

Abstract
Inhalation of crystalline (CS) and amorphous silica (AS) results in human pulmonary inflammation. However, silicosis only develops following CS exposure, and the pathogenic mechanisms are poorly understood. This report describes the differential abilities of CS and AS to directly up-regulate the early inflammatory mediator COX-2, the recently identified prostaglandin E synthase and the downstream mediator PGE2 in primary human lung fibroblasts. Increased COX-2 gene transcription and protein production were demonstrated by ribonuclease protection assay, Western blot analysis, and immunocytochemistry. In each case the ability of AS to induce COX-2 exceeded that of CS. Similarly, downstream of COX-2, production of the anti-fibrotic prostaglandin PGE2, was induced in a dose-dependent fashion, but AS was significantly more potent, (maximal production: CS=4,710 pg/ml and AS=7,651 pg/ml). These increases in COX-2 and PGE2 were preceded by induction of the PGE2 synthase protein, demonstrating the potential role of this novel molecule in silica-mediated inflammation. There was specificity of induction of prostaglandins, as PGF2α, but not PGD2 was induced. Using specific COX-2 inhibitors, increased PG production was shown to be dependent on the COX-2 enzyme. Furthermore stimulation of fibroblasts was particle specific, as silica but not carbon black resulted in fibroblast activation. These results demonstrate that silica can directly stimulate human lung fibroblasts to produce key inflammatory enzymes and prostaglandins. Furthermore they suggest a mechanism to explain the differing fibrogenic potential of CS and AS. The molecules COX-2, prostaglandin E synthase, and PGE2 are identified as effectors in silicosis.
Keywords: Silicosis, Prostaglandin E2, prostaglandin E synthase, inflammation, fibrosis
Introduction
Silicosis is an important occupational lung disease that results from the chronic inhalation of crystalline silica (5; 22). Pathologically, it is characterized by the development of inflammation, silicotic nodules and interstitial scarring. This scarring or fibrosis leads to distortion of the normal lung architecture and disruption of the normal physiological processes of the lung.
There are two distinct types of silica found in nature, crystalline and amorphous silica. They differ significantly in the injury they cause when inhaled. Both crystalline silica and amorphous silica incite pulmonary inflammation, but significantly, only inhalation of crystalline silica ultimately leads to the development of pulmonary fibrosis. It is unclear why there is such a difference in the pathological outcome. Some investigators have suggested that the difference may be related to the physical properties of amorphous and crystalline silica (13; 30). When inhaled crystalline silica particles are ingested by alveolar macrophages, the macrophages are activated and damaged, resulting in ongoing injury and ultimately leading to the development of fibrosis (4). In contrast the amorphous form has a far larger surface area, and hence greater relative solubility within the lung, enabling it to be cleared more easily following inhalation. However, to date little attention has focused on the individual abilities of the crystalline and amorphous forms to induce potentially anti-fibrotic molecules.
Much of the work on the pathogenesis of silicosis, has focused on the role of the alveolar macrophage and to a lesser extent, the alveolar epithelial cell. Certainly, both alveolar macrophages and epithelial cells secrete pro-inflammatory mediators following exposure to silica (21; 28; 32; 39). However, despite the close proximity of fibroblasts to inhaled crystalline silica particles when the epithelial layer has been denuded, the contribution of fibroblasts to silica-induced inflammation and fibrosis has not been well studied (1).
The crucial role of the fibroblast in modulating immune-inflammatory reactions in a number of different tissues and disease states is being increasing recognized (14; 34; 40). When activated, fibroblasts are capable of producing a diverse array of inflammatory mediators, including interleukin-8 (IL-8), monocyte chemoattractant protein–1 (MCP-1), IL-6, cyclooxygenase-2 (COX-2) and transforming growth factor beta (TGF-β). Despite this recognition however, the possibility that fibroblasts can be directly activated by exposure to particulates like silica has received little, if any, attention. This is an area of key importance as fibroblasts may be capable of directing the tissues’ response to injury, either promoting resolution or propelling a cycle of ongoing scarring.
Prostaglandin (PG) E2 is one of the key PGs produced at sites of inflammation and fibrosis. It is secreted by resident fibroblasts, as well as inflammatory cells, in response to early warning signals like TNF-α and IL-1β (8). The prostaglandin synthesis pathway begins with the release of arachidonic acid from phospholipid membranes by the action of phospholipase A2 (PLA2). Archidonic acid is the substrate for two distinct enzymatic pathways, cyclooxygenase (COX) and 5-lipoxygenase. The end-products of the 5-lipoxygenase pathway are leukotrienes whereas the COX pathway gives rise to prostaglandins and thromboxanes. The COX enzymes convert arachidonic acid to the prostaglandin PGH2, which is further processed to individual PGs by specific PG synthases. The COX enzyme exists as two isoforms: COX-1, which is constitutively expressed by fibroblasts in all tissues; and the inducible isoform COX-2 (35). Two forms of PGE synthase have also been described: cytosolic (c) and microsomal (m) PGES. Like COX-1, the cytosolic subtype (cPGES) is generally constitutively expressed, and like COX-2, mPGES is inducible by inflammatory stimuli. mPGES has been shown to be specifically responsible for the up-regulation of PGE2 in some models of inflammation (36). To date there has been little study of the role of these enzymes in lung disease, and none in dust-induced diseases such as silicosis.
The role of PGE2 in regulating inflammatory responses is complex. It has both pro-inflammatory as well as anti-fibrotic properties. PGE2 decreases fibroblast proliferation and collagen gene transcription in vitro (9; 10), and patients with idiopathic pulmonary fibrosis (IPF) have 50% less PGE2 in their bronchoalveolar lavage (BAL) fluid than controls. Similarly, fibroblasts recovered from patients with IPF secrete less PGE2 in response to certain stimuli (16; 37; 38). This strongly suggests that the pathogenesis of this prototypic pulmonary fibrotic disorder, which shares many similarities with silicosis, may be related to an impaired ability to generate PGE2, resulting in the creation of a pro-fibrotic milieu.
This study tests the hypothesis that both crystalline and amorphous silica particles directly activate human lung fibroblasts, but differ in their ability to do so. We demonstrate for the first time that both crystalline and amorphous silica can induce production of key enzymes in the prostaglandin pathway and the downstream prostanoids PGE2 and PGF2α in human primary lung fibroblasts. Interestingly, amorphous silica was found to be considerably more potent than crystalline silica in this regard. These data thus further our understanding of the factors at play during the development of silicosis.
Materials and Methods
Reagents
Crystalline silica (mean size 0.8 microns), amorphous silica and carbon black were a generous gift from Dr. David R. Hemenway (University of Vermont). The selective COX-2 inhibitor SC58125 was purchased from Cayman Chemical Co. (Ann Arbour, MI). Indomethacin was obtained from Sigma Chemical Co. (St. Louis, MI). Monoclonal antibodies against COX-1 and COX-2 for Western blot analysis and immunohistochemistry were purchased from Cayman Chemical Co. The mouse IgG1 was from Caltag Laboratories (Burlingame, CA). A polyclonal antibody against microsomal Prostaglandin E Synthase was purchased from Cayman Chemical Co. The reagents for enzyme immunoassay for detection of PGE2, PGF2α and the enzyme immunoassay kit for detection of PGD2 were purchased from Cayman Chemical Co. The IL-1β ELISA was obtained from R&D Systems (Minneapolis, MN).
Primary Human Lung Fibroblast Culture Conditions
Normal human primary fibroblast cell strains derived by explant technique were maintained in MEM (Life Technologies, Gaithersburg, MD) supplemented with 10% fetal bovine serum (Sigma Aldrich, St. Louis, MO) and penicillin (100 units/ml), streptomycin (100 μg/ml), and amphotericin (0.25 μg/ml) (Life Technologies). These cells were derived from anatomically normal lung from a patient undergoing surgical resection for a benign pulmonary hamartoma. These cells are morphologically consistent with fibroblasts and express the fibroblast markers collagen and vimentin. They do not express CD45, factor VIII or cytokeratin. Cells were incubated at 37 °C in a humidified environment containing 5% CO2 and 95% air. For all experiments, cells were used between passage numbers 2 to 8. Human tissues were obtained under the approval of the University of Rochester Research Subjects Review Board.
Treatment of Primary Lung Fibroblasts with Silica
Before exposure to silica or carbon black, the media was changed to serum free MEM for 48 hours (h) to reduce serum-induced expression of COX-2 (25).
Prior to use, silica and carbon black were baked at 180°C for 2 h to remove any trace of lipopolysaccharide (LPS). They were then tested using a limulus assay (sensitivity of 0.6 EU/ml), to confirm that the depyrogenation had successfully eliminated any LPS contamination. Fibroblasts were grown in 96 well plates (104cells/well) for prostaglandin and cytokine assays, 6 well plates (105 cells/well) for Western blot analysis, and 10 cm dishes (5×105 cells) for RNA harvest. Suspensions of crystalline and amorphous silica (1–100 μg/ml) and carbon black (100 μg/ml) (an alternate particulate) were made in serum-free MEM. Fibroblast viability at these concentrations was assayed using an MTT assay. Interleukin-1β (10 ng/ml) that is known to induce COX-2 was used as a positive control. Serum-free MEM was used as the negative control.
RNA Isolation and RNase Protection Assay
RNA was isolated from the primary fibroblasts 6 h following treatment, using TRI-reagent (Molecular Research Center, Inc. Cincinnati, OH) according to the manufacturer’s instructions. Equal amounts of mRNA were used in the RNase Protection Assays (RPA).
COX-1 and COX-2 antisense probe templates (Cayman Chemical, Ann Arbor, MI) and a human β-actin template (Ambion Inc., Austin, TX) were used to prepare labeled RNA probes with the MAXIscript In Vitro Transcription Kit (Ambion). Full-length RNA transcripts were gel purified from an 8M urea acrylamide gel. The RNase protection assay was performed using the RPA II kit (Ambion). Briefly, RNA was hybridized with labeled probe (8 × 104 cpm) overnight at 42°C. Hybridized samples were digested at 37 °C for 30 minutes (min) with a mixture of RNase A and RNase T1 supplied with the kit. The samples were then separated on a 5% polyacrylamide/8M urea sequencing gel.
Immunocytochemistry
Fibroblasts were seeded (7 × 103 cells/well) onto 8 well chamber slides and grown until sub-confluent. They were serum starved as outlined above, and, following the addition of silica or carbon black, were incubated under standard conditions for 24 h. The cells were fixed with 4% paraformaldehyde, then immunostained with 10 μg/ml of mouse anti-COX-2 monoclonal antibody or isotype control (mouse IgG1) at 4°C overnight. The following day the secondary antibody (a biotinylated horse anti-mouse antibody) was added followed by a streptavidin conjugated-horseradish peroxidase (SA-HRP) (Vector, Burlingame, CA). Immunostaining was visualized with aminoethyl carbazole (AEC, Zymed Laboratories, San Francisco, CA).
Western Blotting for COX-2 and mPGES
Protein was harvested from confluent fibroblasts monolayers by adding ice-cold RIPA lysis buffer (150 mM NaCl, 50 mM Tris, pH 8.0, 1% NP-40, 1 mM EDTA) with added proteinase inhibitor cocktail (Sigma Aldrich, St. Louis, MO). The protein concentration was quantified by bicinchoninic acid (BCA) assay (Pierce, Rockford, IL). The proteins were solubilized by boiling in sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE) sample buffer and separated by SDS-PAGE. Proteins were transferred to nitrocellulose membranes and immunoblotted with the COX-1, COX-2 or mPGES antibodies described above. Proteins were detected with HRP-labeled secondary antibodies and visualized with enhanced chemiluminescence (Perkin Elmer, Boston, MA).
Enzyme Immunoassay for PG Production and Enzyme Linked Immunosorbent Assay for Prostaglandins and Cytokines
Lung fibroblasts were seeded onto flat bottom 96 well plates at a density of 104 cells per well in 200μL of MEM supplemented as described in the culture conditions above. The cells were incubated until approximately 80% confluent. The cells were then serum reduced as previously described. Suspensions of both crystalline and amorphous silica were prepared in serum-free MEM with or without SC58125 (5 μM) (a specific COX-2 inhibitor) and indomethacin (20 μM) (a non-selective inhibitor that blocks the activity of both COX-1 and COX-2) and incubated for 48 hours. The supernatants were assayed for the presence of PGE2, PGF2α, and PGD2 using specific enzyme immunoassay reagents and kits from Cayman Chemical Co. according to the manufacturer’s instructions. IL-1β, MCP-1. IL-6, IL-8 and TGF-β production in the cell supernatants was studied using a commercial ELISA kits (R&D Systems, Minneapolis, MN) according to the manufacturer’s recommendations.
Statistics
All data are expressed as mean ± standard error of the mean (SEM). Statistical significance was assessed using a one-way analysis of variance (ANOVA) test and was recorded at p<0.05. Where significant differences were found, the Tukey-Kramer multiple comparisons post-hoc test was used to identify differences between individual groups. In cases where SEMs were significantly different between groups, a nonparametric ANOVA (Kruskal-Wallis test) was employed, followed by a Dunn’s multiple comparisons post-hoc test, to assess statistical significance.
Results
Crystalline and Amorphous Silica Induce COX-2 but not COX-1 in Primary Human Lung Fibroblasts
Primary human lung fibroblasts were treated with increasing concentrations of silica for 24 hours, and COX-2 expression was evaluated by Western blot. Both amorphous and crystalline silica induced COX-2 in a dose-dependent manner (Figure 1). Interestingly, while amorphous silica strongly induced COX-2 at 10 μg/ml, 10-fold more crystalline silica was required to observe a similar induction of COX-2 protein. Trypan blue exclusion and MTT assays determined that amorphous or crystalline silica were nontoxic to the fibroblasts at concentrations up to 100 μg/ml (data not shown). Carbon black did not induce COX-2 expression, demonstrating that upregulation of COX-2 is not a non-specific response to particulates. COX-1 expression was not affected by silica, consistent with previous reports that COX-1 is constitutively expressed and is not induced by inflammatory stimuli. This demonstrates that the induction of COX-2 is a specific pro-inflammatory response to silica.
Figure 1.
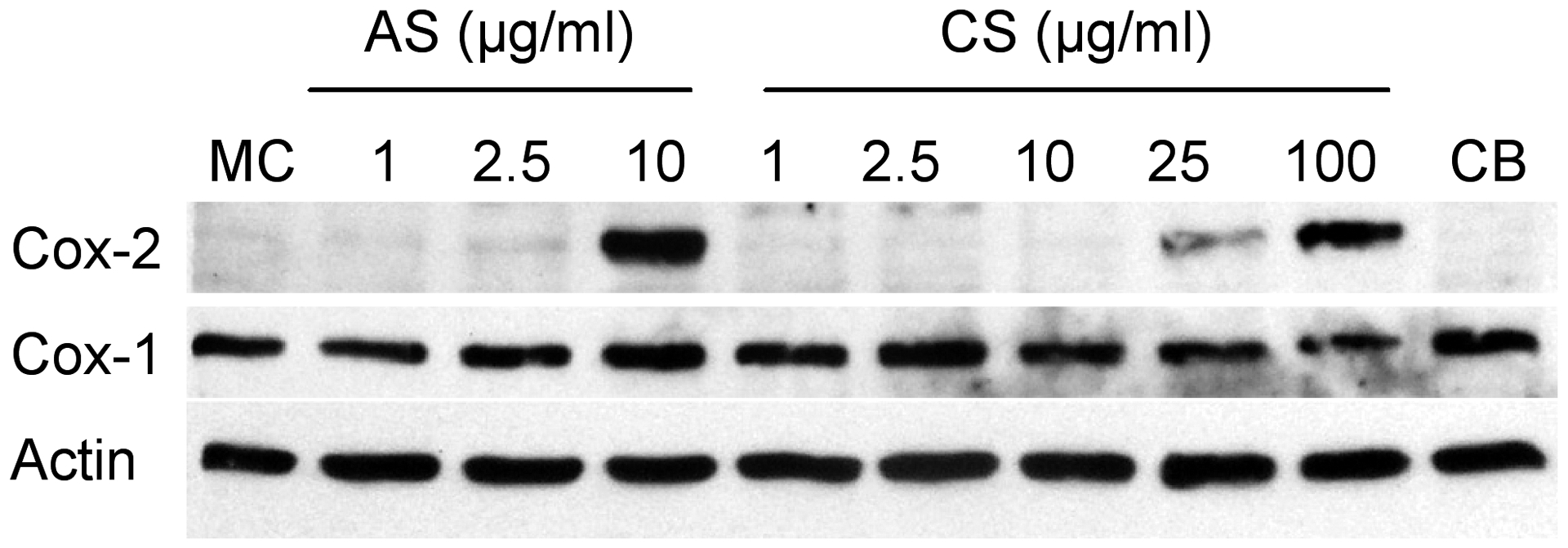
Silica induces COX-2 but not COX-1 protein in human lung fibroblasts. Fibroblasts in culture were treated for 24 h with the indicated concentrations of amorphous (AS) or crystalline (CS) silica. Carbon black (CB, 100 μg/ml) was included as a negative control. Total cell protein was analyzed by Western blot for expression of COX-2, COX-1 and actin (to confirm equal protein loading). Amorphous silica induces COX-2 slightly at 2.5 μg/ml and strongly at 10 μg/ml. 10-fold higher concentrations of crystalline silica are required to produce similar elevations in COX-2 protein. Neither form of silica alters the constitutive expression of COX-1.
Immunocytochemical staining was performed to further delineate and localize the expression of COX-2 protein in the fibroblasts. COX-2 expression is markedly up-regulated following exposure to crystalline silica (100 μg/ml), and amorphous silica (10 μg/ml) (Figure 2). Carbon black did not result in any increased expression of COX-2. This suggested that the COX-2 induction observed with both forms of silica is particle specific, and is not merely induced by all types of particulates. No staining was observed in untreated cells, or any cells stained with the isotype control antibody (data not shown).
Figure 2.

COX-2 protein expression is induced by silica, but not carbon black (CB), in human lung fibroblasts. After 24h of treatment with 10 μg/ml amorphous silica (AS), 100 μg/ml crystalline silica (CS), and 100 μg/ml carbon black (CB), immunocytochemistry was performed for COX-2 protein. Both CS and AS induce COX-2 expression as seen by the deep red cytoplasmic staining. Whereas CB, a relatively inert particle does not. Pictures were taken with a 40x objective.
Silica Induces COX-2 gene expression in primary human lung fibroblasts
To determine if silica directly induces COX gene expression, primary human lung fibroblasts were treated with amorphous and crystalline silica at concentrations of 10 μg/ml and 100 μg/ml respectively, and RNA was harvested and analyzed by ribonuclease protection assay. COX-1 mRNA is expressed constitutively in untreated fibroblasts and was unchanged following treatment with either crystalline or amorphous silica (Figure 3A). COX-2 differs from COX-1 in that it is highly inducible (35). There is no expression of COX-2 mRNA in the untreated human lung fibroblasts (Figure 3B). However, expression is clearly induced following six hours of treatment with either form of silica. In agreement with the Western blot results, 10 μg/ml of amorphous silica was at least as potent a stimulus as 100 μg/ml of crystalline silica.
Figure 3.

Amorphous and crystalline silica induce COX-2 but not COX-1 mRNA in human lung fibroblasts. Total RNA was extracted from fibroblasts after 6 h exposure to selected agents. The RNA was then analyzed by RPA. Lane 1, undigested probe; lane 2, digested probe; lane 3, untreated fibroblasts; lane 4, fibroblasts treated with 100 μg/ml crystalline silica (CS); lane 5, fibroblasts treated with 10 μg/ml amorphous silica (AS). A. Neither form of silica induces COX-1 gene expression. B. Both CS and AS induce COX-2, with AS at least 10 times more potent than CS.
Fibroblast PGE2 production in response to silica is specific and mediated by COX-2
The conversion of arachidonic acid to PGH2 by the COX enzymes is a key rate-limiting step in the production of PGE2, an important immunomodulatory prostaglandin. Production of PGE2 by lung fibroblasts was measured in culture supernatants following stimulation with both forms of silica. Stimulation of fibroblasts with either type of silica resulted in significant production of PGE2 (Figure 4). This increase was both dose (Figure 4) and time dependent (data not shown). Amorphous silica was approximately 7 times more potent than crystalline silica, as stimulation with 100 μg/ml of crystalline silica resulted in PGE2 production (4710 pg/ml) intermediate between 10 and 25 μg/ml amorphous silica (4200 pg/ml and 6500 pg/ml, respectively). Carbon black did not stimulate PGE2 production over background (data not shown).
Figure 4.

PGE2 production is induced in a dose dependent fashion by silica. Fibroblasts were exposed to crystalline (CS) or amorphous (AS) silica at the indicated concentrations; the cell supernatants were harvested after 48 h. and analyzed for PGE2 using an enzyme immunoassay (EIA). MC; media control. The increases in PGE2 production are statistically significant (*p<0.01, **p<0.001). Data represent mean ± SEM of 6 samples per condition. Note the different scales on the y-axis.
To confirm that the induction of PGE2 seen following treatment with silica was dependent upon increased expression of COX-2, fibroblasts were co-cultured with silica and either a specific COX-2 inhibitor (SC58125) or a non-specific COX inhibitor (indomethacin). Each of these inhibitors resulted in complete inhibition of PGE2 production following exposure to amorphous or crystalline silica (Figure 5). Indomethacin is a potent, non-specific inhibitor of both COX-1 and COX-2, and as such, would be expected to completely inhibit the production of PGE2 by inhibiting conversion of arachidonic acid to the precursor PGH2. SC58125 is highly selective for COX-2. The fact that both indomethacin and SC58125 are equally effective in inhibiting silica-induced PGE2 production demonstrates that this increase is dependent on the increased expression of COX-2 induced by silica.
Figure 5.

Silica induced PGE2 production is dependent upon increased COX-2 expression. Suspensions of crystalline (CS) and amorphous silica (AS) and either a non-selective COX inhibitor, indomethacin (20 μM) or a selective COX-2 inhibitor, SC58125 (5 μM) were applied to fibroblasts in culture. IL-1β (10 ng/ml), a cytokine that is known to stimulate COX-2, is used as a positive control. Cell supernatants were harvested after 48 h and PGE2 levels measured using a specific enzyme immunoassay (EIA). Both indomethacin and SC58125 independently attenuated the increase in PGE2 seen with CS (*p<0.005) and AS (**p<0.001). Data represent mean ± SEM of 6 samples per condition.
One route by which silica could induce PGE2 is through IL-1β, a cytokine that has pro-inflammatory and pro-fibrotic properties (18). To determine if silica induced IL-1β expression in lung fibroblasts, cell supernatants were tested for IL-1β production using a specific ELISA. The levels of IL-1β detected in each sample were negligible and no increase was seen with either crystalline or amorphous silica (data not shown). This indicates that increased PGE2 production is not related to increased IL-1β expression.
Amorphous and crystalline silica induce microsomal PGE2 synthase
In addition to the COX enzymes, production of PGE2 is dependent on specific PGE2 synthases to convert PGH2 to PGE2. Two isoforms of PGE synthase have been identified, a microsomal form (mPGES) that is induced by inflammatory mediators and a cytosolic form (cPGES) that is constitutively expressed and not inducible (36). Little is known of the regulation of these specific PG synthases in the lung. To investigate if up-regulation of PGE synthase contributes to increased production of PGE2, Western blot analysis of fibroblasts exposed to crystalline silica (100 μg/ml) and amorphous silica (10 μg/ml) was performed. Both amorphous and crystalline silica induced expression of mPGES, and this expression is persistent, lasting up to 72 hours after treatment (Figure 6). Silica treatment did not induce expression of cPGES (data not shown).
Figure 6.

Crystalline and amorphous silica induce mPGE synthase in primary human lung fibroblasts. Normal pulmonary fibroblasts in culture were exposed to 100 μg/ml crystalline silica (CS), 10 μg/ml amorphous silica (AS), or IL-1β (10 ng/ml) as a positive control. Cell lysates were harvested at 24, 48 and 72 h, and Western blot analysis for mPGES was performed. MC, media control.
Amorphous and crystalline silica induce PGF2α but not PGD2
To examine the specificity of silica-induced induction of various prostaglandins, production of PGF2α and PGD2 were examined in the lung fibroblast supernatants. Both amorphous and crystalline silica stimulated production of PGF2α in a dose-dependent fashion (Figure 7). The levels measured were ten-fold lower than those of PGE2 in the same supernatants. Amorphous silica was approximately 10-fold more potent at stimulating the release of PGF2α, consistent with the results for COX-2 and PGE2. In contrast PGD2 production was negligible in untreated fibroblasts and not induced by crystalline or amorphous silica (data not shown).
Figure 7.

PGF2α production is stimulated by crystalline silica (CS) and amorphous silica (AS). Fibroblasts were exposed to indicated doses of CS and AS. MC, untreated media control. The cell supernatants were harvested after 48 h and analyzed for PGF2α using enzyme immunoassays. The increase in PGF2α production is statistically significant (*p<0.01).
The finding that amorphous silica induces COX-2, PGE2 and PGF2α at 7–10 fold lower concentrations than crystalline silica may help to explain why amorphous and crystalline silica have different effects in vivo. To determine whether these silicas have the capacity to differentially regulate other important pro-inflammatory and pro-fibrotic mediators, supernatants from silica-treated fibroblasts were assayed for IL-6, IL-8, MCP-1 and TGF-β. Neither crystalline nor amorphous silica induced the expression of IL-6, MCP-1 or TGF-β in lung fibroblasts (data not shown). However, amorphous silica strongly induced the production of IL-8 (700 ±166 pg/ml with 10 μg/ml amorphous silica compared to 45 ± 20 pg/ml with 100 μg/ml crystalline silica, p<0.01). This is an interesting finding as IL-8 is a potent neutrophil chemoattractant, which is capable of synergizing with PGE2 to further promote neutrophil chemotaxis.
Discussion
Inhalation of both crystalline and amorphous silica results in pulmonary inflammation. However, only chronic exposure to the crystalline form leads to silicosis, i.e. fibrosis. The mechanisms by which these particles induce pathology remain unclear but little or no attention has been focused on the role of the lung fibroblast, especially primary strains from human lungs. Based on our data and that of others, the fibroblast is now considered a key effector in lung inflammation and fibrosis, capable of producing inflammatory and fibrotic mediators (14; 34; 40).
The data presented here demonstrate that remarkably, both crystalline and amorphous silica directly activate human primary pulmonary fibroblasts to produce COX-2, mPGES and PGE2, independent of production of inflammatory mediators from alveolar macrophages and epithelial cells. Silica induces the early inflammatory response gene COX-2 as determined by Western blot, immunocytochemistry, and RPA (Figures 1–3). Production of the downstream prostaglandin PGE2 was stimulated by silica, and was dependent on the induction of COX-2, as it could be inhibited by the COX inhibitors indomethacin and SC58215 (Figures 4–5). In addition to PGE2, silica stimulated lung fibroblasts to produce elevated levels of PGF2α, but not PGD2 (Figure 7). This is interesting as PGD2 is rapidly converted to 15d-PGJ2 which is reported to have anti-inflammatory actions and which inhibits fibroblast migration, whereas PGF2α functions to promote neutrophil chemotaxis and thus has pro-inflammatory effects (2; 17; 20; 31). It thus appears that as well as COX-2, silica must be acting to induce some of the specific PG synthases but not others. Little is known of the regulation of these synthases in lung structural cells. We therefore examined and demonstrated induction of mPGE synthase in primary human lung fibroblasts treated with silica (Figure 6).
Our data consistently demonstrate that amorphous silica induces COX-2, mPGES, PGE2 and PGF2α more potently than crystalline silica, as approximately 10-fold more crystalline silica is required to observe similar effects. This is a key finding given the fact that while amorphous silica incites pulmonary inflammation, it does not induce fibrosis (13; 30). We speculate that this is due to its greater ability to induce PGE2. An emerging body of evidence suggests that failure of lung fibroblasts to synthesize sufficient PGE2 may be important in helping to promote the development of fibrosis. Clearly, PGE2 has anti-fibrotic properties in vitro as it inhibits fibroblast proliferation and collagen synthesis (3; 6; 9; 10). Furthermore, BAL PGE2 levels are lower in patients with IPF than normal volunteers (4). A number of studies have linked this to a direct failure of fibroblasts derived from patients with pulmonary fibrosis to induce COX-2 and therefore synthesize PGE2 (16; 37; 38). More recently, another study demonstrated that PGE2 can inhibit the phenotypic transition which fibroblasts undergo to myofibroblasts (19). Myofibroblasts are cells that have features intermediate between smooth muscle cells and fibroblasts, and have been identified as the dominant producers of matrix elements like collagen in many fibrotic models (15; 33). Thus, this is a particularly significant discovery and lends further weight to the argument that PGE2 has important anti-fibrotic properties.
It can be argued that prostaglandins also have proinflammatory properties and that increased production of PGs would be expected to lead to increased disease. It has been reported that PLA2 knockout mice are protected from acute lung injury due to sepsis or acid aspiration (24), and are also protected from bleomycin-induced pulmonary fibrosis (23), suggesting that prostanoids have important pro-fibrotic effects. However, it must be noted that eliminating PLA2 will eliminate production of all downstream products of the arachidonic acid pathway, including leukotrienes, thromboxanes and prostaglandins, and this is likely to have complex pleiotropic effects. COX-2 knockout mice develop enhanced fibrosis in response to bleomycin (12; 16); while mice lacking the 5-lipoxygenase gene, which are leukotriene-deficient, are protected from bleomycin-induced fibrosis and express elevated levels of PGE2 in lavage (27). Furthermore, fibroblasts isolated from IPF patients produce elevated levels of the profibrotic thromboxane (TX)A2 and reduced levels of the anti-fibrotic PGI2 (prostacyclin) (7). Thus, it appears that leukotrienes and TXA2 have profibrotic properties, while PGE2 and prostacyclin have antifibrotic properties. In one study, COX-2-deficient mice were protected from enhanced fibrosis by a compensatory upregulation of COX-1 which restored normal PGE2 production (12). While prostaglandins can have both pro- and anti-inflammatory effects, the results discussed above support the hypothesis that PGE2 has antifibrotic effects, and provide the context for our hypothesis that crystalline, but not amorphous silica causes fibrosis in part because amorphous but not crystalline silica is a potent inducer of PGE2 in human lung fibroblasts.
The differential ability of amorphous and crystalline silica to induce fibrosis probably involves both the physical and inflammatory properties of the silicas. Amorphous silica is more soluble and is more easily cleared from the lungs. Although increased IL-8 production leads to greater acute inflammation (13), we hypothesize that increased production of PGE2 prevents this transient inflammatory response from developing into fibrosis. Since crystalline silica particles are far larger and hence less easily cleared from the lung, the epithelial damage they cause is more persistent, while macrophages that ingest crystalline silica are activated and damaged (29). This persistent irritation, combined with insufficient production of PGE2 could allow the establishment of a pro-fibrogenic milieu (16). It should be noted that prominent inflammation does not precede tissue scarring in IPF (11; 26). Perhaps an initial prominent inflammatory response is actually protective against the future development of ongoing scarring and tissue remodeling.
In summary, we demonstrated that both crystalline and amorphous silica are both capable of directly activating human primary pulmonary fibroblasts to upregulate COX-2 and mPGE synthase and to produce elevated levels of PGE2 and PGF2α. Of note, amorphous silica leads to far more intense induction of this pathway. This may help explain the differential abilities of crystalline and amorphous silica to generate a fibrogenic response. These data therefore further our understanding of the potential pathogenic mechanisms of silica-induced pulmonary inflammation and fibrosis. Furthermore, this highlights the importance of pulmonary fibroblast generated PGE2 as a protective element in fibrotic pathologies.
Acknowledgments
The current address for Katherine O’Reilly is Department of Medicine and Therapeutics, Conway Institute of Biomolecular and Biomedical Research, University College Dublin, Belfield, Dublin 4, Ireland
Grants
This work was supported by NIH: HL04492-02, The American Lung Association ALA-DA-004-N, The James P. Wilmot Foundation, Phillip Morris, NIEHS Center Grant P30ES01247 and EPA R827354.
References
- 1.Diseases associated with exposure to silica and nonfibrous silicate minerals. Silicosis and Silicate Disease Committee. Arch Pathol Lab Med 112: 673–720, 1988. [PubMed] [Google Scholar]
- 2.Arnould T, Thibaut-Vercruyssen R, Bouaziz N, Dieu M, Remacle J and Michiels C. PGF(2alpha), a prostanoid released by endothelial cells activated by hypoxia, is a chemoattractant candidate for neutrophil recruitment. Am J Pathol 159: 345–357, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bitterman PB, Wewers MD, Rennard SI, Adelberg S and Crystal RG. Modulation of alveolar macrophage-driven fibroblast proliferation by alternative macrophage mediators. J Clin Invest 77: 700–708, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borok Z, Gillissen A, Buhl R, Hoyt RF, Hubbard RC, Ozaki T, Rennard SI and Crystal RG. Augmentation of functional prostaglandin E levels on the respiratory epithelial surface by aerosol administration of prostaglandin E. Am Rev Respir Dis 144: 1080–1084, 1991. [DOI] [PubMed] [Google Scholar]
- 5.Bowden DH and Adamson IY. The role of cell injury and the continuing inflammatory response in the generation of silicotic pulmonary fibrosis. J Pathol 144: 149–161, 1984. [DOI] [PubMed] [Google Scholar]
- 6.Clark JG, Kostal KM and Marino BA. Modulation of collagen production following bleomycin-induced pulmonary fibrosis in hamsters. Presence of a factor in lung that increases fibroblast prostaglandin E2 and cAMP and suppresses fibroblast proliferation and collagen production. J Biol Chem 257: 8098–8105, 1982. [PubMed] [Google Scholar]
- 7.Cruz-Gervis R, Stecenko AA, Dworski R, Lane KB, Loyd JE, Pierson R, King G and Brigham KL. Altered prostanoid production by fibroblasts cultured from the lungs of human subjects with idiopathic pulmonary fibrosis. Respir Res 3: 17, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dayer JM, Beutler B and Cerami A. Cachectin/tumor necrosis factor stimulates collagenase and prostaglandin E2 production by human synovial cells and dermal fibroblasts. J Exp Med 162: 2163–2168, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elias JA, Rossman MD, Zurier RB and Daniele RP. Human alveolar macrophage inhibition of lung fibroblast growth. A prostaglandin-dependent process. Am Rev Respir Dis 131: 94–99, 1985. [DOI] [PubMed] [Google Scholar]
- 10.Fine A, Poliks CF, Donahue LP, Smith BD and Goldstein RH. The differential effect of prostaglandin E2 on transforming growth factor-beta and insulin-induced collagen formation in lung fibroblasts. J Biol Chem 264: 16988–16991, 1989. [PubMed] [Google Scholar]
- 11.Gauldie J, Kolb M and Sime PJ. A new direction in the pathogenesis of idiopathic pulmonary fibrosis? Respir Res 3: 1, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hodges RJ, Jenkins RG, Wheeler-Jones CP, Copeman DM, Bottoms SE, Bellingan GJ, Nanthakumar CB, Laurent GJ, Hart SL, Foster ML and McAnulty RJ. Severity of lung injury in cyclooxygenase-2-deficient mice is dependent on reduced prostaglandin E(2) production. Am J Pathol 165: 1663–1676, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnston CJ, Driscoll KE, Finkelstein JN, Baggs R, O’Reilly MA, Carter J, Gelein R and Oberdorster G. Pulmonary chemokine and mutagenic responses in rats after subchronic inhalation of amorphous and crystalline silica. Toxicol Sci 56: 405–413, 2000. [DOI] [PubMed] [Google Scholar]
- 14.Jordana M, Sarnstrand B, Sime PJ and Ramis I. Immune-inflammatory functions of fibroblasts. Eur Respir J 7: 2212–2222, 1994. [DOI] [PubMed] [Google Scholar]
- 15.Kawanami O, Jiang HX, Mochimaru H, Yoneyama H, Kudoh S, Ohkuni H, Ooami H and Ferrans VJ. Alveolar fibrosis and capillary alteration in experimental pulmonary silicosis in rats. Am J Respir Crit Care Med 151: 1946–1955, 1995. [DOI] [PubMed] [Google Scholar]
- 16.Keerthisingam CB, Jenkins RG, Harrison NK, Hernandez-Rodriguez NA, Booth H, Laurent GJ, Hart SL, Foster ML and McAnulty RJ. Cyclooxygenase-2 deficiency results in a loss of the anti-proliferative response to transforming growth factor-beta in human fibrotic lung fibroblasts and promotes bleomycin-induced pulmonary fibrosis in mice. Am J Pathol 158: 1411–1422, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kohyama T, Liu XD, Wen FQ, Kim HJ, Takizawa H and Rennard SI. Prostaglandin D2 inhibits fibroblast migration. Eur Respir J 19: 684–689, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Kolb M, Margetts PJ, Anthony DC, Pitossi F and Gauldie J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest 107: 1529–1536, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolodsick JE, Peters-Golden M, Larios J, Toews GB, Thannickal VJ and Moore BB. Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol 29: 537–544, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Lai J, Jin H, Yang R, Winer J, Li W, Yen R, King KL, Zeigler F, Ko A, Cheng J, Bunting S and Paoni NF. Prostaglandin F2 alpha induces cardiac myocyte hypertrophy in vitro and cardiac growth in vivo. Am J Physiol 271: H2197–2208, 1996. [DOI] [PubMed] [Google Scholar]
- 21.Mohr C, Gemsa D, Graebner C, Hemenway DR, Leslie KO, Absher PM and Davis GS. Systemic macrophage stimulation in rats with silicosis: enhanced release of tumor necrosis factor-alpha from alveolar and peritoneal macrophages. Am J Respir Cell Mol Biol 5: 395–402, 1991. [DOI] [PubMed] [Google Scholar]
- 22.Morgan A, Moores SR, Holmes A, Evans JC, Evans NH and Black A. The effect of quartz, administered by intratracheal instillation, on the rat lung. I. The cellular response. Environ Res 22: 1–12, 1980. [DOI] [PubMed] [Google Scholar]
- 23.Nagase T, Uozumi N, Ishii S, Kita Y, Yamamoto H, Ohga E, Ouchi Y and Shimizu T. A pivotal role of cytosolic phospholipase A(2) in bleomycin-induced pulmonary fibrosis. Nat Med 8: 480–484, 2002. [DOI] [PubMed] [Google Scholar]
- 24.Nagase T, Uozumi N, Ishii S, Kume K, Izumi T, Ouchi Y and Shimizu T. Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nat Immunol 1: 42–46, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Noguchi K, Shitashige M, Endo H, Kondo H, Yotsumoto Y, Izumi Y, Nitta H and Ishikawa I. Involvement of cyclooxygenase-2 in serum-induced prostaglandin production by human oral gingival epithelial cells. J Periodontal Res 36: 124–130, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Pardo A and Selman M. Idiopathic pulmonary fibrosis: new insights in its pathogenesis. Int J Biochem Cell Biol 34: 1534–1538, 2002. [DOI] [PubMed] [Google Scholar]
- 27.Peters-Golden M, Bailie M, Marshall T, Wilke C, Phan SH, Toews GB and Moore BB. Protection from pulmonary fibrosis in leukotriene-deficient mice. Am J Respir Crit Care Med 165: 229–235, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Piguet PF, Collart MA, Grau GE, Sappino AP and Vassalli P. Requirement of tumour necrosis factor for development of silica-induced pulmonary fibrosis. Nature 344: 245–247, 1990. [DOI] [PubMed] [Google Scholar]
- 29.Privalova LI, Katsnelson BA, Sharapova NY and Kislitsina NS. On the relationship between activation and breakdown of macrophages in the pathogenesis of silicosis (an overview). Med Lav 86: 511–521, 1995. [PubMed] [Google Scholar]
- 30.Reuzel PG, Bruijntjes JP, Feron VJ and Woutersen RA. Subchronic inhalation toxicity of amorphous silicas and quartz dust in rats. Food Chem Toxicol 29: 341–354, 1991. [DOI] [PubMed] [Google Scholar]
- 31.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M and Santoro MG. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature 403: 103–108, 2000. [DOI] [PubMed] [Google Scholar]
- 32.Schmidt JA, Oliver CN, Lepe-Zuniga JL, Green I and Gery I. Silica-stimulated monocytes release fibroblast proliferation factors identical to interleukin 1. A potential role for interleukin 1 in the pathogenesis of silicosis. J Clin Invest 73: 1462–1472, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sime PJ, Xing Z, Graham FL, Csaky KG and Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest 100: 768–776, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith RS, Smith TJ, Blieden TM and Phipps RP. Fibroblasts as sentinel cells. Synthesis of chemokines and regulation of inflammation. Am J Pathol 151: 317–322, 1997. [PMC free article] [PubMed] [Google Scholar]
- 35.Smith WL, Meade EA and DeWitt DL. Pharmacology of prostaglandin endoperoxide synthase isozymes-1 and −2. Ann N Y Acad Sci 714: 136–142, 1994. [DOI] [PubMed] [Google Scholar]
- 36.Stichtenoth DO, Thoren S, Bian H, Peters-Golden M, Jakobsson PJ and Crofford LJ. Microsomal prostaglandin E synthase is regulated by proinflammatory cytokines and glucocorticoids in primary rheumatoid synovial cells. J Immunol 167: 469–474, 2001. [DOI] [PubMed] [Google Scholar]
- 37.Vancheri C, Sortino MA, Tomaselli V, Mastruzzo C, Condorelli F, Bellistri G, Pistorio MP, Canonico PL and Crimi N. Different expression of TNF-alpha receptors and prostaglandin E(2)Production in normal and fibrotic lung fibroblasts: potential implications for the evolution of the inflammatory process. Am J Respir Cell Mol Biol 22: 628–634, 2000. [DOI] [PubMed] [Google Scholar]
- 38.Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM and Peters-Golden M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Invest 95: 1861–1868, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams AO, Ward JM, Li JF, Jackson MA and Flanders KC. Immunohistochemical localization of transforming growth factor-beta 1 in Kaposi’s sarcoma. Hum Pathol 26: 469–473, 1995. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Cao HJ, Graf B, Meekins H, Smith TJ and Phipps RP. CD40 engagement up-regulates cyclooxygenase-2 expression and prostaglandin E2 production in human lung fibroblasts. J Immunol 160: 1053–1057, 1998. [PubMed] [Google Scholar]