


Keywords: active zone, Lambert-Eaton myasthenic syndrome, MCell modeling, neuromuscular junction
Abstract
Lambert-Eaton myasthenic syndrome (LEMS) is an autoimmune-mediated neuromuscular disease thought to be caused by autoantibodies against P/Q-type voltage-gated calcium channels (VGCCs), which attack and reduce the number of VGCCs within transmitter release sites (active zones; AZs) at the neuromuscular junction (NMJ), resulting in neuromuscular weakness. However, patients with LEMS also have antibodies to other neuronal proteins, and about 15% of patients with LEMS are seronegative for antibodies against VGCCs. We hypothesized that a reduction in the number of P/Q-type VGCCs alone is not sufficient to explain LEMS effects on transmitter release. Here, we used a computational model to study a variety of LEMS-mediated effects on AZ organization and transmitter release constrained by electron microscopic, pharmacological, immunohistochemical, voltage imaging, and electrophysiological observations. We show that models of healthy AZs can be modified to predict the transmitter release and short-term facilitation characteristics of LEMS and that in addition to a decrease in the number of AZ VGCCs, disruption in the organization of AZ proteins, a reduction in AZ number, a reduction in the amount of synaptotagmin, and the compensatory expression of L-type channels outside the remaining AZs are important contributors to LEMS-mediated effects on transmitter release. Furthermore, our models predict that antibody-mediated removal of synaptotagmin in combination with disruption in AZ organization alone could mimic LEMS effects without the removal of VGCCs (a seronegative model). Overall, our results suggest that LEMS pathophysiology may be caused by a collection of pathological alterations to AZs at the NMJ, rather than by a simple loss of VGCCs.
NEW & NOTEWORTHY We used a computational model of the active zone (AZ) in the mammalian neuromuscular junction to investigate Lambert-Eaton myasthenic syndrome (LEMS) pathophysiology. This model suggests that disruptions in presynaptic active zone organization and protein content (particularly synaptotagmin), beyond the simple removal of presynaptic calcium channels, play an important role in LEMS pathophysiology.
INTRODUCTION
Lambert-Eaton myasthenic syndrome (LEMS) is an autoimmune-mediated neuromuscular disease that reduces transmitter release from the neuromuscular junction (NMJ), resulting in severe muscle weakness (measured in patients as a smaller than normal compound muscle action potential; CMAP). A common diagnostic marker for LEMS is a reduced resting CMAP amplitude and a CMAP increase or increment recorded from the patient’s hand muscle following a short (10 s) exercise period, which can result in an increase in the resting CMAP amplitude of between 60% and 1,000%, depending on the patient (1, 2). This CMAP increment is caused by short-term synaptic facilitation of neuromuscular synapses that brings additional muscle fibers to threshold that were initially not generating a nerve-evoked muscle action potential (AP) due to LEMS pathology. The reduction in neurotransmitter release and increase in short-term synaptic facilitation is typically ascribed to antibodies targeting the presynaptic P/Q-type voltage-gated calcium channels (VGCCs) that are primarily responsible for the calcium influx that triggers transmitter release in the active zones (AZs) of mammalian NMJs. However, antibodies to other neuronal proteins are also known to be present in LEMS and may contribute to the disease pathology (3, 4). Furthermore, antibodies to VGCCs are not detected in 10%–15% of patients with LEMS (5–9). These patients with seronegative LEMS have similar clinical presentations to the patients with seropositive LEMS that express the P/Q-type VGCC antibodies (1, 8). Although patients with seronegative LEMS do not produce antibodies to the P/Q-type VGCCs, the repeated injection of mice with seronegative LEMS patient serum can passively transfer LEMS symptoms to the mice, suggesting that seronegative LEMS is still antibody mediated (8). These findings suggest that seronegative LEMS may be caused by antibodies targeting other proteins important to the release of neurotransmitter in the AZ.
Synaptotagmin-1 and synaptotagmin-2 (syt1 and syt2) are both found in mouse NMJs (10) and are known to be the calcium-sensing proteins responsible for fast synchronous release of neurotransmitter (11). Antibodies to synaptotagmin have been found in the serum of patients with LEMS (9, 12). These antibodies are thought to be directed against synaptotagmin-1 and/or 2, and it is unknown if other synaptotagmin types are affected (i.e., there are no studies to date specifically addressing the potential for synaptotagmin-7 antibodies in LEMS). Thus, it has been hypothesized that patient-derived synaptotagmin-1 and/or -2 autoantibodies could be at least partially responsible for seronegative LEMS. This hypothesis is supported by the finding that rats injected with synaptotagmin antibodies developed electrophysiological characteristics of LEMS (13). In addition, synaptotagmin-2 knockout mice display similar electrophysiological characteristics to LEMS (10). Finally, there is a report of a rare genetic alteration of synaptotagmin-2 in human patients that causes symptoms similar to LEMS (14).
We previously developed computational models of an AZ in the healthy frog NMJ (15, 16) and have recently developed a preliminary model of AZs in the healthy mouse NMJ (17). Here, we further develop our computational model of AZs in the healthy mouse NMJ, and then utilize this model to investigate possible mechanisms of LEMS-induced changes to the AZ. We use the known magnitude of LEMS-induced reduction in transmitter release and increase in short-term synaptic facilitation as targets for our model output. We find that simple removal of presynaptic AZ VGCCs is not sufficient to reproduce LEMS effects. Rather, the removal of VGCCs must be accompanied by a disruption in the organization of the remaining VGCCs. We also show that loss of some AZs and the upregulation of L-type VGCCs can contribute to LEMS effects. Furthermore, removal of some of the synaptotagmin fast calcium sensors (either alone or in combination with disruption of AZ structure) can also predict these LEMS effects, providing computational evidence in support of the synaptotagmin-autoantibody hypothesis of seronegative LEMS.
METHODS
Intracellular Recordings
To measure the magnitude of transmitter release, intracellular recordings were made from the mouse epitrochleoanconeus (ETA) neuromuscular preparation in accordance with procedures approved by the University of Pittsburgh Institutional Animal Care and Use Committee as previously described (2). The extracellular saline contained 150 mM NaCl, 5 mM KCl, 11 mM dextrose, 10 mM HEPES, 1 mM MgCl2, 2 mM CaCl2, pH = 7.3–7.4. The nerve was stimulated with a suction electrode and muscle contractions were blocked by exposure to 1 μM μ-conotoxin GIIIB (Alomone Labs, Jerusalem, Israel). Microelectrode recordings were performed using ∼40–60 MΩ borosilicate electrodes filled with 3 M potassium acetate. Spontaneous miniature synaptic events (mEPPs) were collected for 1–2 min in each muscle fiber, and then 10–30 nerve-evoked synaptic events (EPPs) were collected with an interstimulus interval of 5 s. All recordings were corrected for nonlinear summation (18). To calculate quantal content, the average EPP amplitude was divided by the average mEPP amplitude recorded from each NMJ. This ratio estimates the average number of quanta (packages of neurotransmitter stored in synaptic vesicles) that are released following each presynaptic action potential. To evaluate the effects on short-term synaptic plasticity, a train of four EPPs with an interstimulus interval of 20 ms (50 Hz) was collected in each muscle fiber. Data were collected from the same NMJ before and 30–60 min after exposure to 25 nM ω-Agatoxin IVA to block a fraction of presynaptic VGCCs. Data were collected using an Axoclamp 900A and digitized at 10 kHz for subsequent analysis using pClamp 10 software (Molecular Devices, Sunnyvale, CA).
LEMS Passive Transfer to Mice
We used a LEMS passive transfer mouse model as previously reported (2, 19–26). The collection of serum from a patient with LEMS was performed following the guidelines set forth by the University of Pittsburgh Institutional Review Board. Serum from patient “GS” was used for all LEMS passive transfer data reported here and was collected using plasmapheresis. Patient GS is a patient with paraneoplastic LEMS with small cell lung cancer and metastases to mediastinal nodes that have been treated with six cycles of chemotherapy (carboplatin and etoposide). As has been previously reported, a LEMS diagnosis increases survival in patients with small lung cancer (27, 28). Consistent with these reports, GS is now >10 yr postdiagnosis with no evidence of cancer recurrence. However, clinical weakness persists, and GS presents with areflexia that recovers after 10 s of exercise. Limb weakness is symmetrical and unchanged since diagnosis with a Medical Research Council sum score (29) of 46/60. Autoantibodies were detected to voltage-gated calcium channels (128 pM; control <30 pM), but not to voltage-gated potassium channels or acetylcholine receptors. Compound muscle action potential (CMAP) measured from the abductor digiti minimi was reduced (1.3–2.4 mV as compared to normal of 7–8 mV) (30) and showed an augmentation of 200%–225% postexercise. A 3-Hz repetitive nerve stimulation resulted in a CMAP decrement on the 4th stimulation of 20%–30% (normal shows no decrement).
To passively transfer LEMS to mice, adult female BL6/C57 mice (6- to 16-mo old at beginning of passive transfer; weighing 25–32 g; Charles River Laboratories) received one intraperitoneal (ip) injection of LEMS patient serum on day 1, and then an injection of 300 mg/kg of cyclophosphamide on day 2 to suppress the specific immune response to human IgG. This was followed by an intraperitoneal injection of 1.5 mL of LEMS patient serum once per day for 33–35 consecutive days. Control (BL6/C57) mice did not receive any injections. Passive transfer of LEMS was confirmed by measurements of EPP amplitude, mEPP amplitude, and quantal content in ETA nerve-muscle preparations as described earlier. After LEMS passive transfer, EPP size and quantal content were significantly reduced (control mice: mEPP = 0.29 + 0.01 mV, EPP = 28.6 + 1.1 mV, quantal content = 102.1 + 3.9, n = 41; LEMS mice: mEPP = 0.33 + 0.04 mV, EPP = 12.1 + 0.9 mV*, quantal content = 44.2 + 4.6*, n = 18; * = significantly different from control, Student’s t test).
Immunohistochemical Analysis
Tissue was collected from control mice and from LEMS mice following the passive transfer protocol. The gastrocnemius muscle was dissected and postfixed with 2% paraformaldehyde in PBS for 20 min at room temperature. The muscle was then cryoprotected in 20% sucrose/PBS at 4°C overnight and then embedded in optimal cutting temperature compound (Fisher Healthcare). Longitudinal sections (25-µm thick) were cut using a cryostat and blocked with 2% bovine serum albumin, 2% normal goat serum, and 0.1% Triton X-100 in PBS. The sections were incubated with the Synaptotagmin-2 primary antibody (1:4,000; Synaptic Systems, No. 105225) at 4°C overnight, washed with PBS, and incubated with the Alexa Fluor 546-conjugated anti-guinea pig secondary antibody (1:1,000; Invitrogen, No. A-11074) and α-Bungarotoxin, Alexa Fluor 488 conjugate (Invitrogen, No. B13422) for 2 h at room temperature. The sections were washed with PBS and mounted in ProLong Gold antifade reagent (Invitrogen, No. P36934) with a No. 1.5 cover glass (VWR, No. 48393-230).
Synaptotagmin-2 Signal Intensity Quantification
Images were obtained using a Leica TCS SP8 confocal microscope with a ×63 objective lens (HC PL APO CS2 ×63/1.40 OIL) and immersion oil (Leica Type F, refractive index 1.518). Confocal images were obtained by collecting a z-stack and creating a maximum intensity projection of all the scanned slices of the synapse. To facilitate a comparison of the staining intensity for the synaptotagmin-2 antibody in control and LEMS tissue, paired cryostat sections (control and LEMS) were mounted on the same slide and stained together. Then, imaging laser intensity, zoom, and hybrid photodetector gain was maintained at constant values for all images. The NMJs were identified using α-bungarotoxin to label the acetylcholine receptors.
Image analysis was performed using ImageJ. For each synapse, a binary mask encompassing the NMJ was generated using the bungarotoxin (BTX) co-stain. For most synapses analyzed, the mask was generated automatically using the “Make Binary” function in ImageJ. Some synapses were not amenable to automatic mask generation, so for these synapses, the mask was manually drawn using the BTX co-stain. The average intensity of both BTX and syt2 within the mask was then determined for each synapse in both the wild type (WT) and LEMS conditions.
To determine if LEMS caused a dispersal of syt2 outside the bounds of the NMJ (as defined by the BTX co-stain), we measured the syt2 signal in an annulus surrounding the original BTX-defined mask. To do this, we dilated all the original masks by 10 pixels (690 nm) using the “Dilate” function in ImageJ. We determined the integrated intensity of the enlarged mask and then subtracted the integrated intensity of the original mask to determine the average intensity just within the annulus around the original mask.
Monte Carlo Simulations
MCell version 3.4 (www.mcell.org) was used to study synchronous vesicle release at the mouse NMJ. MCell is a stochastic particle-based diffusion-reaction simulator that can model biological systems with arbitrarily complex three-dimensional (3-D) geometries (31–33). For each simulation, 6,000 separate MCell seeds were run. During each run, an AP (or a train of APs) caused VGCCs to open according to a Markov-chain ion channel gating scheme (see Fig. 1D). Calcium was then released from open VGCCs and diffused into the model terminal. As they diffused, the calcium ions bound to either calcium sensors located on synaptic vesicles or to calcium buffer molecules evenly distributed throughout the model terminal. Our models are based on previously developed MCell models of the frog AZ (15, 16) and a recently developed preliminary model of mouse AZs (17). We then further constrained these models with several new parameters such as the predicted number of synaptotagmin sensors at the base of docked synaptic vesicles (35), on- and off-rates for synaptotagmin 1 and synaptotagmin 7 (36–38) and VGCC gating (using a model with two open states) (39). Key parameters for our MCell model are shown in Table 1, and our MCell mouse NMJ AZ model has been deposited in GitHub: https://github.com/r0zita/mcell_mouse_mdl_ctrl.
Figure 1.
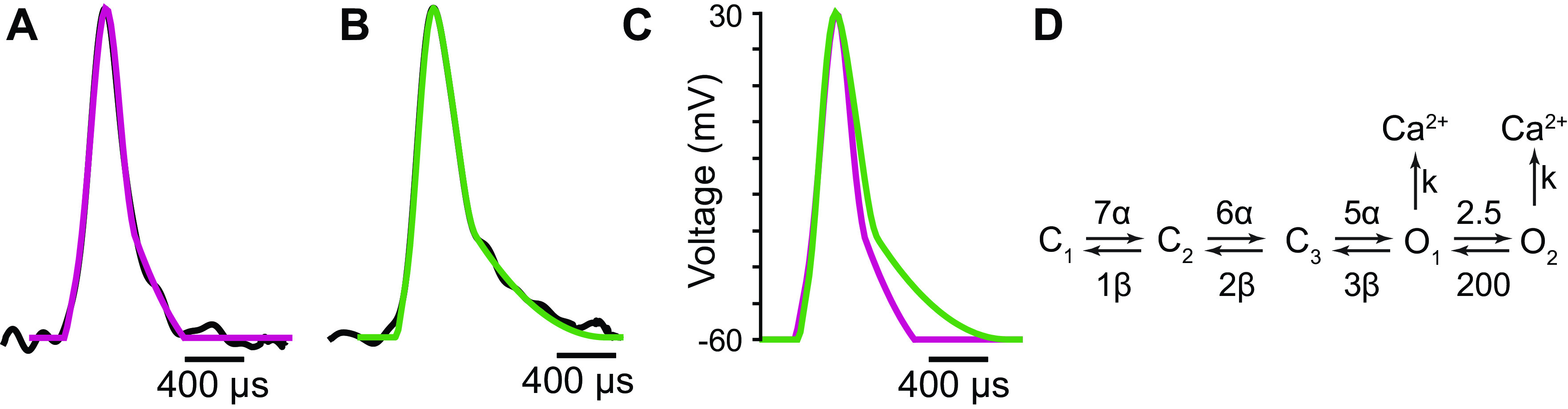
AP waveforms and voltage-gated calcium channel (VGCC) kinetics used in the MCell models. A: the normalized average of AP waveforms recorded from mouse neuromuscular junctions (NMJs) (black) and the computational AP waveform used in MCell simulations (magenta). B: the normalized average of AP waveforms recorded from mouse NMJs in the presence of 1.5 μM 3,4-diaminopyridine (3,4-DAP) (black) and the computational AP waveform used to simulate 3,4-DAP in MCell (green). C: a comparison of the computational AP waveforms used in control (magenta) and 3,4-DAP (green) simulations. D: the Markov-chain ion channel gating scheme used to determine the behavior of VGCCs in our simulation. Average AP waveforms in A and B are from Ginebaugh et al. (34).
Table 1.
Summary of model input parameters
| Input Parameter | Description | References |
|---|---|---|
| AZ ultrastructure | Average dimensions of nerve terminal segment, vesicle diameter, location, and number. The healthy mouse model consisted of six AZs placed 500 nm apart, and each AZ contained two docked synaptic vesicles. | (21, 40–42) |
| Diffusion coefficient for free calcium | D = 6 × 10−6 cm2 s−1 | (43, 44) |
| Action potential | AP waveform inputs based on recently published AP waveforms recorded from the NMJ via voltage imaging. We used the average AP waveforms from both control and 3,4-DAP recordings and prepared them for use in our simulations | (34, 45) |
| Calcium channel kinetic properties | Three closed states, two open states: C0C1C2O1O2; Conductance of open state: 2.4 pS. We fit the average calcium currents to the previously-published recordings from HEK293 cells expressing P/Q type VGCCs. | (15, 26, 39, 46) |
| Calcium channel rate constants | The rate constants α, β, and k in our kinetic scheme | (15, 16) |
| Reversal potential | the reversal potential for calcium used in the calculation of k is +60 mV | (47) |
| Calcium channel distribution | Two docked synaptic vesicles with one row of two VGCCs placed on each side of vesicle. | (21, 42) |
| Calcium buffer binding-site properties | Concentration: 2 mM (∼106 sites); kon = 1 × 108 M−1·s−1, koff = 10,000 s−1, mean Ca2+ dwell time = 1/koff = 100 μs | (48–52) |
| Calcium binding sites arrangement | The number of synaptotagmin sensors at the base of docked synaptic vesicles is likely to be six protein densities (syt1/2 proteins) placed symmetrically around the bottom of the vesicle. | (35) |
| The vesicle fusion mechanism | The activated vesicular fusion mechanism is probabilistic as determined by the Metropolis-Hastings sampling protocol with the free energy barrier of Eb = 40 kBT. | (16, 53, 54) |
| Calcium binding rates | The calcium-binding rates for the syt1/2-like sensor were set to kon = 2.2 × 107 M−1·s−1 and koff = 910 s−1, and the calcium-binding rates for the syt7-like sensor were set to kon = 1 × 107 M−1·s−1 and koff = 15 s−1. | (36–38) |
| Extracellular calcium concentration | 1.8 mM | (17) |
| Calcium buffer concentration | 2 mM | (16) |
AZ, active zone; 3,4-DAP, 3,4-diaminopyridine; NMJ, neuromuscular junction; VGCC, voltage-gated calcium channel.
Action Potentials and VGCCs
To further constrain our model, we used AP waveform inputs based on recently published AP waveforms recorded from the mouse NMJ via voltage imaging (45). We used the average AP waveforms from both control and 3,4-diaminopyridine (3,4-DAP) recordings and prepared them for use in our simulations as previously described (34). In brief, the average AP waveforms were normalized to a peak voltage of 30 mV and a resting potential of −60 mV. We then removed fluctuations caused by noise in the experimental data at the onset of the rising edge of each of the AP waveforms by replacing the first 10% of the rising edge with a line that had the same slope as the next 10% of the rising edge. We then removed fluctuations in the falling edge of each AP waveform by fitting a second-degree polynomial through the last 30% of the falling edge and then replacing that 30% with the polynomial (Fig. 1, A–C).
The kinetics of VGCC gating were based on the Markov-chain model used in our excess-calcium-binding-site model of the frog NMJ (15), which consisted of three closed states and one open state. However, because prior research has shown that VGCC gating is best fit by a model with two open states (39), we altered the previous VGCC gating scheme to contain two open states and re-parameterized it to fit the average calcium currents from previously published recordings from HEK293 cells expressing P/Q type VGCCs (26) (Fig. 1D). The rate constants α, β, and k were the same as used in previous models (15, 16) except the reversal potential for calcium used in the calculation of k was +60 mV rather than the +50 mV value that was used in those previous models (47).
Mouse MCell Model and Vesicle Fusion
The NMJ ultrastructure and placement of mouse NMJ AZs in our MCell model were the same as previously described in our preliminary mouse model (17). The healthy mouse model consisted of six AZs placed 500 nm apart, and each AZ contained two docked synaptic vesicles (Fig. 2A). All distances between channels and vesicles are taken from Fukuoka et al. (21).
Figure 2.

Diagrams of the healthy mouse active zone (AZ) MCell model. A: visualization of the overall geometry of the health mouse AZ MCell model. Six AZs are modeled, and each AZ consists of voltage-gated calcium channels (VGCCs) (small black dots) and two docked synaptic vesicles (black spheres). B: a diagram of the calcium sensors on the underside of each synaptic vesicle. Each vesicle has six synaptotagmin-1 and synaptotagmin-2 (syt1/2)-like sensors, consisting of five calcium ion binding sites each. A syt1/2-like sensor is activated when at least two of its five sites are bound to calcium. Each vesicle also has 18 syt7-sensors, positioned in a single annulus around the outside of the syt1/2-like sensors. Each syt-7 like sensors only contain one calcium-binding site and are activated when the site is bound by a calcium ion. Adapted from Laghaei et al. (17). C: a schematic of a single AZ, showing the positions for transmembrane proteins based on freeze-fracture data (black and white circles). The schematic shows two docked synaptic vesicles (gray circles) with a row of two VGCCs placed on each side (black circles). The white circles are unidentified AZ proteins that do not contribute to the model output.
The vesicle fusion mechanism was similar to our previously described mechanism (16). In brief, the bottom of each vesicle contained two different types of calcium sensors. The first type was a fast sensor that represented synaptotagmin-1 or synaptotagmin-2 (syt1/2). The second was a slow second-sensor binding site. Since the publication of the NMJ AZ model that contained a second-sensor binding site (16), it was revealed that this second sensor is likely to be synaptotagmin-7 (syt7) (55). Thus, to be more consistent with the literature, we altered the calcium binding on- and off-rates of both the syt1/2-like and syt7-like sensors based on a previous simulation of phospholipid binding of both sty1 and syt7 (36). The calcium-binding rates for the syt1/2-like sensor were set to kon = 2.2 × 107 M−1·s−1 and koff = 910 s−1, and the calcium-binding rates for the syt7-like sensor were set to kon = 1 × 107 M−1·s−1 and koff = 15 s−1.
Recent literature has revealed that there are likely to be six protein densities (thought to contain syt1/2 proteins) surrounding the bottom of synaptic vesicles (35). Thus, we rearranged the placement of calcium-sensing syt1/2-like particles on the bottom of each vesicle to match the literature (Fig. 2B) and placed 6 syt1/2-like sensors symmetrically around the bottom of the vesicle. Since less is known about the syt7 proteins, we kept our syt7 placement similar to previous computational models (16) but placed 18 syt7-like sensors (rather than 16 as used in the previous model) around the bottom of the vesicle to be more symmetrical with the 6 syt1/2-like sensors (creating a 3:1 ratio of syt7 to syt1/2 sensors). The precise details of synaptotagmin-mediated vesicle fusion are still unknown, and refinements in the density and distribution of syt7 proteins, and their specific role at the NMJ would require additional experiments that are beyond the scope of this study. Regarding calcium-mediated interactions with synaptotagmin proteins, this is thought to be a complicated two-step process with calcium binding to C2 domains and then the syt—calcium ion complex binding to lipid (56–61), however, for the purpose of our simulations reported here, we have simplified this to a single on and off rate (see Table 1). Our simplified approach has been shown to closely model transmitter release at the NMJ (16).
When bound by a sufficient number of calcium ions, the calcium sensors were considered activated and contributed to increasing the probability of vesicular fusion. Vesicles fuse in a probabilistic manner, with fusion for each vesicle determined with the Metropolis–Hastings sampling protocol (62, 63) using the probability . Here, Eb = 40 kBT is the free energy barrier preventing vesicle fusion (53, 54), nS1 and nS7 are the number of syt1/2-like sensors and syt7-like sensors that have been activated by calcium, and ΔES1 and ΔES7 are the energy each active calcium sensor contributes to overcoming the 40 kBT free energy barrier.
Experimental Design and Statistical Analysis
For experimental data, all statistics were performed using GraphPad Prism software (v.9). The p- and n values are reported in the text. All data in the graphs are shown as the means ± SE. For computer simulation studies, 6,000 separate MCell seeds were run to generate reliable averages for simulation outputs. For mouse electrophysiology experiments measuring endplate potential amplitude, data were collected from 18 synapses from two mice. For immunohistochemical studies, we imaged a total of 33 synapses from three LEMS model mice and the same number from aged-matched control mice (untreated). We confirmed that the data were normally distributed and then performed an unpaired Students t test for significance with an α < 0.05.
RESULTS
Constraining the Healthy Mouse AZ Model
Our MCell model of the healthy mouse AZ was initially based on previous MCell models of the frog AZ (15, 16), a preliminary model of the mouse AZ (17), and experimental and biophysical data (see methods). The only free parameters in our model were the number and location of VGCCs as well as the energy contribution of the syt1/2-like and syt7-like calcium binding sites that contribute to overcoming the 40 kBT free energy barrier for synaptic vesicle fusion. We had previously reported simulation results for models with varying numbers of sensor sites and fusion mechanisms (15, 16) and used these results to constrain model parameters in this study. We also tested a wide variety of spatial configurations for P/Q-type VGCCs based on the previously reported organization of presynaptic proteins demonstrated in freeze-fracture electron microscopy (21) and found that a four-channel VGCC AZ model (Fig. 3) with a binding energy of 15 for syt1/2-like sensors and 8 for syt7-like sensors was able to predict a variety of electrophysiological results. This model was able to match the experimental probability of release per AZ of 0.22 (17), a slight depression over short trains of stimuli (Fig. 3A), a paired-pulse facilitation that is relatively unchanged by changes in inter-spike interval (Fig. 3B), and a vesicle release latency following an AP (Fig. 3C) that is in agreement with previously published experimental data (65). In addition, our model predicted a log-log ratio between extracellular calcium and transmitter release (also known as the calcium-release ratio, or CRR; Fig. 3D) of ∼3, which is consistent with experimental data (66–71).
Figure 3.
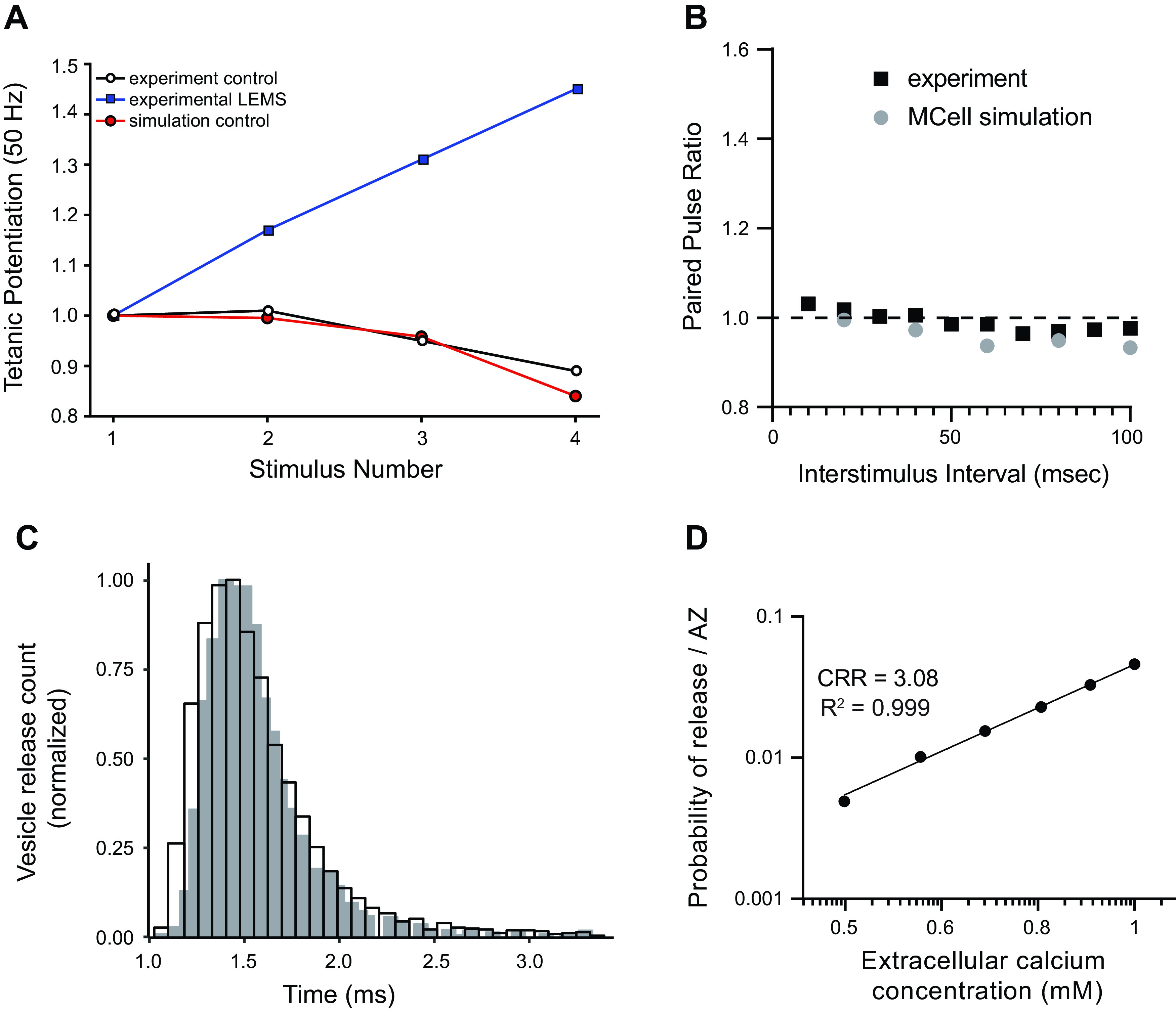
Validation of our healthy mouse active zone (AZ) MCell model. A: plot of tetanic potentiation at 50 Hz. Our MCell model of the healthy mouse AZs (red circles) closely matched experimental results (open circles). The tetanic potentiation seen in Lambert-Eaton myasthenic syndrome (LEMS)-model mice (blue squares) is added for comparison. Experimental results for both LEMS and control tetanic potentiation were adapted from Tarr et al. (26). B: plot of the short-term plasticity measured during a pair of action potentials expressed as the ratio of the magnitude of transmitter release after the second pulse divided by the magnitude after the first pulse (paired pulse ratio) as the interstimulus interval is changed from 10 to 90 ms. The outputs from the MCell model (gray circles) and the experimental data (black squares) both predict little change in transmitter release over differing interstimulus interval. Experimental data are adapted from Laghaei et al. (17). C: the predicted latency of vesicle release following a single presynaptic AP is plotted based on our MCell model (open bars) in comparison with the time course of release measured at the mouse neuromuscular junction (NMJ) (64) (gray bars). D: plot of changes in transmitter release magnitude in our MCell model as the extracellular calcium concentration was varied. The log-log best fit line for the data is shown with a calcium release ratio (CRR; slope of the line) of 3.08 and an R2 value of 0.999.
Our healthy AZ model places VGCCs only ∼15 nm away from the nearest synaptic vesicle (and the associated syt1/2 calcium sensors), which is similar to the estimated distance between VGCCs and calcium sensors in fast-spiking hippocampal GABAergic interneurons (72, 73) and the mature calyx of Held (74, 75). Thus, our VGCC placement agrees with the hypothesis that VGCCs are tightly coupled to docked synaptic vesicles in the NMJ to mediate fast synchronous transmitter release (76).
Constructing an MCell Model of LEMS-Modified AZs
We next sought to create a MCell model that would fit the experimental electrophysiological characteristics seen in LEMS-model mice by altering our model of healthy mouse AZs described earlier. Initially, we altered our model of healthy mouse AZs in an iterative manner based on the experimental evidence that LEMS reduces the number of P/Q-type VGCCs in the AZs (22, 24), causes a compensatory upregulation of L-type VGCC subtypes outside of the AZs (24, 25), reduces the number of AZs (19), and disrupts the organization of AZ proteins in many of the remaining AZs (21). The experimental results we sought to predict with our model were a 50%–75% reduction in quantal content compared with healthy control mice, and an increase in facilitation over short trains of stimuli (2, 20, 25, 26, 77).
Removal of P/Q-Type Calcium Channels Alone Is Insufficient to Reproduce LEMS Electrophysiology
Perineural recordings of calcium currents at the NMJs of ex vivo nerve-muscle preparations from healthy and LEMS-model mice indicate that calcium current from P/Q-type VGCCs is only reduced by ∼30%–40% in LEMS-model mice (22, 24). We found that randomly removing ∼40% (9 of 24) of the P/Q-type VGCCs in our MCell model only reduced transmitter release by 47%. This is close to the 50%–75% reduction of transmitter release seen in LEMS-model mice but did not result in the magnitude of short-term synaptic plasticity observed in LEMS (Fig. 4). These model predictions were similar to the recorded effects of blocking 40%–60% of transmitter release using 25 nM ω-Agatoxin IVA in freshly dissected NMJs from a healthy mouse (Fig. 4D). Removing additional VGCCs resulted in a much greater reduction in calcium current than the experimentally measured value of a 30%–40% reduction (22, 24), but still did not result in the magnitude of short-term synaptic plasticity present in LEMS (Fig. 4C). It is clear from our ex vivo recordings of transmitter release, and our model predictions, that a simple removal or block of P/Q-type VGCCs is not sufficient to reproduce LEMS effects.
Figure 4.
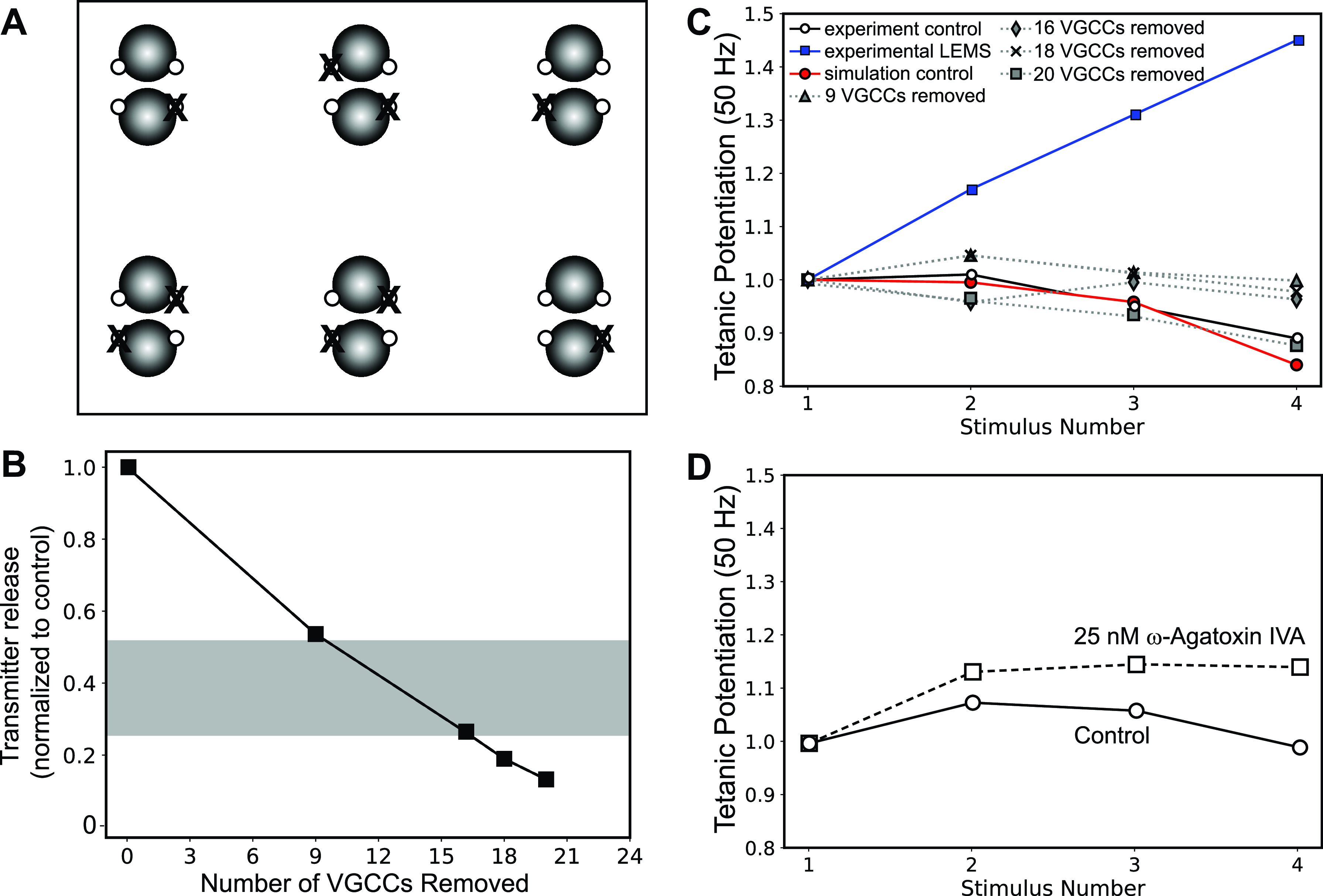
The effects on transmitter release magnitude and short-term plasticity after simply removing presynaptic voltage-gated calcium channels (VGCCs). A: MCell model diagram depicting six active zones (AZs) that contain docked synaptic vesicles (black spheres), VGCCs (open dots), and the random removal of nine VGCCs (X). B: plot of transmitter release (normalized to the control healthy model) predicted by the MCell model as various numbers of VGCCs are removed. The gray bar indicates the range of transmitter release reduction observed in the mouse passive transfer model of Lambert-Eaton myasthenic syndrome (LEMS) adapted from Refs. 2, 20, 25, 26, and 77. C: plot of tetanic potentiation at 50 Hz in healthy control experimental results (open circles), LEMS passive transfer model mice (blue squares), our MCell model of the healthy mouse AZs (red circles), and after removing 9, 16, 18, and 20 VGCCs from our model of 6 AZs that contains a total of 24 VGCCs (dotted lines). Experimental results for both LEMS and control tetanic potentiation were adapted from Tarr et al. (26). D: experimental recording of short-term synaptic plasticity (tetanic potentiation) from healthy control neuromuscular junctions (NMJs) (open circles) and after blocking 40%–60% of transmitter release magnitude using 25 nm ω-Agatoxin IVA (open squares).
Disrupting AZ Structural Organization Decreases Transmitter Release and Increases Facilitation
Not only does LEMS reduce the current through P/Q-type VGCCs, but freeze-fracture electron microscopy reveals that LEMS also disrupts the normally well-organized structure of the AZs, reducing the number of well-organized AZs by 70%–85% (19, 21). These studies also found that LEMS causes a large number of clusters of intramembranous particles to appear [which are likely highly unorganized AZs (21)], outnumbering the remaining well-organized AZs. We first investigated the impact of structural disorganization of the AZs alone in our model by moving the position of P/Q-type VGCCs relative to docked synaptic vesicles. We found that simply moving the VGCCs 5, 10, or 15 nm further away from the synaptic vesicles in the AZ, without removing any VGCCs, reduced transmitter release by 22%, 40%, or 56% respectively (Fig. 5, A and B). Although moving the location of VGCCs had similar effects on the magnitude of transmitter release as removing calcium channels, moving the location of VGCCs had a much larger impact on short-term synaptic facilitation than simply removing VGCCs (Fig. 5C). These results predict that the disorganization of the AZs structure plays an important role in determining the LEMS pathological phenotype with respect to the control of transmitter release at the NMJ (reduced magnitude of transmitter release and a large increase in short-term synaptic facilitation).
Figure 5.

The effects on transmitter release magnitude and short-term plasticity after simply moving presynaptic voltage-gated calcium channels (VGCCs) away from docked synaptic vesicles. A: MCell model diagram depicting six active zones (AZs) that contain docked synaptic vesicles (black spheres), VGCCs (open dots), and the movement of all VGCCs away from synaptic vesicles by 5, 10, and 15 nm. B: plot of transmitter release (normalized to the control healthy model) predicted by the MCell models shown in A as VGCCs are moved 5, 10, or 15 nm away from docked synaptic vesicles. The gray bar indicates the range of transmitter release reduction observed in the mouse passive transfer model of Lambert-Eaton myasthenic syndrome (LEMS) adapted from Refs. 2, 20, 25, 26, and 77. C: plot of tetanic potentiation at 50 Hz in healthy control experimental results (open circles), LEMS passive transfer model mice (blue squares), our MCell model of the healthy mouse AZs (red circles), and after moving VGCCs 5, 10, or 15 nm within our model of six AZs that contains a total of 24 VGCCs (dotted lines). Experimental results for both LEMS and control tetanic potentiation were adapted from Tarr et al. (26).
Based on the results regarding moving the position of VGCCs, we sought to use this approach in combination with a reduction in the number of P/Q-type channels in the AZ to create a more complete LEMS MCell model. To create this model, we randomly removed 9 of 24 P/Q-type VGCCs in our six AZ model and then moved the remaining P/Q-type VGCCs an additional 5, 10, or 15 nm away from the AZ (Fig. 6A). We found that this model resulted in a 57%, 69%, and 77% reduction in transmitter release compared with control when VGCCs were moved 5, 10, and 15 nm respectively (Fig. 6B), which is a close fit for the experimentally measured values of transmitter release recorded from LEMS-model mice. When 9/24 VGCCs were removed and the remaining VGCCs were moved 15 nm away from the AZ, we also observed a large increase in short-term synaptic facilitation to a level similar to the facilitation observed in LEMS passive transfer experiments in mice (Fig. 6C).
Figure 6.

The effects on transmitter release magnitude and short-term plasticity after a combination of moving presynaptic voltage-gated calcium channels (VGCCs) away from docked synaptic vesicles and randomly removing 9 of 24 VGCCs from the six active zone (AZ) model. A: MCell model diagram depicting six AZs that contain docked synaptic vesicles (black spheres), VGCCs (open dots), and the movement of all VGCCs away from synaptic vesicles by 5, 10, and 15 nm, coupled with the random removal of nine VGCCs (X). B: plot of transmitter release (normalized to the control healthy model) predicted by the MCell models shown in A. The gray bar indicates the range of transmitter release reduction observed in the mouse passive transfer model of Lambert-Eaton myasthenic syndrome (LEMS) adapted from Refs. 2, 20, 25, 26, and 77. C: plot of tetanic potentiation at 50 Hz in healthy control experimental results (open circles), LEMS passive transfer model mice (blue squares), our MCell model of the healthy mouse AZs (red circles), and after moving VGCCs 5, 10, or 15 nm and randomly removing nine VGCCs within our model of six AZs that contains a total of 24 VGCCs (dotted lines). Experimental results for both LEMS and control tetanic potentiation were adapted from Tarr et al. (26).
Building a More Complete LEMS Model
Previously, it has been reported that LEMS-mediated changes at the synapse include not only the loss of VGCCs and the disruption in the organization of the AZ proteins, but also fewer total AZs (19, 21), as well as a compensatory upregulation in the expression of L-type VGCCs (25). Although it is known that L-type VGCCs can be upregulated in LEMS passive transfer mouse models and contribute to the regulation of transmitter release, little is known about their number and location relative to the AZs. However, it is hypothesized that they do not localize with the AZs because they lack the necessary synaptic protein interaction site (78–80). Furthermore, buffer experiments have shown that the contribution of L-type VGCCs to transmitter release in LEMS is blocked by low concentrations of fast calcium buffer, placing them outside of the AZ (25, 81). Therefore, we sought to add L-type VGCCs to a model of the LEMS synapse that included fewer total AZs, as well as removing some P/Q-type VGCCs and disrupting the organization of the remaining AZs. To accomplish this more complete LEMS model, we removed two of the six AZs in our model, and to model an increase in L-type current and its effect on transmitter release, we simply placed additional VGCCs on each side of the remaining AZs an additional 25 nm beyond the double rows of proteins in our AZ model (Fig. 7A). In our MCell models, we used the same channel gating scheme (Fig. 1D) for all VGCCs, but for simplicity we will herein refer to VGCCs in our model placed inside the AZs as P/Q-type and VGCCs placed further outside the AZs as L-type. Cav1 (L-type) channels expressed in neurons have been shown to have slightly slower activation kinetics as compared with Cav2 (N and P/Q-type) (82), which may reduce the percentage of available channels activated during brief action potentials. As such, we may have slightly overestimated the impact of L-type channels in our models. To quantify the changes in the magnitude of transmitter released during a single action potential, we compared the total release from the entire segment of the nerve terminal shown in Fig. 7A (4 AZs) with the total transmitter release from a control segment of the nerve terminal shown in Fig. 2A (6 AZs). We found that within our modified LEMS synapse model with fewer VGCCs and disrupted AZ organization, the removal of two of six AZs and the addition of L-type VGCCs resulted in a 54%, 58%, or 64% reduction in transmitter release from the nerve terminal segment in Fig. 7A when P/Q-type VGCCs were moved 5, 10, or 15 nm respectively, as compared with healthy control nerve terminal segment shown in Fig. 2A. These predicted reductions in transmitter release levels were all within the range of the experimentally measured reductions in quantal content in LEMS (Fig. 7B). We also observed an increase in facilitation from this model, but the magnitude was less than that observed in electrophysiological recordings form LEMS passive transfer NMJs (Fig. 7C). These results show that removing some P/Q-type VGCCs, disrupting AZ organization, removing some AZs, and positioning two additional L-type VGCCs adjacent to remaining AZs was not sufficient to predict all of the transmitter release effects observed in LEMS. More information about the L-type channel expression and position within the nerve terminal might be required to more accurately predict their contribution to the pathophysiology in LEMS. However, it is also possible that LEMS patient serum contains antibodies to other presynaptic proteins that contribute to the LEMS pathophysiology.
Figure 7.

The effects on transmitter release magnitude and short-term plasticity after a combination of moving presynaptic voltage-gated calcium channels (VGCCs) away from docked synaptic vesicles, removing two active zones (AZs), randomly removing 6 of 16 remaining P/Q-type VGCCs, and adding 2 L-type VGCCs outside the remaining AZs within our model. A: MCell model diagram depicting four AZs (after removing 2; large “X”) that contain docked synaptic vesicles (black spheres), VGCCs (open dots), and the movement of all VGCCs away from synaptic vesicles by 5, 10, and 15 nm, coupled with the random removal of 6 VGCCs (X), and the addition of 2 L-type VGCCs (black dots) outside of the remaining AZs. B: plot of total transmitter release from the entire nerve terminal segment containing four disease model AZs (normalized to the control healthy model nerve terminal segment that contains 6 healthy AZs) predicted by the MCell models shown in A. The gray bar indicates the range of transmitter release reduction observed in the mouse passive transfer model of Lambert-Eaton myasthenic syndrome (LEMS) adapted from Refs. 2, 20, 25, 26, and 77. C: plot of tetanic potentiation at 50 Hz in healthy control experimental results (open circles), LEMS passive transfer model mice (blue squares), our MCell model of the healthy mouse AZs (red circles), and after moving VGCCs 5, 10, or 15 nm, randomly removing six VGCCs from the remaining four AZs, and the addition of L-type VGCCs outside the AZ area (dotted lines). Experimental results for both LEMS and control tetanic potentiation were adapted from Tarr et al. (26).
Passive Transfer of LEMS to Mice Results in Reduced Synaptotagmin-2 Immunoreactivity at the NMJ
In addition to VGCCs, patients with LEMS have been shown to produce antibodies against synaptotagmin (9, 12), which is known to be a critical presynaptic regulator of transmitter release. We examined the effects of passive transfer of LEMS to mice on synaptotagmin immunoreactivity at the mouse ETA NMJ. We initially tested antibodies to syt1 and syt2 on control NMJs but found that the syt1 antibody did not provide a reliable stain of the presynaptic motor nerve terminal (data not shown). On the other hand, the syt2 antibody provided a robust and reliable stain for the presynaptic nerve terminal (Fig. 8). Therefore, for this study, we restricted our analysis to staining using the syt2 antibody. When comparing control and LEMS passive transfer tissue, syt2 immunoreactivity was significantly reduced by ∼39% within the NMJ (Fig. 8) as defined by overlap with the counterstain α-bungarotoxin-Alexa 594, which defines the postsynaptic acetylcholine receptor region [syt2 values in arbitrary units (means ± SE): control = 60.4 ± 22.85, n = 33 NMJs; LEMS = 36.8 ± 12.8, n = 32 NMJs]. In addition, at LEMS NMJs there was significant syt2 staining that was not confined to the α-bungarotoxin-Alexa 594 stained NMJ region, but this was not present to the same degree in control NMJs (Fig. 8; syt2 values outside of BTX stained regions of the NMJ in arbitrary units (means ± SE: control = 18.23 ± 5.87, n = 33 NMJs; LEMS = 22.65 ± 5.15, n = 32 NMJs). The significance of this extrasynaptic staining is unknown but may represent some aspect of LEMS pathology in mice. Since this staining was outside the AZs, it was not something that our models could address. Overall, these data confirm that LEMS patient antibodies can reduce NMJ syt2 immunoreactivity in mouse passive transfer neuromuscular tissue. Finally, we also observed a significant decrease in BTX intensity at LEMS NMJs relative to control NMJs [BTX values in arbitrary units (means ± SE): control = 83.91 ± 4.86, n = 33 NMJs; LEMS = 58.83 ± 2.25, n = 32 NMJs]. These data confirm that LEMS patient antibodies can reduce NMJ syt2 immunoreactivity in mouse passive transfer neuromuscular tissue and lead us to examine the impact of reduced fast synaptotagmin calcium sensors within our MCell AZ models of LEMS. This change in BTX staining intensity was unexpected since most patients with LEMS are not thought to have compensation in postsynaptic sensitivity (83), although rare cases have been reported of antibodies to postsynaptic acetylcholine receptors, which is often interpreted as a mixed diseases presentation of LEMS with myasthenia gravis (84–88).
Figure 8.
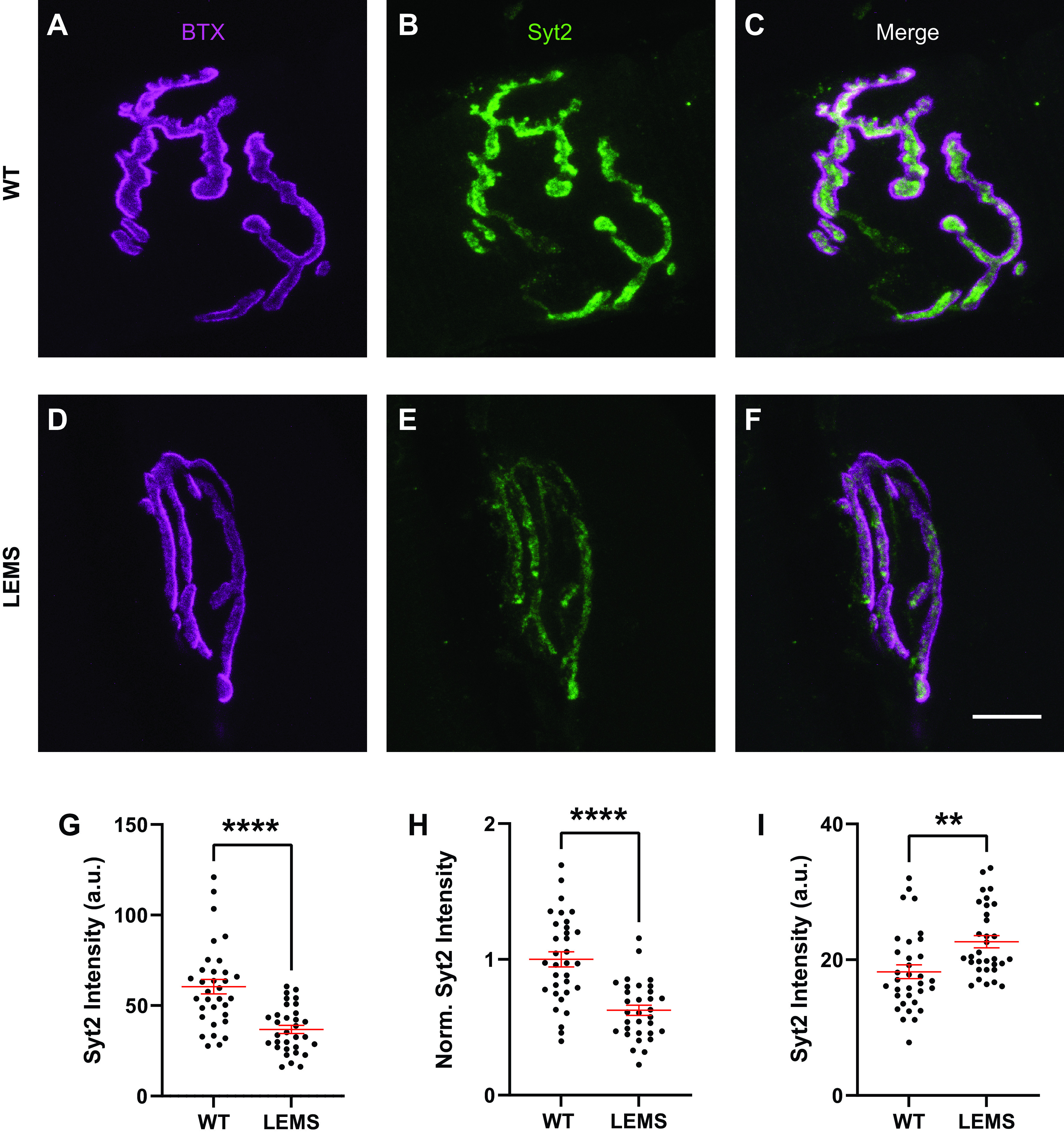
Synaptotagmin 2 (syt2) immunoreactivity in control mouse neuromuscular junctions (NMJs) and in Lambert-Eaton myasthenic syndrome (LEMS) passive transfer model mouse NMJs. Sample staining of control NMJ showing bungarotoxin (BTX) (A), syt2 (B), and the merge (C). Sample staining of LEMS NMJ showing BTX (D), syt2 (E), and the merge (F). G: comparison of syt2 intensity between control and LEMS shows a significant reduction in LEMS NMJs. H: comparison of normalized syt2 intensity between control and LEMS NMJs also shows a significant reduction in LEMS NMJs. All data points are normalized to the average syt2 intensity of the control NMJs stained on the same day. I: comparison of syt2 intensity outside the boundary of the BTX stain shows a significant increase in LEMS NMJs in a 10 pixel perimeter, which is equal to a 690 nm perimeter around BTX stained NMJs. Data in G, H, and I are means ± SE; **P < 0.01, ****P < 0.0001. Scale bar in F is 10 µm and applies to A–F.
Loss of Fast-Synaptotagmin-like Sensors Can Predict the LEMS Phenotype in Our AZ Models
To examine the impact of reduced fast-synaptotagmin sensors in silico, we reduced the number of fast syt1/2-like calcium sensors in our healthy mouse model to test our prediction that loss of syt1/2-like calcium sensors was sufficient to predict the transmitter release and short-term synaptic plasticity changes seen in LEMS-model mice. We sequentially reduced the number of syt1/2 sensors on the bottom of the docked synaptic vesicles in our model and found that removing four or five out of the six syt1/2-like calcium sensors (Fig. 9, A and B) was sufficient to match the magnitude of transmitter release deficit seen in LEMS model mice (Fig. 9C). However, removing four of the six syt1/2-like calcium sensors resulted in a greater increase to short-term synaptic facilitation than seen in LEMS-model mice (Fig. 9D). Removing five or six of the six syt1/2-like calcium sensors resulted in a much higher level of short-term synaptic facilitation (data not shown), due to the increased reliance on the slower synaptotagmin 7 sensors for transmitter release. Although we were not able to predict some of the typical seropositive LEMS-mediated electrophysiological results by simply removing syt1/2, these results may need to be interpreted based on the differences between patients with seropositive and seronegative LEMS clinical data (1). Seronegative patients do not have as great a reduction in the CMAP (a measure of the number of muscle cells that are firing action potentials after nerve stimulation), which could reflect a smaller reduction in transmitter release than is present in seropositive patients (1). Furthermore, seronegative patients have a smaller incremental response to muscle contraction (postexercise facilitation) and a smaller incremental response to high-frequency stimulation of the motor nerve (1). As such, in seronegative patients, short-term synaptic facilitation might be smaller. Taking these observations into consideration, the removal of one or two syt1/2 calcium sensors on each docked synaptic vesicle in our MCell model, which results in a smaller reduction in the magnitude of transmitter release and a smaller increase in short-term synaptic facilitation than is expected for the seropositive condition (Fig. 9), might be considered a reasonable fit for the seronegative condition.
Figure 9.
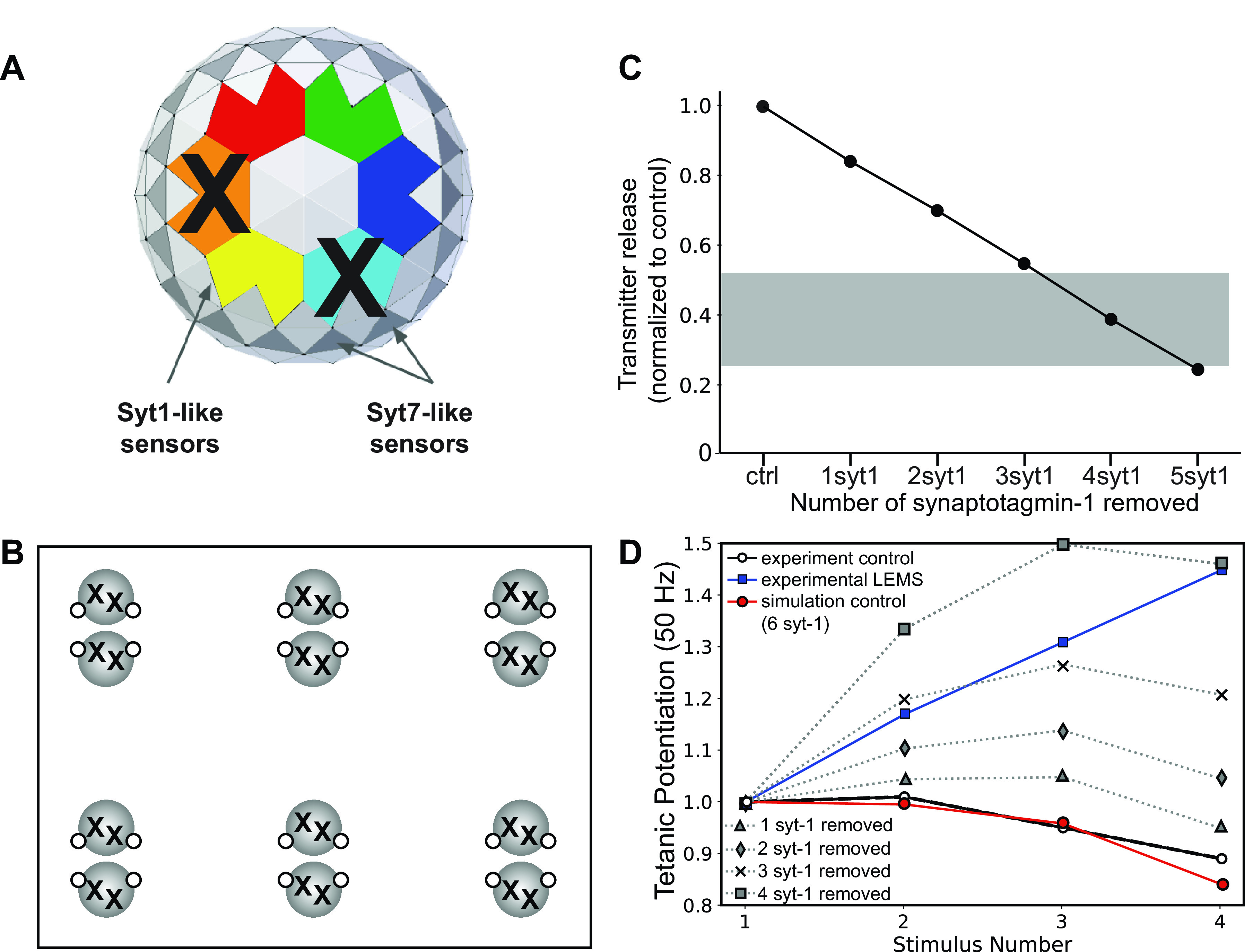
The effects on transmitter release magnitude and short-term plasticity of sequentially removing synaptotagmin-1/2 (syt1/2) calcium sensor proteins from the bottom of the docked synaptic vesicles in our six active zone (AZ) model. A: MCell model diagram depicting the removal of two syt1/2 proteins from the bottom of a synaptic vesicle (X) in our model. B: MCell model diagram depicting six AZs that contain docked synaptic vesicles (black spheres), voltage-gated calcium channels (VGCCs) (open dots), and the removal of two syt1/2 proteins (X) from each docked synaptic vesicle. C: plot of transmitter release (normalized to the control healthy model) predicted by the MCell models shown in B. The gray bar indicates the range of transmitter release reduction observed in the mouse passive transfer model of Lambert-Eaton myasthenic syndrome (LEMS) adapted from Refs. 2, 20, 25, 26, and 77. D: plot of tetanic potentiation at 50 Hz in healthy control experimental results (open circles), LEMS passive transfer model mice (blue squares), our MCell model of the healthy mouse AZs (red circles), and after removing various numbers of syt1/2 proteins from docked synaptic vesicles (dotted lines). Experimental results for both LEMS and control tetanic potentiation were adapted from Tarr et al. (26).
Because all patients with LEMS, including seronegative patients, have autoantibodies to a variety of AZ proteins, we next investigated the impact of the removal of syt1/2 in combination with a disruption in AZ structure that might be mediated by autoantibodies to other presynaptic proteins (Fig. 10). We found that we could closely predict LEMS-like effects on the magnitude of transmitter release and short-term synaptic facilitation if we moved AZ calcium channels 5, 10, or 15 nm away from synaptic vesicles while also removing between one and three syt-1 proteins from our models (Fig. 10, B–G). Again, if seronegative patients have smaller effects on the magnitude of transmitter release and short-term synaptic facilitation, as compared with seropositive patients, then our model results after removing only one syt1/2 calcium sensor and moving AZ P/Q-type calcium channels only 5 nm away from docked synaptic vesicles might be considered a reasonable fit based on clinical observations (1). Taken together, these results suggest that antibodies against fast synaptotagmin sensors (or syt1/2 since we did not distinguish between these two triggers for fast synchronous transmitter release in our models) may be sufficient to cause LEMS-like effects on transmitter release, either alone or when combined with mild AZ disruption, even in the absence of anti-VGCC antibodies.
Figure 10.

The effect on transmitter release magnitude and short-term plasticity of simultaneously removing synaptotagmin-1/2 (syt1/2) proteins and moving voltage-gated calcium channels (VGCCs) in our six active zone (AZ) model. A: MCell model diagram depicting six AZs that contain docked synaptic vesicles (black spheres), VGCCs (open dots), the removal of two syt1/2 proteins (X) from each docked synaptic vesicle, and the movement of VGCCs away from docked synaptic vesicles. B–D: plots of transmitter release (normalized to the control healthy model) predicted by the movement of VGCCs 5, 10, or 15 nm away from docked synaptic vesicles in combination with removing various numbers of syt1/2 proteins from docked synaptic vesicles. The gray bar indicates the range of transmitter release reduction observed in the mouse passive transfer model of Lambert-Eaton myasthenic syndrome (LEMS) adapted from Refs. 2, 20, 25, 26, and 77. E–G: plots of tetanic potentiation at 50 Hz in healthy control experimental results (open circles), LEMS passive transfer model mice (blue squares), our MCell model of the healthy mouse AZs (red circles), and after moving VGCCs 5, 10, or 15 nm and removing various numbers of syt1/2 proteins from docked synaptic vesicles (dotted lines). The effects of removing 1–3 syt1/2 proteins are shown as removing additional syt1/2 proteins resulted in tetanic potentiation that far exceeded the experimental LEMS results (blue squares). Experimental results for both LEMS and control tetanic potentiation were adapted from Tarr et al. (26).
A LEMS Model That Includes All Presynaptic Changes Investigated Earlier
Patients with LEMS are likely to have autoantibodies to several presynaptic proteins, and this could include P/Q-type VGCCs and syt1/2, although a comprehensive characterization of all of the antigens in patients with LEMS has not been carried out. We considered it possible that a model that included reductions in VGCCs and syt1/2 might recapitulate the pathophysiology of LEMS more accurately than our previous models. Therefore, we next created a model that combined all our previously investigated LEMS-induced changes, including the removal of two of six AZs, a reduction in P/Q-type VGCCs among the remaining AZ, disorganization in AZ structure, the addition of L-type VGCCs outside of the remaining AZs, and a reduction in the number of syt1/2 calcium sensors on docked synaptic vesicles (Fig. 11). Using this approach, we performed a parameter sweep of both distance of VGCC disruption as well as the number of syt1/2 calcium sensors removed. We found that several different combinations of these parameters could closely predict LEMS effects on both transmitter release magnitude and short-term synaptic facilitation (Fig. 11, B–G), suggesting that variation of these parameters in LEMS-model mice could explain both LEMS pathophysiology and the range of experimental results seen in LEMS-model mice in response to serum from different patients with LEMS.
Figure 11.
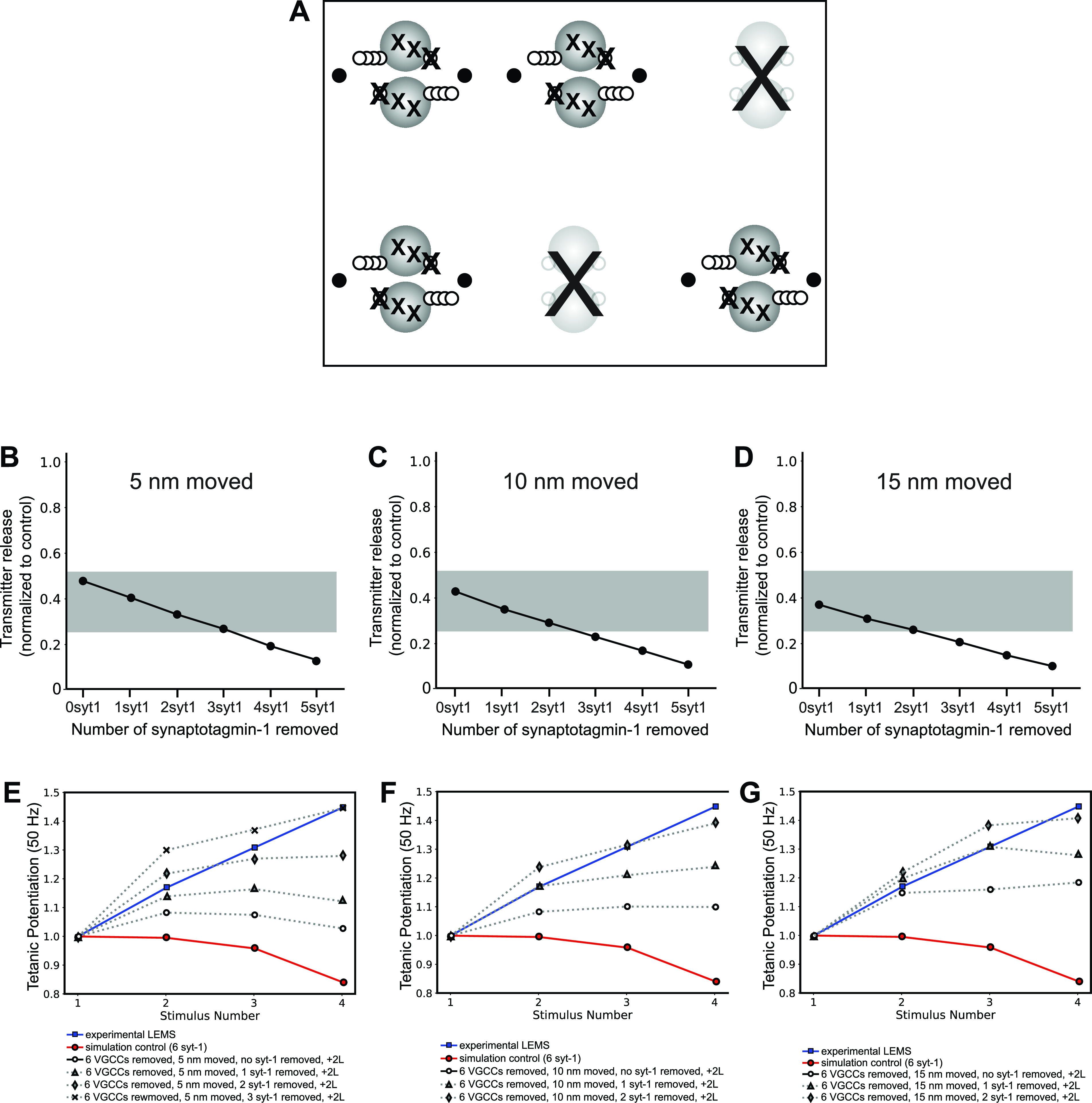
The effects on transmitter release magnitude and short-term plasticity in a Lambert-Eaton myasthenic syndrome (LEMS) model that incorporates all of the hypothesized changes to the neuromuscular junction (NMJ) active zones (AZs). A: MCell model diagram depicting the removal of 2 AZs (large “X”), removing 6/16 P/Q-type voltage-gated calcium channels (VGCCs) (medium “X”) from the remaining AZs, moving the remaining P/Q-type VGCCs away from docked synaptic vesicles (5, 10, or 15 nm; open circles), removing various numbers of synaptotagmin-1/2 (syt1/2) proteins from the bottom of the docked synaptic vesicles (small “X”), and adding 2 L-type VGCCs outside of the remaining AZs (black dots). B–D: plot of transmitter release from the entire nerve terminal segment containing four disease model AZs (normalized to the control healthy model nerve terminal segment containing 6 healthy AZs) predicted by the movement of VGCCs 5, 10, or 15 nm away from docked synaptic vesicles in combination with the other changes described in A. The gray bar indicates the range of transmitter release reduction observed in the mouse passive transfer model of LEMS adapted from Refs. 2, 20, 25, 26, 77. E–G: plot of tetanic potentiation at 50 Hz in healthy control experimental results (open circles), Lambert-Eaton myasthenic syndrome (LEMS) passive transfer model mice (blue squares), our MCell model of the healthy mouse AZs (red circles), and after moving VGCCs 5, 10, 15 nm and the other LEMS-induced changes described in A (dotted lines). The effects of removing greater than three syt1/2 proteins are shown because removing additional syt1/2 proteins resulted in tetanic potentiation that far exceeded the experimental LEMS results (blue squares). Experimental results for both LEMS and control tetanic potentiation were adapted from Tarr et al. (26).
In Silico LEMS AZ Models Predict the Effects of the Currently Approved Treatment for LEMS
The current FDA-approved treatment for LEMS is 3,4-diaminopyridine (3,4-DAP) (89–91). 3,4-DAP is a potassium channel antagonist that indirectly increases calcium flux into the nerve terminal by broadening the AP, which increases the probability that VGCCs in the terminal open during an AP (45). It has been shown to be effective in increasing transmitter release in LEMS-model mice (26) and partially alleviating muscle weakness in patients with LEMS (92–95). However, 3,4-DAP has dose-limiting side effects, and its effectiveness can vary greatly between patients such that up to 70% of patients with LEMS require other treatments, such as immunotherapy, in combination with 3,4-DAP (96, 97).
To our knowledge, no studies to date have investigated potential mechanistic differences in the effectiveness of 3,4-DAP on seropositive and seronegative LEMS. However, one study on patients with a congenital syt2 mutation found that treatment with 3,4-DAP provided only slight improvements in symptoms with no measurable change in initial CMAP, although slight improvements were detected with single-fiber EMG (98). In comparison, 3,4-DAP was shown to increase CMAP in patients with seropositive LEMS (99). Thus, we used our seronegative and seropositive LEMS models to investigate whether the particular LEMS-induced changes in the AZ could play a role in determining the effectiveness of 3,4-DAP treatment. We tested the effects of a clinically relevant concentration (1.5 µm) of 3,4-DAP on our MCell models by replacing the AP waveform in the simulations (Fig. 1A) with the average AP waveform from voltage-imaging recordings of the presynaptic terminal of the mouse NMJ in the presence of 1.5 µm 3,4-DAP (Fig. 1, B and C) (45).
Experiments on LEMS-model mice generated using patient serum from seropositive patients found that 1.5 µm 3,4-DAP increased the quantal content of the LEMS-weakened NMJ terminals 1.84-fold (26). This result was accurately recapitulated by our best seropositive LEMS MCell models (Fig. 11), which predicted a 1.80- to 1.82-fold increase in transmitter release from the 1.5 µm 3,4-DAP-modified AP waveform compared with the control AP waveform. The seronegative MCell models (which included the same number of VGCCs as control, though these channels were displaced and there were fewer syt1/2-like sensors; Fig. 10) also saw similar increases in transmitter release from the 1.5 µm 3,4-DAP-modified AP waveform compared with the control AP waveform (1.81-fold increase). These results suggest that current FDA-approved treatments for LEMS will work for both seronegative and seropositive patients.
DISCUSSION
Here, we have refined a computational model of a healthy mouse NMJ AZ. We then explored the impact of modifying this model based on physiological data collected in the mouse passive transfer model for the neurological disease LEMS, which attacks the AZ. We demonstrated that by rearranging our healthy mouse model based on findings from experimental studies in LEMS-model mice, we were able to construct models of seropositive and seronegative LEMS-modified AZs that predicted the electrophysiological effects of LEMS (reductions in transmitter release and enhanced short-term synaptic facilitation). Finally, we tested the impact of AP waveforms broadened by 3,4-DAP and found that our seronegative and seropositive LEMS models predicted similar responses to this FDA-approved treatment (3,4-DAP).
Disruption of the AZ Structure Is Essential to Reproduce the Pathophysiology of Seropositive LEMS
The electrophysiological effects of LEMS on NMJ transmitter release are often ascribed entirely to a loss of P/Q-type VGCCs. However, this simplified notion of LEMS ignores freeze-fracture electron microscopy findings that reveal a significant dysregulation in the organization of transmembrane proteins within many of the AZs. Early freeze-fracture electron microscopy studies in human patients with LEMS (19) and LEMS-model mice that had been injected with low doses of human LEMS serum daily for several weeks (19) resulted in a large increase in the number of disorganized clusters of intramembranous particles at LEMS NMJs. A later study, which instead created LEMS-model mice by injecting a high dose of IgG three times daily for two days, again found an increase in the number of disorganized clusters as well as the presence of partially disrupted “abnormal” AZs. The authors of this study suggested that these abnormal AZs were an intermediate stage between the normal well-organized AZ and the highly disorganized cluster, and this implied that the clusters were actually highly disorganized AZs (21).
Our model suggested that removing 40% of the P/Q-type VGCCs [as suggested by recordings of calcium currents in LEMS-model mice (22, 24)] only reduced transmitter release by 47%. This is a lesser reduction of transmitter release than the 50%–75% reduction seen in LEMS (25). We also found that moving the location of VGCCs within the AZ by 5, 10, or 15 nm away from the docked synaptic vesicles created a greater reduction in transmitter release than removing 40% of the VGCCs. Based on these computational results and the level of disorganization seen in freeze-fracture electron microscopy of LEMS NMJs, we hypothesize that the disorganization of the normally well-ordered structure of AZs, beyond the simple removal of VGCCs, is an essential component of the pathology of seropositive LEMS.
LEMS and Short-Term Synaptic Plasticity
LEMS is characterized by an increase or increment in the compound muscle action potential (CMAP) recorded from a patient’s hand muscle following a short exercise period. This CMAP increment is caused by short-term synaptic facilitation at the NMJ. Following LEMS passive transfer to mice and subsequent electrophysiological recordings from NMJs, the specific magnitude and time course of the development of tetanic potentiation can vary (25, 26, 100). This variability in the time course of tetanic potentiation can be affected by a variety of factors, including the particular patient serum used for the passive transfer to mice (as different patients have different titers of specific autoantibodies), the number of days of injection (which could alter the specific alterations to the AZ), and the frequency and duration of tetanic nerve stimulation. The variability in tetanic potentiation recorded from dissected muscles from mice after LEMS passive transfer is consistent with the variability in the CMAP increment recorded from different patients (1, 26). In this report, we compared tetanic potentiation in our model results with prior data reported from our laboratory (26). These data were chosen as they were representative of what has been reported by others, but in comparing model results after various AZ manipulations, an exact match with the magnitude and time course of tetanic potentiation was not considered essential. The goal of these modeling studies was to closely approximate the general effects on short-term synaptic plasticity previously recorded from the NMJ after LEMS passive transfer in mice.
VGCC Antibodies and LEMS
Our results, along with the findings of other studies on seronegative LEMS, suggest that anti-VGCC antibodies are not necessary for LEMS. In addition, other evidence suggests that anti-VGCC antibodies alone may also not be sufficient for LEMS. Recent studies have found that tests for the presence of P/Q- and N-type VGCC antibodies have low sensitivity for detecting LEMS and produce a large number of false positives (101–103). Furthermore, rats injected with peptides or recombinant proteins to various VGCC domains were reported to have a reduction in quantal content, but only by ∼30% (104, 105), and this result has been difficult to reproduce in other laboratories (personal communication). These findings suggest a possibility that although antibodies to P/Q-type VGCCs play an important role in a majority of LEMS cases, they may require cooperation with antibodies targeting other AZ proteins to cause the clinical symptoms of LEMS.
The reported compensatory expression of L-type VGCCs in LEMS model mice that contribute to the control of transmitter release at diseased synapses (25) has several implications. First, L-type VGCCs are not expected to localize within the AZ because these VGCCs do not contain the synaptic protein interaction site thought to be important for AZ localization (78). Their increased presence in the nerve terminal near AZs may lead to a slower contribution of extra calcium ions following action potential activity due to this more distant location and their slower kinetics of gating (82). Our models predicted only a minor influence of these channels on transmitter release (compare Figs. 6 and 7).
The multi-antibody hypothesis for LEMS may be supported by the disruption of the normally well-organized structure of the intramembranous particles comprising AZs seen in human patients with LEMS and LEMS passive-transfer mice (19, 21). The AZ contains numerous interconnected structural and functional proteins (106, 107), and it is possible that autoantibody mediated removal of P/Q-type VGCCs could result in damage to the AZ structure that would result in some level of functionally significant disorganization. Furthermore, both P/Q-type VGCC knockout mice and N-type VGCC knockout mice have shown a reduction in the number of AZs (108). However, the LEMS-mediated disruption in the organization of AZs is perhaps a much greater level of damage to the AZ structure than one would expect from the removal of the number of P/Q-type VGCCs necessary to cause the reduction in P/Q-type calcium current seen in LEMS (22, 24).
Interestingly, the protein laminin β2 has been found to be essential for the expression of P/Q-type VGCCs in the AZs (71), and the interaction of laminin β2 and VGCCs is essential for the organization of the AZs (108, 109). Antibodies against the laminin β2-binding domain on P/Q-type VGCCs have been identified in patients with LEMS (110, 111). If an autoantibody attack on P/Q-type VGCCs can impact other AZ proteins attached to the VGCCs and subsequent disruption of the AZ organization, then there is also the possibility that antibody-mediated removal of other AZ proteins could indirectly result in the disorganization of VGCCs. In addition, antibodies to synaptotagmin proteins have also been identified in LEMS and may lead to a disruption of AZ organization. The results of our modeling motivate an in-depth characterization of antibodies present in patients with LEMS (both seronegative and seropositive) and the impact of these antibodies on AZ organization.
DATA AVAILABILITY
Data will be made available upon reasonable request.
GRANTS
This work was supported by National Science Foundation (NSF) Collaborative Research in Computational Neuroscience Award 2011616. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant Number ACI-1548562. Specifically, it used the Bridges-2 system, which is supported by NSF Award Number ACI-1928147, at the Pittsburgh Supercomputing Center (PSC).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.P.G., Y.B., R.L., T.B.T., and S.D.M. conceived and designed research; S.P.G., Y.B., R.L., G.M., C.J.W., T.B.T., C.K., S.R., and S.D.M. performed experiments; S.P.G., Y.B., R.L., G.M., C.J.W., T.B.T., C.K., S.R., and S.D.M. analyzed data; S.P.G., Y.B., R.L., G.M., C.J.W., T.B.T., C.K., and S.D.M. interpreted results of experiments; S.P.G., Y.B., R.L., T.B.T., and S.D.M. prepared figures; S.P.G. drafted manuscript; Y.B., R.L., S.R., and S.D.M. edited and revised manuscript; S.P.G., Y.B., R.L., G.M., C.J.W., T.B.T., C.K., S.R., and S.D.M. approved final version of manuscript.
REFERENCES
- 1. Oh SJ, Hatanaka Y, Claussen GC, Sher E. Electrophysiological differences in seropositive and seronegative Lambert-Eaton myasthenic syndrome. Muscle Nerve 35: 178–183, 2007. doi: 10.1002/mus.20672. [DOI] [PubMed] [Google Scholar]
- 2. Tarr TB, Malick W, Liang M, Valdomir G, Frasso M, Lacomis D, Reddel SW, Garcia-Ocano A, Wipf P, Meriney SD. Evaluation of a novel calcium channel agonist for therapeutic potential in Lambert-Eaton myasthenic syndrome. J Neurosci 33: 10559–10567, 2013. doi: 10.1523/JNEUROSCI.4629-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Benatar M, Blaes F, Johnston I, Wilson K, Vincent A, Beeson D, Lang B. Presynaptic neuronal antigens expressed by a small cell lung carcinoma cell line. J Neuroimmunol 113: 153–162, 2001. doi: 10.1016/s0165-5728(00)00431-8. [DOI] [PubMed] [Google Scholar]
- 4. Takamori M. Lambert-Eaton myasthenic syndrome: search for alternative autoimmune targets and possible compensatory mechanisms based on presynaptic calcium homeostasis. J Neuroimmunol 201–202: 145–152, 2008. doi: 10.1016/j.jneuroim.2008.04.040. [DOI] [PubMed] [Google Scholar]
- 5. Kesner VG, Oh SJ, Dimachkie MM, Barohn RJ. Lambert-Eaton myasthenic syndrome. Neurol Clin 36: 379–394, 2018. doi: 10.1016/j.ncl.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lennon VA, Kryzer TJ, Griesmann GE, O'Suilleabhain PE, Windebank AJ, Woppmann A, Miljanich GP, Lambert EH. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med 332: 1467–1474, 1995. doi: 10.1056/NEJM199506013322203. [DOI] [PubMed] [Google Scholar]
- 7. Motomura M, Lang B, Johnston I, Palace J, Vincent A, Newsom-Davis J. Incidence of serum anti-P/O-type and anti-N-type calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. J Neurol Sci 147: 35–42, 1997. doi: 10.1016/s0022-510x(96)05303-8. [DOI] [PubMed] [Google Scholar]
- 8. Nakao YK, Motomura M, Fukudome T, Fukuda T, Shiraishi H, Yoshimura T, Tsujihata M, Eguchi K. Seronegative Lambert-Eaton myasthenic syndrome: study of 110 Japanese patients. Neurology 59: 1773–1775, 2002. doi: 10.1212/01.wnl.0000037485.56217.5f. [DOI] [PubMed] [Google Scholar]
- 9. Takamori M, Takahashi M, Yasukawa Y, Iwasa K, Nemoto Y, Suenaga A, Nagataki S, Nakamura T. Antibodies to recombinant synaptotagmin and calcium channel subtypes in Lambert-Eaton myasthenic syndrome. J Neurol Sci 133: 95–101, 1995. doi: 10.1016/0022-510x(95)00162-u. [DOI] [PubMed] [Google Scholar]
- 10. Pang ZP, Melicoff E, Padgett D, Liu Y, Teich AF, Dickey BF, Lin W, Adachi R, Sudhof TC. Synaptotagmin-2 is essential for survival and contributes to Ca2+ triggering of neurotransmitter release in central and neuromuscular synapses. J Neurosci 26: 13493–13504, 2006. doi: 10.1523/JNEUROSCI.3519-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pang ZP, Sun J, Rizo J, Maximov A, Sudhof TC. Genetic analysis of synaptotagmin 2 in spontaneous and Ca2+-triggered neurotransmitter release. EMBO J 25: 2039–2050, 2006. doi: 10.1038/sj.emboj.7601103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takamori M, Komai K, Iwasa K. Antibodies to calcium channel and synaptotagmin in Lambert-Eaton myasthenic syndrome. Am J Med Sci 319: 204–208, 2000. doi: 10.1097/00000441-200004000-00002. [DOI] [PubMed] [Google Scholar]
- 13. Takamori M, Hamada T, Komai K, Takahashi M, Yoshida A. Synaptotagmin can cause an immune-mediated model of Lambert-Eaton myasthenic syndrome in rats. Ann Neurol 35: 74–80, 1994. doi: 10.1002/ana.410350112. [DOI] [PubMed] [Google Scholar]
- 14. Herrmann DN, Horvath R, Sowden JE, Gonzalez M, Sanchez-Mejias A, Guan Z, Whittaker RG, Almodovar JL, Lane M, Bansagi B, Pyle A, Boczonadi V, Lochmuller H, Griffin H, Chinnery PF, Lloyd TE, Littleton JT, Zuchner S. Synaptotagmin 2 mutations cause an autosomal-dominant form of Lambert-Eaton myasthenic syndrome and nonprogressive motor neuropathy. Am J Hum Genet 95: 332–339, 2014. [Erratum in Am J Hum Genet 95: 472, 2014]. doi: 10.1016/j.ajhg.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dittrich M, Pattillo JM, King JD, Cho S, Stiles JR, Meriney SD. An excess-calcium-binding-site model predicts neurotransmitter release at the neuromuscular junction. Biophys J 104: 2751–2763, 2013. doi: 10.1016/j.bpj.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ma J, Kelly L, Ingram J, Price TJ, Meriney SD, Dittrich M. New insights into short-term synaptic facilitation at the frog neuromuscular junction. J Neurophysiol 3: 71–87, 2015. doi: 10.1152/jn.00198.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Laghaei R, Ma J, Tarr TB, Homan AE, Kelly L, Tilvawala MS, Vuocolo BS, Rajasekaran HP, Meriney SD, Dittrich M. Transmitter release site organization can predict synaptic function at the neuromuscular junction. J Neurophysiol 119: 1340–1355, 2018. doi: 10.1152/jn.00168.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McLachlan EM, Martin AR. Non-linear summation of end-plate potentials in the frog and mouse. J Physiol 311: 307–324, 1981. doi: 10.1113/jphysiol.1981.sp013586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fukunaga H, Engel AG, Lang B, Newsom-Davis J, Vincent A. Passive transfer of Lambert-Eaton myasthenic syndrome with IgG from man to mouse depletes the presynaptic membrane active zones. Proc Natl Acad Sci USA 80: 7636–7640, 1983. doi: 10.1073/pnas.80.24.7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lang B, Molenaar PC, Newsom-Davis J, Vincent A. Passive transfer of Lambert-Eaton myasthenic syndrome in mice: decreased rates of resting and evoked release of acetylcholine from skeletal muscle. J Neurochem 42: 658–662, 1984. doi: 10.1111/j.1471-4159.1984.tb02733.x. [DOI] [PubMed] [Google Scholar]
- 21. Fukuoka T, Engel AG, Lang B, Newsom-Davis J, Prior C, Wray DW. Lambert-Eaton myasthenic syndrome. I. Early morphological effects of IgG on the presynaptic membrane active zones. Ann Neurol 22: 193–199, 1987. doi: 10.1002/ana.410220203. [DOI] [PubMed] [Google Scholar]
- 22. Smith DO, Conklin MW, Jensen PJ, Atchison WD. Decreased calcium currents in motor nerve terminals of mice with Lambert-Eaton myasthenic syndrome. J Physiol 487: 115–123, 1995. doi: 10.1113/jphysiol.1995.sp020865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen JC, Wiley AA, Ho TY, Frankshun AL, Hord KM, Bartol FF, Bagnell CA. Transient estrogen exposure from birth affects uterine expression of developmental markers in neonatal gilts with lasting consequences in pregnant adults. Reproduction 139: 623–630, 2010. doi: 10.1530/REP-09-0454. [DOI] [PubMed] [Google Scholar]
- 24. Xu YF, Hewett SJ, Atchison WD. Passive transfer of Lambert-Eaton myasthenic syndrome induces dihydropyridine sensitivity of ICa in mouse motor nerve terminals. J Neurophysiol 80: 1056–1069, 1998. doi: 10.1152/jn.1998.80.3.1056. [DOI] [PubMed] [Google Scholar]
- 25. Flink MT, Atchison WD. Passive transfer of Lambert-Eaton syndrome to mice induces dihydropyridine sensitivity of neuromuscular transmission. J Physiol 543: 567–576, 2002. doi: 10.1113/jphysiol.2002.021048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tarr TB, Lacomis D, Reddel SW, Liang M, Valdomir G, Frasso M, Wipf P, Meriney SD. Complete reversal of Lambert-Eaton myasthenic syndrome synaptic impairment by the combined use of a K+ channel blocker and a Ca2+ channel agonist. J Physiol 592: 3687–3696, 2014. doi: 10.1113/jphysiol.2014.276493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maddison P, Newsom-Davis J, Mills KR, Souhami RL. Favourable prognosis in Lambert-Eaton myasthenic syndrome and small-cell lung carcinoma. Lancet 353: 117–118, 1999. doi: 10.1016/S0140-6736(05)76153-5. [DOI] [PubMed] [Google Scholar]
- 28. Lipka AF, Boldingh MI, van Zwet EW, Schreurs MWJ, Kuks JBM, Tallaksen CM, Titulaer MJ, Verschuuren J. Long-term follow-up, quality of life, and survival of patients with Lambert-Eaton myasthenic syndrome. Neurology 94: e511–e520, 2020. [Erratum in Neurology 97: 1053, 2021]. doi: 10.1212/WNL.0000000000008747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Turan Z, Topaloglu M, Ozyemisci Taskiran O. Medical Research Council-sumscore: a tool for evaluating muscle weakness in patients with post-intensive care syndrome. Crit Care 24: 562, 2020. doi: 10.1186/s13054-020-03282-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim DH. Ulnar nerve conduction study of the first dorsal interosseous muscle in Korean subjects. Ann Rehabil Med 35: 658–663, 2011. doi: 10.5535/arm.2011.35.5.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bartol TM Jr, Land BR, Salpeter EE, Salpeter MM. Monte Carlo simulation of miniature endplate current generation in the vertebrate neuromuscular junction. Biophys J 59: 1290–1307, 1991. doi: 10.1016/S0006-3495(91)82344-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kerr RA, Bartol TM, Kaminsky B, Dittrich M, Chang JC, Baden SB, Sejnowski TJ, Stiles JR. Fast Monte Carlo simulation methods for biological reaction-diffusion systems in solution and on surfaces. SIAM J Sci Comput 30: 3126, 2008. doi: 10.1137/070692017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stiles JR, Van Helden D, Bartol TM Jr, Salpeter EE, Salpeter MM. Miniature endplate current rise times less than 100 microseconds from improved dual recordings can be modeled with passive acetylcholine diffusion from a synaptic vesicle. Proc Natl Acad Sci USA 93: 5747–5752, 1996. doi: 10.1073/pnas.93.12.5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ginebaugh SP, Cyphers ED, Lanka V, Ortiz G, Miller EW, Laghaei R, Meriney SD. The frog motor nerve terminal has very brief action potentials and three electrical regions predicted to differentially control transmitter release. J Neurosci 40: 3504–3516, 2020. doi: 10.1523/JNEUROSCI.2415-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Radhakrishnan A, Li X, Grushin K, Krishnakumar SS, Liu J, Rothman JE. Symmetrical arrangement of proteins under release-ready vesicles in presynaptic terminals. Proc Natl Acad Sci USA 118: e2024029118, 2021. doi: 10.1073/pnas.2024029118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jackman SL, Regehr WG. The mechanisms and functions of synaptic facilitation. Neuron 94: 447–464, 2017. doi: 10.1016/j.neuron.2017.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sugita S, Shin OH, Han W, Lao Y, Sudhof TC. Synaptotagmins form a hierarchy of exocytotic Ca(2+) sensors with distinct Ca(2+) affinities. EMBO J 21: 270–280, 2002. doi: 10.1093/emboj/21.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bhalla A, Tucker WC, Chapman ER. Synaptotagmin isoforms couple distinct ranges of Ca2+, Ba2+, and Sr2+ concentration to SNARE-mediated membrane fusion. Mol Biol Cell 16: 4755–4764, 2005. doi: 10.1091/mbc.e05-04-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. DeStefino NR, Pilato AA, Dittrich M, Cherry SV, Cho S, Stiles JR, Meriney SD. (R)-Roscovitine prolongs the mean open time of unitary N-type calcium channel currents. Neuroscience 167: 838–849, 2010. doi: 10.1016/j.neuroscience.2010.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heuser JE, Reese TS, Dennis MJ, Jan Y, Jan L, Evans L. Synaptic vesicle exocytosis captured by quick freezing and correlated with quantal transmitter release. J Cell Biol 81: 275–300, 1979. doi: 10.1083/jcb.81.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pawson PA, Grinnell AD, Wolowske B. Quantitative freeze-fracture analysis of the frog neuromuscular junction synapse. II. Proximal-distal measurements. J Neurocytol 27: 379–391, 1998. doi: 10.1023/a:1006995010453. [DOI] [PubMed] [Google Scholar]
- 42. Nagwaney S, Harlow ML, Jung JH, Szule JA, Ress D, Xu J, Marshall RM, McMahan UJ. Macromolecular connections of active zone material to docked synaptic vesicles and presynaptic membrane at neuromuscular junctions of mouse. J Comp Neurol 513: 457–468, 2009. doi: 10.1002/cne.21975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Donahue BS, Abercrombie RF. Free diffusion coefficient of ionic calcium in cytoplasm. Cell Calcium 8: 437–448, 1987. doi: 10.1016/0143-4160(87)90027-3. [DOI] [PubMed] [Google Scholar]
- 44. Hrabetova S, Masri D, Tao L, Xiao F, Nicholson C. Calcium diffusion enhanced after cleavage of negatively charged components of brain extracellular matrix by chondroitinase ABC. J Physiol 587: 4029–4049, 2009. doi: 10.1113/jphysiol.2009.170092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ojala KS, Ginebaugh SP, Wu M, Miller EW, Ortiz G, Covarrubias M, Meriney SD. A high affinity, partial antagonist effect of 3,4-diaminopyridine mediates action potential broadening and enhancement of transmitter release at NMJs. J Biol Chem 296: 100302, 2021. doi: 10.1016/j.jbc.2021.100302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Church PJ, Stanley EF. Single L-type calcium channel conductance with physiological levels of calcium in chick ciliary ganglion neurons. J Physiol 496: 59–68, 1996. doi: 10.1113/jphysiol.1996.sp021665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Campbell DL, Giles WR, Shibata EF. Ion transfer characteristics of the calcium current in bull-frog atrial myocytes. J Physiol 403: 239–266, 1988. doi: 10.1113/jphysiol.1988.sp017248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Matveev V, Sherman A, Zucker RS. New and corrected simulations of synaptic facilitation. Biophys J 83: 1368–1373, 2002. doi: 10.1016/S0006-3495(02)73907-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Matveev V, Zucker RS, Sherman A. Facilitation through buffer saturation: constraints on endogenous buffering properties. Biophys J 86: 2691–2709, 2004. doi: 10.1016/S0006-3495(04)74324-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gilmanov IR, Samigullin DV, Vyskocil F, Nikolsky EE, Bukharaeva EA. Modeling of quantal neurotransmitter release kinetics in the presence of fixed and mobile calcium buffers. J Comput Neurosci 25: 296–307, 2008. doi: 10.1007/s10827-008-0079-5. [DOI] [PubMed] [Google Scholar]
- 51. Xu T, Naraghi M, Kang H, Neher E. Kinetic studies of Ca2+ binding and Ca2+ clearance in the cytosol of adrenal chromaffin cells. Biophys J 73: 532–545, 1997. doi: 10.1016/S0006-3495(97)78091-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yazejian B, Sun XP, Grinnell AD. Tracking presynaptic Ca2+ dynamics during neurotransmitter release with Ca2+-activated K+ channels. Nat Neurosci 3: 566–571, 2000. doi: 10.1038/75737. [DOI] [PubMed] [Google Scholar]
- 53. Li F, Pincet F, Perez E, Eng WS, Melia TJ, Rothman JE, Tareste D. Energetics and dynamics of SNAREpin folding across lipid bilayers. Nat Struct Mol Biol 14: 890–896, 2007. doi: 10.1038/nsmb1310. [DOI] [PubMed] [Google Scholar]
- 54. Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science 316: 1205–1208, 2007. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- 55. Jackman SL, Turecek J, Belinsky JE, Regehr WG. The calcium sensor synaptotagmin 7 is required for synaptic facilitation. Nature 529: 88–91, 2016. doi: 10.1038/nature16507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chapman ER, Jahn R. Calcium-dependent interaction of the cytoplasmic region of synaptotagmin with membranes. Autonomous function of a single C2-homologous domain. J Biol Chem 269: 5735–5741, 1994. [PubMed] [Google Scholar]
- 57. Chapman ER, Davis AF. Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. J Biol Chem 273: 13995–14001, 1998. doi: 10.1074/jbc.273.22.13995. [DOI] [PubMed] [Google Scholar]
- 58. van den Bogaart G, Meyenberg K, Diederichsen U, Jahn R. Phosphatidylinositol 4,5-bisphosphate increases Ca2+ affinity of synaptotagmin-1 by 40-fold. J Biol Chem 287: 16447–16453, 2012. doi: 10.1074/jbc.M112.343418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Desai RC, Vyas B, Earles CA, Littleton JT, Kowalchyck JA, Martin TF, Chapman ER. The C2B domain of synaptotagmin is a Ca(2+)-sensing module essential for exocytosis. J Cell Biol 150: 1125–1136, 2000. doi: 10.1083/jcb.150.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tran HT, Anderson LH, Knight JD. Membrane-binding cooperativity and coinsertion by C2AB tandem domains of synaptotagmins 1 and 7. Biophys J 116: 1025–1036, 2019. doi: 10.1016/j.bpj.2019.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bowers MR, Reist NE. The C2A domain of synaptotagmin is an essential component of the calcium sensor for synaptic transmission. PLoS One 15: e0228348, 2020. doi: 10.1371/journal.pone.0228348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hastings WK. Monte-Carlo sampling methods using Markov chains and their applications. Biometrika 57: 97–109, 1970. doi: 10.1093/biomet/57.1.97. [DOI] [Google Scholar]
- 63. Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E. Equation of state calculations by fast computing machines. J Chem Phys 21: 1087–1092, 1953. doi: 10.1063/1.1699114. [DOI] [Google Scholar]
- 64. Bukharaeva EA, Samigullin D, Nikolsky EE, Magazanik LG. Modulation of the kinetics of evoked quantal release at mouse neuromuscular junctions by calcium and strontium. J Neurochem 100: 939–949, 2007. doi: 10.1111/j.1471-4159.2006.04282.x. [DOI] [PubMed] [Google Scholar]
- 65. Wang X, Pinter MJ, Rich MM. Ca2+ dependence of the binomial parameters p and n at the mouse neuromuscular junction. J Neurophysiol 103: 659–666, 2010. doi: 10.1152/jn.00708.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Urbano FJ, Piedras-Renteria ES, Jun K, Shin HS, Uchitel OD, Tsien RW. Altered properties of quantal neurotransmitter release at endplates of mice lacking P/Q-type Ca2+ channels. Proc Natl Acad Sci USA 100: 3491–3496, 2003. doi: 10.1073/pnas.0437991100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nishimura M. Zinc competitively inhibits calcium-dependent release of transmitter at the mouse neuromuscular junction. Pflugers Arch 410: 623–626, 1987. doi: 10.1007/BF00581322. [DOI] [PubMed] [Google Scholar]
- 68. Nishimura M, Awano H, Yagasaki O. Sodium salicylate facilitates calcium-dependent release of transmitter at mouse neuromuscular junctions. Br J Pharmacol 97: 1239–1245, 1989. doi: 10.1111/j.1476-5381.1989.tb12584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nishimura M, Tsubaki K, Yagasaki O, Ito K. Ryanodine facilitates calcium-dependent release of transmitter at mouse neuromuscular junctions. Br J Pharmacol 100: 114–118, 1990. doi: 10.1111/j.1476-5381.1990.tb12061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Urbano FJ, Rosato-Siri MD, Uchitel OD. Calcium channels involved in neurotransmitter release at adult, neonatal and P/Q-type deficient neuromuscular junctions (Review). Mol Membr Biol 19: 293–300, 2002. doi: 10.1080/0968768021000035087. [DOI] [PubMed] [Google Scholar]
- 71. Chand KK, Lee KM, Schenning MP, Lavidis NA, Noakes PG. Loss of beta2-laminin alters calcium sensitivity and voltage-gated calcium channel maturation of neurotransmission at the neuromuscular junction. J Physiol 593: 245–265, 2015. doi: 10.1113/jphysiol.2014.284133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Arai I, Jonas P. Nanodomain coupling explains Ca(2)(+) independence of transmitter release time course at a fast central synapse. eLife 3: e04057, 2014. doi: 10.7554/eLife.04057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bucurenciu I, Kulik A, Schwaller B, Frotscher M, Jonas P. Nanodomain coupling between Ca2+ channels and Ca2+ sensors promotes fast and efficient transmitter release at a cortical GABAergic synapse. Neuron 57: 536–545, 2008. doi: 10.1016/j.neuron.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 74. Chen Z, Das B, Nakamura Y, DiGregorio DA, Young SM Jr.. Ca2+ channel to synaptic vesicle distance accounts for the readily releasable pool kinetics at a functionally mature auditory synapse. J Neurosci 35: 2083–2100, 2015. doi: 10.1523/JNEUROSCI.2753-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nakamura Y, Harada H, Kamasawa N, Matsui K, Rothman JS, Shigemoto R, Silver RA, DiGregorio DA, Takahashi T. Nanoscale distribution of presynaptic Ca(2+) channels and its impact on vesicular release during development. Neuron 85: 145–158, 2015. doi: 10.1016/j.neuron.2014.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Stanley EF. The nanophysiology of fast transmitter release. Trends Neurosci 39: 183–197, 2016. doi: 10.1016/j.tins.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 77. Lang B, Newsom-Davis J, Prior C, Wray D. Antibodies to motor nerve terminals: an electrophysiological study of a human myasthenic syndrome transferred to mouse. J Physiol 344: 335–345, 1983. doi: 10.1113/jphysiol.1983.sp014943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Catterall WA. Interactions of presynaptic Ca2+ channels and snare proteins in neurotransmitter release. Ann NY Acad Sci 868: 144–159, 1999. doi: 10.1111/j.1749-6632.1999.tb11284.x. [DOI] [PubMed] [Google Scholar]
- 79. Mochida S, Westenbroek RE, Yokoyama CT, Zhong H, Myers SJ, Scheuer T, Itoh K, Catterall WA. Requirement for the synaptic protein interaction site for reconstitution of synaptic transmission by P/Q-type calcium channels. Proc Natl Acad Sci USA 100: 2819–2824, 2003. doi: 10.1073/pnas.262787699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sheng ZH, Westenbroek RE, Catterall WA. Physical link and functional coupling of presynaptic calcium channels and the synaptic vesicle docking/fusion machinery. J Bioenerg Biomembr 30: 335–345, 1998. doi: 10.1023/a:1021985521748. [DOI] [PubMed] [Google Scholar]
- 81. Flink MT, Atchison WD. Ca2+ channels as targets of neurological disease: Lambert-Eaton syndrome and other Ca2+ channelopathies. J Bioenerg Biomembr 35: 697–718, 2003. doi: 10.1023/b:jobb.0000008033.02320.10. [DOI] [PubMed] [Google Scholar]
- 82. Helton TD, Xu W, Lipscombe D. Neuronal L-type calcium channels open quickly and are inhibited slowly. J Neurosci 25: 10247–10251, 2005. doi: 10.1523/JNEUROSCI.1089-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rich MM. The control of neuromuscular transmission in health and disease. Neuroscientist 12: 134–142, 2006. doi: 10.1177/1073858405281898. [DOI] [PubMed] [Google Scholar]
- 84. Katz JS, Wolfe GI, Bryan WW, Tintner R, Barohn RJ. Acetylcholine receptor antibodies in the Lambert-Eaton myasthenic syndrome. Neurology 50: 470–475, 1998. doi: 10.1212/wnl.50.2.470. [DOI] [PubMed] [Google Scholar]
- 85. Newsom-Davis J, Leys K, Vincent A, Ferguson I, Modi G, Mills K. Immunological evidence for the co-existence of the Lambert-Eaton myasthenic syndrome and myasthenia gravis in two patients. J Neurol Neurosurg Psychiatry 54: 452–453, 1991. doi: 10.1136/jnnp.54.5.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Oh SJ, Sher E. MG and LEMS overlap syndrome: case report with electrophysiological and immunological evidence. Clin Neurophysiol 116: 1167–1171, 2005. doi: 10.1016/j.clinph.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 87. Lee JH, Shin HY, Kim SM, Sunwoo IN. A case of Lambert-Eaton myasthenic syndrome with small-cell lung cancer and transient increase in anti-acetylcholine-receptor-binding antibody titer. J Clin Neurol 8: 305–307, 2012. doi: 10.3988/jcn.2012.8.4.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Matsumoto H, Ohtomo G, Akahori T, Hashida H. Lambert-Eaton myasthenic syndrome with anti-acetylcholine receptor antibody and anterior mediastinal tumor. J Gen Fam Med 18: 282–284, 2017. doi: 10.1002/jgf2.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Oh SJ. Amifampridine to treat Lambert-Eaton myasthenic syndrome. Drugs Today (Barc) 56: 623–641, 2020. doi: 10.1358/dot.2020.56.10.3137144. [DOI] [PubMed] [Google Scholar]
- 90. Voelker R. Drug approved for rare muscle weakening syndrome. JAMA 321: 239, 2019. doi: 10.1001/jama.2018.21321. [DOI] [PubMed] [Google Scholar]
- 91. Yoon CH, Owusu-Guha J, Smith A, Buschur P. Amifampridine for the management of Lambert-Eaton myasthenic syndrome: a new take on an old drug. Ann Pharmacother 54: 56–63, 2020. doi: 10.1177/1060028019864574. [DOI] [PubMed] [Google Scholar]
- 92. Sanders DB. 3,4-Diaminopyridine (DAP) in the treatment of Lambert-Eaton myasthenic syndrome (LEMS). Ann NY Acad Sci 841: 811–816, 1998. doi: 10.1111/j.1749-6632.1998.tb11022.x. [DOI] [PubMed] [Google Scholar]
- 93. Sanders DB, Juel VC, Harati Y, Smith AG, Peltier AC, Marburger T, Lou JS, Pascuzzi RM, Richman DP, Xie T, Demmel V, Jacobus LR, Ales KL, Jacobus DP; Dapper Study Team. 3,4-Diaminopyridine base effectively treats the weakness of Lambert-Eaton myasthenia. Muscle Nerve 57: 561–568, 2018. doi: 10.1002/mus.26052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Shieh P, Sharma K, Kohrman B, Oh SJ. Amifampridine phosphate (Firdapse) is effective in a confirmatory phase 3 clinical trial in LEMS. J Clin Neuromuscul Dis 20: 111–119, 2019. doi: 10.1097/CND.0000000000000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Strupp M, Teufel J, Zwergal A, Schniepp R, Khodakhah K, Feil K. Aminopyridines for the treatment of neurologic disorders. Neurol Clin Pract 7: 65–76, 2017. doi: 10.1212/CPJ.0000000000000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Maddison P, Lang B, Mills K, Newsom-Davis J. Long term outcome in Lambert-Eaton myasthenic syndrome without lung cancer. J Neurol Neurosurg Psychiatry 70: 212–217, 2001. doi: 10.1136/jnnp.70.2.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol 10: 1098–1107, 2011. doi: 10.1016/S1474-4422(11)70245-9. [DOI] [PubMed] [Google Scholar]
- 98. Whittaker RG, Herrmann DN, Bansagi B, Hasan BA, Lofra RM, Logigian EL, Sowden JE, Almodovar JL, Littleton JT, Zuchner S, Horvath R, Lochmuller H. Electrophysiologic features of SYT2 mutations causing a treatable neuromuscular syndrome. Neurology 85: 1964–1971, 2015. doi: 10.1212/WNL.0000000000002185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wirtz PW, Verschuuren JJ, van Dijk JG, de Kam ML, Schoemaker RC, van Hasselt JG, Titulaer MJ, Tjaden UR, den Hartigh J, van Gerven JM. Efficacy of 3,4-diaminopyridine and pyridostigmine in the treatment of Lambert-Eaton myasthenic syndrome: a randomized, double-blind, placebo-controlled, crossover study. Clin Pharmacol Ther 86: 44–48, 2009. doi: 10.1038/clpt.2009.35. [DOI] [PubMed] [Google Scholar]
- 100. Hewett SJ, Atchison WD. Specificity of Lambert-Eaton myasthenic syndrome immunoglobulin for nerve terminal calcium channels. Brain Res 599: 324–332, 1992. doi: 10.1016/0006-8993(92)90408-2. [DOI] [PubMed] [Google Scholar]
- 101. Abboud H, Rossman I, Mealy MA, Hill E, Thompson N, Banerjee A, Probasco J, Levy M. Neuronal autoantibodies: differentiating clinically relevant and clinically irrelevant results. J Neurol 264: 2284–2292, 2017. doi: 10.1007/s00415-017-8627-4. [DOI] [PubMed] [Google Scholar]
- 102. Albadareen R, Gronseth G, Goeden M, Sharrock M, Lechtenberg C, Wang Y. Paraneoplastic autoantibody panels: sensitivity and specificity, a retrospective cohort. Int J Neurosci 127: 531–538, 2017. doi: 10.1080/00207454.2016.1207644. [DOI] [PubMed] [Google Scholar]
- 103. Di Lorenzo R, Mente K, Li J, Shayya L, Rae-Grant A, Li Y, Jammoul A. Low specificity of voltage-gated calcium channel antibodies in Lambert-Eaton myasthenic syndrome: a call for caution. J Neurol 265: 2114–2119, 2018. doi: 10.1007/s00415-018-8959-8. [DOI] [PubMed] [Google Scholar]
- 104. Komai K, Iwasa K, Takamori M. Calcium channel peptide can cause an autoimmune-mediated model of Lambert-Eaton myasthenic syndrome in rats. J Neurol Sci 166: 126–130, 1999. doi: 10.1016/s0022-510x(99)00125-2. [DOI] [PubMed] [Google Scholar]
- 105. Takamori M, Iwasa K, Komai K. Antigenic sites of the voltage-gated calcium channel in Lambert-Eaton myasthenic syndrome. Ann NY Acad Sci 841: 625–635, 1998. doi: 10.1111/j.1749-6632.1998.tb10994.x. [DOI] [PubMed] [Google Scholar]
- 106. Sudhof TC. The presynaptic active zone. Neuron 75: 11–25, 2012. doi: 10.1016/j.neuron.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Sudhof TC. A molecular machine for neurotransmitter release: synaptotagmin and beyond. Nat Med 19: 1227–1231, 2013. doi: 10.1038/nm.3338. [DOI] [PubMed] [Google Scholar]
- 108. Chen J, Billings SE, Nishimune H. Calcium channels link the muscle-derived synapse organizer laminin β2 to Bassoon and CAST/Erc2 to organize presynaptic active zones. J Neurosci 31: 512–525, 2011. doi: 10.1523/JNEUROSCI.3771-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Nishimune H, Sanes JR, Carlson SS. A synaptic laminin-calcium channel interaction organizes active zones in motor nerve terminals. Nature 432: 580–587, 2004. doi: 10.1038/nature03112. [DOI] [PubMed] [Google Scholar]
- 110. Rogers RS, Nishimune H. The role of laminins in the organization and function of neuromuscular junctions. Matrix Biol 57–58: 86–105, 2017. doi: 10.1016/j.matbio.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Takamori M, Maruta T, Komai K. Lambert-Eaton myasthenic syndrome as an autoimmune calcium-channelopathy. Neurosci Res 36: 183–191, 2000. doi: 10.1016/s0168-0102(99)00135-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request.