

Summary
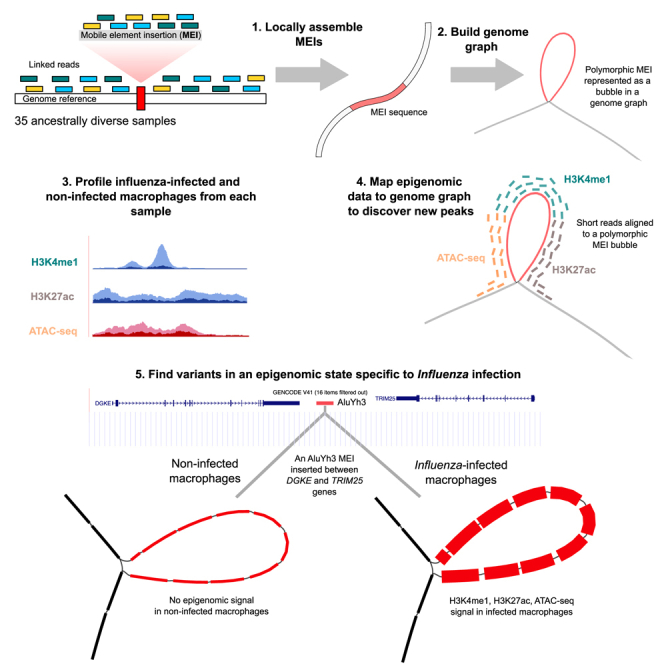
Genetic variants, including mobile element insertions (MEIs), are known to impact the epigenome. We hypothesized that genome graphs, which encapsulate genetic diversity, could reveal missing epigenomic signals. To test this, we sequenced the epigenome of monocyte-derived macrophages from 35 ancestrally diverse individuals before and after influenza infection, allowing us to investigate the role of MEIs in immunity. We characterized genetic variants and MEIs using linked reads and built a genome graph. Mapping epigenetic data revealed 2.3%–3% novel peaks for H3K4me1, H3K27ac chromatin immunoprecipitation sequencing (ChIP-seq), and ATAC-seq. Additionally, the use of a genome graph modified some quantitative trait loci estimates and revealed 375 polymorphic MEIs in an active epigenomic state. Among these is an AluYh3 polymorphism whose chromatin state changed after infection and was associated with the expression of TRIM25, a gene that restricts influenza RNA synthesis. Our results demonstrate that graph genomes can reveal regulatory regions that would have been overlooked by other approaches.
Keywords: epigenomics, mobile elements, pangenomics, influenza
Graphical abstract
Highlights
-
•
We built a genome graph for 35 individuals with transposable element polymorphisms
-
•
Mapping epigenomic data to the genome graph revealed between 2% and 3% novel peaks
-
•
Some new peaks were near immune genes and impacted quantitative trait loci estimates
-
•
We found peaks within reconstructed polymorphisms such as in an AluYh3 near TRIM25
Groza et al. created a cohort genome graph that represents the genetic variation in 35 ancestrally diverse individuals. They used the genome graph to reveal novel peaks that were hidden by reference bias and to find the epigenomic state of mobile element insertions that are eQTLs for genes that regulate antiviral innate immunity. Their finding also suggests that genome graphs may improve hQTL and caQTL discovery.
Introduction
Structural variants (SVs) contribute the largest number of variable nucleotides in an individual,1 have larger effect sizes on gene expression,2 and are associated with functionally relevant epigenetic differences between humans and chimpanzees.3 A particular class of SVs, mobile element insertions (MEIs), likely influence the epigenome since fixed mobile elements are known to harbor transcription factor binding sites4,5 and have contributed primate-specific regulatory regions.6 The epigenetic features that occur on SVs are not immediately accessible when mapping to a linear and incomplete reference genome7,8 but could potentially be accessed using a graph genome. Indeed, using the personal genome of a single individual, we have shown previously that genome graphs can recover epigenomic signals in genetically variable regions of the genome.9 Genome graph approaches have also been used to find differential CpG methylation within SVs in twelve medaka fish genomes.10
Obtaining accurate maps of SVs with short-read libraries can be challenging for several reasons.11 First, repeats are abundant in eukaryotic genomes, and resolving variation in these regions can be more difficult since read mapping is often ambiguous. Second, mapping short reads reveals only the break points of insertions and does not provide their actual sequence without assembling the reads into larger contigs. Third, short-read assembly algorithms cannot distinguish between highly similar sequences and tend to collapse copy-number variation. To mitigate these shortcomings, paired-end and linked-read libraries12 have been developed. Linked-read libraries go further than paired-end libraries by labeling each read with a barcode that represents the DNA fragments from which it originates. This provides long-range positional information in regions of the human genome that cannot be reached by short reads alone. Linked reads have been used to genotype,13,14,15 identify SVs,16,17,18 detect MEI polymorphisms,19 and assemble genomes.20,21
To test the ability of graph genomes and local de novo assembly to recover the epigenomic state of SVs, we used data obtained from monocyte-derived macrophages from 35 individuals of African and European descent before and after in vitro influenza virus (IAV) infection.22 This included whole-genome sequencing (WGS) data together with H3K4me1 and H3K27ac chromatin immunoprecipitation sequencing (ChIP-seq), ATAC-seq, and RNA-seq data to characterize the transcriptome and the chromatin state. Given that macrophages are important effector cells of the innate immune response23 and that mobile elements have been found to be co-opted in innate immunity,24 we decided to also apply these methods on the epigenome of genetic variants and MEIs in the response to IAV infection. First, we developed a new approach to build a genome graph that includes MEIs by resolving the sequence of insertions using locally assembled linked reads (Figure S1). We validated the method by generating ChIP-seq and ATAC-seq data from the NA12878 benchmark genome25 following the protocols used in Aracena et al.22 Next, we generated linked-read data for the 35 individuals in the IAV-infected cohort and applied our new approach to build a graph that includes SNPs, insertions or deletions (indels), and MEIs. Using this genome graph, we showed that we could identify regulatory sequences that would have been missed otherwise.
Results
Adding MEIs to the NA12878 genome graph reveals additional epigenomic signal
We chose the NA12878 genome to develop and benchmark our approach since WGS, linked reads, and a haplotype-resolved assembly were already available.25 First, we ran MELT and ERVcaller on paired-end WGS data to identify and genotype 2,175 Alu, 351 LINE1, 106 SVA, and 6 ERV insertions (Figure 1A). Of these calls, 66% (1,738) were previously observed by Ebert et al.26 MELT and ERVcaller could only predict the breakpoints of MEIs, and to obtain the sequence of these insertions, we developed a local linked-read assembly tool, which we called BarcodeAsm (STAR Methods). By applying this tool, we successfully assembled the sequences of 1,054 Alu, 117 LINE1, 17 SVA, and 35 ERV instances (Figure 1B), sometimes recovering both copies of homozygous insertions corresponding to 963 loci (Figure 1A, right). We also assembled an excess of ERV-annotated insertions that tend to be shorter and more divergent in their sequences from consensus ERVs (Figure S2). Next, to validate these assembled insertions, we aligned them against the haplotype-resolved assembly of NA12878 and calculated the proportion of their length that matched (Figure 1C). Despite expecting some errors in our local assembly and in the de novo assembly, we found that 925 loci (96.1%) had a match over 95% of their length. These matches were also equally distributed between the two haplotypes of the de novo assembly, which is consistent with the phasing of the assembly. We also validated 6 MEIs by Sanger sequencing (STAR Methods). When comparing the Sanger sequence27 with the assembled sequence, three MEIs showed 100% similarity. Two MEIs showed similarity, 99.4% and 91.4%, respectively, due to uncalled “N” nucleotides in the Sanger sequence. Only one MEI showed 46.5% similarity due a stretch of low-complexity “CT” repeat in the MEI that aligns poorly.
Figure 1.

Adding MEIs to the NA12878 genome graph reveals additional epigenomic signal
(A) The number of MEI breakpoints, or transposable element (TE), detected using ERVcaller and MELT and the full-length MEIs recovered by BarcodeAsm.
(B) The reassembled families ordered by frequency in the NA12878 genome. Homozygous insertions that are assembled twice are double counted.
(C) The spans of MEI contigs that could be matched and confirmed in the haplotype-resolved assembly of NA12878.
(D) Change in the number of true positive alignments relative to the previous genome in increasingly complete genomes of NA12878, starting with the reference as the baseline.
(E) Proportion and number of peaks on MEI (full line) that were called at least once, twice, and three times in the replicates. Proportion also shown for all peaks (dashed lined).
(F) MEI and reference allele coverage at the peak calls that overlap MEIs, stratified by MEI (color) and replicate (shape).
Previously, we introduced an axis that orders reference genomes according to how similar they are to the truth.9 This axis ranged from the least accurate sequence (the reference genome) to the most representative (the de novo assembly). We now define multiple genome graphs as intermediate stages along this axis: a +SNVs graph containing only SNVs, a +indels graph containing SNVs and indels (137 bp maximum length, 3 bp mean length), and a +MEIs graph containing SNVs, indels, and MEIs (6,074 bp maximum length, 288 bp mean length; STAR Methods). We also created a de novo graph with all the variants called in the NA12878 haplotype-resolved assembly. Next, we simulated 600 million standard WGS reads from this de novo graph to be used as a set of true alignments and also realigned them to the genomes along this axis and compared the positions (STAR Methods). As expected, we observed that the number of true positive alignments increased as the graphs became more complete (Figure 1D). The +SNVs graph (with SNVs) correctly finds around more true alignments (+0.062 per SNV) compared to the reference graph. The +indels graph (with indels), adds true alignments (+0.063 per indel) over SNVs alone. The +MEIs graph (with 963 MEIs) adds another true alignments (+14.5 per MEI) on top of SNVs and indels. These results recapitulate findings in Garrison et al.,28 where allelic bias is small in SNVs and short indels but much larger in longer indels. Therefore, the +MEIs graph reliably maps reads to MEIs while maintaining a false positive rate that is lower than the reference graph (Figure S3). Finally, the de novo graph outperforms our best genome by true alignments since it represents even more SVs.
Having established that our genome graph recovers more true mappings, we looked for MEIs that support active histone marks and chromatin accessibility signals. To mimic the data obtained from Aracena et al.,22 we used cells derived from NA12878 and generated three replicates for H3K4me1 and H3K27ac ChIP-seq and ATAC-seq (STAR Methods). These data were then mapped to the MEI graph, and we called peaks using Graph Peak Caller.29 We observed 58 H3K4me1, 22 H3K27ac, and 19 ATAC-seq peaks that overlapped MEIs in at least one of the replicates (Figure 1E). Similar to other peaks, roughly half were observed in all three replicates. This proportion of unique peaks among replicates is inflated by the large range of library sizes, the smallest library counting 118 million reads and the largest 378 million reads (Table S1). Most loci were covered on both the MEI and the reference allele, and a smaller subset were covered on only one of the alleles (Figure 1F). Of the MEIs previously confirmed with Sanger sequencing, we successfully applied ChIP-qPCR to one MEI (STAR Methods) and confirmed that it is indeed marked by H3K27ac (Figure S4). While the number of events is small in a single genome, they show that using a graph, we can profile the chromatin in regions that are missing from the reference.
Linked reads recover the sequences of polymorphic MEIs in a cohort
We looked to extend the method and apply it to a cohort with the 35 individuals that were exposed to IAV infection.22 First, using MELT and ERVcaller on the WGS data, we identified and genotyped 7362 Alu, 1344 LINE1, 649 SVA, and 19 ERV insertion loci (Figure 2A). Next, we generated linked-read data from the same individuals and introduced a population consensus approach before attempting to identify the final MEI sequence at each locus with BarcodeAsm (STAR Methods). Using this approach, we were able to assemble and annotate the insertions for 5140 Alu, 316 LINE1, 94 SVA, and 48 ERV (Figure 2A right panel). Again, we obtained an excess of ERV-annotated insertions that are shorter and more divergent from consensus ERVs (Figure S5A). Looking closer, Alu-, L1-, and SVA-annotated insertions are closer to consensus than ERV sequences (Figure S5B), indicating that RepeatMasker is less reliable in annotating some ERV subsequences. Nonetheless, we kept these variants as incidental findings in the local assembly windows.
Figure 2.

BarcodeAsm recovers the sequences of polymorphic MEIs in a cohort
(A) The number and family of MEIs genotyped from short-read sequencing data using ERVcaller and MELT in the entire cohort.
(B and C) The lengths of assembled MEIs (B) below 800 bp and (C) above 800 bp.
(D) The reassembled families ordered by frequency in the cohort.
(E) Number of MEIs inserted in enhancers or exons.
(F) The observed population structure in MEI genotypes as projected by principal-component analysis.
The population consensus approach allowed us to recover a larger fraction (60% versus 37%) of MEIs because the number of attempts to reassemble a locus is equal to the frequency of the MEI allele in the cohort. Consequently, while singleton MEIs are the most numerous, they are also the least likely to be assembled (Figure S6). As expected, the length distributions of the consensus insertions identified include the Alu peak at 300 bp and a long tail associated with longer truncated and full-length MEIs such as the LINE1 (Figures 2B and 2C). The resulting multiple sequence alignments showed few ambiguous nucleotides, suggesting that the consensus insertions are representative of most samples (Figure S7A). When we ordered mobile element families based on abundance (Figure 2D), we observed unsurprisingly that AluY families were the most common among Alus and that the L1HS subfamily was ranked highly.30,31 Similarly, the human-specific SVA_F subfamily32 was found to be the most abundant among the SVA families in our dataset.
Next, we looked at the distance between MEI polymorphisms and the nearest exon, enhancer, and repetitive sequences in the reference genome (STAR Methods). We found that 87 polymorpisms, mostly Alus, were within the bodies of exons (Figure 2E), which is notable since Alus are associated with alternative transcription events.33 We also observed 135 polymorphisms located within enhancers (Figure 2E) and that more than half of the polymorphisms (2,941) were nested within a repetitive sequence. Overall, the distribution of these MEIs around exons, enhancers, and other repeats is similar to those in the 1000 Genomes Project34 (Figures S7B–S7D). Lastly, we checked if the insertions that we reconstructed with BarcodeAsm and their genotype would recapitulate the known African and European ancestry structure of the cohort. We found two clusters in the principal-component analysis (Figure 2F) that were consistent with the genetic ancestry and what we see from SNV calls from WGS data (Figure S8). Overall, this confirms that we were able to recover a high-quality set of MEIs for our cohort together with their assembled sequences.
Genome graphs increase the number of peak calls and impact quantitative trait locus (QTL) estimates
We wanted to explore the extent to which a cohort genome graph would impact read mapping and peak calling for epigenomic datasets. As expected,34 we observed more variants relative to the reference in samples of African ancestry (Figure 3A). We note that multiple ancestries may contribute to a genome. Genomes labeled as African ancestry in this cohort feature 13%–57% European ancestry, and genomes labeled as European ancestry feature 0.001%–0.69% African ancestry.22 Combining all these variants, we built a genome graph containing a total of SNVs, insertions, deletions, and MEIs. We then mapped ChIP-seq and ATAC-seq datasets before and after IAV infection22 and observed a decrease in unmapped reads and an increase in perfectly mapped reads that can reach up to 0.15% of reads in a dataset (Figure 3B). Next, we called peaks using the reference genome and the cohort genome graph, treating the infected and non-infected datasets of each genome as independent samples. Peaks called with both the cohort and the reference graph in the same genome and in the same condition were called common peaks, while those called only in the reference graph are ref only and those called only in the cohort graph are graph only. Among H3K4me1 samples, we observed an average of 4,700 (2.5%) graph-only peaks and 2,200 (1.2%) ref-only peaks per sample (Figures 3C and S9). We have shown before in Groza et al.9 that graph-only peaks are enriched in reads that were previously unmapped in the reference genome, as expected from previous applications of genome graphs.28,35 These additional reads push peaks above the significance threshold.9 Graph-only peaks being approximately twice as numerous as ref only is also consistent with what was observed previously.9 For H3K27ac, we counted 1,800 (3.0%) graph-only peaks and 1,100 (1.9%) ref-only peaks (Figure S10) on average. Among ATAC-seq datasets, graph-only events averaged 4,000 (2.3%) peaks in flu-infected samples and ref-only events averaged 2,400 (1.4%) peaks per sample. In non-infected samples, the same numbers and proportions are roughly halved (Figure S11). We suspect that this is linked to cell death in flu-infected samples introducing cell-free DNA and more background in the ATAC-seq library preparation. Consistent with this hypothesis, infected samples show an excess of low-quality peaks relative to non-infected samples (Figures S12A and S12B). We successfully applied ChIP-qPCR (STAR Methods) to 5H3K27ac graph-only peaks in the NA12878 cell line and confirmed that 4 of these calls are indeed above the background (Figure S4).
Figure 3.

Genome graphs with millions of variants impact downstream results
(A) Number of SNVs, insertions, deletions, and MEIs in the cohort graph for each sample.
(B) Changes in the distribution of read mapping quality when using a cohort genome graph relative to the reference genome. Each point is an individual sample.
(C) The number of H3K4me1 graph-only, ref-only, and common peaks between the cohort and the reference genome graphs, stratified between flu-infected and non-infected (NI) read sets.
(D) Inverse cumulative distributions describing how many peaks are observed in more than a number of samples. Curves that are expected by chance are also shown (dashed lines).
(E) Immune-related Gene Ontology descriptions of genes within 10 Kbp of graph-only peaks. One gene may contribute multiple descriptions.
(F) Distributions showing how caQTL effect sizes and p values (infected condition) changed when we used a genome graph instead of the reference genome. First and second vertical lines mark the 99th and 99.9th percentiles.
To confirm whether graph-only peaks were associated with sequence variants, we estimated the influence of genotype on common and graph-only peaks by logistic regression on SNPs and indels within the peaks while controlling for peak width (Table S2). We found that the log odds for a peak to be graph only increased by 0.11–0.19 when SNPs were present and by 0.45–0.56 when indels were present. Overall, this confirms that indels have a stronger influence on peak calling compared with SNPs, as expected from previous results linking longer alleles with more allelic bias when aligning reads to reference genomes.28 A similar analysis could not be performed with MEIs because of the small numbers. This multi-sample epigenomic dataset was also an opportunity to better understand how reliable the graph-only peaks were compared with common peaks. We contrasted the population frequency of graph-only, ref-only, and common peaks with a peak replication cumulative distribution curve (STAR Methods). H3K4me1, H3K27ac, and ATAC peak sets generated very similar curves, with common peaks having the longest tails, followed by graph-only and ref-only peaks, with the curves expected by chance decaying the fastest (Figures 3D, S13A, and S13B). Under the random simulations, none of the peaks were observed in more than 20 datasets, but a proportion of graph-only peaks were replicated in more than 40 datasets (each individual has a non-infected and an infected dataset).
To confirm the relevance of the graph-only peaks, we retrieved the ontological descriptions of genes within 10 Kbp of a peak (STAR Methods). Indeed, there are many instances where H3K27ac, H3K4me1, and ATAC graph-only peaks appear near genes that are involved in the immune response (Figure 3E). For example, 281, 203, and 249 genes related to “positive regulation of NF-κB” had graph-only peaks for H3K4me1, H3K27ac, and ATAC, respectively. When checking for enrichment against the GO:Biological Process database, we found that graph-only peaks, like common peaks, were similarly functionally enriched for immune biological processes (Figure S14). When genes near common peaks are used as a background for genes near graph-only peaks, there is no statistically significant enrichment of pathways.
Finally, the use of a genome graph could affect methods to identify QTLs, which aim to characterize the impact of genetic variants.36 To measure this, we mapped caQTLs (chromatin accessibility QTLs) and hQTLs (H3K4me1 and H3K27ac histone modification QTLs) using reference-based and graph-based read-count estimates in a set of consensus peaks across all samples (STAR Methods). We measured how the effect sizes and p values change when using the graph genome instead of the reference genome (in the same condition) and found that while most QTLs remained the same, some do change. For example, when mapping caQTLs, the estimated effect size changes by 1.4 or more for one QTL in 1,000, and the observed p value (as ) changes by 7.9 or more (Figure 3F; see Figure S15 for all marks and conditions). The mapping of H3K4me1-QTLs and H3K27ac-QTLs is similarly affected (Figure S15). We checked the genes that are nearby these top changing QTLs and found enrichment in pathways, some related to immunity, relative to QTLs that remain the same (Figure S16). For example, the gene HLA-DQA1 is near such changing QTLs for H3K4me1, H3K27ac, and ATAC-seq. Therefore, removing reference bias from the analysis of epigenomic data using genome graphs can reveal novel peaks and improve QTL discovery.
Genome graphs measure epigenomic signal on MEIs
We wanted to focus next on the MEIs that were assembled and introduced in the cohort genome graph. First, we took advantage of the genotypes of the individuals in the cohort to see how specific read mapping was in these repetitive and polymorphic sequences. We did this by aligning WGS reads to the genome graph and regenotyping the MEIs in each sample (STAR Methods). Reassuringly, we find that the graph genotypes are highly consistent ( score 0.93 over all samples) with the calls made by MELT and ERVcaller (Figure 4A). When genotyping, the graph recapitulates between 1,274 and 1,563 genotypes per sample and only misses 60–95 polymorphisms. The graph also gains between 110 and 196 polymorphisms per sample. These gained genotypes are not necessarily false positives and could be explained by an increase in sensitivity since the exact location and sequence of these polymorphisms were known a priori with the graph genotyping algorithm but needed to be found de novo by MELT and ERVcaller. Having confirmed that read mapping was reliable in MEIs, we extracted H3K4me1 and H3K27ac ChIP-seq and ATAC-seq peaks overlapping MEIs (Figures 4B, S17A, and S17B). The peaks were labeled either as reference peaks, biallelic peaks, or MEI peaks (binomial test, STAR Methods). Notably, we found 714 MEI peaks for H3K4me1, 191 for H3K27ac, and 316 for ATAC-seq. As an additional negative control, we repeated the same with peaks in MEI loci for which the samples were homozygous references. We found that reads in these peaks were overwhelmingly assigned to the reference allele (Figures S17C–S17E).
Figure 4.

Genome graphs measure epigenomic signal on MEIs
(A) Assembled MEIs that were regenotyped using the cohort genome graph. Graph genotypes are compared with the previous genotypes that were called with ERVcaller and MELT.
(B) Partitioning of reads between the reference and alternative allele in peaks that overlap heterozygous or homozygous MEIs.
(C) TE families that support peaks in at least one sample. Singletons not shown.
(D) Number of graph-only peaks that overlap MEIs in each sample, with total number across genomes annotated at the top.
(E) A graph region that represents an MEI locus. The black nodes represent the reference sequence, and the red nodes represent the Alu insertion carried by some samples. The numbers show the average number of H3K4me1 reads that were mapped to each nucleotide over the span of the node. Nodes are at most 32 bp long.
MEI peaks for H3K4me1 and H3K27ac were mostly from AluY families or other Alu elements (Figure 4C). L1 (L1HS, L1PA2) and ERV (LTR5_Hs) polymorphisms also support a small number of peaks. Furthermore, we detected 30 SVA polymorphisms in open chromatin states (Figure 4C). Notably, Chen et al.37 found that SVA families are overrepresented in open chromatin in macrophages infected with influenza and are variable between individuals (Figure S18). Consistent with this, the overwhelming majority of ATAC-seq peaks that lie on SVA polymorphisms were detected in the infected condition (92 of 108 peaks, 85.2%). Finally, we counted how many of these peaks were detected with the graph genome and would have been missed by a traditional approach. In total, we tallied 366 H3K4me1, 161 H3k27ac, and 320 ATAC graph-only peaks (Figure 4D) on MEIs. These graph-only peaks on MEIs were supported by 168 Alus, 30 SVAs, and 8 ERVs. Altogether, 22% H3K4me1 MEI peaks, 44.5% H3K27ac MEI peaks, and 44% ATAC MEI peaks were graph only. On the other hand, MEIs rarely disrupt peaks in the reference graph (Figure S19). We show one Alu polymorphism in the graph that supports a H3K4me1 graph-only peak (Figure 4E) and its linear surjection (Figure S20).
Cohort data reveal MEIs that act as potential enhancers
So far, we have focused on detecting single MEI alleles that support chromatin marks in individual genomes. Next, we wanted to obtain an overview of MEIs at the level of the entire cohort. In total, we identified 375 MEIs that carry at least one epigenomic peak. Specifically, 218 MEIs support H3K4me1 peaks, 90 support H3K27ac peaks, and 210 support ATAC peaks (Figure 5A), many of which were found in more than one sample (Figure S21). Among the H3K4me1 MEI peaks, 58 were unique to flu-infected samples, 41 were unique to non-infected samples, and 119 were found in both conditions (Figure 5B). Peaks in H3K27ac and ATAC-seq were also well balanced between the two conditions. Ordering MEIs by allele frequency shows variable levels of occupancy by peaks (Figure 5A, right side).
Figure 5.

Cohort data reveal MEIs that act as potential enhancers
(A) Summary of MEIs (rows) that support H3K4me1, H3K27ac, and ATAC peaks in the cohort samples (columns). Occupancy is the ratio between samples that support peaks on the MEI (N, red) and those that carry the MEI (N, blue).
(B) Venn diagrams showing MEI peaks that are shared between flu-infected and NI conditions.
(C) Upset plot describing the number of MEIs that support a combination of H3K4me1, H3K27ac, and ATAC peaks, annotated by transposable element class.
(D) The annotated positions of genome-wide peaks and MEI peaks in the genome, with uniformly and randomly sampled genome positions for comparison. Peaks near genes are within 10 Kbp of a gene boundary.
Next, we looked for MEIs that carry combinations of epigenomic marks that are characteristic of enhancer sequences (Figure 5C). For example, 56 MEIs supported both H3K4me1 and ATAC, a combination that suggests poised enhancers in open chromatin. Similarly, 16 instances showed H3K27ac and ATAC and 26 polymorphisms were marked by H3K4me1, H3K27ac, and ATAC, which is strong evidence for active enhancers in open chromatin. There is a noticeable number of SVA elements showing ATAC peaks, second only to Alu elements. We find that peaks on MEIs follow the expected distribution of the epigenomic mark relative to exons, introns, genes, and enhancers (Figure 5D) and are not randomly distributed around the genome (see the expected random distribution). Additionally, we find dozens of MEI loci that are in the vicinity of genes associated with “viral processes,” “immune system processes,” the “inflammatory response,” and the “positive regulation of NF-κB” (Figure S22). For example, an Alu polymorphism that supports H3K27ac peaks (Figure S23A) in 21 samples is immediately upstream of CD300E, an immune-activating receptor gene.38 This frequent MEI peak is also located within DNase and transcription factor clusters (Figure S23B), which is further evidence for active chromatin.
Finally, since Alu polymorphisms could alter gene transcript levels,39 we asked if any MEIs were gene expression QTLs (eQTLs), separately in each infected or non-infected condition. We mapped MEI-eQTLs and found 18 MEIs in the flu-infected condition and 34 MEIs in the non-infected condition that were associated with gene expression (false discovery rate [FDR] lower than ; STAR Methods). In total, there are also 354 MEIs that are caQTLs/hQTLs for at least one mark or condition (Figure S24). Of the MEI-eQTLs, 3 have a supporting caQTL or hQTL in the flu-infected condition and 9 in the non-infected condition (Table S3). In particular, in the flu-infected condition, we detected an AluYh3 MEI that is an eQTL for DGKE and TRIM25, a gene that restricts influenza RNA synthesis,40 and is also a QTL for 2 H3K4me1 peaks, 2 H3K27ac peaks, and 3 ATAC peaks (Table S4). We show the average read depth at this locus in flu-infected samples that carry the MEI (Figure 6A), in flu-infected samples that do not carry the MEI (Figure 6B), in non-infected samples that carry the MEI (Figure 6C), and in non-infected samples that do not carry the MEI (Figure 6D). The more traditional linear projection of the average read depth at this locus confirms that the signal is higher in flu-infected samples that carry the MEI when using an accurate genome graph (Figures 6E and 6F). Note that there is an unavoidable amount of distortion when projecting graph alignments over long polymorphisms back to the reference genome, where reads partially in the insertion are truncated and reads fully in the insertion are not visible in the projection. Overall, this MEI exists in a flu-specific active chromatin state, is associated with TRIM25 and DGKE gene expression, and would have been missed with the linear reference genome.
Figure 6.

Example of flu-specific and genotype-specific peak on an MEI
(A–D) Average H3K27ac read depth in the locus of an AluYh3 MEI-eQTL in flu-infected samples that carry the insertion, (B) in flu-infected samples that do not carry the MEI, (C) in NI samples that carry the MEI, and (D) in NI samples that do not carry the MEI. The read depths of homozygous nodes were halved before averaging. Reads below a MAPQ of 10 were not counted.
(E) A genome browser view of the read depth after projecting alignments onto the linear genome, contrasting the alignments to the graph genome and the reference genome.
(F) Nearby genes that are associated with this MEI-eQTL (DGKE, TRIM25). The gray strips denote the position of the MEI.
Discussion
We have constructed a genome graph to encapsulate the genetic diversity of a cohort of 35 individuals by resequencing their genome with linked reads. These data allowed us to reliably discover and phase SNVs, small indels, and a large number of MEIs, the latter being through the development of a local assembly method.
We showed that our cohort genome graph improves read mapping for epigenomic data. This led to a net increase in the number of peak calls, which are replicated across samples and occur in functionally interesting loci such as in the vicinity of immune genes. We also see ref-only peaks that emerge when pileups shrink in the graph and fall below the significance threshold, primarily because the reads move to a better-matching position in the graph or because the locus is less mappable due to another similar sequence in the graph. In addition, we showed that using a genome graph had an impact on the discovery of histone and chromatin accessibility QTLs in polymorphic regions.
Furthermore, we used the cohort genome graph to regenotype 5,598 MEIs using short-read WGS data and reliably assign epigenomic signals to MEI alleles. While rare, we sometimes observed heterozygous MEIs where the majority of the epigenomic signal was either on the insertion or the reference allele, revealing homologous loci that are in different chromatin states. This shows that genome graphs can be exploited to search for an epigenomic signal that is specific to alleles in a cohort of individuals. Meanwhile, current methods that measure allelic-specific signals are limited in their ability to represent complex SVs or relate genomes within a population to one another. Therefore, genome graphs in combination with biological replicates to reduce noise could prove a powerful framework to study the chromatin state of polymorphic SVs in a large number of genomes.
Finally, with the recent completion of the first full human genome,41 there is a general appreciation of the need to use a new baseline for genomic and epigenomic analysis. While this genome is complete, it still represents only one possible haplotype in the human population. Therefore, graph genomes are needed to represent many haplotypes to capture genetic diversity. The Human Pangenome Reference Consortium published the first draft human pangenome,42 and new approaches are needed to demonstrate the benefits of these richer references. This human pan-genome graph is developed from a set of high-quality and diverse genome assemblies and enables access to a more comprehensive set of SVs. The hope is that similar analyses as were done here could be employed on a multitude of cell types and phenotypes to identify functionally relevant SVs without the challenges associated with the construction of cohort-specific genome graphs.
Limitations of the study
While we assembled and included 60% of all genotyped MEIs, these were skewed toward the shorter insertions and missed a significant portion of larger events. Indeed, out of the 1,344 L1 polymorphisms detected in the cohort, 1,048 (78%) could not be assembled and integrated into the genome graph because of the limitations of the short linked-reads technology. We anticipate that long-read technologies26 will be needed to construct more complete genome graphs that represent even more complex SVs.
Therefore, our results were restricted to regions where sequence variants were added the graph, which, based on our current approach, remain relatively small (SNPs, indels, and short MEIs). This means that our gains were mostly associated with narrow regions marked by histone modifications or chromatin accessibility. We anticipate that adding larger and more complex structural variations to the graph, such as segmental duplications, will reveal even more epigenomic features that are present but difficult to measure using traditional approaches.
Finally, much of the human pangenome is composed of sequences with lower mappability, which makes mapping short-read data from ChIP-seq and ATAC-seq in these regions less reliable. This limitation could be addressed by developing epigenomic assays on top of long-read sequencing technologies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| H3K27ac | Diagenode | Cat# C15410196; RRID: AB_2637079 |
| H3K4me1 | Cell Signaling | Cat# CST5326; RRID: AB_10695148 |
| Deposited data | ||
| WGS | European Genome-Phenome Archive | EGA: EGAD00001008359 |
| ChIP-seq and ATAC-seq | European Genome-Phenome Archive | EGA: EGAD00001008422 |
| NA12878 ChIP-seq and ATAC-seq | Gene Expression Omnibus | GEO: GSE225708 |
| Processed data | Zenodo | https://doi.org/10.5281/zenodo.6525192 |
| Validation data | Zenodo | https://doi.org/10.5281/zenodo.7429679 |
| Experimental models: Cell lines | ||
| GM12878 lymphoblastoid cell line | Coriell Institute for Medical Research | |
| Software and algorithms | ||
| BarcodeAsm | Zenodo | https://doi.org/10.5281/zenodo.5510086 |
| Vg | Github | https://github.com/vgteam/vg |
| Graph Peak Caller | Github | https://github.com/uio-bmi/graph_peak_caller |
| Figure and analysis code | Zenodo | https://doi.org/10.5281/zenodo.6525192 |
Resource availability
Lead contact
Guillaume Bourque is the lead contact author and may be reached at guil.bourque@mcgill.ca.
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
ATAC-seq and ChIPmentation library preparation
ATAC-Seq library preparation was performed according to the Omni-ATAC protocol.46 50,000 GM12878 cultured cells were resuspended in 1 ml of cold ATAC-seq resuspension buffer (RSB; 10 mM Tris-HCl pH 7.4, 10 mM NaCl, and 3 mM MgCl2 in water). Cells were centrifuged at 500 g for 5 min in a pre-chilled (4°C) fixed-angle centrifuge. After centrifugation, supernatant was aspirated and cell pellets were then resuspended in 50 l of ATAC-seq RSB containing 0.1% IGEPAL, 0.1% Tween-20, and 0.01% digitonin by pipetting up and down three times. This cell lysis reaction was incubated on ice for 3 min. After lysis, 1 ml of ATAC-seq RSB containing 0.1% Tween-20 (without IGEPAL and digitonin) was added, and the tubes were inverted to mix. Nuclei were then centrifuged for 10 min at 500 rcf in a pre-chilled (4°C) fixed-angle centrifuge. Supernatant was removed and nuclei were resuspended in 50 L transposition mix (2x TD Buffer, 100 nM final transposase, 16.5 L PBS, 0.5 L 1% digitonin, 0.5 L 10% Tween-20, 5 L H2O ). Transposition reactions were incubated at 37°C for 30 min in a thermomixer with shaking at 1000 rpm. Reactions were cleaned up with Zymo DNA Clean and Concentrator 5 columns. Primers (i5 and i7) were added by amplification (12 cycles) using NEBNext 2x MasterMix. Sequencing of the ATAC-Seq libraries was performed on the Illumina NovaSeq 6000 system using 100-bp paired-end sequencing.
ChIPmentation library preparation was performed on 5 million cross-linked cells (1% formaldehyde). After cell lysis, sonication of nuclei was performed on a BioRuptor UCD-300 targeting 150-500 bp size. Immunoprecipitation and library preparation for the histone marks H3K27ac (Diagenode antibody, cat # C15410196) and H3K4me1 (Cell Signaling antibody, cat # CST5326) was performed following the Auto-ChIPmentation protocol for Histones (Diagenode inc, Denville, USA) according to the manufacturer’s indications. Sequencing of the ChIPmentation libraries was performed on the Illumina NovaSeq 6000 system using 100-bp paired-end sequencing.
Whole genome sequencing with linked reads and genotyping
High molecular weight DNA was extracted using the MagAttract HMW kit from Qiagen and quantified using the Qubit dsDNA HS assay kit (ThermoFisher) for 35 samples. In order to generate the linked read libraries 1ng of HMW DNA was loaded on a 10x Chromium device using 10x Genome Sequencing Solution v2 reagents (10X Genomics). Illumina compatible libraries were prepared as per the 10x Genomics protocol and loaded as one library per lane on the Illumina HiSeqX instrument for 150bp paired-end sequencing. SNVs and indels were called using the longranger pipeline.
Validation of the MEIs by Sanger sequencing
Genomic DNA extraction was performed on 2 million GM12878 cells using the DNeasy Bood & Tissue kit (Qiagen). The genomic DNA was quantified using the Qubit 2.0 Fluorometer (Thermo Fisher Scientific).
In order to perform PCR amplification of the MEIs, a primer set for each region was designed using the Primer-BLAST tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast). The UCSC BLAT tool (https://genome.ucsc.edu/cgi-bin/hgBlat) was used to ensure that the primers were unique sequences, and in silico PCR was performed using the UCSC In-Silico PCR tool (https://genome.ucsc.edu/cgi-bin/hgPcr) to ensure that the primers were specific to the region to be amplified.
For each primer set, the PCR reaction was performed in a final volume of 25 L. The PCR mix contained 60 ng of genomic DNA, 0.25 L of Phusion High-Fidelity DNA polymerase (Thermo Fisher Scientific), 5 L of 5x Phusion HF Buffer Polymerase, 0.5 L of 10 mM dNTPs, 0.75 L of DMSO, 2.5 L of 5 M forward and reverse primers (final concentration: 0.5 M), and 11.5 L H2O. The PCR parameters were as follows : initial denaturation at 98° C for 30 s, 30 cycles of amplification (denaturation at 98° C for 10 s, annealing at 60° C, 65° C or 68° C (depending on the primer set used) for 30 s and extension at 72° C for 30 s) and final extension at 72° C for 5 min. The detection of each PCR amplicon was done by electrophoresis on 1.0% agarose gel. The PCR amplicons were Sanger sequenced at Centre d’expertise et de services Genome Québec, Montreal, Canada. The chromatogram trace analysis and the consensus sequence assembly were conducted with the Benchling software (https://www.benchling.com). The alignment of the consensus sequence with the template sequence was done with the EMBOSS Water tool (https://www.ebi.ac.uk/Tools/psa/emboss_water). The percentage of similarity between the consensus sequence and the template sequence was used as a metric to validate each MEI.
Validation of the H3K27ac peaks by ChIP-qPCR
ChIP (Chromatin immunoprecipitation) library preparation was performed on 5 million GM12878 cross-linked cells (1% formaldehyde). After cell lysis, sonication of nuclei was done on a BioRuptor UCD-300 targeting 150-500 bp size. The ChIP reaction was performed with H3K27ac antibody (Diagenode, cat # C15410196) on the Diagenode SX-8G IP-Star Compact robot using the Diagenode automated Ideal Kit reagents (C01010011). 5% of the sonicated chromatin volume used in the ChIP reaction was set aside as Input DNA sample. Reverse cross-linking of the ChIP’ed and Input DNA samples took place on a heat block at 65° C for 4 h, followed by a treatment with 2 L RNAse cocktail at 65° C for 30 min and 2 L Proteinase K at 65° C for 30 min. The ChIP’ed and Input DNA samples were then purified with a Qiagen MiniElute PCR purification kit as per the manufacturers’ protocol.
In order to perform qPCR amplification of the peak regions, a primer set for each region was designed using the Primer-BLAST tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast). The UCSC BLAT tool (https://genome.ucsc.edu/cgi-bin/hgBlat) was used to ensure that the primers were unique sequences, and in silico PCR was performed using the UCSC In-Silico PCR tool (https://genome.ucsc.edu/cgi-bin/hgPcr) to ensure that the primers were specific to the region to be amplified.
For each primer set, the qPCR reaction was performed in duplicate in a final volume of 15 L. The qPCR mix contained 4.5 L of ChIP’ed DNA, Input DNA or H2O (blank), 7.5 L of 2x SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) and 1.5 L of 5 M forward and reverse primers (final concentration: 0.5 M). The qPCR parameters were as follows: initial denaturation at 98° C for 2 min, 45 cycles of amplification (denaturation at 98° C for 5 s, annealing at 60° C for 30 s and extension at 72° C for 20 s). The qPCR product specificity was assessed by a melting curve analysis, which confirmed that a single melting peak was produced for each amplicon. The similarity between the experimental and predicted melting profiles was also confirmed with the uMelt tool (https://dna-utah.org/umelt/quartz). For larger amplicons (MEIs peak regions), since the melting curve analysis is not appropriate given the multiple melting peaks that can be generated by longer sequences, the product specificity was assessed by the validation of the size of the amplicon by electrophoresis on 1.0% agarose gel. The ChIP-qPCR data was normalized by the Percent Input method, which is an expression of the ratio of the number of target sequences measured in the ChIP’ed DNA sample to the number of targeted sequences in the Input DNA sample47:
where is the number of quantification cycles for the ChIP’ed DNA sample, is the number of quantification cycles for the Input DNA sample and DF (dilution factor) is the ratio of the quantity of sonicated DNA used in the ChIP reaction to the quantity of sonicated DNA set aside as Input DNA.
Method details
Locally assembling linked reads with BarcodeAsm
For the purpose of locally assembling linked reads, we wrote BarcodeAsm.43 The inputs to BarcodeAsm are a BED file describing the regions to be locally assembled and a BAM file that was aligned with lariat.17 This BAM file must be provided twice, once sorted by position and once sorted by barcode (with bxtools).
BarcodeAsm uses the position sorted BAM file to identify barcodes that are present in the target assembly window. Then it moves to the barcode sorted BAM file to retrieve all the reads that are tagged by the previously identified barcodes.
BarcodeAsm also allows filtering reads recovered from outside the local window (800 bp for Alus, 8000 bp for other TEs) by mapping quality. We introduced this feature because we expect reads that belong to novel transposable element insertions to be unmapped or mapped to the wrong copy, have very low mapping quality, or be multi-mapped.
Next, the fermi-lite48 library assembles the resulting collection of reads and creates a unitig graph, from which the contigs associated with the assembly window are extracted. Finally, BarcodeAsm aligns the contigs to the local window with minimap2.49 A select set of fermi-lite assembly parameters and minimap2 alignment parameters are exposed via the command line and can be adjusted to each application.
Reassembling transposable element insertions
ERVcaller50 and MELT51 were used to genotype novel insertions of Alu, LINE1, SVA, and ERV transposable elements using short reads. To further recover potential insertions within the same type of reference repeats (nested TE insertions), candidate nested insertions that were detected in other public datasets were not removed. For each genotype, local genomic windows were centered on the insertion site. These windows are 800 bp in length for the short Alu elements and 8 Kbp for the longer transposable elements. To optimize the outcomes of the assembly, BarcodeAsm was run separately on the short ( 300 bp) and long insertions ( 300 bp) with different parameters. For Alus, a minimum read overlap of 30 bp, a maximum mapping quality of 10 (as assigned by lariat), and a minimum k-mer frequency of 2 were required. For the larger MEIs, a minimum read overlap of 35, a maximum mapping quality of 20, and a minimum k-mer frequency of 8 were used instead. These parameters were found through a grid search approach and are expected to vary with different datasets.
For the NA12878 benchmark, the SRA accession numbers SRA: ERR174324, ERR174325 to SRA: ERR174341 were merged in order to genotype MEIs. MEIs were assembled from the public NA12878 linked reads hosted at support.10xgenomics.com/genome-exome/datasets/2.0.0/NA12878_WGS" title="https://support.10xgenomics.com/genome-exome/datasets/2.0.0/NA12878_WGS">https://support.10xgenomics.com/genome-exome/datasets/2.0.0/NA12878_WGS. Insertions were extracted directly from the BarcodeAsm output using scripts/alignment_to_vcf.py to create a VCF file.
For the larger cohort, a consensus sequence approach was used to take advantage of multiple MEI copies in the population (see BarcodeAsm/scripts/). Here, contigs that contain insertions are selected but not immediately used to recover an insertion. Instead, scripts/msa.py generates a multiple alignment using MUSCLE and calculates a consensus contig for a particular locus across samples. This consensus contig is aligned back to the local window to call the consensus insertion and to create a multi-sample VCF file using scripts/extract_consensus.py. For NA12878, the assembled contigs were validated by matching them against its haplotype resolved de novo assembly25 using minimap2 -H49 and selecting the haplotype with the best mapping quality.
Annotating assembled insertions
To annotate the insertions (without any flanking sequences), we used RepeatMasker with the Dfam52 database and the longest annotation was selected for each insertion. For each assembled TE polymorphism, the distance to the nearest enhancer in the GeneHancer annotation53 and the distance to the nearest exon in the GENCODE annotation54 were calculated. The same was done with the MEIs from the 1000 Genome Project34 and with an equal number of random positions sampled uniformly from the genome. The population structure of the MEI genotypes were compared to the population structure of WGS variants by running principal component analysis with SNPRelate.55
Creating and benchmarking genome graphs
The genomes graphs were generated with vg construct28 on VCF file containing SNVs, indels and MEIs. For the benchmark genome graph, the NA12878 Platinum callset56 was merged with the MEI VCF file. For the cohort genome graph, SNPs and indels were called using the LongRanger pipeline21 independently for each sample were merged with the population MEIs to create a multi-sample VCF file. A Nextflow script to generate the genome graphs from these inputs and the b37 reference genome is found in pop_graph.nf. The sensitivity and specificity of the resulting genome graphs was checked by aligning a matching but separate WGS dataset of the same samples (downsampling the merged read set by 5x in the case of NA12878) and removing non-specific alignments with vg filter -r 0.90 -fu -m 1 -q 10 -D 999. After, the assembled MEI snarls were genotyped with vg call -m2,457 and the graph genotypes were compared to ERV and MELT genotypes.
To evaluate the impact of the genome graphs on alignment, a WGS read set was simulated from the diploid assembly of NA12878. To achieve this, a genome graph was created from hg19 using the structural variant sequences called by the authors directly from the chromosome scale assembly. Paired-end reads were simulated using vg sim from this graph with a fragment size of 2000 bp, to ensure that we can access at least some of the long copy number variants that have low mappability. First, this read set was aligned to the hg19 reference graph. Then, the alignment of the simulated reads was repeated on increasingly complete genome graphs, first by including only SNVs, then indels and lastly MEIs. Finally, the simulated reads were aligned to the NA12878 de novo genome graph, which is the true genome by construction. The previous alignments are compared against the simulated alignment using vg gamcompare -r 100, where 100 is the maximum distance for two alignments to be considered mapped to the same position. Smaller values lead to more different mappings. We selected this parameter empirically by gradually increasing it until the number of different mappings between the two compared graph genomes stops decreasing.
Evaluating the impact of genome graphs on peaks
In parallel, ChIP-seq and ATAC-seq data was aligned to the reference graph to obtain reference peaks. The reference peaks were intersected with the graph peaks, which requires projecting the graph peaks onto the reference genome using graph_peak_caller peaks_to_linear. The projection may deform the exact location of peaks that overlap indels, and projects peaks wholly in an insertion onto the break-point of the insertion. These deformations are handled conservatively by considering peaks that overlap by even 1bp to be the same peak. The projected peaks were categorized into common peaks, graph-only and ref-only peaks. Common peaks are found with both the reference and cohort genome graph. Graph-only peaks and ref-only peaks are only found in the cohort graph or the reference graph respectively. A logistic regression model including peak width, the presence of an indel and the presence of a SNP was fitted using cv.glnmnet (see peak_variants.R).58 The number of common and graph-only peaks were balanced by subsampling common peaks. We report the cross-validation mean coefficients and median AUC. To summarize the population properties of peaks, curves were generated for common, graph-only and ref-only peaks (see Rscripts/peak_replication.R). The width of each peak was fixed to 200 bp and the proportion of samples that have a peak at the same location was calculated. The resulting curve is the inverse cumulative distribution of peak frequencies. A permutation simulation was performed to obtain the inverse cumulative distribution that is expected from random overlaps. In the simulation, a new peak set of the same size is randomly sampled for each individual from the set of all peaks and the overlaps are used to recompute the inverse cumulative distribution. The simulation was run 100 times and the average inverse cumulative distribution is reported.
QTL mapping
We filtered to exclude non-autosomal and non-biallelic variants. Additionally, we removed SNPs and MEIs that had a call rate of 90% across all samples, that deviated from Hardy–Weinberg equilibrium at , and with minor allele frequency less than 5%. This resulted in 7,383,243 SNPs and 1222 MEIs used for QTL mapping. We used the R package MatrixeQTL59 to examine the associations between SNP genotypes and chromatin accessibility, H3K4me1 and H3K27ac histone marks using both graph and reference read counts. We also mapped MEI-eQTLs to find association between mobile element insertion genotypes and gene expression counts (derived from alignments to the reference genome with STAR60). In each case, we calculated age and batch corrected expression matrices. Here, batch is a categorical variable. We calculated normalization factors to scale the raw library sizes using calcNormFactors in edgeR (v 3.28.1)61 and used the voom function in limma (v 3.42.2)62 to apply these factors to estimate the mean-variance relationship and convert raw read counts to values. We then fit a model using mean-centered age and admixture (see STAR Methods in Aracena et al. 202222), removing batch effects using ComBat from the sva Bioconductor package.63 In this study, global ancestry estimates were used solely to correct for population structure in QTL mapping and nothing else. We then regressed out age effects, resulting in the age and batch corrected expression matrices used as inputs for MatrixeQTL. To increase the power to detect cis-QTL, we accounted for unmeasured-surrogate confounders by performing principal component analysis (PCA) on the age and batch corrected expression matrices.64,65 The number of PCs chosen for each data type empirically led to the identification of the largest QTL in each condition and are reported in Table S5.
Mapping was performed combining individuals in order to increase power, thus, we included the first eigenvector obtained from a PCA on the SNP data as a covariate in our linear model in order to account for population structure. Local associations (i.e., putative cis QTL) were tested against all SNPs and MEIs located within the peak or gene or 100 Kbp upstream and downstream of each peak or gene. We recorded the strongest association (minimum p-value) for each peak/gene, which we used as statistical evidence for the presence of at least one QTL for that peak/gene. We permuted the genotypes ten times, re-performed the linear regressions, and recorded the minimum p-value for each peak/gene for each permutation. We used the R package qvalue66 to estimate FDR. In all cases, we assume that alleles affect phenotype in an additive manner.
Functional enrichment analysis
To find if peaks are enriched in functional pathways, the names of genes that are within 10 Kbp of a peak were compiled into gene sets (see pathways.R). A gene may appear only once in a gene set, even if it is nearby several peaks. This was done separately for the flu-infected and non-infected conditions and for common and graph-only H3K4me1, H3K27ac and ATAC peaks. Peaks that occur only in one sample within a condition were excluded. Then, gene sets were checked for functional enrichment67,68 in Biological Process Gene Ontology (GO:BP) terms with gprofiler2.69 We also calculated the gene ratio of enriched terms, which is the share of genes in the list associated with a GO term.
In a similar analysis, we compiled gene sets that are within 10 Kbp of a peak that is under the control of a QTL, separately for H3K4me1-QTLs, H3K27ac-QTLs and ca-QTLs, and separately in the infected and non-infected condition. We checked for GO enrichment of genes near hQTL/caQTLs that change relative to genes near QLTs that remain the same when using the genome graph. Specifically, we defined the QTLs that change to be those that are above the 99th percentile of absolute differences in p-value or effect size when using the genome graph. Those that are below the 99th percentile are considered to remain the same.
Evaluating peaks on genome graphs
H3K27ac, H3K4me1 ChIP-seq and ATAC-seq libraries were aligned to the genome graphs and peaks were called with Graph Peak Caller29 to obtain graph peaks. All the snarls in the genome graph were genotyped with vg call -m2,4 in order to partition the reads between the reference and alternative alleles in peaks that overlap a polymorphism. This information is available in the AD (alternate allele depth) and DP (site depth) tags in the VCF output. As before, non-specific alignments were removed. This approach was validated by listing peaks that overlap polymorphic loci in homozygous reference samples to create a negative control peak set, which should not show any coverage on the alternate allele.
Since the MEI allele is longer than the reference allele, we scaled down read counts (see Rscripts/allele_support.R) on the longer allele according to:
where denotes the read count on the long allele, the read length and the length of the long allele. This allows us to perform statistical tests that require count data in a way that is adjusted for allele length, such as the binomial test found in AlleleSeq.70
Further, peaks were binned according to the partitioning of reads between the reference and alternative alleles using a two sided binomial test with . Peaks that were skewed towards the MEI allele (p-value ) are placed in the MEI support bin. Peaks that show roughly equal read coverage on both alleles (p-value ) are labeled as biallelic. When reads are skewed towards the reference allele (p-value ), the peak belongs to the reference support bin. We also counted the MEIs that support combinations of marks to support their functional relevance.71,72,73,74.
Acknowledgments
This work was supported by a Canada Institute of Health Research (CIHR) program grant (CEE-151618) for the McGill Epigenomics Mapping Center, which is part of the Canadian Epigenetics, Environment and Health Research Consortium (CEEHRC) Network. G.B. is supported by a Canada Research Chair Tier 1 award, an FRQ-S, and a Distinguished Research Scholar award, and the Canadian Center for Computational Genomics (C3G) is supported by a Genome Canada Genome Technology Platform grant. This research was enabled in part by support provided by Calcul Quebec and the Digital Research Alliance of Canada. C.G. is supported by an NSERC PGSD award.
Author contributions
G.B., L.B.B., and T.P. conceived the study. G.B. directed the study. K.A.A. performed the QTL discovery. X.C. genotyped the MEIs. A.P. and M.-M.S. performed experimental work and molecular validation of results. A.P. contributed to bioinformatic processing of data. C.G. implemented software and led the computational analysis of results. C.G. and G.B. wrote and edited the manuscript, with feedback from authors.
Declaration of interests
The authors declare no competing interests.
Published: April 7, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100294.
Supplemental information
Document S3. Article plus supplemental information
Data and code availability
-
•
The source code of BarcodeAsm is available on the Zenodo repository.43
-
•
Additional code and processed data to reproduce the analysis, figures and manuscript are available on the Zenodo repository.44
-
•
Data for the Sanger sequencing and qPCR validation are available on the Zenodo repository.27
-
•
Code for QTL mapping is available on the Zenodo repository.45
-
•
WGS, ChIP-seq and ATAC-seq data generated for this study has been deposited at European Genome-Phenome Archive (EGA) under accession numbers EGA: EGAD00001008359 and EGA: EGAD00001008422 and Gene Expression Omnibus (GEO) under GEO: GSE225708.
References
- 1.Sudmant P.H., Rausch T., Gardner E.J., Handsaker R.E., Abyzov A., Huddleston J., Zhang Y., Ye K., Jun G., Fritz M.H.Y., et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526:75–81. doi: 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiang C., Scott A.J., Davis J.R., Tsang E.K., Li X., Kim Y., Hadzic T., Damani F.N., Ganel L., et al. GTEx Consortium The impact of structural variation on human gene expression. Nat. Genet. 2017;49:692–699. doi: 10.1038/ng.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhuo X., Du A.Y., Pehrsson E.C., Li D., Wang T. Epigenomic differences in the human and chimpanzee genomes are associated with structural variation. Genome Res. 2020;31:279–290. doi: 10.1101/gr.263491.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bourque G., Leong B., Vega V.B., Chen X., Lee Y.L., Srinivasan K.G., Chew J.-L., Ruan Y., Wei C.-L., Ng H.H., et al. Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res. 2008;18:1752–1762. doi: 10.1101/gr.080663.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang T., Zeng J., Lowe C.B., Sellers R.G., Salama S.R., Yang M., Burgess S.M., Brachmann R.K., Haussler D. Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc. Natl. Acad. Sci. USA. 2007;104:18613–18618. doi: 10.1073/pnas.0703637104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacques P.É., Jeyakani J., Bourque G. The majority of primate-specific regulatory sequences are derived from transposable elements. PLoS Genet. 2013;9:e1003504. doi: 10.1371/journal.pgen.1003504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daron J., Slotkin R.K. EpiTEome: simultaneous detection of transposable element insertion sites and their DNA methylation levels. Genome Biol. 2017;18:91. doi: 10.1186/s13059-017-1232-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gershman A., Sauria M.E.G., Hook P.W., Hoyt S., Razaghi R., Koren S., Altemose N., Caldas G.V., Vollger M.R., Logsdon G., et al. Epigenetic patterns in a complete human genome. bioRxiv. 2021 doi: 10.1101/2021.05.26.443420. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groza C., Kwan T., Soranzo N., Pastinen T., Bourque G. Personalized and graph genomes reveal missing signal in epigenomic data. Genome Biol. 2020;21:124. doi: 10.1186/s13059-020-02038-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leger A., Brettell I., Monahan J., Barton C., Wolf N., Kusminski N., Herder C., Aadepu N., Becker C., Gierten J., et al. Genomic variations and epigenomic landscape of the Medaka Inbred Kiyosu-Karlsruhe (MIKK) panel. bioRxiv. 2021 doi: 10.1101/2021.05.17.444424. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Treangen T.J., Salzberg S.L. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 2011;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitzman J.O. Haplotypes drop by drop. Nat. Biotechnol. 2016;34:296–298. doi: 10.1038/nbt.3500. [DOI] [PubMed] [Google Scholar]
- 13.Chu C., Borges-Monroy R., Viswanadham V.V., Lee S., Li H., Lee E.A., Park P.J. Comprehensive identification of transposable element insertions using multiple sequencing technologies. Nat. Commun. 2021;12:3836. doi: 10.1038/s41467-021-24041-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spies N., Weng Z., Bishara A., McDaniel J., Catoe D., Zook J.M., Salit M., West R.B., Batzoglou S., Sidow A. Genome-wide reconstruction of complex structural variants using read clouds. Nat. Methods. 2017;14:915–920. doi: 10.1038/nmeth.4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meleshko D., Marks P., Williams S., Hajirasouliha I. Detection and assembly of novel sequence insertions using Linked-Read technology. bioRxiv. 2019 doi: 10.1101/551028. Preprint at. [DOI] [Google Scholar]
- 16.Garcia S., Williams S., Xu A.W., Herschleb J., Marks P., Stafford D., Church D.M. Linked-Read sequencing resolves complex structural variants. bioRxiv. 2017 doi: 10.1101/231662. Preprint at. [DOI] [Google Scholar]
- 17.Bishara A., Liu Y., Weng Z., Kashef-Haghighi D., Newburger D.E., West R., Sidow A., Batzoglou S. Read clouds uncover variation in complex regions of the human genome. Genome Res. 2015;25:1570–1580. doi: 10.1101/gr.191189.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marks P., Garcia S., Barrio A.M., Belhocine K., Bernate J., Bharadwaj R., Bjornson K., Catalanotti C., Delaney J., Fehr A., et al. Resolving the full spectrum of human genome variation using linked-reads. bioRxiv. 2018 doi: 10.1101/230946. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wildschutte J.H., Baron A., Diroff N.M., Kidd J.M. Discovery and characterization of Alu repeat sequences via precise local read assembly. Nucleic Acids Res. 2015;43:10292–10307. doi: 10.1093/nar/gkv1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weisenfeld N.I., Kumar V., Shah P., Church D.M., Jaffe D.B. Direct determination of diploid genome sequences. Genome Res. 2017;27:757–767. doi: 10.1101/gr.214874.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ott A., Schnable J.C., Yeh C.-T., Wu L., Liu C., Hu H.-C., Dalgard C.L., Sarkar S., Schnable P.S. Linked read technology for assembling large complex and polyploid genomes. BMC Genom. 2018;19:651. doi: 10.1186/s12864-018-5040-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aracena K.A., Lin Y.-L., Luo K., Pacis A., Gona S., Mu Z., Yotova V., Sindeaux R., Pramatarova A., Simon M.-M., et al. Epigenetic variation impacts ancestry-associated differences in the transcriptional response to influenza infection. bioRxiv. 2022 doi: 10.1101/2022.05.10.491413. Preprint at. [DOI] [PubMed] [Google Scholar]
- 23.Medzhitov R., Janeway C. Innate immunity. N. Engl. J. Med. 2000;343:338–344. doi: 10.1056/NEJM200008033430506. [DOI] [PubMed] [Google Scholar]
- 24.Chuong E.B., Elde N.C., Feschotte C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science. 2016;351:1083–1087. doi: 10.1126/science.aad5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garg S., Fungtammasan A., Carroll A., Chou M., Schmitt A., Zhou X., Mac S., Peluso P., Hatas E., Ghurye J., et al. Chromosome-scale, haplotype-resolved assembly of human genomes. Nat. Biotechnol. 2021;39:309–312. doi: 10.1038/s41587-020-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ebert P., Audano P.A., Zhu Q., Rodriguez-Martin B., Porubsky D., Bonder M.J., Sulovari A., Ebler J., Zhou W., Serra Mari R., et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science. 2021;372:eabf7117. doi: 10.1126/science.abf7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Groza C., Simon M.-M., Pramatarova A. Genome Graphs Detect Human Polymorphisms in Active Epigenomic States during Influenza Infection: Validation. Zenodo. 2022 doi: 10.5281/zenodo.7429679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garrison E., Sirén J., Novak A.M., Hickey G., Eizenga J.M., Dawson E.T., Jones W., Garg S., Markello C., Lin M.F., et al. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol. 2018;36:875–879. doi: 10.1038/nbt.4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grytten I., Rand K.D., Nederbragt A.J., Storvik G.O., Glad I.K., Sandve G.K. Graph peak caller: calling ChIP-seq peaks on graph-based reference genomes. PLoS Comput. Biol. 2019;15:e1006731. doi: 10.1371/journal.pcbi.1006731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deininger P. Alu elements: know the SINEs. Genome Biol. 2011;12:236. doi: 10.1186/gb-2011-12-12-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hermant C., Torres-Padilla M.-E. TFs for TEs: the transcription factor repertoire of mammalian transposable elements. Genes Dev. 2021;35:22–39. doi: 10.1101/gad.344473.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bantysh O.B., Buzdin A.A. Novel family of human transposable elements formed due to fusion of the first exon of gene MAST2 with retrotransposon SVA. Biochemistry. 2009;74:1393–1399. doi: 10.1134/S0006297909120153. [DOI] [PubMed] [Google Scholar]
- 33.Kim S., Cho C.-S., Han K., Lee J. Structural variation of Alu element and human disease. Genomics Inform. 2016;14:70–77. doi: 10.5808/GI.2016.14.3.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.The 1000 Genomes Project Consortium. Auton A., Abecasis G.R., Altshuler D.M., Durbin R.M., Abecasis G.R., Bentley D.R., Chakravarti A., Clark A.G., Donnelly P., et al. A global reference for human genetic variation. Nature. 2015;526:68. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martiniano R., Garrison E., Jones E.R., Manica A., Durbin R. Removing reference bias and improving indel calling in ancient DNA data analysis by mapping to a sequence variation graph. Genome Biol. 2020;21:250. doi: 10.1186/s13059-020-02160-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abiola O., Angel J.M., Avner P., Bachmanov A.A., Belknap J.K., Bennett B., Blankenhorn E.P., Blizard D.A., Bolivar V., Brockmann G.A., et al. The nature and identification of quantitative trait loci: a community’s view. Nat. Rev. Genet. 2003;4:911–916. doi: 10.1038/nrg1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X., Pacis A.S., Aracena K.A., Gona S., Kwan T., Groza C., Lin Y.L., Sindeaux R.H.M., Yotova V., Pramatarova A., et al. Transposable elements are associated with the variable response to influenza infection. bioRxiv. 2022 doi: 10.1101/2022.05.10.491101. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Isobe M., Izawa K., Sugiuchi M., Sakanishi T., Kaitani A., Takamori A., Maehara A., Matsukawa T., Takahashi M., Yamanishi Y., et al. The CD300e molecule in mice is an immune-activating receptor. J. Biol. Chem. 2018;293:3793–3805. doi: 10.1074/jbc.RA117.000696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Payer L.M., Steranka J.P., Kryatova M.S., Grillo G., Lupien M., Rocha P.P., Burns K.H. Alu insertion variants alter gene transcript levels. Genome Res. 2021;31:2236–2248. doi: 10.1101/gr.261305.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meyerson N.R., Zhou L., Guo Y.R., Zhao C., Tao Y.J., Krug R.M., Sawyer S.L. Nuclear TRIM25 specifically targets influenza virus ribonucleoproteins to block the onset of RNA chain elongation. Cell Host Microbe. 2017;22:627–638.e7. doi: 10.1016/j.chom.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nurk S., Koren S., Rhie A., Rautiainen M., Bzikadze A.V., Vollger M.R., Altemose N., Uralsky L., Gershman A., Mikheenko A., et al. The complete sequence of a human genome. Science. 2022;376:44–53. doi: 10.1126/science.abj6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang T., Antonacci-Fulton L., Howe K., Lawson H.A., Lucas J.K., Phillippy A.M., Popejoy A.B., Asri M., Carson C., Chaisson M.J.P., et al. The Human Pangenome Project: a global resource to map genomic diversity. Nature. 2022;604:437–446. doi: 10.1038/s41586-022-04601-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cristian G. 2021. Cgroza/BarcodeAsm: Publication Version. [DOI] [Google Scholar]
- 44.Groza C. Genome Graphs Detect Human Polymorphisms in Active Epigenomic States during Influenza Infection: Code and Processed Data. Zenodo. 2022 doi: 10.5281/zenodo.6525192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aracena K. 2021. Katiearacena/Groza_et_al_mapping: for_preprint. [DOI] [Google Scholar]
- 46.Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B., et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017;14:959–962. doi: 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Solomon E.R., Caldwell K.K., Allan A.M. A novel method for the normalization of ChIP-qPCR data. MethodsX. 2021;8:101504. doi: 10.1016/j.mex.2021.101504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H. Exploring single-sample SNP and INDEL calling with whole-genome de novo assembly. Bioinformatics. 2012;28:1838–1844. doi: 10.1093/bioinformatics/bts280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen X., Li D. ERVcaller: identifying polymorphic endogenous retrovirus and other transposable element insertions using whole-genome sequencing data. Bioinformatics. 2019;35:3913–3922. doi: 10.1093/bioinformatics/btz205. [DOI] [PubMed] [Google Scholar]
- 51.Gardner E.J., Lam V.K., Harris D.N., Chuang N.T., Scott E.C., Pittard W.S., Mills R.E., 1000 Genomes Project Consortium. Devine S.E. the mobile element locator tool (MELT): population-scale mobile element discovery and biology. Genome Res. 2017;27:1916–1929. doi: 10.1101/gr.218032.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hubley R., Finn R.D., Clements J., Eddy S.R., Jones T.A., Bao W., Smit A.F.A., Wheeler T.J. The Dfam database of repetitive DNA families. Nucleic Acids Res. 2016;44:D81–D89. doi: 10.1093/nar/gkv1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fishilevich S., Nudel R., Rappaport N., Hadar R., Plaschkes I., Iny Stein T., Rosen N., Kohn A., Twik M., Safran M., et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database. 2017;2017:bax028. doi: 10.1093/database/bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frankish A., Diekhans M., Ferreira A.-M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J., et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766–D773. doi: 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng X., Levine D., Shen J., Gogarten S.M., Laurie C., Weir B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28:3326–3328. doi: 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eberle M.A., Fritzilas E., Krusche P., Källberg M., Moore B.L., Bekritsky M.A., Iqbal Z., Chuang H.-Y., Humphray S.J., Halpern A.L., et al. A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res. 2017;27:157–164. doi: 10.1101/gr.210500.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hickey G., Heller D., Monlong J., Sibbesen J.A., Sirén J., Eizenga J., Dawson E.T., Garrison E., Novak A.M., Paten B. Genotyping structural variants in pangenome graphs using the vg toolkit. Genome Biol. 2020;21:35. doi: 10.1186/s13059-020-1941-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Friedman J., Hastie T., Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 59.Shabalin A.A. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–1358. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smyth G.K., Gentleman R., Carey V., Dudoit S., Irizarry R., Huber W. Bioinformatics and computational biology solutions using r and bioconductor. Springer; 2005. Limma: linear models for microarray data; pp. 397–420. [Google Scholar]
- 63.Leek J.T., Johnson W.E., Parker H.S., Jaffe A.E., Storey J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pickrell J.K., Marioni J.C., Pai A.A., Degner J.F., Engelhardt B.E., Nkadori E., Veyrieras J.-B., Stephens M., Gilad Y., Pritchard J.K. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barreiro L.B., Tailleux L., Pai A.A., Gicquel B., Marioni J.C., Gilad Y. Deciphering the genetic architecture of variation in the immune response to Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. USA. 2012;109:1204–1209. doi: 10.1073/pnas.1115761109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Storey J.D., Bass A.J., Dabney A., Robinson D. Qvalue: Q-Value Estimation for False Discovery Rate Control. Bioconductor. 2021 [Google Scholar]
- 67.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The gene ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gene Ontology Consortium The gene ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021;49:D325–D334. doi: 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kolberg L., Raudvere U., Kuzmin I., Vilo J., Peterson H. gprofiler2 – an R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Res. 2020;9:ELIXIR-709. doi: 10.12688/f1000research.24956.2. version 2; peer review: 2 approved. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rozowsky J., Abyzov A., Wang J., Alves P., Raha D., Harmanci A., Leng J., Bjornson R., Kong Y., Kitabayashi N., et al. AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol. Syst. Biol. 2011;7:522. doi: 10.1038/msb.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A., et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zentner G.E., Tesar P.J., Scacheri P.C. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011;21:1273–1283. doi: 10.1101/gr.122382.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rada-Iglesias A., Bajpai R., Swigut T., Brugmann S.A., Flynn R.A., Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Daugherty A.C., Yeo R.W., Buenrostro J.D., Greenleaf W.J., Kundaje A., Brunet A. Chromatin accessibility dynamics reveal novel functional enhancers in C. elegans. Genome Res. 2017;27:2096–2107. doi: 10.1101/gr.226233.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S3. Article plus supplemental information
Data Availability Statement
-
•
The source code of BarcodeAsm is available on the Zenodo repository.43
-
•
Additional code and processed data to reproduce the analysis, figures and manuscript are available on the Zenodo repository.44
-
•
Data for the Sanger sequencing and qPCR validation are available on the Zenodo repository.27
-
•
Code for QTL mapping is available on the Zenodo repository.45
-
•
WGS, ChIP-seq and ATAC-seq data generated for this study has been deposited at European Genome-Phenome Archive (EGA) under accession numbers EGA: EGAD00001008359 and EGA: EGAD00001008422 and Gene Expression Omnibus (GEO) under GEO: GSE225708.