

Abstract
Background:
CLN3 is an autosomal recessive lysosomal disorder with intracellular accumulation of ceroid-lipofuscins. CLN3 classically has onset around 4–6 years of age involving vision loss, followed by developmental regression and seizures. Symptoms are progressive and result in premature death. Because treatments are under development, here we explore magnetic resonance spectroscopy (MRS) measurements of metabolite levels in the brain as a potential objective outcome measures.
Methods:
Individuals with genetically confirmed CLN3 were enrolled from October 2017-November 2021 in a prospective natural history study (NCT033007304). Baseline concentrations of brain metabolites measured by MRS were compared to concurrently collected dimensional assessment measures: Vineland-3 Adaptive Behavior Composite (ABC) score, verbal intelligence quotient (VIQ), and the Physical, Capability with actual vision, and Clinical global impression of change sub-domains of the Unified Batten Disease Rating Scale (UBDRS).
Results:
27 participants with typical CLN3 presentation (15F:12M; ages 6.0–20.7 years) completed MRS, ABC, and UBDRS; 20 (12F:8M; ages 6.5–20.7 years) also completed the VIQ assessment. N-acetyl aspartate [B(95% CI)=−0.61(−0.78;−0.45)] and glutamine/glutamate/GABA [B(95% CI)=−0.82(−1.04;−0.6)] in the parietal gray matter (PGM) decreased across the ages. The strongest correlations between MRS metabolite measurements and the clinical severity assessments were found with N-acetyl aspartate [VIQ (ρ=0.58), Vineland-3 ABC (ρ=0.59), UBDRS |ρ| range=(0.57;0.7)] and glutamine/glutamate/GABA [VIQ (ρ=0.57), Vineland-3 ABC (ρ=0.60), UBDRS |ρ| range=(0.59;0.77)] measured in the midline PGM. These correlations were accounted for when age was considered.
Conclusions:
Based on their correlations to established assessments, NAA and glutamine/glutamate/GABA measured in the midline parietal gray matter may be useful indicators of CLN3 disease state. In a clinical trial, divergence of the MRS measurements and clinical severity markers from age may be useful as surrogate measures for treatment responses.
Keywords: Juvenile neuronal ceroid lipofuscinosis, neurodevelopment, NAA, Glx
INTRODUCTION
CLN3 disease, resulting from disease-associated variants in both copies of the CLN3 gene [1], is an autosomal recessive neuronal ceroid lipofuscinosis (NCL) disorder. The 13 types of NCL have similar clinical presentations but differ in typical age of onset, which ranges from infant to adult. The common histopathological findings encompass intracellular inclusions (typically in the lysosomes) that contain carbohydrates, lipids, proteins, and autofluorescent materials [2, 3]. CLN3 disease (OMIM 204200), also known as Juvenile NCL or Juvenile Batten disease, is the most common type of NCL. It presents around 4–6 years of age with visual impairment as the typical first symptom. Progression to blindness is accompanied by neurocognitive decline, behavioral dysregulation, seizures, and motor disabilities [4–7].
Current research efforts are directed toward elucidating the function of CLN3 protein and the underlying pathophysiology of CLN3 disease [8], and toward discovering a treatment for the neurodegenerative process [3, 9]. Neurodegeneration in CLN3 disease eventually results in brain atrophy [10–13]. It is unclear whether brain atrophy as measured by brain MRI is sufficiently sensitive for clinical trials of short duration or for clinical trials involving younger children. Alternatively, brain metabolite levels measured by proton magnetic resonance spectroscopy (MRS) have been shown in an infantile form of NCL (CLN1 disease) to track with disease course [14, 15]. Recent cross-sectional analysis of neurofilament light chain level, a protein whose level has been demonstrated to be high in conditions of neuronal damage in adults [16, 17], showed higher expression in cerebrospinal fluid in pediatric participants with CLN3 as compared to a similar age non-CLN3 cohort [18]. Higher neurofilament light chain level correlated with lower N-acetylaspartate+N-acetylaspartyl glutamate level in the parietal gray matter.
Here, we used MRS to measure levels of metabolites that are markers of neuronal health or inflammation in the brain of individuals with CLN3 disease and to correlate them to other measures indicative of disease state. In this cross-sectional analysis, we aimed to explore at a cohort level whether:
brain metabolite levels quantified by MRS that are markers for healthy neurons or inflammation worsen across age, and
deviation of these metabolite levels from normal correlate to scores on the Unified Batten Disease Rating Scale (UBDRS), a validated outcome measure of CLN3 disease severity [19, 20], and to standardized measures of neurodevelopment ability such as verbal IQ and the Vineland Adaptive Behavior Composite (ABC) assessment.
METHODS
Enrollment.
Participants in the NICHD Institutional Review Board-approved protocol NCT033007304 included individuals of any age, sex, race, ethnicity, or disease severity who had bi-allelic disease-associated variants in CLN3, clinical signs and symptoms consistent with CLN3 disease, and the ability to travel to the study site without increased risk to their health. CLN3 gene variants were determined based on reports from CLIA-certified laboratories. We obtained written consent from capable individuals at least 18 years of age, assent from capable individuals at least 7 years of age, and written consent from parents or guardians as appropriate.
Disease severity assessment.
We evaluated disease severity at the baseline visit using the UBDRS [20, 21] (version 12/20/2017). We made use of the 3 subsections of this rating scale that other authors [20, 22] have found to be most useful from their cumulative experience: physical assessment, capability assessment, and clinician global impression of change (CGI). The physical assessment rates 28 neurological items on a scale from 0 (normal) to 4 (severely impaired), resulting in a scale of 0–112 points where higher score indicates more severe impairment. This assessment was carried out by a pediatric neurologist (AGS). The capability (given actual vision) assessment rates 5 activities of daily living on a total scale of 0–14 points where a lower score indicates more severe impairment. This assessment was carried out by a psychologist (AET). The CGI is a 7-item provider’s rating of overall disease state (score range 6–35) that includes factors considered in the other subsections, with a higher score indicating greater severity. This assessment was carried out by a geneticist (ADD). Scores were adjusted for missing items as described in a previous publication [18].
Neurodevelopmental assessment.
Based on the participant’s age at the baseline visit, we used either the Wechsler Intelligence Scale for Children-Fifth Edition (WISC-V) [23] or the Wechsler Adult Intelligence Scale, Fourth Edition (WAIS-IV) [24] to assess verbal IQ as described [25] (population mean = 100±15, test floor = 40). Similar to previous investigations of CLN3 [26, 27], we limited our IQ assessment to verbal IQ because visual impairment precluded assessment of the nonverbal IQ in many participants. We also used the Vineland Adaptive Behavior Scale, Third Edition [28] to assess global functioning. Both verbal IQ and Vineland ABC were assessed by a psychologist (AET).
Brain Imaging and Spectroscopy.
All participants included in this report completed the MRI and MRS baseline exam under sedation. The examinations were performed on a 3T Philips Achieva scanner using an 8-channel SENSE head coil. A standard clinical brain MRI (without a contrast agent) was performed; the results of those exams have been reported elsewhere [29]. Quantitative single-voxel 1H-MRS was performed on the left centrum semiovale (LCSO) and midline parietal gray matter (PGM). Voxels were graphically prescribed from sagittally-acquired 3D-TFE images reformatted into three planes. 1H-MRS acquisition was performed with PRESS localization, CHESS water suppression, TE=38 ms, TR=2000 ms, and NEX=128. An unsuppressed water spectrum (TR=5000 ms, TE=38 ms, NEX=16) was also acquired for each voxel. Identical gain and shim settings were used for both spectra from each voxel so that metabolite concentrations could be determined. To correct for cerebral spinal fluid (CSF) included within the voxels, we acquired a heavily T2-weighted image with location and slice thickness corresponding to the location of each MRS voxel (FSE; ETL=8; TE=500 ms; TR=3000 ms), and a phantom containing water was placed beside the head and included in the field-of-view of the CSF correction image.
Post-processing of the spectra was performed using LCModel [30], followed by correction for estimated tissue water and T1 of the metabolites within tissue [14]. Because only one echo time was acquired, no correction was made for T2 decay of the metabolites. We analyzed creatine, myo-inositol, choline-containing compounds (“choline”), N-acetylaspartate+N-acetylaspartyl glutamate (NAA), and glutamate+glutamine+gamma-aminobutyric acid (“Glx”). Post-processing for correction of CSF partial volume was done as described in other reports [14, 31, 32].
To account for normal age-related evolution of metabolite levels, we utilized reference (expected) values from 38 individuals aged 1–42 years (interquartile range: 3.6–23.3 years) to generate a reference curve for each metabolite at each location. Similarly-acquired and post-processed data from healthy children are not available; instead, children in the reference group were asymptomatic or neurologically pre-symptomatic participants in other protocols at our institution who were scanned and post-processed with the same method as the CLN3 participants. Adults in the reference group were healthy volunteers. The reference curve for each metabolite was generated by fitting a model of the form y=AxB (where x is age and y is metabolite concentration) to the reference data. The difference from the reference curve value was used in all analyses.
Statistical analysis.
Descriptive analysis included pairwise Spearman correlations amongst all key variables: metabolite concentration (difference from expected for age), behavioral variables (VIQ, ABC, UBDRS scores), and age. Each of the five metabolites at both locations was modeled as a function of mean-centered chronological age in a series of general linear models, secondarily interacting age with genotype to explore possible differences. To quantify the relationship between metabolite concentration and behavior, we evaluated VIQ, ABC, and UBDRS scores in separate general linear models as a function of metabolite concentration, with age and genotype as covariates. While sibships did exist in the data, due to the small sample size we assumed independence of observations. Following the guidelines of the American Statistical Association, we report uncorrected p-values alongside their test statistics [33].
RESULTS
Cohort characteristics.
A total of 33 participants enrolled in the study and have baseline assessments between October 2017 – November 2021. Of these, six participants are excluded from further analyses: four (13.2–80 years of age) presented at enrolment with vision-only symptom, one was pre-symptomatic, and one did not have brain imaging. The remaining 27 participants (15F, 12M; ages 6–20.7 years) completed MRS, UBDRS, and Vineland ABC assessment; 21 of these participants (12F, 9M; ages 6.5–20.7 years) also completed the verbal IQ assessment. Of the six who did not complete the verbal IQ assessment, four did not use English as the primary language, one could not score above the floor of the test, and one had post lumbar puncture headache and could not cooperate. Twelve participants (ages 6–16.6 years) were homozygous for the common c.461−280_677+382del (966-bp deletion) variant, 10 (ages 6.5–17.5 years) were compound heterozygous for the 966-bp deletion and another variant, and five (ages 6.8–20.7 years) had other non 966-bp deletion genotypes. The cohort included four sib pairs (Table 1 and Supplemental Table 1).
Table 1. Summary statistics for the clinical measures.
ABC: Vineland-3 adaptive behavior composite. CGI: clinician global impression of change. IQR = interquartile range. SD: standard deviation. UBDRS: Unified Batten Disease Rating Scale. VIQ: verbal intelligence quotient.
| n | Mean±SD | Range | Median[IQR] | |
|---|---|---|---|---|
| Age (years) | 27 | 11.0±4.1 | 6.0–20.7 | 9.9 [7.6, 14.8] |
| VIQ | 21 | 74.9±19.2 | 45–106 | 70 [59, 93] |
| Vineland-3 ABC | 27 | 58.6±21.7 | 20–84 | 66 [38.5, 78.5] |
| UBDRS Physical | 27 | 18.0±18.6 | 2–64 | 10 [4.5, 21.5] |
| UBDRS Capability | 27 | 6.9±2.8 | 1–14 | 6 [5.5, 9] |
| UBDRS CGI | 27 | 15.9±5.1 | 9.3–24.5 | 16.3 [11.1, 19.8] |
Age progression of metabolites.
Figure 1 shows plots of the metabolite measurements (difference from expected for age, in mM) at baseline visit versus age and Table 2 contains the results of linear modeling. In the PGM, NAA [B(95%CI):−0.61(−0.78;−0.45)], Glx [B(95%CI):−0.82(−1.04;−0.6)], and creatine [B(95%CI):−0.27(−0.39;−0.15)] deficits (relative to expected) increased across age while choline and myo-inositol remained close to expected. In the LCSO, NAA deficit [B(95%CI):−0.25(−0.37;−0.13)] also increased, but there were trends for increasingly elevated creatine and choline. As a secondary analysis, we explored differences in age progression as a function of genotype. Given the small number of measurements in the “other” category, we focused on the comparison of the 966-bp deletion compound heterozygotes and homozygotes to age-expected values. The effect of age on aberrant metabolite concentration was similar between these genotypic groups for most features, with the exception of myo-inositol in the LCSO. For this metabolite, difference from expected increased with age among the 966-bp deletion compound heterozygotes (B[SE] = 0.35[0.08], p = .0004) but decreased with age among the 966-bp deletion homozygotes (B[SE] = −0.16[0.06], p = 0.02) (comparison, t=4.9, p < .0001) (Supplementary Figure 1 illustrates the regression lines within genotype).
Fig. 1. Age progression of metabolites relative to normative values.
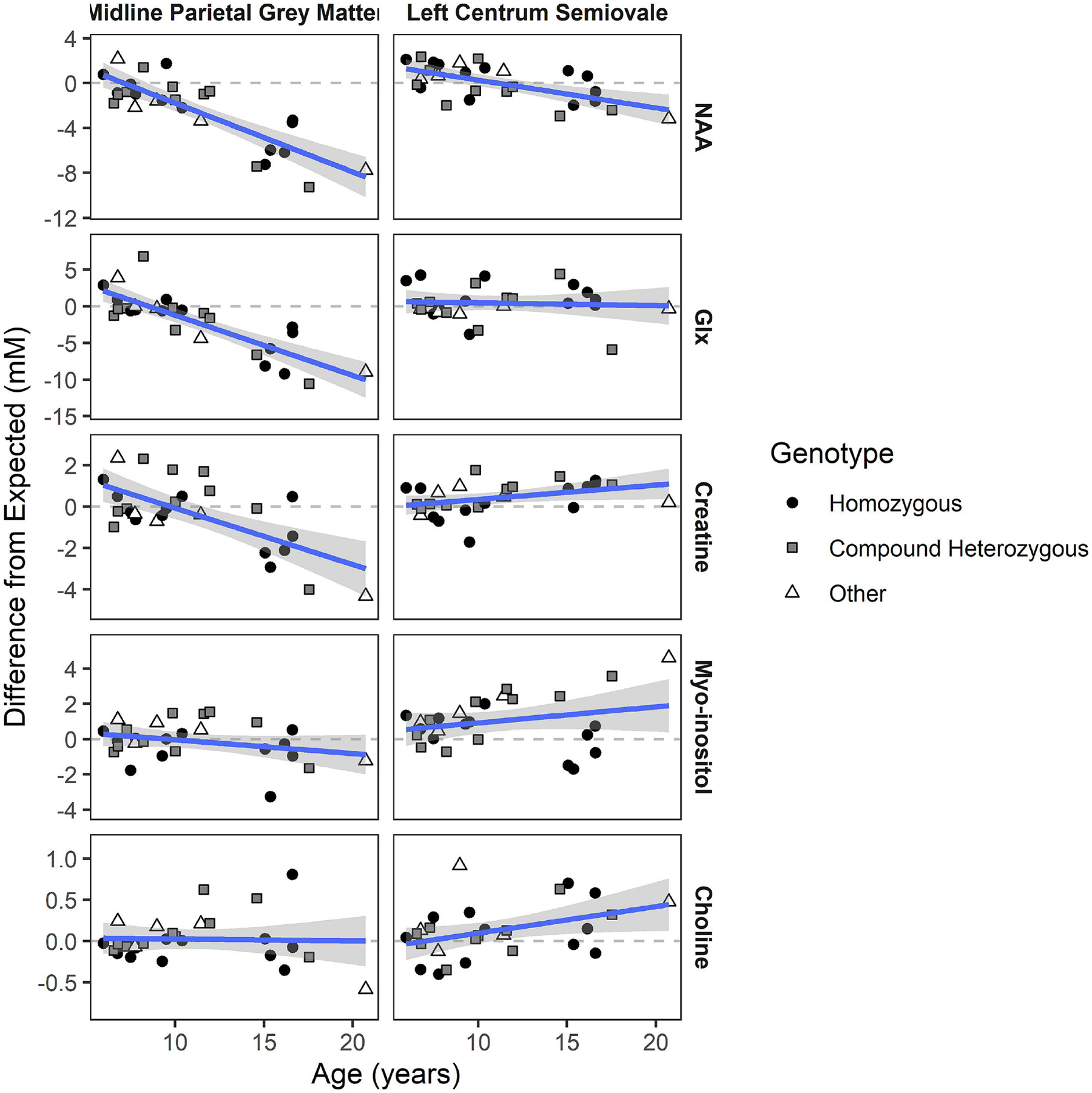
NAA: N-acetyl aspartic acid. Glx: Glu+Gln+GABA Regression line shows average effect of age on difference in concentration from age expectation, which is illustrated by the dotted line at zero (shaded area is 95% CI). Individual data points are coded by genotype. Slopes and test statistics for age are listed in Table 1. Homozygous: biallelic 966-bp deletion variants. Compound heterozygous: 966-bp deletion in trans with a non 966-bp deletion variant. Other: biallelic non 966-bp variants.
Table 2. Results of linear modeling of age progression of MRS metabolites relative to normative values.
Slope of age is the expected difference in metabolite concentration (in mM) for a one-year difference in age. CI: confidence interval. DF: degree of freedom. Glx: glutamine/glutamate/gamma-aminobutyric acid. NAA: n-acetylaspartate/n-acetylaspartyl glutamate. LCSO: left centrum semiovale. PGM: parietal gray matter.
| Region | Metabolite | Slope of Age [95% CI] | Test Statistic for Age (DF = 25) |
|---|---|---|---|
| LCSO | Creatine | 0.07 [0, 0.14] | t=2.02, p=0.05 |
| Myo-inositol | 0.09 [−0.05, 0.23] | t=1.31, p=0.20 | |
| Choline | 0.03 [0, 0.06] | t=2.21, p=0.04 | |
| NAA | −0.25 [−0.37, −0.13] | t=−3.96, p<.001 | |
| Glx | −0.04 [−0.27, 0.19] | t=−0.32, p=0.75 | |
| PGM | Creatine | −0.27 [−0.39, −0.15] | t=−4.48, p<.001 |
| Myo-inositol | −0.08 [−0.18, 0.02] | t=−1.52, p=0.14 | |
| Choline | 0 [−0.03, 0.03] | t=−0.16, p=0.87 | |
| NAA | −0.61 [−0.78, −0.45] | t=−7.44, p<.001 | |
| Glx | −0.82 [−1.04, −0.6] | t=−7.36, p<.001 |
Relationship of clinical assessments to metabolite concentration differences.
The second set of analyses concerned the relationship between baseline clinical phenotypic measurements (VIQ, ABC, and UBDRS variables) and metabolite concentration differences. Descriptive statistics (Spearman correlations) are illustrated in Figure 2. We noted very strong correlations amongst these clinical variables (|ρ| ranging 0.70–0.88).
Fig. 2. Spearman correlations amongst target variables.
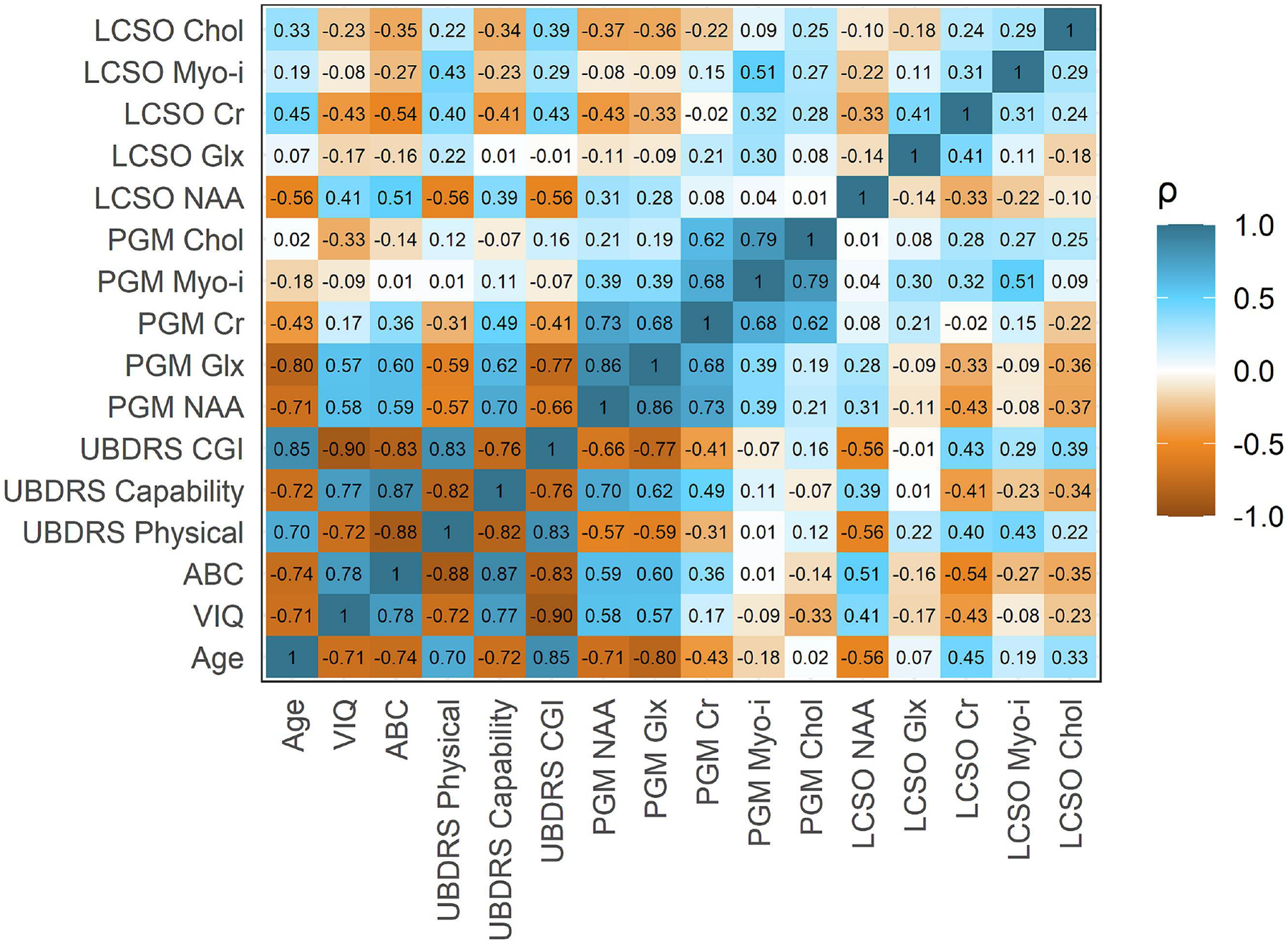
ABC: adaptive behavior composite. CGI: clinician global impression of change. Chol: choline. Cr: creatine. Glx: Glu+Gln+GABA. LCSO: left centrum semiovale. myo-i: myo-inositol. NAA: N-acetyl aspartic acid. PGM: parietal gray matter. UBDRS: Unified Batten Disease Rating Scale. VIQ: verbal intelligence quotient.
Left centrum semiovale (LCSO).
Within the LCSO, creatine excess [VIQ (ρ=−0.43), ABC (ρ=−0.54), UBDRS |ρ| ranges = 0.40;0.43] and NAA deficit [VIQ (ρ=0.41), ABC (ρ=0.51), UBDRS |ρ| ranges = 0.39;0.56] were most robustly correlated with severity. The linear models revealed that age accounts for the association of most metabolite differences with severity in these measures (Figure 3 and Supplemental Table 2). The exception was LCSO myo-inositol excess, which remained associated with the UBDRS Physical score [B(95% CI)=3.98 (0.64;7.31); p=0.03] even after accounting for age and genotype. Due to the preceding exploratory analyses suggesting a differential age progression of LCSO myo-inositol between genotypes, we performed additional modeling of the relationship of this metabolite to severity as a function of genotype. For the UBDRS Physical outcome only, we noted a possible difference between compound heterozygous and homozygous genotypes (t=2.5, p=.02). For the homozygous genotype, greater excess of LCSO myo-inositol was associated with lower UBDRS Physical scores (B[SE]=−4.28[2.93], p=0.16) The effect was in the opposite direction for the compound heterozygous genotype (B[SE]=3.30[3.44], p=0.35).
Fig. 3. Linear modeling of phenotypic features by MRS metabolite, controlling for age and genotype.
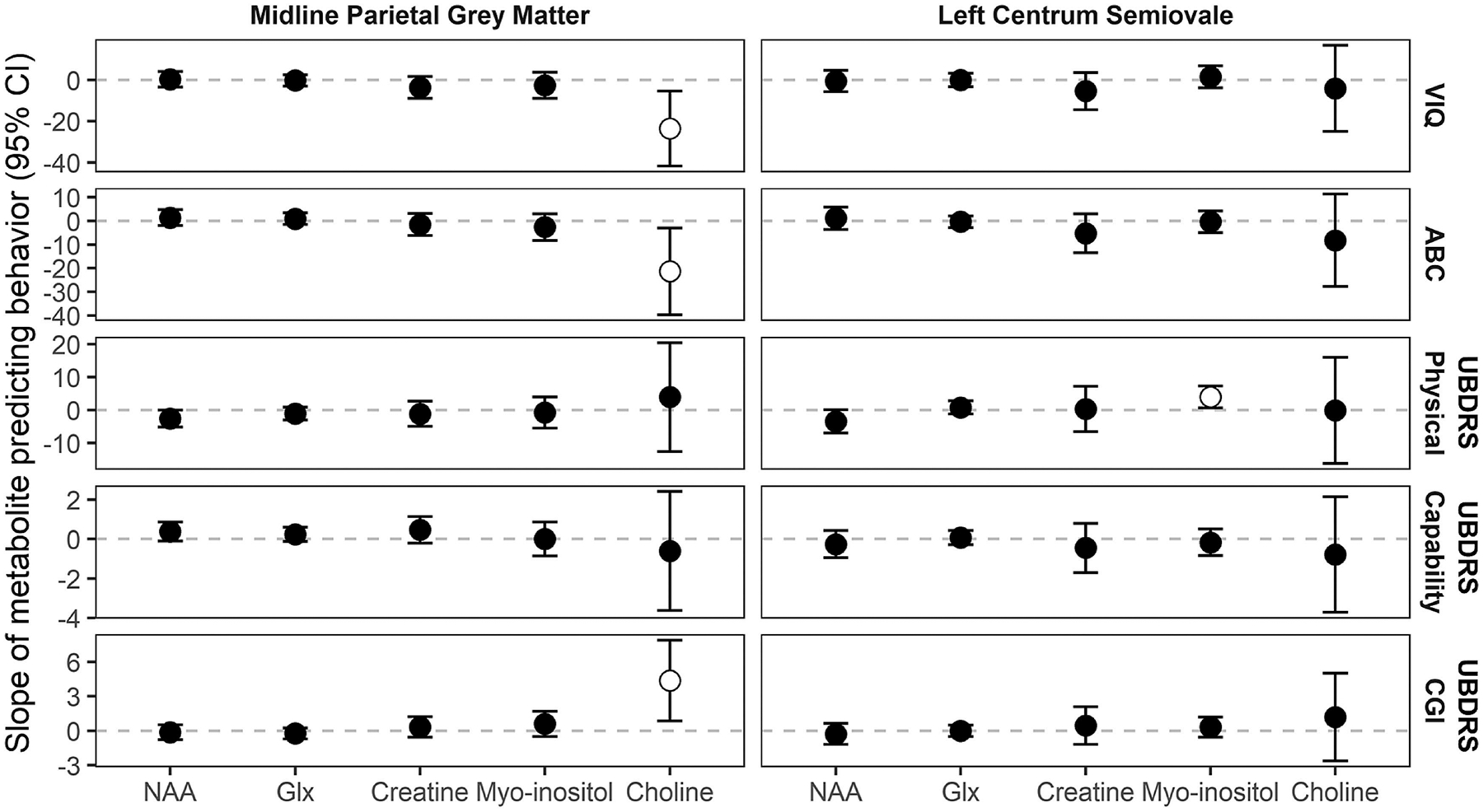
The slope of metabolite (i.e., expected change in behavior score for each one-unit change in metabolite difference from normative value) is plotted with its 95% confidence interval; where the 95% confidence interval does not include zero the slope marker is white filled. ABC: adaptive behavior composite. CGI: clinician global impression of change. Glx: Glu+Gln+GABA. NAA: N-acetyl aspartic acid. PGM: parietal gray matter. Physical, Capability, CGI: Unified Batten Disease Rating Scale sub-domains. VIQ: verbal intelligence quotient.
Midline parietal gray matter (PGM).
Within the PGM, NAA [VIQ (ρ=0.58), ABC (ρ=0.59), UBDRS |ρ| ranges = 0.57;0.70] and Glx [VIQ (ρ=0.57), ABC (ρ=0.60), UBDRS |ρ| ranges = 0.59;0.77] were most strongly correlated with severity in measures (Figure 2). In all cases, greater deficit of NAA or Glx was correlated with greater severity. As in the LCSO, most of the correlations between metabolite and severity were accounted for by age in the linear model (Figure 3 and Supplemental Table 2). However, PGM choline, which was not strongly correlated with severity in the descriptive analysis, did have unique associations with VIQ (t=−2.54, p=0.02), ABC (t=−2.28, p=0.03), and UBDRS CGI (t=2.44, p=0.02) after adjusting for age and genotype. In each case, greater excess of choline in the PGM was associated with more severe disease state, according to these measures.
DISCUSSION
In this report we describe cross-sectional data from our assessment of individuals with CLN3 disease in a natural history study. The assessment included outcome measures pertinent to assessment of disease state that we intend to use to monitor progression during the course of the study. In addition to using three sub-domains of the established UBDRS, the Vineland-3 composite score, and verbal IQ for clinical assessment of neurodevelopment and disease state, we also used proton MR spectroscopy to measure five MRS-visible metabolites at two locations in the brain. Of the MRS-visible metabolites measured, the cross-sectional results support the validity of measuring levels of NAA and Glx in the midline parietal gray matter (PGM), and NAA in the left centrum semiovale (LCSO) as promising quantitative markers of disease state in CLN3 disease. These metabolite measurements decreased across age and accounted for the same pathophysiology that age does in correlating with the clinical assessments of neurodevelopment (VIQ and adaptive behavior) and disease state (UBDRS).
The LCSO voxel placement corresponds anatomically to the left centrum semiovale; and although located in white matter, is adjoined on three sides by Brodmann area 4, the primary motor cortex. The U-fibers related to primary motor functions pass through this location, as does a sizeable fraction of the corticospinal tract, as well as the crossing fibers to the contralateral motor areas that go via the corpus callosum. While intended to give us a general sense of the state of the white matter, the LCSO voxel is biased toward primary motor functions. The PGM voxel placement corresponds anatomically to the precuneus (medial portion of the superior parietal lobule) on both sides of the interhemispheric fissure. This voxel is predominantly composed of Brodmann area 7 with a small contribution from Brodmann area 5, both of which are involved in visual-motor coordination and proprioception; there are also small contributions from portions of Brodmann areas 1, 2, and 3, all primary somatosensory cortex. The PGM voxel location is biased toward motor, somatosensory, and visual functions. This may contribute to the trend of PGM metabolite levels having stronger relationships with the other outcome measures than those in the LCSO.
Comparisons of individuals compound heterozygous versus homozygous for the common 966-bp deletion variant suggested genotypic differences for myo-inositol, a metabolite other than choline that can be related to inflammation. Myo-inositol is a glia-specific marker [34] that is elevated by gliosis or inflammation [35]. In the LCSO, the change in this metabolite across age and in relationship to the UBDRS Physical assessment differ, with excess increasing with older age and more severe disease state in compound heterozygotes but having the opposite relationship in homozygotes. In infantile forms of NCL (CLN1 and CLN2), MRS markers associated with neuroinflammation (choline and myo-inositol) were elevated during the early stages of the disease, consistent with the proposed disease mechanisms [14, 36]. Immune and inflammatory processes are hypothesized to contribute to the neurologic phenotype of CLN3, based on increased immunoreactive signals in rat brain treated ex vivo with sera from affected versus unaffected individuals and increased markers for fibrosis (glial fibrillary acidic protein and F4/80) in Cln3−/− mice [37, 38]. The LCSO myo-inositol observations in this study may suggest pathophysiological evolution or timing differences in the white matter of 966-bp deletion compound heterozygotes as compared to homozygotes. We note that this observation was made from a small number of data points. Thus, additional samples and longitudinal observations will be needed to confirm and decipher this further.
Development of therapies for ultrarare pediatric diseases is limited by scarcity of affected individuals for randomized controlled study designs and paucity of feasible and accessible outcome measures[39, 40]. Identification of appropriate outcome measures to be used in designing clinical trials is helpful. Imaging-based outcome measures would optimally be quantitative, reproducible, reflective of disease pathology, sufficiently sensitive to identify changes over a short time frame, generalizable, and accessible. MR spectroscopy meets these criteria [39] and has been used to evaluate markers of disease progression in other neurometabolic conditions [14, 15, 41–44].
Previously-reported MRS results in humans with CLN3 disease involve very small numbers of subjects and variously show normal [15, 45, 46], lower [15, 45], and progressively decreasing [15] NAA level in the thalami compared to controls, any of which could be consistent with our findings in the PGM. Previously-reported results in humans with CLN1 disease demonstrated a consistently increasing deficit with increasing age in markers relating to healthy neuron metabolism (NAA and Glx) in both gray matter and white matter [14]. Our findings demonstrated similar correlations for NAA and Glx deficits in the PGM for both age and clinical assessments. In the LCSO, we found a strong correlation between NAA deficits and the clinical assessments. Finding that NAA and Glx deficits correlate with age and/or severity is consistent with definitive evidence of neuronal loss in CLN3 from histopathologic studies [10] and measurements of brain volume by imaging [11, 12].
As an assessment of disease status, proton MR spectroscopy has the advantage of being quantitative and reproducible, and it can be performed as an add-on procedure during a clinical MRI exam of the brain. However, it requires off-line processing, so that the results are not immediately available. It also requires comparison to a reference group, and references for pediatric ages are not readily available. Due to lack of ability to cooperate, most patients who are very young or have significant intellectual disability require sedation for an MRI/MRS exam. It is not clear at this time whether the result for an individual patient at a given time point is a clinically useful number, or if the numbers are meaningful only in aggregate. Our study was biased toward inclusion of participants healthy enough to travel to our study site and complete the itinerary for the visit, and therefore we may be lacking in individuals at both the pre-symptomatic (undiagnosed) and advanced ends of the spectrum. We expect that as this study progresses, longitudinal data and a larger cohort will provide further insights into the reproducibility, applicability, and sensitivity of these findings.
Data from this cohort show strong correlations between established severity markers (Vineland ABC, verbal IQ, and three sub-domains of the UBDRS) and MRS-measured NAA and Glx levels in the PGM, and NAA level in the LCSO. Statistically, these correlations account for the same contribution made by considering the age of the participants; therefore, age seems to be the dominant variable predicting disease severity in the population. These findings support use of these MRS-based measures of disease state in clinical trials, and longitudinal data will be helpful in confirming this. In a clinical trial, divergence of the MRS measurements and clinical severity markers from age may be useful as surrogate measures for treatment responses.
Supplementary Material
Acknowledgments
We thank the study participants and their families, as well as the family advocacy organizations (Beyond Batten Disease Foundation, Batten Disease Support Research and Advocacy Foundation) for the motivation and inspiration. Kisha Jenkins assisted in data collection and management. Sandra McKee, Andy Yan, Claudia Stephenson, and R. Reese Baehr performed the MRI and MRS exams.
Funding:
The NIH Intramural Research Program of NICHD [ZIA HD008989 (FDP), ZIA HD009001-01(ADD)], NIH Clinical Center, NINDS, NIMH [1ZICMH002961 (AET)], and an NIH Clinical Center Bench-to-Bedside Award (FDP) supported this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Ethical Standards: This study was performed in line with the principles of the Declaration of Helsinki. The National Institutes of Health Institutional Review Board approved the studies. All participants or parents/guardians gave their informed consent, and assent if age appropriate, prior to their inclusion in the study.
Competing Interests: ADD and FDP have collaborative research and development agreement with Amicus Therapeutics, Inc. and Beyond Batten Disease Foundation. The remaining authors report no conflicts of interest.
Data and Code Availability:
Data and code will be provided by the authors upon reasonable requests.
REFERENCES
- [1].“Isolation of a novel gene underlying Batten disease, CLN3. The International Batten Disease Consortium,” Cell, vol. 82, no. 6, pp. 949–57, Sep 22 1995, doi: 10.1016/0092-8674(95)90274-0. [DOI] [PubMed] [Google Scholar]
- [2].Haltia M and Goebel HH, “The neuronal ceroid-lipofuscinoses: a historical introduction,” Biochim Biophys Acta, vol. 1832, no. 11, pp. 1795–800, Nov 2013, doi: 10.1016/j.bbadis.2012.08.012. [DOI] [PubMed] [Google Scholar]
- [3].Rosenberg JB, Chen A, Kaminsky SM, Crystal RG, and Sondhi D, “Advances in the Treatment of Neuronal Ceroid Lipofuscinosis,” Expert Opin Orphan Drugs, vol. 7, no. 11, pp. 473–500, 2019, doi: 10.1080/21678707.2019.1684258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Adams HR, Mink JW, and G. University of Rochester Batten Center Study, “Neurobehavioral features and natural history of juvenile neuronal ceroid lipofuscinosis (Batten disease),” J Child Neurol, vol. 28, no. 9, pp. 1128–36, Sep 2013, doi: 10.1177/0883073813494813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Preising MN, Abura M, Jager M, Wassill KH, and Lorenz B, “Ocular morphology and function in juvenile neuronal ceroid lipofuscinosis (CLN3) in the first decade of life,” Ophthalmic Genet, vol. 38, no. 3, pp. 252–259, May-Jun 2017, doi: 10.1080/13816810.2016.1210651. [DOI] [PubMed] [Google Scholar]
- [6].Abdennadher M et al. , “Seizure phenotype in CLN3 disease and its relation to other neurologic outcome measures,” J Inherit Metab Dis, Feb 7 2021, doi: 10.1002/jimd.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hildenbrand H et al. , “Characterizing upper limb function in the context of activities of daily living in CLN3 disease,” Am J Med Genet A, vol. 185, no. 5, pp. 1399–1413, May 2021, doi: 10.1002/ajmg.a.62114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mirza M et al. , “The CLN3 gene and protein: What we know,” Mol Genet Genomic Med, vol. 7, no. 12, p. e859, Dec 2019, doi: 10.1002/mgg3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Johnson TB, Cain JT, White KA, Ramirez-Montealegre D, Pearce DA, and Weimer JM, “Therapeutic landscape for Batten disease: current treatments and future prospects,” Nat Rev Neurol, vol. 15, no. 3, pp. 161–178, Mar 2019, doi: 10.1038/s41582-019-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Anderson GW, Goebel HH, and Simonati A, “Human pathology in NCL,” Biochim Biophys Acta, vol. 1832, no. 11, pp. 1807–26, Nov 2013, doi: 10.1016/j.bbadis.2012.11.014. [DOI] [PubMed] [Google Scholar]
- [11].Autti T, Raininko R, Santavuori P, Vanhanen SL, Poutanen VP, and Haltia M, “MRI of neuronal ceroid lipofuscinosis. II. Postmortem MRI and histopathological study of the brain in 16 cases of neuronal ceroid lipofuscinosis of juvenile or late infantile type,” Neuroradiology, vol. 39, no. 5, pp. 371–7, May 1997, doi: 10.1007/s002340050427. [DOI] [PubMed] [Google Scholar]
- [12].Tokola AM, Salli EK, Aberg LE, and Autti TH, “Hippocampal volumes in juvenile neuronal ceroid lipofuscinosis: a longitudinal magnetic resonance imaging study,” Pediatr Neurol, vol. 50, no. 2, pp. 158–63, Feb 2014, doi: 10.1016/j.pediatrneurol.2013.10.013. [DOI] [PubMed] [Google Scholar]
- [13].Hochstein JN et al. , “Natural history of MRI brain volumes in patients with neuronal ceroid lipofuscinosis 3: a sensitive imaging biomarker,” Neuroradiology, vol. 64, no. 10, pp. 2059–2067, Oct 2022, doi: 10.1007/s00234-022-02988-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Baker EH, Levin SW, Zhang Z, and Mukherjee AB, “Evaluation of disease progression in INCL by MR spectroscopy,” Ann Clin Transl Neurol, vol. 2, no. 8, pp. 797–809, Aug 2015, doi: 10.1002/acn3.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Brockmann K, Pouwels PJ, Christen HJ, Frahm J, and Hanefeld F, “Localized proton magnetic resonance spectroscopy of cerebral metabolic disturbances in children with neuronal ceroid lipofuscinosis,” Neuropediatrics, vol. 27, no. 5, pp. 242–8, Oct 1996, doi: 10.1055/s-2007-973772. [DOI] [PubMed] [Google Scholar]
- [16].Bridel C et al. , “Diagnostic Value of Cerebrospinal Fluid Neurofilament Light Protein in Neurology: A Systematic Review and Meta-analysis,” JAMA Neurol, vol. 76, no. 9, pp. 1035–1048, Sep 1 2019, doi: 10.1001/jamaneurol.2019.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Skillback T et al. , “CSF neurofilament light differs in neurodegenerative diseases and predicts severity and survival,” Neurology, vol. 83, no. 21, pp. 1945–53, Nov 18 2014, doi: 10.1212/WNL.0000000000001015. [DOI] [PubMed] [Google Scholar]
- [18].Dang Do AN et al. , “Neurofilament light chain levels correlate with clinical measures in CLN3 disease,” Genet Med, vol. 23, no. 4, pp. 751–757, Apr 2021, doi: 10.1038/s41436-020-01035-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kwon JM et al. , “Quantifying physical decline in juvenile neuronal ceroid lipofuscinosis (Batten disease),” (in eng), Neurology, vol. 77, no. 20, pp. 1801–1807, 2011, doi: 10.1212/WNL.0b013e318237f649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Masten MC et al. , “The CLN3 Disease Staging System: A new tool for clinical research in Batten disease,” Neurology, vol. 94, no. 23, pp. e2436–e2440, Jun 9 2020, doi: 10.1212/WNL.0000000000009454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Marshall FJ et al. , “A clinical rating scale for Batten disease: reliable and relevant for clinical trials,” Neurology, vol. 65, no. 2, pp. 275–9, Jul 26 2005, doi: 10.1212/01.wnl.0000169019.41332.8a. [DOI] [PubMed] [Google Scholar]
- [22].Kuper WFE et al. , “Motor function impairment is an early sign of CLN3 disease,” Neurology, vol. 93, no. 3, pp. e293–e297, Jul 16 2019, doi: 10.1212/WNL.0000000000007773. [DOI] [PubMed] [Google Scholar]
- [23].Wechsler D, “Wechsler intelligence scale for children–Fifth Edition (WISC-V),” Bloomington, MN: Pearson, 2014. [Google Scholar]
- [24].Wechsler D, “WAIS-IV administration and scoring manual (Canadian),” San Antonio, TX: The Psychological Corporation, 2008. [Google Scholar]
- [25].Dang Do AN et al. , “Use of the Vineland-3, a measure of adaptive functioning, in CLN3,” Am J Med Genet A, vol. 188, no. 4, pp. 1056–1064, Apr 2022, doi: 10.1002/ajmg.a.62607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Adams HR et al. , “Cross-validation of the Vineland-III with independent assessments of cognition and adaptive skills in CLN3 disease,” (in English), Mol Genet Metab, vol. 129, no. 2, pp. S17–S17, Feb 2020, doi: 10.1016/j.ymgme.2019.11.014. [DOI] [Google Scholar]
- [27].Lamminranta S et al. , “Neuropsychological test battery in the follow-up of patients with juvenile neuronal ceroid lipofuscinosis,” Journal of Intellectual Disability Research, vol. 45, no. 1, pp. 8–17, 2001. [DOI] [PubMed] [Google Scholar]
- [28].Sparrow S, Cicchetti D, and Saulnier C, “Vineland adaptive behavior scales–third edition (Vineland-3),” Circle Pines, MN: American Guidance Service, 2016. [Google Scholar]
- [29].Abdennadher M et al. , “Seizure phenotype in CLN3 disease and its relation to other neurologic outcome measures,” Journal of inherited metabolic disease, vol. 44, no. 4, pp. 1013–1020, Jul 2021, doi: 10.1002/jimd.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Provencher SW, “Automatic quantitation of localized in vivo 1H spectra with LCModel,” NMR Biomed, vol. 14, no. 4, pp. 260–4, Jun 2001, doi: 10.1002/nbm.698. [DOI] [PubMed] [Google Scholar]
- [31].Baker EH, Basso G, Barker PB, Smith MA, Bonekamp D, and Horska A, “Regional apparent metabolite concentrations in young adult brain measured by (1)H MR spectroscopy at 3 Tesla,” J Magn Reson Imaging, vol. 27, no. 3, pp. 489–99, Mar 2008, doi: 10.1002/jmri.21285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Horska A, Calhoun VD, Bradshaw DH, and Barker PB, “Rapid method for correction of CSF partial volume in quantitative proton MR spectroscopic imaging,” Magn Reson Med, vol. 48, no. 3, pp. 555–8, Sep 2002, doi: 10.1002/mrm.10242. [DOI] [PubMed] [Google Scholar]
- [33].Wasserstein R, Schirm A, and Lazar N, “Moving to a World Beyond “p < 0.05”,” The American Statistician, vol. 73, sup1, pp. 1–19, 2019, doi: 10.1080/00031305.2019.1583913. [DOI] [Google Scholar]
- [34].Brand A, Richter-Landsberg C, and Leibfritz D, “Multinuclear NMR studies on the energy metabolism of glial and neuronal cells,” (in eng), Developmental neuroscience, Research Support, Non-U.S. Gov’t vol. 15, no. 3–5, pp. 289–98, 1993. [Online]. Available: http://www.ncbi.nlm.nih.gov/pubmed/7805581. [DOI] [PubMed] [Google Scholar]
- [35].Bitsch A et al. , “Inflammatory CNS demyelination: histopathologic correlation with in vivo quantitative proton MR spectroscopy,” (in eng), AJNR. American journal of neuroradiology, Research Support, Non-U.S. Gov’t vol. 20, no. 9, pp. 1619–27, Oct 1999. [Online]. Available: http://www.ncbi.nlm.nih.gov/pubmed/10543631. [PMC free article] [PubMed] [Google Scholar]
- [36].Seitz D, Grodd W, Schwab A, Seeger U, Klose U, and Nagele T, “MR imaging and localized proton MR spectroscopy in late infantile neuronal ceroid lipofuscinosis,” AJNR Am J Neuroradiol, vol. 19, no. 7, pp. 1373–7, Aug 1998. [Online]. Available: https://www.ncbi.nlm.nih.gov/pubmed/9726485. [PMC free article] [PubMed] [Google Scholar]
- [37].Chattopadhyay S et al. , “An autoantibody inhibitory to glutamic acid decarboxylase in the neurodegenerative disorder Batten disease,” Hum Mol Genet, vol. 11, no. 12, pp. 1421–31, Jun 1 2002, doi: 10.1093/hmg/11.12.1421. [DOI] [PubMed] [Google Scholar]
- [38].Lim MJ et al. , “Distinct patterns of serum immunoreactivity as evidence for multiple brain-directed autoantibodies in juvenile neuronal ceroid lipofuscinosis,” Neuropathol Appl Neurobiol, vol. 32, no. 5, pp. 469–82, Oct 2006, doi: 10.1111/j.1365-2990.2006.00738.x. [DOI] [PubMed] [Google Scholar]
- [39].Dickson PI et al. , “Research challenges in central nervous system manifestations of inborn errors of metabolism,” Mol Genet Metab, vol. 102, no. 3, pp. 326–38, Mar 2011, doi: 10.1016/j.ymgme.2010.11.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Shapiro E et al. , “Neurocognitive clinical outcome assessments for inborn errors of metabolism and other rare conditions,” Mol Genet Metab, vol. 118, no. 2, pp. 65–9, Jun 2016, doi: 10.1016/j.ymgme.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].De Stefano N et al. , “Guidelines for using proton MR spectroscopy in multicenter clinical MS studies,” Neurology, vol. 69, no. 20, pp. 1942–52, Nov 13 2007, doi: 10.1212/01.wnl.0000291557.62706.d3. [DOI] [PubMed] [Google Scholar]
- [42].Krivitzky L, Babikian T, Lee HS, Thomas NH, Burk-Paull KL, and Batshaw ML, “Intellectual, adaptive, and behavioral functioning in children with urea cycle disorders,” Pediatr Res, vol. 66, no. 1, pp. 96–101, Jul 2009, doi: 10.1203/PDR.0b013e3181a27a16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lally PJ et al. , “Magnetic resonance spectroscopy assessment of brain injury after moderate hypothermia in neonatal encephalopathy: a prospective multicentre cohort study,” Lancet Neurol, vol. 18, no. 1, pp. 35–45, Jan 2019, doi: 10.1016/S1474-4422(18)30325-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Peters C et al. , “Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999,” Blood, vol. 104, no. 3, pp. 881–8, Aug 1 2004, doi: 10.1182/blood-2003-10-3402. [DOI] [PubMed] [Google Scholar]
- [45].Lauronen L et al. , “Delayed classic and protracted phenotypes of compound heterozygous juvenile neuronal ceroid lipofuscinosis,” Neurology, vol. 52, no. 2, pp. 360–5, Jan 15 1999, doi: 10.1212/wnl.52.2.360. [DOI] [PubMed] [Google Scholar]
- [46].Sitter B et al. , “High-resolution magic angle spinning and 1H magnetic resonance spectroscopy reveal significantly altered neuronal metabolite profiles in CLN1 but not in CLN3,” J Neurosci Res, vol. 77, no. 5, pp. 762–9, Sep 1 2004, doi: 10.1002/jnr.20123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data and code will be provided by the authors upon reasonable requests.