

Abstract
The COVID-19 pandemic caused by SARS-CoV-2 virus is an ongoing global health burden. Severe cases of COVID-19 and the rare cases of COVID-19 vaccine-induced-thrombotic-thrombocytopenia (VITT) are both associated with thrombosis and thrombocytopenia; however, the underlying mechanisms remain inadequately understood. Both infection and vaccination utilize the spike protein receptor-binding domain (RBD) of SARS-CoV-2. We found that intravenous injection of recombinant RBD caused significant platelet clearance in mice. Further investigation revealed the RBD could bind platelets, cause platelet activation, and potentiate platelet aggregation, which was exacerbated in the Delta and Kappa variants. The RBD–platelet interaction was partially dependent on the β3 integrin as binding was significantly reduced in β3−/− mice. Furthermore, RBD binding to human and mouse platelets was significantly reduced with related αIIbβ3 antagonists and mutation of the RGD (arginine-glycine-aspartate) integrin binding motif to RGE (arginine-glycine-glutamate). We developed anti-RBD polyclonal and several monoclonal antibodies (mAbs) and identified 4F2 and 4H12 for their potent dual inhibition of RBD-induced platelet activation, aggregation, and clearance in vivo, and SARS-CoV-2 infection and replication in Vero E6 cells. Our data show that the RBD can bind platelets partially though αIIbβ3 and induce platelet activation and clearance, which may contribute to thrombosis and thrombocytopenia observed in COVID-19 and VITT. Our newly developed mAbs 4F2 and 4H12 have potential not only for diagnosis of SARS-CoV-2 virus antigen but also importantly for therapy against COVID-19.
Introduction
Coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has reached to >651 million cases and has caused >6.6 million deaths as of December 2022. Despite its respiratory origin, systemic thromboembolic complications are a common feature of critically ill patients [1–3]. Pulmonary embolism [4], stroke [5], and myocardial infarction [6] are often reported complications of hospitalized patients. COVID-19 also continues to birth new variants that modulate associated cardiovascular complications. The Delta variant in particular has been linked with a higher risk of mortality, intensive care unit admission, hospitalization [7–9], and more serious blood clots [10,11]. However, the role of platelets in COVID-19 thrombosis and the effect of its variants have not been adequately explored.
Platelets are small anucleate blood cells known for their critical roles in thrombosis and hemostasis in mammals [12–16]. The life spans of platelets are relatively short, around 7 to 10 d in humans and 4 to 5 d in mice. Platelets are removed from circulation by the reticuloendothelial system, in which autoantibody-targeted platelets are predominantly cleared in the spleen, while aged/desialylated platelets are likely cleared in the liver by Kupffer cells [17–22]. Notably, hospitalized COVID-19 patients generally present with hyperactive platelets that may account for associated thrombosis and disseminated intravascular coagulation [23–25]. Platelet hyperactivity can also translate to consumption and clearance causing thrombocytopenia [17,26,27], a complication also reported in COVID-19 patients and is associated with a worse prognosis [28–30]. Interestingly, COVID-19 vaccinations that utilize the spike protein receptor-binding domain (RBD) have also been associated with reports of thrombosis and thrombocytopenia, known as vaccine-induced thrombotic thrombocytopenia (VITT) [31–35]. However, the platelet clearance mechanism in both COVID-19 and VITT remains unclear. Understanding the complexity of platelet hyperactivity and activation in COVID-19 and VITT is vitally important to help guide prophylactic efforts and treatment for critically ill patients.
There are some reports [36–38] that SARS-CoV-2 directly interacts with platelets, but the mechanism has not been well explored. Although angiotensin-converting enzyme-2 (ACE2) is the dominant receptor of the spike protein RBD, the expression of ACE2 on platelets remains controversial [23,25,36,37,39–41], perhaps in part due to genetic differences among patients and varying methodologies. Nevertheless, SARS-CoV-2–platelets interactions independent of ACE2 have been reported [37,41], indicating alternative cognate receptors [42–46]. Interestingly, the SARS-CoV-2 spike protein RBD contains the integrin-binding RGD (arginine-glycine-aspartate) motif that is absent in other coronaviruses [46–48].
Integrins are a large family of adhesion molecules, and 24 integrins have been identified [49,50]. There are 2 reported β3-containing integrins, αIIbβ3 and αVβ3. Different from αVβ3 integrin that might be expressed on various cells, αIIbβ3 is almost exclusively expressed on platelets and their precursor megakaryocytes. The αIIbβ3 receptor is the dominant β3-containing integrin expressed on platelets (~80,000 copies per platelet), compared to negligible quantities of αVβ3 integrin [50,51]. Structural and functional analyses of αIIbβ3 in human and some vertebrates show significant conservation in human and mouse. The primary structure of αIIbβ3 in human (UniProt IDs: αIIb, P08514; β3, P05106) and mouse (UniProt IDs: αIIb, Q9QUM0; β3, O54890) are 80.4% and 90.3% homologous, respectively [49,52]. Some studies reported that the RGD motif in the RBD can bind to integrins αVβ3 and αVβ6 and inconsistent findings concerning α5β1 [53–55]. In silico sequence analysis also suggests a potential interaction with integrin αIIbβ3 [46,47]; however, to our knowledge, there is no solid report that provides direct evidence for this interaction.
In the present study, we discovered that the SARS-CoV-2 spike protein RBD can bind platelets partially through integrin αIIbβ3 (GPIIbIIIa) and cause platelet activation and clearance, providing novel mechanisms of thrombosis and thrombocytopenia in COVID-19 and VITT.
Results
The RBD of the spike protein can induce platelet clearance in vivo
Since thrombocytopenia is associated with severe cases of COVID-19 and VITT, we first explored whether the RBD could induce platelet clearance in vivo. We generated recombinant RBD in a mammalian cell culture system linked with a rabbit Fc tag (RBD-rFc) for optimal stability and expression. We then intravenously injected different doses (0.25, 0.5, and 1.0 μg/g) of RBD-rFc into 6-week-old female CD1 mice and recorded the platelet counts at 0, 1, 3, 8, 24, and 48 h postinjection as we did for murine models of thrombocytopenia [17,56,57]. Platelet counts at the 1st, 3rd, and 8th hour after injection of 1.0 μg/g RBD-rFc decreased significantly and was most pronounced at the 3rd hour (RBD versus control, 1 h: *P < 0.05; 3 h: ***P < 0.001; 8 h: **P < 0.01). Conversely, the lower doses of 0.25 μg/g and 0.5 μg/g did not significantly (only trends) decrease platelet counts, indicating that lower doses of RBD were not sufficient to cause thrombocytopenia (Fig. 1A and Fig. S1A).
Fig. 1.

The RBD-rFc induces platelet clearance in vivo. (A) CD1 mouse platelets were counted at different time intervals (0, 1, 3, 8, 24, and 48 h) after I.V. injection with either a PBS control or RBD-rFc (PBS, n = 8; 0.25 μg/g RBD-rFc, n = 4; 0.5 μg/g RBD-rFc, n = 4; 1 μg/g RBD-rFc, n = 11). (B) A total of 2 × 108 Far Red-labeled platelets were incubated with 2.5 μg/ml RBD-rFc for 30 min and then I.V. injected into CD1 mice. Circulating platelets were quantified at different time intervals (0, 1, 3, and 24 h). All the data were expressed as means ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test was applied to evaluate the difference between RBD group and control group. *P < 0.05, **P < 0.01, ***P < 0.001, versus control at the same time point. *P < 0.05, **P < 0.01, versus baseline.
To confirm that the RBD induced platelet clearance instead of platelet sequestration, labeled mouse platelets were incubated with RBD-rFc for 30 min, then intravenously (I.V.) injected into CD1 mice, and quantified at 0, 1, 3, and 24 h posttransfusion. We found the RBD caused clearance of transfused platelet that did not recover up to 24 h posttransfusion (Fig. 1B), confirming that the drop in platelet count was due to clearance rather than sequestration. To exclude the possibility that the decrease in mouse platelet count was caused by the rabbit Fc tag attached to the RBD, RBD without a tag was generated after tobacco etch virus (TEV) protease cleavage of an N-terminal His-tag. Even after removal of the Fc tag, RBD-induced thrombocytopenia persisted, especially at 1 and 3 h postinjection (RBD versus control, 1 h: *P < 0.05; 3 h: **P < 0.01) (Fig. S1B).
The SARS-CoV-2 spike protein RBD binds platelet β3 integrin
To investigate how the RBD induced platelet clearance in vivo, we assessed its binding to platelets by flow cytometry. We found that RBD binds to both human and mouse platelets (Fig. 2A to I) and that RBD (20, 50, 100, and 200 μg/ml) could bind to human platelets in a dose-dependent manner. Platelet expression of ACE2 has been a contentious topic. We assessed ACE2 presence on human platelets with 5 different anti-ACE2 antibodies via western blot. Our results show positive bands in nonreducing protein samples with an approximate molecular weight of 250 kDa (Fig. S2), indicating that dimeric ACE2 may distribute on the human platelets. However, we still cannot exclude the possibility that these antibodies may cross-react with other mimotopes.
Fig. 2.

The RBD can bind to platelets partially via integrin αIIbβ3. The binding of RBD (10 μg/ml) to healthy human donor (A to C) and mouse (E to H) washed platelets were analyzed by flow cytometry using Alexa Fluor 647-labeled anti-RBD mAb 4H12. The binding of RBD-rFC to human (D) and mouse (I) platelets was analyzed by flow cytometry using FITC-labeled anti-rabbit Fc antibody. A total of 5 × 105 platelets were incubated with wild-type RBD-rFc or RBD-RGE-rFc (RBD with RGE mutation). (G) RBD binding to integrin β3 knockout, and wild-type BALB/c mice platelets were analyzed by flow cytometry using Alexa Fluor 647-labeled anti-RBD mAb 4H12. All the data are displayed as means ± SEM. Two-way ANOVA Tukey’s multiple comparisons test was applied to evaluate the difference between groups. *P < 0.05, **P < 0.01, ***P < 0.0001. (J) Direct binding analysis of labeled RBD as a function of integrin αIIbβ3 concentration in vitro utilizing normalized mean fluorescence anisotropy of Alexa Fluor 488 in binding buffer. (K) Direct binding kinetics characterization of spike-αIIbβ3 using BLI in binding buffer (20 mM tris [pH 7.4], 137 mM NaCl, 1 mM MgCl2, 1 mM MnCl2, 1 mM CaCl2, and 50% glycerol) plus 1X Octet Kinetics Buffer in a concentration range from 68 to 512 nM (red sensograms) at 25 °C. Mean control sensogram (black) is 2% BSA in binding buffer.
The RBD contains an RGD (arginine-glycine-aspartate) tripeptide motif that is known to bind integrins, which are highly expressed on platelets [50,58]. Interestingly, we found that the RBD binds to β3−/− mouse platelets significantly less relative to wild-type platelets (Fig. 2G). Preincubation of platelets with β3 antagonist eptifibatide or anti-β3 monoclonal antibody (mAb) M1 blocks RBD binding to human and murine platelets, respectively (Fig. 2C and H). We then mutated the RGD motif to RGE (arginine-glycine-glutamate), which is known to abrogate integrin binding, and found that the RGD/E mutation reduced RBD binding to platelets (Fig. 2D and I). We further examined direct binding of RBD with recombinant αIIbβ3 in vitro using a fluorescence anisotropy assay and quantified the dissociation constant (Kd) of (96 ± 8) nM and a binding stoichiometry value of 1.3 at 25 °C (Fig. 2J). Also, we examined direct binding kinetics of the full-length spike protein to quantify the affinity parameters of spike–αIIbβ3. Our global association and dissociation analyses of the obtained sensograms quantifies the kon and koff rates (Table S1) with a Kd value of (47 ± 2) nM for recombinant spike and purified αIIbβ3 binding at 25 °C (Fig. 2K). Moreover, our calorimetry study confirmed a sigmoidal thermogram, evident for the direct interactions of spike–αIIbβ3 integrin at 25 °C (Fig. S3). Taken together, the spike protein and RBD bind platelets partially via the RGD motif–αIIbβ3 integrin interaction.
The RBD induces platelet activation and potentiates platelet aggregation in vitro
We hypothesized the RBD binding to platelets may directly induce platelet clearance by promoting activation and aggregation. To investigate this, we incubated human platelets with RBD and evaluated a panel of platelet activation markers by flow cytometry. These include P-selectin, the prototypical marker of platelet degranulation; Annexin V, to detect exposed phosphatidylserine involved in cell-based thrombin generation [59,60]; platelet activation complex-1 (PAC-1) mAb/fibrinogen binding; and Ricinus communis agglutinin-1 (RCA-1) binding, an emerging platelet activation marker that detects desialylation [17,61], which has been demonstrated by us and others to trigger liver-mediated platelet clearance [17,22,62]. We found the RBD increases P-selection expression, Annexin V binding, RCA-1 binding, and PAC-1/fibrinogen binding to platelets (RBD versus control, *P < 0.05, Fig. 3A to E and Fig. S4). We also demonstrated that the RBD potentiates platelet aggregation in human platelets with low-dose thrombin, adenosine diphosphate (ADP), and collagen (Fig. 3F to H and Fig. S5A and S5B). As expected, the RBD without a tag also potentiated platelet aggregation (Figs. S5C and D and S6), and the RBD-induced platelet activation and enhanced aggregation were attenuated by β3 antagonist eptifibatide (Fig. S5E to G).
Fig. 3.

The RBD induces human platelet activation and potentiates platelet aggregation in vitro. (A to E) Human platelet activation (P-selection expression; PAC-1, fibrinogen binding), apoptosis (Annexin V binding) and desialylation (RCA-1 binding) were detected via flow cytometry. RBD without tag: 50 μg/ml, n = 3. All flow cytometry data are expressed as fold change from control group. (F to H) Human gel-filtered platelet (preincubated with 200 μg/ml RBD-rFc) aggregation was stimulated by 0.02 U/ml thrombin, or 2 μM ADP with fibrinogen, or 2 μg/ml collagen. n = 5 to 9. All the data were expressed as means ± SEM (***P < 0.001, **P < 0.01, *P < 0.05).
The κ and δ variants have greater potential to bind, activate, and aggregate platelets
We constructed 3 RBD mutants of B.1.617.1 (Kappa strain), B.1.617.2 (Delta strain), and B.1.617.2.1 (Delta Plus (+) strain) and compared their effect on platelet activation and aggregation relative to wild-type RBD. Flow cytometry results showed that all 3 RBD variants bind to human platelets (Fig. 4A) and induce platelet activation (Fig. 4B to F). Furthermore, we found trending evidence for more pronounced platelet activation (Fig. 4B to F) and platelet aggregation potentiation (Fig. 4G to I) induced by the κ and δ variants, which was greatest in the κ variant. Together, these data indicate that the κ and δ variants induce stronger platelet activation, which is consistent with clinical reports of severe thrombosis in patients infected with these variants [11,63,64].
Fig. 4.

The κ, δ, and δ+ variants have enhanced platelet binding, activation, and potentiation of platelet aggregation. (A) The binding of RBD-rFc variants (200 μg/ml) to human healthy donors’ platelets was analyzed by flow cytometry using FITC-labeled anti-rabbit IgG (Fc specific). (B to F) Human platelet activation (P-selection expression/PAC-1 or fibrinogen binding), apoptosis (Annexin V binding), and desialylation (RCA-1 binding) were detected via flow cytometry, n = 5 to 10. (G to I) Human gel-filtered platelet (preincubated with 200 μg/ml RBD-rFc or RBD-rFc variants) aggregation was stimulated by 0.02 U/ml thrombin, or 2 μM ADP with addition of fibrinogen or 2 μg/ml collagen. All the data were expressed as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Novel anti-RBD mAbs inhibit RBD-induced activation, aggregation, and clearance in vivo
We generated a panel of anti-RBD mouse mAbs using the hybridoma technique (Table). We selected 4F2 and 4H12 for further study based on their strong inhibition of the RBD–ACE2 interaction. Interestingly, 4F2 and 4H12 also inhibited RBD binding to human platelets (Fig. 5A) and suppressed RBD-induced platelet clearance in vivo (Fig. 5I). 4F2 and 4H12 also alleviated RBD-induced human platelet activation (Fig. 5B to F) and RBD-potentiated platelet aggregation in vitro (Fig. 5G and H), while the mAbs themselves did not induce platelet activation/aggregation. 4F2 and 4H12 could still recognize and neutralize the κ variant, inhibiting its binding to human platelets (Fig. S7A) and alleviating its platelet activation in vitro (Fig. S7B to D). The other 3 anti-RBD mAbs 4H10/2B5/4E10 also alleviated RBD-induced human platelet activation in vitro (Table).
Fig. 5.
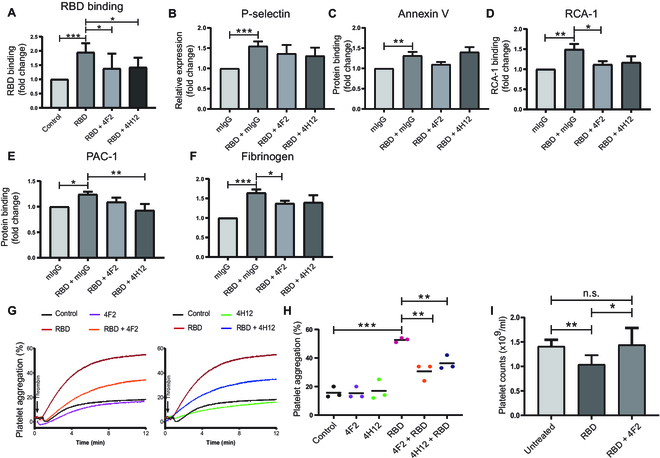
The novel anti-RBD mAbs 4F2 and 4H12 inhibit RBD-induced platelet activation and RBD-potentiated platelet aggregation. (A) The inhibitive effect of anti-RBD mAbs (50 μg/ml) on RBD-rFc (200 μg/ml) binding to human washed platelets was analyzed by flow cytometry using FITC-labeled anti-rabbit IgG (Fc specific). (B to F) The inhibitive effect of anti-RBD mAbs 4F2 and 4H12 (50 μg/ml) on RBD-induced human platelet activation (P-selection expression/PAC-1 or fibrinogen binding), apoptosis (Annexin V binding), and desialylation (RCA-1 binding) was detected via flow cytometry. (G and H) Human gel-filtered platelet (preincubated with rFC control, 200 μg/ml RBD-rFc, or 50 μg/ml 4F2 + 200 μg/ml RBD-rFc) aggregation was stimulated by 0.02 U/ml thrombin. (I) CD1 mouse platelets were counted at 0 h (Untreated) and 3 h after I.V. injection with RBD-rFc or RBD-rFc + 4F2 (RBD-rFc incubated with 4F2 before injection, n = 3). RBD-rFc: 1 μg/g, 4F2: 1 μg/g. All data are expressed as means ± SEM. n.s. denotes no significance. *P < 0.05, **P < 0.01, ***P < 0.001.
Table.
Characterization of our novel anti-RBD antibodies on RBD-induced platelet activation.
| Clone name | IgG subtype | Inhibit RBD–ACE2 binding | Inhibit RBD-PLT binding | Inhibit P-selectin | Inhibit PAC-1 | Inhibit fibrinogen |
|---|---|---|---|---|---|---|
| 4F2 | IgG 1 | Strong | Yes | Yes (*) | Yes | Yes (*) |
| 4H10 | IgG 2a | Weak | Yes | Yes | Yes (*) | Yes (**) |
| 2B5 | IgG 2a | Weak | Yes | Yes | Yes (*) | Yes (*) |
| 4E10 | IgG 2b | Weak | Yes | Yes | Yes | Yes |
| 4H12 | IgG 1 | Strong | Yes | Yes | Yes (**) | Yes |
*P < 0.05, **P < 0.01, RBD + anti-RBD mAb vs. RBD + mIgG control.
Antibodies 4F2 and 4H12 dose-dependently inhibit SARS-CoV-2 infection of Vero E6 cells
To investigate whether our anti-RBD 4F2 and 4H12 antibodies could interfere with SARS-CoV-2 virus infection, we infected Vero E6 cells with SARS-CoV-2/SB2 at a multiplicity of infection (MOI) of 2 [65]. The viral envelope gene was quantified by reverse transcription real-time quantitative polymerase chain reaction (RT-qPCR) assay (Fig. 6A) as a marker for viral replication. SARS-CoV-2 infection of Vero E6 cells was significantly inhibited 15 h after introduction of cells to media preincubated with virus and either 4F2 or 4H12 dose-dependently (Fig. 6A). In line with reduced viral load, we observed a significant reduction in SARS-CoV-2 titers in 4F2/4H12 pretreated Vero E6 cells (*P < 0.05; Fig. 6B) in a dose-dependent manner to near undetectable levels. Cytotoxicity studies revealed that neither 4F2 nor 4H12 was toxic to Vero E6 cells (Fig. 6C). Next, we examined the expression of viral and entry proteins. Interestingly, we saw 4F2 and 4H12 dose-dependently decreased spike and nucleocapsid protein expression but increased/regained ACE2 protein expression (Fig. 6D to G), which is consistent with the earlier reports that SARS-CoV-2 binding leads to ACE2–SARS-CoV-2 complex internalization and lysosomal degradation [66,67]. As expected, transmembrane protease serine 2 expression was independent of SARS-CoV-2 virus infection and 4F2 or 4H12 treatment (Fig. 6D). Collectively, these data demonstrate that 4F2 and 4H12 can inhibit SARS-CoV-2 virus infection and provide support for the study of anti-RBD therapies to treat COVID-19.
Fig. 6.

Anti-RBD mAbs 4F2 and 4H12 inhibit SARS-CoV-2 infection in a dose-dependent manner. (A) Different concentrations of anti-RBD mAb 4F2 or 4H12 were mixed with MOI 2 of SARS-CoV-2 for 30 min and then added to the culture medium of Vero E6 cells (the inoculum was not removed). Vero E6 cells were recovered 15 h postinfection and viral RNA was assayed by RT-qPCR. Dotted horizontal lines indicate the limit of detection (LOD). (B) Virus progeny was evaluated for viable virus in a TCID50 assay. (C) The addition of 4F2 or 4H12 was not toxic to the Vero E6 cells. (D) Detection of SARS-CoV-2 entry proteins and viral proteins in cells lysate by western blot. Glyceraldehyde-3-phosphate dehydrogenase was used as a loading control. (E to G) Densitometric analysis of protein bands. Data are represented as means ± SEM. *P < 0.05, **P < 0.01, versus SARS-CoV-2 group.
Discussion
COVID-19 continues as a global health threat, in part due to the constant emergence of novel variants. COVID-19 patients face the risk of life-threatening thrombotic events including heart attack and stroke; however, the mechanism remains inadequately understood. We herein demonstrated that the SARS-CoV-2 spike protein RBD causes significant platelet clearance in vivo. Further investigation revealed the RBD bound platelets partially via an interaction between its RGD motif and the αIIbβ3 integrin. RBD–platelet binding could induce platelet activation and potentiate platelet aggregation, which was amplified in the Delta and Kappa variants. Notably, the RBD also induced platelet desialylation, suggesting liver-mediated platelet clearance. Lastly, we generated a panel of novel anti-RBD mAbs, including 4F2 and 4H12, that inhibited RBD-induced platelet activation and clearance and also attenuated SARS-CoV-2 infection of Vero E6 cells. These findings reveal a novel mechanism of RBD-mediated thrombosis and platelet clearance with implications for both COVID-19 and VITT and also provide justification for the use of anti-RBD mAbs for treating COVID-19.
Several viruses are reported to interact with integrins [68,69]. More recently, the RGD motif in SARS-CoV-2 RBD is reported to interact with integrins αVβ3, αVβ6, and α5β1 [53–55]. In this report, our data highlights the virus RBD also binds to the αIIbβ3 integrin on platelets via its RGD motif. RBD platelet binding was reduced with αIIbβ3 antagonists eptifibatide and mAb M1, confirming its binding site within the αIIbβ3 ligand-binding pocket. Although structural studies suggest steric hindrance of the RGD motif in the full-length spike protein conformation, it is possible that RGD exposure increases with dynamic structural changes. We propose that thiol-isomerase activity of the β integrin PSI domain may help prime the spike protein for an integrin interaction, especially given its cysteine-rich nature [58,70]. Interestingly, however, we found the RBD–platelet interaction could not be completely inhibited by either αIIbβ3 antagonists or in β3−/− mice, indicating additional receptors are/may be involved. Our western blot showed positive bands for ACE2 in nonreducing protein samples at approximately 250 kDa, suggesting dimeric ACE2 may distribute on human platelets. However, we interpret these results cautiously due to the possibility of antibody cross-reactivity. Binding to additional candidate receptors including c-mpl and CD147 are worth future investigation [25,44,71].
Platelet hyperactivation is well reported in COVID-19. Immune dysregulation such as cytokine storm is an indirect mechanism that is thought to, at least partially, contribute to platelet activation in COVID-19. Our study describes a direct mechanism of RBD-induced platelet activation. Although the mechanism of platelet activation is currently unclear, interactions with ACE2, toll-like receptors, and other binding partners may synergistically contribute to this event [25]. It is also possible that the RGD motif in the RBD contributes to platelet activation, particularly in a multivalent and full-length spike protein conformation. Given the high copy number of spike protein on the virus surface, we cannot exclude the virus; otherwise, this may bridge multiple platelets via RBD–αIIbβ3 interaction, causing platelet aggregation and contributing to microthrombi formation in COVID-19.
COVID-19 patients also exhibit hypercoagulability, yet the SARS-CoV-2 virus does not appear to have intrinsic procoagulant effects [72]. Platelet-monocyte aggregates bearing tissue factor are reported to drive coagulation in COVID-19 [28]. We show the RBD induces P-selectin expression, which promotes the formation of tissue-factor-bearing platelet-monocyte and other leukocyte aggregates. The RBD also induced platelet phosphatidylserine exposure, which harbors coagulation factors and promotes coagulation via cell-based thrombin generation [13,59,70]. Subsequent thrombin may feedback to further activate platelets, creating a vicious positive feedback loop. Taken together, the RBD may directly activate platelets and indirectly orchestrate hypercoagulation to synergistically contribute to thrombotic incidences including deep vein thrombosis, pulmonary emboli, lung microcirculatory thrombi, and arterial thrombosis such as stroke. Thus, although anticoagulant therapy has been a primary treatment against COVID-19 [73], our work provides strong rationale for further investigation of antiplatelet therapy as a potential upstream target.
The Delta (L452R and T478K) and Kappa (L452R and E484Q) variants each bear 2 mutations in the RBD. Our data shows that the Delta and Kappa RBD variants bound to platelets and induced activation and potentiated platelet aggregation significantly more than the original strain. It is possible that these mutations in the RBD may lead to structural alterations that enhance exposure of the RGD integrin-binding motif to promote its interaction with platelets. Recently, Omicron has succeeded as the most common variant and has 15 mutations in RBD region and ~32 mutations in the spike protein. Although these structural changes bolster affinity for the ACE2 receptor [74], the effect on its accessibility for the αΙΙbβ3 integrin is unclear and is worth further investigation.
The prevailing view for VITT has been that anti-RBD antibodies cross-react with platelet factor 4 to create a large immunogenic complex capable of inducing platelet activation via the FcγRIIA [34,75]. This mechanism is reminiscent of heparin-induced thrombocytopenia but occurs in the absence of heparin. Our finding of direct RBD-induced platelet activation may contribute by providing the initial wave of platelet factor 4 release. We also cannot exclude the possibility that administration of the RBD-containing spike protein in adenovirus-based vaccines directly causes VITT. Indeed, our data shows that RBD injection over a concentration threshold could elicit significant platelet clearance. Given the variable RBD concentration derived from DNA/RNA COVID-19 vaccines, it is possible that high RBD concentrations from vaccination may account for some incidences of platelet clearance or even VITT. Thus, future research investigating the correlation between plasma RBD concentration and incidence of VITT merits further study.
Our group previously reported platelet desialylation as a novel mechanism of liver-mediated platelet clearance [14,17,61]. Since the RBD induced significant platelet desialylation, we suspect that the liver may be a key site of RBD-induced platelet clearance. The effect of excessive hepatocellular platelet clearance on liver function is unclear. However, liver injury is reported in both COVID-19 [76] and VITT [77]. Given the importance of the hepatocytes in the production of coagulation factors, an altered function may contribute to coagulopathy, a common symptom in advanced COVID-19 [78]. Furthermore, hepatocytes are also key producers of the platelet regulator thrombopoietin [79,80], thus, hepatocellular dysfunction may impair platelet production and exacerbate thrombocytopenia. On the other hand, our group has also discovered immunosuppression linked with liver-mediated platelet clearance (manuscript submitted to Nature Communications). Whether RBD-mediated platelet clearance in the liver can modulate immune response [81–85] or down-regulate coagulation factor and thrombopoietin production requires further investigation.
We found that RBD-induced platelet clearance in vivo and platelet activation and aggregation in vitro were susceptible to the therapeutic effects of our novel mAbs F42 and 4H12. Moreover, 4F2 and 4H12 also demonstrated the capacity to neutralize the RBD–ACE2 interaction. To our knowledge, this is the first reported therapeutic agent capable of inhibiting SARS-CoV-2 infection, thrombosis, and thrombocytopenia.
In summary, our data reveal that the RBD directly binds to platelets and that this binding is partially mediated by RGD–integrin αIIbβ3 interaction. RBD binding causes platelet activation and clearance, which provide new insights into the mechanisms of thrombosis and thrombocytopenia that are observed in COVID-19 and VITT. We also cannot exclude that this interaction may contribute to the virus internalization into platelets, which may contribute to immune evasion and hematological dissemination to new sites of infection. Furthermore, our newly developed anti-RBD mAbs 4F2 and 4H12 can ameliorate RBD-induced platelet activation/clearance and therefore may have the potential not only for diagnosis of SARS-CoV-2 virus antigen but importantly also for treatment of COVID-19 and its complications.
Materials and Methods
Blood collection and platelet isolation
All blood collection procedures were approved by the Research Ethics Board of St. Michael’s Hospital (Toronto, ON, Canada) and carried out as previously described [15,58,86]. Venous blood samples from healthy human donors were collected with informed consent into BD Vacutainer plastic blood collection tubes with 3.2% sodium citrate. Mice were anesthetized using 2.5% avertin via intraperitoneal injection, and whole blood was collected into 3.2% sodium citrate via retro-orbital bleeding. Platelet-rich plasma (PRP) was separated by centrifugation (300×g, 10 min). Platelets were washed (1,050×g, 15 min with 60 nM prostaglandin E1 [P5515, Sigma]) in modified Tyrode’s buffer (134 mM NaCl, 2.9 mM KCl, 0.34 mM Na2HPO4, 12 mM NaHCO3, 20 mM Hepes, 1 mM MgCl2, and 5 mM glucose, pH 7.35). Then, platelets were counted with a Z2 Series Coulter Counter (Beckman Coulter, USA).
Mice
Integrin β3−/− mice were kindly provided by Dr. Richard O. Hynes from the Massachusetts Institute of Technology and further backcrossed with BALB/c mice [87,88]. Six-week-old female BALB/c mice and CD1 mice were purchased from Charles River Canada. All mice were housed and bred in the St. Michael’s Hospital Research Vivarium. The animal procedures were approved by the Animal Care Committee and in compliance with the Guidelines of the Canadian Council of Animal Care.
Cell lines
Vero E6 cells (African green monkey cells; CRL-1586, American Type Culture Collection) were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 1 × l-glutamine and penicillin/streptomycin (Corning).
SARS-CoV-2 virus
SARS-CoV-2 SB3-TYAGNC was generously provided by Dr. Samira Murabeka from Sunnybrook Research Institute, Toronto. The virus was isolated from a nasopharyngeal sample of a patient in Canada. Supernatant from the cell lysate was used to determine virus titers (50% tissue culture infectious dose [TCID50]/ml) according to the Spearman and Karber method as outlined previously [65].
SARS-CoV-2 virus infection
All work involving live SARS-CoV-2 was performed in the Combined Containment Level 3 Unit (C-CL3 Unit) of the Temerty Faculty of Medicine at the University of Toronto in accordance with institutional biosafety requirements. Vero E6 cells were seeded in 48-well plates (5 × 104 cells per well) (Sarstedt) in DMEM containing 10% fetal bovine serum. Twenty-four hours postseeding, different concentrations of our newly developed antibody 4F2/4H12 were mixed with SARS-CoV-2 at an MOI of 2 in a final volume of 100 μl of DMEM per well at 37 °C. After 30 min, Vero E6 cells were infected with mixes containing antibody 4F2/4H12 and SARS-CoV-2 (the inoculum was not removed). Fifteen hours postinfection, supernatants were removed, and cells were recovered with viral RNA quantified by RT-qPCR.
RT-qPCR
Samples were extracted using the QiaAmp Viral mini kit (52906, QIAGEN) according to the manufacturer’s instructions. RT-qPCR was performed using E-gene SARS-CoV-2 following guidelines by the World Health Organization (https://www.who.int). The RT-PCR assays were performed by using Luna Universal qPCR Master Mix (E3005L, New England Biolabs) based on the manufacturer’s instructions. Two separate gene targets were employed for detecting the envelope (E) gene. The E-gene CAGGTACGTTAATAGTTAATAGCGT and ATATTGCAGCAGTACGCACACA were used as forward and reverse primers. qPCR was performed on an Applied Biosystems QuantStudio 7 Flex Real-Time PCR System. The reaction conditions were 1 cycle of denaturation (60 °C for 10 min, then 95 °C for 2 min), followed by 40 amplification cycles (95 °C for 10 s and 60 °C for 15 s). QuantStudio Real-Time PCR software was used for analysis.
Construction of RBD recombinant proteins
SARS-Cov-2 spike protein subunit 1 (RBD residues, V327 to T531) was cloned in AbVec2.0-IGHG1 plasmid (80795, Addgene) and fused with glycine serine linker (Gly4 S)1 and mouse/rabbit Fc-tag on C-terminus. An N-terminal 10× His-tag was fused with (G3S1)1, TEV cleavage site and RBD residue V327 to T531. Unless stated otherwise, the RBD and full spike protein sequences used in this study were derived from the original SARS-CoV-2 virus. Kappa variant (L452R, E484Q)/Delta variant (L452R, T478K)/Delta+ variant (K417N, L452R, T478K) RBD rabbit Fc-tagged constructs and RBD (RGE mutation) rabbit Fc-tagged mutant (D405E) were generated using an In-Fusion Cloning Kit (Takara Bio, Inc.). RBD without a tag was generated after TEV protease cleavage using an N-terminal His-tag RBD construct. These RBD constructs were transfected in Expi293F cells (3 × 106 viable cells/ml) in Expi293 Expression Medium (Thermo Fisher Scientific) using FectoPRO transfection reagent (Polyplus-transfection) at 37 °C, 8% CO2 under 120 rpm shaking conditions for 3 to 4 d. RBD proteins were purified using Protein A agarose or HisPur nickel nitrilotriacetic acid resin (Thermo Fisher Scientific). Proteins were buffer exchanged with phosphate-buffered saline (PBS) (10 mM sodium phosphate buffer, pH 7.4, 2.7 mM KCl, and 137 mM NaCl) and stored at −80 °C until use.
Development and purification of mAbs against RBD
Female BALB/c mice 6 to 8 weeks old were immunized with 30 to 50 μg of RBD recombinant protein mixed with TiterMax (Sigma-Aldrich, Canada) 3 times as we previously described [17,58,89]. The immunized spleen cells were then fused with mouse myeloma cells (P3X63Ag8.653, American Type Culture Collection), and hybridomas were selected by HAT medium (Sigma-Aldrich, Canada). Positive hybridomas were identified using enzyme-linked immunosorbent assay and subcloned through limit dilution. Hybridomas that secret antibodies were cultured in sera-free medium for large-scale antibody production. The mAbs were isolated/purified using Protein-G agarose beads (Thermo Fisher Scientific).
RBD injection and platelet count assay
We injected different doses (0.25, 0.5, and 1.0 μg/g) of RBD into CD1 mice via tail vein injection and counted the number of platelets at 0, 1, 3, 8, 24, and 48 h postinjection. CD1 mice were bled via the saphenous vein, and platelet count procedures were performed as we previously described [17,56,57]. Briefly, 10 μl of whole blood per mouse was mixed with 240 μl of PBS-EDTA, spun down (150×g, 3 min), then 50 μl of supernatant was mixed with 10 ml of Isotone II Diluent and platelets were counted with a Z2 Series-Coulter-Counter. We examined the effect of RBD-rFc on platelet counts applying a dose-response assay by quantifying the rate of change in platelet counts as a function of elapsed time and the amount of administered dose. Then, we quantified the effective concentration value for RBD-Fc utilizing a dose-response analysis model in OriginPro (OriginLab, 2016).
Flow cytometry analysis
For analysis of labeled transfused platelets [17], CD1 mouse platelets were labeled with Far Red dye (Thermo Fisher Scientific) for 30 min at 37 °C and washed with PBS buffer. Labeled platelets (108 counts) were then incubated with RBD for 30 min and then transfused via tail vein into syngeneic mice. Mice were bled at 0 h (baseline, following platelet transfusion), 1, 3, 24 h posttransfusion via retro-orbital bleeding into PBS-EDTA and centrifuged at 150×g, 3 min. The supernatant was then analyzed via flow cytometry to detect the percentage of fluorescent positive platelets. For analysis of platelet activation, human platelet surface P-selectin was detected with anti-human CD62P (P-Selectin) fluorescein isothiocyanate (FITC) antibody (304904, Biolegend); GPIIbIIIa activation was detected with anti-human CD41/CD61 PAC-1 Alexa Fluor 647 antibody (362806, Biolegend) for human platelets. Annexin V, fibrinogen, and RCA-1 binding were detected with Alexa Fluor 647 Annexin V (640912, Biolegend), Alexa Fluor 647 Fibrinogen (F35200, Thermo Fisher Scientific) and fluorescein labeled RCA-1 (FL-1081-5, Vector Labs), respectively, as we previously described [26,61,90–92]. For analysis of RBD binding, RBD (with rabbit Fc tag) binding was detected with anti-rabbit immunoglobulin G (IgG) [Fc specific]-fluorescein antibody (1:500, SAB3700850, Sigma). Anti-RBD antibody 4H12 was labeled with an antibody labeling kit (Cat. No. A20181, ThermoFisher) according to the product instructions and RBD binding was detected using Alexa Fluor 647 4H12 in a dilution of 1:100. A total of 10,000 platelet events were acquired and data were analyzed via FlowJo 7.6 (Becton, Dickinson and Company, NJ, USA).
Direct binding analysis of RBD-αIIbβ3 in vitro
The recombinant monomeric RBD protein (22 kDa) was expressed and purified. Then, the purification tag was cleaved as described above. The covalent conjugation of the N-terminal RBD monomer to a fluorescein derivative label was performed through an amine-tetrafluorophenyl reaction in PBS at 25 °C (Alexa Fluor 488, Invitrogen). The labeled RBD was further purified and concentrated in the binding buffer (20 mM tris, [pH 7.4], 137 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, and 1 mM MnCl2). We measured the fluorescence anisotropy (r) of 5 nM labeled RBD in a titration with purified αIIbβ3 protein (GPIIbIIIa, Innovative Research) in the binding buffer at 25 °C using a Cary Eclipse spectrofluorometer (λex = 495 nm and λem = 517 nm) as previously described [93]. The results from 3 trials were averaged, normalized, and plotted as a function of αIIbβ3 concentration. To quantify the dissociation constant (Kd) values, the binding curve was fit to a 1-site binding function as described elsewhere [94].
Western blot
Total proteins of platelets were extracted by RIPA Lysis and Extraction Buffer (89900, Thermo Fisher Scientific). The proteins of platelets or Vero E6 cells or purified RBD were separated by 8% bis-tris sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (1620115, Bio-Rad). The blots were blocked with 5% fat-free milk (sc-2325, Santa Cruz Biotechnology) for 1 h at room temperature and then with an anti-ACE2 antibody (1:1,000, ab15348/ab272500/ab108252, Abcam; MA5-32307, Thermo Fisher Scientific; 21115-1-AP, Proteintech), anti-nucleocapsid antibody (40143-R001, Cell Signaling), anti-spike antibody (ZMS1076, Sigma), anti-transmembrane protease serine 2 antibody (AB92323, Abcam). or anti-glyceraldehyde-3-phosphate dehydrogenase antibody (5174, Cell Signaling) overnight at 4 °C. The membranes were then incubated with horse radish-conjugated goat anti-mouse IgG (1:3,000, 62-6520, Thermo Fisher Scientific) or anti-rabbit IgG (1:10,000, 111-035-003, Jackson ImmunoResearch) for 1.5 h at room temperature and visualized with enhanced chemiluminescence (ECL) substrate (32106, Thermo Fisher Scientific) and Bio-Rad ChemiDoc imaging system. Densitometry was performed using Image Lab software (Bio-Rad).
Platelet aggregation assay
PRP or human gel-filtered platelets were prepared as we previously described [95–98]. Gel-filtered platelets were isolated from PRP using a Sepharose 2B chromatography column with PIPES buffer. Platelet count was adjusted to 2 × 108 counts/ml and preincubated with 200 μg/ml RBD or RBD variants with or without 50 μg/ml anti-RBD antibodies. Platelet aggregation was induced by 0.02 to 0.04 U/ml thrombin, 2 μM ADP with the addition of fibrinogen, 2 μg/ml collagen with 2 mM CaCl2. Platelet aggregation was measured in a Chrono-Log Model 700 aggregometer (Chrono-Log, USA). and data were collected and recorded for at least 8 min by Aggrolink8 software.
Biolayer interferometry
Recombinant spike protein samples with a C-terminal 8× His-tag were prepared as described in Construction of RBD recombinant proteins. Purified spike protein samples (0.35 μM) were immobilized onto hydrated nickel nitrilotriacetic acid biosensor probes employing an 8-channel Octet RH16 biolayer interferometry instrument and tilted-bottom microplates in the supplied Octet Kinetics Buffer (Sartorius, Germany). Probes were quenched with SuperBlock buffer (Thermo Fisher Scientific) and equilibrated in binding buffer (20 mM tris [pH 7.4], 137 mM NaCl, 1 mM MgCl2, 1 mM MnCl2, 1 mM CaCl2, and 50% [v/v] glycerol) plus 1X Octet Kinetics Buffer shaken at 1,000 rpm to obtain a baseline response for 3 min at 25 °C. The wavelength shifts corresponding to the association rates (kon) were measured by immersing the loaded biosensors into wells containing platelet-derived purified human αIIbβ3 protein (Innovative Research) in binding buffer at a concentration range from zero to 0.51 μM. The dissociation rates (koff) were measured by transferring the loaded biosensors into wells containing binding buffer. The effect of binding buffer on the threshold of detection was optimized, and raw data were subtracted from a reference loaded biosensor in the binding buffer. A triplicated sample of 0.3 mM bovine serum albumin (2% BSA) in binding buffer was used as a nonspecific binding control. All concentrations were verified by using NanoDrop (Thermo Fisher Scientific ND-One). The acquired wavelength shifts from a minimum of 3 trials were fit to a global binding model and analyzed to quantify binding parameters using the supplied Octet software package and GraphPad Prism.
Isothermal titration calorimetry
Purified spike protein samples (12 μM) were titrated in 1 μM purified human αIIbβ3 integrin samples, and the heat of binding was measured utilizing an isothermal titration calorimetry (ITC) instrument (Microcal ITC-200). Samples were degassed before use with a MicroCal Thermo Vac unit for 5 min. Titrations were performed with αIIbβ3 integrin in the cell and recombinant spike protein serving as the titrant in the syringe. All experiments were performed in 20 mM tris (pH 7.4), 100 mM NaCl, 1 mM CaCl2, 1 mM MnCl2, 1 mM MgCl2, 0.05% NaN3, and 10% (v/v) glycerol at 25 °C. Each trial consisted of an initial delay of 60 s, first injection of 0.2 μl and 300-s delay. A subsequent 18 injections were 2 μl, spaced at 300-s intervals. The first point was removed from all data sets due to different injection volume and delay parameters. The acquired ITC data were subtracted from a titration of spike protein in a blank buffer control. The analyzed thermogram was fit to a 1-site binding model using the manufacturer provided Origin 7 software.
Statistical analysis
All the data were analyzed using GraphPad Prism 8.0 (GraphPad Software Inc., USA), and presented as mean ± standard error of mean (SEM). Statistical significance was determined using an unpaired Student t test (2-tailed) for comparisons between 2 groups. One-way analysis of variance (ANOVA) followed by Dunnett’s and Tukey’s multiple comparisons tests were used to compare the statistical significances among multiple groups. A P value < 0.05 was considered statistically significant.
Acknowledgments
We would like to thank Dr. R. O. Hynes for his generous gift of integrin β3−/− mice. We thank Dr. P. Johnson and Dr. L Donaldson (York University, Toronto, ON) for the use of their lab facilities and the fluorescence spectrophotometer. We thank the Hospital for Sick Children (Toronto, ON, Canada) for the use of Structural & Biophysical Core Facility. We would like to acknowledge the Keenan Research Centre for Biomedical Science Core Facilities use at St. Michael's Hospital (Toronto, ON, Canada). Funding: This study was supported by the Canadian Institutes of Health Research (CIHR) Foundation grant (389035) and CCOA Therapeutics Inc. research fund to Dr. H.N. CIHR grant for COVID-19 (OV3-170344, SBC-171482, and VS1-175560) to Dr. H.Z. D.T.M., Z.C., and DK are recipients of the Queen Elizabeth II (QE-II) Graduate Scholarship, Ontario, Canada. S.S. is a recipient of the Canadian Blood Services postdoctoral award. D.T.M. is a recipient of a Graduate Scholarship, Department of Physiology, University of Toronto. Z.C. is the recipient of the Canadian Blood Services Graduate Scholarship, Ontario, Canada. C.S. is a recipient of a postdoctoral Mitacs award, University of Toronto. Z.L. is a recipient of Mitacs Accelerate Postdoctoral Fellowship. LL is a recipient of a scholarship from the University of Chinese Academy of Sciences. X.W. is a recipient of Killam Research Fellowship from the Canadian Council for the Arts. D.K. is also the recipient of the St. Michael’s Hospital Research Training Centre Scholarship and the 2021-2022 Vanier Canada Graduate Scholarships (Vanier CGS) , and V.P. is the recipient of the CGS awarded by the CIHR. Author contributions: X.M., J.L., A.A.S., and D.T.M. carried out in vitro experiments, analyzed data, and wrote the manuscript. G.Z., Z.L., X.L., Z.C., and D.K. carried out in vivo experiments and analyzed data. P.B., C.Z., and H.S. constructed RBD/variants recombinant proteins. L.L., S.S., C.J.K., and V.P. carried out in vitro experiments and analyzed data. G.Z. developed the polyclonal and monoclonal antibodies, and S.M., J.L., Y.L., and G.Z. designed the in vitro SARS-CoV-2 infection model. C.S., J.D., Z.R., J.Z., T.N., P.C., X.W., K.A.C., H.Z., and O.R. analyzed data and contributed to the preparation of the manuscript. H.N. is the principal investigator who designed the project and the related experiments, analyzed the data, and prepared the manuscript. Competing interests: The work of RBD variants proteins construction/preparation and anti-RBD monoclonal antibody generation was supported by CCOA Therapeutics Inc.
Data Availability
The data of this study are available from the first and corresponding authors upon request.
Supplementary Materials
Fig. S1. The RBD can induce platelet clearance in vivo.
Fig. S2. Dimeric ACE2 may distribute on human platelets.
Fig. S3. ITC thermograms demonstrate full-length spike protein binding to αIIbβ3 integrin.
Fig. S4. The RBD can induce human platelet activation.
Fig. S5. The RBD can potentiate human gel-filtered platelet aggregation.
Fig. S6. The RBD potentiate human PRP aggregation in vitro.
Fig. S7. 4F2 and 4H12 alleviate κ variant RBD-induced human platelet activation in vitro.
Table S1. Direct interaction kinetics parameters for biolayer interferometry analyses of full-length spike protein and purified human αIIbβ3 in vitro.
References
- 1.Gu SX, Tyagi T, Jain K, Gu VW, Lee SH, Hwa JM, Kwan JM, Krause DS, Lee AI, Halene S, et al. Thrombocytopathy and endotheliopathy: Crucial contributors to COVID-19 thromboinflammation. Nat Rev Cardiol. 2021;18(3):194–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, Merdji H, Clere-Jehl R, Schenck M, Gandet FF, et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020;46(6):1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liao D, Zhou F, Luo L, Xu M, Wang H, Xia J, Gao Y, Cai L, Wang Z, Yin P, et al. Haematological characteristics and risk factors in the classification and prognosis evaluation of COVID-19: A retrospective cohort study. Lancet Haematol. 2020;7(9):e671–e678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ciceri F, Beretta L, Scandroglio AM, Colombo S, Landoni G, Ruggeri A, Peccatori J, D’Angelo A, De Cobelli F, Rovere-Querini P, et al. Microvascular COVID-19 lung vessels obstructive thromboinflammatory syndrome (MicroCLOTS): An atypical acute respiratory distress syndrome working hypothesis. Crit Care Resusc. 2020;22(2):95–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boeckh-Behrens T, Golkowski D, Ikenberg B, Schlegel J, Protzer U, Schulz C, Novotny J, Kreiser K, Zimmer C, Hemmer B, et al. COVID-19-associated large vessel stroke in a 28-year-old patient : NETs and platelets possible key players in acute thrombus formation. Clin Neuroradiol. 2021;31(2):511–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Modin D, Claggett B, Sindet-Pedersen C, Lassen MCH, Skaarup KG, Jensen JUS, Fralick M, Schou M, Lamberts M, Gerds T, et al. Acute COVID-19 and the incidence of ischemic stroke and acute myocardial infarction. Circulation. 2020;142(21):2080–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Funk T, Pharris A, Spiteri G, Bundle N, Melidou A, Carr M, Gonzalez G, Garcia-Leon A, Crispie F, O’Connor L, et al. Characteristics of SARS-CoV-2 variants of concern B.1.1.7, B.1.351 or P.1: Data from seven EU/EEA countries, weeks 38/2020 to 10/2021. Euro Surveill. 2021;26(16):Article 2100348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheikh A, McMenamin J, Taylor B, Robertson C, Public Health Scotland and the EAVE II Collaborators . SARS-CoV-2 Delta VOC in Scotland: Demographics, risk of hospital admission, and vaccine effectiveness. Lancet. 2021;397(10293):2461–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisman DN, Bogoch I, Lapointe-Shaw L, McCready J, Tuite AR. Risk factors associated with mortality among residents with coronavirus disease 2019 (COVID-19) in long-term care facilities in Ontario, Canada. JAMA Netw Open. 2020;3(7):Article e2015957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Callaway E. Delta coronavirus variant: Scientists brace for impact. Nature. 2021;595(7865):17–18. [DOI] [PubMed] [Google Scholar]
- 11.Manzur-Pineda K, O’Neil CF, Bornak A, Lalama MJ, Shao T, Kang N, Kennel-Pierre S, Tabbara M, Velazquez OC, Rey J. COVID-19-related thrombotic complications experience before and during Delta wave. J Vasc Surg. 2022;76(5):1374–1382.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jackson SP. Arterial thrombosis--insidious, unpredictable and deadly. Nat Med. 2011;17(11):1423–1436. [DOI] [PubMed] [Google Scholar]
- 13.Xu XR, Carrim N, Neves MAD, McKeown T, Stratton TW, Coelho RMP, Lei X, Chen P, Xu J, Dai X, et al. Platelets and platelet adhesion molecules: Novel mechanisms of thrombosis and anti-thrombotic therapies. Thromb J. 2016;14(Suppl 1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J, Sullivan JA, Ni H. Pathophysiology of immune thrombocytopenia. Curr Opin Hematol. 2018;25(5):373–381. [DOI] [PubMed] [Google Scholar]
- 15.Xu XR, Wang Y, Adili R, Ju L, Spring CM, Jin JW, Yang H, Neves MAD, Chen P, Yang Y, et al. Apolipoprotein A-IV binds αIIbβ3 integrin and inhibits thrombosis. Nat Commun. 2018;9(1):Article 3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu XR, Zhang D, Oswald BE, Carrim N, Wang X, Hou Y, Zhang Q, Lavalle C, McKeown T, Marshall AH, et al. Platelets are versatile cells: New discoveries in hemostasis, thrombosis, immune responses, tumor metastasis and beyond. Crit Rev Clin Lab Sci. 2016;53(6):409–430. [DOI] [PubMed] [Google Scholar]
- 17.Li J, van der Wal DE, Zhu G, Xu M, Yougbare I, Ma L, Vadasz B, Carrim N, Grozovsky R, Ruan M, et al. Desialylation is a mechanism of fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat Commun. 2015;6:7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aslam R, Kapur R, Segel GB, Guo L, Zufferey A, Ni H, Semple JW. The spleen dictates platelet destruction, anti-platelet antibody production, and lymphocyte distribution patterns in a murine model of immune thrombocytopenia. Exp Hematol. 2016;44(10):924–930.e1. [DOI] [PubMed] [Google Scholar]
- 19.Grozovsky R, Giannini S, Falet H, Hoffmeister KM. Novel mechanisms of platelet clearance and thrombopoietin regulation. Curr Opin Hematol. 2015;22(5):445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quach ME, Dragovich MA, Chen W, Syed AK, Cao W, Liang X, Deng W, de Meyer SF, Zhu G, Peng J, et al. Fc-independent immune thrombocytopenia via mechanomolecular signaling in platelets. Blood. 2018;131(7):787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang Y, Tang Y, Hoover C, Kondo Y, Huang D, Restagno D, Shao B, Gao L, Michael McDaniel J, Zhou M, et al. Kupffer cell receptor CLEC4F is important for the destruction of desialylated platelets in mice. Cell Death Differ. 2021;28(11):3009–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deppermann C, Kratofil RM, Peiseler M, David BA, Zindel J, Castanheira FVES, van der Wal F, Carestia A, Jenne CN, Marth JD, et al. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J Exp Med. 2020;217(4):Article e20190723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, Limami Y, Zaid N, Sadki K, Ben el Haj R, et al. Platelets can associate with SARS-Cov-2 RNA and are hyperactivated in COVID-19. Circ Res. 2020;127(11):1404–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fard MB, Fard SB, Ramazi S, Atashi A, Eslamifar Z. Thrombosis in COVID-19 infection: Role of platelet activation-mediated immunity. Thromb J. 2021;19(1):Article 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campbell RA, Boilard E, Rondina MT. Is there a role for the ACE2 receptor in SARS-CoV-2 interactions with platelets? J Thromb Haemost. 2021;19(1):46–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li C, Piran S, Chen P, Lang S, Zarpellon A, Jin JW, Zhu G, Reheman A, van der Wal DE, Simpson EK, et al. The maternal immune response to fetal platelet GPIbα causes frequent miscarriage in mice that can be prevented by intravenous IgG and anti-FcRn therapies. J Clin Invest. 2011;121(11):4537–4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Callum JL, Lin Y, Zhou Y, Zhu G, Ni H. Severe platelet desialylation in a patient with glycoprotein Ib/IX antibody-mediated immune thrombocytopenia and fatal pulmonary hemorrhage. Haematologica. 2014;99(4):e61–e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CRR, Righy C, Franco S, Souza TML, Kurtz P, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood. 2020;136(11):1330–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mei H, Luo L, Hu Y. Thrombocytopenia and thrombosis in hospitalized patients with COVID-19. J Hematol Oncol. 2020;13(1):161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang X, Yang Q, Wang Y, Wu Y, Xu J, Yu Y, Shang Y. Thrombocytopenia and its association with mortality in patients with COVID-19. J Thromb Haemost. 2020;18(6):1469–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greinacher A, Thiele T, Warkentin TE, Weisser K, Kyrle PA, Eichinger S. Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N Engl J Med. 2021;384(22):2092–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schultz NH, Sørvoll IH, Michelsen AE, Munthe LA, Lund-Johansen F, Ahlen MT, Wiedmann M, Aamodt AH, Skattør TH, Tjønnfjord GE, et al. Thrombosis and thrombocytopenia after ChAdOx1 nCoV-19 vaccination. N Engl J Med. 2021;384(22):2124–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scully M, Singh D, Lown R, Poles A, Solomon T, Levi M, Goldblatt D, Kotoucek P, Thomas W, Lester W. Pathologic antibodies to platelet factor 4 after ChAdOx1 nCoV-19 vaccination. N Engl J Med. 2021;384(23):2202–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huynh A, Kelton JG, Arnold DM, Daka M, Nazy I. Antibody epitopes in vaccine-induced immune thrombotic thrombocytopaenia. Nature. 2021;596(7873):565–569. [DOI] [PubMed] [Google Scholar]
- 35.Kelton JG, Arnold DM, Nazy I. Lessons from vaccine-induced immune thrombotic thrombocytopenia. Nat Rev Immunol. 2021;21(12):753–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, Liu M, Zhao X, Xie Y, Yang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. 2020;13(1):Article 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koupenova M, Corkrey HA, Vitseva O, Tanriverdi K, Somasundaran M, Liu P, Soofi S, Bhandari R, Godwin M, Parsi KM, et al. SARS-CoV-2 initiates programmed cell death in platelets. Circ Res. 2021;129(6):631–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cox D. Targeting SARS-CoV-2-platelet interactions in COVID-19 and vaccine-related thrombosis. Front Pharmacol. 2021;12:Article 708665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, Petrey AC, Tolley ND, Guo L, Cody M, et al. Platelet gene expression and function in patients with COVID-19. Blood. 2020;136(11):1317–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sahai A, Bhandari R, Koupenova M, Freedman J, Godwin M, McIntyre T, Chung M, Iskander JP, Kamran H, Hariri E, et al. SARS-CoV-2 receptors are expressed on human platelets and the effect of aspirin on clinical outcomes in COVID-19 patients. Research Square. 2020. https://www.researchsquare.com/article/rs-119031/v1
- 41.Shen S, Zhang J, Fang Y, Lu S, Wu J, Zheng X, Deng F. SARS-CoV-2 interacts with platelets and megakaryocytes via ACE2-independent mechanism. J Hematol Oncol. 2021;14(1):Article 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, Kurt-Jones EA, Ravid K, Freedman JE. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood. 2014;124(5):791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Banerjee M, Huang Y, Joshi S, Popa GJ, Mendenhall MD, Wang QJ, Garvy BA, Myint T, Whiteheart SW. Platelets endocytose viral particles and are activated via TLR (toll-like receptor) signaling. Arterioscler Thromb Vasc Biol. 2020;40(7):1635–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang K, Chen W, Zhang Z, Deng Y, Lian JQ, du P, Wei D, Zhang Y, Sun XX, Gong L, et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther. 2020;5(1):Article 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vankadari N, Wilce JA. Emerging WuHan (COVID-19) coronavirus: Glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes Infect. 2020;9(1):601–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Makowski L, Olson-Sidford W, W-Weisel J. Biological and clinical consequences of integrin binding via a rogue RGD motif in the SARS CoV-2 spike protein. Viruses. 2021;13(2):146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sigrist CJ, Bridge A, Le Mercier P. A potential role for integrins in host cell entry by SARS-CoV-2. Antivir Res. 2020;177:Article 104759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beddingfield BJ, Iwanaga N, Chapagain PP, Zheng W, Roy CJ, Hu TY, Kolls JK, Bix GJ. The integrin binding peptide, ATN-161, as a novel therapy for SARS-CoV-2 infection. JACC Basic Transl Sci. 2021;6(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hynes RO. Integrins: Bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687. [DOI] [PubMed] [Google Scholar]
- 50.Ni H, Freedman J. Platelets in hemostasis and thrombosis: Role of integrins and their ligands. Transfus Apher Sci. 2003;28(3):257–264. [DOI] [PubMed] [Google Scholar]
- 51.Xin H, Huang J, Song Z, Mao J, Xi X, Shi X. Structure, signal transduction, activation, and inhibition of integrin αIIbβ3. Thromb J. 2023;21(1):Article 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hynes RO. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell. 1992;69(1):11–25. [DOI] [PubMed] [Google Scholar]
- 53.Liu J, Lu F, Chen Y, Plow E, Qin J. Integrin mediates cell entry of the SARS-CoV-2 virus independent of cellular receptor ACE2. J Biol Chem. 2022;298(3):Article 101710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bugatti A, Filippini F, Bardelli M, Zani A, Chiodelli P, Messali S, Caruso A, Caccuri F. SARS-CoV-2 infects human ACE2-negative endothelial cells through an αvβ3 integrin-mediated endocytosis even in the presence of vaccine-elicited neutralizing antibodies. Viruses. 2022;14(4):705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Norris EG, Pan XS, Hocking DC. Receptor binding domain of SARS-CoV-2 is a functional αv-integrin agonist. J Biol Chem. 2023;299(3):Article 102922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma L, Simpson E, Li J, Xuan M, Xu M, Baker L, Shi Y, Yougbaré I, Wang X, Zhu G, et al. CD8+ T cells are predominantly protective and required for effective steroid therapy in murine models of immune thrombocytopenia. Blood. 2015;126(2):247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Webster ML, Sayeh E, Crow M, Chen P, Nieswandt B, Freedman J, Ni H. Relative efficacy of intravenous immunoglobulin G in ameliorating thrombocytopenia induced by antiplatelet GPIIbIIIa versus GPIbalpha antibodies. Blood. 2006;108(3):943–946. [DOI] [PubMed] [Google Scholar]
- 58.Zhu G, Zhang Q, Reddy EC, Carrim N, Chen Y, Xu XR, Xu M, Wang Y, Hou Y, Ma L, et al. The integrin PSI domain has an endogenous thiol isomerase function and is a novel target for antiplatelet therapy. Blood. 2017;129(13):1840–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roberts HR, Hoffman M, Monroe DM. A cell-based model of thrombin generation. Semin Thromb Hemost. 2006;32(Suppl 1):32–38. [DOI] [PubMed] [Google Scholar]
- 60.Hou Y, Carrim N, Wang Y, Gallant RC, Marshall A, Ni H. Platelets in hemostasis and thrombosis: Novel mechanisms of fibrinogen-independent platelet aggregation and fibronectin-mediated protein wave of hemostasis. J Biomed Res. 2015;29(6):437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tao L, Zeng Q, Li J, Xu M, Wang J, Pan Y, Wang H, Tao Q, Chen Y, Peng J, et al. Platelet desialylation correlates with efficacy of first-line therapies for immune thrombocytopenia. J Hematol Oncol. 2017;10(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li MF, Li XL, Fan KL, Yu YY, Gong J, Geng SY, Liang YF, Huang L, Qiu JH, Tian XH, et al. Platelet desialylation is a novel mechanism and a therapeutic target in thrombocytopenia during sepsis: An open-label, multicenter, randomized controlled trial. J Hematol Oncol. 2017;10(1):104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fluss G, Cranwell V, Rao A, Lee JS, Elshafey R, Wallack M, Finlay D. Covid-19 Delta variant resulting in multi system thromboembolic disease. Ann Vasc Surg Brief Rep Innov. 2022;2(3):Article 100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cohen CT, Riedl RA, Gowda ST, Sartain SE, Bashir DA. Pulmonary embolism in pediatric and adolescent patients with COVID-19 infection during the SARS-CoV-2 delta wave. Pediatr Blood Cancer. 2022;69(8):Article e29721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Banerjee A, Nasir JA, Budylowski P, Yip L, Aftanas P, Christie N, Ghalami A, Baid K, Raphenya AR, Hirota JA, et al. Isolation, sequence, infectivity, and replication kinetics of severe acute respiratory syndrome coronavirus 2. Emerg Infect Dis. 2020;26(9):2054–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu Y, Zhu Q, Fox DM, Gao C, Stanley SA, Luo K. SARS-CoV-2 down-regulates ACE2 through lysosomal degradation. Mol Biol Cell. 2022;33(14):Article ar147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46(4):586–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li C, Li J, Ni H. Crosstalk between platelets and microbial pathogens. Front Immunol. 2020;11:1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hussein HA, Walker LR, Abdel-Raouf UM, Desouky SA, Montasser AK, Akula SM. Beyond RGD: Virus interactions with integrins. Arch Virol. 2015;160(11):2669–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.MacKeigan DT, Ni T, Shen C, Stratton TW, Ma W, Zhu G, Bhoria P, Ni H. Updated understanding of platelets in thrombosis and hemostasis: The roles of integrin PSI domains and their potential as therapeutic targets. Cardiovasc Hematol Disord Drug Targets. 2020;20(4):260–273. [DOI] [PubMed] [Google Scholar]
- 71.Mungmunpuntipantip R, Wiwanitkit V. Pattern of molecular mimicry between spike protein of SARS CoV2 and human thrombopoietin in beta, delta and omicron variants: A basic pathophysiological process of COVID-19 related thrombocytopenia. Am J Blood Res. 2022;12(2):60–63. [PMC free article] [PubMed] [Google Scholar]
- 72.Wool GD, Miller JL. The impact of COVID-19 disease on platelets and coagulation. Pathobiology. 2021;88(1):15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sholzberg M, Tang GH, Rahhal H, Hamzah MA, Kreuziger LB, Áinle FN, Alomran F, Alayed K, Alsheef M, Sumait FA, et al. Effectiveness of therapeutic heparin versus prophylactic heparin on death, mechanical ventilation, or intensive care unit admission in moderately ill patients with covid-19 admitted to hospital: RAPID randomised clinical trial. BMJ. 2021;375:Article n2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lupala CS, Ye Y, Chen H, Su XD, Liu H. Mutations on RBD of SARS-CoV-2 omicron variant result in stronger binding to human ACE2 receptor. Biochem Biophys Res Commun. 2022;590:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ruggeri ZM, Ruf W. Is VITT really a HIT. Nat Immunol. 2021;22(11):1352–1353. [DOI] [PubMed] [Google Scholar]
- 76.Jothimani D, Venugopal R, Abedin MF, Kaliamoorthy I, Rela M. COVID-19 and the liver. J Hepatol. 2020;73(5):1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Efe C, Kulkarni AV, Terziroli Beretta-Piccoli B, Magro B, Stättermayer A, Cengiz M, Clayton-Chubb D, Lammert C, Bernsmeier C, Gül Ö, et al. Liver injury after SARS-CoV-2 vaccination: Features of immune-mediated hepatitis, role of corticosteroid therapy and outcome. Hepatology. 2022;76(6):1576–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McConnell MJ, Kawaguchi N, Kondo R, Sonzogni A, Licini L, Valle C, Bonaffini PA, Sironi S, Alessio MG, Previtali G, et al. Liver injury in COVID-19 and IL-6 trans-signaling-induced endotheliopathy. J Hepatol. 2021;75(3):647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xu M, Li J, Neves MAD, Zhu G, Carrim N, Yu R, Gupta S, Marshall J, Rotstein O, Peng J, et al. GPIbα is required for platelet-mediated hepatic thrombopoietin generation. Blood. 2018;132(6):622–634. [DOI] [PubMed] [Google Scholar]
- 80.Karakas D, Xu M, Ni H. GPIbα is the driving force of hepatic thrombopoietin generation. Res Pract Thromb Haemost. 2021;5(4):Article e12506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li J, Sullivan JA, Ni H. Is platelet desialylation a novel biomarker and therapeutic target in immune thrombocytopenia? J Cell Immunol. 2019;2(1):6–14. [Google Scholar]
- 82.Li C, Chen P, Vadasz B, Ma L, Zhou H, Lang S, Freedman J, Ni H. Co-stimulation with LPS or poly I:C markedly enhances the anti-platelet immune response and severity of fetal and neonatal alloimmune thrombocytopenia. Thromb Haemost. 2013;110(6):1250–1258. [DOI] [PubMed] [Google Scholar]
- 83.Semple JW, Italiano JE Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11(4):264–274. [DOI] [PubMed] [Google Scholar]
- 84.Chen ZY, Oswald BE, Sullivan JA, Dahmani FZ, Pasman Y, Liu Z, Chen P, Ni H. Platelet physiology and immunology: Pathogenesis and treatment of classical and non-classical fetal and neonatal alloimmune thrombocytopenia. Ann Blood. 2019;4(29):19. [Google Scholar]
- 85.Koupenova M, Livada AC, Morrell CN. Platelet and megakaryocyte roles in innate and adaptive immunity. Circ Res. 2022;130(2):288–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li BX, Dai X, Xu XR, Adili R, Neves MAD, Lei X, Shen C, Zhu G, Wang Y, Zhou H, et al. In vitro assessment and phase I randomized clinical trial of anfibatide a snake venom derived anti-thrombotic agent targeting human platelet GPIbα. Sci Rep. 2021;11(1):Article 11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ni H, Chen P, Spring CM, Sayeh E, Semple JW, Lazarus AH, Hynes RO, Freedman J. A novel murine model of fetal and neonatal alloimmune thrombocytopenia: Response to intravenous IgG therapy. Blood. 2006;107(7):2976–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tiller H, Killie MK, Chen P, Eksteen M, Husebekk A, Skogen B, Kjeldsen-Kragh J, Ni H. Toward a prophylaxis against fetal and neonatal alloimmune thrombocytopenia: Induction of antibody-mediated immune suppression and prevention of severe clinical complications in a murine model. Transfusion. 2012;52(7):1446–1457. [DOI] [PubMed] [Google Scholar]
- 89.Reheman A, Yang H, Zhu G, Jin W, He F, Spring CM, Bai X, Gross PL, Freedman J, Ni H. Plasma fibronectin depletion enhances platelet aggregation and thrombus formation in mice lacking fibrinogen and von Willebrand factor. Blood. 2009;113(8):1809–1817. [DOI] [PubMed] [Google Scholar]
- 90.Yang H, Lang S, Zhai Z, Li L, Kahr WHA, Chen P, Brkić J, Spring CM, Flick MJ, Degen JL, et al. Fibrinogen is required for maintenance of platelet intracellular and cell-surface P-selectin expression. Blood. 2009;114(2):425–436. [DOI] [PubMed] [Google Scholar]
- 91.Shen C, Liu M, Xu R, Wang G, Li J, Chen P, Ma W, Mwangi J, Lu Q, Duan Z, et al. The 14-3-3ζ-c-Src-integrin-β3 complex is vital for platelet activation. Blood. 2020;136(8):974–988. [DOI] [PubMed] [Google Scholar]
- 92.Yang Y, Shi Z, Reheman A, Jin JW, Li C, Wang Y, Andrews MC, Chen P, Zhu G, Ling W, et al. Plant food delphinidin-3-glucoside significantly inhibits platelet activation and thrombosis: Novel protective roles against cardiovascular diseases. PLoS One. 2012;7(5):Article e37323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shoara AA, Churcher ZR, Steele TWJ, Johnson PE. Analysis of the role played by ligand-induced folding of the cocaine-binding aptamer in the photochrome aptamer switch assay. Talanta. 2020;217:Article 121022. [DOI] [PubMed] [Google Scholar]
- 94.Shoara AA, Slavkovic S, Donaldson LW, Johnson PE. Analysis of the interaction between the cocaine-binding aptamer and its ligands using fluorescence spectroscopy. Can J Chem. 2017;95(12):1253–1260. [Google Scholar]
- 95.Ya F, Xu XR, Tian Z, Gallant RC, Song F, Shi Y, Wu Y, Wan J, Zhao Y, Adili R, et al. Coenzyme Q10 attenuates platelet integrin αIIbβ3 signaling and platelet hyper-reactivity in ApoE-deficient mice. Food Funct. 2020;11(1):139–152. [DOI] [PubMed] [Google Scholar]
- 96.Ya F, Xu XR, Shi Y, Gallant RC, Song F, Zuo X, Zhao Y, Tian Z, Zhang C, Xu X, et al. Coenzyme Q10 upregulates platelet cAMP/PKA pathway and attenuates integrin αIIbβ3 signaling and thrombus growth. Mol Nutr Food Res. 2019;63(23):Article e1900662. [DOI] [PubMed] [Google Scholar]
- 97.Yang H, Reheman A, Chen P, Zhu G, Hynes RO, Freedman J, Wagner DD, Ni H. Fibrinogen and von Willebrand factor-independent platelet aggregation in vitro and in vivo. J Thromb Haemost. 2006;4(10):2230–2237. [DOI] [PubMed] [Google Scholar]
- 98.Wang Y, Reheman A, Spring CM, Kalantari J, Marshall AH, Wolberg AS, Gross PL, Weitz JI, Rand ML, Mosher DF, et al. Plasma fibronectin supports hemostasis and regulates thrombosis. J Clin Invest. 2014;124(10):4281–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The RBD can induce platelet clearance in vivo.
Fig. S2. Dimeric ACE2 may distribute on human platelets.
Fig. S3. ITC thermograms demonstrate full-length spike protein binding to αIIbβ3 integrin.
Fig. S4. The RBD can induce human platelet activation.
Fig. S5. The RBD can potentiate human gel-filtered platelet aggregation.
Fig. S6. The RBD potentiate human PRP aggregation in vitro.
Fig. S7. 4F2 and 4H12 alleviate κ variant RBD-induced human platelet activation in vitro.
Table S1. Direct interaction kinetics parameters for biolayer interferometry analyses of full-length spike protein and purified human αIIbβ3 in vitro.
Data Availability Statement
The data of this study are available from the first and corresponding authors upon request.