

Keywords: COUP-TF2, congenital heart disease, endothelial dysfunction, NR2F2, pulmonary arterial hypertension
Abstract
NR2F2 is expressed in endothelial cells (ECs) and Nr2f2 knockout produces lethal cardiovascular defects. In humans, reduced NR2F2 expression is associated with cardiovascular diseases including congenital heart disease and atherosclerosis. Here, NR2F2 silencing in human primary ECs led to inflammation, endothelial-to-mesenchymal transition (EndMT), proliferation, hypermigration, apoptosis-resistance, and increased production of reactive oxygen species. These changes were associated with STAT and AKT activation along with increased production of DKK1. Co-silencing DKK1 and NR2F2 prevented NR2F2-loss-induced STAT and AKT activation and reversed EndMT. Serum DKK1 concentrations were elevated in patients with pulmonary arterial hypertension (PAH) and DKK1 was secreted by ECs in response to in vitro loss of either BMPR2 or CAV1, which are genetic defects associated with the development of PAH. In human primary ECs, NR2F2 suppressed DKK1, whereas its loss conversely induced DKK1 and disrupted endothelial homeostasis, promoting phenotypic abnormalities associated with pathologic vascular remodeling. Activating NR2F2 or blocking DKK1 may be useful therapeutic targets for treating chronic vascular diseases associated with EC dysfunction.
NEW & NOTEWORTHY NR2F2 loss in the endothelial lining of blood vessels is associated with cardiovascular disease. Here, NR2F2-silenced human endothelial cells were inflammatory, proliferative, hypermigratory, and apoptosis-resistant with increased oxidant stress and endothelial-to-mesenchymal transition. DKK1 was induced in NR2F2-silenced endothelial cells, while co-silencing NR2F2 and DKK1 prevented NR2F2-loss-associated abnormalities in endothelial signaling and phenotype. Activating NR2F2 or blocking DKK1 may be useful therapeutic targets for treating vascular diseases associated with endothelial dysfunction.
INTRODUCTION
Chicken ovalbumin upstream promoter transcription factor 2 (COUP-TF2; NR2F2), a member of the steroid/thyroid nuclear receptor superfamily, participates in cellular differentiation, metabolism, neurogenesis, organogenesis, fertility, and inflammation (1). In the developing mouse embryo, NR2F2 has been implicated in cardiac development, angiogenesis, and artery/vein identity (2–4). Although these studies suggest that NR2F2 is primarily a venous marker that suppresses notch-mediated activation of pro-arterial gene expression, subsequent work has indicated a more complex role for NR2F2. Arterial marker expression was NR2F2 dependent in mouse embryonic stem cell-derived CD31+ cells (5). In zebrafish, nr2f2 knockdown reduced the expression of a subset of venous markers without affecting notch signaling or pro-arterial gene expression (6). Similar to zebrafish and in contrast to murine venous endothelium, NR2F2 heterodimerizes with PROX1 and has little influence on notch signaling during human lymphatic endothelial cell (EC) development (7). Furthermore, NR2F2 expression is not limited to human venous endothelium but instead has been found in human atria, coronary arteries, aorta (8), vascular smooth muscle (2), and immune cells (9), while a global histochemical approach identified NR2F2 expression in human tissues associated with the genitourinary, endocrine, respiratory, cardiac and immune systems (10). As such, our understanding of how NR2F2 affects cell fate and behavior is evolving, but remains incomplete.
Targeted deletion of Nr2f2 in mice is lethal by embryonic day 10. Although these embryos exhibit poorly developed atria and cardinal veins with enlarged hemorrhagic vessels, hypomorphic mutants are born with atrioventricular septal and valvular defects and abnormal coronary morphogenesis (3, 4). In an endothelial-specific knockout model, deletion of Nr2f2 leads to embryonic lethality attributable to vascular defects including hypoplastic endocardial cushions, widespread hemorrhage, and thin, dilated vessels (4). In humans, NR2F2 missense variants or 15q terminal deletions encompassing NR2F2 have been predominately associated with atrial or ventricular septal defects either alone or coupled with other congenital heart defects including aortic stenosis and coarctation of the aorta (8, 11–13). Notably, congenital heart disease (CHD) ascribed to excess flow and shear stress in the pulmonary vasculature is associated with the development of pulmonary arterial hypertension (PAH), but the occurrence of this complication is highly variable and difficult to predict (14). Although PAH is reversible following defect repair in some cases, repair does not uniformly prevent or reverse PAH despite similar flow abnormalities and clinical characteristics. This suggests that some patients might harbor unappreciated intrinsic defects in endothelial function that combined with aberrant flow may contribute to the development of PAH. In vitro, loss of NR2F2 in ECs has been linked to the induction of pro-atherogenic and pro-inflammatory genes and endothelial-to-mesenchymal transition (EndMT) (15). We hypothesized that NR2F2 loss in the endothelium from diverse adult vasculature beds may result in a dysfunctional endothelial phenotype prone to inflammation and pathologic vascular remodeling. If so, endothelial deficiency of NR2F2 may contribute to the development of diverse vascular diseases such as atherosclerosis and PAH. Understanding the role of NR2F2 in regulating EC homeostasis is the first step toward potential therapeutic applications. Here, the consequences of NR2F2 loss were investigated in multiple EC types for changes in 1) global gene expression, 2) inflammatory signaling, 3) EndMT, 4) proliferation, 5) migration, 6) apoptosis-resistance, and 7) reactive oxygen species production.
MATERIALS AND METHODS
Cell Culture and Treatments
Normal human lung microvascular endothelial cells (LMVECs) and human coronary artery endothelial cells (CAECs; Lonza, Walkersville, MD) were grown in endothelial cell growth medium (EGM-2MV or EGM-2; Lonza). Normal human pulmonary artery endothelial cells (PAECs) were grown on collagen-coated plates (Corning; Corning, NY). All primary cells were grown for a maximum of four passages and cells from multiple donors were used in all phenotypic assays. Cell donor characteristics are provided in Supplemental Table S1 (all Supplemental Material is available at http://doi.org/10.6084/m9.figshare.22009427). Human EA.hy926 cells (ATCC, Manassas, VA) were grown in DMEM supplemented with 10% heat-inactivated FBS (Life Technologies, Grand Island, NY). Recombinant human IFNα and γ (Millipore, Burlington, MA) as well as IFN β and DKK1 (PeproTech; East Windsor, NJ) were prepared according to the manufacturer’s directions. Cells were exposed to equivalent volumes of vehicle in each experiment.
NR2F2 Silencing
Prior to transfection (24 h), cells were seeded onto 35-mm plates. For primary endothelial cells, cells were transiently transfected with 10 nM of control siRNA, siRNA against NR2F2 or siRNA against BMPR2, CAV1 or DKK1 using Dharmafect 1. In double knockdown experiments, cells were transiently transfected with 20 nM of control siRNA, or 10 nM of control siRNA with 10 nM of siRNA against NR2F2 or DKK1 or 10 nM each NR2F2 and DKK1 siRNA. For the EA.hy926 line, cells were transiently transfected with 10 nM of an siRNA control (Dharmacon, Lafayette, CO) or siRNA against NR2F2 (Life Technologies) using lipofectamine RNAiMAX (Life Technologies). Cells were exposed to equivalent volumes of vehicle in each experiment.
Quantitative Real-Time PCR
Cells were seeded onto 35-mm plates and transiently transfected with siRNAs as described in NR2F2 Silencing. Post-transfection (48–72 h) cells were treated with vehicle or IFN (α, β, γ) as indicated for 4 h. Pulmonary arterial endothelial cells (PAECs) from idiopathic PAH and healthy control donors were provided by the Pulmonary Hypertension Breakthrough Initiative (PHBI) Network (Supplemental Table S2). These PAECs were isolated from vessels with diameters less than 1,000 μm and total RNA was purified from cells between passages 4 and 5. RNA was extracted using RNeasy Mini kits (Qiagen). Total RNA was quantified with a NanoDrop spectrophotometer and reverse transcription was performed with iScript cDNA Reverse Transcription kit (BioRad; Hercules, CA) using a thermal cycler. Gene expression assays were performed by quantitative real-time PCR (RT-PCR) using SYBR Green primers (Supplemental Table S3) on a ViiA7 cycler. Target gene expression was normalized to GAPDH or β-actin.
Oligonucleotide Microarrays
LMVECs and CAECs were transfected with siRNA as described in NR2F2 Silencing. Total RNA was isolated using RNeasy Mini kits. Quality of total RNA was evaluated using RNA 6000 Nano LabChip (Agilent 2100 Bioanalyzer). All samples had intact 18S and 28S ribosomal RNA bands with RNA integrity number values from 8.8 to 10 and RNA 260/280 ratios between 1.9 and 2.0. Gene expression levels were determined using Primeview Human Gene Expression Arrays. Total RNA (100 ng) was reverse transcribed and labeled (GeneChip 3′ IVT PLUS kit) according to the manufacturer’s protocols. Labeled and fragmented cRNA was hybridized for 16 h and arrays were stained and washed in the Affymetrix Fluidics Station 400 and scanned (Affymetrix 7G). Raw and robust multiarray average (RMA) normalized data were submitted to the National Center for Biotechnology Information Gene Expression Omnibus database (NCBI GEO; https://www.ncbi.nlm.nih.gov/projects/geo/index.cgi; accession no. GSE188465, GSE188466, GSE188467) and all data are MIAME compliant.
TRANSFAC Analysis
The “no set” was selected based on 1) a P value >0.1 for the interaction term (siCTRL vs. siNR2F2), 2) an overall mean probe intensity >3.5, and 3) a mean standard error <0.0092. For the TRANSFAC analysis, a transcriptional window of −500/+100 was used along with a P value cutoff of 0.01.
Luciferase Reporter Assay
Luciferase reporter assays were performed on EA.hy926 cells transfected with firefly luciferase reporter (pGL4.11[luc2P]; Promega) construct driven by a portion of the DKK1 promoter (−2,000/+100 from the transcriptional start site). Briefly, 5 × 104 cells were seeded on 12-well plates in normal growth medium without antibiotics for 24 h before transient siRNA transfection in normal growth medium without antibiotics. Cells were then transiently transfected with 20 nM control siRNA as described in NR2F2 Silencing using RNAiMAX. Post recovery (24 h), cells were transfected with 200 ng/well of the DKK1 promoter reporter along with 100 ng/well pGL4.74[hRluc TK] as a transfection efficiency control. Following recovery (24 h), luciferase reporter activity was measured using a dual-luciferase reporter assay system (Promega) on a GloMax (Promega).
Western Blotting
Post-transfection (48–72 h), cells were lysed on ice with RIPA buffer supplemented with Halt Protease and phosphatase inhibitors (Life Technologies). Lysates were cleared by centrifugation (13,000 g for 10 min at 4°C). Samples were boiled for 5 min in NuPAGE LDS sample buffer and NuPAGE reducing agent (Life Technologies). Lysates were run on 4%–12% Bis-Tris gels and then transferred using an iBlot at 20 V for 7 min (Life Technologies). Blots were blocked with ECL Primer blocking agent (GE Healthcare; Chicago, IL) and stained with antibodies (Supplemental Table S4) overnight at 4°C and washed three times for 10 min each with 0.1% Tween-20 in PBS. Blots were then incubated with HRP-conjugated goat anti-rabbit or goat anti-mouse antibody (Cell Signaling; Danvers, MA) for 1 h at room temperature (RT), re-washed as above and developed with an ECL substrate using the ChemiDoc MP Imaging System (BioRad). Image Lab software (v6.1; BioRad) was used for densitometry analysis.
ELISAs
Cell culture or human plasma samples obtained from healthy donors and PAH patients (Supplemental Table S5) were centrifuged at 1,000 g for 15 min at 4°C and then subjected to ELISA for human DKK1 (1:16–1:32 dilution for cell culture supernatants, undiluted for human plasma samples), CCL5 (undiluted), CXCL10 (1:30 dilution; R&D Systems; Minneapolis, MN), or IL6 (1:2 dilution; BioTechne; Minneapolis, MN). All subjects were enrolled in NIH institutional review board approved protocols (99-CC-0168 and 13-CC-0012, respectively) and all participants provided written informed consent before participation in the study and the investigation conformed to the principles outlined in the Declaration of Helsinki. Registry to Evaluate Early And Long-term PAH Disease Management (REVEAL) 2.0 (16) and REVEAL Lite 2 scores (17) were determined for each patient at the time of sample collection. Etiology of PAH, age, sex, estimated glomerular filtration rate, New York Heart Association (NYHA) functional class, heart rate, systolic blood pressure, all-cause hospitalizations within the past 6 mo, 6-min walk distance, NT-proBNP, presence of a pericardial effusion on echocardiogram, and the diffusing capacity for carbon monoxide were available for all 15 patients. Hemodynamic measures by right heart catheterization are reported from the date closest to blood sample collection. Data from right heart catheterizations are only included in REVEAL 2.0 scores when completed within 12 mo of sample collection.
Immunofluorescence
PAECs (5–6 × 104) were seeded onto collagen-coated 8 chamber slides (Sigma-Aldrich; St. Louis, MO) and subjected to siRNA transfection following 24 h recovery as described in NR2F2 Silencing. After 48 h, cells were washed with basal medium and fixed with 4% formaldehyde for 20 min at RT, blocked with goat serum (Sigma-Aldrich) with 0.3% Triton X-100 for 40 min at RT, then washed three times with PBS supplemented with 0.1% BSA. Slides were then incubated with primary antibodies overnight at 4°C then washed three times as above, incubated with secondary antibodies for 1 h, washed three times as above and mounted using mounting medium with DAPI (Sigma-Aldrich). Stained cells were examined using epifluorescence (DMi8 epifluorescence; Leica, IL) at ×20 magnification and analyzed using ImageJ. Specificity was determined with duplicate wells stained with secondary antibody alone.
Proliferation Analysis
PAECs (1 × 106) were seeded onto collagen-coated T75 flasks and subjected to siRNA transfection following 24 h recovery as described in NR2F2 Silencing. After 72 h, cells were harvested and re-plated on collagen-coated 96-well plates at 4 × 103 cells/well in 200 μL of medium. The day 0 plate was harvested 4 h after cell seeding to show initial cell density. Cells were then fed with fresh media every 48 h. Remaining cells were harvested at days 1–4 as follows: medium was aspirated and wells washed with PBS. Microplates were subsequently frozen at −70°C until the time of analysis. For analysis, plates were thawed at RT, then 200 μL of the CyQUANT GR dye/cell-lysis buffer (LifeTechnologies) was added to each well, mixed gently, and incubated for 5 min at RT, protected from light. Cell lysate (150 μL) was transferred to an OptiPlate and fluorescence was measured using a microplate reader. For the CellTiter-Glo assay, PAECs (1 × 104) were seeded onto collagen-coated 96-well clear bottom white-walled optical plates (LifeTechnologies) and treated with recombinant human DKK1 as above. After 48 h, 100 μL of CellTiter-Glo 2.0 reagent was dispensed into each well and luminescence was recorded after 10 min RT incubation.
Apoptosis-Resistance Assays
LMVEC apoptosis was evaluated by Caspase-Glo 3/7 Assay (Promega; Madison, WI). LMVECs were grown and subjected to NR2F2 knockdown by siRNA as described in NR2F2 Silencing for 48 h, then seeded on 96-well plates at 5,000 cells/well. LMVECs were then challenged with serum and growth factor withdrawal for 24 h to trigger apoptosis.
Migration Assay
Cell migration assays were performed as described previously (18) following siRNA transfection as described in NR2F2 Silencing for 48 h.
Lactate Assay
For lactate analysis, PAECs were cultured in 6-well plates and transfected as described in NR2F2 Silencing after 24 h recovery. Post-transfection (48 h), cell culture supernatants were collected, diluted 1:40, and subjected to Lactate-Glo (Promega) as per manufacturer recommendations.
CellROX
PAECs (5 × 104) were seeded onto collagen-coated 12-well plates and subjected to siRNA transfection following 48 h recovery as described in NR2F2 Silencing. Cells were then treated with vehicle (ethanol) or menadione (1 μM, 1 h; Sigma). CellROX Deep Red (Life Technologies) was then added at a final concentration of 5 μM for 30 min at 37°C in the dark along with Hoerscht 33342 (10 μM; Life Technologies). Cells were washed three times with PBS and stained cells were examined using an epifluorescence (DMi8 epifluorescence; Leica, IL) at ×20 magnification. Images were analyzed using ImageJ.
TransAM
PAECs (1 × 106) were seeded onto collagen-coated 100-mm plates and subjected to siRNA transfection following 48 h recovery as described in NR2F2 Silencing. Nuclear extracts were prepared using the Nuclear Extract kit (Active Motif) and quantified using the Pierce BCA Protein Assay Kit. Nuclear extracts (5 μg/well) were then subjected to a TransAM assay using a TransAM STAT Family kit per manufacturer’s guidelines.
Statistical Analysis
Signal intensity values for the oligonucleotide arrays were normalized and log2 transformed using the robust multiarray average (RMA) method in Thermo Fisher Transcriptome Analysis Console (v. 4.0.1.36). Probe set IDs were annotated using PrimeView Human Gene Expression Array annotation file v. 36 (release date 03/30/16). The transformed data matrix with the resulting signal intensity values for each of 49,372 probe sets was imported to R and subjected to principal components analysis to visualize the relative location of chips in a low-dimensional space allowing for the detection of outliers or other relevant patterns. A linear model with fixed effects for replication (experiment day) and knockdown were fit to the expression data for each gene using the Bioconductor R package limma (19). As part of each linear model analysis, P values were obtained for the knockdown effect and differentially expressed transcripts were selected by the post hoc t test for the comparison of siCTRL and siNR2F2 groups. All P values are two-sided and false discovery rate (FDR) adjustment was used to declare significance. For quantitative RT-PCR and IFN-stimulated reporter activity, results were normalized to 100% of that seen in the absence of stimulus (vehicle only). Data are presented as either treatment versus vehicle or siRNA knockdown versus siRNA control as normalized mean fold changes ± geometric standard deviation. The raw data for the Ct values for the target genes were normalized by the Ct values of the housekeeping gene. A linear model with appropriate blocking was used to test the effects of nominal factors and their interactions. Post hoc contrasts of interest were tested directly when significant interactions precluded the reporting of main effects. Paired t tests were only used for two-condition comparisons, as appropriate. All P values are two-sided and P < 0.05 was considered significant. All statistical analyses were carried out on normalized data using JMP v. 14 (SAS Institute Inc., Cary, NC).
RESULTS
Global Gene Expression Profiling of NR2F2 Silencing and Interferon-Biased Endothelial Inflammation
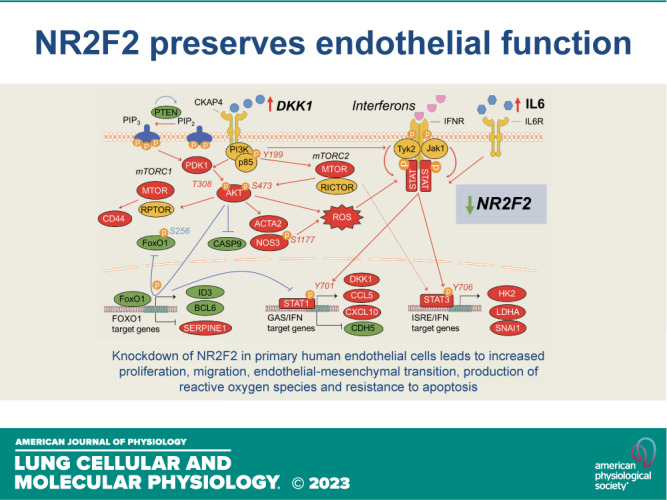
NR2F2 is expressed in normal, primary human LMVECs (Fig. 1A). The global effect of NR2F2 loss was assessed by microarray profiling following siRNA targeting wherein NR2F2 protein levels were reduced by ≥85% (Fig. 1A). Selection of differentially regulated transcripts [false discovery rate (FDR) <1% and fold change (FC) >1.5] following NR2F2 knockdown identified 814 annotated, nonredundant transcripts of which 380 were upregulated and 434 were downregulated (Supplemental Table S6). An analysis of overrepresented canonical pathways among differentially regulated genes following NR2F2 knockdown using Ingenuity Pathway Analysis (IPA) identified interferon (IFN) signaling, STAT3 signaling, T-helper (Th)-1 and Th2 activation and notch signaling (Fig. 1B). In addition, an analysis of transcription factor binding sites among genes affected by NR2F2 silencing indicated an over-representation of IFN regulatory factors (IRFs), BCL6, HMGA1, NF-κB, and sox-related factor sites that are implicated in a wide variety of biologic processes including inflammation, immune response, proliferation, apoptosis, migration, and cardiac development (Table 1). In agreement with these findings, NR2F2-silenced LMVECs displayed a strong inflammatory signature by IPA, and also by Gene Set Enrichment Analysis (GSEA), which evaluates all interrogated transcripts independent of specific selection criteria (Fig. 1C; Supplemental Table S7). Collectively, these NR2F2 loss-induced expression changes reflect the upregulation of many cytokines, chemokines, and adhesion molecules (Supplemental Table S7). Moreover in vivo, this endothelial inflammatory response would likely recruit and activate circulating immune cells (Fig. 1D).
Figure 1.
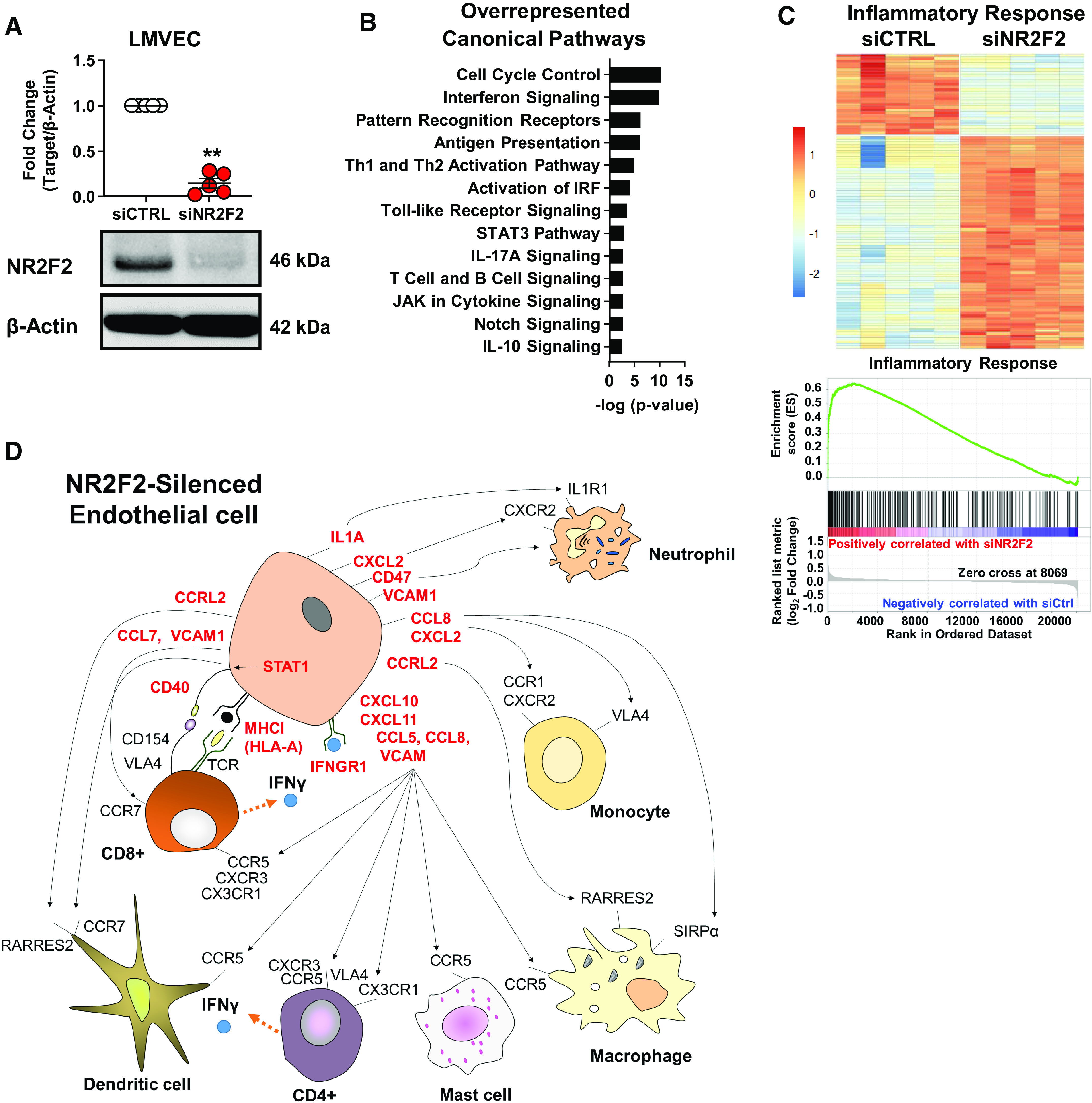
NR2F2 silencing activates an inflammatory response and corresponding gene signature. Human primary lung microvascular endothelial cells (LMVECs) were transiently transfected with either control (siCTRL) or NR2F2 siRNA (siNR2F2) for 48 h. A: Western blots demonstrate NR2F2 protein levels after knockdown. Quantification of NR2F2 protein normalized to β-actin and expressed as means ± SD fold-change from siCTRL (n = 5). **P ≤ 0.01 vs. siCTRL by paired t test. B: RNA from cells transfected as above was subjected to microarray using Affymetrix PrimeView human arrays. Ingenuity Pathway Analysis (IPA) of microarray results using a false discovery rate (FDR) ≤1% and fold change (FC) >1.5 (814 annotated, nonredundant transcripts; Supplemental Table S6) revealed canonical pathways significantly associated with NR2F2 knockdown using a P ≤ 0.05, plotted on the x-axis as −log(P value). C: heatmap (top) of IPA-annotated inflammatory response genes (n = 104; P = 4.80E−16; Supplemental Table S7) among the differentially regulated transcripts in NR2F2-silenced LMVECs. Gene Set Enrichment Analysis also identified inflammatory response as an overrepresented hallmark gene set in NR2F2 loss affected target genes (bottom; normalized enrichment score = 2.74; FDR < 0.0001). The running enrichment score (y-axis) and position of gene set members on the rank-ordered list (x-axis) is shown. D: schematic of cytokines, chemokines, and adhesion molecules upregulated (red; FDR ≤ 1% and FC > 1.5) in LMVECs as determined by genome-wide, microarray profiling following NR2F2 loss (Supplemental Table S6); see Supplemental Table S7 for full annotations and their potential impact on immune cell recruitment and activation.
Table 1.
Transcription factor binding sites among promoters of differentially regulated gene set following NR2F2 knockdown in primary human lung microvascular endothelial cells
| Matrix Name | Associated Transcription Factor(s) | Yes/Noa | Matched Promoter P Value | Signaling Pathways Affected |
|---|---|---|---|---|
| V$IRF1_Q5 | IRF factors* | 21.76 | 3.39E-06 | Inflammation |
| V$BCL6_Q3_01 | BCL6 factors | 6.74 | 2.82E-02 | Apoptosis |
| V$HMGIY_Q3 | HMGA factors | 5.70 | 9.28E-03 | apoptosis, wnt signaling |
| V$CDX2_01 | CDX group | 4.49 | 8.55E-03 | Proliferation |
| V$RELA_Q6 | NF-κB-related factor | 4.14 | 2.39E-03 | inflammation, apoptosis |
| V$SOX2_Q3_01 | Sox-related factors* | 3.52 | 6.44E-03 | wnt signaling, migration |
| V$NFY_Q3 | NF-Y | 3.41 | 9.06E-07 | Proliferation |
| V$AIRE_01 | AIRE | 3.11 | 3.73E-03 | immune response |
| V$E2F_Q6_01 | E2F related factors* | 3.03 | 1.65E-03 | apoptosis, proliferation |
| V$HOMEZ_01 | HOMEZ | 2.33 | 3.36E-03 | cardiac development |
| V$BLIMP1_Q4 | Blimp-1* | 2.24 | 6.21E-03 | Inflammation |
| V$ERALPHA_01 | ER group | 2.12 | 2.71E-02 | apoptosis, proliferation |
| V$HMX1_02 | HMX group | 1.85 | 7.90E-03 | apoptosis, wnt signaling, migration |
Yes/no score, ratio of transcription factor binding sites of a particular matrix in the set of upregulated genes in NR2F2 knockdown from primary human microvascular cells from the lung relative to control siRNA (yes set) compared with the background set of genes (no set). IRF, IFN-regulatory factors; HMGA, High Mobility Group at-Hook; CDX, caudal type homeobox 1; Sox, SRY-box transcription factor; NF-Y, nuclear transcription factor Y; blimp-1, B lymphocyte-induced maturation protein-1; ER, estrogen receptor; HMX, H6 family homeobox 1* denotes transcription factors differentially affected by NR2F2-silencing in the associated microarray data (Supplemental Table S6).
Notably, IPA identified IFN and STAT signaling as canonical pathways (Fig. 1B) and IFNs (α, β, γ), STAT1, IRF1, and IRF7 as significant upstream regulators (Table 2) indicating that the inflammatory response associated with NR2F2 knockdown was strongly IFN-biased. IFNα was the most significant upstream activator predicted by IPA (P = 4.71E−65) and the predicted influence of NR2F2 silencing on IFNα network genes was found by both IPA (Fig. 2A, top; Supplemental Table S8) and GSEA (Fig. 2A, bottom). Consistent with constitutive activation of the IFN/STAT pathway, immunoblotting of STAT1 and phosphorylated STAT1 (pSTAT1 Y701) revealed increased STAT1 expression and activation in NR2F2-silenced LMVECs (Fig. 2B). In addition, IFN target gene expression including STAT1, CCL5, CXCL10, CCL7, and CXCL11 was increased in NR2F2-silenced LMVECs in the absence and presence of exogenous IFNα/β stimulation (Fig. 2C).
Table 2.
Significant upstream regulators of target genes differentially affected by siNR2F2 loss in primary, human lung microvascular endothelial cells
| Upstream Regulator | Activation z-Score | P |
|---|---|---|
| IFNα | 7.300 | 4.71E-65 |
| IFNβ | 6.392 | 1.52E-32 |
| IFNγ | 10.153 | 9.09E-64 |
| IFNλ | 7.414 | 1.31E-60 |
| IFNAR | 6.256 | 1.02E-37 |
| STAT1 | 6.868 | 3.05E-48 |
| IRF1 | 5.959 | 4.13E-43 |
| IRF7 | 7.343 | 2.61E-43 |
Figure 2.
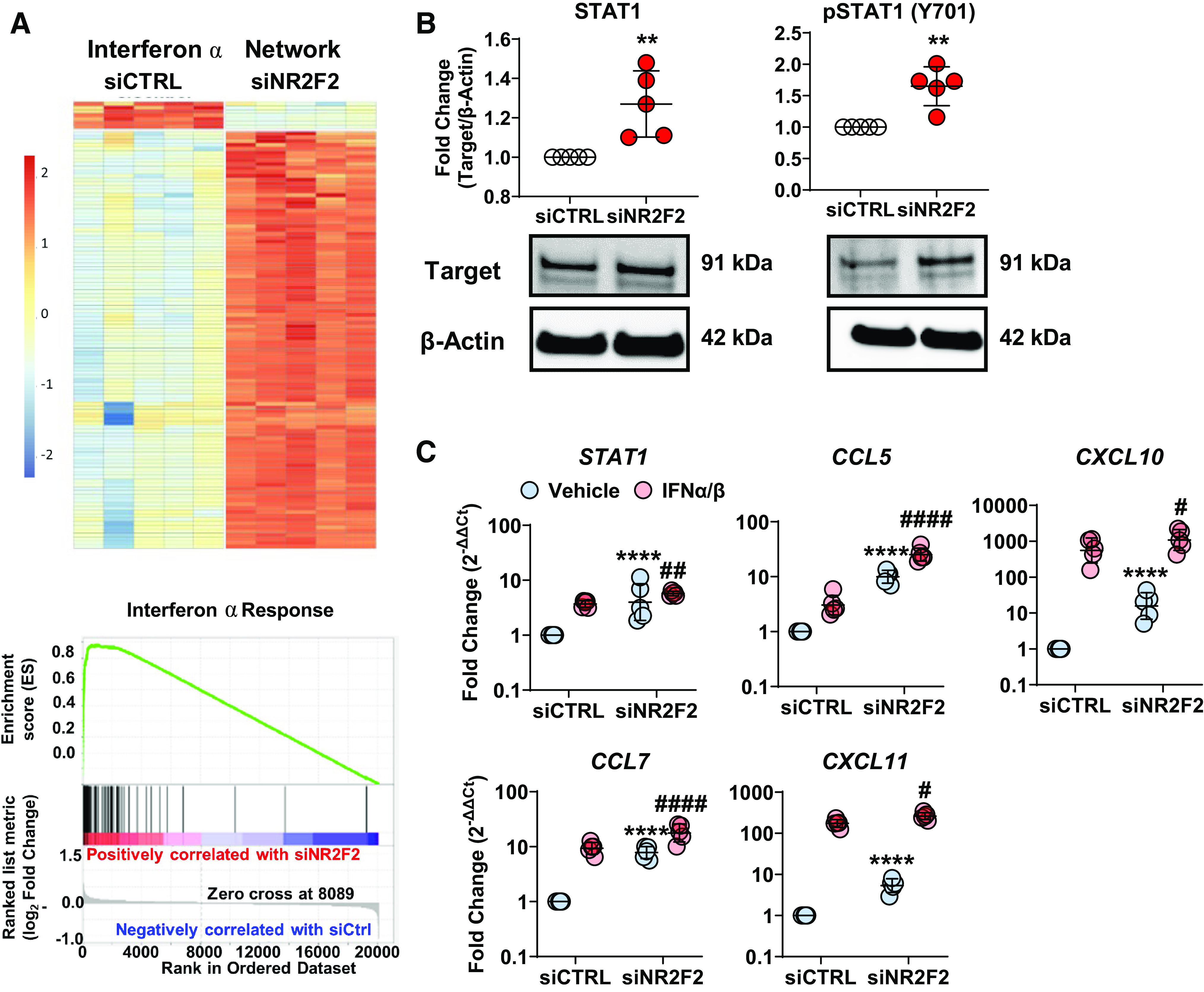
Upregulation of IFN pathway signaling and gene targets following NR2F2 silencing. Human primary human lung microvascular endothelial cells (LMVECs) were transiently transfected with either control (siCTRL) or NR2F2 siRNA (siNR2F2) for 48 h. A: RNA from cells transfected as above was subjected to microarray using Affymetrix PrimeView human arrays. Heatmaps of differentially expressed IFNα target genes, as annotated by Ingenuity Pathway Analysis (IPA), are shown (top; red = upregulated transcripts; blue = downregulated transcripts). IPA analysis predicted that IFNα targets were activated (115 unique transcripts; P = 5.25E−64; Supplemental Table S8). Gene Set Enrichment Analysis also identified IFNα as a key regulator of target genes affected by NR2F2 loss (bottom; normalized enrichment score = 3.37; false discovery rate < 0.001). The running enrichment score (y-axis) and position of gene set members on the rank-ordered list (x-axis) is shown. B: STAT1 expression and activation (i.e., phosphorylation at Y701) was increased in NR2F2 knockdown cell lysates (n = 5). C: effect of NR2F2 knockdown without or with IFNα/IFNβ exposure (4 h) on expression of IFN regulated genes, STAT1, CCL5, CXCL10, CCL7, and CXCL11, in LMVECs (n = 5). Data are presented as means ± SD (or geometric mean for quantitative RT-PCR) **P ≤ 0.01, ****P ≤ 0.0001 siNR2F2 vs. siCTRL; #P ≤ 0.05, ##P ≤ 0.01, ####P ≤ 0.0001 siNR2F2 vs. siCTRL (IFNα/IFNβ) by paired t test (B) or two-way ANOVA (C).
Since NR2F2 expression is necessary for normal coronary artery morphogenesis (4) and has been linked to inflammation in atherosclerosis (15), as has interferon signaling (20), it was pertinent to assess whether the effects of NR2F2 silencing in LMVECs were also seen in normal, primary human CAECs. Genome-wide expression profiling of CAECs following knockdown of NR2F2 (>87%; Supplemental Fig. S1A) led to the differential expression (FDR <1%; FC > 1.5) of 767 annotated, nonredundant gene transcripts of which 378 were upregulated and 389 were downregulated (Supplemental Table S9). Notably, 55% of the gene signature associated with NR2F2 loss in CAECs was identical to and directionally concordant with that seen in LMVECs (Supplemental Fig. S1B). Similar to our results in LMVEC, activation of IFN/STAT signaling was the predominant feature of NR2F2 silencing in CAECs (Supplemental Fig. S1, C and D).
Next, human EA.hy926 cells, which express NR2F2 (Supplemental Fig. S2A), were used to generate stable lines expressing luciferase reporter genes driven by IFNα/IFNβ or IFNγ response elements in order to test whether NR2F2 loss was associated with the activation of IFN response elements in target promoters. Although NR2F2 silencing alone activated the IFNα/β reporter, knockdown also significantly augmented the IFNα/β- and IFNγ-induced luciferase activity of both IFN reporters, respectively (Supplemental Fig. S2B). Recapitulating the gene signatures from LMVECs and CAECs, NR2F2 knockdown in EA.hy926 cells similarly upregulated STAT1, pSTAT1 (Y701), and IFN target genes including CCL5, IFITM1, and CCL7 (Supplemental Fig. S2, C and D).
STAT activation, IFN signaling, and endothelial inflammation have been implicated in PAH pathogenesis using blood outgrowth ECs and pulmonary artery ECs (PAECs) (21, 22), as well as in the pathogenesis of Eisenmenger syndrome (23). Similarly, a meta-analysis of genome-wide expression profiling studies in PAH revealed enrichment of interferon (α, β, γ) signaling as well as IRF and STAT binding sites among top-upregulated PAH genes (24). Therefore, it was of interest to determine whether NR2F2-silenced PAECs also exhibited increased STAT activity and IFN signaling similar to that seen in other ECs. Recapitulating our results from other ECs, knockdown of NR2F2 in PAECs (>85%; Supplemental Fig. S3A) resulted in STAT1 activation (Supplemental Fig. S3B), increased STAT1 DNA binding (Supplemental Fig. S3C) as well as the induction of prototypic IFN target genes across all four of these human EC-types (Table 3). Increases in CCL5, CXCL10, and IL6 following NR2F2 knockdown were also confirmed in PAEC culture supernatants by ELISA (Supplemental Fig. S3D). Taken together, these results implicate NR2F2 as a regulator of IFN signaling in ECs from both large and small vessels and that loss of NR2F2 primes ECs for an exaggerated response to exogenous IFN stimulation.
Table 3.
Changes in IFN regulated gene expression following NR2F2 silencing in various types of endothelial cells
| LMVEC |
CAEC |
PAEC |
EA.hy926 |
|||||
|---|---|---|---|---|---|---|---|---|
| Gene Symbol | Fold-Changea | P Value | Fold-Changea | P Value | Fold-Changea | P Value | Fold-Changea | P Value |
| CCL5 | 10.0 | <0.001 | 3.2 | <0.001 | 3.9 | 0.012 | 3.5 | <0.001 |
| CCL7 | 7.8 | <0.001 | 3.2 | 0.026 | 2.3 | 0.052 | 2.7 | <0.001 |
| CD40 | 1.5 | 0.006 | 1.9 | <0.001 | 1.8 | 0.002 | NT | |
| CD274 | 2.5 | 0.005 | 1.9 | 0.003 | 1.5 | 0.230 | 2.0 | <0.001 |
| CXCL10 | 15.7 | <0.001 | 6.6 | <0.001 | 3.0 | 0.028 | 0.8 | 0.002 |
| CXCL11 | 5.4 | <0.001 | 4.5 | 0.002 | 0.6 | 0.002 | 3.2 | <0.001 |
| IFITM1 | 1.5 | <0.001 | 2.6 | 0.001 | 1.3 | 0.346 | 4.7 | <0.001 |
Results are shown as fold changes of siNR2F2 vs. siCTRL. LMVEC, lung microvascular endothelial cell; CAEC, coronary artery endothelial cell; PAEC, pulmonary artery endothelial cell; NT, not tested, P < 0.05 bolded.
Phenotypic Effects of NR2F2 Silencing in Endothelial Cells
Inflammation, including increased IFNγ signaling, has been linked to EndMT through alteration of TGFβ or hypoxic signaling mechanisms, each of which has been associated with cardiovascular disease (25). In addition, NR2F2 loss has been shown to induce EndMT in human saphenous vein ECs (15), as well as alter EndMT in vivo (4). Therefore, we examined relative changes in the expression of endothelial and mesenchymal markers in NR2F2-silenced PAECs. NR2F2 silencing led to decreased expression of endothelial markers including CDH5 (vascular endothelial cadherin), VWF and CD31 (also known as PECAM1; Supplemental Fig. S3E). Conversely, there was a concurrent increase in the expression of mesenchymal markers such as ACTA2 (α-smooth muscle actin), CD44, and S100A4 (fibroblast-specific protein-1). Consistent with changes in gene expression, immunofluorescence of EndMT markers in NR2F2-silenced PAECs revealed decreased expression of CDH5, VWF, and CD31 coupled with increased expression of ACTA2, SNAI1/SLUG, and CD44 (Fig. 3A). Likewise, CDH5 protein expression was decreased, while ACTA2, CD44, and SNAI1 levels increased by Western blot of whole cell lysates (Supplemental Fig. S3F). Similar to PAECs, EndMT markers were altered in NR2F2-silenced LMVECs (Supplemental Fig. S4A).
Figure 3.
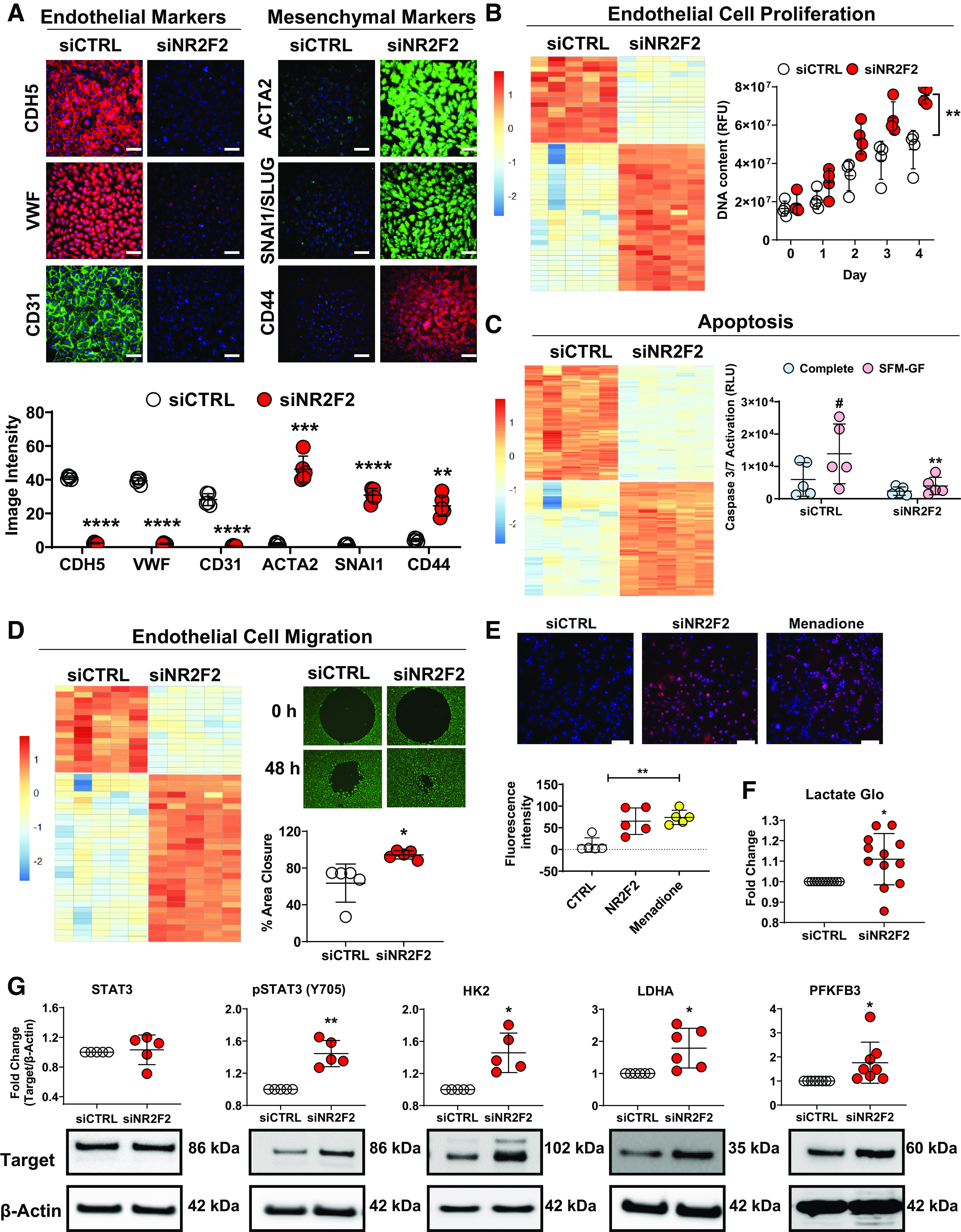
NR2F2-silenced human pulmonary artery endothelial cells (PAECs) and lung microvascular endothelial cells (LMVECs) exhibit endothelial to mesenchymal transition with increased proliferation and migration, resistance to apoptosis and altered metabolism. PAECs were transiently transfected with either control (siCTRL) or NR2F2 siRNA (siNR2F2) for 72 h. A: NR2F2 loss led to the downregulation of endothelial [CDH5 (VE-cadherin), VWF and CD31 (PECAM1)] and upregulation of mesenchymal [ACTA2 (α-smooth muscle actin), SNAI1/SLUG and CD44] marker by immunofluorescent microscopy (n = 5; top). Fluorescence intensity is quantified (bottom). Scale bar, 100 µm. B: heatmap (left) of Ingenuity Pathway Analysis (IPA)-annotated genes related to proliferation (n = 48; P = 2.61E−11; Supplemental Table S10) among the differentially regulated transcripts in NR2F2-silenced LMVECs (814 annotated, nonredundant transcripts; false discovery rate ≤1% and fold-change >1.5; Supplemental Table S6). NR2F2 knockdown in PAECs (72 h) also increased proliferation as assessed by CyQUANT cell proliferation assay (right; n = 4). C: heatmap (left) of IPA-annotated genes related to apoptosis (n = 293; P value 1.21E−32; Supplemental Table S10) among the gene expression changes in LMVECs. Following NR2F2 knockdown in LMVECs (48 h), LMVECs were resistant to apoptosis as assessed by caspase 3/7 reporter assay (right) following 24 h of continued growth in serum-free media without growth factors (SFM-GF; n = 5). D: heatmap (left) of IPA-annotated genes related to endothelial cell migration (n = 44; P = 3.16E−9; Supplemental Table S10) among the gene expression changes in LMVECs. NR2F2 knockdown in LMVECs (48 h) resulted in hypermigration as assessed by cell migration assay. After NR2F2 knockdown, LMVECs were transferred to 96-well plates containing a circular insert and percent area closure was quantified 48 h later (n = 5). E: PAECs were transiently transfected with either control (siCTRL) or NR2F2 siRNA (siNR2F2) for 48 h and stained with CellROXDeep Red and Hoescht (blue; 10 μM; upper). Nontransfected duplicate wells were treated with menadione (1 μM), an oxidant stress inducer, and stained as above. Scale bar, 100 µm. Fluorescence intensity was quantified (n = 5; lower). F: following transfection as above, PAEC cell culture supernatants (n = 11) were subjected to Lactate-Glo assay. G: Western blots of whole cell lysates from PAECs, transfected as above, for total and phosphorylated STAT3 (Y706; n = 5), HK2 (n = 5), LDHA (n = 6), and PFKFB3 (n = 8). Data are presented as means ± SD. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001 siNR2F2 vs. siCTRL; #P ≤ 0.05, SFM-GF vs. complete medium by paired t test (A, D, F, G) or two-way ANOVA (B, C) or one-way ANOVA (E).
EndMT promotes a proliferative and hypermigratory cellular phenotype (26) that has been linked to the initiation of atherosclerosis (25, 27) as well as vascular remodeling and late-stage obliterative lesions in PAH and CHD-PAH (28–30). Importantly, overexpression of NR2F2 repressed proliferation in human umbilical vein ECs (HUVECs) (31) while mice with cardiomyocyte-specific NR2F2 ablation exhibit increased atrial size due to cardiomyocyte proliferation (2). Similarly, in NR2F2-silenced LMVECs, thematic analysis of differentially regulated genes using IPA, identified signatures for cell proliferation (P = 2.61E−11), apoptosis (P = 1.21E−32), and migration (P = 3.16E−9; Fig. 3, B–D; Supplemental Table S10). To confirm these findings, phenotypic assays for proliferation, migration, and apoptosis were performed. PAECs were found to proliferate more rapidly following NR2F2 knockdown (43%–60% increase in DNA content 96–168 h post-transfection, Fig. 3B). Similarly, NR2F2 knockdown in LMVECs led to an apoptosis-resistant phenotype tolerant of serum and growth factor withdrawal (Fig. 3C). Furthermore, NR2F2-silenced LMVECs exhibited increased migration with 94.4 ± 1.5% area closure over 48 h compared with 63.6 ± 4.6% closure in control siRNA transfected cells (Fig. 3D).
Since the production of reactive oxygen species (ROS) has been linked to the promotion of EndMT (32, 33) as well as proliferation and migration (34), ROS levels were measured following NR2F2 silencing in PAEC. Importantly, PAECs exhibited increased production of ROS following NR2F2 knockdown (Fig. 3E). Since ROS has been shown to be associated with metabolic regulation (35), increased glycolysis in pulmonary hypertension (36) as well as STAT signaling (37), lactate levels were measured following NR2F2 silencing in PAEC. NR2F2 silencing in PAECs appeared to increase glycolysis as evidenced by increased lactate levels (Fig. 3F) as well as increased STAT3 activation (measured by tyrosine 705 phosphorylation; Fig. 3G), an inducer of glycolysis (38). Expression levels of glycolytic genes were also increased including HK2, LDHA, and PFKFB3 (Fig. 3G). These effects on STAT3 activation and HK2 expression were similarly recapitulated in NR2F2-silenced LMVECs (Supplemental Fig. S4B). Taken together, NR2F2-silenced ECs undergo EndMT and exhibit a proliferative, hypermigratory, and apoptosis-resistant phenotype coupled with oxidative stress and increased glycolytic gene expression
Aberrant AKT/FOXO1 Signaling in NR2F2-Silenced Endothelial Cells
Sustained JAK/STAT signaling has been shown to require AKT activation (39), a pro-proliferative pathway that facilitates EndMT (28). AKT activation has likewise been implicated in ROS production and glycolysis (36). Therefore, we examined whether AKT signaling was altered in NR2F2-silenced ECs. Analysis of cell lysates from PAECs confirmed that NR2F2 silencing resulted in AKT activation (measured by phosphorylation at serine 473; Fig. 4A). In addition, FOXO1, a proliferation inhibitor that is a downstream target of AKT and STAT3, exhibited increased phosphorylation at serine 256 following NR2F2-silencing, which has been shown to inhibit its transcriptional activity by blocking nuclear localization (40). Increased phosphorylation of NOS3 at serine 1177 (eNOS; Fig. 4B), another direct target of AKT (41), was further evidence of AKT pathway activation as a consequence of NR2F2 silencing. AKT activation along with NOS3 phosphorylation and inhibition of FOXO1 was recapitulated in NR2F2-silenced LMVECs (Supplemental Fig. S4B).
Figure 4.
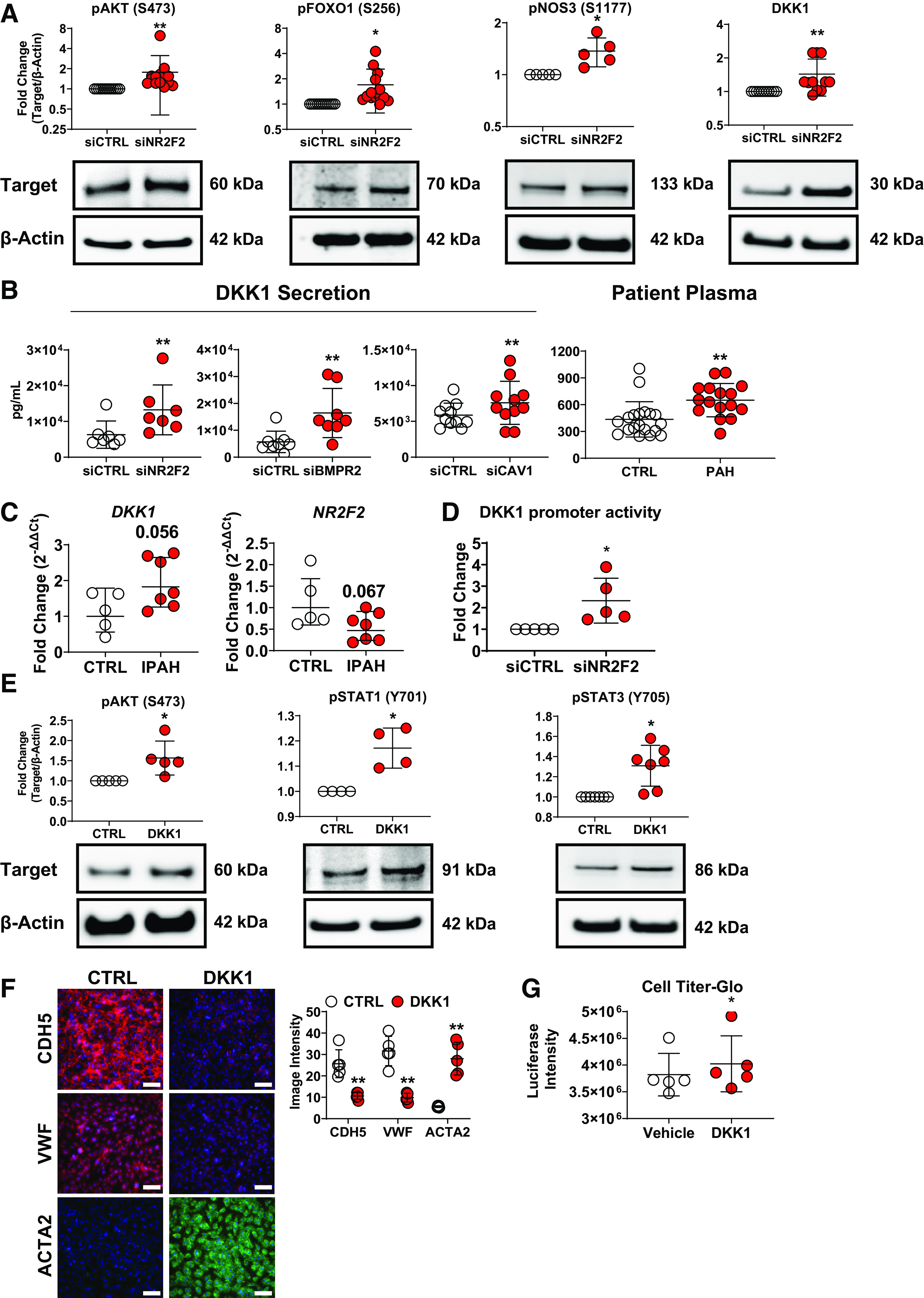
NR2F2 silencing in human pulmonary artery endothelial cells (PAECs) activates AKT and upregulates DKK1, a cytokine increased in BMPR2- and CAV1-silenced ECs and in patients with pulmonary arterial hypertension (PAH). PAECs were transiently transfected with either control (siCTRL) or NR2F2 siRNA (siNR2F2) for 48 h. A: Western blots and quantification of whole cell lysates consistently demonstrated activation of AKT (phosphorylation at S473) following knockdown of NR2F2 in pulmonary artery ECs (PAECs; n = 13). In addition, phosphorylation of AKT targets was increased, including FOXO1 at S256 (n = 14), an inhibitory event, and NOS3 at S1177 (n = 5). Increased expression of DKK1, is shown in Western blots of NR2F2-silenced PAECs (n = 11). B: DKK1 levels were also increased in culture supernatants from NR2F2-silenced PAECs (n = 7) as measured by ELISA. Notably, increased DKK1 secretion was also detected in both BMPR2- (n = 8) and CAV1-silenced (n = 11) PAEC models of PAH-associated EC dysfunction. Finally, plasma DKK1 levels (far right) were significantly higher in PAH patients (n = 16) compared with healthy controls (CTRL; n = 19; Supplemental Table S5). C: comparison of gene expression between PAECs from idiopathic PAH (IPAH) patients (n = 7) vs. unused donor lung controls (n = 5) by quantitative RT-PCR (DKK1 and NR2F2). Data are presented as geometric means. P values shown are for unpaired t tests of patients with IPAH vs. unused donor lung controls. D: luciferase activity of EA.hy926 cells expressing an luciferase reporter driven by a portion of the DKK1 promoter (−2,000/+100) following NR2F2 knockdown (48 h; n = 5). E: Western blots and quantification of whole cell lysates demonstrated activation of AKT (phosphorylation at S473), STAT1 (Y701) and STAT3 (Y706) following incubation of PAECs with recombinant human DKK1 (500 ng/mL; 48 h; n = 5). F: incubation of PAEC with recombinant DKK1 or vehicle control (CTRL) as above led to the downregulation of endothelial (CDH5 [VE-cadherin]) and VWF and upregulation of mesenchymal (ACTA2 [α-smooth muscle actin] marker expression by immunofluorescent microscopy (n = 4; top). Scale bar, 100 µm. G: incubation with recombinant DKK1 increased proliferation in PAECS (48 h) as assessed by CellTiter-Glo 2.0 cell proliferation assay (right; n = 5). Data are presented as means ± SD. *P ≤ 0.05, **P ≤ 0.01 siNR2F2 vs. siCTRL by paired t test (unpaired for patient samples).
Since NR2F2 loss induced AKT activation in human ECs, our genome-wide NR2F2-silenced expression profiles were reassessed for potential upstream regulators of AKT. DKK1, a secreted protein involved in heart development (42) and ischemic cardiac injury (43), was upregulated in our global gene analysis, as well as in a similar global gene analysis of BMPR2-silenced PAECs, an established in vitro model of PAH pathobiology (18). In addition to functioning as a Wnt/β-catenin pathway inhibitor, DKK1 can alternatively activate AKT signaling (44) and induce EndMT (27). Increased expression of DKK1 following NR2F2-silencing was confirmed by Western blotting in PAECs (Fig. 4A) and LMVECs (Supplemental Fig. S4B). Secretion of DKK1 was likewise increased in cell supernatants from NR2F2-silenced PAECs and also in BMPR2- and CAV1-silenced PAECs (Fig. 4B), genetic defects strongly associated with PAH. Notably, BMPR2 (18) and CAV1 loss (30, 45) have both been associated with a proliferative, hypermigratory, and apoptosis-resistant PAEC phenotype analogous to NR2F2-silenced ECs and typical of lung vascular cells isolated from late-stage PAH patients (30). As such, DKK1 levels were increased in plasma from PAH patients versus healthy controls (Fig. 4B, Supplemental Table S5). Similarly, mRNA levels of DKK1 were increased while NR2F2 mRNA was decreased in PAECs isolated from idiopathic patients with PAH versus PAECs isolated from donor lung controls (Fig. 4C). Furthermore, the TRANSFAC database identified NR2F2, IRF1, IRF2, IRF4, STAT1, and STAT3 binding sites in the DKK1 promoter consistent with the direct transcriptional regulation of DKK1 by NR2F2 and/or IFN regulatory factors (Supplemental Table S11). In order to determine whether NR2F2 directly influences DKK1 expression, a portion of the DKK1 promoter (−2,000/+100) was cloned into a luciferase reporter construct and activity was assessed in EA.hy926 cells following NR2F2 knockdown. In NR2F2-silenced cells, DKK1 promoter reporter activity is increased compared to siRNA control transfected cells (Fig. 4D), suggesting loss of NR2F2 influences DKK1 expression at the promoter level.
To test whether DKK1 plays a causative role in aberrant EC signaling, the effect of recombinant DKK1 on ECs was examined. Analysis of cell lysates from DKK1-treated PAECs confirmed that DKK1 activated AKT (measured by phosphorylation at serine 473) as well as STAT1 (Y701) and STAT3 (Y705; Fig. 4E). DKK1-treated PAECs also exhibited altered expression of the EndMT markers CDH5, VWF, and ACTA2 and increased cellular proliferation similar to NR2F2-deficient cells (Fig. 4, F and G). Next, DKK1 and NR2F2 were co-silenced in PAECs (Fig. 5A) to further examine the contribution of DKK1 to aberrant EC signaling following NR2F2 loss. In contrast to NR2F2 knockdown alone where STAT1 and STAT3 DNA binding is increased, NR2F2- and DKK1-co-silenced PAECs exhibited reduced DNA binding of both STAT1 and STAT3 (Fig. 5B). Consistent with these results, NR2F2- and DKK1-co-silencing also attenuated NR2F2 loss-associated CCL5 and CXCL10 secretion, indicative of decreased IFN signaling (Fig. 5C). Co-silencing of NR2F2 and DKK1 also abrogated NR2F2 loss-induced AKT activation and NOS3 phosphorylation (S1177). Evidence of EndMT was also partially reversed by NR2F2 and DKK1 co-silencing (Fig. 5, D and E), consistent with previous findings that DKK1 acts as an EndMT enhancer (46). Collectively, these results indicate an upstream role for DKK1 in AKT/STAT activation and the inflammatory, dysfunctional phenotype of NR2F2-silenced ECs.
Figure 5.
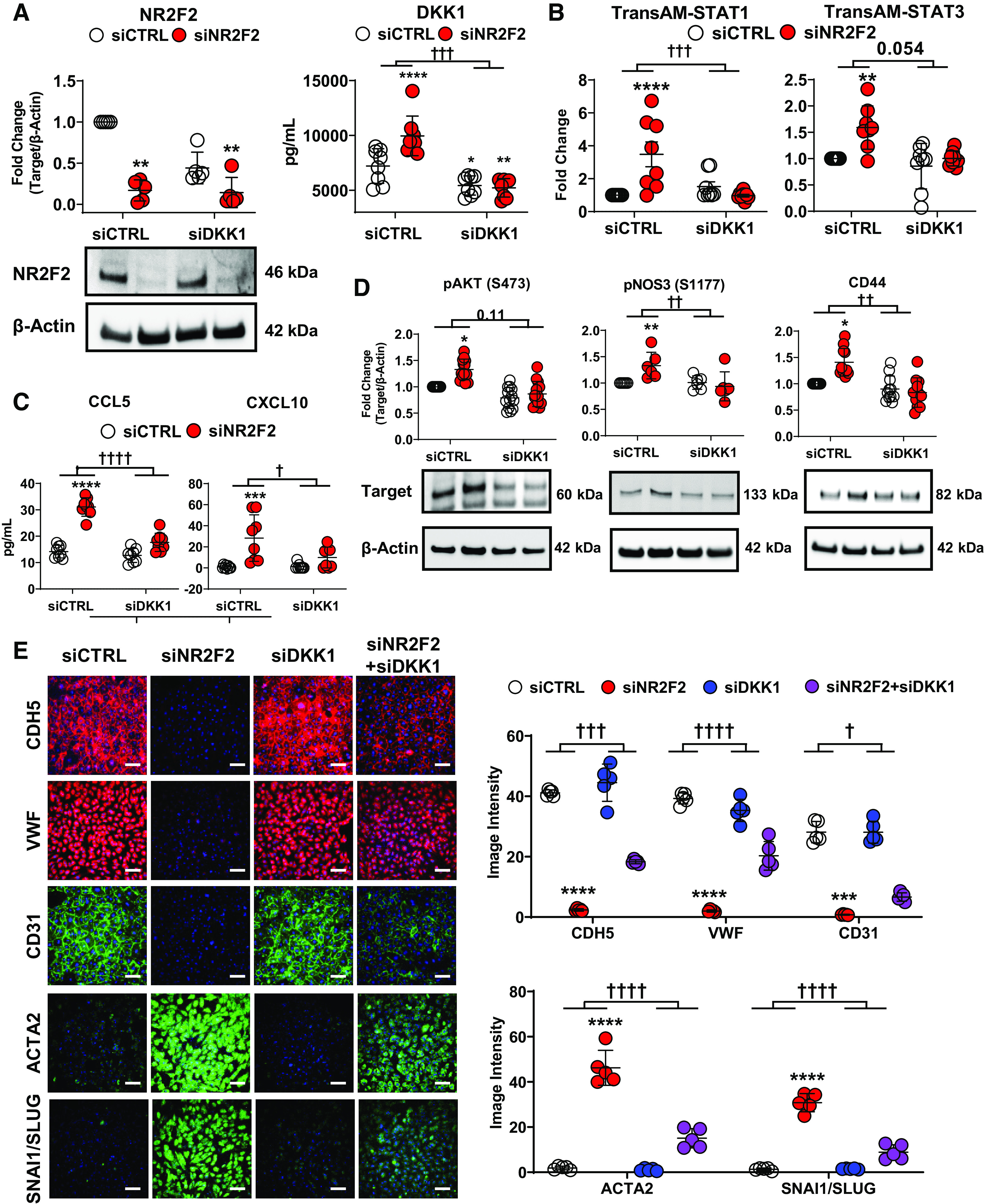
Co-silencing of DKK1 attenuates the effects of NR2F2 silencing on STAT and STAT target genes in pulmonary artery endothelial cells (PAECs) and partially reverses NR2F2 knockdown-induced endothelial to mesenchymal transition marker expression. PAECs were transiently transfected with either control (siCTRL) or NR2F2 siRNA (siNR2F2) for 48 h. A: knockdown of NR2F2 (n = 5) and DKK1 (n = 8) in PAECs was confirmed by Western blot and ELISA, respectively. B: co-silencing NR2F2 and DKK1 reduced STAT1 and STAT3 DNA binding as measured by TransAM in NR2F2-deficient PAECs (n = 8 for both). C: similarly, co-silencing NR2F2 and DKK1 as above completely blocked the upregulation of IFN pathway targets CCL5 and CXCL10 in NR2F2-deficient PAECs as determined by ELISA (n = 8 for both). D: knockdown of DKK1 blocked AKT phosphorylation at S473 (n = 13), NOS3 phosphorylation at S1177 (n = 6), and CD44 upregulation (n = 11) in NR2F2-deficient PAECs. E: NR2F2 loss led to the downregulation of endothelial (CDH5 [VE-cadherin], VWF and CD31 [PECAM1]) and upregulation of mesenchymal (ACTA2 [α-smooth muscle actin] and SNAI1/SLUG) marker expression, as determined by immunofluorescent microscopy, that was partially reversed by co-silencing of DKK1 (n = 5). Fluorescence intensity is quantified to the right. Scale bar, 100 µm. Data are presented as means ± SD. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001 siNR2F2 vs. siCTRL; †P ≤ 0.05, ††P ≤ 0.01, †††P ≤ 0.001, ††††P ≤ 0.0001 for the interaction effect of NR2F2 silencing vs. DKK1- and NR2F2-co-silencing by two-way ANOVA.
DISCUSSION
NR2F2 silencing in human ECs from diverse vascular beds led to a pro-inflammatory, proliferative, hypermigratory, and apoptosis-resistant cellular phenotype that exhibited EndMT and increased ROS production. These abnormalities correspond to prototypic pathological mechanisms closely associated with CHD (47, 48), pulmonary vascular disease (49), and atherosclerosis (25). Conspicuously, the transcriptomic signature of NR2F2 deficiency in human ECs was indicative of an IFN inflammatory response with evidence of both STAT and AKT activation similar to that seen with CAV1 silencing (45). Finally, silencing DKK1, a Wnt/β-catenin pathway inhibitor that also activates AKT through activation of its receptor CKAP4 (44), blocked NR2F2 loss-associated AKT and STAT activation. Collectively, this investigation suggests that defects in NR2F2 abundance or function, in addition to known effects on cardiovascular embryogenesis, may result in a pro-inflammatory endothelium predisposed to pathologic vascular remodeling and the development of vascular disease including but not limited to PAH. In CHD associated with NR2F2 mutations, this endothelial dysfunction, separate from flow abnormalities and shear stress, may increase the risk of progressive, pulmonary plexogenic arteriopathy, even after successful repair.
Loss of NR2F2 in venous ECs was previously shown to increase inflammatory gene expression (including CXCL10, CXCL11, and CCL5) and promote leukocyte adhesion (15). In HUVECs, NR2F2 loss produced an IFN inflammatory response (7), also found here in HUVEC-derived EA.hy926 cells and consistent with our findings in arterial and microvascular lung ECs, as well as CAECs. Using in vitro data, NR2F2 binding sites have been described in JAK/STAT regulated gene promoters including STAT1, STAT3, and IRF7 (Supplemental Table S11) (50), implicating NR2F2 in the direct regulation of IFN target genes. Here, NR2F2 silencing in human primary LMVECs, PAECs, and CAECs, as well as in an immortalized human EC line results in an inflammatory response characterized by the activation of STAT1 and induction of IFN target genes including IL6, CXCL10, and CCL5 (Figs. 1 and 2; Supplemental Figs. S1–S3).
Interestingly, JAK/STAT signaling pathways were previously shown to be enriched in lymphoblastoid cell lines derived from individuals with cardiac malformations compared to those without heart defects (51). Activation of JAK/STAT signaling has also been implicated in PAH pathogenesis (24, 45), balloon injury-induced arterial remodeling, and neointimal formation (52), as well as aberrant EC proliferation and migration (53, 54). In addition, patients with PAH have elevated circulating levels of IL6, CXCL10, and CCL5 and exhibit inflammatory infiltrates adjacent to pathognomonic plexiform lesions (49, 55, 56). Lung tissue from patients with PAH exhibits an increased expression of a subunit of the IFN α/β receptor IFNAR1, while IFNAR1 knockout mice were protected from the effects of hypoxia on right heart function, vascular remodeling, and elevated levels of endothelin 1 (EDN1) (21). These studies also demonstrated that IFN-stimulated pulmonary vascular cells strongly release CXCL10 and EDN1, and that serum levels of IFNs, as well as CXCL10 and EDN1 are increased in patients with systemic sclerosis-associated PAH. Inflammation and IFN signaling pathways have likewise been implicated in EndMT (27), a process linked to both CHD and atherosclerosis (27).
Moreover, increased expression of mesenchymal markers including ACTA2, CD44, and SNAI1 have also been seen in the plexiform lesions of patients with PAH (57, 58). In BMPR2-deficient PAECs and in ECs from patients with PAH, which undergo EndMT, this process was linked to elevated HMGA1 (59). Notably, genes differentially regulated in NR2F2-silenced ECs, shown here to undergo EndMT (Fig. 3A; Supplemental Fig. S2, C and D), were enriched for HMGA1 binding sites (Table 1). In vivo, NR2F2-mutant hearts have increased ACTA2 (3), a mesenchymal marker, while Nr2f2+/- vessels have reduced CD31, an endothelial marker, again suggesting a role for NR2F2 loss in the induction of EndMT (60). In contrast, the formation of hypoplastic endocardial cushions was attributed to reduced EndMT and proliferation in endothelial-specific NR2F2 hypomorphic mutant mice where SNAI1 expression was decreased (4). Although SNAI1 and other mesenchymal markers were spontaneously upregulated in NR2F2-silenced PAECs and LMVECs (Fig. 3A, Supplemental Fig. S3E), Cui et al. (15) reported that NR2F2-loss associated EndMT required BMP4 in human saphenous vein ECs. Notably, BMP4 is more highly expressed by arterial ECs than venous ECs and is induced by NR2F2 loss (15). Likewise, BMP4 induction of EndMT is Notch-dependent (61) and Notch signaling is also more highly activated in arterial ECs compared with venous ECs (62).
EndMT, Notch signaling, and ROS have been linked with metabolic changes (63, 64). In mice, NR2F2 modulates the expression of genes involved in lipid metabolism and glucose tolerance (65–67), and NR2F2 silencing in human ECs upregulates glucose transporters (68). Similarly, NR2F2 repression has been linked to the induction of glycolytic and lipogenic pathways in mice, both of which are implicated in PAH pathogenesis (69, 70). Increased expression of PFKFB3, a PI3K/AKT target induced in NR2F2-silenced PAECs (Fig. 3F), destabilizes plaques and promotes the progression of atherosclerosis (71), but may also contribute to the development of PAH by promoting endothelial glycolysis and proliferation (69, 72). Likewise, increased STAT3 phosphorylation, seen here following NR2F2 silencing in human ECs (Fig. 3F), has been implicated in the pathogenesis of atherosclerosis (73) and also found in plexiform lesions and PAECs isolated from patients with idiopathic PAH (22). Importantly, STAT3 activation affects EC proliferation as well as mitochondrial function through its direct regulation of mitochondrial genes (74). STAT3 is also a regulator of insulin signaling which in turn has been shown to both repress NR2F2 expression and activate PI3K/AKT signaling (75, 76).
Furthermore, repression of FOXO1 and activation of STAT3 have each been linked to the migratory, proliferative, and apoptosis-resistant phenotype of the late-stage PAH endothelium (77, 78). As shown here after NR2F2 loss (Fig. 3, B–D), PAECs from patients with idiopathic PAH (IPAH) or following BMPR2 or CAV1 silencing are proliferative, hypermigratory, and resistant to apoptosis (18, 22, 45). Impaired endothelial apoptosis has similarly been associated with intimal proliferation in patients with PAH secondary to CHD (49). While the effects of NR2F2 silencing on proliferation vary among studies using different cell types and culture conditions, here in primary human ECs, we found that NR2F2 loss resulted in a markedly similar phenotype to that seen in PAECs deficient in either BMPR2 or CAV1. For CAV1 loss, AKT activation and NOS3 phosphorylation, as shown here in NR2F2-silenced ECs, were directly implicated in the shift toward a pathogenic PAH-like cellular phenotype (45).
Notably, AKT can activate STAT independent of JAK to reinforce STAT signaling (39). DKK1, an AKT activator (44), was upregulated in NR2F2-silenced PAECs, but also in in vitro models of PAH based on BMPR2- or CAV1-silencing. DKK1 could therefore be linked to NOS3 through studies indicating that NR2F2 loss increased NOS3 in human ECs (68) and AKT-dependent phosphorylation of NOS3 increased proliferation in embryonic ECs (79). Furthermore, increased plasma DKK1 expression has been linked to endothelial activation, EndMT, and invasiveness (80), as well as poor cardiovascular outcomes (43) and inflammatory diseases including atherosclerosis (46). Similarly, transgenic overexpression of DKK1 in zebrafish results in cardiomyocyte proliferation (81). Furthermore, DKK1 injected into zebrafish embryos blocks endocardial cushion formation (82), mimicking observations in mouse NR2F2 ablation models (2, 4). Here, incubation with recombinant DKK1 activated AKT, STAT1 and 3 (Fig. 4E), altered the expression of EndMT markers (Fig. 4F) and led to increased proliferation similar to NR2F2 loss (Fig. 4G). Conversely, co-silencing DKK1 mitigated the IFN-biased inflammatory response induced by NR2F2 loss (STAT1 and STAT3 DNA binding; CCL5 and CXCL10 secretion), blocked AKT activation and NOS3 phosphorylation, and reversed cellular markers of EndMT (CD44, CDH5, VWF, CD31, ACTA2, and SNAI1/SLUG; Fig. 5).
In conclusion, loss of NR2F2 caused a distinctive form of endothelial dysfunction in multiple human EC types as evidenced by an IFN-biased inflammatory response with activation of both AKT and STAT signaling. This resulted in a proliferative, hypermigratory, apoptosis-resistant phenotype coupled with oxidant stress as well as increased lactate production and the expression of glycolytic genes. Collectively, these effects are highly consistent with current notions of cardiovascular disease pathogenesis. Determining whether NR2F2 agonists or inhibition of DKK1/AKT signaling can be therapeutically targeted in cardiovascular disease warrants further study. Furthermore, this study provides a basis for the variable development of PAH in CHD with similar clinical flow/shear stress characteristics, sometimes despite successful surgical repair, wherein an underlying dysfunctional endothelium may be a contributing factor.
The authors acknowledge that there are limitations to the current study. These studies have used human primary cells ex vivo grown under standard culture conditions. Following passaging, these cells could differ phenotypically from endothelium in vivo. Biological samples from patients harboring NR2F2 mutations could not be obtained due to the rarity of these genetic defects. Further studies to examine endothelial-specific NR2F2 conditional knockout mice for phenotypes resembling pulmonary arterial hypertension with subsequent treatment with DKK1 inhibitors would support our findings, but lie outside the scope of this study.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Tables S1–S11 and Supplemental Figs. S1–S5 (Western blotting data and data regarding antibody specificity are provided as Supplemental Fig. S5): https://doi.org/10.6084/m9.figshare.22009427.
GRANTS
This work was supported by intramural funds from the National Institutes of Health Clinical Center.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.J.D., J.M.K., Y.D., A.F.S., M.A.S., J.M.E., and R.L.D. conceived and designed research; E.J.D., L.-Y.C., K.S.A., G.A.F., A.K., S.G., K.A.J., M.E.H., A.B.S., and C.S.C. performed experiments; E.J.D., L.-Y.C., K.S.A., G.A.F., C.Y.D., A.K., C.S.C., and J.M.E. analyzed data; E.J.D., L.-Y.C., A.K., and C.S.C. interpreted results of experiments; E.J.D., C.S.C., and J.M.E. prepared figures; E.J.D. drafted manuscript; E.J.D., L.-Y.C., K.S.A., G.A.F., C.Y.D., A.K., S.G., K.A.J., M.E.H., A.B.S., C.S.C., J.M.K., Y.D., A.F.S., M.A.S., J.M.E., and R.L.D. edited and revised manuscript; E.J.D., L.-Y.C., K.S.A., G.A.F., C.Y.D., A.K., S.G., K.A.J., M.E.H., A.B.S., C.S.C., J.M.K., Y.D., A.F.S., M.A.S., J.M.E., and R.L.D. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank patients and volunteers whose participation made this work possible. Pulmonary artery endothelial cells used in this study were provided by the Pulmonary Hypertension Breakthrough Initiative (PHBI) Network. Funding for the PHBI is provided under a National Heart Lung and Blood Institute R24 grant (HL123767) and by the Cardiovascular Medical Research and Education Fund (CMREF). The authors also thank Kelly Byrne for assistance with editing and formatting of the figures and manuscript. The opinions expressed in this article are those of the authors and do not represent any position or policy of the NIH, the US Department of Health and Human Services, or the US Government.
Present addresses: K. A. Johnston, Des Moines University Medicine and Health Sciences, Des Moines, IA 50312, United States; M. E. Hicks, Lewis Katz School of Medicine at Temple University, Philadelphia, PA 19140, United States; A. B. Sandler, George Washington University School of Medicine and Health Sciences, Washington, DC 20010, United States; Y. Ding, Dept. of Pathophysiology and Key Laboratory of Applied Pharmacology, Weifang Medical University, Weifang, Shandong 261053, People’s Republic of China.
REFERENCES
- 1. Planchais J, Boutant M, Fauveau V, Qing LD, Sabra-Makke L, Bossard P, Vasseur-Cognet M, Pégorier JP. The role of chicken ovalbumin upstream promoter transcription factor II in the regulation of hepatic fatty acid oxidation and gluconeogenesis in newborn mice. Am J Physiol Endocrinol Physiol 308: E868–E878, 2015. doi: 10.1152/ajpendo.00433.2014. [DOI] [PubMed] [Google Scholar]
- 2. Wu SP, Cheng CM, Lanz RB, Wang T, Respress JL, Ather S, Chen W, Tsai SJ, Wehrens XH, Tsai MJ, Tsai SY. Atrial identity is determined by a COUP-TFII regulatory network. Dev Cell 25: 417–426, 2013. doi: 10.1016/j.devcel.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pereira FA, Qiu Y, Zhou G, Tsai MJ, Tsai SY. The orphan nuclear receptor COUP-TFII is required for angiogenesis and heart development. Genes Dev 13: 1037–1049, 1999. doi: 10.1101/gad.13.8.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lin FJ, You LR, Yu CT, Hsu WH, Tsai MJ, Tsai SY. Endocardial cushion morphogenesis and coronary vessel development require chicken ovalbumin upstream promoter-transcription factor II. Arterioscler Thromb Vasc Biol 32: e135–e146, 2012. doi: 10.1161/ATVBAHA.112.300255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Masuda S, Matsuura K, Anazawa M, Iwamiya T, Shimizu T, Okano T. Formation of vascular network structures within cardiac cell sheets from mouse embryonic stem cells. Regen Ther 2: 6–16, 2015. doi: 10.1016/j.reth.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Swift MR, Pham VN, Castranova D, Bell K, Poole RJ, Weinstein BM. SoxF factors and Notch regulate nr2f2 gene expression during venous differentiation in zebrafish. Dev Biol 390: 116–125, 2014. doi: 10.1016/j.ydbio.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aranguren XL, Beerens M, Coppiello G, Wiese C, Vandersmissen I, Lo Nigro A, Verfaillie CM, Gessler M, Luttun A. COUP-TFII orchestrates venous and lymphatic endothelial identity by homo- or hetero-dimerisation with PROX1. J Cell Sci 126: 1164–1175, 2013. doi: 10.1242/jcs.116293. [DOI] [PubMed] [Google Scholar]
- 8. Al Turki S, Manickaraj AK, Mercer CL, Gerety SS, Hitz MP, Lindsay S, , et al. Rare variants in NR2F2 cause congenital heart defects in humans. Am J Hum Genet 98: 592, 2016. doi: 10.1016/j.ajhg.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hermann-Kleiter N, Baier G. Orphan nuclear receptor NR2F6 acts as an essential gatekeeper of Th17 CD4+ T cell effector functions. Cell Commun Signal 12: 38, 2014. doi: 10.1186/1478-811X-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suzuki T, Moriya T, Darnel AD, Takeyama J, Sasano H. Immunohistochemical distribution of chicken ovalbumin upstream promoter transcription factor II in human tissues. Mol Cell Endocrinol 164: 69–75, 2000. doi: 10.1016/s0303-7207(00)00242-2. [DOI] [PubMed] [Google Scholar]
- 11. Nakamura E, Makita Y, Okamoto T, Nagaya K, Hayashi T, Sugimoto M, Manabe H, Taketazu G, Kajino H, Fujieda K. 5.78 Mb terminal deletion of chromosome 15q in a girl, evaluation of NR2F2 as candidate gene for congenital heart defects. Eur J Med Genet 54: 354–356, 2011. doi: 10.1016/j.ejmg.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 12. Qiao XH, Wang Q, Wang J, Liu XY, Xu YJ, Huang RT, Xue S, Li YJ, Zhang M, Qu XK, Li RG, Qiu XB, Yang YQ. A novel NR2F2 loss-of-function mutation predisposes to congenital heart defect. Eur J Med Genet 61: 197–203, 2018. doi: 10.1016/j.ejmg.2017.12.003. [DOI] [PubMed] [Google Scholar]
- 13. Upadia J, Gonzales PR, Robin NH. Novel de novo pathogenic variant in the NR2F2 gene in a boy with congenital heart defect and dysmorphic features. Am J Med Genet A 176: 1423–1426, 2018. doi: 10.1002/ajmg.a.38700. [DOI] [PubMed] [Google Scholar]
- 14. D'Alto M, Mahadevan VS. Pulmonary arterial hypertension associated with congenital heart disease. Eur Respir Rev 21: 328–337, 2012. doi: 10.1183/09059180.00004712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cui X, Lu YW, Lee V, Kim D, Dorsey T, Wang Q, Lee Y, Vincent P, Schwarz J, Dai G. Venous endothelial marker COUP-TFII regulates the distinct pathologic potentials of adult arteries and veins. Sci Rep 5: 16193, 2015. doi: 10.1038/srep16193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Benza RL, Gomberg-Maitland M, Elliott CG, Farber HW, Foreman AJ, Frost AE, McGoon MD, Pasta DJ, Selej M, Burger CD, Frantz RP. Predicting survival in patients with pulmonary arterial hypertension: the REVEAL risk score calculator 2.0 and comparison with ESC/ERS-based risk assessment strategies. Chest 156: 323–337, 2019. doi: 10.1016/j.chest.2019.02.004. [DOI] [PubMed] [Google Scholar]
- 17. Benza RL, Kanwar MK, Raina A, Scott JV, Zhao CL, Selej M, Elliott CG, Farber HW. Development and validation of an abridged version of the REVEAL 2.0 risk score calculator, REVEAL Lite 2, for use in patients with pulmonary arterial hypertension. Chest 159: 337–346, 2021. doi: 10.1016/j.chest.2020.08.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Awad KS, Elinoff JM, Wang S, Gairhe S, Ferreyra GA, Cai R, Sun J, Solomon MA, Danner RL. Raf/ERK drives the proliferative and invasive phenotype of BMPR2-silenced pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 310: L187–L201, 2016. doi: 10.1152/ajplung.00303.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: e47, 2015. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moss JW, Ramji DP. Interferon-γ: promising therapeutic target in atherosclerosis. World J Exp Med 5: 154–159, 2015. doi: 10.5493/wjem.v5.i3.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. George PM, Oliver E, Dorfmuller P, Dubois OD, Reed DM, Kirkby NS, Mohamed NA, Perros F, Antigny F, Fadel E, Schreiber BE, Holmes AM, Southwood M, Hagan G, Wort SJ, Bartlett N, Morrell NW, Coghlan JG, Humbert M, Zhao L, Mitchell JA. Evidence for the involvement of type I interferon in pulmonary arterial hypertension. Circ Res 114: 677–688, 2014. doi: 10.1161/CIRCRESAHA.114.302221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Masri FA, Xu W, Comhair SA, Asosingh K, Koo M, Vasanji A, Drazba J, Anand-Apte B, Erzurum SC. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 293: L548–L554, 2007. doi: 10.1152/ajplung.00428.2006. [DOI] [PubMed] [Google Scholar]
- 23. Ramakrishnan S, Kukreti BB, Ramakrishnan L, Salahuddin S, Pendharkar A, Karthikeyan G, Bhargava B, Juneja R, Seth S, Kothari SS, Saxena A, Bahl VK. Inflammatory markers are elevated in Eisenmenger syndrome. Pediatr Cardiol 34: 1791–1796, 2013. doi: 10.1007/s00246-013-0715-3. [DOI] [PubMed] [Google Scholar]
- 24. Elinoff JM, Mazer AJ, Cai R, Lu M, Graninger G, Harper B, Ferreyra GA, Sun J, Solomon MA, Danner RL. Meta-analysis of blood genome-wide expression profiling studies in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 318: L98–L111, 2020. doi: 10.1152/ajplung.00252.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G, Baker AH. Endothelial to mesenchymal transition in cardiovascular disease: JACC state-of-the-art review. J Am Coll Cardiol 73: 190–209, 2019. doi: 10.1016/j.jacc.2018.09.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arciniegas E, Frid MG, Douglas IS, Stenmark KR. Perspectives on endothelial-to-mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 293: L1–L8, 2007. doi: 10.1152/ajplung.00378.2006. [DOI] [PubMed] [Google Scholar]
- 27. Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman KR, d'Escamard V, Li JR, Hadri L, Fujitani K, Moreno PR, Benard L, Rimmele P, Cohain A, Mecham B, Randolph GJ, Nabel EG, Hajjar R, Fuster V, Boehm M, Kovacic JC. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun 7: 11853, 2016. [Erratum in Nat Commun 8: 14710, 2017]. doi: 10.1038/ncomms11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nath AK, Brown RM, Michaud M, Sierra-Honigmann MR, Snyder M, Madri JA. Leptin affects endocardial cushion formation by modulating EMT and migration via Akt signaling cascades. J Cell Biol 181: 367–380, 2008. doi: 10.1083/jcb.200708197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pascall E, Tulloh RM. Pulmonary hypertension in congenital heart disease. Future Cardiol 14: 343–353, 2018. doi: 10.2217/fca-2017-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Evans CE, Cober ND, Dai Z, Stewart DJ, Zhao YY. Endothelial cells in the pathogenesis of pulmonary arterial hypertension. Eur Respir J 58: 2003957, 2021. doi: 10.1183/13993003.03957-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yamazaki T, Yoshimatsu Y, Morishita Y, Miyazono K, Watabe T. COUP-TFII regulates the functions of Prox1 in lymphatic endothelial cells through direct interaction. Genes Cells 14: 425–434, 2009. doi: 10.1111/j.1365-2443.2008.01279.x. [DOI] [PubMed] [Google Scholar]
- 32. Thuan DTB, Zayed H, Eid AH, Abou-Saleh H, Nasrallah GK, Mangoni AA, Pintus G. A potential link between oxidative stress and endothelial-to-mesenchymal transition in systemic sclerosis. Front Immunol 9: 1985, 2018. doi: 10.3389/fimmu.2018.01985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Montorfano I, Becerra A, Cerro R, Echeverría C, Sáez E, Morales MG, Fernández R, Cabello-Verrugio C, Simon F. Oxidative stress mediates the conversion of endothelial cells into myofibroblasts via a TGF-β1 and TGF-β2-dependent pathway. Lab Invest 94: 1068–1082, 2014. doi: 10.1038/labinvest.2014.100. [DOI] [PubMed] [Google Scholar]
- 34. Frey RS, Ushio-Fukai M, Malik AB. NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxid Redox Signal 11: 791–810, 2009. doi: 10.1089/ars.2008.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res 122: 877–902, 2018. doi: 10.1161/CIRCRESAHA.117.311401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rafikov R, Sun X, Rafikova O, Louise Meadows M, Desai AA, Khalpey Z, Yuan JX, Fineman JR, Black SM. Complex I dysfunction underlies the glycolytic switch in pulmonary hypertensive smooth muscle cells. Redox Biol 6: 278–286, 2015. doi: 10.1016/j.redox.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zuo L, Rose BA, Roberts WJ, He F, Banes-Berceli AK. Molecular characterization of reactive oxygen species in systemic and pulmonary hypertension. Am J Hypertens 27: 643–650, 2014. doi: 10.1093/ajh/hpt292. [DOI] [PubMed] [Google Scholar]
- 38. Wang R, Wang M, Ye J, Sun G, Sun X. Mechanism overview and target mining of atherosclerosis: Endothelial cell injury in atherosclerosis is regulated by glycolysis (review). Int J Mol Med 47: 65–76, 2021. doi: 10.3892/ijmm.2020.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nguyen H, Ramana CV, Bayes J, Stark GR. Roles of phosphatidylinositol 3-kinase in interferon-gamma-dependent phosphorylation of STAT1 on serine 727 and activation of gene expression. J Biol Chem 276: 33361–33368, 2001. doi: 10.1074/jbc.M105070200. [DOI] [PubMed] [Google Scholar]
- 40. Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta 1813: 1978–1986, 2011. doi: 10.1016/j.bbamcr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 41. Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399: 601–605, 1999. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 42. Guo Y, Dorn T, Kühl SJ, Linnemann A, Rothe M, Pfister AS, Vainio S, Laugwitz KL, Moretti A, Kühl M. The Wnt inhibitor Dkk1 is required for maintaining the normal cardiac differentiation program in Xenopus laevis. Dev Biol 449: 1–13, 2019. doi: 10.1016/j.ydbio.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ueland T, Åkerblom A, Ghukasyan T, Michelsen AE, Becker RC, Bertilsson M, Himmelmann A, James SK, Siegbahn A, Storey RF, Kontny F, Aukrust P, Wallentin L; PLATO Investigators. Admission levels of DKK1 (Dickkopf-1) are associated with future cardiovascular death in patients with acute coronary syndromes. Arterioscler Thromb Vasc Biol 39: 294–302, 2019. doi: 10.1161/ATVBAHA.118.311042. [DOI] [PubMed] [Google Scholar]
- 44. Kimura H, Fumoto K, Shojima K, Nojima S, Osugi Y, Tomihara H, Eguchi H, Shintani Y, Endo H, Inoue M, Doki Y, Okumura M, Morii E, Kikuchi A. CKAP4 is a Dickkopf1 receptor and is involved in tumor progression. J Clin Invest 126: 2689–2705, 2016. doi: 10.1172/JCI84658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gairhe S, Awad KS, Dougherty EJ, Ferreyra GA, Wang S, Yu ZX, Takeda K, Demirkale CY, Torabi-Parizi P, Austin ED, Elinoff JM, Danner RL. Type I interferon activation and endothelial dysfunction in caveolin-1 insufficiency-associated pulmonary arterial hypertension. Proc Natl Acad Sci USA 118: e2010206118, 2021. doi: 10.1073/pnas.2010206118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chae WJ, Bothwell ALM. Dickkopf1: an immunomodulatory ligand and Wnt antagonist in pathological inflammation. Differentiation 108: 33–39, 2019. doi: 10.1016/j.diff.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mou SS, Haudek SB, Lequier L, Peña O, Leonard S, Nikaidoh H, Giroir BP, Stromberg D. Myocardial inflammatory activation in children with congenital heart disease. Crit Care Med 30: 827–832, 2002. doi: 10.1097/00003246-200204000-00018. [DOI] [PubMed] [Google Scholar]
- 48. Aggarwal S, Gross C, Fineman JR, Black SM. Oxidative stress and the development of endothelial dysfunction in congenital heart disease with increased pulmonary blood flow: lessons from the neonatal lamb. Trends Cardiovasc Med 20: 238–246, 2010. doi: 10.1016/j.tcm.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lévy M, Maurey C, Celermajer DS, Vouhe PR, Danel C, Bonnet D, Israel-Biet D. Impaired apoptosis of pulmonary endothelial cells is associated with intimal proliferation and irreversibility of pulmonary hypertension in congenital heart disease. J Am Coll Cardiol 49: 803–810, 2007. doi: 10.1016/j.jacc.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 50. Erdős E, Bálint BL. COUP-TFII is a modulator of cell-type-specific genetic programs based on genomic localization maps. J Biotechnol 301: 11–17, 2019. doi: 10.1016/j.jbiotec.2019.05.305. [DOI] [PubMed] [Google Scholar]
- 51. Ripoll C, Rivals I, Ait Yahya-Graison E, Dauphinot L, Paly E, Mircher C, Ravel A, Grattau Y, Bléhaut H, Mégarbane A, Dembour G, de Fréminville B, Touraine R, Créau N, Potier MC, Delabar JM. Molecular signatures of cardiac defects in Down syndrome lymphoblastoid cell lines suggest altered ciliome and Hedgehog pathways. PLoS One 7: e41616, 2012. doi: 10.1371/journal.pone.0041616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Seki Y, Kai H, Shibata R, Nagata T, Yasukawa H, Yoshimura A, Imaizumi T. Role of the JAK/STAT pathway in rat carotid artery remodeling after vascular injury. Circ Res 87: 12–18, 2000. doi: 10.1161/01.res.87.1.12. [DOI] [PubMed] [Google Scholar]
- 53. Gomez D, Reich NC. Stimulation of primary human endothelial cell proliferation by IFN. J Immunol 170: 5373–5381, 2003. doi: 10.4049/jimmunol.170.11.5373. [DOI] [PubMed] [Google Scholar]
- 54. Roger I, Milara J, Montero P, Cortijo J. The role of JAK/STAT molecular pathway in vascular remodeling associated with pulmonary hypertension. Int J Mol Sci 22: 4980, 2021. doi: 10.3390/ijms22094980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, Scheed A, Ritter C, Dahal BK, Vater A, Klussmann S, Ghofrani HA, Weissmann N, Klepetko W, Banat GA, Seeger W, Grimminger F, Schermuly RT. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 186: 897–908, 2012. doi: 10.1164/rccm.201202-0335OC. [DOI] [PubMed] [Google Scholar]
- 56. Mamazhakypov A, Viswanathan G, Lawrie A, Schermuly RT, Rajagopal S. The role of chemokines and chemokine receptors in pulmonary arterial hypertension. Br J Pharmacol 178: 72–89, 2021. doi: 10.1111/bph.14826. [DOI] [PubMed] [Google Scholar]
- 57. Ohta-Ogo K, Hao H, Ishibashi-Ueda H, Hirota S, Nakamura K, Ohe T, Ito H. CD44 expression in plexiform lesions of idiopathic pulmonary arterial hypertension. Pathol Int 62: 219–225, 2012. doi: 10.1111/j.1440-1827.2011.02779.x. [DOI] [PubMed] [Google Scholar]
- 58. Jonigk D, Golpon H, Bockmeyer CL, Maegel L, Hoeper MM, Gottlieb J, Nickel N, Hussein K, Maus U, Lehmann U, Janciauskiene S, Welte T, Haverich A, Rische J, Kreipe H, Laenger F. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am J Pathol 179: 167–179, 2011. doi: 10.1016/j.ajpath.2011.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hopper RK, Moonen JR, Diebold I, Cao A, Rhodes CJ, Tojais NF, Hennigs JK, Gu M, Wang L, Rabinovitch M. In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial-to-mesenchymal transition via HMGA1 and its target slug. Circulation 133: 1783–1794, 2016. doi: 10.1161/CIRCULATIONAHA.115.020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Takamoto N, Kurihara I, Lee K, Demayo FJ, Tsai MJ, Tsai SY. Haploinsufficiency of chicken ovalbumin upstream promoter transcription factor II in female reproduction. Mol Endocrinol 19: 2299–2308, 2005. doi: 10.1210/me.2005-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Choi S, Yu J, Park A, Dubon MJ, Do J, Kim Y, Nam D, Noh J, Park KS. BMP-4 enhances epithelial mesenchymal transition and cancer stem cell properties of breast cancer cells via Notch signaling. Sci Rep 9: 11724, 2019. doi: 10.1038/s41598-019-48190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sissaoui S, Yu J, Yan A, Li R, Yukselen O, Kucukural A, Zhu LJ, Lawson ND. Genomic characterization of endothelial enhancers reveals a multifunctional role for NR2F2 in regulation of arteriovenous gene expression. Circ Res 126: 875–888, 2020. doi: 10.1161/CIRCRESAHA.119.316075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bi P, Kuang S. Notch signaling as a novel regulator of metabolism. Trends Endocrinol Metab 26: 248–255, 2015. doi: 10.1016/j.tem.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cho JG, Lee A, Chang W, Lee MS, Kim J. Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front Immunol 9: 294, 2018. doi: 10.3389/fimmu.2018.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Crowther LM, Wang SC, Eriksson NA, Myers SA, Murray LA, Muscat GE. Chicken ovalbumin upstream promoter-transcription factor II regulates nuclear receptor, myogenic, and metabolic gene expression in skeletal muscle cells. Physiol Genomics 43: 213–227, 2011. doi: 10.1152/physiolgenomics.00195.2010. [DOI] [PubMed] [Google Scholar]
- 66. Myers SA, Wang SC, Muscat GE. The chicken ovalbumin upstream promoter-transcription factors modulate genes and pathways involved in skeletal muscle cell metabolism. J Biol Chem 281: 24149–24160, 2006. doi: 10.1074/jbc.M601941200. [DOI] [PubMed] [Google Scholar]
- 67. Li L, Xie X, Qin J, Jeha GS, Saha PK, Yan J, Haueter CM, Chan L, Tsai SY, Tsai MJ. The nuclear orphan receptor COUP-TFII plays an essential role in adipogenesis, glucose homeostasis, and energy metabolism. Cell Metab 9: 77–87, 2009. doi: 10.1016/j.cmet.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Brunssen C, Korten S, Brux M, Seifert S, Roesler J, Bornstein SR, Morawietz H, Goettsch W. COUP-TFII is regulated by high glucose in endothelial cells. Horm Metab Res 42: 81–87, 2010. doi: 10.1055/s-0029-1241862. [DOI] [PubMed] [Google Scholar]
- 69. Cao Y, Zhang X, Wang L, Yang Q, Ma Q, Xu J, Wang J, Kovacs L, Ayon RJ, Liu Z, Zhang M, Zhou Y, Zeng X, Xu Y, Wang Y, Fulton DJ, Weintraub NL, Lucas R, Dong Z, Yuan JX, Sullivan JC, Meadows L, Barman SA, Wu C, Quan J, Hong M, Su Y, Huo Y. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc Natl Acad Sci USA 116: 13394–13403, 2019. doi: 10.1073/pnas.1821401116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Talati M, Hemnes A. Fatty acid metabolism in pulmonary arterial hypertension: role in right ventricular dysfunction and hypertrophy. Pulm Circ 5: 269–278, 2015. doi: 10.1086/681227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Poels K, Schnitzler JG, Waissi F, Levels JHM, Stroes ESG, Daemen M, Lutgens E, Pennekamp AM, De Kleijn DPV, Seijkens TTP, Kroon J. Inhibition of PFKFB3 hampers the progression of atherosclerosis and promotes plaque stability. Front Cell Dev Biol 8: 581641, 2020. doi: 10.3389/fcell.2020.581641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rodríguez-García A, Samsó P, Fontova P, Simon-Molas H, Manzano A, Castaño E, Rosa JL, Martinez-Outshoorn U, Ventura F, Navarro-Sabaté À, Bartrons R. TGF-β1 targets Smad, p38 MAPK, and PI3K/Akt signaling pathways to induce PFKFB3 gene expression and glycolysis in glioblastoma cells. FEBS J 284: 3437–3454, 2017. doi: 10.1111/febs.14201. [DOI] [PubMed] [Google Scholar]
- 73. Chen Q, Lv J, Yang W, Xu B, Wang Z, Yu Z, Wu J, Yang Y, Han Y. Targeted inhibition of STAT3 as a potential treatment strategy for atherosclerosis. Theranostics 9: 6424–6442, 2019. doi: 10.7150/thno.35528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Marshall JD, Bazan I, Zhang Y, Fares WH, Lee PJ. Mitochondrial dysfunction and pulmonary hypertension: cause, effect, or both. Am J Physiol Lung Cell Mol Physiol 314: L782–L796, 2018. doi: 10.1152/ajplung.00331.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Moh A, Zhang W, Yu S, Wang J, Xu X, Li J, Fu XY. STAT3 sensitizes insulin signaling by negatively regulating glycogen synthase kinase-3β. Diabetes 57: 1227–1235, 2008. doi: 10.2337/db06-1582. [DOI] [PubMed] [Google Scholar]
- 76. Ashraf UM, Sanchez ER, Kumarasamy S. COUP-TFII revisited: its role in metabolic gene regulation. Steroids 141: 63–69, 2019. doi: 10.1016/j.steroids.2018.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Savai R, Al-Tamari HM, Sedding D, Kojonazarov B, Muecke C, Teske R, Capecchi MR, Weissmann N, Grimminger F, Seeger W, Schermuly RT, Pullamsetti SS. Pro-proliferative and inflammatory signaling converge on FoxO1 transcription factor in pulmonary hypertension. Nat Med 20: 1289–1300, 2014. doi: 10.1038/nm.3695. [DOI] [PubMed] [Google Scholar]
- 78. Zhu M, Liu X, Wang Y, Chen L, Wang L, Qin X, Xu J, Li L, Tu Y, Zhou T, Sang A, Song E. YAP via interacting with STAT3 regulates VEGF-induced angiogenesis in human retinal microvascular endothelial cells. Exp Cell Res 373: 155–163, 2018. doi: 10.1016/j.yexcr.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 79. Gentile C, Muise-Helmericks RC, Drake CJ. VEGF-mediated phosphorylation of eNOS regulates angioblast and embryonic endothelial cell proliferation. Dev Biol 373: 163–175, 2013. doi: 10.1016/j.ydbio.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Johansson M, Giger FA, Fielding T, Houart C. Dkk1 controls cell-cell interaction through regulation of non-nuclear β-catenin pools. Dev Cell 51: 775–786.e3, 2019. doi: 10.1016/j.devcel.2019.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Peng X, Lai KS, She P, Kang J, Wang T, Li G, Zhou Y, Sun J, Jin D, Xu X, Liao L, Liu J, Lee E, Poss KD, Zhong TP. Induction of Wnt signaling antagonists and p21-activated kinase enhances cardiomyocyte proliferation during zebrafish heart regeneration. J Mol Cell Biol 13: 41–58, 2021. [Erratum in J Mol Cell Biol 13: 921, 2022]. doi: 10.1093/jmcb/mjaa046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hurlstone AF, Haramis AP, Wienholds E, Begthel H, Korving J, Van Eeden F, Cuppen E, Zivkovic D, Plasterk RH, Clevers H. The Wnt/β-catenin pathway regulates cardiac valve formation. Nature 425: 633–637, 2003. doi: 10.1038/nature02028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Tables S1–S11 and Supplemental Figs. S1–S5 (Western blotting data and data regarding antibody specificity are provided as Supplemental Fig. S5): https://doi.org/10.6084/m9.figshare.22009427.
Data Availability Statement
Data will be made available upon reasonable request.