

Structured Summary
Background:
Patients with gastroparesis and related disorders have symptoms including early satiety, postprandial fullness, and bloating. Buspirone, a 5-HT1 receptor agonist, may improve fundic accommodation.
Aim:
To determine if buspirone treatment improves early satiety and postprandial fullness in patients with symptoms of gastroparesis.
Methods:
This 4-week multicenter clinical trial randomised patients with symptoms of gastroparesis and moderate to severe symptoms of fullness (Gastroparesis Cardinal Symptom Index (GCSI) early satiety/postprandial fullness subscore (ES/PPF)) to buspirone (10 mg orally) or placebo three times per day. The primary outcome was change in the ES/PPF from baseline to 4-weeks. Primary analysis was per protocol intention-to-treat ANCOVA of between-group baseline vs. 4-week differences (DoD) in ES/PPF adjusted for baseline ES/PPF. Results are reported using both nominal and Bonferroni (BF) P-values.
Results and Conclusions:
96 patients (47 buspirone, 49 placebo; 92% female, 50% delayed gastric emptying, 39% diabetic) were enrolled. There was no between-groups difference in 4-week ES/PPF primary outcome: −1.16±1.25(SD) on buspirone vs. −1.03±1.29(SD) on placebo (mean DoD: −0.11 [95%CI:−0.68, 0.45]; P=0.69). Buspirone performed better than placebo in patients with severe to very severe bloating at baseline compared to patients with none to moderate: (ES/PPF DoD=−0.65 vs 1.58, PTX*GROUP=0.003;PBF=0.07). Among individual GCSI symptoms, only bloating appeared to improve with buspirone vs. placebo.
Conclusions:
Patients with moderate to severe early satiety/postprandial fullness and other symptoms of gastroparesis did not benefit from buspirone treatment to improve the ES/PPF primary outcome compared with placebo. There was a suggestion of benefit with buspirone in patients with more severe bloating.
Trial registration.
Keywords: gastroparesis, idiopathic gastroparesis, gastric emptying, early satiety, buspirone
Introduction
Patients with gastroparesis have symptoms of early satiety, postprandial fullness, bloating, nausea, vomiting, and/or abdominal pain (1,2). Symptoms of gastroparesis are not well correlated with gastric emptying results (3). Some patients have symptoms similar to gastroparesis but normal gastric emptying, a group that corresponds closely to the Rome III definition of functional dyspepsia - postprandial distress subtype (FD-PDS) (4).
With food ingestion, the proximal stomach normally relaxes, with an increase in volume, allowing it to accommodate the meal without a major increase in intragastric pressure (5,6). This is followed by progressive tonic contraction to deliver food into the distal stomach. With impaired accommodation, there is increased pressure in the upper stomach which compromises the ability of the upper stomach to act as a reservoir for ingested food, resulting in rapid transit of food into the distal stomach (7). The impaired accommodation with increase in intragastric pressure results in early satiety in both functional dyspepsia (8) and idiopathic gastroparesis (9).
The proximal gastric fundus tone is mediated by a balance of excitatory cholinergic and inhibitory non-adrenergic, non-cholinergic neural input. Other neurotransmitters may also play a role as demonstrated by the effects of the anxiolytic drug buspirone, a 5-HT1 receptor agonist, on gastric fundal relaxation (10). Buspirone improved gastric accommodation in a small dose-finding study of healthy participants, slowing solid and liquid gastric emptying at the 20 mg dose, but not at lower doses (11). In another small cross-over study of patients with functional dyspepsia, buspirone 10 mg three times daily (t.i.d.) over 4-weeks improved gastric accommodation and symptoms of early satiety, postprandial fullness, and bloating but did not alter the rate of gastric emptying of solids or sensitivity to gastric distension although it slightly delayed gastric emptying of liquids (12).
Given this background, the National Institutes of Diabetes and Digestive and Kidney Diseases (NIDDK) Gastroparesis Clinical Research Consortium (GpCRC) conducted a randomised placebo-controlled treatment trial to test whether 4-weeks of treatment with buspirone improves symptoms of early satiety and postprandial fullness based on the Gastroparesis Cardinal Symptom Index (GCSI) early satiety/postprandial fullness subscore (ES/PPF). Secondary objectives were to determine whether buspirone improves overall and other gastroparesis symptoms, gastric physiology (gastric emptying, intragastric meal distribution (IMD), and gastric capacity) and other participant clinical characteristics (anxiety, depression, quality of life) and to determine the safety and tolerability of buspirone in patients with gastroparesis symptoms.
METHODS
Study Design
The GpCRC conducted this multi-center, randomised, double-masked, placebo-controlled, parallel treatment groups phase 2 trial to determine whether buspirone, 10 mg three times per day (t.i.d.), can improve early satiety and postprandial fullness in patients with symptoms of gastroparesis and with at least moderately severe symptoms of early satiety and/or postprandial fullness. We hypothesised buspirone treatment would reduce mean early satiety/postprandial fullness symptom severity subscore (ES/PPF) over 4-weeks compared to treatment with placebo. After enrollment, patients were randomised to either treatment with buspirone or to a matching placebo for 4-weeks followed by a 2-week post-treatment washout period. The primary outcome for the study was the 4-week change (week 4 minus baseline) in the 4-item early satiety/postprandial fullness subscore (ES/PPF) (average of stomach fullness, inability to finish a normal-sized meal, feeling excessively full after meals, and loss of appetite symptom scores) from the GCSI questionnaire.
Study Subjects
Adult patients, aged 18 years or older, with moderate to severe early satiety and postprandial fullness along with other symptoms suggestive of a gastric origin (nausea, fullness, bloating and epigastric pain) for at least 3 months, who had a normal upper endoscopy within 2 years of registration were enrolled into this study from August 2019 through February 2022. The Patient Assessment of Upper Gastrointestinal Symptom Severity Index (PAGI-SYM) questionnaire at enrollment had to have the 9-item GCSI total score > 2.0 and an ES/PPF ≥ 3.0. Patients on stable doses of gastrointestinal drugs including metoclopramide or erythromycin were not excluded. Patients could have delayed or normal gastric emptying on a 4-hour gastric emptying scintigraphy test and be of either diabetic or idiopathic etiology. Patients were excluded if they had post-surgical gastroparesis, currently used opiate narcotic analgesics more than 3 days per week or monoamine oxidase inhibitors, benzodiazepines, warfarin, haloperidol or drugs to inhibit seizures within the prior month; intolerance for buspirone; if pregnant or breast feeding; HbA1c of 10% or more. Patients with history of chronic kidney disease, impaired renal function, significant cardiac disorders, or hepatic injury as defined by alanine aminotransferase elevation of greater than twice Upper Limit of Normal or Child-Pugh score of 10 or greater were excluded from this trial.
Randomisation
Patients meeting eligibility criteria and providing written consent were randomly assigned (1:1) to either buspirone (10 mg t.i.d.) or to a matching placebo using a computer-generated, centrally-administered procedure developed and managed by the Scientific Data Research Center (SDRC). A permuted block randomisation scheme stratified by clinic was used to ensure that the two treatment groups were balanced by calendar time of enrollment and by clinic. Sequentially numbered bottles contained each patient’s treatment. Patients, investigators and clinical site staff were masked to treatment assignment. Two statisticians at the SDRC were unblinded during the trial in order to produce biannual DSMB reports; however, aliases were used to label treatments groups.
Study visits
During screening, surveys were administered to each patient to assess upper gastrointestinal symptoms (PAGI-SYM which includes the GCSI (13,14), Gastrointestinal Symptom Rating Scale (GSRS) (15)), psychological well-being (Hospital Anxiety and Depression Scale (HADS; cut-points defined as <8 (none), 8–14 (mild/moderate, >14 (severe) (16,17), Patient Health Questionnaire 15 Somatic Symptom Severity Scale (PHQ15) (18)), and quality of life (QOL), both disease-specific and general health (Patient Assessment of Upper Gastrointestinal Disorders-Quality of Life (PAGI-QOL) (19), Health Survey Short Form 36 version (SF36 v2) (20)). The daily diary version of the GCSI (ANMS GCSI-DD) (21), to be completed by the patient each night, was distributed. At the initial visit, a medical history was taken and concomitant medicines and diseases were reviewed by the physician. A physical examination including anthropometric measures and a resting electrocardiogram (ECG) was recorded. Fasting blood samples were collected for complete blood count, complete metabolic panel, thyroid stimulating hormone test.
Patients underwent a combined electrogastrography (EGG) and water load satiety test (WLST) test (22,23). In the fasting condition, a 10-minute baseline EGG was obtained. Then, the patient drank cool water “until completely full” for 5 minutes from an opaque 1-liter cup. This was followed by another 30-minute EGG, with recordings at 0, 10, 20, and 30 minutes after ingestion of the water. For the WLST, a volume of water drank <238 mL was considered a low-volume of water (23). For the EGG, each of the % power readings were centrally reviewed and validated in a blinded fashion, and these were the values used for analysis.
On another day, patients underwent the 4-hour gastric emptying scintigraphy (GES) test using a low-fat, egg-white Eggbeaters® meal with imaging at 0, 1, 2, 4 hours after meal ingestion (24,25). The meal includes liquid egg white radiolabeled with 0.5–1 mCI Tc-99m sulfur colloid in order to analyze solid phase gastric emptying. Delayed gastric emptying was present if gastric retention was >60% at 2 hours and/or >10% at 4 hours. Rapid gastric emptying defined as gastric emptying scintigraphy of < 30% retention at 1 hour. In addition, the regional intragastric meal distribution was assessed over time, using a modification of the method reported by Piessevaux et al (26) to assess the intragastric meal distribution (IMD) as an estimation of fundic accommodation (FA) (27). Abnormal IMD suggesting impaired fundic accommodation was defined as IMD <0.568 ratio of the proximal gastric counts to the total gastric counts immediately postprandially and normal IMD values were ≥0.568 (27). The EGG and GES procedures were rescheduled if a patient was diabetic and their glucose prior to the test was >270 mg/dL.
After randomisation, patients were seen at 2-week intervals for a total of 4 weeks on treatment and a final post-washout visit at 6-weeks to assess symptoms and safety of the drug. At each follow-up visit, medical history was updated and reviewed for concomitant medications and comorbidities, adverse events were collected and review of study drug adherence was reported. A physical examination including anthropometric measures was performed. The PAGI-SYM, HADS, PHQ-15 surveys were administered at all follow-up visits, and the daily diary forms for the prior two weeks were collected. The Clinical Patient Grading Assessment Scale (CPGAS) was used at follow-up visits to help assess the therapeutic response of the patient (28). For females of child-bearing potential, a pregnancy test was done at each visit prior to procedures and drug distribution.
The 4-week visit also included the patient undergoing both the 4-hour GES and electrogastrography (EGG) with WLST in order to assess the change after 4-weeks of treatment in either percent retention, the volume of water a patient could consume, and the patterns of stomach rhythm. In addition, all of the questionnaires administered at baseline were reassessed, the routine lab tests (except TSH) and the ECG were repeated for safety.
The 6-week visit mirrored the 4-week visit except with no procedures, and no QOL assessment except for the disease-specific PAGI-QOL.
As part of the patient visit, reminders of routine standard of care advice that may benefit the patient was given. If a patient developed a treatment-related adverse event (defined using CTCAE v5.0 grade 3 or above) on the 10 mg t.i.d. dose, the study medication was stopped after assessment by the site study investigator. If the study investigator determined the patient was intolerant of the study medication, the patient was withdrawn from the study medication, but continued to be followed to the end of the trial. Rescue medications (promethazine, ondansetron) and a patient’s routine drug regimen were allowed during the trial.
Patients were asked to return their remaining study drug at the end of week 4. Adherence to treatment was assessed by capsule count remaining subtracted from the 120 capsules dispensed at randomization. The number of days from baseline to the 4-week visit, multiplied by 3 (capsules per day) is the expected amount required for the study. Adherence was defined as taking the prescribed dose 80% of the time.
Primary and Secondary Outcomes
The primary outcome was defined as the change at 4-weeks from baseline in the early satiety/postprandial fullness (ES/PPF) subscore determined by averaging 4 items of the PAGI-SYM questionnaire: stomach fullness, inability to finish a normal-sized meal, feeling excessively full after meals, and loss of appetite.
Secondary outcomes included the PAGI-SYM GCSI items as well as severity of upper and lower abdominal pain, constipation, diarrhea, and the 7 symptoms of Gastroesophageal reflux disease (GERD) subscore changes at 4-weeks from baseline. An overall symptom severity, GCSI total score, was computed as the average of the following PAGI-SYM subscores: nausea/vomiting (3 items), fullness/early satiety (4 items), bloating (2 items). If the change in the GCSI total score was −1 or less, then a binary outcome indicative of substantial symptom improvement was computed. The patients also rated their symptoms over the prior 24-hours from 0 (no symptoms) to 4 (very severe) each night using the GCSI-DD daily diary. The fullness and early satiety items were averaged to determine the fullness subscore. In addition, an item rating the overall symptoms over the day was included. In order to compute the exploratory daily-diary outcomes, the average of the 7 days of baseline severities for each item was subtracted from the average severities in each item over the last 7 days of the trial.
Statistical Methods
The primary analysis was made on “intention-to-treat” basis, including all patients randomised into the trial. The treatment effect was estimated as an analysis of covariance (ANCOVA) model, regressing the change from baseline at 4-weeks in the ES/PPF on an indicator variable for the treatment group and the baseline value of the outcome. Per-protocol, if more than 10% of the patients were missing the 4-week outcome, multiple imputation under the missing at random assumption modeling missing data on treatment group and baseline value of the outcome using 50 datasets was performed. The 95% confidence limits reported are the results of the imputation analysis (29,30).
The main secondary outcome was the change at 4-weeks in the total GCSI score, both as a continuous and binary improvement variable, analyzed using complete-case as an ANCOVA model. The treatment effect in the binary overall symptom improvement was analyzed using a logistic regression model regressing improvement, the treatment group variable and the baseline value of the total GCSI.
All remaining continuous secondary outcomes were analyzed using complete-case ANCOVA models, regressing the change from baseline to follow-up on treatment group and baseline value of the outcome. All P-values were two-sided and reported as nominal. Bonferroni (BF) multiplicity adjusted levels of significance for alpha 0.05 were computed for comparison for the secondary nonexploratory and subgroup outcomes reported.
Pre-specified subgroup variation in the primary outcome between treatment groups using baseline subgroups was done using ANCOVA models regressing the indicator variable for treatment group on the fullness/early satiety subscore and adjusting for the subscore baseline value, separately within each stratum of the subgroup. P for the interaction of treatment by subgroup was derived from Wald’s test after regressing one or more indicator variables for the subgroup and for the treatment group on the ES/PPF, adjusting for baseline value of ES/PPF within each stratum of the subgroup. Subgroups included demographics, BMI (<25 vs ≥25 kg/m2), etiology, gastric retention (delay vs normal), fundic accommodation (impaired vs normal), baseline medication use, symptom severity (severe/very severe vs none to moderate), any depression or anxiety vs none, low water intake volume vs normal and adherence (adherent vs not).
Sensitivity tests for the primary outcome included complete-case ANCOVA analysis of the primary outcome, as well as changes at week-4 for each individual symptom included in the ES/PPF, ANCOVA analysis of the primary outcome for those patients adhering to treatment, change at 4-weeks in the fullness subscore from the Daily-Diary report, along with the change in the 2 ANMS GCSI-DD items included in the subscore. In addition, the patterns of mean change from baseline at each visit (screening, 2, 4, 6 weeks) and two-sided confidence intervals were plotted by treatment group, for both the primary outcome and the GCSI-DD fullness subscore. The significance of the overall treatment effect of change over time was computed from generalized estimating equations (GEE) linear regression, modeling change as a function of treatment group, visit code indicator, baseline value of the outcome, and a treatment group by visit code interaction term.
Continuous safety outcomes, analyzed with ANCOVA as described above, included mean changes at 4-weeks in laboratory measures, BMI, weight, waist circumference, blood pressure and ECG QTc interval. Adverse events were classified using the CTCAE v5 classification system and reported by treatment group by severity, relatedness, number per person, and body classification and also presented as event rates along with the number of hospitalizations and emergency room visits. P’s were computed by either Fisher’s exact test, a two-sided Binomial probability test with probability of success=0.5, or as an event rate, using an exact Poisson test.
The planned sample size was 108 participants with equal assignment for 2 groups (54 per group). The study had 90% power to detect a mean 0.5 change from baseline between treatment groups for the primary outcome using a 2-sided ANCOVA regression of the outcome on the baseline value and treatment group indicator and assuming a 0.05 Type 1 error (2-sided) rate and 10% loss in follow-up. The Minimum Clinically Important Difference (MCID) was determined to be a change in primary outcome of 0.5 subscore points (0.62 SD) in buspirone compared to placebo (14).
Statistical analyses for this study were generated using SAS (SAS/STAT version 9.4, SAS Institute Inc. Cary, NC, USA) and Stata/MP (StataCorp. 2019. Stata Statistical Software: Release 16 (64-bit), College Station, TX: StataCorp LLC).
Study Oversight and Safety
The trial was designed by the GpCRC investigators and the SDRC and approved by the GpCRC Steering Committee, local institutional review boards (IRB) and a central DSMB appointed by NIDDK. Performance and safety data were monitored biannually by the DSMB and members of the NIDDK. All patients provided written consent prior to the study enrollment. Adverse events from treatment were monitored and recorded throughout the trial and those with severity grade of 3 (severe) or higher were reviewed by the independent medical safety officer. The drug and placebo used in the study were provided by the NIDDK GpCRC. All authors had access to the study data and reviewed and approved the final manuscript.
Results
Patients
A total of 96 patients, 47 in the buspirone group and 49 in the placebo group, were enrolled across 6 tertiary clinic sites in the U.S from 27 August 2019 to 28 February 2022, which resulted in 89% of the sample size achieved (Figure 1). Eighty-one percent of the patients completed the primary outcome at 4-weeks (39 in buspirone, 39 in placebo) and 76 (80%) of patients completed the last 7 days of the 4-week GCSI-DD (35 buspirone, 41 placebo, P=0.57); a primary outcome was not assessed for 18 (19%) patients. Two patients (1 buspirone, 1 placebo P=1.00) had a treatment interruption of 1–2 days before resuming therapy, a total of 15 patients discontinued treatment before the end of the trial (9 buspirone, 6 placebo, P=0.41). Of these 15 patients discontinuing, 7 were due to a physician-directed decision due to patient safety or intolerability of the treatment, and 8 patients refused to continue in the trial or did not contact the clinic, nor respond to the site calling, texting or mailing a reminder card or the questionnaires. Adherence using the capsule count was 60% in the buspirone group vs 59% in the placebo group (P=1.00). All of the baseline characteristics were balanced across treatment groups (Table 1).
Figure 1. CONSORT Patient Flow for BESST trial.
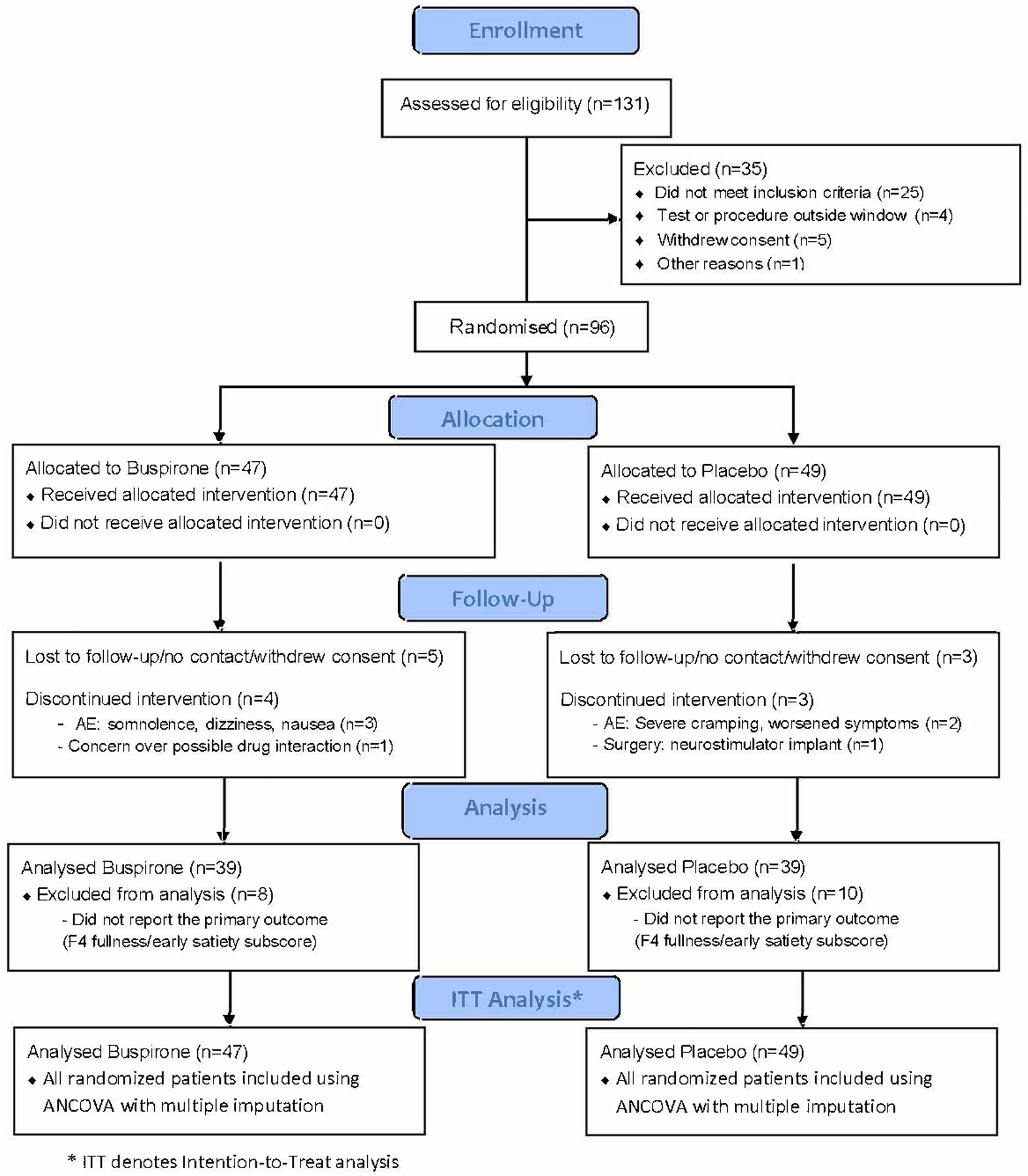
Of 131 patients assessed for eligibility, 96 patients were randomised; 47 to buspirone, and 49 to placebo.
Table 1.
Baseline characteristics of the study population by treatment group
| Mean (SD)* | |||
|---|---|---|---|
| Characteristic | Buspirone (N=47) |
Placebo (N=49) |
Total (N=96) |
| Demographic/lifestyle/anthropometric | |||
| Age (years) | 43.0 (15.8) | 44.2 (15.0) | 43.6 (15.3) |
| Women, No. (%) | 43 (91%) | 45 (92%) | 88 (92%) |
| Hispanic, No. (%) | 13 (28%) | 17 (35%) | 30 (31%) |
| Race, No. (%) | |||
| White | 43 (91%) | 41 (85%) | 84 (88%) |
| Black | 2 (4%) | 5 (10%) | 7 (7%) |
| Other† | 2 (4%) | 2 (4%) | 4 (4%) |
| Married, No. (%) | 16 (34%) | 21 (43%) | 37 (39%) |
| Diabetes type 1 or type 2†, No. (%) | 16 (34%) | 21 (43%) | 37 (39%) |
| Diabetes type 1 | 3 (6%) | 6 (12%) | 9 (9%) |
| Diabetes type 2 | 13 (28%) | 15 (31%) | 28 (29%) |
| Body mass index (BMI) (kg/m2), mean (SD) | 29.5 (7.5) | 29.8 (7.3) | 29.7 (7.4) |
| Overweight to obese (BMI ≥ 25 kg/m2) | 31 (66%) | 37 (76%) | 68 (71%) |
| Weight (kg), mean (SD) | 78.1 (21.2) | 80.2 (22.2) | 79.2 (21.6) |
| Waist circumference (cm), mean (SD) | 98.1 (22.2) | 98.2 (19.6) | 98.1 (20.8) |
| Medications taken in past month, No. (%) | |||
| Proton pump inhibitors | 35 (74%) | 34 (69%) | 69 (72%) |
| Anxiolytic | 1 (2%) | 0 (0%) | 1 (1%) |
| Prokinetic | 12 (26%) | 13 (27%) | 25 (26%) |
| Antiemetic | 31 (66%) | 25 (51%) | 56 (58%) |
| Narcotic (3X/week or less) | 2 (4%) | 4 (8%) | 6 (6%) |
| Anti-depressant | 22 (47%) | 22 (45%) | 44 (46%) |
| Neuropathic or pain modulator, anti-seizure, or other psychiatric medication | 28 (60%) | 31 (63%) | 59 (61%) |
| Gastroparesis symptoms inventories | |||
| PAGI-SYM Severity index (symptoms each scored 0 to 5, none to very severe over past 2 weeks) | |||
| Gastroparesis Cardinal Symptom Index (GCSI), total score | 3.6 (0.7) | 3.5 (0.6) | 3.5 (0.6) |
| Nausea/vomiting severity subscale | 2.5 (1.3) | 2.4 (1.3) | 2.5 (1.3) |
| Nausea severity | 3.6 (1.3) | 3.6 (1.2) | 3.6 (1.3) |
| Retching severity | 2.1 (1.7) | 1.8 (1.6) | 2.0 (1.6) |
| Vomiting severity | 1.9 (1.8) | 1.8 (1.7) | 1.8 (1.7) |
| Fullness/ early satiety subscale | 4.0 (0.6) | 4.0 (0.6) | 4.0 (0.6) |
| Stomach fullness severity | 4.2 (0.8) | 4.2 (0.7) | 4.2 (0.7) |
| Unable to finish meal severity | 4.0 (1.0) | 4.0 (0.9) | 4.0 (0.9) |
| Excessive fullness severity | 4.2 (0.9) | 4.3 (0.7) | 4.3 (0.8) |
| Loss of appetite severity | 3.6 (1.3) | 3.5 (1.2) | 3.5 (1.2) |
| Bloating subscale | 4.2 (1.0) | 3.9 (1.0) | 4.1 (1.0) |
| Bloating severity | 4.4 (0.9) | 4.0 (1.1) | 4.2 (1.0) |
| Stomach distension severity | 4.0 (1.2) | 3.9 (1.1) | 4.0 (1.1) |
| Upper abdominal pain subscale | 3.3 (1.3) | 3.0 (1.2) | 3.1 (1.2) |
| Gastroesophageal Reflux (GERD) subscale | 2.1 (1.4) | 2.3 (1.2) | 2.2 (1.3) |
| Predominant symptom cluster†, No. (%) | |||
| Excessive fullness | 4 (9%) | 13 (27%) | 17 (18%) |
| Early satiety or loss of appetite | 2 (4%) | 4 (8%) | 6 (6%) |
| Bloating or distended stomach | 14 (30%) | 10 (21%) | 24 (26%) |
| Nausea or vomiting or retching | 15 (33%) | 13 (27%) | 28 (30%) |
| Upper abdominal pain or discomfort | 5 (11%) | 1 (2%) | 6 (6%) |
| Lower abdominal pain or discomfort | 4 (9%) | 1 (2%) | 5 (5%) |
| Gastrointestinal Symptom Rating Scale (GSRS) (items coded 0 to 7, no to very severe discomfort) | |||
| Total score, mean (SD) | 3.8 (1.0) | 3.7 (1.1) | 3.8 (1.0) |
| Patient Health Questionnaire somatization severity scale (PHQ-15) (0 to 30, 30 items rated not (0) to very bothered a lot (3)) | |||
| Total score, mean (SD) | 14.7 (5.0) | 14.9 (4.1) | 14.8 (4.5) |
| ANMS Gastroparesis Cardinal Symptom Index Daily Diary (GCSI-DD) (all items scored 0 to 4, none to very severe) | |||
| Fullness/early satiety subscale | 2.62 (0.94) | 2.68 (0.86) | 2.65 (0.90) |
| Early satiety severity | 2.56 (0.78) | 2.59 (0.86) | 2.57 (0.94) |
| Excessive fullness severity | 2.40 (0.98) | 2.77 (0.98) | 2.74 (0.94) |
| Depression, Anxiety & Quality of Life (QOL) | |||
| PAGI-QOL disease-specific QOL (items coded 0 to 5, none to all of the time in past 2 weeks) | |||
| Total score, mean (SD) | 2.3 (1.1) | 2.4 (1.1) | 2.4 (1.1) |
| Hospital Anxiety and Depression Scale (HADS) (each of 16 items scored 1 to 4, with higher total scores indicating more severe), mean (SD) | |||
| HADS-A total score | 9.3 (4.5) | 9.1 (4.8) | 9.2 (4.6) |
| HADS-D total score | 6.7 (5.1) | 6.8 (4.7) | 6.8 (4.9) |
| SF-36 Quality of Life (QOL) (0 to 100, low to high over the past 4 weeks), mean (SD) | |||
| Physical component summary score | 33.7 (8.2) | 33.3 (9.0) | 33.5 (8.6) |
| Mental component summary score | 37.9 (15.6) | 41.3 (13.8) | 39.6 (14.7) |
| Physiological tests | |||
| Gastric emptying scintigraphy (GES)† | |||
| Not delayed gastric emptying‡ | 20 (43%) | 25 (51%) | 45 (47%) |
| Delayed gastric emptying‡, No. (%) | 26 (55%) | 22 (45%) | 48 (50%) |
| Rapid gastric emptying‡, No. (%) | 1 (2%) | 2 (4%) | 3 (3%) |
| Intra-gastric meal distribution (IMD) category | |||
| Normal (0.644 – 1.00) | 17 (87%) | 25 (76%) | 52 (81%) |
| Borderline (0.568 – 0.643) | 0 (0%) | 3 (9%) | 3 (5 %) |
| Impaired (<0.568) | 4 (13%) | 5 (15%) | 9 (14%) |
| Water load test | |||
| Volume water consumed (mL), median (Q1,Q3) | 415.9 (217.0) | 401.7 (198.0) | 408.5 (206.3) |
| Consumed low volume (< 238 mL), No. (%) | 9 (20%) | 10 (20%) | 19 (20%) |
Data are mean (SD), unless otherwise noted.
There were no significant differences by treatment group due to chance of the 57 baseline characteristics analyzed (denoted by an asterisk (*)). P value (2-sided) determined using Fisher’s exact test for categorical variables and a t-test for continuous variables.
Other race: 1 Placebo participant reported American indian,1 Buspirone participant reported Asian, 1 Placebo participant reported Pacific Islander, 1 Buspirone subject reported mixed race, 1 Buspirone subject did not report a race; PAGI-SYM predominant symptom: 1 Buspirone, 1Placebo subject did not report a predominant symptom; for gastric emptying scintigraphy (GES): 2 Buspirone, 3 Placebo participants did not have GES recorded at 1 hour or 4 hours, 1 Buspirone subject did not have GES recorded at 2 hours; 1 Buspirone, 1 Placebo subject did not have EGG and satiety results; 64 total patients had an evaluable IMD result, 21 Buspirone did not have, 19 Placebo did not have.
Delayed gastric emptying defined as gastric emptying scintigraphy of > 60% retention at 2 hours OR > 10% retention at 4 hours; rapid gastric emptying defined as gastric emptying scintigraphy of < 30% retention at 1 hour.
Of the 96 enrolled patients, the average age was 43.6±15.3 years, the average BMI was 29.7±7.4 kg/m2 with 88 patients (92%) female and 27 patients (39%) had diabetes (Table 1). Delayed gastric emptying was present in 48 patients (50%). Comorbidities were recorded in 99% of patients; these included 68% patients with GERD, 39% had hypertension, 39% major depression or anxiety, 33% migraine headache, 17% any autoimmune disorder, and 13% with fibromyalgia. Forty percent of patients did not have anxiety and 56% did not have depression based on the HADS scale <8 points. Mean SF-36 mental and physical QOL was low (39.6±14.7, 33.5±8.6), respectively. Eight-one percent of patients had normal IMD value and 20% drank a low volume of water at baseline for the WLST.
The average GCSI total score was 3.5±0.6 with subscores of 4.0±0.6 for fullness/early satiety subscore, 2.5±1.3 for nausea/vomiting subscore, and 4.1±1.0 for bloating subscore (Table 1). Upper abdominal pain subscore was 3.1±1.2. Predominant symptoms were nausea/vomiting/retching in 28 (30%), bloating or distended stomach in 24 (26%), and excessive fullness in 17 (18%).
Primary Outcome Measure: GCSI ES/PPF Subscore
Patients treated with buspirone, as well as those treated with placebo, had a decrease in the ES/PPF over the 4 weeks of treatment (Table 2). The overall symptomatic improvement in the ES/PPF did not differ between the two treatment groups: −1.16±1.25 in the buspirone group compared to −1.03±1.29 for the placebo group (adjusted mean difference of differences (DoD), −0.11 [95% CI, 0.68, 0.45]; P=0.69). The sensitivity analysis using complete-case duplicated the result: DoD= −0.13 [−0.65, 0.39] P=0.62 (Table 2).
Table 2.
Changes in Early satiety/Postprandial fullness subscore: Primary outcome with sensitivity analysis and Secondary Outcomes
| Outcomes: Changes from baseline at F4 |
Mean differences from baseline to 4-weeks: Baseline adjusted* [absolute (SD)] |
Adjusted mean difference of differences (Buspirone -Placebo) (95% CI)† |
||
|---|---|---|---|---|
| Buspirone | Placebo | P | ||
| Primary analysis ‡ | ||||
| Change from baseline in Early Satiety/Postprandial subscore (ES/PPF) (ITT) | ||||
| No. of patients randomised | 47 | 49 | ||
| Means | −1.16 (1.25) | −1.03 (1.19) | −0.11 (−0.68,0.45) | 0.69 |
| Sensitivity analyses: | ||||
| Change from baseline in ES/PPF (Completers) | ||||
| No. evaluable patients | 39 | 39 | ||
| Means | −1.16 (1.25) | −1.03 (1.19) | −0.13 (−0.65,0.39) | 0.62 |
| Change in baseline ES/PPF in Adherent Patients | ||||
| No. adherent patients with outcome | 23 | 29 | 0.62 | |
| Means | −1.35 (1.23) | −1.09 (1.07) | −0.25 (−0.89, 0.38) | |
| Secondary outcomes of early satiety/postprandial fullness | ||||
| PAGI-SYM Severity index
(0=none to 5=very severe): Changes from baseline |
||||
| Stomach fullness | −1.16 (1.55) | −1.07 (1.34) | −0.09 (−0.69, 0.50) | 0.76 |
| Excessive fullness after meal | −1.14 (1.56) | −0.99 (1.30) | −0.15 (−0.76, 0.47) | 0.64 |
| Inability to finish the meal | −1.27 (1.55) | −1.12 (1.63) | −0.14 (−0.79, 0.50) | 0.66 |
| Loss of appetite | −1.09 (1.64) | −0.96 (1.62) | −0.12 (−0.75, −0.50) | 0.70 |
| Exploratory outcomes of ES/PPF | ||||
| ANMS Gastroparesis Cardinal Symptom Index Daily Diary (GCSI-DD) (all items scored 0 to 4, none to very severe) | ||||
| Early satiety/Postprandial fullness subscore | −0.71 (1.03) | −0.39 (0.71) | −0.32 (−0.71, 0.06) | 0.10 |
| Feeling excessively full after meals | −0.82 (1.03) | −0.31 (0.84) | −0.51 (−0.92, −0.11) | 0.01 |
| Unable to finish a normal-sized meal | −0.62 (1.10) | −0.46 (0.76) | −0.16 (−.061, 0.43) | 0.43 |
| Change in baseline GCSI-DD ES/PPF in Adherent Patients | ||||
| No. evaluable patients | 22 | 23 | ||
| Means | −0.70 (0.90) | −0.28 (0.64) | −0.42 (−0.88, −0.03) | 0.07 |
Mean differences adjusted for the baseline value of the outcome and treatment group indicator are the Least Squares means (LS-means) (i.e., the predicted population margins) computed using a multivariable ANCOVA model regressing the primary and sensitivity outcomes on treatment group and baseline value.
Adjusted mean difference of differences (DoD) from baseline were computed using ANCOVA, regressing change from baseline to 4-weeks on treatment group and baseline value of the outcome. Wald 95% Confidence Limits (CI) are reported; P (2-sided) determined from a Wald Chi-Square test. Bonferroni p-value for the 10 sensitivity outcomes is < 0.005; Benjamini-Hochberg p-value must be < 0.04. No result was below the limits of the adjusted significance levels for multiple comparisons.
The primary outcome is change in the early satiety/postprandial subscore (ES/PPF) from baseline to 4 weeks. This subscore is calculated by averaging the scores for 4 PAGI-SYM questionnaire items: stomach fullness, inability to finish a normal-sized meal, feeling excessively full after meals, and loss of appetite.
There were 18/96 (19%) randomised patients without the outcome at f4; therefore, the adjusted mean DoD was determined from 50 multiple imputation data sets for the 18 patients with missing primary outcome per protocol.
Changes in the ES/PPF by treatment group over time did not show any difference between placebo and buspirone groups from baseline, at week 2, 4 and 6 (Pthrough week 4=0.66) (Figure 2A).
Figure 2. Changes from baseline over time for buspirone and placebo treated patients in symptoms measured by the PAGI-SYM. A. ES/PPF Subscore, B. GCSI C. nausea/vomiting subscore, D. bloating subscore, E. upper abdominal pain subscore, and F. Gastroesophageal Reflux Disease subscore.
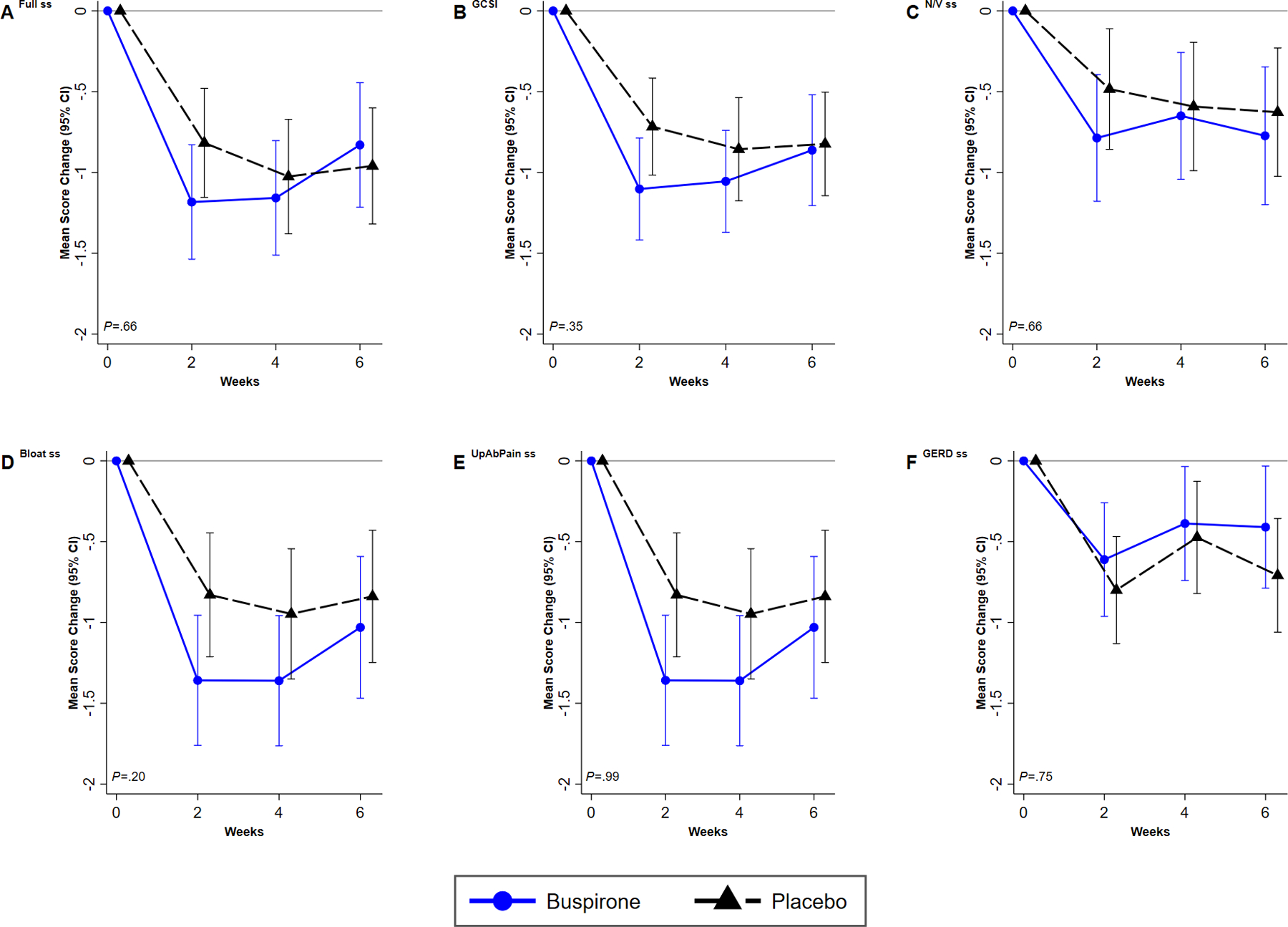
Description: Graphs plotting the time course of adjusted mean change from baseline for each outcome score at each visit and two-sided 95% confidence intervals by treatment group across visit (screening, 2, 4, 6 weeks) are presented. The significance of the overall treatment effect of change over time was computed from generalized estimating equations (GEE) linear regression, modeling change as a function of treatment group, visit code indicator, baseline value of the outcome, and a treatment group by visit code interaction term. The P’s are reported for the trend from baseline to end-of treatment at week 4. Results for buspirone are plotted in blue solid lines, and for placebo in black dashed lines. Number of patients at visit 0, 2, 4, 6, respectively, are: Buspirone: 47, 39, 39, 33; Placebo: 49, 43, 39, 38.
The treatment effect on the primary outcome by various subgroups of patient baseline characteristics is reported in Table 3 and Figure 3. There was an improvement in ES/PPF subscore in the buspirone treated patients compared to placebo in patients with severe to very severe bloating at baseline (DoD=−0.65 [−1.25, −0.05], P=0.03, interaction P=0.003; PBF=0.07) compared to change in ES/PPF subscore for buspirone compared to placebo for the patients with none to moderate bloating at baseline (DoD=1.58 (0,36, 2.82), P=0.01). There was tendency for patients to do better with buspirone in diabetic patients (reductions of 1.80 vs 1.03, interaction P=0.09, PBF=1.00), patients not on narcotics (reductions of 1.22 vs 0.94, interaction P=0.05, PBF=1.00), and in patients with low volume consumed during the WLST (reductions of 1.48 vs 0.02) DoD=−1.46 [−2.22, −0.70], P=0.003; PBF=0.07, interaction P=0.19, PBF=1.00). There was no significant difference in response of patients with abnormally low IMD (suggesting impaired fundic accommodation) vs normal IMD. There were no differences in the treatment effect by white race, Hispanic ethnicity or any baseline medication use, or adherence subgroups.
Table 3.
Subgroup variation in the 4-week change from baseline in early satiety/postprandial fullness subscore between treatment groups using prespecified baseline post-randomization subgroups
| Baseline Subgroup§ | Adjusted Early satiety/fullness change from baseline to 4-weeks* | Difference of differences (Buspirone -Placebo) (95% CI)† |
Interaction P‡ | ||||
|---|---|---|---|---|---|---|---|
| Buspirone | Placebo | ||||||
| N | Mean ) (SD) |
N | Mean ) (SD) |
P | |||
| Overall | 39 | −1.17 (1.19) | 39 | −1.03 (1.19) | −0.13 (−0.67, 0.41) | 0.63 | |
| Clinic | 0.86 | ||||||
| 1 | 10 | −0.43 | 7 | −0.75 | 0.33 (−0.59,1.24) | 0.46 | |
| 2 | 1 | −1.25 | 2 | −0.38 | −0.88 (−47.64, 45.89) | 0.85 | |
| 3 | 11 | −1.18 | 17 | −1.13 | −0.05 (−1.12,1.03) | 0.93 | |
| 4 | 0 | n/c | 0 | n/c | n/c | n/c | |
| 5 | 10 | −1.87 | 6 | −1.29 | −0.61 (−1.98, 0.77) | 0.36 | |
| 6 | 7 | −1.19 | 7 | −0.99 | −0.11 (−1.23, 1.02) | 0.84 | |
| Demographic | |||||||
| Gender | |||||||
| Female | 36 | −1.08 | 36 | −0.99 | −0.10 (−0.61, 0.49) | 0.74 | 0.34 |
| Male | 3 | −2.17 | 3 | −1.58 | −0.58 (−3.49, 2.32) | 0.61 | |
| Age (years) | 0.15 | ||||||
| < 50 | 24 | −1.00 | 24 | −1.21 | 0.21 (−0.53, 0.95) | 0.57 | |
| ≥ 50 | 15 | −1.42 | 15 | −0.76 | −0.68 (−1.52, 0.17) | 0.11 | |
| Race | 0.67 | ||||||
| White | 37 | −1.10 | 34 | −0.92 | −0.24 (−0.81, 0.34) | 0.42 | |
| Non-white | 2 | −1.38 | 5 | −1.80 | 0.43 (−2.20, 3.05) | 0.70 | |
| Hispanic Ethnicity | 0.80 | ||||||
| Hispanic | 8 | −1.40 | 15 | −1.03 | −0.37 (−1.53, 0.78) | 0.51 | |
| Not Hispanic | 31 | −1.10 | 24 | −1.03 | −0.07 (−0.74, 0.59) | 0.83 | |
| Clinical | |||||||
| BMI group (kg/m2) | 0.43 | ||||||
| < 25 (low-normal) | 14 | −0.75 | 9 | −0.75 | 0.0 (−1.01, 1.01) | 1.00 | |
| ≥ 25 (overweight/ obese) | 25 | −1.40 | 30 | −1.12 | −0.28 (−0.96, 0.39) | 0.40 | |
| Etiology | 0.09 | ||||||
| Diabetic | 11 | −1.80 | 18 | −1.03 | −0.77 (−1.66, 0.12) | 0.09 | |
| Idiopathic | 28 | −0.92 | 21 | −1.04 | 0.12 (−0.61, 0.84) | 0.75 | |
| Scintigraphic gastric emptying (GES) ¶ | |||||||
| Gastric retention | 0.25 | ||||||
| Delayed retention | 19 | −1.09 | 17 | −1.40 | 0.30 (−0.42,1.22) | 0.40 | |
| Not delayed | 20 | −1.24 | 22 | −0.75 | −0.49 (−1.32, 0.34) | 0.24 | |
| Intragastric meal distribution | |||||||
| Low | 1 | −0.75 | 4 | −0.5 | −0.25 (−3.73, 3.23) | 0.83 | 0.77 |
| Normal | 14 | −1.14 | 18 | −1.29 | 0.15 (−70, −1.85) | 0.72 | |
| Medication use (prior month) | |||||||
| Antiemetic | |||||||
| Any antiemetic | 26 | −1.19 | 19 | −1.14 | −0.05 (−0.81, 0.72) | 0.90 | 0.66 |
| None | 13 | −1.12 | 20 | −0.93 | −0.19 (−1.07, 0.68) | 0.66 | |
| Any 5-HT3 receptor agonist | 18 | −1.25 | 10 | −1.10 | −0.15 (−1.22,0.92) | 0.78 | 0.81 |
| No use | 21 | −1.09 | 29 | −1.01 | −0.09 (−0.77, 0.60) | 0.80 | |
| Prokinetic use | 10 | −1.38 | 9 | −1.08 | −0.29 (−1.79, 1.21) | 0.69 | 0.52 |
| No use | 29 | −1.09 | 30 | −1.02 | −0.08 (−0.67, 0.51) | 0.79 | |
| Sx modulator (TCA) | 4 | −0.88 | 2 | −0.50 | −0.38 (−2.35, 1.60) | 0.63 | 0.93 |
| No use | 35 | −1.20 | 37 | −1.06 | −0.14 (−0.73, 0.45) | 0.64 | |
| Proton pump inhibitor use | 28 | −1.17 | 25 | −1.05 | −0.12 (−0.84, 0.59) | 0.74 | 0.79 |
| No use | 11 | −1.16 | 14 | −1.00 | −0.16 (−1.07, 0.75) | 0.72 | |
| Narcotic use | 2 | −0.13 | 3 | −2.08 | 1.95 (−2.42, 6.34) | 0.25 | 0.05 |
| Not used | 37 | −1.22 | 36 | −0.94 | −0.28 (−0.84, 0.28) | 0.32 | |
| Symptoms | |||||||
| Nausea | 0.83 | ||||||
| Severe/very severe | 26 | −1.25 | 23 | −1.04 | −0.21 (−0.91, 0.50) | 0.56 | |
| None to moderate | 13 | −1.00 | 16 | −1.02 | 0.02 (−0.95, 0.98) | 0.97 | |
| Vomiting | 0.79 | ||||||
| Severe/very severe | 8 | −1.34 | 9 | −0.94 | −0.40 (−1.76, 0.96) | 0.54 | |
| None to moderate | 31 | −1.12 | 30 | −1.06 | −0.06 (−0.69, 0.56) | 0.84 | |
| Stomach fullness | 0.38 | ||||||
| Severe/very severe | 33 | −1.27 | 33 | −1.02 | −0.26 (−0.87, 0.36) | 0.41 | |
| None to moderate | 6 | −0.58 | 6 | −1.13 | 0.54 (−0.81, 1.89) | 0.39 | |
| Early satiety | 0.19 | ||||||
| Severe/very severe | 25 | −1.28 | 29 | −1.31 | 0.03 (−0.66, 0.72) | 0.93 | |
| None to moderate | 14 | −0.96 | 10 | −0.23 | −0.74 (−1.59, 0.12) | 0.09 | |
| Bloating | 0.003 | ||||||
| Severe/very severe | 33 | −1.36 | 26 | −0.71 | −0.65 (−1.25, −0.05) | 0.03 | |
| None to moderate | 6 | −0.08 | 13 | −1.67 | 1.58 (0.36, 2.82) | 0.01 | |
| Abdominal pain | 0.96 | ||||||
| Severe/very severe | 17 | −1.40 | 14 | −1.21 | −0.18 (−1.29, 0.92) | 0.74 | |
| None to moderate | 22 | −0.99 | 25 | −0.93 | −0.06 (−0.66, 0.54) | 0.84 | |
| Anxiety & Depression (HADS) | 0.90 | ||||||
| Any anxiety (8+) | 23 | −1.29 | 23 | −1.03 | −0.26 (−1.00, 0.47) | 0.47 | |
| No anxiety (<8) | 14 | −1.05 | 14 | −1.00 | −0.05 (−1.05, 0.94) | 0.84 | |
| 0.53 | |||||||
| Any depression (8+) | 18 | −1.15 | 15 | −1.03 | −0.12 (−0.95, 0.71) | 0.77 | |
| No depression (<8) | 19 | −1.25 | 22 | −1.01 | −0.24 (−1.07, 0.59) | 0.56 | |
| Water load test | 0.19 | ||||||
| Low volume (<238 ml) | 5 | −1.48 | 5 | −0.02 | −1.46 (−2.22, −0.70) | 0.003 | |
| Volume ≥ 238 ml | 21 | −1.33 | 23 | −1.20 | −0.14 (−0.99, 0.72) | 0.75 | |
| Adherence to prescribed dose ║ | |||||||
| Dose by capsule count | |||||||
| Adherent | 23 | −1.35 | 29 | −1.09 | −0.25 (−0.92, 0.41) | 0.45 | 0.72 |
| Non-adherent | 10 | −0.90 | 16 | −0.86 | −0.04 (−1.07, 0.99) | 0.94 | |
| Dose by ANMS Daily Diary self-report |
|||||||
| Adherent | 19 | −1.41 | 21 | −1.01 | −0.40 (−1.19, 0.39) | 0.32 | 0.22 |
| Non-adherent | 10 | −71 | 10 | −1.59 | 0.88 (−0.59, 2.36) | 0.22 | |
Mean differences adjusted for the baseline value of the outcome and treatment group indicator are the Least Squares means (LS-means) (i.e., the predicted population margins) computed using a generalized linear ANCOVA model of the primary outcome on treatment group and baseline value, and holding independent variables at their means.
The mean difference of differences (DoD) between Buspirone and Placebo, 95% confidence interval and P (2-sided) were derived from an ANCOVA, regressing an indicator for treatment group on fullness/early satiety subscore, adjusting for the baseline value of fullness/early satiety subscore, within each stratum of the subgroup.
The P for the interaction of treatment by subgroup was derived from Wald’s test after regressing one or more indicator variables for the subgroup and for treatment group on fullness/early satiety subcore, adjusting for baseline value of fullness/early satiety subscore within each stratum of the subgroup. Bonferroni adjusted P-value for comparison of subgroup variation is 0.0002; No P-values were significant by Bonferroni-Hochberg corrected significance level, either.
Excludes observations with missing subgroup data.
Delayed gastric emptying defined as gastric emptying scintigraphy of > 60% retention at 2 hours OR >10% retention at 4 hours, which includes the rapid emptiers.
Adherence defined as patient taking the treatment medication 80% of the days during the treatment period using the Drug Dispensing case-report form ((capsules dispensed – capsules returned)/3*No. days in the treatment)*100 and by the ANMS Daily-Dairy GCSI form ((capsules taken/3*No. days in diary during tx)*100.
Figure 3. Subgroup variation in the 4-week change from baseline in early satiety/postprandial fullness subscore between treatment groups using prespecified baseline post-randomization subgroups.
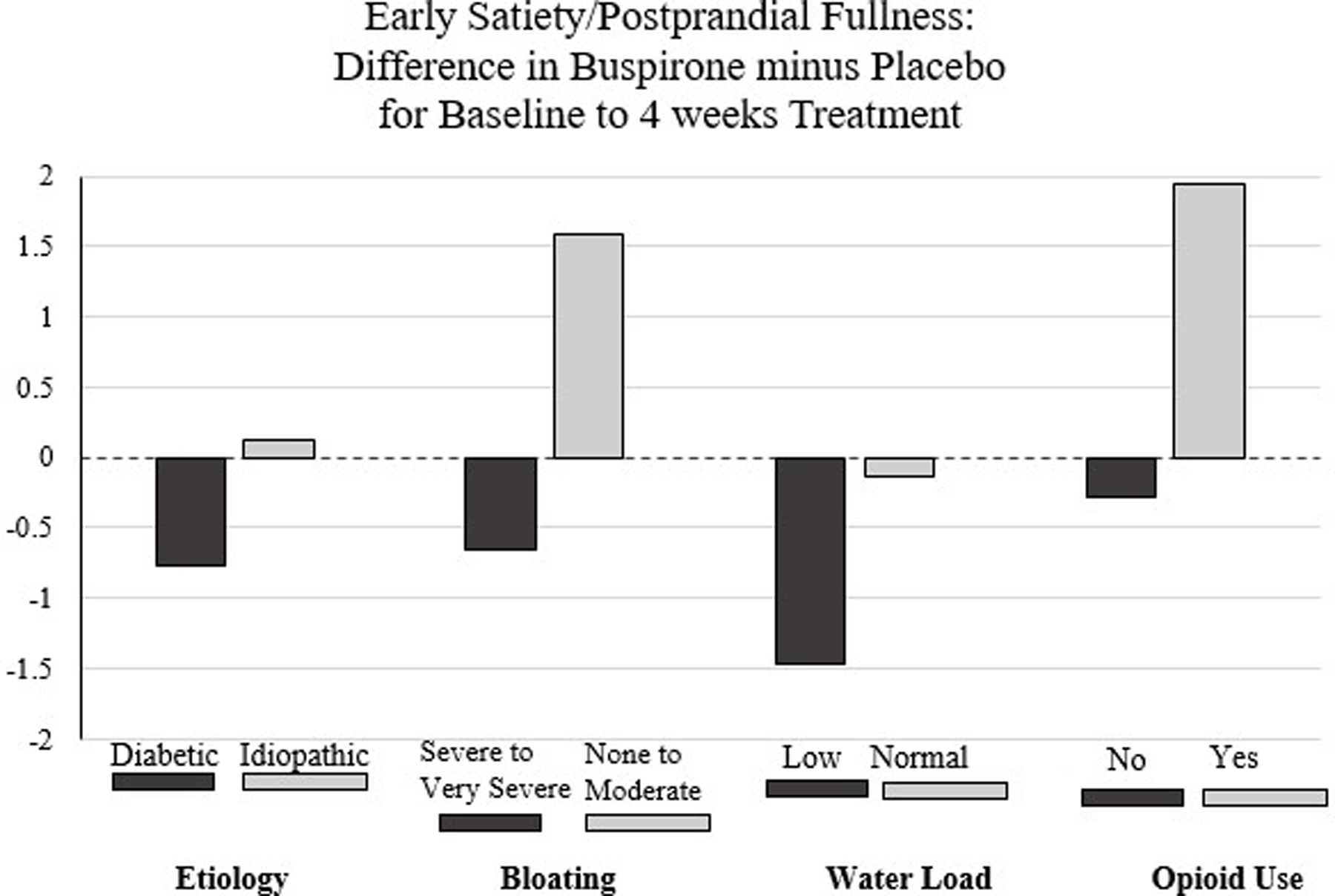
Subgroups displayed are: 1) Etiology: Diabetic vs Idiopathic, 2) Bloating: Severe/very severe vs none to moderate, 3) Water Load Satiety Test volume consumed: Low vs Normal, 4) Narcotic use 3+ times/week or less: No use vs used.
The use of rescue medication over the 4 weeks of the trial were compared by treatment group. Rescue medications were used by 35 patients in the buspirone group and 37 patients in the placebo group, with no differences in any rescue medication by treatment. For instance, antiemetics were used by 57% of the buspirone patients and 59% of the placebo patients with ondansetron used by 40% of the buspirone group and 35% of the placebo group (P=0.81).
Secondary and Exploratory Outcomes
The treatment group comparisons of changes from baseline to 4-weeks for secondary outcomes are shown in Table 4. The exploratory outcomes treatment differences, comprising changes from baseline in 7 PAGI-SYM additional individual items, the 5 GSRS symptom clusters, the 6 GCSI-DD symptoms (excluding fullness and early satiety) and episodes of vomiting, and the baseline and average post-satiety average percent power in 4 EGG frequency regions are reported in Table 5.
Table 4.
Changes in secondary outcomes by treatment
|
Secondary Outcomes:
Changes from baseline at F4 |
Mean differences from baseline to 4-weeks: Baseline adjusted* [absolute (SD)] |
Adjusted mean difference of differences or Odds Ratio (Buspirone -Placebo) (95% CI)† |
||
|---|---|---|---|---|
| Buspirone | Placebo | P‡ | ||
| No. evaluable patients‡ | 39 | 39 | ||
| Gastroparesis symptoms inventories | ||||
| PAGI-SYM Severity index (0=none to 5=very severe) |
||||
| Gastroparesis Cardinal Symptom Index (GCSI) score | −1.06 (1.16) | −0.86 (1.00) | −0.20 (−0.66, 0.27) | 0.40 |
| GCSI 1+ improvement§ (n, %) | 19 (49%) | 16 (42%) | 1.31 (0.53,3.21) | 0.56 |
| Nausea/vomiting severity subscore | −0.65 (1.40) | −0.60 (1.26) | −0.05 (−0.58, 0.48) | 0.86 |
| Nausea severity | −0.75 (1.40) | −0.70 (1.51) | −0.06 (−0.64, 0.53) | 0.85 |
| Vomiting severity | −0.71 (1.81) | −0.57 (1.47) | −0.14 (−0.76, 0.47) | 0.65 |
| Bloating subscore | −1.36 (1.42) | −0.95 (1.27) | −0.41 (−0.99, 0.16) | 0.16 |
| Bloating severity | −1.34 (1.46) | −0.69 (1.18) | −0.65 (−1.23, −0.08) | 0.03 |
| Upper abdominal pain subscore | −0.78 (1.55) | −0.95 (1.63) | 0.17 (−0.44, 0.77) | 0.59 |
| Upper abdominal pain severity | −0.69 (1.73) | −0.93 (1.63) | 0.24 (−0.39, 0.88) | 0.46 |
| GERD subscore | −0.36 (1.44) | −0.45 (1.01) | 0.09 (−0.42, 0.59) | 0.73 |
| Gastrointestinal Symptom Rating Scale (GSRS) (0=no to 7=very severe discomfort) |
||||
| Total score | −0.63 (1.09) | −0.49 (0.90) | −0.14 (−0.55, 0.27) | 0.50 |
| Clinical Patient Grading Assessment Score (CPGAS)(−3 to 3, where −3=very considerably worse to 0=no change to 3=completely better) | ||||
| Change at f4 | 0.69 (1.03) | 0.37 (1.10) | 0.32 (−0.15, 0.79) | 0.18 |
| Patient Health Questionnaire somatization severity score (0 to 30, 30 items rated not (0) to very bothered a lot (3)) |
||||
| Total somatization score | −0.99 (3.88) | −2.00 (3.93) | 1.01 (−0.49, 2.51) | 0.19 |
| Depression, Anxiety & Quality of Life (QOL) | ||||
| PAGI-QOL disease-specific QOL (items coded 0 to 5, none to all of the time in past 2 weeks) | ||||
| Total score | 0.43 (0.81) | 0.64 (0.95) | −0.21 (−0.57, 0.15) | 0.25 |
| Hospital Anxiety and Depression Scale (HADS) (each of 16 items scored 1 to 4, with higher total scores indicating more severe) | ||||
| HADS-A total score | −1.27 (4.82) | −2.03 (3.94) | 0.76 (−1.02, 2.54) | 0.40 |
| HADS-D total score | 0.42 (4.06) | −1.43 (3.91) | 1.86 (0.33, 3.38) | 0.02 |
| SF-36 Quality of Life (QOL) (0 to 100, low to high over past 4 weeks) | ||||
| Physical component summary score | 1.10 (7.18) | 3.17 (5.95) | −2.07 (−4.91, 0.76) | 0.15 |
| Mental component summary score | 1.19 (10.54) | 3.17 (11.83) | −1.98 (−6.20, 2.24) | 0.36 |
| Physiologic tests | ||||
| Gastric emptying scintigraphy (GES)§ | ||||
| Percent gastric retention at: | ||||
| 2 hours (%) | 6.58 (20.1) | 10.08 (28.1) | −3.50 (−13.76, 6.76) | 0.50 |
| 4 hours (%) | 4.24 (21.19) | 2.87 (21.8) | 1.37 (−7.51, 10.25) | 0.76 |
| Diagnosis at f4: | ||||
| Delay (vs Normal emptying) | 18 (70%) | 19 (58%) | 0.79 (0.45, 1.38) | 0.40 |
| Intra-gastric meal distribution (IMD)§ | ||||
| Change in IMD at f4 | −0.04 (0.13) | 0.02 (0.13) | −0.05 (−0.12, (0.01) | 0.10 |
| Classification at f4: | ||||
| Abnormal (vs Normal IMD) | 2 (12%) | 1 (4%) | 0.46 (0.11, 1.84) | 0.27 |
| Water load test | ||||
| Volume water consumed (mL) | −43.3 (219.1) | −21.8 (145.6) | −21.51 (−99.4, 56.4) | 0.29 |
| Low volume consumed (<238 mL) at f4 visit§ | 5 (19%) | 4 (13%) | 1.15 (0.24, 10.26) | 0.89 |
Adjusted mean difference of differences (DoD) from baseline were computed using ANCOVA, regressing change from baseline to 4-weeks on treatment group and baseline value of the outcome. Wald 95% Confidence Limits (CI) are reported; P (2-sided) determined from a Wald Chi-Square test.
Absolute mean differences (SD) from baseline to f4 by treatment group for each outcome were calculated from t-test. Mean differences adjusted for the baseline value of the outcome and treatment group indicator are the Least Squares means (LS-means) (i.e., the predicted population margins) computed using a generalized linear ANCOVA model of the primary outcome on treatment group and baseline value.
Adjusted mean difference of differences (DoD) from baseline were computed using ANCOVA, regressing change from baseline to f4 on treatment group and baseline value of the outcome. Wald 95% Confidence Limits (CI) are reported; P (2-sided) determined from a Wald Chi-Square test. There are 25 protocol defined secondary outcomes. To adjust for multiple comparisons using the Bonferroni correction, the null hypothesis of no difference between the treatment groups would not be rejected with α=0.05 unless the p-value is ≤ 0.002; no P-values were significant by Bonferroni-Hochberg corrected significance levels, either.
There are 1 Buspirone not reporting GERD severities; 1 Placebo patient not reporting GSRS, PAGI-QOL, HADS. GES analysis: No. patients in analyses: 26 (Buspirone, 33 (Placebo); WLST analysis: No. patients in analyses: 26 (Buspirone), 26 (Placebo); IMD analysis: No. patients in analyses: 17 (Buspirone, 24 (Placebo)
Substantial symptomatic improvement defined as a decrease of at least 1 point from baseline in GCSI score at F4. No. patients in analyses: 39 (Buspirone), 38 (Placebo).Odds ratio, 95% confidence intervals and P from a logistic regression of outcome on treatment group for symptomatic improvement in GCSI, and a logistic regression of outcome on treatment group adjusted for the baseline value for GES and IMD are reported in the table.
Table 5.
Changes in exploratory outcomes
|
Exploratory Secondary Outcomes:
Changes from baseline at F4 |
Mean differences from baseline to 4-weeks: Baseline adjusted* [absolute (SD)] |
Adjusted mean difference of differences or Odds ratio (Buspirone -Placebo) (95% CI)†§ |
||
|---|---|---|---|---|
| Buspirone | Placebo | P | ||
| No. evaluable patients‡ | 40 | 39 | ||
| Gastroparesis symptoms inventories | ||||
| PAGI-SYM Severity index symptom severities (0=none to 5=very severe) |
||||
| Retching severity | −0.46 (1.93) | −0.70 (1.51) | 0.16 (−0.49, 0.81) | 0.64 |
| Stomach distension severity | −1.38 (1.58) | −1.20 (1.58) | −0.18 (−0.83, 0.47) | 0.59 |
| Upper abdominal discomfort | −0.87 (1.50) | −1.00 (1.61) | 0.12 (−0.49, 0.73) | 0.69 |
| Lower abdominal pain severity | −0.31 (1.93) | −0.66 (1.39) | 0.35 (−0.26, 0.96) | 0.26 |
| Lower abdominal discomfort | −0.48 (1.89) | −0.77 (1.51) | 0.29 (−0.36, 0.94) | 0.38 |
| Constipation severity | −0.22 (1.61) | −0.57 (1.58) | 0.35 (−0.27, 0.97) | 0.27 |
| Diarrhea severity | −0.54 (1.77) | −0.51 (1.64) | −0.03 (−0.71, 0.64) | 0.91 |
| Gastrointestinal Symptom Rating Scale (GSRS) subscores (0=no to 7=very severe discomfort) |
||||
| Reflux score | −0.72 (1.50) | −0.67 (1.56) | −0.04 (−0.64, 0.56) | 0.89 |
| Abdominal pain score | −0.97 (1.40) | −0.54 (1.55) | −0.42 (−0.98, 0.13) | 0.14 |
| Indigestion score | −0.67 (1.40) | −0.48 (1.13) | −0.19 (−0.70, 0.32) | 0.46 |
| Diarrhea score | −0.57 (1.98) | −0.35 (1.35) | −0.21 (−0.83, 0.40) | 0.50 |
| Constipation score | −0.23 (1.61) | −0.53 (1.82) | 0.29 (−0.34, 0.93) | 0.37 |
| ANMS Gastroparesis Cardinal Symptom Index Daily Diary (GCSI-DD) (all items scored 0 to 4, none to very severe) | ||||
| GCSI total score | −0.73 (0.94) | −0.28 (0.56) | −0.45 (−0.79, −0.12) | 0.007 |
| Nausea severity | −0.65 (0.98) | −0.18 (0.76) | −0.48 (−0.86, −0.09) | 0.01 |
| Bloating severity | −0.82 (0.97) | −0.27 (0.87) | −0.55 (−0.93, −0.17) | 0.004 |
| Upper abdominal pain | −0.79 (0.93) | −0.34 (1.06) | −0.45 (−0.87, −0.04) | 0.03 |
| Vomiting (No. of episodes) | −0.001 (1.33) | 0.37 (2.16) | −0.37 (−1.18, 0.43) | 0.36 |
| Overall symptom severity | −0.58 (1.00) | −0.26 (0.66) | −0.32 (−0.68, 0.04) | 0.08 |
| Physiologic test: | ||||
| Electrogastrogram (EGG) results‖ | ||||
| Average power in frequency region, % | ||||
| Bradygastria (1-<2.5 cpm) | ||||
| Baseline | 2.63 (25.78) | −2.52 (17.71) | 5.15 (−3.89, 14.19) | 0.26 |
| 0–30 post-satiety | 3.98 (15.80) | 0.36 (17.97) | 3.62 (−3.00, 10.25) | 0.28 |
| Normogastria (2.5-<3.8 cpm) | ||||
| Baseline | −0.23 (11.73) | −1.13 (12.03) | 0.90 (−4.38, 6.189) | 0.74 |
| 0–30 post-satiety | −1.71 (10.19) | −1.87 (7.24) | −0.52 (−4.20, 3.17) | 0.78 |
| Tachygastria (3.8–10 cpm) | ||||
| Baseline | 0.10 (16.76) | 1.72 (11.06) | −1.62 (−6.16, 2.93) | 0.49 |
| 0–30 post-satiety | 0.92 (8.96) | −0.59 (11.87) | 1.15 (−3.07, 6.10) | 0.52 |
| Duodenal (>10–15 cpm) | ||||
| Baseline | −1.92 (15.51) | 1.75 (10.87) | −3.66 (−9.58, 2.25) | 0.22 |
| 0–30 post-satiety | −0.76 (8.26) | 0.44 (11.92) | −1.20 (−4.47, 2.07) | 0.47 |
Mean differences adjusted for the baseline value of the outcome and treatment group indicator are the Least Squares means (LS-means) (i.e., the predicted population margins) computed using a generalized linear ANCOVA model of the primary outcome on treatment group and baseline value.
Adjusted mean difference of differences (DoD) from baseline were computed using ANCOVA, regressing change from baseline to 4-weeks on treatment group and baseline value of the outcome. Wald 95% Confidence Limits (CI) are reported; P (2-sided) determined from a Wald Chi-Square test.
Number of evaluable patients (complete-case analysis) will vary with each outcome depending on the questionnaire or procedure. No. patients with 4-week satiety and EGG data: 26 Buspirone, 26 placebo. For DD analysis: 76 total patients (41 P, 35 B).
Odds ratio, 95% confidence intervals and P from an exact logistic regression of outcome on treatment group adjusted for the baseline value. For the 29 outcomes, the Bonferroni level of significance would be P≤0.0017; no P-values were significant by Bonferroni-Hochberg corrected significance levels either.
Electrogastrogram (EGG) results for average percent power have been validated by central review.
PAGI-SYM and GCSI
Individual GCSI and PAGI-SYM assessment of the mean differences over 4-weeks for the cardinal symptoms and subscores by treatment are reported in Tables 2 and 4.
The GCSI decreased 1.06 in the buspirone group compared to 0.86 in the placebo group resulting in no treatment effect (DoD=−0.20) (Table 4 and Figure 2B). Using the overall symptom improvement variable (GCSI improvement of 1 or greater) that we have used in our prior studies (4,31), there were no differences in this response rate in the buspirone patients (49%) compared to the placebo patients (42%) resulting in an OR=1.31 [0.53, 3.21], P=0.56.
Figure 2 shows changes in the 5 PAGI-SYM symptom subscores during treatment visits at 2 and 4 weeks, and the 6-week post-treatment visit. Changes in the ES/PPF subscore by treatment group over time did not show any difference between placebo and buspirone groups from baseline, at week 2, 4 and 6 (Pthrough week 4=0.66) (Figures 2C, 2D, 2E, 2F).
Of the four individual symptoms making up the early satiety/postprandial fullness subscore (fullness, excessive fullness, not able to finish meal, loss of appetite symptom scores), there were mean differences between the treatment groups of 0.09, 0.15, 0.14, 0.12 respectively; P values of 0.64 to 0.76 for the DoD, respectively (Table 2).
The largest differences between treatment groups was in the individual bloating severity score with reduction in the buspirone group of 1.34 compared to 0.69 in placebo patients; DoD=−0.65 [−1.23, −0.08], P=0.03, PBF=0.78 (Table 4). None of the other PAGI-SYM symptoms showed any treatment effect. There were no treatment group differences in overall GCSI (either continuous or substantial improvement) and for nausea, vomiting, retching, bloating subscore, upper abdominal pain subscore or the GERD subscore. In addition, there were no treatment group differences in the GSRS total or symptom clusters, anxiety total score, overall quality of life (physical and mental components) (Table 4).
CPGAS
The study captured the patients’ evaluation of their symptom relief using CPGAS, rated from −3 (very considerably worse) to 3 (completely better). CPGAS did not show a significant treatment difference (Table 4); the mean adjusted difference at 4-weeks did show some improvement in the symptoms for buspirone compared to placebo: DoD=0.32 [−0.15, 0.79], P=0.18.
HADS
Buspirone did not improve the anxiety score as assessed with HADS (Table 4). There was a significant improvement in HADS depression in the placebo group compared to the buspirone group, DoD=1.86 (0.33, 3.38), P=0.02, PBF=0.50.
Exploratory Outcomes: ANMS GCSI-DD
The exploratory daily diary measure GCSI-DD produced some favorable results for buspirone. The GCSI-DD total score, bloating, postprandial fullness, nausea, and upper abdominal pain showed greater adjusted mean reductions in the buspirone group compared to placebo at the end of trial, though were greater than BF significance levels. The GCSI-DD total score resulted in DoD=−0.45 [−0.79, −0.12], P=0.007, PBF=0.20, and bloating severity adjusted mean DoD=−0.55 [−0.93, −0.17], P=0.004, PBF=0.12 (Table 5). The GCSI-DD effect sizes resulted in greater reductions when comparing buspirone to placebo, the largest reduction being 0.51 for the “feeling excessively full after meals” (Tables 2, 5).
Physiologic testing
There were no treatment group differences in gastric emptying scintigraphy, the volume of water consumed during WLST or in any of the EGG results. Buspirone tended to produce greater fundic accommodation as indicated by a change in the IMD-0 at 4 weeks in compared to placebo (−0.04 vs 0.02; P=0.10) (Tables 4, 5).
Adverse events
There was a total of 21 adverse events in 20 patients over the course of the treatment (12 in buspirone group and 8 in placebo group, P=0.37). The overall event rate per person-month was 0.18 in placebo and 0.23 in placebo, P=0.62). The AEs were predominantly in gastrointestinal (e.g., diarrhea, abdominal pain, worse nausea), infections (e.g., upper respiratory, enterocolitis, surgical wound infections) and the nervous system (e.g., dizziness possibly related to the drug, somnolence impairing activities) body classes, with no differences between treatment group. There were no treatment group differences in the body classification classes, number of events per person, the severity categories, or in the physician judgment of drug relatedness. There were two serious adverse events (1 buspirone, 1 placebo, P=1.00). Each of the SAEs was judged not to be related to treatment; the placebo event was due to an elected surgical procedure for a neurostimulator implantation, and the buspirone event was due to an allergic reaction to mannitol interacting with patient’s other drugs. There were no deaths, 1 hospitalization (0.02 events/patient-month in buspirone and 0 in placebo, P=0.98) and 1 ER visit (0.02 event rate) in buspirone group and 0 in placebo, P=0.98. Reviewing the changes in laboratory and other safety outcomes, no differences or concerns occurred comparing the treatment groups.
Discussion
This randomized, double-blind, placebo controlled trial investigated the symptomatic benefits of buspirone in patients with symptoms of gastroparesis with at least moderate severity of early satiety and postprandial fullness. Both the buspirone treated group and placebo treated group had similar reductions in the primary outcome (ES/PPF) and GCSI. Overall, for our primary outcome for this study, the average reduction in the early satiety/postprandial fullness subscore with buspirone over 4-weeks was not significantly different from the placebo treated group.
Although our study did not support our primary outcome that buspirone would improve ES/PPF to a greater extent compared to placebo, this does not necessarily mean that buspirone is of no therapeutic value in this condition, as several of our secondary outcomes were nominally significant (though not significant after multiple comparison adjustments). Subgroups of patients may benefit from the use of buspirone for their symptoms (Figure 3). Post-hoc analysis suggested that patients with severe bloating at baseline had much larger reductions in symptoms with buspirone compared to the patients with none to moderate bloating. Although non-significant findings, buspirone may also have better effects improving ES/PPF subscore in other groups of patients: those diagnosed with diabetes, non-narcotic medication users, and those with low volume consumed during WLST at enrollment. The improvement of patients with severe bloating is attractive for further study. Bloating is a particularly difficult symptom to treat. Our prior GpCRC gastroparesis registry study found that 41% of patients with gastroparesis have severe or very severe bloating (32)
In an exploratory fashion, this study assessed symptoms on a daily basis using the ANMS GCSI-DD for which the symptoms of bloating and postprandial fullness as well as nausea and abdominal pain were nominally significantly improved with buspirone treatment compared to placebo treatment. The mean adjusted group differences for bloating and GCSI were −0.55 and −0.45, respectively, suggesting a positive benefit of buspirone. These trial results suggest using the daily recording of symptoms may be a more sensitive measure of gastroparesis symptoms than the 2-week recall GCSI used for the primary outcome. The FDA now is suggesting using daily assessment of symptoms (33).
In this study, we studied patients with symptoms of gastroparesis and having at least moderate symptoms of early satiety and postprandial fullness, as these appear to symptoms of impaired accommodation (8). However, only 20% of our patients had objective dysfunction of the proximal stomach as measured by scintigraphic intragastric meal distribution or decreased volume of water consumed during the WLST. This study evaluated three physiological parameters for predicting the response of buspirone and studied if these were improved by buspirone treatment: gastric emptying, intragastric meal distribution, and water load satiety testing. Patients with delayed gastric emptying had similar results as patients with normal gastric emptying. Our study also assessed gastric emptying changes with buspirone and found no effect of buspirone. Some studies report that buspirone can impede gastric emptying, at least gastric emptying of liquids (12). Buspirone had been shown in prior studies to improve fundic accommodation (12). In our study, patients with low IMD suggesting more rapid transit into the antrum from impaired fundic accommodation tended to improve with buspirone. Buspirone tended to produce greater fundic accommodation as indicated by a change in the IMD-0 at 4 weeks in compared to placebo. The third physiologic measure assessed in this study was the volume of water consumed during the 5-minute WLST, which assesses gastric capacity. Patients consuming a low volume of water at enrollment tended to have a better improvement in ES/PPF compared to patients consuming normal quantity of water of water. There was no significant improvement in the volume of water consumed with buspirone treatment. Future studies may want to specifically study the group of patients with impaired accommodation, perhaps using low volume of water consumed during the WLST.
Buspirone is approved for treatment of anxiety. We used the HADS to monitor both anxiety and depression during the study. Buspirone treatment did not improve the anxiety score. Interestingly, there was an improvement in HADS depression in the placebo group compared to the buspirone group; the drug significantly negated the placebo effect on depression.
Our study in patients with symptoms of gastroparesis can be compared to that of Tack et al reporting on the effects of buspirone in patients with functional dyspepsia; which was a much smaller trial using a crossover study design (12). Their conclusions were that in patients with FD, 4-weeks of administration of buspirone improved symptoms and gastric accommodation, compared with placebo, whereas gastric emptying of liquids was delayed. Our placebo-controlled study with parallel treatment groups did not confirm improvement in symptoms or change in gastric emptying or IMD using twice the number of patients. We used different procedures to assess gastric emptying and fundic accommodation. We studied patients with symptoms of gastroparesis, allowing both delayed and normal gastric emptying. Although we did not collect the ROME IV classification data in our study, the vast majority of our patients probably also had functional dyspepsia, as described in our registries of patients with symptoms of gastroparesis (2). We did allow patients to take a small amount of opioids (<3 days per week), the use of narcotics during the trial was minimal and balanced between treatment groups.
The study was stopped prematurely, prior to enrollment of the 108 patients which was the calculated sample size needed. The study was performed during the years of the COVID pandemic, which affected recruitment during this time period. Other factors that impeded enrollment included the common use of symptom modulators in this patient group, as well as the common use of buspirone, a prescription available drug. It is unlikely enrolling more patients would have allowed us to meet our primary objective based on the conditional power analysis. Other possible limitations of our study include poor adherence to the study medication during the study as only 60% patients were taking at least 80% of their medication. This low adherence was not different between treatment groups and may reflect the severity of symptoms in our patient population. In addition, not all the patients completed the primary outcome assessment at the end of the study, generally due to the Covid-19 clinic restrictions. For these patients, we imputed values using an analysis method assuming that the missing data is at random. This method allows the use of a continuous change measure; while setting all patients’ without an outcome as “failure” would require the use of a binary outcome not specified in the protocol, and would likely ”bias” the results against the test treatment which might result in a fallacious finding that buspirone made outcomes significantly worse, which is not what the data show.
In summary, this randomized, double-blind, placebo controlled trial investigated the symptomatic benefits of buspirone in patients with symptoms of gastroparesis with at least moderate to severe symptoms of early satiety and postprandial fullness. Patients did not benefit from 4-weeks of buspirone treatment to improve in the ES/PPF primary outcome compared with placebo. In secondary post hoc analyses, there was a suggestion of benefit with buspirone in these patients with more severe bloating. While more evidence is needed, buspirone may be considered for treating patients with moderate to severe fullness symptoms and severe bloating. Further study in a larger group of patients, perhaps targeting diabetic patients with severe to very severe bloating symptoms or impaired fundic accommodation is warranted.
Funding:
The NIH/NIDDK Gastroparesis Clinical Research Consortium (GpCRC) is supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (grants U01DK073975 [Parkman], U01DK073983 [Pasricha], U01DK074007 [Abell], U01DK073974 [Koch], U01DK074035 [McCallum], U01DK112193 [Kuo], U01DK074008 [Tonascia]).
Footnotes
ClinicalTrials.gov number, NCT03587142
Competing Interests: the authors have no competing interests.
References
- 1.Parkman HP, Hasler WL, Fisher RS. American Gastroenterological Association technical review on the diagnosis and treatment of gastroparesis. Gastroenterology 2004;127:1592–1622. [DOI] [PubMed] [Google Scholar]
- 2.Parkman HP, Hallinan EK, Hasler WL, et al. ; NIDDK Gastroparesis Clinical Research Consortium (GpCRC). Early satiety and postprandial fullness in gastroparesis correlate with gastroparesis severity, gastric emptying, and water load testing. Neurogastroenterol Motil 2017. Apr;29(4). doi: 10.1111/nmo.12981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stanghellini V, Tack J. Gastroparesis: separate entity or just a part of dyspepsia? Gut 2014;63:1972–8. [DOI] [PubMed] [Google Scholar]
- 4.Pasricha PJ, Colvin R, Yates K, Hasler WL, Abell TL, Ünalp-Arida A, Nguyen L, Farrugia G, Koch KL, Parkman HP, Snape WJ, Lee L, Tonascia J, Hamilton F and The NIDDK Gastroparesis Clinical Research Consortium (GpCRC). Clinical characteristics of patients with chronic unexplained nausea and vomiting and normal gastric emptying. Clin Gastroenterol Hepatol 2011;9:567–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelly KA. Gastric emptying of liquids and solids: roles of proximal and distal stomach. Am J Physiol 1980;239:G71–6. [DOI] [PubMed] [Google Scholar]
- 6.Azpiroz F Control of gastric emptying by gastric tone. Dig Dis Sci 1994;39:18S–9S. [DOI] [PubMed] [Google Scholar]
- 7.Troncon LE, Bennett RJ, Ahluwalia NK, Thompson DG. Abnormal intragastric distribution of food during gastric emptying in functional dyspepsia patients. Gut 1994;35:327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tack J, Piessevaux H, Coulie B, et al. Role of impaired gastric accommodation to a meal in functional dyspepsia. Gastroenterology 1998;115:1346–1352. [DOI] [PubMed] [Google Scholar]
- 9.Karamanolis G, Caenepeel P, Arts J, Tack J. Determinants of symptom pattern in idiopathic severely delayed gastric emptying: gastric emptying rate or proximal stomach dysfunction? Gut 2007;56:29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Youn YUH, Choi EJ, Lee YH, Oshima T, Miwa H, Park H. The effects of 5-hydroxytryptamine1a receptor agonist, buspirone on the gastric fundus accommodation in an animal model using guinea pigs. Neurogastroenterol Motil 2015;27:532–41. [DOI] [PubMed] [Google Scholar]
- 11.van Oudenhove L, Kindt S, Vos R, Coulie B, Tack J. Influence of buspirone on gastric sensorimotor function in man. Aliment Pharmacol Ther 2008;28:1326–1333. [DOI] [PubMed] [Google Scholar]
- 12.Tack J, Janssen P, Masaoka T, Farré R, Van Oudenhove L. Efficacy of buspirone, a fundus-relaxing drug, in patients with functional dyspepsia. Clin Gastroenterol Hepatol 2012;10:1239–45. [DOI] [PubMed] [Google Scholar]
- 13.Rentz AM, Kahrilas P, Stanghellini V et al. Development and psychometric evaluation of the patient assessment of upper gastrointestinal symptom severity index (PAGI-SYM) in patients with upper gastrointestinal disorders. Qual Life Res 2004;13:1737–49. [DOI] [PubMed] [Google Scholar]
- 14.Revicki DA, Rentz AM, Dubois D, Kahrilas P, Stanghellini V, Talley NJ, Tack J. Gastroparesis Cardinal Symptom Index (GCSI): Development and validation of a patient reported assessment of severity of gastroparesis symptoms. Qual Life Research 2004;13:833–844. [DOI] [PubMed] [Google Scholar]
- 15.Svedlund J, Sjodin I, Dotevall G. GSRS--a clinical rating scale for gastrointestinal symptoms in patients with irritable bowel syndrome and peptic ulcer disease. Dig Dis Sci 1988;33:129–34. [DOI] [PubMed] [Google Scholar]
- 16.Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand 1983;67:361–370. [DOI] [PubMed] [Google Scholar]
- 17.Bjelland I, Dahl AA, Haug TT, Neckelmann D. The validity of the Hospital Anxiety and Depression Scale. An updated literature review. J Psychosom Res 2002;52:69–77. [DOI] [PubMed] [Google Scholar]
- 18.Kroenke K, Spitzer RL, Williams JB. The PHQ-15: validity of a new measure for evaluating the severity of somatic symptoms. Psychosom Med 2002;64:258–266. [DOI] [PubMed] [Google Scholar]
- 19.De La Loge C, Trudeau E, Marquis P, Kahrilas P, Stanghellini V, Talley NJ, Tack J, Revicki DA, Rentz AM, Dubois D. Cross cultural development and validation of a patient self-administered questionnaire to access quality of life in upper gastrointestinal disorders: the PAGI-QOL. Qual Life Res 2004;13:1751–1762. [DOI] [PubMed] [Google Scholar]
- 20.Ware JE Jr, Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med Care 1992;30:473–483. [PubMed] [Google Scholar]
- 21.Revicki DA, Camilleri M, Kuo B, Szarka LA, McCormack J, Parkman HP. Evaluating symptom outcomes in gastroparesis clinical trials: validity and responsiveness of the Gastroparesis Cardinal Symptom Index-Daily Diary (GCSI-DD). Neurogastroenterol Motil 2012;24:456–63. [DOI] [PubMed] [Google Scholar]
- 22.Koch KL, Hong SP, Xu L. Reproducibility of gastric myoelectrical activity and the water load test in patients with dysmotility-like dyspepsia symptoms and in control subjects. J Clin Gastro 2000;31:125–9. [DOI] [PubMed] [Google Scholar]
- 23.Koch KL, Van Natta M, Parkman HP, Grover M, Abell TL, McCallum RW, Shaltout HA, Sarosiek I, Farrugia G, Shulman RJ, Tonascia J, Miriel L, Hamilton F, Pasricha PJ; Gastroparesis Clinical Research Consortium. Effect of liquid and solid test meals on symptoms and gastric myoelectrical activity in patients with gastroparesis and functional dyspepsia. Neurogastroenterol Motil 2022. Apr 11:e14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tougas G, Eaker EY, Abell TL, et al. Assessment of gastric emptying using a low fat meal: Establishment of international control values. Am J Gastroenterol 2000;95:1456–1462. [DOI] [PubMed] [Google Scholar]
- 25.Abell TL, Camilleri M, Donohoe K, et al. Consensus recommendations for gastric emptying scintigraphy: a joint report of the American Neurogastroenterology and Motility Society and the Society of Nuclear Medicine. Am J Gastroenterol 2008;103:753–763. [DOI] [PubMed] [Google Scholar]
- 26.Piessevaux H, Tack J, Walrand S, Pauwels S, Geubel A. Intragastric distribution of a standardized meal in health and functional dyspepsia: correlation with specific symptoms. Neurgastroenterol Motil 2003;15:447–455. [DOI] [PubMed] [Google Scholar]
- 27.Orthey P, Yu D, Van Natta ML, Ramsey FV, Diaz JR, Bennett PA, Iagaru AH, Fragomeni RS, McCallum RW, Sarosiek I, Hasler WL, Farrugia G, Grover M, Koch KL, Nguyen L, Snape WJ, Abell TL, Pasricha PJ, Tonascia J, Hamilton F, Parkman HP, Maurer AH; NIH Gastroparesis Consortium. Intragastric Meal Distribution During Gastric Emptying Scintigraphy for Assessment of Fundic Accommodation: Correlation with Symptoms of Gastroparesis. J Nucl Med 2018;59:691–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simmons K, Parkman HP. Granisetron Transdermal System Improves Refractory Nausea and Vomiting in Gastroparesis. Dig Dis Sci 2014;59:1231–1234. [DOI] [PubMed] [Google Scholar]
- 29.Carpenter JR, Kenward MG. 2013. Multiple Imputation and its Application Chichester, UK: Wiley. [Google Scholar]
- 30.Little RJA, Rubin DB. 2020. Statistical Analysis with Missing Data 3rd ed. Hoboken, NJ: Wiley. [Google Scholar]
- 31.Pasricha PJ, Yates KP, Nguyen L, et al. Outcomes and Factors Associated With Reduced Symptoms in Patients with Gastroparesis. Gastroenterology 2015;149:1762–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hasler WL, Wilson LA, Parkman HP, Nguyen L, Abell TL, Koch KL, Pasricha PJ, Snape WJ, Farrugia G, Lee L, Tonascia J, Unalp-Arida A, Hamilton F; NIDDK Gastroparesis Clinical Research Consortium (GpCRC). Bloating in gastroparesis: severity, impact, and associated factors. Am J Gastroenterol 2011;106:1492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hui R Gastroparesis: Clinical evaluation of drugs for treatment: Guidance for industry. Food and Drug Administration Center for Drug Evaluation and Research (CDER) July 2015.