

Abstract
The genomic processes enabling speciation and species coexistence in sympatry are still largely unknown. Here we describe the whole-genome sequencing and assembly of 3 closely related species from the butterfly genus Morpho: Morpho achilles (Linnaeus, 1758), Morpho helenor (Cramer, 1776), and Morpho deidamia (Höbner, 1819). These large blue butterflies are emblematic species of the Amazonian rainforest. They live in sympatry in a wide range of their geographical distribution and display parallel diversification of dorsal wing color pattern, suggesting local mimicry. By sequencing, assembling, and annotating their genomes, we aim at uncovering prezygotic barriers preventing gene flow between these sympatric species. We found a genome size of 480 Mb for the 3 species and a chromosomal number ranging from 2n = 54 for M. deidamia to 2n = 56 for M. achilles and M. helenor. We also detected inversions on the sex chromosome Z that were differentially fixed between species, suggesting that chromosomal rearrangements may contribute to their reproductive isolation. The annotation of their genomes allowed us to recover in each species at least 12,000 protein-coding genes and to discover duplications of genes potentially involved in prezygotic isolation like genes controlling color discrimination (L-opsin). Altogether, the assembly and the annotation of these 3 new reference genomes open new research avenues into the genomic architecture of speciation and reinforcement in sympatry, establishing Morpho butterflies as a new eco-evolutionary model.
Keywords: sympatric speciation, reinforcement, mimicry, structural variant, inversions, gene duplication, wing color pattern, evolutionary convergence: genomic divergence, karyotype
Key Points.
We sequenced, assembled, annotated and karyotyped the genomes of three mimetic and sympatric Morpho species.
We found inversions on the Z sex chromosome that were differentially fixed between species and uncovered duplications of genes involved in vision.
We discuss the potential implications of these findings in preventing gene flow between close sympatric species.
Introduction
Chromosomal rearrangements are likely to play a major role in both adaptation and speciation processes [1, 2]. Inversions, for instance, can favor the emergence of adaptive syndromes by locking together coadapted allelic variations [3]. Chromosomal rearrangements have also been suggested to contribute to reproductive isolation between species by promoting divergent adaptation or by bringing together genetic incompatibilities [4]. Nevertheless, the role of structural variants in these evolutionary processes is still largely unknown. Recently developed sequencing and assembly methods now provide access to complete genomes, therefore opening the investigation of structural variation within and among species (see [5] for a review).
Here, we focus on emblematic species of the Amazonian rainforest, the blue Morpho. We describe the whole-genome sequences of 3 closely related Morpho species living in sympatry for a large range of their geographical distribution (Fig. 1): Morpho helenor, Morpho achilles, and Morpho deidamia [6], thereby developing relevant resources to study the evolution of barriers to gene flow in sympatry. In Lepidoptera, specialization toward host plant has been shown to be a major factor affecting species diversification [7]. Such ecological specialization may favor speciation and coexistence in sympatry and may stem from the evolution of gustatory receptors enabling plant recognition by females [8].
Figure 1:

Geographical distribution of the 3 neotropical species M. helenor (green areas), M. achilles (blue areas), and M. deidamia (red areas). M. helenor has the widest distribution, from Central America to southern Brazil, while M. achilles and M. deidamia are restricted to the Amazonian basin. The 3 species are in sympatry throughout the Amazonian rainforest, including French Guiana (marked with the yellow star), where the samples studied here were collected.
The evolution of visual [9] and olfactory signals [10] between species may also limit gene flow between sympatric species of Lepidoptera. In the 3 Morpho species studied here, both males and females display conspicuous iridescent blue color patterns on the dorsal side of their wings, combined with cryptic brownish color on the ventral side [11]. Such a combination of dorsoventral patterns, associated with a fast and erratic flight, is thought to contribute to the high escape abilities from predators of these butterflies, promoting color pattern convergence between sympatric species (i.e., escape mimicry, [12]). Parallel geographic variation of dorsal wing color pattern has indeed been detected in the 3 Morpho species studied here, suggesting local convergence promoted by predators’ behavior [13]. Given the key role of color pattern in both sexual selection and species recognition in diurnal butterflies, such a resemblance is thought to enhance reproductive interference between sympatric species [14]. Behavioral experiments carried out in the wild revealed that males from the 3 mimetic Morpho species are indeed attracted by both intra- and interspecific wing patterns [15]. Despite this heterospecific attraction of males at long distances, RAD-sequencing markers revealed a highly limited gene flow between these 3 sympatric species [15]. This might be due to the differences in the timing of daily activities observed between these sympatric species, limiting heterospecific encountering [15]. This divergence in daily phenology may contribute to the initiation of speciation or to the reinforcement of prezygotic barriers to heterospecific matings.
Genetic incompatibilities may also contribute to speciation and reinforcement processes by generating postzygotic barriers. For instance, variation in chromosome numbers has been shown to correlate with speciation rate in Lepidoptera [16]. Similarly, chromosomal inversions may fuel the speciation process: by capturing genetic variations, inversions may lead to increased genetic divergence between species. Such divergence may lead to maladaption in hybrids and further limit gene flow between species living in sympatry.
By relying on both karyotype data and PacBio-Hifi sequencing, we generated de novo genome assemblies for 3 sympatric species of Morpho butterflies. The divergence between the 2 sister species M. helenor and M. achilles was estimated to occur about 3.91 million years (My) ago, while the divergence between these 2 sister species and M. deidamia was estimated to circa 16.68 My ago [17], enabling to compare the genome divergence in sympatry at different time scales. We then investigated the structural variants and variation in genes potentially contributing to prezygotic isolation among these species. We aim to shed light on the genomic processes involved in sympatric speciation and reinforcement as well as detecting chromosomal rearrangements. We also provide their mitochondrial genomes, study their transposable element (TE) contents, and annotate the genomes. These genomic resources will open new research avenues into the understanding of adaptive processes, such as convergence evolution of color pattern or divergence in visual systems, as well as speciation and coexistence of sister species in sympatry, establishing Morpho butterflies as a new eco-evolutionary model.
Material and Methods
Butterfly sampling
Males from the species M. helenor (n = 1), M. achilles (n = 4), and M. deidamia (n = 2) were caught with a handnet at the Patawa waterfall, located in the Kaw mountain area of French Guiana (GPS location: 4.54322; −52.15832) to perform DNA extractions. In these species, males typically patrol in river beds and are easy to catch, while females are more rarely encountered. We therefore focused on males only. Because in butterflies sex is controlled by a ZW sex chromosome system (females being the heterogametic sex), we were thus able to access the Z sex chromosome but not the W chromosome.
Karyotype study
Cytogenetic techniques were applied to wild-caught males (M. helenor, n = 3; M. achilles, n = 4; and M. deidamia, n = 2) that were collected at the abovementioned location in 2019. Their testicles were dissected and processed shortly after capture following the protocol described in [18]. The obtained cell suspension was conserved in fixative at about 4°C. The cell spreading and staining were then performed as described in [18]. The chromosome staining relied on the Giemsa method.
DNA extractions and genome sequencing
Live butterflies (M. helenor, n = 1; M. achilles, n = 4; and M. deidamia, n = 2) captured in 2021 at the same site in French Guiana were killed in the lab and their bodies immediately placed in liquid nitrogen. The DNA extraction was carried out the following day using the Qiagen Genomic-tip 100/G kit and following supplier instructions. The extracted DNA of a single male from each species was used (see Supplementary Fig. S1 for pictures of the wings of the sequenced specimens). Library preparation and sequencing were performed at the GeT-PlaGe core facility (INRAe Toulouse) according to the manufacturer’s instructions, “Procedure and Checklist Preparing HiFi SMRTbell Libraries Using SMRTbell Express Template Prep Kit 2.0.” At each step, DNA was quantified using the Qubit dsDNA HS Assay Kit (Life Technologies). DNA purity was tested using the nanodrop (Thermo Fisher) and size distribution and degradation assessed using the Femto pulse Genomic DNA 165-kb Kit (Agilent). Purification steps were performed using AMPure PB beads (Pacific Biosciences). Next, 15 μg DNA was purified and then sheared at 15 kb (speed 31 and 32) with the Megaruptor3 system (Diagenode). Using the SMRTbell Express Template prep kit 2.0, a single-strand overhang removal, a DNA and END damage repair step was performed on 10 μg of sample. Blunt hairpin adapters were then ligated to the library, which was treated with an exonuclease cocktail to digest unligated DNA fragments. A size selection step using a 10-kb cutoff was performed on the BluePippin Size Selection system (Sage Science) with the “0.75 percent DF Marker S1 6-10 kb vs3 Improved Recovery” protocol. Using Binding kit 2.2 and sequencing kit 2.0, the primer V5 annealed and polymerase 2.2 bounded library was sequenced by diffusion loading onto 1 SMRTcells per sample on a SequelII instrument at 80 pM with a 2-hour preextension and a 30-hour movie.
K-mer analysis, genome size, and heterozygosity estimation
We used Jellyfish (v.2.3.0) [19] to perform a k-mer analysis on each PacBio dataset with a k-mer size of 21. For each dataset, k-mers were counted and aggregated (jellyfish count option) and histograms were generated using the “-histo” command. The resulting histograms allowed the estimation of genome length and heterozygosity with GenomeScope version 2.0 [20] using the web application.
Nuclear and mitochondrial genome assembly
For the assembly of the nuclear genomes, we compared 3 long-read assembly tools: IPA-Improved Phased Assembler (v1.0.3-0) (https://github.com/PacificBiosciences/pbipa), Flye (v2.9) [21], and Hifiasm (v0.16.1 with the option -l3 to purge all types of haplotigs in the most aggressive way) [22]. For each assembler, we estimated basic assembly statistics such as scaffold count, contig count, and N50 using the “stats.sh” program from the BBMap v38.93 package [23]. The completeness of each assembly was assessed using BUSCO v5.2.2 and MetaEuk for gene prediction against the  database [24]. We retained the Hifiasm assembly because it had the highest BUSCO score, the highest contiguity (N50), and the longest contig. Despite the high level of purging performed by Hifiasm, the species (M. helenor and M. achilles, respectively) retained a high level of duplicates in the BUSCO score. To remove false haplotypic duplications in these 2 species, we used Purge_dups v1.2.5, setting the cutoffs manually (with calcuts -l 5 -m 33 -u 135 for M. helenor and calcuts -l 10 -m 45 -u 145 for M. achilles) [25]. The completeness of the purged genomes was then reassessed using BUSCO.
database [24]. We retained the Hifiasm assembly because it had the highest BUSCO score, the highest contiguity (N50), and the longest contig. Despite the high level of purging performed by Hifiasm, the species (M. helenor and M. achilles, respectively) retained a high level of duplicates in the BUSCO score. To remove false haplotypic duplications in these 2 species, we used Purge_dups v1.2.5, setting the cutoffs manually (with calcuts -l 5 -m 33 -u 135 for M. helenor and calcuts -l 10 -m 45 -u 145 for M. achilles) [25]. The completeness of the purged genomes was then reassessed using BUSCO.
The mitochondrial genome of each species was assembled and circularized using Rebaler (https://github.com/rrwick/Rebaler) directly from the PacBio Hifi reads and using the mitochondrial genome of the closely related species Pararge aegeria as a reference.
Annotation of repetitive regions
The annotation of repetitive regions in the 3 species was performed following 2 main steps. First, we used RepeatModeler v2.0.2a [26] with the option -s (slow search) and -a (to get a.align output file) to create de novo libraries of repetitive elements for each species. The library was then used to hardmask the corresponding genome assembly using RepeatMasker 4.1.2.p1 [26]. A summary of the repeated elements was generated with the script “buildSummary.pl” included in RepeatMasker.
Genome annotation
Each of the 3 genomes was independently annotated using MAKER v2.31.10 [27], following the protocol given in [28]. In short, MAKER is usually run several times successively and uses the gene models generated in 1 round to train ab initio gene predictors and improve the initial gene models in the next round (see below). We used the abovementioned hardmasked genomes and carried out their annotation using the proteomes of 3 closely related species, namely, P. aegeria [29], Maniola hyperantus [30], and Bicyclus anynana [31]. For each species, the output files were merged into a gff3 file that was then used to generate the necessary files to train SNAP (version 2006-07-28), an ab initio gene-finding program [32]. A second run of MAKER with the abovementioned gff3 file and the .hmm file provided by SNAP resulted in a second gff3 file that was used to train SNAP a second time. A third round of MAKER with the second gff3 and .hmm files was followed by the training of Augustus (3.3.3), another gene prediction tool [33], with the third gff3 file. A final round of MAKER with the third gff3 file and the files generated by Augustus led to the fourth and last gff3 file, containing all the genome features for each species.
Protein-Protein BLAST 2.9.0+ (-evalue 1e-6 -max_hsps 1 -max_target_seqs 1) was then used to assess putative protein functions in each Morpho species by comparing the protein sequences given by MAKER to the protein sequences from the annotated genomes of Maniola jurtina [29], P. aegeria [29], B. anynana [31], and Spodoptera littoralis specifically for the detection of OR (olfactory receptor) sequences [34]. We used BUSCO to assess the completeness of the proteome with the protein mode and the 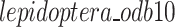 database on the annotated gene set produced by MAKER [24].
database on the annotated gene set produced by MAKER [24].
Phylogenetic analysis
To specifically compare the exon sequences of the opsins detected in the Morpho genomes to the opsins described in other Lepidoptera, we retrieved the coding sequences of opsins from NCBI and used the software Mega v.11 [35] to build a maximum likelihood tree and compute the associated bootstrap values.
Regarding the OR repertoire in the 3 Morpho species, we curated the sequences obtained by blast comparison of the MAKER-annotated genes on the reference genome of S. littoralis, as a number of sequences showed incorrect lengths (<300 or >500 amino acids). We used exonerate version 2.4.0 [36] with the options -maxintron 2000 independently in each Morpho species. The exonerate alignment files and the assemblies were used with InsectOR (http://caps.ncbs.res.in/gws_ors/), a website specifically designed to help predict OR genes from insect genomes, with the option HMMSEARCH against 7tm_6 [37, 38]. The sequences uncovered with insectOR for each Morpho species were aligned with the ORs of S. littoralis using MAFFT [39], and we generated a maximum likelihood phylogenetic tree with IQ-TREE version 2.2.0 [40] with the options -bb 1000 and -nt AUTO.
Synteny and rearrangement detection
To assess variation in chromosome-scale synteny, we compared the assemblies of each Morpho to the assembly of M. jurtina, the closest relative of Morpho with a karyotype of 29 chromosomes and for which a high-quality chromosome-level assembly (based on N50 values and BUSCO score, accession ID GCF_905333055.1) is available [29]. We used MUMmer 3.23 [41] to align the masked assembled genomes of M. helenor, M. achilles, and M. deidamia to the M. jurtina genome. The output produced by MUMmer is an ASCII delta file that was then filtered and parsed using the utility programs delta-filter and show-coords from MUMmer. Synteny was visualized with the MUMmer results in R with the packages circlize v 0.4.12 [42] and Paletteer (https://github.com/EmilHvitfeldt/paletteer) using the Rscript from [43] described here: https://github.com/bioinfowheat/Polygonia_calbum_genomics/blob/7c75aac624157faa3ab229e3fc1e0e315302194d/synteny/circlePlot_nucmerOutput.R, removing short contigs, short alignments (less than 200 bp), and low-identity alignments (less than 90% identity).
In order to detect potential genome rearrangements between Morpho and closely related species, we estimated the whole-genome collinearity between the Morpho assemblies and 5 closely related Nymphalidae species whose genomes exhibit good-quality assemblies in the NCBI genome database: M. jurtina (GCA_905333055.1), P. aegeria (GCA_905333055.1), Erebia ligea (GCA_923060345.2), Melanargia galathea (GCA_920104075.1), and Lasiommata megera (GCA_928268935.1) using D-GENIES [44]. Paired alignments between a Morpho species and 1 Nymphalidae species were performed using the minimap2 aligner [45] in D-GENIES, treating each Morpho species genome as the query and the Nymphalidae species genome as the target reference. We also used D-GENIES to pair-compare the genomes of the 3 Morpho species. As D-GENIES revealed differences between Morpho species in the contig corresponding to the Z chromosome (see Results), we used SyRI [46] to study in detail the rearrangements in the sequences of this contig between the 3 species. We generated paired alignments of the Z contig with minimap2 and ran SyRI with the option -c on.sam files. SyRI requires that the 2 compared genomes represent the same strand, and in the case of M. achilles, the orientation of the sequence produced by Hifiasm was complementary to the sequences of M. helenor and M. deidamia. We then reverse-complemented this sequence in order to make the alignments. All the genomic structures predicted by SyRI were plotted using plotsr [47].
Results
Comparing karyotypes between species
First, we characterized the karyotypes of the 3 studied species (see Supplementary Fig. S2 to visualize the chromosomes). In M. helenor, the detected number of diploid chromosomes ranged from 54 to 56 in the different replicates of mitoses, with a discreet mode at 2n = 56. This variation is probably due to technical difficulties. The presence of n = 28 bivalents in metaphase confirmed the diploid number of 2n = 56 chromosomes. In M. achilles, 4 specimens had the same modal chromosome counts: mitoses, 2n = 56 chromosomes; pachynema, n = 28 bivalents; metaphase I, n = 28 bivalents; and metaphase II, n = 28 chromosomes with 2 chromatids. Surprisingly, the karyotype of the last male was quite different, with a modal number of 84 mitotic chromosomes. Interestingly, there was the same number (n = 28) of elements as above at the pachynema stage, indicating that they were trivalents. They were thicker than bivalents, and a more careful analysis showed the recurrent asynapsis of 1 of the 3 chromosomes (Supplementary Fig. S3). No “normal” metaphase I or II was observed. It was concluded that this specimen was triploid with 3n = 84 and probably sterile. In M. deidamia, the diploid chromosome number had a discreet mode of 2n = 54, suggesting a slightly smaller number of chromosome pairs (n = 27) in this more distantly related species. Our result is consistent with the modal number of chromosomes in the Morphinae (n = 28) described in previous karyotypic studies conducted in 8 Morpho species [48], where the reported number of chromosomes was also n = 28 for both M. helenor and M. achilles.
GenomeScope analyses suggested very high levels of heterozygosity for the 3 species (Table 1). In all of them, the N50 and contig sizes were generally larger in the assemblies produced by Hifiasm than in IPA and Flye assemblies (see Supplementary Table S1). The BUSCO scores revealed a very high percentage of duplicated sequences, especially in the assemblies produced by IPA and Flye. The use of purge_dups strongly reduced the number of duplicates, the estimated size of the genome, and the number of final contigs (see Supplementary Fig. S4 and Supplementary Table S1). Hifiasm and the posttreatment with Purge_dups v1.2.5 gave an assembly of 143 contigs for M. helenor (size of the longest contig: 42,411,663 bp), of 32 contigs for M. achilles (size of the longest contig: 24,854,087 bp), and of 58 contigs for M. deidamia (size of the longest contig: 22,518,629 bp) (Supplementary Table S1). The Rebaler pipeline identified a circular mitochondrial genome of 15,336 bp for the species M. helenor, 15,340 bp for M. achilles, and 15,196 bp for M. deidamia.
Table 1:
Genome heterozygosity estimated with GenomeScope and Genome statistics for the assemblies of 3 Morpho species using different computational methods. Assemblies were purged using purge_dups. Statistics were obtained with BBMap. The assembly produced with Hifiasm for the individual M. deidamia was not purged with purge_dups as BUSCO results on the preliminary assembly revealed a very low duplicate content.
| M. helenor | M. achilles | M. deidamia | |
|---|---|---|---|
| Heterozygosity (%) | 3.35 | 2.78 | 1.68 |
| Assembly method | |||
| Hifiasm | |||
| Total contigs | 143 | 32 | 58 |
| Genome size | 470.254 Mb | 478.514 Mb | 489.914 Mb |
| N50 | 12 Mb | 12 Mb | 13 Mb |
| IPA | |||
| Total contigs | 128 | 56 | 47 |
| Genome size | 473.620 Mb | 493.177 Mb | 481.177 Mb |
| N50 | 17 Mb | 14 Mb | 13 Mb |
| Flye | |||
| Total contigs | 134 | 114 | 291 |
| Genome size | 466.515 Mb | 477.638 Mb | 484.463 Mb |
| N50 | 21 Mb | 20 Mb | 34 Mb |
Annotation of repetitive region
In each of the 3 species of Morpho, we annotated around 50% of the genome as repeated elements (Supplementary Fig. S5). In M. helenor, 241,166,073 bp (51.28% of the genome) corresponded to repeated elements, 261,488,514 bp (54.65% of the genome) in M. achilles, and 255,779,512 bp (52.75% of the genome) in M. deidamia. The repetitive elements categories are shown in Supplementary Fig. S5. For the 3 species, long interspersed nuclear elements (LINEs) accounted for the largest percentage (between 13.53% and 17.22% ) of the repeated elements in the genomes.
Genome annotation
We recovered 12,651, 12,978, and 12,093 protein-coding genes in the genomes of M. helenor, M. achilles, and M. deidamia, respectively. These values were comparable to what was found in Maniola hyperantus (13,005 protein-coding genes) and P. aegeria (13,515 protein-coding genes) but were lower than in M. jurtina (13,777 protein-coding genes) and B. anynana (14,413 protein-coding genes). BUSCO results for the proteome and transcriptome are presented as supplementary material (see Supplementary Figs. S5 and S6). In order to assess if the annotations were complete, we estimated in each species the percentage of proteins with a Pfam domain as this value has been found to vary between 57% and 75% in eukaryotes [49]. This value ranged from 65.50% in M. achilles to 71.32% in M. helenor with an intermediate value of 70.42% in M. deidamia, thus showing that the annotations were of good quality. Proteome completeness using BUSCO was also high. From a set of 5,286 single-copy orthologs from the Lepidoptera lineage, the proteome completeness varied between 69% and 79% depending on the species (Supplementary Fig. S5). We were thus able to further investigate gene families that could be involved in prezygotic isolation through duplication or loss events. This includes genes having a role in vision (L opsin) but also chemosensory genes such as odorant and gustatory receptors that reflect the degree of species specialization.
Duplications in opsin genes
Vision in butterflies notably relies on opsins, for which 3 major types of molecules have been described depending on their wavelength of peak absorbance: in the ultraviolet (UV, 300–400 nm), blue (B, 400–500 nm), and long-wavelength (L, 500–600 nm) part of the visible spectrum. Opsins are encoded by UV, B, and L opsin genes. We investigated the number of copies for each opsin gene in the 3 Morpho species. We consistently found 1 copy of the UV opsin gene and 2 copies of the B opsin genes in the 3 Morpho species. Duplications of L opsin were observed in M. achilles, M. deidamia, and M. helenor. In the other reference genomes, M. jurtina, B. anynana, and P. aegeria, a single copy of the UV opsin gene, the B opsin gene, and the L opsin gene was found. By comparing the L opsin sequences using a maximum likelihood tree based on the exon sequences (Fig. 2), we showed that the duplications observed in Morpho butterflies probably occurred independently from previously described duplications that happened in other clades of Lepidoptera. The phylogenetic relationships between the copies in the 3 species reveal that the duplications observed in the 3 Morpho species probably occurred before their speciation (Fig. 2). The detection of the different copies in different species within the Morpho genus and in closely related genus is now required to precisely characterize the evolutionary origin of these duplications.
Figure 2:

Maximum likelihood tree of L-opsin exon sequences detected in the genomes of M. helenor, M. achilles, and M. deidamia and other butterflies species, with bootstrap values. The colored dots indicate the putative locations of the duplication events on the tree: the putative origin of duplications of the L-opsin observed within the genus Morpho appear in blue, while the duplications that occurred in the Hermeuptychia hermes clade and in the Papilio clade appear in yellow and orange, respectively.
Odorant and gustatory receptors
In order to estimate the number of OR and GR genes in the 3 Morpho species, we blasted our MAKER-annotated genes on the reference genome of S. littoralis. In this moth species, 60 OR and 16 GR genes were curated [50]. Interestingly, we recovered only 31 OR genes including Orco in M. helenor, 32 in M. achilles, and 36 in M. deidamia, while we found 14 GR genes in M. helenor and 16 in M. achilles and M. deidamia. With insectOR, we found 36 OR genes including Orco in M. helenor, 37 in M. achilles, and 38 in M. deidamia, confirming the major loss of ORs in our 3 Morpho species. For comparison, we blasted against the same reference genome of S. littoralis the annotated sequences of the 3 outer Lepidopteran species used in the previous analyses and uncovered a much higher number of OR and GR genes with 61 OR and 28 GR in M. jurtina, 60 OR and 35 GR in B. anynana, and 50 OR and 20 GR in P. aegeria, respectively. The drastic reduction of chemosensory receptors, particularly in the number of OR genes in the 3 Morpho species, could potentially reflect a higher degree of specialization to their respective biochemical environment. A phylogenetic analysis of Morpho ORs along with those of S. littoralis, the sole Lepidopteran species for which a considerable number of ORs were functionally deorphanized and divided into 3 chemical classes (aromatics, terpenes, and aliphatics) as described in [34], showed that the loss of ORs in Morpho was not clustered around a particular set of genes (Supplementary Fig. S9). Further functional characterization coupled with precise ecological investigations are therefore needed to understand the loss of ORs in the Morpho genus.
Synteny and rearrangement detection
Conserved synteny with other Lepidoptera species
We found a high concordance between the n = 29 chromosomes of M. jurtina and the contigs of the 3 Morpho species (Fig. 3). The MUMmer alignment and the postalignment treatment to remove short contigs and low-identity alignments reduced the assembly to 27 contigs containing 97% of the total genome for M. helenor (removing 117 short contigs from the original assembly), 29 contigs (98% of the genome) for M. achilles (3 contigs removed), and 27 for M. deidamia (31 contigs removed) (Fig. 3).
Figure 3:

Synteny between the chromosome-assembled genome of M. jurtina (colored chromosomes) and the genome assemblies of the species M. helenor (A), M. achilles (B), and M. deidamia (C). Equivalent chromosomes/contigs are linked by same color ribbons. Chromosome Z for each species is labeled in red. Single chromosomes in Morpho that are not assigned to a single chromosome in M. jurtina are labeled in blue.
The synteny plot between M. helenor and M. jurtina showed 27 contigs for M. helenor, 1 contig less than expected based on its karyotype of n = 28. In the plot, 1 single contig (ptg000028l) was assigned to 2 different chromosomes from the M. jurtina assembly (chromosomes 2 and 6, NC_060030.1 and NC_060034.1). Contig ptg000028l is twice the size of any other contig found in the 3 Morpho species analyzed here. Based on the differences between the number of contigs and the karyotype and the unexpectedly big size of the contig ptg000028l, we believe the difference in chromosome number between M. jurtina and M. helenor can be explained by an overassembly of the genome of M. helenor by Hifiasm, which assigned 1 single contig to 2 different chromosomes from the M. jurtina assembly (Fig. 3). In M. deidamia, the Hifiasm assembly showed a single contig, ptg000008l (size 20.29 Mb), containing chromosomes NC_060051.1 and NC_060052.1 (sizes 10.05 Mb and 9.43 Mb, respectively) from M. jurtina. Because the number of contigs recovered for this species is in accordance with the karyotype of n = 27, the differences between M. deidamia and M. jurtina suggest that in this case, chromosomes NC_060051.1 and NC_060052.1 in M. jurtina may have fused to form contig ptg000008l in M. deidamia. Other rearrangement in this species compared to M. jurtina seems to be the contig ptg0000161l, which appears to contain small portions of chromosomes NC_060054.1 and NC_060055.1 from M. jurtina.
For the 3 Morpho species, we were able to identify a single contig corresponding to the chromosome Z (NC_060058.1) in M. jurtina (contig ptg000030l in M. helenor, contig ptg000024l in M. achilles, and contig ptg000019l in M. deidamia).
We also found a high level of collinearity between the genomes of the 3 Morpho species and the 5 Nymphalidae species used for comparisons. The alignment between M. jurtina and the 3 Morpho species (Fig. 3) was very similar to the alignments obtained for the other Nymphalidae (Supplementary Fig. S6) and confirmed that the assembly of the genome of M. helenor by Hifiasm might have merged together 2 chromsomes: the single contig ptg000028l was scattered into 2 chromosomes in the other Nymphalidae. Although collinearity was generally high, we detected some putative inversions located in regions that varied among pairs for the 3 Morpho species in comparison with the Nymphalidae (see Supplementary Fig. S6). Interestingly, the contig corresponding to the chromosome Z was the only one consistently showing inversions in the pairwise genome-wide alignments (see Supplementary Fig. S6).
Inversions in the Z chromosome between the 3 sympatric Morpho species
Pairwise whole-genome alignments of the 3 Morpho species showed a very high similarity between genomes (see Supplementary Fig. S7). The only contig that differed between species was the one corresponding to the Z chromosome. SyRI identified 1 inversion of 1.6 Mb between M. helenor and M. deidamia, 5 inversions (comprising 1 of more than 1.8 Mb) between M. helenor and M. achilles, and 2 between M. deidamia and M. achilles with 1 of 1.6 Mb (Fig. 4). Interestingly, the inversion found in M. deidamia when compared to M. achilles or M. helenor has the same size and is located in exactly the same position of the chromosome (from bp 1567583 to 3192401), suggesting that this inversion is ancestral to the speciation of M. achilles and M. helenor. In the case of M. achilles versus M. helenor, 2 inversions were found flanking the site of the putative ancient inversion, and a bigger inversion was found at the end of the chromosome (Fig. 4).
Figure 4:
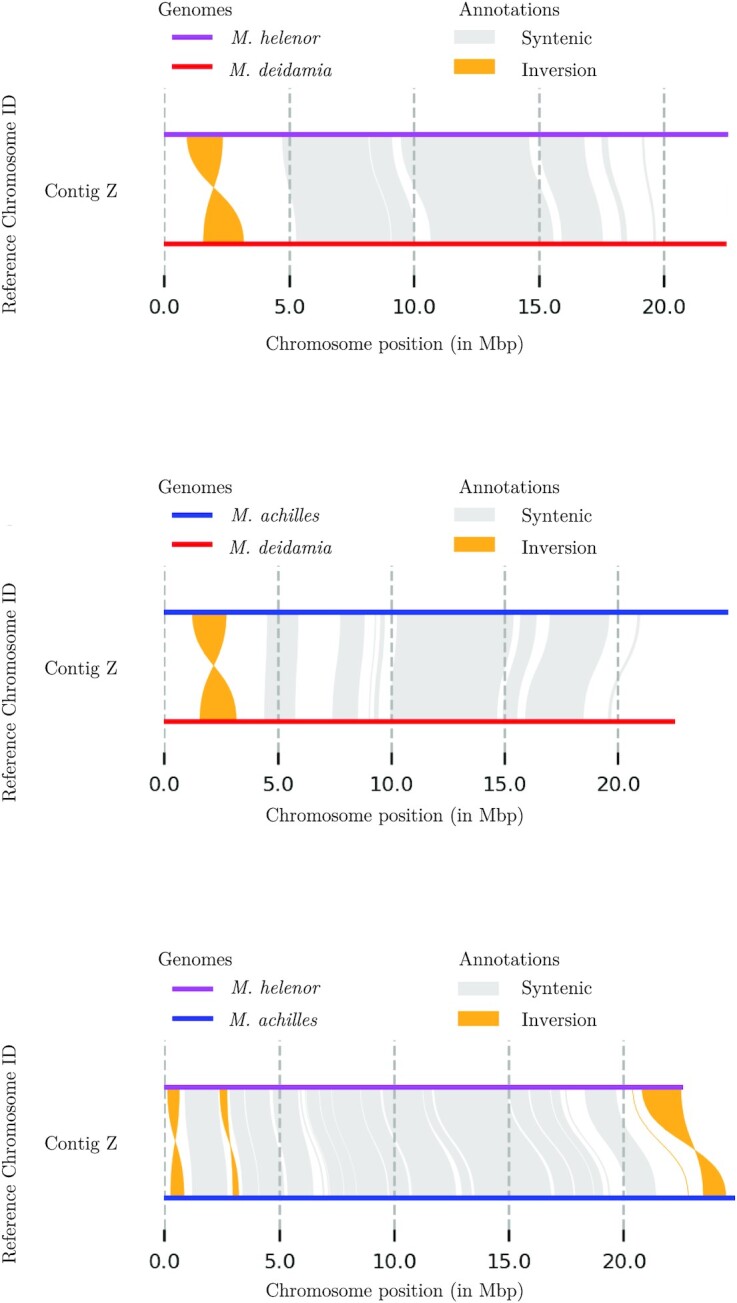
Synteny and rearrangement (SyRI) plot of the paired comparisons for the Z contig between the 3 Morpho species. Upper figure: M. helenor and M. deidamia; middle: M. deidamia and M. achilles; lower: M. helenor and M. achilles.
Discussion
Assembly of heterozygous Lepidoptera genomes with a high proportion of repeated elements
We generated de novo, reference-quality genome assemblies for 3 emblematic species of Amazonian butterflies: M. helenor, M. achilles, and M. deidamia. Our results indicate genome sizes comprised between 470 Mb and 489 Mb, similar to most of the closely related Nymphalidae species sequenced so far, for example, B. anynana (475 Mb), P. aegeria (479 Mb), or M. jurtina (429 Mb). This is also close to the 479 Mb estimated from phylogenetic comparison using the taxon-centered database “Genomes on a Tree” (GoaT) [51]. The final number of contigs within each of the 3 species ranged from 27 to 29, close to the number of chromosome pairs observed in our cytogenetics study. The number of chromosomes found in those French Guiana samples (i.e., in the subspecies M. helenor helenor and M. achilles achilles) is consistent with those found in other subspecies of both species in previous studies [48]. The available sequenced species of Nymphalidae that are closely related to the genus Morpho also generally show 29 pairs of chromosomes (28 autosomes, plus Z and W sex chromosomes), which is close to the chromosomal numbers observed in the 3 Morpho species studied here. The mapping between the assemblies of Morpho species to the chromosome-level assembly of M. jurtina and the posttreatment to eliminate small contigs allowed us to identify between 27 and 29 contigs in Morpho that were homologous to M. jurtina chromosomes, including the contig corresponding to the Z chromosome. This suggests a high conservation of chromosomal synteny among closely related Nymphalidae species, which is consistent with the high level of synteny observed throughout the whole Lepidoptera clade [52]. In the 3 species, genome heterozygosity was very high (from 1.68% in M. deidamia to 3.35% in M. helenor), and heterozygosity presents a major challenge in de novo assembly of diploid genomes. Indeed, levels of heterozygosity of 1% or above are considered “moderate to high,” and most assemblers struggle when 2 divergent haplotypes are sequenced together, as heterozygosity may impair the distinction of different alleles at the same locus from paralogs at different loci [53]. Then, final assemblies of heterozygous genomes are expected to be of poor quality, highly fragmented, and containing redundant contigs [54]. Hifiasm generated the most completely haplotype-resolved assemblies; nevertheless, the level of heterozygosity clearly impacted the quality of the assemblies, and a posttreatment to remove duplicated sequences was necessary for the 2 most heterozygous genomes (M. helenor and M. achilles), showing the difficulty that heterozygosity still imposes on long-read heterozygosity-aware assemblers. Such a high heterozygosity has been observed in other genomes of Lepidoptera [31] and can be a signature of high effective population sizes. The wide Amazonian distribution of these species and their flight activity could contribute to such a high level of genetic diversity within the population, because elevated dispersal contributes to increased gene flow within each species throughout their geographic range. Our results also showed that around 50% of the genomes of the sequenced Morpho were composed of repeated elements, a very high proportion as compared to other genomes of Lepidoptera. In Lepidoptera, TE content has been found to be correlated with genome size [55], but in the case of the 3 Morpho species studied here, the repeat content is higher than for other species with similar genome sizes such as the Bombyx mori moth, with a genome size estimated at 530 Mb and a TE content of 35% [56] or the more closely related species B. anynana with a genome size of 475 Mb and a repeat content of 26% [31].
Structural variations between genomes of sympatric species
The karyotype and assembly analyses suggest some differences in chromosome number between the 3 sympatric Morpho species studied here, particularly between M. deidamia (27 chromosome pairs) versus M. helenor and M. achilles (28 chromosome pairs). Differences in chromosome numbers and other chromosomal rearrangements may strongly affect reproductive barriers. Two groups of models have been proposed to explain how chromosomal rearrangements prevent gene flow and contribute to species maintenance and speciation. First, hybrid-sterility models suggest reduced fertility or viability in individuals heterozygous for chromosomal rearrangements. These models are considered inconsistent and difficult to evaluate [4]. More recently, suppressed-recombination models propose that chromosomal rearrangements permit speciation in sympatry because they reduce recombination between chromosomes carrying different rearrangements [4]. Indeed, in Lepidoptera, differences in chromosome number are proposed to be an important mechanism leading to species diversification in Agrodiaetus, Erebia, and Lysandra butterflies [57–59].
Besides differences in chromosome numbers, we systematically found inversions in the contig corresponding to the Z chromosome when comparing the genomes of Morpho to the other Nymphalidae and between the 3 different Morpho species. Inversions are also a type of chromosomal rearrangement known to occur throughout evolution and are considered an important mechanism for speciation, particularly for species living in sympatry [1, 4]. Empirically and theoretically, it has been suggested that inversions may have contributed to speciation in sympatry in different groups of animals. In 2 ascidian species of the genus Ciona and in insects like Drosophila, inversions may promote speciation by reduction of the fitness or by causing sterility of heterozygotes [60, 61]. In the Anopheles gambiae species complex, inversions may allow for ecotypic differentiation and niche partitioning, leading to different sympatric and genetically isolated populations [62]. In groups like paserine birds where sexual differentiation is controlled by a ZW sex chromosome system (females being the heterogametic sex), inversions in the Z chromosome in particular seem to explain speciation in sympatry between close species. Cytological data show that across the Passeriformes, the Z chromosome has accumulated more inversions than any other autosome and that the inversion fixation rate on the Z chromosome is 1.4 times greater than the average autosome. Interestingly, inversions on the Z chromosome are significantly more common in sympatric than in allopatric closely related clades [63, 64].
In Lepidoptera, the role of inversions in speciation in sympatry has been studied in the species Heliconius melpomene and Heliconius cydno, 2 sympatric species that can hybridize (although rarely) in the wild [65]. The analyses of the genomic differences between the 2 species showed some small inversions (less than 50 kb), and there was no evidence for a reduction of recombination in hybrids, suggesting that in this case, inversions were not involved in the maintenance of the species barriers, and other processes such as strong mate preference could prevent hybridization in the wild [65]. In the Morpho species studied here, however, we found inversions between Morpho Z chromosomes that were longer than 1.5 Mb. Models suggest that to be associated with adaptive traits or species barriers, inversions should typically be megabases long in order to be fixed in populations [65]. The position of the inversion in the Z contig when comparing M. helenor or M. achilles to M. deidamia is at the exact same place in M. deidamia’s genome, suggesting that this specific inversion likely occurred before the speciation between M. achilles and M. helenor. When comparing M. helenor to M. achilles, we found 2 different smaller inversions that are not found in M. deidamia and that are close to the putative ancestral inversion region, suggesting that these 2 smaller inversions could have appeared after the speciation between M. achilles and M. helenor. At the moment, we do not know if the inversions segregate at different frequencies in the Morpho populations or if they are fixed. Population analyses are needed to answer this question and to enlighten what evolutionary forces could be acting to maintain them. The copy number variation detected in genes involved in color perception (i.e., L opsin) may also play a significant role in reproductive isolation in these sympatric species. For instance, the 3 copies of L opsins found in the Papilio genus (Fig. 2) have been found to also show subfunctionalization and neofunctionalization [66]. The duplication followed by genetic divergence observed in these 3 mimetic Morpho species may improve their visual discrimination capacities and facilitate species recognition, therefore reinforcing barriers to gene flow in sympatry. Genes potentially involved in color pattern variations (e.g., bric-a-brac or bab) may also play a role in prezygotic isolation, but they were not thoroughly investigated here as their functional evolution involves changes in regulatory sequences rather than events of duplication or gene loss [67]. Interestingly, an orthologous search of the putative proteic sequences of each Morpho species against those of M. jurtina allowed us to uncover different copy numbers of the gene bric-a-brac, which play a significant role in differences of UV iridescence between males of 2 incipient species of sulfur butterflies [68]. The copy responsible for the presence/absence of UV iridescence is located on the Z chromosome, and in the 3 Morpho species, we found 1 or more copies of bric-a-brac on the contigs that correspond to the Z chromosome: M. deidamia had 1 copy of bric-a-brac, while M. helenor and M. achilles displayed 2 copies of this gene. It seems, however, that the second copy in M. helenor and M. achilles corresponds to truncated copies of bric-a-brac. While this is certainly the sign of an ancient duplication followed by a pseudogenization event, this could lead to further investigations of putative functions of the truncated copies. It is worth noting that variations in the number of bab copies were also observed in the 3 reference genomes used for the blast: M. jurtina had 2 copies on the Z chromosome (including a truncated copy), B. anynana had only 1 copy, and P. aegeria had none. The investigation of gene levels of polymorphism on the Z chromosomes would also be of great interest as genes linked to the Z chromosome are often among the most divergent between closely related species [69].
Altogether, the assembly and annotation of these 3 mimetic species of Morpho butterflies reveal differences in chromosome numbers, the presence of several Mb-long inversions in the Z chromosome, and copy number variation and genetic divergence among copies of genes that may play a significant role in reproductive isolation. Our study thus open new avenues into the investigation of the ecological and genomic factors involved in sympatric speciation and its reinforcement.
Supplementary Material
Charlotte Julie Wright, MPhil -- 12/8/2022 Reviewed
Niklas Wahlberg -- 12/15/2022 Reviewed
Acknowledgement
The authors thank Patrick Blandin for his continuous support on their Morpho studies; Mélanie McClure, Mathieu Chouteau, Camille Le Roy, and Ombeline Sculfort for their help during fieldwork in French Guiana; Elise Gay, Romuald Laso-Jadart, Pierre Lesturgie, Christelle Fraisse, Clément Gilbert, and Quentin Rougemont for help with some scripts and for discussions of previous versions of the manuscript; Charlotte J. Wright and Niklas Wahlberg for their careful reading of our manuscript and their many insightful comments and suggestions; and the Plateforme de Calcul Intensif et Algorithmique PCIA, Muséum national d’histoire naturelle, Centre national de la recherche scientifique, UAR 2700 2AD, CP 26, and the Genotoul bioinformatics platform Toulouse Occitanie (Bioinfo Genotoul, https://doi.org/10.15454/1.5572369328961167E12).
Contributor Information
Héloïse Bastide, IDEEV, Bât. 680,12, 91190 Gif Sur Yvette, France.
Manuela López-Villavicencio, Institut de Systématique, Evolution et Biodiversité (UMR 7205 CNRS/MNHN/SU/EPHE/UA), Muséum National d’Histoire Naturelle–CP50, 75005 Paris, France.
David Ogereau, IDEEV, Bât. 680,12, 91190 Gif Sur Yvette, France.
Joanna Lledo, GeT-PlaGe, Bât G2, INRAe, 31326 Castanet-Tolosan Cedex, France.
Anne-Marie Dutrillaux, Institut de Systématique, Evolution et Biodiversité (UMR 7205 CNRS/MNHN/SU/EPHE/UA), Muséum National d’Histoire Naturelle–CP50, 75005 Paris, France.
Vincent Debat, Institut de Systématique, Evolution et Biodiversité (UMR 7205 CNRS/MNHN/SU/EPHE/UA), Muséum National d’Histoire Naturelle–CP50, 75005 Paris, France.
Violaine Llaurens, Institut de Systématique, Evolution et Biodiversité (UMR 7205 CNRS/MNHN/SU/EPHE/UA), Muséum National d’Histoire Naturelle–CP50, 75005 Paris, France.
Data Availability
Fastq files, genome assemblies, assembly methods, and collection information were uploaded at the ENA website (https://www.ebi.ac.uk/ena/browser/home) under the project number PRJEB56642. Genome accession numbers are ERZ14213098 for M. helenor, ERZ14213099 for M. achilles, and ERZ14213100 for M. deidamia. Other data further supporting this work are openly available in the GigaScience repository, GigaDB [70–72].
Additional Files
Supplementary Table S1. Genome statistics for the assemblies of 3 Morpho species before the use of Purge_dups and using different assemblers. Statistics were obtained with BBMap.
Supplementary Fig. S1. Pictures of dorsal and ventral sides of the wings of the sequenced specimens of M. helenor, M. achilles, and M. deidamia sampled in French Guiana.
Supplementary Fig. S2. Pictures of caryotypes obtained from specimens of M. helenor, M. achilles, and M. deidamia sampled in French Guiana. Note that the chromosome number differs between M. deidamia and the other 2 species.
Supplementary Fig. S3. Picture of the putative triploid caryotype obtained from a specimen of M. achilles. There was the same number (n = 28) of elements as in the other M. achilles individuals at the pachynema stage. However, these elements were thicker than bivalents, and a more careful analysis showed the recurrent asynapsis of 1 of the 3 chromosomes (see arrows).
Supplementary Fig. S4. BUSCO assessment of the genome assembly completeness of Morpho species assembled using IPA, Flye, and Hifiasm. Results correspond to the BUSCO score before (top) and after (bottom) the use of purge_dups.
Supplementary Fig. S5. BUSCO analysis of gene annotation of Morpho species. BUSCO was run in “protein” mode on the annotated gene set produced by MAKER.
Supplementary Fig. S6. Proportion of each class of transposable elements (TEs) in the assembled genomes of the 3 Morpho species.
Supplementary Fig. S7. D-GENIES dot plots comparing the genome assemblies of 3 Morpho species against the assembly of the species M. jurtina. Chromosomes of M. jurtina are on the x-axis while the y-axis shows the scaffolds of (upper) M. helenor, (middle) M. achilles, and (bottom) M. deidamia. The alignment between the Z chromosome of M. jurtina and Morpho corresponds to the square in the upper right corner. Here we show only the plot between Morpho and M. jurtina; comparisons with 4 other species of Nymphalidae are available upon request.
Supplementary Fig. S8. D-GENIES dot plots comparing the genome assemblies of pairs of Morpho genomes. The red arrows indicate the inversions (in the scaffolds corresponding to the Z chromosome).
Supplementary Fig. S9. Phylogenetic tree by maximum likelihood of ORs built from amino acid sequences of M. helenor “MhelOR” (red), M. achilles “MachOR” (green), M. deidamia “MdeiOR” (blue), and S. littoralis “SlitOR.” The latest were colored following their chemical class (magenta: aromatics; cyan: terpenes; orange: aliphatics; black: unclassified) as defined in [34].
Authors’ Contributions
V.L. and V.D. collected the butterflies in the wild. A.M.D. performed the karyotype analyses. V.L. and D.O. extracted the DNA. J.L. performed the PacBio sequencing. M.L.V. and D.O. performed the genome assembly. M.L.V. performed the TE analyses. M.L.V. and H.B. performed the structural variation detection. H.B. performed the genome annotations. V.L. supervised the whole project. All authors contributed to the writing of the manuscript.
Competing interests
The authors do not declare any competing interests.
Ethical Approval
All butterflies used in this study were sampled in French Guiana and used by researchers from French institutions. Following the recommendation of the Nagoya protocol, our results were presented to the Université Antilles/Guyane in French Guiana during a seminar by V.L. V.L. and V.D. also produced a popularization movie and performed several talks in public school from French Guiana, ensuring feedback from our research on natural resources toward local populations.
Funding
This work was funded by 3 programs provided to V.L. as a principal investigator: the MITI program from the CNRS, the ATM program from the Museum National d’Histoire Naturelle (Paris, France), and the ANR-T-ERC-OUTOFTHEBLUE from the French National Research Agency.
References
- 1. Kirkpatrick M, Barton N. Chromosome inversions, local adaptation and speciation. Genetics. 2006;173(1):419–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. De-Kayne R, Selz OM, Marques DA et al. Genomic architecture of adaptive radiation and hybridization in Alpine whitefish. Nat Commun. 2022;13(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoffmann AA, Sgrò CM, Weeks AR. Chromosomal inversion polymorphisms and adaptation. Trends Ecol Evol. 2004;19(9):482–8. [DOI] [PubMed] [Google Scholar]
- 4. Faria R, Navarro A. Chromosomal speciation revisited: rearranging theory with pieces of evidence. Trends Ecol Evol. 2010;25(11):660–9. [DOI] [PubMed] [Google Scholar]
- 5. Mérot C, Oomen RA, Tigano A, et al. A roadmap for understanding the evolutionary significance of structural genomic variation. Trends Ecol Evol. 2020;35(7):561–72. [DOI] [PubMed] [Google Scholar]
- 6. Blandin P, Purser B. Evolution and diversification of neotropical butterflies: insights from the biogeography and phylogeny of the genus Morpho Fabricius, 1807 (Nymphalidae: Morphinae), with a review of the geodynamics of South America. Trop Lepidoptera Res. 2013; 23: 62–85. [Google Scholar]
- 7. Allio R, Nabholz B, Wanke S et al. Genome-wide macroevolutionary signatures of key innovations in butterflies colonizing new host plants. Nat Commun. 2021;12(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Briscoe AD, Macias-Munoz A, Kozak KM, et al. Female behaviour drives expression and evolution of gustatory receptors in butterflies. PLoS Genet. 2013;9(7):e1003620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Naisbit RE, Jiggins CD, Mallet J. Disruptive sexual selection against hybrids contributes to speciation between Heliconius cydno and Heliconius melpomene. Proc R Soc Lond Ser B Biol Sci. 2001;268(1478):1849–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smadja C, Butlin R. On the scent of speciation: the chemosensory system and its role in premating isolation. Heredity. 2009;102(1):77–97. [DOI] [PubMed] [Google Scholar]
- 11. Debat V, Berthier S, Blandin P et al. Why are Morpho blue?In: Biodiversity and Evolution. Elsevier: London SW19 4EU UK.2018;139–74. [Google Scholar]
- 12. Pinheiro C, Freitas A, Campos V, et al. Both palatable and unpalatable butterflies use bright colors to signal difficulty of capture to predators. Neotrop Entomol. 2016;45(2):107–13. [DOI] [PubMed] [Google Scholar]
- 13. Llaurens V, Le Poul Y, Puissant A et al. Convergence in sympatry: evolution of blue-banded wing pattern in Morpho butterflies. J Evol Biol. 2021;34(2):284–95. [DOI] [PubMed] [Google Scholar]
- 14. Boussens-Dumon G, Llaurens V. Sex, competition and mimicry: an eco-evolutionary model reveals unexpected impacts of ecological interactions on the evolution of phenotypes in sympatry. Oikos. 2021;130(11):2028–39. [Google Scholar]
- 15. Le Roy C, Roux C, Authier E et al. Convergent morphology and divergent phenology promote the coexistence of Morpho butterfly species. Nat Commun. 2021;12(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Vos JM, Augustijnen H, Bätscher L, et al. Speciation through chromosomal fusion and fission in Lepidoptera. Phil Trans R Soc B. 2020;375(1806):20190539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chazot N, Blandin P, Debat V, et al. Punctuational ecological changes rather than global factors drive species diversification and the evolution of wing phenotypes in Morpho butterflies. J Evol Biol. 2021;34(10):1592–607. [DOI] [PubMed] [Google Scholar]
- 18. McClure M, Dutrillaux B, Dutrillaux AM, et al. Heterozygosity and chain multivalents during meiosis illustrate ongoing evolution as a result of multiple holokinetic chromosome fusions in the genus Melinaea (Lepidoptera, Nymphalidae). Cytogenet Genome Res. 2017;153(4):213–22. [DOI] [PubMed] [Google Scholar]
- 19. Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27(6):764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ranallo-Benavidez TR, Jaron KS, Schatz MC. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 2020;11(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kolmogorov M, Yuan J, Lin Y et al. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37(5):540+. [DOI] [PubMed] [Google Scholar]
- 22. Cheng H, Concepcion GT, Feng X et al. Haplotype-resolved de novo assembly using phased assembly graphs with Hifiasm. Nat Methods. 2021;18(2):170+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bushnell B. BBMap: a fast, accurate, splice-aware aligner 2014. https://www osti gov/se rvlets/purl/1241166 [Last accessed 12 June 2020].
- 24. Manni M, Berkeley MR, Seppey M et al. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol. 2021;38(10):4647–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guan D, McCarthy SA, Wood J, et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 2020;36(9):2896–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Flynn JM, Hubley R, Goubert C et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci U S A. 2020;117(17):9451–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cantarel BL, Korf I, Robb SMC et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18(1):188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Muller H, Ogereau D, Da Lage JL, et al. Draft nuclear genome and complete mitogenome of the Mediterranean corn borer, Sesamia nonagrioides, a major pest of maize. G3 (Bethesda). 2021;11(7):1–8. 10.1093/g3journal/jkab155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ellis EA, Storer CG, Kawahara AY. De novo genome assemblies of butterflies. Gigascience. 2021;10(6):giab041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mead D, Saccheri I, Yung CJ, et al. The genome sequence of the ringlet, Aphantopus hyperantus Linnaeus 1758. Wellcome Open Res. 2021;6(165):165. [Google Scholar]
- 31. Nowell RW, Elsworth B, Oostra V et al. A high-coverage draft genome of the mycalesine butterfly Bicyclus anynana. Gigascience. 2017;6(7):gix035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. König S, Romoth LW, Gerischer L et al. Simultaneous gene finding in multiple genomes. Bioinformatics. 2016;32(22):3388–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. De Fouchier A, Walker III WB, Montagné N, et al. Functional evolution of Lepidoptera olfactory receptors revealed by deorphanization of a moth repertoire. Nat Commun. 2017;8(1):15709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tamura K, Stecher G, Kumar S. MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol. 2021;38(7):3022–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Slater GSC, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005;6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7(10):e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Finn RD, Coggill P, Eberhardt RY et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44(D1):D279–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Minh BQ, Schmidt HA, Chernomor O et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37(5):1530–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kurtz S, Phillippy A, Delcher AL et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gu Z, Gu L, Eils R, et al. Circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30(19):2811–2. [DOI] [PubMed] [Google Scholar]
- 43. Celorio-Mancera MdlP, Rastas P, Steward RA, et al. Chromosome level assembly of the comma butterfly (Polygonia c-album). Genome Biol Evol. 2021;13(5):evab054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cabanettes F, Klopp C. D-GENIES: dot plot large genomes in an interactive, efficient and simple way. PeerJ. 2018;6:e4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Goel M, Sun H, Jiao WB et al. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 2019;20(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goel M, Schneeberger K. plotsr: visualizing structural similarities and rearrangements between multiple genomes. Bioinformatics. 2022;38(10):2922–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brown KS, Freitas AV, Von Schoultz B et al. Chromosomal evolution of South American frugivorous butterflies in the Satyroid clade (Nymphalidae: Charaxinae, Morphinae and Satyrinae). Biol J Linnean Soc. 2007;92(3):467–81. [Google Scholar]
- 49. Yandell M, Ence D. A beginner’s guide to eukaryotic genome annotation. Nat Rev Genet. 2012;13(5):329–42. [DOI] [PubMed] [Google Scholar]
- 50. Walker WB, Roy A, Anderson P et al. Transcriptome analysis of gene families involved in chemosensory function in Spodoptera littoralis (Lepidoptera: Noctuidae). BMC Genomics. 2019;20(1):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gomes Sotero-Caio C, Challis R, Kumar S et al. Genomes on a Tree (GoaT): a centralized resource for eukaryotic genome sequencing initiatives. Biodiversity Inform Sci Stand. 2021;5:e74138. [Google Scholar]
- 52. d’Alencon E, Sezutsu H, Legeai F et al. Extensive synteny conservation of holocentric chromosomes in Lepidoptera despite high rates of local genome rearrangements. Proc Natl Acad Sci U S A. 2010;107(17):7680–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Asalone KC, Ryan KM, Yamadi M et al. Regional sequence expansion or collapse in heterozygous genome assemblies. PLoS Comput Biol. 2020;16(7):1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pryszcz LP, Gabaldon T. Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 2016;44(12):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Talla V, Suh A, Kalsoom F, et al. Rapid increase in genome size as a consequence of transposable element hyperactivity in Wood-White (Leptidea) butterflies. Genome Biol Evol. 2017;9(10):2491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Osanai-Futahashi M, Suetsugu Y, Mita K, et al. Genome-wide screening and characterization of transposable elements and their distribution analysis in the silkworm, Bombyx mori. Insect Biochem Mol Biol. 2008;38(12):1046–57. [DOI] [PubMed] [Google Scholar]
- 57. Lucek K. Evolutionary mechanisms of varying chromosome numbers in the radiation of erebia butterflies. Genes. 2018;9(3):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Talavera G, Lukhtanov VA, Rieppel L et al. In the shadow of phylogenetic uncertainty: The recent diversification of Lysandra butterflies through chromosomal change. Mol Phylogenet Evol. 2013;69(3):469–78. [DOI] [PubMed] [Google Scholar]
- 59. Lukhtanov V, Kandul N, Plotkin J, et al. Reinforcement of pre-zygotic isolation and karyotype evolution in Agrodiaetus butterflies. Nature. 2005;436(7049):385–9. [DOI] [PubMed] [Google Scholar]
- 60. Noor M, Grams K, Bertucci L, et al. Chromosomal inversions and the reproductive isolation of species. Proc Natl Acad Sci U S A. 2001;98(21):12084–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Satou Y, Sato A, Yasuo H et al. Chromosomal inversion polymorphisms in two sympatric ascidian lineages. Genome Biol Evol. 2021;13(6):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Coluzzi M, Sabatini A, della Torre A et al. A polytene chromosome analysis of the Anopheles gambiae species complex. Science. 2002;298(5597):1415–8. [DOI] [PubMed] [Google Scholar]
- 63. Hooper DM, Price TD. Chromosomal inversion differences correlate with range overlap in passerine birds. Nat Ecol Evol. 2017;1(10):1526–34. [DOI] [PubMed] [Google Scholar]
- 64. Hooper DM, Griffith SC, Price TD. Sex chromosome inversions enforce reproductive isolation across an avian hybrid zone. Mol Ecol. 2019;28(6):1246–62. [DOI] [PubMed] [Google Scholar]
- 65. Davey JW, Barker SL, Rastas PM et al. No evidence for maintenance of a sympatric Heliconius species barrier by chromosomal inversions. Evol Lett. 2017;1(3):138–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Arikawa K. Spectral organization of the eye of a butterfly, Papilio. J Comp Physiol A Neuroethol Sensory Neural Behav Physiol. 2003;189(11):791–800. [DOI] [PubMed] [Google Scholar]
- 67. Rebeiz M, Williams TM. Using Drosophila pigmentation traits to study the mechanisms of cis-regulatory evolution. Curr Opin Insect Sci. 2017;19:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ficarrotta V, Hanly JJ, Loh LS, et al. A genetic switch for male UV iridescence in an incipient species pair of sulphur butterflies. Proc Natl Acad Sci U S A. 2022;119(3):e2109255118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cong Q, Zhang J, Shen J et al. Speciation in North American Junonia from a genomic perspective. Syst Entomol. 2020;45(4):803–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bastide H, López-Villavicencio M, Ogereau D et al. Genomic data of the Amazonian blue butterfly, Morpho achilles. GigaScience Database. 2023. 10.5524/102366. [DOI]
- 71. Bastide H, López-Villavicencio M, Ogereau D, et al. Genomic data of the Amazonian blue butterfly, Morpho helenor. GigaScience Database. 2023. 10.5524/102367. [DOI]
- 72. Bastide H, López-Villavicencio M, Ogereau D et al. Genomic data of the Amazonian blue butterfly, Morpho deidamia. GigaScience Database. 2023. 10.5524/102368. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Bastide H, López-Villavicencio M, Ogereau D et al. Genomic data of the Amazonian blue butterfly, Morpho achilles. GigaScience Database. 2023. 10.5524/102366. [DOI]
- Bastide H, López-Villavicencio M, Ogereau D, et al. Genomic data of the Amazonian blue butterfly, Morpho helenor. GigaScience Database. 2023. 10.5524/102367. [DOI]
- Bastide H, López-Villavicencio M, Ogereau D et al. Genomic data of the Amazonian blue butterfly, Morpho deidamia. GigaScience Database. 2023. 10.5524/102368. [DOI]
Supplementary Materials
Charlotte Julie Wright, MPhil -- 12/8/2022 Reviewed
Niklas Wahlberg -- 12/15/2022 Reviewed
Data Availability Statement
Fastq files, genome assemblies, assembly methods, and collection information were uploaded at the ENA website (https://www.ebi.ac.uk/ena/browser/home) under the project number PRJEB56642. Genome accession numbers are ERZ14213098 for M. helenor, ERZ14213099 for M. achilles, and ERZ14213100 for M. deidamia. Other data further supporting this work are openly available in the GigaScience repository, GigaDB [70–72].