

Abstract
Emphysema, a component of chronic obstructive pulmonary disease (COPD), is characterized by irreversible alveolar destruction that results in a progressive decline in lung function. This alveolar destruction is caused by cigarette smoke, the most important risk factor for COPD. Only 15-20% of smokers develop COPD, suggesting that unknown factors contribute to disease pathogenesis. We postulate that the aryl hydrocarbon receptor (AHR), a receptor/transcription factor highly expressed in the lungs, may be a new susceptibility factor whose expression protects against COPD. Here, we report that Ahr-deficient mice chronically exposed to cigarette smoke develop airspace enlargement concomitant with a decline in lung function. Chronic cigarette smoke exposure also increased cleaved caspase-3, lowered SOD2 expression and altered MMP9 and TIMP-1 levels in Ahr-deficient mice. We also show that people with COPD have reduced expression of pulmonary and systemic AHR, with systemic AHR mRNA levels positively correlating with lung function. Systemic AHR was also lower in never-smokers with COPD. Thus, AHR expression protects against the development of COPD by controlling interrelated mechanisms involved in pathogenesis of this disease. This study identifies the AHR as a new, central player in the homeostatic maintenance of lung health, providing a foundation for the AHR as a novel therapeutic target and/or predictive biomarker in chronic lung disease.
1. INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is characterized by progressive and irreversible airflow obstruction. Worldwide, COPD affects an estimated 380 million people and is the third leading cause of death (1). Despite the significant disease burden of COPD, few-to-no effective therapeutic options exist to stop or slow its progression, in part because the mechanistic basis of this disease remains unknown. COPD is an umbrella term that encompasses chronic bronchitis and emphysema (alveolar airspace enlargement). The emphysema component of COPD is proposed to be mediated by several inter-dependent mechanisms including chronic inflammation, oxidative stress, a protease:anti-protease imbalance and accelerated cell death in the lungs (2). How these multiple pathogenic processes are regulated at the molecular level in COPD is unknown but is important to consider, especially as only 15-20% of all individuals who smoke develop clinically-relevant COPD. This suggests that unknown molecular mediators contribute to COPD pathogenesis (3, 4).
We postulate that the aryl hydrocarbon receptor (AHR) may be a new susceptibility factor whose expression protects against the development of COPD. The AHR is a transcription factor that mediates the deleterious effects of the man-made toxicant dioxin. After binding dioxin, the AHR translocates to the nucleus and forms a heterodimer with the AHR nuclear transporter (ARNT). This AHR•ARNT complex binds to DNA sequences termed the dioxin response element (DRE), initiating the transcription of target genes such as cytochrome P450 (CYP) CYP1A1 and the AHR repressor (AHRR), a negative regulator of the AHR pathway that competes with AHR for ARNT binding (5). Following gene induction, the AHR dissociates from ARNT, is shuttled back to the cytoplasm and degraded by the 26S ubiquitin-proteasome system (6). Although the AHR has largely been associated with xenobiotic metabolism leading to toxicity, a broad range of biochemical and genetic studies have now revealed that the AHR has important endogenous functions, including control over cell proliferation, differentiation, migration and survival (7–10). We have published that the AHR controls acute cigarette smoke-induced pulmonary inflammation in vivo and controls oxidative stress and cell death pathways in vitro (8, 11, 12). There is also low AHR expression in many human disease, including inflammatory bowel disease (IBD) (13) and Behcet’s disease (14). Thus, lower-than-normal AHR levels raise the possibility that reduced AHR expression may predispose some smokers to developing COPD, suggesting that AHR may offer protection in the lung against the development of COPD.
Our data herein illustrate that the AHR controls the development of COPD through the integration of multiple pathogenic mechanisms, including suppression of inflammation and control over anti-oxidant proteins and protease levels. We also show that there is a significant reduction in pulmonary and systemic AHR in COPD, and that there is a positive correlation between both AHR and ARNT expression with lung function in people with COPD. Collectively, these data suggest a protective role for the AHR in the pathogenesis of smoke-induced emphysema/COPD. Given that no effective therapeutic options currently exist to stop or slow disease progression, these findings could provide the basis for the development of new therapeutic agents or biomarkers for COPD.
2. Materials and Methods
2.1. Animals
AHR-knockout (Ahr−/−) mice (strain B6.129-Ahrtm1Bra) were purchased from the Jackson Laboratory, bred and maintained as previously described (15). This strain carries a targeted deletion of exon 2 of the Ahr gene and was backcrossed for 12 generations onto C57BL/6.. A breeding scheme of heterozygous Ahr+/− to Ahr−/− mice are used, rendering mice of the Ahr+/− genotype as littermate controls. Ahr+/− mice are phenotypically indistinguishable from wild-type (Ahr+/+) mice and there are no differences between C57BL/6 and Ahr+/− mice (11). Finally, Ahr+/+ or Ahr+/− mice do not exhibit any difference in AHR protein expression in lungs/lung cells (Figure 1) (16) or differ in the ability to be activated by AhR ligands or cigarette smoke (11, 15). Therefore Ahr+/− mice were used as controls in this study. All animal procedures were approved by the McGill University Animal Care Committee (Protocol Number: 5933).
FIGURE 1.
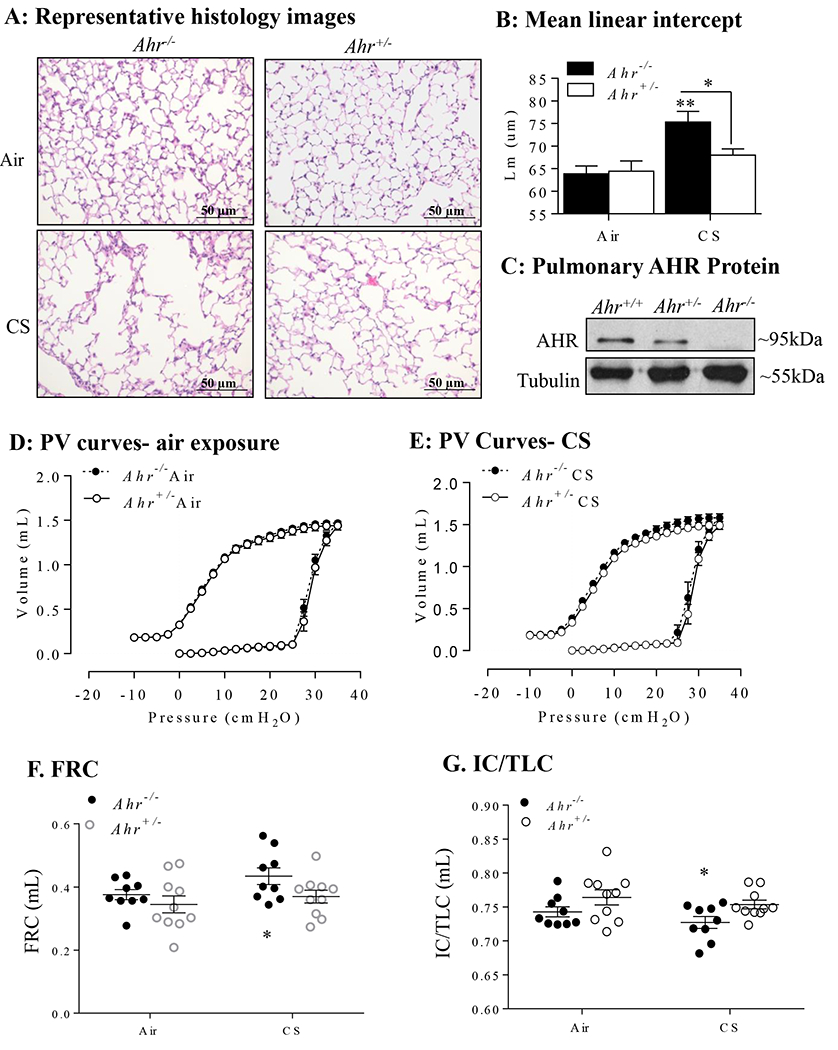
AHR deficiency drives the development of cigarette smoke (CS)-induced emphysema in vivo. A: Representative histology images: Representative images from the lung parenchyma of Ahr−/− and Ahr+/− mice exposed to a 4-month CS regime are shown (magnification = 20x). B: Mean linear intercept: Lm was used to quantify airspace enlargement in the lung parenchyma. There was a significant increase in Lm in CS-exposed Ahr−/− mice compared to air-exposed mice (**p < 0.01) as well as CS-exposed Ahr+/− mice (*<0.05). Results are expressed as mean ± SEM, n=6-8 mice per group. C: Pulmonary AHR protein: Representative western blot showing that AHR protein levels are not different between Ahr−/− and Ahr+/− mice. Lung volume curves are shown comparing air-exposed (D) and CS-exposed (E) Ahr+/− and Ahr−/− mice. There were significant changes in functional residual capacity (FRC) (F), and the ratio of inspiratory capacity to the total lung capacity (IC/TLC) (G) in CS-exposed Ahr−/− mice (*p< 0.05). Results are expressed as mean ± the SEM, n= 9-10 mice per group.
2.2. Cigarette Smoke Exposure
Mice (age 8-12 weeks) were exposed to cigarette smoke using a SCIREQ® InExpose Exposure System (SCIREQ, Montreal, QC) for 8 weeks or 4 months as previously described (15). Briefly, research cigarettes (3R4F; University of Kentucky, Lexington, KY) were smoked in groups of 4 at 1 puff/cigarette/15 seconds for a total of 1 hour (daily for 5 days per week). Mice received whole-body exposure of mainstream smoke diluted with air. The amount of smoke particulates was monitored using a MicroDust Pro (Casella CEL; Buffalo, NY) and maintained at a cumulative particulate density (CPD) of 300g/L. Both male and female mice were used unless otherwise indicated. Twenty-four hours after the final exposure, mice were euthanized by exsanguination. Lung tissue was collected for protein/western blot analysis and/or measurement of airspace enlargement.
2.3. Mean Linear Intercept (Lm)
Following formalin fixation, the left lung was embedded in paraffin, sectioned and stained with hematoxylin-eosin (H&E). Airspace enlargement was assessed by calculating the mean linear intercept (Lm) as described (17).
2.4. Lung Function Assessment
Each mouse was administered xylazine IP (10 mg/kg) 5 min before being anaesthetized with 50 mg/kg of sodium pentobarbital. Mechanical ventilation was set at a frequency of 150 breaths/min, a tidal volume of 10 mL/kg, a 2:3 inspiratory:expiratory ratio and a 3 cm H2O positive end expiratory ratio. Rocuronium (0.2 mg/kg) was then administered, followed by a deep lung inflation maneuver to 30 cm H2O and a 2-3 min equilibration period. Next, deep lung inflation and a stepwise pressure-volume (PV) curve, both set to a final pressure of 35 cm H2O, were run and the mechanical ventilation was switched from room air to 100% oxygen for 5 min to degas the lungs by oxygen absorption prior to the construction of a full-range PV curve (18).
2.5. Western Blot
Lung tissue was homogenized in RIPA buffer supplemented with protease inhibitor cocktail (Roche, Indianapolis, IN). Ten micrograms of total protein were separated on SDS-PAGE gels, electroblotted onto polyvinylidene difluoride (PVDF) membrane (Bio-Rad Laboratories, Hercules, CA) and blocked with 5% non-fat dry milk in 0.1% Tween 20 (in PBS). Antibodies against AHR (1:1000, Enzo Life Sciences), cleaved-casepase-3 (1:1000; Cell Signaling), SOD2 (1:1000; R&D Systems), HO-1, MMP9 (1:1000; Abcam), TIMP-1 (1:1000; Abcam), tubulin (1:10,000; Sigma-Aldrich) and total actin (1:50,000, EMD Millipore) were used to assess changes in relative expression. Proteins were visualized using HRP-conjugated secondary antibodies (1:10,000) followed by enhanced chemiluminescence (ECL) and imaged using a ChemiDoc™ XRS+ System (Bio-Rad Laboratories; Hercules, CA).
2.6. Lung Tissue Expression Quantitative Loci (eQTL) Study
Gene expression profiling was performed using an Affymetrix custom array (GPL10379), which contained 51,627 non-control probesets and data were normalized using RMA (19). Genotyping was performed using the Illumina Human1M-Duo BeadChip array. Genotype imputation was undertaken using the 1000G reference panel. Following standard microarray and genotyping quality controls, data from 1,111 patients were available including 409 from Laval, 363 from Groningen and 339 from UBC. Table 1 contains patient characteristics for the Laval cohort; as lung specimens for this cohort were obtained from patients that underwent surgery for lung cancer, post-bronchodilator (BD) spirometry was not performed. Association testing for each variant with mRNA expression in either cis (within 1 Mb of transcript start site) or in trans (all other combinations) was undertaken separately for each study sample, after which the results were meta-analyzed using inverse variance weighting. A genome-wide 10% false discovery rate (FDR) was applied to this analysis. A subset of these subjects was used to test for the association of ARNT gene with lung function. The lung tissue mRNA association testing with lung function parameters was restricted to 727 individuals who did not have any significant respiratory diseases, other than smoking-related conditions, and for whom lung function measurements were available. Gene expression data are available on GEO under accession number GSE23546.
TABLE 1.
Clinical characteristics of the lung eQTL study (Laval cohort).
| Variable | Control (n = 168) | COPD (n = 217) | p-value |
|---|---|---|---|
| Age (years) | 60.3 ± 10.6 | 65.6 ± 8.8 | 2.50E-07 |
|
| |||
| Male (n) | 86 | 131 | 0.09 |
|
| |||
| FEV1 % predicted | 90.8 ± 16.3 | 72.6 ± 17.4 | <2.2E-16 |
|
| |||
| FVC % predicted | 91.9 ± 15.8 | 88.2 ± 16.2 | 0.03 |
|
| |||
| FEV1/FVC | 75.5 ± 4.2 | 61.3 ± 7.9 | <2.2E-16 |
|
| |||
| Smoking (%) | |||
| Smoker | 28 (16.7) | 59 (27.2) | 0.0001 |
| Former | 117 (69.6) | 151 (69.6) | |
| Never | 23 (13.7) | 7 (3.2) | |
Data presented as the mean ± SD
2.7. Multiplex Immunohistochemistry (mIHC)
Human subjects were approved by the Research Ethics Board of St Joseph’s Healthcare Hamilton (00-1839). Subject characteristics are in Table 2; post-BD spirometry lung function values are indicated. Lung tissue was fixed in 10% formalin and embedded in paraffin. Five-μm thick sections were first incubated in primary AHR antibody (Abcam #2770; 1:100) for 16 min at 37°C, followed by incubation in secondary antibody OMap anti-Ms HRP (Roche #760-4310) for 16 min at 37°C. Sections were next incubated in primary vimentin antibody (Cell Signaling #3932; 1:50) for 16 min at 37°C, followed by incubation in secondary antibody UltraMap Anti Rb Alk Phos (Roche #760-431) for 16 min at 37°C. Lastly, sections were incubated in pre-diluted primary cytokeratin-19 antibody (Roche #760-4281) for 16 min at 37°C, followed by incubation in secondary antibody OMap anti-Ms HRP (Roche #760-4310) for 16 min at 37°C. Sections were counter-stained using hematoxylin. Detection was accomplished using a 3,3’-diaminobenzidine (DAB) detection kit.
TABLE 2.
Clinical characteristics of the St Joseph’s Healthcare Hamilton subjects.
| Variable | Non-Smoker (11) | At Risk (11) | COPD (13) | p-value |
|---|---|---|---|---|
| Age (years) | 68.1 ± 10.3 | 57.8 ± 8.7 | 65 ± 10.7 | Non-smoker vs smoker p=0.056 Non-smoker vs COPD p=0.733 Smoker vs COPD p=0.20 |
| Male (n) | 5 | 6 | 7 | N/A |
| Female (n) | 6 | 5 | 6 | N/A |
| FEV1/FVC (%) | 80.5 ± 8.2 | 75.5 ± 6.6 | 57.7 ± 10.4 | Non-smoker vs smoker p=0.28 Non-smoker vs COPD p<0.0001 Smoker vs COPD p=0.0001 |
| Pack Years* | 0 ± 0 | 39.5 ± 16.2 | 48.1 ± 21.6 | Non-smoker vs smoker p<0.0001 Non-smoker vs COPD p<0.001 Smoker vs COPD p=0.39 |
Data presented as the mean ± SD.
2.8. Droplet Digital PCR (ddPCR)
Analysis of systemic AHR expression was analyzed by ddPCR using biological samples from the CanCOLD cohort; CanCOLD has been described in detail (20). Briefly, CanCOLD is a population-based longitudinal study that aims to understand the heterogeneity of COPD presentation and disease progression. Subjects were sampled from the general population and data collected includes pulmonary function and cardiorespiratory exercise tests, CT scans and blood sampling (20). Patient characteristics of the samples analyzed in this study are in Table 3, which includes lung function, smoking status, emphysema and bronchiolitis score assessed by CT, as well as co-morbidities. Peripheral blood was collected using PAXgene blood RNA tubes (PreAnalytiX GmbH, Hombrechtikon, Germany) at initial visit and frozen at −80°C until analysis (21). Total RNA was isolated using a PAXgene blood RNA kit (PreAnalytiX GmbH) according to the supplier protocol. RNA was reverse transcribed to cDNA, diluted and primers added in QX200 ddPCR Eva Green® Super-mix (Bio-Rad) as previously described (22). Samples were then partitioned into 20,000 droplets and PCR amplification was performed using the QX100 Droplet Digital™ PCR System (Bio-Rad). Primer sequences for human AHR are: AGAGGCTCAGGTTATCAGTTT(f) and AGTCCATCGGTTGTTTTTT (r). Primer sequences for β-actin are CTACCATGAGCTGCGTGTG (f) and TGGGGTGTTGAAGGTCTC (r). Gene expression data was normalized to β-actin.
TABLE 3.
Clinical characteristics of CanCOLD subjects
| Variable | Total N=63 | Normal N=16 | At Risk N=15 | COPD N=32 | p-values |
|---|---|---|---|---|---|
| Age, in year [mean(sd)] | 67.00 ± 9.17 | 67.75 ± 5.84 | 65.53 ± 8.46 | 67.31 ± 10.86 | 0.974 |
| Sex, male gender, n (%) | 27 (42.86) | 4 (25.00) | 5 (33.33) | 18 (56.25) | 0.087 |
| Tobacco smoking status, n (%) | |||||
| Never | 26 (41.27) | 16 (100.00)a | - | 10 (31.25)a | <0.001* |
| Ex-smokers | 27 (42.86) | - | 13 (86.67)a | 14 (43.75)a | <0.001* |
| Current smokers | 10 (15.87) | - | 2 (13.33) | 8 (25.00) | 0.066 |
| Cigarette smoker pack-years | 15.16 ± 22.36 | - | 13.40 ± 10.61a | 23.57 ± 27.49a | <0.001* |
| Post-bronchodilator spirometry | |||||
| FEV1, L | 2.32 ± 0.68 | 2.38 ± 0.48 | 2.60 ± 0.60 | 2.16 ± 0.76 | 0.18 |
| FEV1, % predicted | 91.73 ± 22.10 | 101.28 ± 11.33a | 100.18 ± 11.11b | 82.72 ± 26.52ab | 0.002* |
| FEV1/FVC, % | 68.99 ± 12.70 | 76.39 ± 4.22a | 78.61 ± 4.36b | 60.52 ± 12.68ab | <0.001* |
| Emphysema score | 0.89 ± 1.89 | 0.00 | 0.15 ± 0.55a | 1.63 ± 2.37a | 0.001* |
| Emphysema (score ≥ 1) | 15 (24.59) | 0 (0.0)a | 1 (7.69)ab | 14 (43.75)ab | <0.001* |
| Bronchiolitis (score ≥ 2) | 7 (11.48) | 0 (0.0) | 0 (0.0) | 7 (21.88) | 0.194 |
| Self-reported comorbidities, n (%) | |||||
| Bronchiectasis | 2 (3.2) | 0 (0.0) | 0 (0.0) | 2 (6.3) | 0.738 |
| Pneumonia | 8 (12.7) | 0 (0.0) | 1 (6.7) | 7 (21.9) | 0.078 |
| Osteoporosis | 12 (19.0) | 1 (6.3) | 3 (20.0) | 8 (25.0) | 0.344 |
| Any Musculoskeletal | 23 (36.5) | 2 (12.5)a | 9 (60.0)a | 12 (37.5) | 0.023* |
| Angina | 5 (7.9) | 0 (0.0) | 1 (6.7) | 4 (12.5) | 0.419 |
| Hypertension | 20 (31.7) | 5 (31.3) | 7 (46.7) | 8 (25.0) | 0.33 |
| CVD (excluding Hypertension) | 13 (20.6) | 2 (12.5) | 2 (13.3) | 9 (28.1) | 0.426 |
| Depression | 3 (4.8) | 1 (6.3) | 1 (6.7) | 1 (3.1) | 0.8 |
| Cataract | 12 (19.0) | 5 (31.3) | 2 (13.3) | 5 (15.6) | 0.48 |
| Glaucoma | 7 (11.1) | 2 (12.5) | 2 (13.3) | 3 (9.4) | 0.883 |
| Diabetes | 5 (7.9) | 2 (12.5) | 1 (6.7) | 2 (6.3) | 0.831 |
| Asthma | 18 (28.6) | 2 (12.5)a | 2 (13.3) | 14 (43.8)a | 0.029* |
Data presented as the mean ± SD.
means with same letters are significantly different from each other after Tukey adjustment for multiple comparisons (p<0.05).
2.9. Bronchial Airway Epithelial Gene Expression Study
In the study by Steiling et al. (23), bronchial brushings were obtained from 6th to 8th generation bronchi from heavy current and ex-smokers who participated in an early lung cancer detection study at the British Columbia Cancer Agency between June 2000 and May 2009 (24) (25). In this dataset, gene expression profiling of the bronchial specimens was performed in 238 current and former smokers with (n = 87) and without COPD (n = 151). RNA was processed and hybridized to Affymetrix Human Gene 1.0 ST Arrays (Affymetrix, Santa Clara, CA). Gene expression estimates were derived using the RMA algorithm (19). Microarray data have been deposited in GEO under accession number GSE37147.
2.10. GWAS of Lung Function in the UK Biobank
To test if eQTLs for the ARNT gene were associated with lung function in large scale human genetic studies, we used summary statistics from the UK Biobank GWAS of lung function measures in the general population (n= 48,943 individuals) (26). GWAS summary data were available through approved access to the UK Biobank.
2.11. Single Cell RNA-sequencing Analysis
Raw single-cell RNA-seq expression matrices for cells from 18 COPD and 28 lungs from (27) were filtered using SCANY software (28). Cells with less than 200 expressing genes or more than 40% of mitochondrial reads were filtered. Genes that were only expressed in less than three cells were also removed. Genes were further filtered if they exhibited low dispersion (< 0.15) or very low-level expression (<0.0125). Gene expression was then converted to log2 space (log2 expression + 1).
2.12. Statistical Analyses
Statistical differences between group mean values were determined by 2-way ANOVA followed by the Tukey’s multiple comparisons test unless otherwise indicated. Systemic AHR mRNA expression in CanCOLD subjects were analyzed using a Kruskal-Wallis non-parametric one-way ANOVA (2+ comparison groups) or a Komologrov-Smirnov non-parametric t-test (2 comparison groups). Statistical analysis was conducted for CanCOLD subjects (Table 3) using SAS v9.4 (SAS Institute Inc., Cary, NC). These subject data were analyzed either using a one-way ANOVA (for a normal distribution) or a Kruskal-Wallis test, and a chi-squared test for category variables. Statistical analysis was conducted for St Joseph’s Healthcare Hamilton subjects (Table 2) using a Kruskal-Wallis non-parametric one-way ANOVA. Genetic associations in the eQTL study (Laval cohort- Table 1) were evaluated using chi-square tests in PLINK. A P-value < 0.05 was considered significant.
3. RESULTS
3.1. AHR deficiency promotes the development of cigarette smoke-induced emphysema and reduces lung function in mice
As chronic exposure to cigarette smoke is the main risk factor for the development of COPD, we first utilized our preclinical mouse model to causally address the contribution of the AHR to the development of airspace enlargement (Lm), a surrogate marker of emphysema. Ahr+/− and Ahr−/− mice were exposed to cigarette smoke for 4 months and Lm was calculated. Only Ahr−/− mice had a significant increase in Lm from chronic cigarette smoke exposure (Figure 1A–1B); this increase was also significant compared to smoke-exposed Ahr+/− mice. Verification of pulmonary AHR protein expression is shown in Figure 1C; note that there is no difference in AHR levels between Ahr+/− and Ahr−/− mice. We also assessed if chronic cigarette smoke exposure led to corresponding alterations in lung function using flexiVent. The parameters evaluated were derived from PV curves obtained using a full volume lung maneuver. PV curves are virtually super-imposable between the air-exposed Ahr+/− and Ahr−/− mice (Figure 1D). In Ahr−/− mice exposed to cigarette smoke for 4 months, there is an upward/leftward shift of the PV curve compared to the smoke-exposed Ahr+/− mice (Figure 1E). This shift in the PV curve of the smoke-exposed Ahr−/− mice is consistent with the PV curve shift in patients with emphysema due to the loss of elastic tissue and parenchymal destruction (29). From these curves, we calculated the functional residual capacity (FRC). FRC is a parameter of lung function that increases in emphysema as a result of elastic destruction (29). The FRC was significantly increased in the smoke-exposed Ahr−/− mice (Figure 1F). The ratio of the inspiratory capacity to the total lung capacity (IC/TLC) is also reduced in emphysema (30) and was significantly reduced in the smoke-exposed Ahr−/− mice (Figure 1G). Taken together, these data support that the AHR protects against the development of a cigarette smoke-induced emphysema-like phenotype, as evidenced by alterations in lung structure and function.
3.2. AHR deficiency exacerbates pathogenic mechanisms implicated in the development and progression of COPD
The pathogenesis of emphysema is mediated by a variety of inter-related mechanisms, including chronic inflammation, heightened oxidative stress, a protease-anti-protease imbalance and accelerated death of lung cells (2). We have previously published that the AHR suppresses pulmonary inflammation in response to cigarette smoke (11, 15). This inflammatory response is believed to underlie COPD pathogenesis, as it is postulated that oxidants released by recruited immune cells, along with oxidants in cigarette smoke cause accelerated cell death (i.e. apoptosis) that leads to emphysema (31, 32). We have also previously shown in vitro that the AHR suppresses smoke-induced oxidative stress, in part, due to the upregulation of key anti-oxidant proteins such as superoxide dismutase-2 (SOD2), a mitochondrial enzyme that converts superoxide anions to hydrogen peroxide and water and thus protects cells against oxidative damage leading to apoptosis (8). Whether dysregulation of anti-oxidant proteins/apoptosis also occurs in the lungs of Ahr−/− mice exposed chronically to smoke has not been examined. Therefore, to assess whether the AHR is a global regulator of these pathogenic mechanisms, we also now evaluated select anti-oxidants, activated caspase-3 and as well as MMP levels in mice exposed to cigarette smoke for 8 weeks. Analyses of samples from this exposure time allowed us to compare whether dysregulation of these mechanisms occurs prior to airspace enlargement in Ahr−/− mice. Our novel data show that there is significant induction of cleaved caspase-3 in response to an 8-week smoke exposure regime in Ahr−/− mice compared to smoke-exposed Ahr+/− mice (Figure 2A). Although cigarette smoke significantly increased the expression of SOD2 in both Ahr+/− and Ahr−/− mice, the level of induction was significantly less in the lungs of Ahr−/− mice (Figure 2B). There was no difference in the induction of heme oxygenase 1 (HO-1), a cytoprotective enzymes that catalyze the rate-limiting step in the degradation of heme, between smoke exposed Ahr+/− and Ahr−/− mice (Figure 2C).
FIGURE 2.
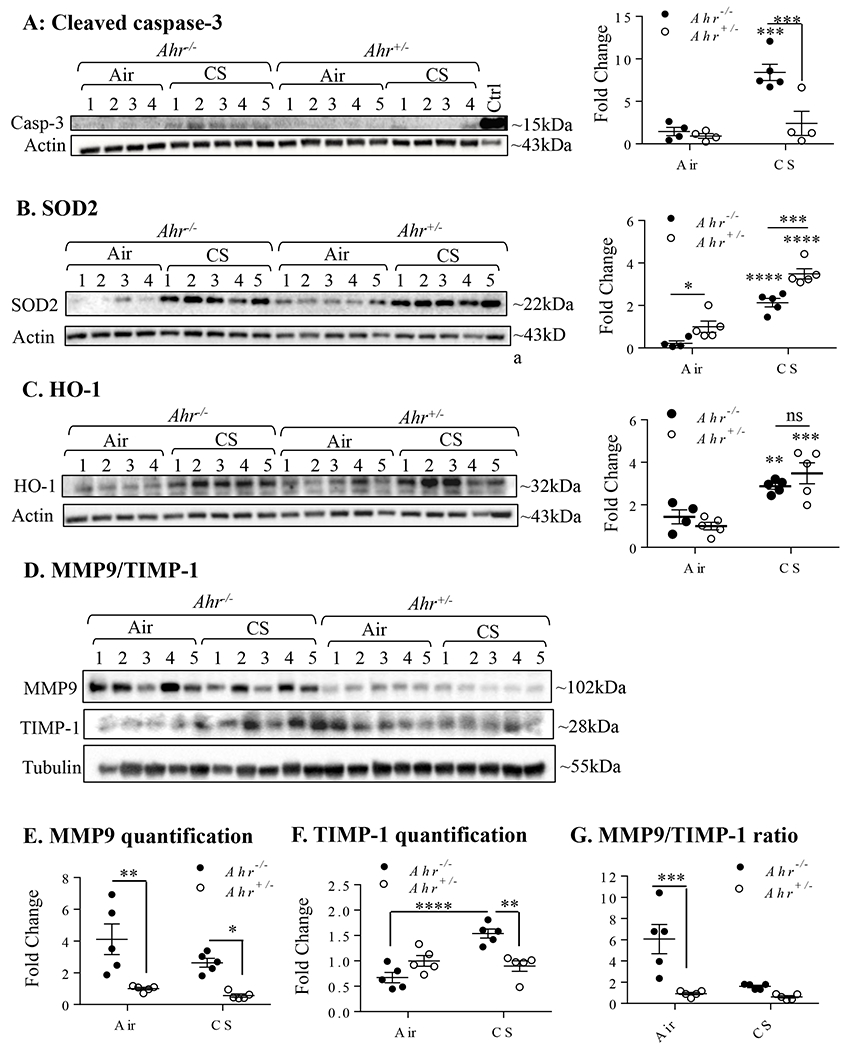
AHR deficiency results in a dysregulated anti-oxidant and apoptotic response as well as a protease:anti-protease imbalance. Ahr+/− and Ahr−/− mice were exposed to CS for 8 weeks and protein expression analyzed by western blot. A: Cleaved caspase-3-There was a significant increase in cleaved caspase-3 in Ahr−/− mice exposed to CS (**p< 0.001); this increase in cleaved-caspase-3 was significantly higher compared to smoke-exposed Ahr+/− mice. B: SOD2-there was a significant increase in the protein expression of SOD2 in Ahr+/− and Ahr−/− mice exposed to CS (****p < 0.0001); the increase in smoke-exposed Ahr−/− mice was higher than Ahr+/− mice (***p < 0.001). C: HO-1-there was an increase in HO-1 expression in the lungs of smoke-exposed Ahr+/− and Ahr−/− mice (**p<0.01; ***p < 0.001); there was no difference between the genotypes (ns). D: MMP9/TIMP-1-Western blot of MMP9 and TIMP-1 in lung homogenates; number of mice is indicated. E: MMP9 quantification- There was significantly more MMP9 as baseline in the lungs of Ahr−/− mice that did not change significantly change in response to CS (*p<0.05; ***p < 0.01); levels remained higher compared to Ahr+/− mice. F: TIMP-1- TIMP-1 expression significantly increased only in CS-exposed Ahr−/− mice (**p<0.01; ***p < 0.001). G: MMP9: TIMP-1- The ratio of MMP9:TIMP-1 is significantly higher in the air-exposed Ahr−/− mice relative to the air-exposed Ahr+/− mice. Results are expressed as mean ± SEM.
Lastly, a protease/antiprotease imbalance within the lungs is also a mechanism thought to underlie the pathogenesis of emphysema, with an increased matrix metalloproteinase 9 (MMP9) to tissue inhibitor of metalloproteinase 1 (TIMP-1) ratio commonly observed in patients with emphysema (2). Therefore, MMP9 and TIMP-1 protein expression were assessed by western blot in whole lung homogenate from Ahr−/− and Ahr+/− mice. MMP9 expression was significantly elevated in the lungs of both the air and smoke-exposed Ahr−/− mice relative to the Ahr+/− mice (Figure 2D- and 2E) while TIMP-1 expression was only elevated in the smoke-exposed Ahr−/− mice (Figure 2D and 2F). This resulted in a significantly higher MMP9:TIMP1 ratio in the lungs of the air-exposed Ahr−/− mice relative to the Ahr+/− mice (Figure 2G). Collectively, these preclinical data support that the AHR controls multiple pathogenic mechanisms associated with the development of a COPD-like phenotype, pointing towards its role in the homeostatic regulation of lung health in response to cigarette smoke.
3.3. Correlation of lung function with expression of AHR and ARNT in COPD subjects
There is a reduction in AHR expression in several human diseases that have an underlying inflammatory etiology (13, 14). This, combined with our experimental data showing that absence of the AHR results in heightened cigarette smoke-induced inflammation and development of a COPD-like phenotype, led us to wonder if AHR expression was altered in people with COPD.
To determine this, we analyzed AHR expression using biological samples obtained from COPD and control subjects from four separate cohorts (Tables 1–3) and as in (27). In our first cohort (Table 1), analysis revealed that there was significantly less AHR mRNA in the lungs of COPD subjects compared to subjects without COPD (i.e. smokers) (Figure 3A). In the second cohort (Table 2), we found that there appeared to be less intense staining with an anti-AHR antibody in COPD-derived lung specimens as determined by multiplex IHC compared to the relatively uniform staining seen in lung samples from Non-smokers as well as smokers without COPD (Figure 3B–3C). The reduction in staining intensity was particularly noticeable in the bronchial epithelium of COPD subjects compared to At Risk smokers (Figure 3C). This suggests that AHR protein expression may be reduced t in the lungs of those with COPD relative to both non-smokers and smokers without COPD (“At Risk”).
FIGURE 3.
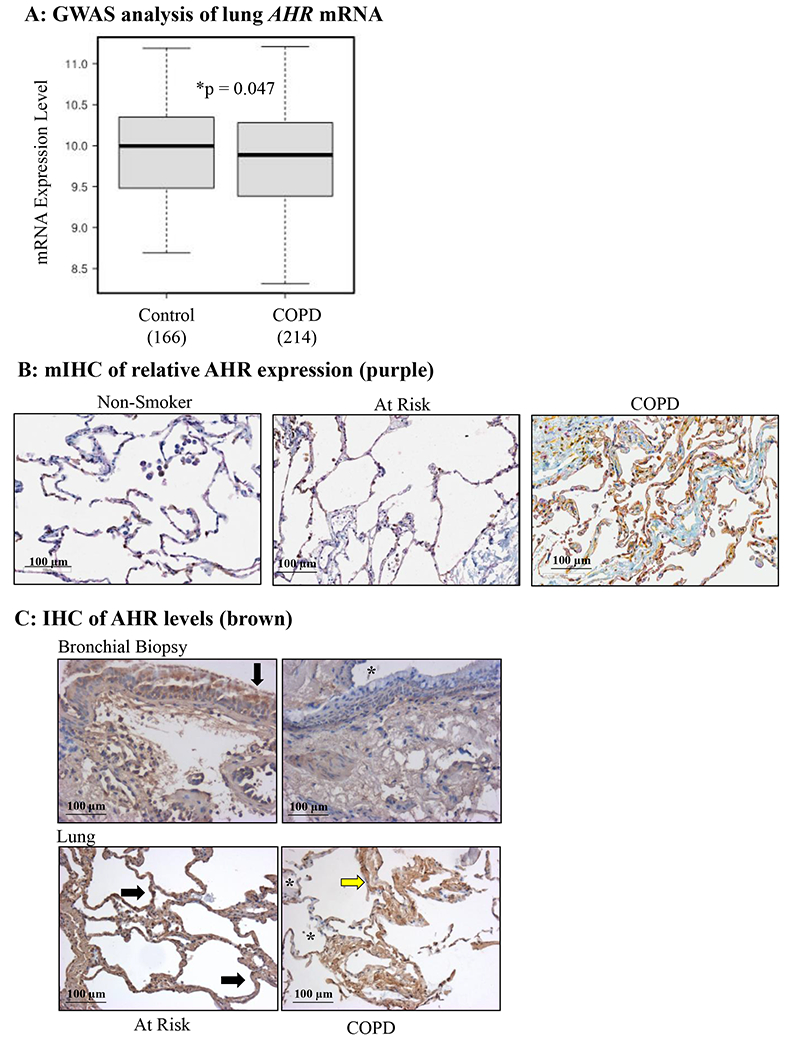
AHR expression is reduced in lung tissue from human COPD subjects. A: There was significantly less AHR mRNA in the lungs of COPD subjects (n=214) compared to subjects without COPD (Control) (n=166). B: mIHC was used to detect AHR (purple), vimentin (fibroblasts; yellow) and cytokeratin-19 (epithelial cells; brown) in human lungs. Note the relative decrease in AHR (purple) in COPD subjects relative to Non-smokers and those At Risk (smokers without COPD); representative images are shown. C: Bronchial biopsy (top panels) and lung tissue (bottom panels)- there is less intense AHR expression (brown) in lungs tissue from COPD subjects compared to those considered At Risk (smokers); regions of noticeably less AHR are denoted by an asterisk (*). Representative images are shown.
We also analyzed scRNA-seq data from control and COPD subjects using data available from (27). After preprocessing, we obtained 165,755 cells in total, of which 58,604 were COPD cells (35.45%); the remainders were Control cells. Based on the processed and normalized expression matrix, we generated a dot plot (Figure 4A), a matrix plot (Figure 4B) and a bar plot (Figure 4C) to compare the difference between AHR in COPD and Control cells. We found that AHR expression level is significantly lower (one-sided Mann-Whitney U-test; P-value= 1.38e-12) in COPD cells compared with Control cells. We also annotated the cell type for all cells we obtained using the same strategy described as in the original study (27), and examined the percentage difference of AHR positive cells between COPD and Control cells for each cell type. We found that the largest positive difference is observed in alveolar type II epithelial (ATII) cells (more AHR positive cells in COPD vs. Control), while the most negative difference (fewer AHR positive cells in COPD vs. Control) is in mast cells and to a lesser extent myofibroblasts (Figure 4D).
FIGURE 4.
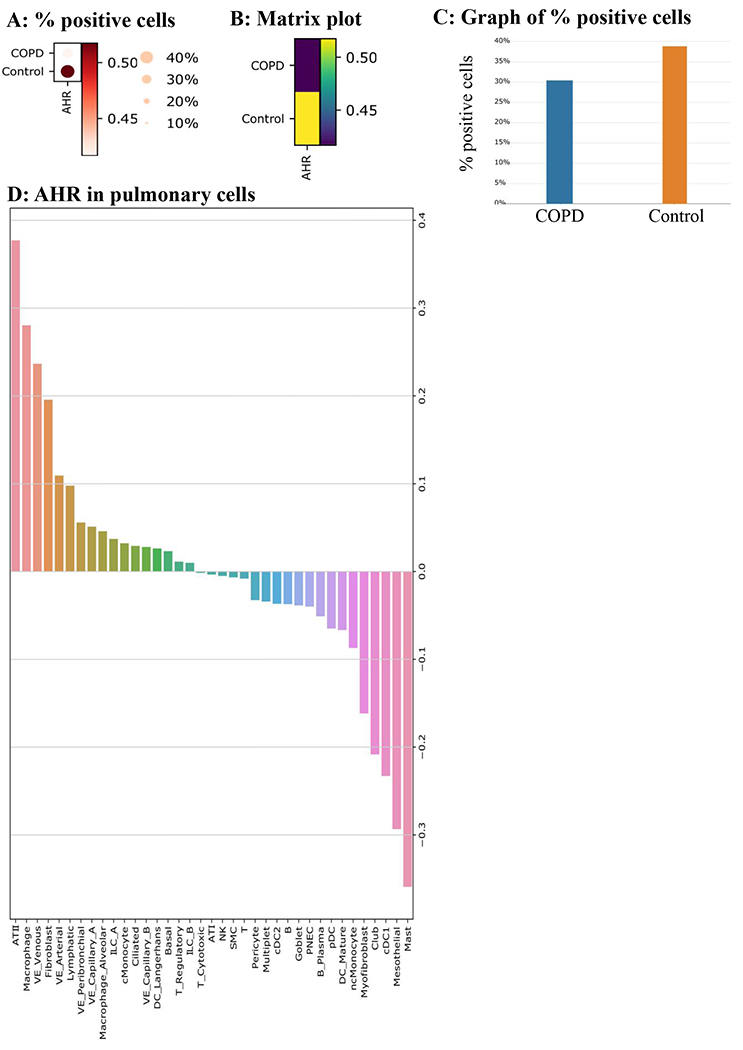
AHR expression is significantly lower in COPD cells compared to Control cells. A: % positive cells- AHR is lower in COPD, as indicated in a dot plot. The size of the dot represents the % of cells with positive AHR expression. The grade of the dot color represents the AHR expression level. B: Matrix Plot- A matrix plot for AHR in COPD vs. Control. C: Graph of % positive cells- COPD has 30.5% AHR positive cells, while 38.9% of Control cells are AHR positive. D. Percent difference of AHR positive cells between COPD and Control for each pulmonary cell type. The most considerable positive difference is observed in ATII cell type (many more AHR positive cells in COPD vs. Control), while the largest negative difference (much less AHR positive cells in COPD vs. Control) is found in mast cells.
Finally, in subjects participating in CanCOLD (20) (Table 3), we found that there was significantly less systemic AHR mRNA in subjects with COPD relative to At Risk subjects (smokers; Figure 5A) as well as in COPD-Never Smokers (no history of smoking) (Figure 5B). Subjects with an emphysema score of 1 or more as assessed by computed tomography (CT) scan also have significantly less AHR mRNA relative to the At Risk subjects (Figure 5C). Lastly, systemic AHR mRNA levels were positively correlated with the lung function parameter FEV1/FVC (%) (Figure 5D).
FIGURE 5.
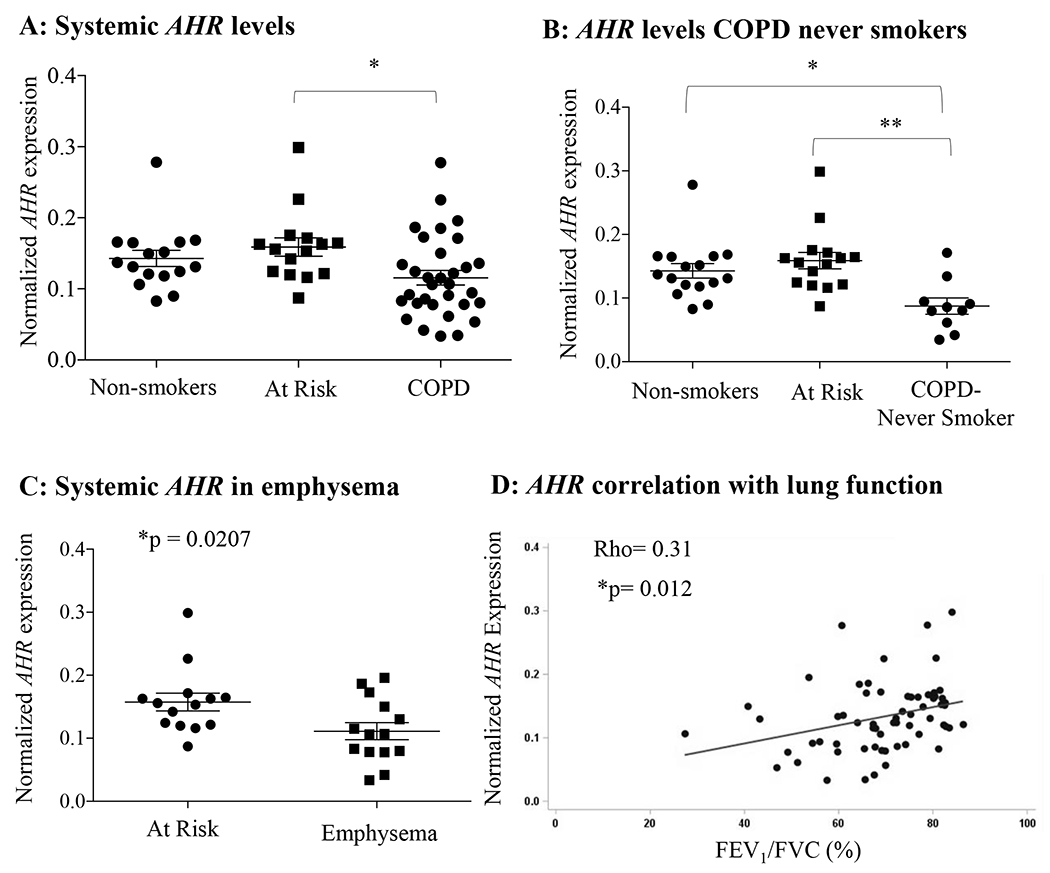
AHR expression is significantly reduced in the blood of human COPD subjects from the CanCOLD cohort. A: There was significantly less systemic AHR mRNA in COPD subjects relative to the “At Risk” subjects (*p< 0.05). B: AHR mRNA expression was significantly less in COPD subjects with no history of smoking (COPD-Never smokers) relative to both Non-smokers and At Risk control subjects (*p < 0.05, **p < 0.01). Results are expressed as mean ± SEM. C: AHR mRNA expression was significantly less in COPD subjects that had also been diagnosed with emphysema via CT (Emphysema) relative to At Risk control subjects. Results are expressed as mean ± SEM. D: There was a significant positive correlation between systemic AHR mRNA expression and lung function (FEV1/FVC) as assessed using a Pearson’s correlation coefficient.
In addition, ARNT expression was positively associated with FEV1 after adjusting for age, gender and smoking status (beta =0.0056, P=0.0005). In human airway epithelium, ARNT expression was also positively associated with FEV1 (beta=0.002, P= 0.00001). In lung tissue, ARNT is under genetic control with the top eQTL SNP rs72237606 (eQTL P= 7.38E-59). This same SNP was also positively associated with FEV1 in the UK Biobank GWAS (P=1.24E-05), suggesting a correlation between the expression of ARNT in lung tissue and lung function. Overall, these data show that there is dysregulation of AHR and ARNT expression in COPD and that there is a positive correlation with their expression and lung function.
4. DISCUSSION
COPD is characterized by abnormal airway inflammation and irreversible airflow obstruction. While the main treatment option for targeting inflammation is glucocorticoids, these medications have little effect on the inflammatory response caused by cigarette smoke (33, 34). Other approaches to reduce inflammation have included targeting specific mediators involved in the chemotaxis and accumulation of neutrophils (e.g. CXCL8, TNF-α) or downstream signal transduction pathways (e.g. p38) known to be activated by cigarette smoke, but these have either had little clinical benefit or have been associated with significant side-effects (35, 36). Thus, the development of safe and effective therapies with disease-modifying effects for smokers with and without obstructive lung disease remains a priority. Cigarette smoke is a complex mixture that activates numerous biological pathways in the lung and other organs to ultimately contribute to the pathophysiology of COPD. However, we speculated that the AHR is an essential protein in the homeostatic control in the lung, capable of mitigating the damaging effects of tobacco smoke. Herein, we show that the AHR protects against emphysematous lung destruction and preserves lung function of mice in response to chronic smoke exposure. We also show that the AHR controls other key pathogenic mechanisms involved in COPD development, including antioxidant levels and MMP9 expression. Thus, defining the AHR as an important protein that integrates multiple protective mechanisms in the lung may enable the development of more effective approaches to treat complex diseases such as COPD.
Our data support that the AHR protects against the development of the emphysema component of COPD. The fact that Ahr−/− mice have extensive lung parenchymal destruction after only a 4-month smoke-exposure is striking, as the Ahr−/− mice are on the C57BL/6 background, a strain that requires ~6 months of cigarette smoke exposure to develop the features of emphysema (37). Airspace enlargement was not evident in air-only Ahr−/− mice, suggesting that the emphysematous changes in response to cigarette smoke are not attributable to impaired lung development or a congenital defect due to absence of the AHR. However, our observation of a significantly elevated MMP9:TIMP1 ratio in the air-only Ahr−/− mice suggests that this protease:antiprotease imbalance may predispose these mice to the development of emphysema upon exposure to subsequent pulmonary insults, such as cigarette smoke. In addition to the lung structural damage, smoke-exposed Ahr−/− mice also exhibited emphysema-like changes in lung function, including a significant increase in the FRC and a significant decrease in the IC/TLC ratio. This is in accordance with lung function in humans with emphysema, where lung parenchymal destruction results in an outward recoil of the lung, leading to hyperinflation, air trapping and an increased FRC (29). Moreover, a reduced IC/TLC ratio is associated with worse clinical outcomes in COPD patients as well as with more severe disease, increased exacerbations and increased mortality. Overall, our data suggest that the AHR globally protects the lungs from the deleterious effects of smoking.
It could be that AHR dysregulation (altered function and/or reduced expression) may predispose a susceptible individual to the development of emphysema and consequent deterioration in lung function. In line with this notion is our findings that lung and systemic AHR expression is reduced in COPD subjects-even those that are never-smokers- and that low AHR levels correlate with worse lung function. Why there is a correlation between systemic AHR levels and lung function is not clear but could reflect an overall reduction in AHR due to genetic factors. Given the now-recognized homeostatic role for the AHR in inflammation, cell proliferation, differentiation and survival, including our own data, this reduction in AHR may therefore alter lung function/disease through perturbation of multiple mechanisms. In these analyses, it was important to distinguish if the reduced AHR expression in COPD was a consequence of smoking, given that reduced pulmonary AHR protein expression occurs from smoke exposure both in vitro and in vivo (15, 38) due to AHR degradation by the proteasome. In assessment of systemic and pulmonary AHR expression, our data show that AHR expression was reduced in COPD subjects relative to subjects without COPD who were current smokers. We also observed that there was significantly less systemic AHR in COPD never-smokers. Numerous studies now show that never-smokers comprise a substantial proportion of people with COPD (39–41), with Tan et al showing a COPD prevalence in never smokers of 6.4% in the general population (40). COPD_ in never-smokers could be due to age, exposure to other environmental toxicants or existing conditions (e.g. asthma). Our data that there is reduced AHR in individuals with clinically-defined COPD further supports that AHR is important in preventing a COPD-disease phenotype that is independent of smoking. Finally, AHR levels were also decreased at the mRNA level. This is an important distinction because ligand-induced proteasomal degradation of the AHR protein has little effect on AHR gene expression. The sum of these data support that the reduced AHR protein expression observed in COPD is not a consequence of smoking, but rather a reflection of the disease state. Although the focus of this manuscript was on AHR and COPD, airflow obstruction in non-smokers can occur from other conditions including asthma, bronchiectasis and obliterative bronchiolitis. It is therefore interesting to speculate that the AHR may play a more global role in preserving lung function outside COPD.
One of the strengths of this study is the consistency in which we observed a decrease in AHR expression among the different cohorts of subjects. Although the cohorts differed substantially in the patient population, biological samples collected (lung versus blood), number of participants, and clinical information available, our collective data show that in people diagnosed with COPD according to current GOLD guidelines, AHR expression is significantly reduced. This suggests an important role for the AHR in the pathogenesis of COPD, which we were able to demonstrate using Ahr-deficient mice. However, this study is not without its limitations, including the cross-sectional nature in which these data were obtained. It is also possible that additional variables in these cohorts contribute to alterations in AHR levels. For example, the presence of comorbidities may also impact AHR, as reduced expression has been observed in other chronic disease (13). Our results may also be influenced by the presence of other lung conditions such as asthma, bronchiectasis and/or bronchiolitis. It is possible therefore that reduced AHR expression is more broadly applicable to lung diseases associated with inflammation and obstruction; further studies would be needed address its expression in this context. Finally, it is possible that medication use by the subjects in these cohorts is impacting AHR expression. Notable to this is downregulation of the AHR protein in response to dexamethasone in vitro in non-pulmonary cells (42, 43). The extent to which glucocorticoids (or other medications) is impacting AHR levels in COPD remains to be determined. Nonetheless, in spite of these limitations, the totality of our data from different cohorts of subjects strongly support that there is reduced AHR in COPD.
Cigarette smoke is also a mixture of gases and particulate matter composed of metals (iron, nickel), gases, biological agents (endotoxins) and organic chemicals such as polychlorinated dibenzodioxins ([PCDDs] and polycyclic aromatic hydrocarbons [PAHs]) (44–47). Many of these organic chemicals are ligands of the AHR (48, 49). Perhaps unsurprisingly, there is considerable overlap in cellular signaling pathways that are modulated by both cigarette smoke and AHR; these include nuclear factor-κB (NF-κB), human antigen R (HuR) and nuclear factor erythroid 2-related factor 2 (NRF2). The NF-κB family is comprised of five members: RelA (p65), RelB, c-Rel, NF-κB1 (p50) and NF-κB2 (p52) (9). Cigarette smoke potently activates the canonical NF-κB pathway (50), whereby RelA interacts with p50, which are sequestered in the cytoplasm via the inhibitory protein TκBα. Phosphorylation of IκBα leads to its degradation via the proteasome, allowing RelA and p50 heterodimers to translocate to the nucleus for subsequent activation of target genes involved in inflammation and cell survival, among others. However, the AHR can physically interact with RelB (51), a component of the alternative NF-κB pathway that exerts anti-inflammatory and anti-apoptotic control. Although AhR-RelB dimers can initiate transcription of target genes via binding to a unique and DRE-independent response element termed the RelBAHRE (51), we have shown that AHR-mediated suppression of smoke-induced neutrophilia in the murine lung is associated with the nuclear retention of RelB (15). These findings raise the possibility that the AHR-mediated nuclear retention of RelB may represent one way through which the AHR can suppress acute CS-induced inflammation. Another example of an AHR-dependent protein is the regulation of HuR, an RNA-binding protein that functions to stabilize target mRNA when localized to the cytoplasm. The AHR attenuates smoke-induced cyclooxygenase-2 (COX-2) expression via the nuclear sequestration of HuR, resulting in the destabilization and degradation of Cox-2 mRNA in vitro (52). Moreover, another established target gene of HuR is Mmp9 (53). Thus, it is not unreasonable to postulate that similar to COX-2, perhaps the elevated protease expression (i.e. increased MMP9:TIMP1 ratio) we observed in the lungs of the Ahr−/− mice is a consequence of the cytoplasmic shuttling of the HuR, resulting in enhanced Mmp9 mRNA stability and expression. It should also be considered whether the AHR may be interacting with NRF2, a master regulator of antioxidants. This consideration is relevant based on our finding that the upregulation of SOD2 is impaired in smoke-exposed Ahr−/− mice (12). Although the AHR and NRF2 regulate several common antioxidant genes such as Nqo1, we speculate that the AhR regulation of antioxidant defenses in the murine lung is likely independent of NRF2. This assertion is supported by previous reports demonstrating no difference in either the expression or nuclear localization of NRF2 between AHR-deficient and expressing lung structural cells (8). However, it may be that dampened upregulation of the mitochondrial antioxidant SOD2 in the lungs of the smoke-exposed Ahr−/− mice is an indirect result of mitochondrial dysfunction that ensues as a consequence of AHR ablation. In addition to interaction with numerous cellular proteins, the AHR may control the pathogenesis of COPD is via regulation of noncoding (nc) RNA. One type of ncRNA is miRNA, which target mRNA for degradation. Changes in miRNA levels in response to cigarette smoke exposure is well-noted in vitro as well as in vivo and in individuals with COPD (54–57). We have shown that the AHR controls the expression of a number of these miRNAs, including miR-196a, miR-96, miR-137 and miR-133b (48). In line with this, the attenuation of smoke-induced apoptosis is mediated by the AHR-dependent regulation of miR-196a (7) and control over inflammation is related to expression of miR-96 (48). While it remains to be established how the AHR controls diverse pathogenic mechanisms such as inflammation, oxidative stress and cell death, it seems plausible that the AhR exerts its protective effects via non-canonical interactions with other cellular proteins and/or control over epigenetic pathways implicated in disease development.
Since its discovery as the receptor responsible for the induction of CYP1A1 expression in response to dioxin (58), the endogenous role of the AHR has remained enigmatic. The relatively high expression of the AHR in the human lung, coupled with the identification of endogenous AHR ligands, raises the possibility that the AhR plays important physiological roles independent of its response to dioxin. Within the lung- an organ continuously exposed to the external environment- we establish herein that the AHR is a critical component of the immune response to cigarette smoke. Not only do we report that lung and systemic AHR expression is reduced in COPD subjects, and that low AHR levels correlate with worse lung function but we also establish that low AHR expression is causally implicated in COPD pathogenesis by demonstrating that absence of the AHR drives the development of smoke-induced emphysema. Collectively our data position the AHR as a central player in the homeostatic maintenance of lung health(59). These findings lay the foundation for the eventual use of the AHR as a novel therapeutic target or biomarker in identifying those individuals susceptible to the development of COPD.
ACKNOWLEDGEMENTS
This work was supported by the Canada Foundation for Innovation (CFI), Natural Sciences and Engineering Research Council of Canada (NSERC), and the Canadian Institutes for Health Research (CIHR). Y.B. holds a Canada Research Chair in Genomics of Heart and Lung Diseases. B.M.S. and C.J.B. were supported by a salary award from the Fonds de recherche du Quebec-Sante (FRQ-S). This work was supported in part by National Institutes of Health grants HL075432 and HL120908 to R.P.P. and P.J.S. Grant support for R.P.P. and P.J.S. was also provided by P30ES001247, RO1HL120908 and T32HL066988. A.R. is employed by SCIREQ Inc., a commercial entity involved in a subject area related to the content of this article. SCIREQ is an emka Technologies company. P.N. is supported by a Frederick E. Hargreave Teva Innovation Chair in Airway Diseases. MO is a Scholar with the Michael Smith Foundation for Health Research and is a fellow of the Parker B Francis Foundation. The authors would like to thank the staff at the Respiratory Health Network Tissue Bank of the FRQS for their valuable assistance with the lung eQTL dataset at Laval University. We also acknowledge the assistance of Katherine Radford and the Division of Thoracic Surgeon and the Department of Pathology of McMaster University and St Joseph’s Healthcare Hamilton. The mIHC images were acquired through the Histopathology Platform of the RI-MUHC.
Abbreviations
- AhR
Aryl hydrocarbon receptor
- AhRR
Aryl hydrocarbon receptor repressor
- ANOVA
Analysis of variance
- ARNT
AhR nuclear translocator
- ATII
alveolar type II epithelial
- BAL
Bronchoalveolar lavage
- CanCOLD
Canadian chronic obstructive lung disease
- COPD
Chronic obstructive pulmonary disease
- CPD
Cumulative particulate density
- CS
Cigarette smoke
- CYP
Cytochrome P450
- DAB
3,3’- diaminobenzidine
- ddPCR
Digital droplet polymerase chain reaction
- DRE
Dioxin response element
- ECL
Enhanced chemiluminescence
- eQTL
Expression quantitative trait loci
- FBGC
Foreign Body Giant Cell
- FDR
False Discovery Rate
- FEV1
Forced expiratory volume in 1 second
- FRC
Functional residual capacity
- FVC
Forced vital capacity
- GWAS
Genome-wide association study
- H&E
Hematoxylin-Eosin
- HO-1
Heme oxygenase 1
- HRP
Horse radish peroxidase
- IBD
Inflammatory Bowel Disease
- IC/TLC
Inspiratory capacity/ Total lung capacity
- Lm
Mean linear intercept
- mIHC
Multiplex immunohistochemistry
- MMP
Matrix metalloproteinase
- MNGC
Multinucleated giant cell
- PV
Pressure-volume
- PVDF
Polyvinylidene difluoride
- RIPA
Radio-immunoprecipitation assay
- SDS-PAGE
Sodium dodecyl sulfate polyacrylamide gel electrophoresi
- SNP
Single nucleotide polymorphism
- SOD-2
Superoxide dismutase-2
- TIMP
Tissue inhibitor of metalloproteinase
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
REFERENCES
- 1.Adeloye D, Chua S, Lee C, Basquill C, Papana A, Theodoratou E, Nair H, Gasevic D, Sridhar D, Campbell H, Chan KY, Sheikh A, Rudan I, and Global Health Epidemiology Reference, G. (2015) Global and regional estimates of COPD prevalence: Systematic review and meta-analysis. J Glob Health 5, 020415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharafkhaneh A, Hanania NA, and Kim V (2008) Pathogenesis of emphysema: from the bench to the bedside. Proc Am Thorac Soc 5, 475–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Busch R, Hobbs BD, Zhou J, Castaldi PJ, McGeachie MJ, Hardin ME, Hawrylkiewicz I, Sliwinski P, Yim JJ, Kim WJ, Kim DK, Agusti A, Make BJ, Crapo JD, Calverley PM, Donner CF, Lomas DA, Wouters EF, Vestbo J, Tal-Singer R, Bakke P, Gulsvik A, Litonjua AA, Sparrow D, Pare PD, Levy RD, Rennard SI, Beaty TH, Hokanson J, Silverman EK, Cho MH, National Emphysema Treatment Trial, G., Evaluation of, C. L. t. I. P. S. E.-P., International, C. G. N., and Investigators, C. O. (2017) Genetic Association and Risk Scores in a Chronic Obstructive Pulmonary Disease Meta-analysis of 16,707 Subjects. Am J Respir Cell Mol Biol 57, 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hancock DB, Eijgelsheim M, Wilk JB, Gharib SA, Loehr LR, Marciante KD, Franceschini N, van Durme YM, Chen TH, Barr RG, Schabath MB, Couper DJ, Brusselle GG, Psaty BM, van Duijn CM, Rotter JI, Uitterlinden AG, Hofman A, Punjabi NM, Rivadeneira F, Morrison AC, Enright PL, North KE, Heckbert SR, Lumley T, Stricker BH, O’Connor GT, and London SJ (2010) Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet 42, 45–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Evans BR, Karchner SI, Allan LL, Pollenz RS, Tanguay RL, Jenny MJ, Sherr DH, and Hahn ME (2008) Repression of aryl hydrocarbon receptor (AHR) signaling by AHR repressor: role of DNA binding and competition for AHR nuclear translocator. Mol Pharmacol 73, 387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davarinos NA, and Pollenz RS (1999) Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. J Biol Chem 274, 28708–28715 [DOI] [PubMed] [Google Scholar]
- 7.Hecht E, Zago M, Sarill M, Rico de Souza A, Gomez A, Matthews J, Hamid Q, Eidelman DH, and Baglole CJ (2014) Aryl hydrocarbon receptor-dependent regulation of miR-196a expression controls lung fibroblast apoptosis but not proliferation. Toxicol Appl Pharmacol 280, 511–525 [DOI] [PubMed] [Google Scholar]
- 8.Rico de Souza A, Zago M, Pollock SJ, Sime PJ, Phipps RP, and Baglole CJ (2011) Genetic Ablation of the Aryl Hydrocarbon Receptor Causes Cigarette Smoke-induced Mitochondrial Dysfunction and Apoptosis. J Biol Chem 286, 43214–43228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang X, Fan Y, Karyala S, Schwemberger S, Tomlinson CR, Sartor MA, and Puga A (2007) Ligand-independent regulation of transforming growth factor beta1 expression and cell cycle progression by the aryl hydrocarbon receptor. Mol Cell Biol 27, 6127–6139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu R, Zhang L, Hoagland MS, and Swanson HI (2007) Lack of the aryl hydrocarbon receptor leads to impaired activation of AKT/protein kinase B and enhanced sensitivity to apoptosis induced via the intrinsic pathway. J Pharmacol Exp Ther 320, 448–457 [DOI] [PubMed] [Google Scholar]
- 11.Thatcher TH, Maggirwar SB, Baglole CJ, Lakatos HF, Gasiewicz TA, Phipps RP, and Sime PJ (2007) Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB. Am J Pathol 170, 855–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarill M, Zago M, Sheridan JA, Nair P, Matthews J, Gomez A, Roussel L, Rousseau S, Hamid Q, Eidelman DH, and Baglole CJ (2015) The aryl hydrocarbon receptor suppresses cigarette-smoke-induced oxidative stress in association with dioxin response element (DRE)-independent regulation of sulfiredoxin 1. Free Radic Biol Med 89, 342–357 [DOI] [PubMed] [Google Scholar]
- 13.Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, Macdonald TT, Pallone F, and Monteleone G (2011) Aryl Hydrocarbon Receptor-Induced Signals Up-regulate IL-22 Production and Inhibit Inflammation in the Gastrointestinal Tract. Gastroenterology 141, 237–248 e231 [DOI] [PubMed] [Google Scholar]
- 14.Wang C, Ye Z, Kijlstra A, Zhou Y, and Yang P (2014) Decreased expression of the aryl hydrocarbon receptor in ocular Behcet’s disease. Mediators Inflamm 2014, 195094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Souza AR, Zago M, Eidelman DH, Hamid Q, and Baglole CJ (2014) Aryl hydrocarbon receptor (AhR) attenuation of subchronic cigarette smoke-induced pulmonary neutrophilia is associated with retention of nuclear RelB and suppression of intercellular adhesion molecule-1 (ICAM-1). Toxicol Sci 140, 204–223 [DOI] [PubMed] [Google Scholar]
- 16.Baglole CJ, Maggirwar SB, Gasiewicz TA, Thatcher TH, Phipps RP, and Sime PJ (2008) The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-kappaB family member RelB. J Biol Chem 283, 28944–28957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baglole CJ, Liang F, Traboulsi H, Rico de Souza A, Giordano C, Tauer JT, Rauch F, and Petrof BJ (2018) Pulmonary and diaphragmatic pathology in collagen type I alpha1 mutant mice with osteogenesis imperfecta. Pediatr Res 83, 1165–1171 [DOI] [PubMed] [Google Scholar]
- 18.Robichaud A, Fereydoonzad L, Limjunyawong N, Rabold R, Allard B, Benedetti A, Martin JG, and Mitzner W (2017) Automated Full-Range Pressure-Volume Curves in Mice and Rats. J Appl Physiol (1985), jap 00856 02016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, and Speed TP (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 [DOI] [PubMed] [Google Scholar]
- 20.Bourbeau J, Tan WC, Benedetti A, Aaron SD, Chapman KR, Coxson HO, Cowie R, Fitzgerald M, Goldstein R, Hernandez P, Leipsic J, Maltais F, Marciniuk D, O’Donnell D, Sin DD, and Cancold Study G (2014) Canadian Cohort Obstructive Lung Disease (CanCOLD): Fulfilling the need for longitudinal observational studies in COPD. COPD 11, 125–132 [DOI] [PubMed] [Google Scholar]
- 21.Sheridan JA, Zago M, Nair P, Li PZ, Bourbeau J, Tan WC, Hamid Q, Eidelman DH, Benedetti AL, and Baglole CJ (2015) Decreased expression of the NF-kappaB family member RelB in lung fibroblasts from Smokers with and without COPD potentiates cigarette smoke-induced COX-2 expression. Respir Res 16, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor SC, Carbonneau I, Shelton DN, and Boivin G (2015) Optimization of Droplet Digital PCR from RNA and DNA extracts with direct comparison to RT-qPCR: Clinical implications for quantification of Oseltamivir-resistant subpopulations. J Virol Methods 224, 58–66 [DOI] [PubMed] [Google Scholar]
- 23.Steiling K, van den Berge M, Hijazi K, Florido R, Campbell J, Liu G, Xiao J, Zhang X, Duclos G, Drizik E, Si EL, Perdomo C, Dumont C, Coxson HO, Alekseyev YO, Sin D, Pare P, Hogg JC, McWilliams A, Hiemstra PS, Sterk PJ, Timens W, Chang JT, Sebastiani P, O’Connor GT, Bild AH, Postma DS, Lam S, Spira A, and Lenburg ME (2013) A Dynamic Bronchial Airway Gene Expression Signature of Chronic Obstructive Pulmonary Disease and Lung Function Impairment. American Journal of Respiratory and Critical Care Medicine 187, 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tammemagi MC, Lam SC, McWilliams AM, and Sin DD (2011) Incremental Value of Pulmonary Function and Sputum DNA Image Cytometry in Lung Cancer Risk Prediction. Cancer Prevention Research 4, 552–561 [DOI] [PubMed] [Google Scholar]
- 25.McWilliams A, Tammemagi MC, Mayo JR, Roberts H, Liu G, Soghrati K, Yasufuku K, Martel S, Laberge F, Gingras M, Atkar-Khattra S, Berg CD, Evans K, Finley R, Yee J, English J, Nasute P, Goffin J, Puksa S, Stewart L, Tsai S, Johnston MR, Manos D, Nicholas G, Goss GD, Seely JM, Amjadi K, Tremblay A, Burrowes P, MacEachem P, Bhatia R, Tsao M-S, and Lam S (2013) Probability of Cancer in Pulmonary Nodules Detected on First Screening CT. New England Journal of Medicine 369, 910–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wain LV, Shrine N, Artigas MS, Erzurumluoglu AM, Noyvert B, Bossini-Castillo L, Obeidat M, Henry AP, Portelli MA, Hall RJ, Billington CK, Rimington TL, Fenech AG, John C, Blake T, Jackson VE, Allen RJ, Prins BP, Campbell A, Porteous DJ, Jarvelin MR, Wielscher M, James AL, Hui J, Wareham NJ, Zhao JH, Wilson JF, Joshi PK, Stubbe B, Rawal R, Schulz H, Imboden M, Probst-Hensch NM, Karrasch S, Gieger C, Deary IJ, Harris SE, Marten J, Rudan I, Enroth S, Gyllensten U, Kerr SM, Polasek O, Kahonen M, Surakka I, Vitart V, Hayward C, Lehtimaki T, Raitakari OT, Evans DM, Henderson AJ, Pennell CE, Wang CA, Sly PD, Wan ES, Busch R, Hobbs BD, Litonjua AA, Sparrow DW, Gulsvik A, Bakke PS, Crapo JD, Beaty TH, Hansel NN, Mathias RA, Ruczinski I, Barnes KC, Bosse Y, Joubert P, van den Berge M, Brandsma CA, Pare PD, Sin DD, Nickle DC, Hao K, Gottesman O, Dewey FE, Bruse SE, Carey DJ, Kirchner HL, Jonsson S, Thorleifsson G, Jonsdottir I, Gislason T, Stefansson K, Schurmann C, Nadkarni G, Bottinger EP, Loos RJ, Walters RG, Chen Z, Millwood IY, Vaucher J, Kurmi OP, Li L, Hansell AL, Brightling C, Zeggini E, Cho MH, Silverman EK, Sayers I, Trynka G, Morris AP, Strachan DP, Hall IP, and Tobin MD (2017) Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat Genet 49, 416–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M, Duan Q, Arnett HA, Siddiqui A, Washko GR, Homer R, Yan X, Rosas IO, and Kaminski N (2020) Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Science Advances 6, eaba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolf FA, Angerer P, and Theis FJ (2018) SCANPY: large-scale single-cell gene expression data analysis. Genome Biology 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Papandrinopoulou D, Tzouda V, and Tsoukalas G (2012) Lung compliance and chronic obstructive pulmonary disease. Pulm Med 2012, 542769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boschetto P, Quintavalle S, Zeni E, Leprotti S, Potena A, Ballerin L, Papi A, Palladini G, Luisetti M, Annovazzi L, Iadarola P, De Rosa E, Fabbri LM, and Mapp CE (2006) Association between markers of emphysema and more severe chronic obstructive pulmonary disease. Thorax 61, 1037–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, Ifedigbo E, Parameswaran H, Ryter SW, and Choi AM (2010) Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A 107, 18880–18885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Demedts IK, Demoor T, Bracke KR, Joos GF, and Brusselle GG (2006) Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res 7, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Angelis N, Porpodis K, Zarogoulidis P, Spyratos D, Kioumis I, Papaiwannou A, Pitsiou G, Tsakiridis K, Mpakas A, Arikas S, Tsiouda T, Katsikogiannis N, Kougioumtzi I, Machairiotis N, Argyriou M, Kessisis G, and Zarogoulidis K (2014) Airway inflammation in chronic obstructive pulmonary disease. J Thorac Dis 6 Suppl 1, S167–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keatings VM, Jatakanon A, Worsdell YM, and Barnes PJ (1997) Effects of inhaled and oral glucocorticoids on inflammatory indices in asthma and COPD. Am J Respir Crit Care Med 155, 542–548 [DOI] [PubMed] [Google Scholar]
- 35.van der Vaart H, Koeter GH, Postma DS, Kauffman HF, and ten Hacken NH (2005) First study of infliximab treatment in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 172, 465–469 [DOI] [PubMed] [Google Scholar]
- 36.Barnes PJ, and Stockley RA (2005) COPD: current therapeutic interventions and future approaches. Eur Respir J 25, 1084–1106 [DOI] [PubMed] [Google Scholar]
- 37.Guerassimov A, Hoshino Y, Takubo Y, Turcotte A, Yamamoto M, Ghezzo H, Triantafillopoulos A, Whittaker K, Hoidal JR, and Cosio MG (2004) The development of emphysema in cigarette smoke-exposed mice is strain dependent. Am J Respir Crit Care Med 170, 974–980 [DOI] [PubMed] [Google Scholar]
- 38.Martey CA, Baglole CJ, Gasiewicz TA, Sime PJ, and Phipps RP (2005) The aryl hydrocarbon receptor is a regulator of cigarette smoke induction of the cyclooxygenase and prostaglandin pathways in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 289, L391–399 [DOI] [PubMed] [Google Scholar]
- 39.Lamprecht B, McBurnie MA, Vollmer WM, Gudmundsson G, Welte T, Nizankowska-Mogilnicka E, Studnicka M, Bateman E, Anto JM, Burney P, Mannino DM, Buist SA, and Group BCR (2011) COPD in never smokers: results from the population-based burden of obstructive lung disease study. Chest 139, 752–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan WC, Sin DD, Bourbeau J, Hernandez P, Chapman KR, Cowie R, FitzGerald JM, Marciniuk DD, Maltais F, Buist AS, Road J, Hogg JC, Kirby M, Coxson H, Hague C, Leipsic J, O’Donnell DE, Aaron SD, and Can CCRG (2015) Characteristics of COPD in never-smokers and ever-smokers in the general population: results from the CanCOLD study. Thorax 70, 822–829 [DOI] [PubMed] [Google Scholar]
- 41.Nguyen Viet N, Yunus F, Nguyen Thi Phuong A, Dao Bich V, Damayanti T, Wiyono WH, Billot L, Jakes RW, and Kwon N (2015) The prevalence and patient characteristics of chronic obstructive pulmonary disease in non-smokers in Vietnam and Indonesia: An observational survey. Respirology 20, 602–611 [DOI] [PubMed] [Google Scholar]
- 42.Stejskalova L, Rulcova A, Vrzal R, Dvorak Z, and Pavek P (2013) Dexamethasone accelerates degradation of aryl hydrocarbon receptor (AHR) and suppresses CYP1A1 induction in placental JEG-3 cell line. Toxicol Lett 223, 183–191 [DOI] [PubMed] [Google Scholar]
- 43.Vrzal R, Stejskalova L, Monostory K, Maurel P, Bachleda P, Pavek P, and Dvorak Z (2009) Dexamethasone controls aryl hydrocarbon receptor (AhR)-mediated CYP1A1 and CYP1A2 expression and activity in primary cultures of human hepatocytes. Chem Biol Interact 179, 288–296 [DOI] [PubMed] [Google Scholar]
- 44.Valavanidis A, Fiotakis K, and Vlachogianni T (2008) Airborne particulate matter and human health: toxicological assessment and importance of size and composition of particles for oxidative damage and carcinogenic mechanisms. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 26, 339–362 [DOI] [PubMed] [Google Scholar]
- 45.Ghio AJ (2014) Particle exposures and infections. Infection 42, 459–467 [DOI] [PubMed] [Google Scholar]
- 46.Council NR (2010) Review of the Department of Defence Enhanced Particulate Matter Surveillance Program Report. (Defence D. o., ed) p. 85, The National Academies Press, Washington: [PubMed] [Google Scholar]
- 47.Nemmar A, Holme JA, Rosas I, Schwarze PE, and Alfaro-Moreno E (2013) Recent advances in particulate matter and nanoparticle toxicology: a review of the in vivo and in vitro studies. Biomed Res Int 2013, 279371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rogers S, de Souza AR, Zago M, Iu M, Guerrina N, Gomez A, Matthews J, and Baglole CJ (2017) Aryl hydrocarbon receptor (AhR)-dependent regulation of pulmonary miRNA by chronic cigarette smoke exposure. Sci Rep 7, 40539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van den Bogaard EH, Bergboer JG, Vonk-Bergers M, van Vlijmen-Willems IM, Hato SV, van der Valk PG, Schroder JM, Joosten I, Zeeuwen PL, and Schalkwijk J (2013) Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J Clin Invest 123, 917–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martey CA, Pollock SJ, Turner CK, O’Reilly KM, Baglole CJ, Phipps RP, and Sime PJ (2004) Cigarette smoke induces cyclooxygenase-2 and microsomal prostaglandin E2 synthase in human lung fibroblasts: implications for lung inflammation and cancer. Am J Physiol Lung Cell Mol Physiol 287, L981–991 [DOI] [PubMed] [Google Scholar]
- 51.Vogel CF, Sciullo E, Li W, Wong P, Lazennec G, and Matsumura F (2007) RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol Endocrinol 21, 2941–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zago M, Sheridan JA, Nair P, Rico de Souza A, Gallouzi IE, Rousseau S, Di Marco S, Hamid Q, Eidelman DH, and Baglole CJ (2013) Aryl Hydrocarbon Receptor-Dependent Retention of Nuclear HuR Suppresses Cigarette Smoke-Induced Cyclooxygenase-2 Expression Independent of DNA-Binding. PLoS One 8, e74953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Akool el S, Kleinert H, Hamada FM, Abdelwahab MH, Forstermann U, Pfeilschifter J, and Eberhardt W (2003) Nitric oxide increases the decay of matrix metalloproteinase 9 mRNA by inhibiting the expression of mRNA-stabilizing factor HuR. Mol Cell Biol 23, 4901–4916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sheridan JA, Zago M, Nair P, Li PZ, Bourbeau J, Tan WC, Hamid Q, Eidelman DH, Benedetti AL, and Baglole CJ (2015) Decreased expression of the NF-kappaB family member RelB in lung fibroblasts from Smokers with and without COPD potentiates cigarette smoke-induced COX-2 expression. Respir Res In Press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zago M, Rico de Souza A, Hecht E, Rousseau S, Hamid Q, Eidelman DH, and Baglole CJ (2014) The NF-kappaB family member RelB regulates microRNA miR-146a to suppress cigarette smoke-induced COX-2 protein expression in lung fibroblasts. Toxicol Lett 226, 107–116 [DOI] [PubMed] [Google Scholar]
- 56.Izzotti A, Calin GA, Arrigo P, Steele VE, Croce CM, and De Flora S (2009) Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J 23, 806–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pottelberge GR, Mestdagh P, Bracke KR, Thas O, Durme YM, Joos GF, Vandesompele J, and Brusselle GG (2011) MicroRNA expression in induced sputum of smokers and patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 183, 898–906 [DOI] [PubMed] [Google Scholar]
- 58.Poland A, Glover E, and Kende AS (1976) Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J Biol Chem 251, 4936–4946 [PubMed] [Google Scholar]
- 59.Guerrina N, Traboulsi H, Eidelman DH, and Baglole CJ (2018) The Aryl Hydrocarbon Receptor and the Maintenance of Lung Health. Int J Mol Sci 19 [DOI] [PMC free article] [PubMed] [Google Scholar]