the hippocampus [37,38] and proper multipolar-to-bipolar transition in the cortex [39]. Here, we showed that CDK5 mRNA decreased markedly in patients with PD. CDK5 was also reduced in the serum of patients with PD. Consistent with these findings, Cdk5 deficiency in cholinergic neurons triggered dyskinesia-like behaviors. Striatal cholinergic neurons exhibit 2 periodic firing patterns, i.e., rhythmic bursts and single spikes, and spontaneous firing patterns are caused by K+-current-mediated afterhyperpolarization [40]. Here, Cdk5 deficiency had no effect on the spontaneous firing rate in cell-attached voltage-clamp recordings. However, in whole-cell patch-clamp recordings, Cdk5 deficiency increased the frequency and decreased the half-width of APs in striatal cholinergic neurons after current injections, which may have accounted for the dyskinesia-like behaviors observed in ChAT-Cre;Cdk5f/f mice. Hence, our model has the advantage of mimicking, at least in part, the aetiology and symptomatology of dyskinesia-like behaviors in neurological disorders.
How does the reduction in Cdk5 signaling in striatal cholinergic neurons contribute to motor dysfunction? We examined the voltage-gated Na+ and K+ currents, which mediate the formation and process of APs [26], and BK currents, which contribute to repolarization [14]. Here, we found that voltage-gated Na+ and K+ currents were unchanged in Cdk5-deficient cholinergic neurons. BK channels have profound effects on neuronal excitability [41,42]. Numerous studies have indicated that activation of BK channels enhances neuronal excitability [43–45]. In the present study, we used IBTX to segregate BK currents in striatal cholinergic neurons; we found increased BK currents in ChAT-Cre;Cdk5f/f mice. Next, we identified T908 as the phosphorylation site of CDK5 on BK channels. These data suggest that BK channels, which link intracellular molecular signals and neuronal electrical signals [46], are negatively regulated by CDK5 via phosphorylation at T908. Thus, deficit of CDK5 in striatal cholinergic neurons increased BK currents, leading to cholinergic neuron hyperexcitability.
The therapeutic relevance of our findings is highlighted by the amelioration of dyskinesia-like behaviors after local restoration of CDK5 in ChAT-Cre;Cdk5f/f mice. Motor deficits are associated with the emergence of exaggerated β oscillations in PD [47,48]. Here, we show that striatal cholinergic CDK5 regulates motor function through modulating frequency bands (alpha, beta, and gamma) in the corticostriatal circuit. The therapeutic effects of restoring CDK5 on LFPs highlight the potential clinical applications of closed-loop neuromodulation. This application is consistent with the idea that local intervention at the level of the striatum, in both rodents and humans, ameliorates dyskinesia-like phenotypes [49–52]. In future, it would be interesting to further explore whether CDK5-induced phosphorylation of BK channels also involves in dyskinesia-like behaviors of other neurological diseases.
In conclusion, our study identified BK channels in striatal cholinergic neurons as new targets for CDK5 phosphorylation, and we described electrophysiological and molecular mechanisms of Cdk5-deficiency-mediated dyskinesia-like behaviors. The association between CDK5 and its neuron-specific activator molecules p35 and p39 is essential for kinase activation [53,54]. In contexts where high BK current densities in striatal cholinergic neurons are associated with hyperexcitability of cholinergic neurons, the novel CDK5 activator may provide an intervention target to suppress dyskinesia-like behaviors in neurological disorders.
Materials and Methods
Animals
All mice were housed in group and in ventilated cages under a 12-h light/dark schedule and a 22 ± 1 °C temperature to maintain conventional healthy states [55,56]. All experimental protocols in mice were strictly performed in compliance with the guide of Nanjing Medical University Animal Experimentation Committee.
ChAT-Cre mice (stock no. 006410, The Jackson Laboratory) were crossed with Cdk5f/f mice (stock no. 014156, The Jackson Laboratory) [57] to generate ChAT-Cre;Cdk5f/f mice. ChATBAC-eGFP mice (EGFP was expressed in cholinergic neurons under the direction of choline acetyltransferase transcriptional regulatory elements; stock no. 007902, The Jackson Laboratory) were crossed with Cdk5f/f mice and ChAT-Cre;Cdk5f/f mice respectively for the electrophysiology recording of cholinergic neurons.
Patient samples
Patients with PD with motor disorders were enrolled in this study. Serum samples were collected from age- and race-matched controls and patients. All patients and controls were 50 to 80 years old and Asian race. The average age was 67.2 years old for patients with PD and 67.2 years old for HCs. The study with patients is approved by the Ethics Committee of Affiliated Brain Hospital of Nanjing Medical University with number 2021-KY007-01.
The snRNA-seq data analysis
Major cell types from the midbrain of patients with PD and HCs were identified and annotated by interrogating the expression patterns of known marker genes: cholinergic neurons (marked by CHAT and ACHE) and processing snRNA-seq data analysis as reported [58].
Virus vectors
To overexpress Cdk5 specifically in cholinergic neurons of striatum, the pAAV-EF1α-DIO-Cdk5-mCherry (1 × 1012 viral particles/ml) vector was constructed using Cdk5 with a 3×Flag. The pAAV-EF1α-DIO-mCherry (1 × 1012 viral particles/ml) was used as control. The pAAV-EF1α-DIO-Cdk5-mCherry was bilaterally injected into the striatum of ChAT-Cre;Cdk5f/f mice. The pAAV-EF1α-DIO-mCherry was bilaterally injected into the striatum of ChAT-Cre and ChAT-Cre;Cdk5f/f mice. Both of AAV vectors were packaged into AAV2/9 serotype by OBiO Technology (Shanghai, China).
To eliminate the striatal cholinergic neuron CDK5, the rAAV-ChAT-Cre-EGFP (5.1 × 1012 viral particles/ml) or the rAAV-ChAT-EGFP (as control) (2.1 × 1012 viral particles/ml) was bilaterally injected into the striatum of Cdk5f/f mice. These AAV vectors were packaged into AAV2/9 serotype by BrainVTA (Wuhan, China).
Stereotaxic injections
Mice were anesthetized with 1.5% to 2% isoflurane gas/oxygen mixture and fixed to the stereotaxic apparatus [24,59]. An AAV of 1.2 μl was bilaterally injected into the striatum (anterior–posterior, +0.8 mm from bregma; medial–lateral, ±2.0 mm; dorsal–ventral, −3.0 mm) using the stereotaxic frame at postnatal day 60 (P60). The injection speed was 80 nl/min. After the injection, the injector was retained for another 7 min and then slowly withdrawn. The behavioral experiments were conducted 3-week after injection.
Behavioral assays
The mice used for behavioral assays are male at the age of 4 to 6 months; besides, the behavioral experiments on the mice injected with AAVs were performed at 2 to 3 months old.
Rotarod test was performed as previously reported [24]. Briefly, mice were trained to run on an accelerating rotarod for 10 min each day (day 1, 4 rpm/min; day 2, from 4 to 15 rpm/min gradually). After 2 d of training, the mice were tested at 4 to 40 rpm/min 3 times each. The latency of falling off the rod was recorded and averaged out.
Traction test was conducted as previously described [60] to estimate muscle strength and equilibrium. The test included 3 trials. Mice forelimbs were placed on a metallic wire (diameter, 2 mm). The latency of falling from the wire was recorded and averaged out. The maximal test duration was 120 s to avoid fatigue.
Y-maze test was used to examine the spatial working memory of mice. As described previously [59], 3 successive entries were defined as an alternation.
Open-field test was applied to reveal anxiety-like behavior and motor function in a plastic cube box (40 cm × 40 cm) [59]. All mouse movement were recorded by video and analyzed with VideoTrack v3 program (ViewPoint Life Sciences). Total distance of each mouse was calculated to assess general locomotor activity.
Footprint test was done as previously described [61]. Mice were placed in an enclosed walkway (150-cm length × 15-cm width × 30-cm height) on a glass plate and traversed 3 times from one side of the walkway to the other each day. After 2 d of training, the mice footprints were printed touching with the glass plate green light through a high-speed video camera. The time through the walkway, the stride length, and front/hind paw overlap of footprints were analyzed.
Grip strength test was conducted using the grip test system [62]. Mice were placed on a metal grip with 4 limbs or the front limbs and were pulled back by the tail gently until the limbs of mice were disconnected with the grip. Each mouse was tested 3 times. The average values were calculated.
Brain slice preparation and electrophysiologic recording
Mice at the age of P21 to P30 were anesthetized. As described previously [59], the brain was immediately taken away and put into cold artificial cerebrospinal fluid, and then striatum slices were cut using vibratome (Leica VT 1200S). The slices were transferred into the normal artificial cerebrospinal fluid solution for electrophysiology recording [59].
Cell-attached recordings were applied to measure the spontaneous firing APs of cholinergic neurons as described before [63].
Whole-cell recordings were performed from striatum cholinergic neurons with a MultiClamp 700B amplifier as described previously [64]. Glass micropipettes (3 to 8 MΩ) were used to established tight seals at −60mV for cholinergic neurons. Under current-clamp patch, APs, input resistance (Rin), membrane time constant (Tau), and membrane capacitance (Cm) were recorded.
Na+ currents and K+ currents were recorded in voltage-clamp mode. For Na+ currents recording, the bath solution contained tetraethylammonium chloride (10 mM; Aladdin) and CsCl (0.1 mM; Aladdin), the Na+ in intracellular solution was replaced by Cs+. The total K+ currents were recorded in the presence of tetrodotoxin (1 μM; Tocris). Ik currents were recorded at voltages of −120 and +10 mV for 200 ms, followed by −80- to +70-mV voltages, with 10-mV increments, and 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (1 mM; Sigma-Aldrich) was added to electrode internal fluid to inhibit calcium-dependent potassium currents [65]. IA currents were inactivated at voltages of −120 and +10 mV for 200 ms and then calculated by subtracting the remaining K+ currents from total K+ currents.
BK channel activity was recorded under voltage-clamp mode as previously reported [28]. Before and after the application of IBTX (150 nM; APExBIO), currents were respectively generated at voltages from −80 to +120 mV in 10-mV steps. The tetrodotoxin (1 μM) was also added to the bath solution to eliminate spontaneous activity. IBTX-sensitive BK currents were obtained by subtracting currents before the application of IBTX from currents after the application of IBTX.
Calcium currents were recorded according to previously used protocol [66].
Data were analyzed using MATLAB (MathWorks, Natick, MA, USA), Clampfit 10 (Molecular Devices), and MiniAnalysis software (Synaptosoft).
Immunohistochemistry
The immunohistochemistry staining was performed as previously [24]. Briefly, 40-μm-thick brain slices were incubated with 0.1% Triton X-100 in phosphate-buffered saline and blocked with 3% bovine serum albumin in phosphate-buffered saline. Finally, the slices were incubated with primary antibodies as follows: anti-ChAT (Millipore; 1:500) and anti-CDK5 (Cell Signaling Technology; 1:200). Images were captured through a confocal laser scanning microscope (Zeiss LSM 800, Germany).
Cell culture and plasmid
Cell culture and transfection were done as before [24]. N2A cells were obtained from American Type Culture Collection, the cells were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml) at a 37°C incubator with 5% CO2. After cultured for 24 h, the N2A cells were transfected with plasmids using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) for 12 h and replaced with fresh medium for another 48-h culture.
The pcDNA3.1-3X-Flag-Cdk5 was provided by X. Guo (Department of Neurobiology, Nanjing Medical University). Full-length mslo (encoding BK) (NM_001253378.1) cDNA was amplified from the cDNA of primary neuronal cells using the following primer set: sense, 5′- AAACTCGAGATGGCAAACGGTGGCGGCGGCGGCGGCGGC-3′; antisense, 5′- AAAGCGGCCGCCACATTCATCTTCAACTTCTCTGAT-3′. In these primers, Xho I and Not I restriction site sequences have been underlined. The polymerase chain reaction products were cloned into the pEGFP-N1 vector. BK carrying the S716A mutation was amplified using the following primer set: sense, 5′-GGGCATGAGGAACGCGCCCAACACCTC-3′; antisense, 5′-GAGGTGTTGGGCGCGTTCCTCATGCCC-3′. BK carrying the S720A mutation was amplified using the following primer set: sense, 5′-CTCGCCCAACACCGCCCCGAAGCAGAT-3′; antisense, 5′-ATCAGCTTCGGGGCGGTGTTGGGCGAG-3′. BK carrying the T908A mutation was amplified using the following primer set: sense, 5′-CTAATTCCCAAGGATTCGCACCTCCTGGAATGGAC-3′; antisense, 5′-GTCCATTCCAGGCGGTGCGAATCCTTGGGAATTAG-3′. BK carrying the T1112A mutation was amplified using the following primer set: sense, 5′-CCCACCTCAGCGCCCCCAGCCAG-3′; antisense, 5′-CTGGCTGGGGGCGTGAGGTGGG-3′. All constructions were ensured by sequencing.
Immunoprecipitation and Western blot
Immunoprecipitation (IP) and Western blot (WB) were conducted as previously described [24]. Briefly, after transfection, N2A cell lysates were precleared with immunoglobulin G-Agarose beads (Beyotime) for 2 h and incubated supernatant with anti-CDK5 affinity gel beads, or anti-GFP affinity gel beads for overnight at 4 °C on a gentle shaker. Immunoprecipitated samples were washed with lysis buffer and eluted with 2× SDS loading buffer and then subjected to WB analysis. Primary antibodies were used as follows: anti-GFP antibody (Abcam; 1:100 for IP and 1:2,000 for WB), anti-CDK5 (Santa Cruz Biotechnology; 1:50 for IP and 1:1,000 for WB), anti-BK (Neuromab; 1:1,000), anti-Flag (Sigma-Aldrich; 1:5,000), and anti-glyceraldehyde-3-phosphate dehydrogenase (Cell Signaling Technology; 1:5,000).
EMG analysis
The EMG of leg muscles were recorded as described before [67]. The gastrocnemius muscles of mice left hind limbs were exposed via a left hind limb incision. The gastrocnemius muscle was then hung on a pair of paralleled silver electrodes. The spontaneous muscle electrical activity was recorded and amplified with an AD/CD differential amplifier (model DP-304, Warner Instruments, Hamden, CT) [23]. The signals between 100 and 3,000 Hz were filtered and saved for further analysis.
Surgery for implantation of tetrodes
Mice were deeply anesthetized with 1.5% to 2% isoflurane gas/oxygen mixture and fixed on the stereotactic apparatus [56,68]. Then, a hole was drilled over the right arcuate nucleus for implantation (striatum from bregma: anterior–posterior, +0.8 mm; medial-lateral, 0.2 mm). For LFP recording, the tetrodes were implanted into the arc with an average depth of 3 mm to reach the striatum. After tetrodes implantation, the signals were amplified by a 32-channel amplifier and sampled at 20 kHz by a Neuralynx recording system.
LFP recordings
For LFP recordings, signals were amplified (1,000 × gain; Neuralynx), filtered at 0.1 to 250 Hz, and sampled at 1 kHz. The analysis of the LFP signals was conducted by a program written in MATLAB.
Spectral analysis and phase–amplitude coupling
Spectral power was obtained by using MATLAB’s wavelet method (MathWorks), continuous wavelet transform, and Morlet wavelets [69]. Power spectral analysis of different oscillations was performed via the multitaper method in the MATLAB.
Statistical analysis
Data were analyzed by experimenters blinded to the mice genotype. GraphPad Prism 8 was applied for statistical analysis. All data were displayed as means ± SEM. Unpaired 2-tailed Student’s t test was performed for 2 independent group comparisons. One-way analysis of analysis of variance (ANOVA) followed by Tukey’s post hoc test was used for multiple normally distributed data. Two-way ANOVA was used for dataset with 2 factors, followed by Bonferroni’s multiple comparison test. *P < 0.05 was considered to be statistically significant.
Acknowledgments
Funding: This work was supported by the National Key Research and Development Program of China (2022YFE0108600), the State Key Program of National Natural Science Foundations of China (81930103), the National Natural Science Foundations of China (81973300, 82104162, and 82171249). Author contributions: C.T. and P.-X.M. performed most of the experiments and data analyses and wrote the draft manuscript. Q.Z., Y.-H.B., and X.-X.L. fulfilled whole-cell recording partly. R.-X.G. and L.Z. collected human serum and data analyses. Y.-H.W. and Y.S. conducted the immunohistochemical analysis and molecular experiments partly. X.-F.M. and H.-Y.Y. conducted in vivo experiments and data analyses partly. X.C. and Y.-X.Z. performed bioinformatics analysis. T.S. reviewed the manuscript. F.H. and Y.-M.L. were involved in the overall design of the project, individual experiments, data analysis, and the writing of the final manuscript. Competing interests: The authors declare that they have no competing interests.
Data Availability
All data required to support the conclusions are presented in the main text and the Supplementary Materials.
Supplementary Materials
Table S1
Figs. S1 to S5
Movies S1 and S2
References
- 1.Fahn S. Classification of movement disorders. Mov Disord. 2011;26(6):947–957. [DOI] [PubMed] [Google Scholar]
- 2.Harris MK, Shneyder N, Borazanci A, Korniychuk E, Kelley RE, Minagar A. Movement disorders. Med Clin North Am. 2009;93(2):371–388. [DOI] [PubMed] [Google Scholar]
- 3.Prodoehl J, Li H, Planetta PJ, Goetz CG, Shannon KM, Tangonan R, Comella CL, Simuni T, Zhou XJ, Leurgans S, et al. Diffusion tensor imaging of Parkinson's disease, atypical parkinsonism, and essential tremor. Mov Disord. 2013;28(13):1816–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vaillancourt DE, Spraker MB, Prodoehl J, Abraham I, Corcos DM, Zhou XJ, Comella CL, Little DM. High-resolution diffusion tensor imaging in the substantia Nigra of De novo Parkinson disease. Neurology. 2009;72(16):1378–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, Kreitzer AC. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466(7306):622–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Exley R, Cragg SJ. Presynaptic nicotinic receptors: A dynamic and diverse cholinergic filter of striatal dopamine neurotransmission. Br J Pharmacol. 2008;153(Suppl 1):S283–S297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shuen JA, Chen M, Gloss B, Calakos N. Drd1a-Tdtomato bac transgenic mice for simultaneous visualization of medium spiny neurons in the direct and indirect pathways of the basal ganglia. J Neurosci. 2008;28(11):2681–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKinley JW, Shi Z, Kawikova I, Hur M, Bamford IJ, Sudarsana Devi SP, Vahedipour A, Darvas M, Bamford NS. Dopamine deficiency reduces striatal cholinergic interneuron function in models of Parkinson’s disease. Neuron. 2019;103(6):1056–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bohnen NI, Yarnall AJ, Weil RS, Moro E, Moehle MS, Borghammer P, Bedard MA, Albin RL. Cholinergic system changes in Parkinson’s disease: Emerging therapeutic approaches. Lancet Neurol. 2022;21(4):381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connolly BS, Lang AE. Pharmacological treatment of Parkinson disease: A review. JAMA. 2014;311(16):1670–1683. [DOI] [PubMed] [Google Scholar]
- 11.Katzenschlager R, Sampaio C, Costa J, Lees A. Anticholinergics for symptomatic management of Parkinson’s disease. Cochrane Database Syst Rev. 2003;2002(2): Article CD003735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim SA, Kang UJ, McGehee DS. Striatal cholinergic interneuron regulation and circuit effects. Front Synaptic Neurosci. 2014;6(22):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benhamou L, Kehat O, Cohen D. Firing pattern characteristics of tonically active neurons in rat striatum: Context dependent or species divergent? J Neurosci. 2014;34(6):2299–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennett BD, Callaway JC, Wilson CJ. Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J Neurosci. 2000;20(22):8493–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldberg JA, Wilson CJ. Control of spontaneous firing patterns by the selective coupling of calcium currents to calcium-activated potassium currents in striatal cholinergic interneurons. J Neurosci. 2005;25(44):10230–10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tubert C, Taravini IRE, Flores-Barrera E, Sánchez GM, Prost MA, Avale ME, Tseng KY, Rela L, Murer MG. Decrease of a current mediated by Kv1.3 channels causes striatal cholinergic interneuron hyperexcitability in experimental parkinsonism. Cell Rep. 2016;16(10):2749–2762. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y-F, Reynolds JNJ, Cragg SJ. Pauses in cholinergic interneuron activity are driven by excitatory input and delayed rectification, with dopamine modulation. Neuron. 2018;98(5):918–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim SH, Ryan TA. Cdk5 serves as a major control point in neurotransmitter release. Neuron. 2010;67(5):797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou H, Zhang J, Shi H, Li P, Sui X, Wang Y, Wang L. Downregulation of Cdk5 signaling in the dorsal striatum alters striatal microcircuits implicating the Association of Pathologies with circadian behavior in mice. Mol Brain. 2022;15(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chergui K, Svenningsson P, Greengard P. Cyclin-dependent kinase 5 regulates dopaminergic and glutamatergic transmission in the striatum. Proc Natl Acad Sci U S A. 2004;101(7):2191–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bibb JA, Snyder GL, Nishi A, Yan Z, Meijer L, Fienberg AA, Tsai LH, Kwon YT, Girault JA, Czernik AJ, et al. Phosphorylation of Darpp-32 by Cdk5 modulates dopamine signalling in neurons. Nature. 1999;402(6762):669–671. [DOI] [PubMed] [Google Scholar]
- 22.Benavides DR, Quinn JJ, Zhong P, Hawasli AH, DiLeone RJ, Kansy JW, Olausson P, Yan Z, Taylor JR, Bibb JA. Cdk5 modulates cocaine reward, motivation, and striatal neuron excitability. J Neurosci. 2007;27(47):12967–12976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye C, Qiu Y, Zhang F, Chen AD, Zhou H, Wang JJ, Chen Q, Li YH, Kang YM, Zhu GQ. Chemical stimulation of renal tissue induces sympathetic activation and a pressor response via the paraventricular nucleus in rats. Neurosci Bull. 2020;36(2):143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shao LX, Jiang Q, Liu XX, Gong DM, Yin YX, Wu G, Sun NH, Wang CK, Chen QZ, Yu C, et al. Functional coupling of Tmem74 and HCN1 channels regulates anxiety-like behavior in BLA neurons. Mol Psychiatry. 2019;24(10):1461–1477. [DOI] [PubMed] [Google Scholar]
- 25.McCormick DA. From molecules to networks. 3rd ed. Academic Press; 2014. p. 351–376.
- 26.Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8(6):451–465. [DOI] [PubMed] [Google Scholar]
- 27.Li B, Suutari BS, Sun SD, Luo Z, Wei C, Chenouard N, Mandelberg NJ, Zhang G, Wamsley B, Tian G, et al. Neuronal inactivity co-opts LTP machinery to drive potassium channel splicing and homeostatic spike widening. Cell. 2020;181(7):1547–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roshchin MV, Matlashov ME, Ierusalimsky VN, Balaban PM, Belousov VV, Kemenes G, Staras K, Nikitin ES. A BK channel–mediated feedback pathway links single-synapse activity with action potential sharpening in repetitive firing. Sci Adv. 2018;4(7): Article eaat1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gomez K, Calderon-Rivera A, Sandoval A, González-Ramírez R, Vargas-Parada A, Ojeda-Alonso J, Granados-Soto V, Delgado-Lezama R, Felix R. Cdk5-dependent phosphorylation of Cav3.2 T-type channels: Possible role in nerve ligation-induced neuropathic allodynia and the compound action potential in primary afferent C fibers. J Neurosci. 2020;40(2):283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan JS, Olsen JV, Park KS, Li W, Bildl W, Schulte U, Aldrich RW, Fakler B, Trimmer JS. Profiling the phospho-status of the BKCa channel α subunit in rat brain reveals unexpected patterns and complexity. Mol Cell Proteomics. 2008;7(11):2188–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou XB, Wulfsen I, Utku E, Sausbier U, Sausbier M, Wieland T, Ruth P, Korth M. Dual role of protein kinase C on BK channel regulation. Proc Natl Acad Sci U S A. 2010;107(17):8005–8010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tian LJ, Coghill LS, McClafferty H, MacDonald SHF, Antoni FA, Ruth P, Knaus HG, Shipston MJ. Distinct stoichiometry of BKCa channel tetramer phosphorylation specifies channel activation and inhibition by Camp-dependent protein kinase. Proc Natl Acad Sci U S A. 2004;101(32):11897–11902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294(5):1351–1362. [DOI] [PubMed] [Google Scholar]
- 34.Bloem BR, Okun MS, Klein C. Parkinson’s disease. Lancet. 2021;397(10291):2284–2303. [DOI] [PubMed] [Google Scholar]
- 35.Ding J, Guzman JN, Tkatch T, Chen S, Goldberg JA, Ebert PJ, Levitt P, Wilson CJ, Hamm HE, Surmeier DJ. RGS4-dependent attenuation of M4 autoreceptor function in striatal cholinergic interneurons following dopamine depletion. Nat Neurosci. 2006;9(6):832–842. [DOI] [PubMed] [Google Scholar]
- 36.Paz RM, Stahl AM, Rela L, Murer MG, Tubert C. D1/D5 inverse agonists restore striatal cholinergic interneuron physiology in dyskinetic mice. Mov Disord. 2022;37(8):1693–1706. [DOI] [PubMed] [Google Scholar]
- 37.Mitra A, Mitra SS, Tsien RW. Heterogeneous reallocation of presynaptic efficacy in recurrent excitatory circuits adapting to inactivity. Nat Neurosci. 2011;15(2):250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sato S, Xu JQ, Okuyama S, Martinez LB, Walsh SM, Jacobsen MT, Swan RJ, Schlautman JD, Ciborowski P, Ikezu T. Spatial learning impairment, enhanced Cdk5/P35 activity, and downregulation of Nmda receptor expression in transgenic mice expressing tau-tubulin kinase 1. J Neurosci. 2008;28(53):14511–14521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohshima T, Hirasawa M, Tabata H, Mutoh T, Adachi T, Suzuki H, Saruta K, Iwasato T, Itohara S, Hashimoto M, et al. Cdk5 is required for multipolar-to-bipolar transition during radial neuronal migration and proper dendrite development of pyramidal neurons in the cerebral cortex. Development. 2007;134(12):2273–2282. [DOI] [PubMed] [Google Scholar]
- 40.Reynolds JNJ, Hyland BI, Wickens JR. Modulation of an afterhyperpolarization by the substantia nigra induces pauses in the tonic firing of striatal cholinergic interneurons. J Neurosci. 2004;24(44):9870–9877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brenner R, Chen QH, Vilaythong A, Toney GM, Noebels JL, Aldrich RW. BK channel β4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci. 2005;8(12):1752–1759. [DOI] [PubMed] [Google Scholar]
- 42.Gu N, Vervaeke K, Storm JF. BK potassium channels facilitate high-frequency firing and cause early spike frequency adaptation in rat CA1 hippocampal pyramidal cells. J Physiol. 2007;580(3):859–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin M, Hatcher JT, Wurster RD, Chen QH, Cheng ZJ. Characteristics of single large-conductance Ca2+-activated K+ channels and their regulation of action potentials and excitability in parasympathetic cardiac motoneurons in the nucleus ambiguus. Am J Physiol Cell Physiol. 2014;306(2):C152–C166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shruti S, Clem RL, Barth AL. A seizure-induced gain-of-function in BK channels is associated with elevated firing activity in neocortical pyramidal neurons. Neurobiol Dis. 2008;30(3):323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang B, Rothberg BS, Brenner R. Mechanism of increased BK channel activation from a channel mutation that causes epilepsy. J Gen Physiol. 2009;133(3):283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salkoff L, Butler A, Ferreira G, Santi C, Wei A. High-conductance potassium channels of the Slo family. Nat Rev Neurosci. 2006;7(12):921–931. [DOI] [PubMed] [Google Scholar]
- 47.Kondabolu K, Roberts EA, Bucklin M, McCarthy M, Kopell N, Han X. Striatal cholinergic interneurons generate beta and gamma oscillations in the corticostriatal circuit and produce motor deficits. Proc Natl Acad Sci U S A. 2016;113(22):E3159–E3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Asadi A, Madadi Asl M, Vahabie AH, Valizadeh A. The origin of abnormal beta oscillations in the parkinsonian corticobasal ganglia circuits. Parkinsons Dis. 2022;2022:7524066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adam EM, Brown EN, Kopell N, McCarthy MM. Deep brain stimulation in the subthalamic nucleus for Parkinson’s disease can restore dynamics of striatal networks. Proc Natl Acad Sci U S A. 2022;119, 19: Article e2120808119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Okun MS. Deep-brain stimulation for Parkinson’s disease. N Engl J Med. 2012;367(16):1529–1538. [DOI] [PubMed] [Google Scholar]
- 51.Weaver FM, Follett K, Stern M, Hur K, Harris C, Marks WJ Jr, Rothlind J, Sagher O, Reda D, Moy CS, et al. Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson disease: A randomized controlled trial. JAMA. 2009;301(1):63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Katz M, Luciano MS, Carlson K, Luo P, Marks WJ Jr, Larson PS, Starr PA, Follett KA, Weaver FM, Stern MB, et al. Differential effects of deep brain stimulation target on motor subtypes in Parkinson's disease. Ann Neurol. 2015;77(4):710–719. [DOI] [PubMed] [Google Scholar]
- 53.Tsai L-H, Delalle I, Caviness VS Jr, Chae T, Harlow E. P35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371(29):419–423. [DOI] [PubMed] [Google Scholar]
- 54.Kobayashi H, Saito T, Sato K, Furusawa K, Hosokawa T, Tsutsumi K, Asada A, Kamada S, Ohshima T, Hisanaga SI. Phosphorylation of cyclin-dependent kinase 5 (Cdk5) at Tyr-15 is inhibited by Cdk5 activators and does not contribute to the activation of Cdk5. J Biol Chem. 2014;289(28):19627–19636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li ZM, Liu XX, Li C, Wei Z-C, Shi Y, Song H-Y, Chen X, Zhang Y, Li J-W, Zhu R-F, et al. Decreased synapse-associated proteins are associated with the onset of epileptic memory impairment in endothelial Cdk5-deficient mice. MedComm. 2022;3(3): Article e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu XX, Chen XH, Zheng ZW, Jiang Q, Li C, Yang L, Chen X, Mao X-F, Yuan H-Y, Feng L-L, et al. Bod1 regulates the cerebellar IV/V lobe-fastigial nucleus circuit associated with motor coordination. Signal Transduct Target Ther. 2022;7(1):170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu XX, Yang L, Shao LX, He Y, Wu G, Bao Y-H, Lu N-N, Gong D-M, Lu Y-P, Cui T-T, et al. Endothelial Cdk5 deficit leads to the development of spontaneous epilepsy through Cxcl1/Cxcr2-mediated reactive astrogliosis. J Exp Med. 2020;217(1): Article e20180922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smajic S, Prada-Medina CA, Landoulsi Z, Ghelfi J, Delcambre S, Dietrich C, Jarazo J, Henck J, Balachandran S, Pachchek S, et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain. 2022;145(3):964–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tan C, Lu NN, Wang CK, Chen DY, Sun NH, Lyu H, Körbelin J, Shi WX, Fukunaga K, Lu YM, et al. Endothelium-derived semaphorin 3G regulates hippocampal synaptic structure and plasticity via neuropilin-2/PlexinA4. Neuron. 2019;101(5):920–937. [DOI] [PubMed] [Google Scholar]
- 60.Rajsombath MM, Nam AY, Ericsson M, Nuber S. Female sex and brain-selective estrogen benefit α-synuclein tetramerization and the PD-like motor syndrome in 3K transgenic mice. J Neurosci. 2019;39(38):7628–7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crowley ST, Fukushima Y, Uchida S, Kataoka K, Itaka K. Enhancement of motor function recovery after spinal cord injury in mice by delivery of brain-derived neurotrophic factor mRNA. Mol Ther Nucleic Acids. 2019;17:465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benoit B, Meugnier E, Castelli M, Chanon S, Vieille-Marchiset A, Durand C, Bendridi N, Pesenti S, Monternier PA, Durieux AC, et al. Fibroblast growth factor 19 regulates skeletal muscle mass and ameliorates muscle wasting in mice. Nat Med. 2017;23(8):990–996. [DOI] [PubMed] [Google Scholar]
- 63.Cheng J, Umschweif G, Leung J, Sagi Y, Greengard P. HCN2 channels in cholinergic interneurons of nucleus accumbens shell regulate depressive behaviors. Neuron. 2019;101(4):662–672. [DOI] [PubMed] [Google Scholar]
- 64.Lu NN, Tan C, Sun NH, Shao LX, Liu XX, Gao YP, Tao RR, Jiang Q, Wang CK, Huang JY, et al. Cholinergic Grb2-associated-binding protein 1 regulates cognitive function. Cereb Cortex. 2018;28(7):2391–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Itri JN, Michel S, Vansteensel MJ, Meijer JH, Colwell CS. Fast delayed rectifier potassium current is required for circadian neural activity. Nat Neurosci. 2005;8(5):650–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McNally BA, Plante AE, Meredith AL. Diurnal properties of voltage-gated Ca2+ currents in suprachiasmatic nucleus and roles in action potential firing. J Physiol. 2020;598(9):1775–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hadzipasic M, Ni W, Nagy M, Steenrod N, McGinley MJ, Kaushal A, Thomas E, McCormick DA, Horwich AL. Reduced high-frequency motor neuron firing, EMG fractionation, and gait variability in awake walking ALS mice. Proc Natl Acad Sci U S A. 2016;113(47):7600–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun M, Mao XF, Li ZM, Zhu ZH, Gong DM, Lu L, Chen X, Zhang Y, Fukunaga K, Ji Y, et al. Endothelial peroxynitrite causes disturbance of neuronal oscillations by targeting caspase-1 in the arcuate nucleus. Redox Biol. 2021;47:102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mederos S, Sanchez-Puelles C, Esparza J, Valero M, Ponomarenko A, Perea G. GABAergic signaling to astrocytes in the prefrontal cortex sustains goal-directed behaviors. Nat Neurosci. 2021;24(1):82–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Figs. S1 to S5
Movies S1 and S2
Data Availability Statement
All data required to support the conclusions are presented in the main text and the Supplementary Materials.