

Summary
Diverse inbred mouse strains are important biomedical research models, yet genome characterization of many strains is fundamentally lacking in comparison with humans. In particular, catalogs of structural variants (SVs) (variants ≥ 50 bp) are incomplete, limiting the discovery of causative alleles for phenotypic variation. Here, we resolve genome-wide SVs in 20 genetically distinct inbred mice with long-read sequencing. We report 413,758 site-specific SVs affecting 13% (356 Mbp) of the mouse reference assembly, including 510 previously unannotated coding variants. We substantially improve the Mus musculus transposable element (TE) callset, and we find that TEs comprise 39% of SVs and account for 75% of altered bases. We further utilize this callset to investigate how TE heterogeneity affects mouse embryonic stem cells and find multiple TE classes that influence chromatin accessibility. Our work provides a comprehensive analysis of SVs found in diverse mouse genomes and illustrates the role of TEs in epigenetic differences.
Keywords: structural variation, long read sequencing, whole genome assembly, inbred micetransposable elements, embryonic stem cells, chromatin accessibility, mouse genomics, collaborative cross, effects of structural variation
Graphical abstract
Highlights
-
•
Long-read whole-genome assemblies enable resolution of diverse mouse genomes
-
•
A high-quality, sequence-resolved mouse structural variant resource
-
•
Identification and annotation of transposable element variants
-
•
Polymorphic transposable elements promote changes in mESC chromatin accessibility
Ferraj et al. leverage long-read whole-genome sequencing to resolve structural variation and transposable element polymorphisms across genetically diverse mouse genomes. This resource can be used for genotype-phenotype studies and was used here to detect changes in chromatin accessibility associated with polymorphic transposon families between diverse mouse embryonic stem cells.
Introduction
Mice of varying genetic backgrounds are often used for models of human disease and to generate populations of infinitely diverse substrains that exhibit a wide range of genotypic and phenotypic heterogeneity. Such panels include the collaborative cross (CC) and diversity outbred (DO) populations, which are multi-parental groups of recombinant inbred lines and derivative outbred stocks constructed from eight original founder strains.1,2 These reference panels are often used to determine genotype-phenotype relationships and have been invaluable tools for precise genomic mapping of numerous quantitative trait loci, including regions associated with addiction, stem cell pluripotency, and insulin secretion.3,4,5
Discovery of the genetic origins of disease and phenotypes present within diverse populations depends on high-quality reference genomes and precise variant catalogs.6,7,8 For decades now, genetic and genomic studies have depended on the Mus musculus reference genome, which is derived from the popular C57BL/6J strain. Although the C57BL/6J (GRCm39) reference is relatively complete, reliance on an assembly built from one genetic background limits the analysis of diverse strains as sequencing reads from divergent haplotypes are often misplaced or unmapped. This problem has been noted in human research,9 but is compounded in mouse where there is a much larger amount of genetic diversity in laboratory inbred strains than in the human population. Despite recent strain-specific reference genomes,7 much of the variability between strains remains incomplete due to the limitations of short-reads in detecting structural variants (SVs). Regions that remain unresolvable include complex repeats, segmental duplications, and transposable elements (TEs).9 TEs are repeats that comprise approximately 37.5% of the mouse genome and are known to generate extensive genomic variation by moving to new locations by copy and paste mechanisms.10,11 Repetitive sequences can contain important variants affecting genes that potentially lead to phenotypic changes, and, until we can accurately resolve SVs, their impacts on mouse biology remains unknown.
Recent advances in long-read sequencing have enabled accurate resolution of repetitive DNA and large insertion variants12,13,14,15,16,17,18 and have greatly increased sensitivity for SVs over short-read technologies.12,13,14,15,16,17,18 In particular, detailed studies of the same human genomes have revealed that 60%–70% of SVs are missed when relying solely on short-read sequence data, and, for those detected by both platforms, long-read sequences more fully resolve alleles.12,13 Although large-scale long-read sequencing studies in humans have been published,13 efforts to fully resolve and detect SVs across diverse mouse genomes with these technologies are lacking.
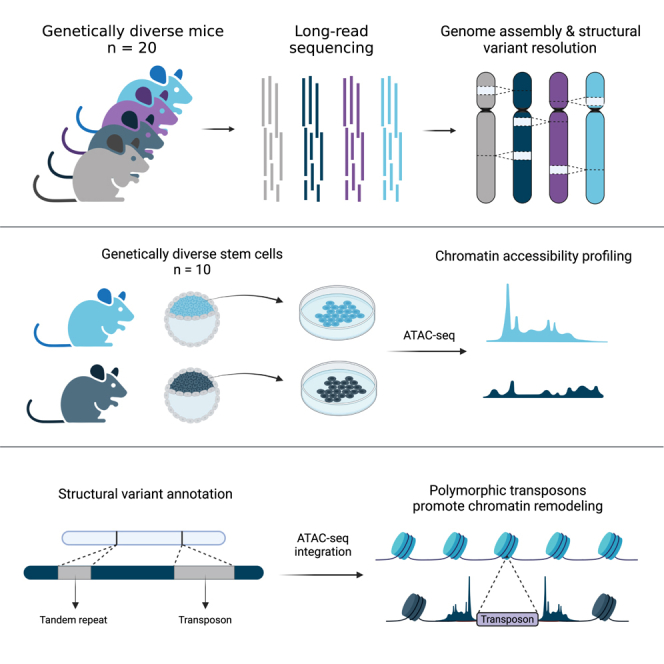
Here, we used Pacific Biosciences (PacBio) long-read whole-genome sequencing to assemble the genomes of 20 diverse inbred laboratory strains of mice. From whole-genome comparisons, we have generated a sequence-resolved callset of 413,758 SVs (54% novel) spanning three subspecies of Mus musculus (domesticus, musculus, and castaneus) with ∼0.5 Myr of divergence, including SVs in regions that cannot be resolved with short-read sequencing. We used this resource to identify and resolve TE polymorphisms present between diverse mice, revealing multiple retrotransposition competent subfamilies, including L1MdTf and IAP elements, which cause widespread changes in mouse embryonic stem cell (mESC) chromatin accessibility and gene expression. We present these data as a comprehensive mouse SV resource that can be used for future genomic studies, aid in modeling and studying the effects of genetic variation, and enhance genotype-to-phenotype research.
Results
Long-read sequencing assemblies of diverse mouse genomes
Whole-genome long-read sequencing data were generated for 20 diverse inbred mouse strains to a minimum of 30-fold coverage. We selected a mixture of classical and wild-derived inbred laboratory strains including the parental founders of the CC (129S1/SvImJ, A/J, CAST/EiJ, NOD/ShiLtJ, NZO/HILtJ, PWK/PhJ, and WSB/EiJ), six resultant CC animals that harbor phenotypic abnormalities of unknown genetic origin (CC005, CC015, CC032, CC055, CC060, and CC074),19 and seven additional strains with distinct genetic backgrounds (BALB/cByJ, BALB/cJ, C3H/HeJ, C3H/HeOuJ, C57BL/6NJ, DBA/2J, and PWD/PhJ). We chose this cohort as it represents a wide variety of commonly used strains with diverse genetic backgrounds, strains with complex phenotypes of unknown cause, and strains interesting for mapping variants responsible for quantitative traits.1,20,21
We generated de novo assemblies for each genome reaching a 6.45 Mbp contig N50 (Table S1) and on average achieving a 534× increase in contig N50 compared with previous short-read assemblies built from the same strains (14 kbp).7 In addition, our assemblies are contained in 143× fewer contigs (average of 2,483 per genome vs. 355,353) with 228 Mbp of additional sequence on average compared with short-read assemblies. Therefore, we have created the most contiguous genome assemblies of diverse mouse genomes produced to date.
SVs are prevalent across mouse genomes
We aligned each de novo assembly to the GRCm39 (mm39, Mus musculus) reference genome and called SVs with the phased assembly variant caller.13 From these strain-specific SV calls, we created a non-redundant variant callset by merging SVs across strains.13 In total, we detected 413,758 SVs that occur at unique sites across the current mouse reference assembly (Table S2), including 244,859 (59%) insertions, 168,652 (41%) deletions, and 247 (0.06%) inversions (Figure 1A). We find that strains from the musculus (PWD/PhJ and PWK/PhJ) and castaneus (CAST/EiJ) subspecies contain an average of 200,401 SVs per genome when compared with the C57BL/6J reference, with strains less evolutionarily diverged from the reference (domesticus subspecies) containing the fewest (60,490) variant calls per animal (Table 1). We calculate a 5.8% false discovery rate from 584 PCR validations (73 SVs across 8 samples) (Table S3). We identify 59,874 (15%) SVs that are unique to a single strain and 1,483 (0.4%) shared variants (Figure 1A) that indicate reference biases or rare SVs found within the reference.9 We also find 268,436 SVs (243 Mbp) specific to one subspecies (Figure 1B), of which 73% are absent from GRCm39 adding 178 Mbp of subspecies-specific sequence not present in the current mouse reference genome.
Figure 1.

Discovery of SVs in diverse mouse genomes
(A) Total number of SVs discovered in each mouse genome when compared with the Mus musculus (GRCm39, C57BL/6J background) reference. Variants are grouped by their frequency within the cohort: shared (present in all strains), major (≥50% of the cohort), minor (<50% exclusive to one strain [unique] or exclusive to one subspecies [subspecies specific]). We merged variants into a non-redundant callset (donut plot), shown along with the proportion of insertion and deletion calls (blue and purple [the 247 inversions are not shown]).
(B) Total number of SVs discovered and shared between each subspecies sequenced (domesticus, musculus, and castaneus).
(C) Length distribution of SVs identified as insertions (blue), deletions (purple), and inversions (green).
(D) Average number of SVs per mouse genome supported by long-read (LR) and short-read (SR) detection, along with the proportion of insertions and deletions that contain tandem repeat sequences (black striped bar).
(E) Total number of SVs from LR and mouse genomes project (MGP) callset, along with the proportion of variants validated by raw long read alignment size differences (red striped bar).
Table 1.
Count of structural variants detected for each mouse genome
| Strain | Total SVs | Insertion | Deletion | Inversion | Bases | Percent genome |
|---|---|---|---|---|---|---|
| 129S1/SvImJ | 59,481 | 31,641 | 27,791 | 49 | 59,583,060 | 2.18 |
| C57BL/6NJ | 7,872 | 6,077 | 1,770 | 25 | 11,249,662 | 0.41 |
| A/J | 57,606 | 30,589 | 26,973 | 44 | 58,753,047 | 2.15 |
| BALB/cByJ | 53,636 | 28,175 | 25,416 | 45 | 56,099,638 | 2.06 |
| BALB/cJ | 53,845 | 28,596 | 25,205 | 44 | 56,083,785 | 2.06 |
| C3H/HeJ | 58,851 | 31,333 | 27,473 | 45 | 58,222,355 | 2.13 |
| C3H/HeOuJ | 59,167 | 31,552 | 27,561 | 54 | 59,774,259 | 2.19 |
| CAST/EiJ | 202,011 | 108,179 | 93,734 | 98 | 163,832,319 | 6.01 |
| CC005/TauUncJ | 67,554 | 36,638 | 30,867 | 49 | 64,084,771 | 2.35 |
| CC015/UncJ | 82,180 | 44,486 | 37,641 | 53 | 75,240,374 | 2.76 |
| CC032/GeniUncJ | 74,412 | 40,356 | 34,002 | 54 | 67,795,817 | 2.48 |
| CC055/TauUncJ | 86,741 | 46,294 | 40,393 | 54 | 82,149,711 | 3.01 |
| CC060/UncJ | 74,115 | 39,672 | 34,395 | 48 | 70,722,386 | 2.59 |
| CC074/UncJ | 75,800 | 40,930 | 34,818 | 52 | 65,807,956 | 2.41 |
| DBA/2J | 59,469 | 31,753 | 27,672 | 44 | 59,156,224 | 2.17 |
| NOD/ShiLtJ | 59,322 | 31,430 | 27,847 | 45 | 57,404,786 | 2.1 |
| NZO/HlLtJ | 60,608 | 32,429 | 28,125 | 54 | 62,428,318 | 2.29 |
| PWD/PhJ | 198,893 | 106,754 | 92,038 | 101 | 157,938,496 | 5.79 |
| PWK/PhJ | 200,301 | 107,968 | 92,223 | 110 | 162,623,052 | 5.96 |
| WSB/EiJ | 82,919 | 42,910 | 39,956 | 53 | 91,213,812 | 3.34 |
Total number of SVs detected in each mouse genome separated by variant type (insertions, deletions, and inversions), the total number of nucleotide bases changed, and the percentage of the genome affected with respect to the GRCm39 reference genome.
These SVs contribute extensively to mouse variation; the 413,758 SVs encompass a total of 356 Mbp of variable sequence, accounting for 13% of the current mouse reference assembly. We find that these SVs contain 4.9× the number of bases affected when compared with previously published single nucleotide variants from diverse mouse genomes.8 The length of SVs varies greatly, and the size of SVs are non-randomly distributed (Figure 1C). An increased number of small SVs (50–100 bp) contain variants with lengths divisible by two resulting from dinucleotide repeat expansions and contractions, as observed previously in humans.13 We also observe peaks in the size distribution consistent with abundant TE polymorphisms (200 bp, short interspersed nuclear elements, or SINEs; 6.3 kbp, long interspersed nuclear elements, or LINEs). Although the majority of variants are under 1 kbp in length (83%), the total sequence content due to SVs is dominated by those 1 kbp or greater, which account for 295 Mbp (81%) of variable sequence. Each species contains an average of 80 Mbp of unique sequence due to SVs. We found that a large portion of SVs (201,342, 49%) were found within tandemly repeated regions.
When compared with human genomes, which have an average of 24,653 SVs per individual,13 we find 60,490 SVs per domesticus genome (2.3× human), indicating greater diversity from SVs between mouse genomes than human genomes. Because they are determined by comparing de novo assemblies to a domesticus reference, the number of SVs per genome is even greater in musculus (199,597, 8.1× human) and castaneus (202,011, 8.2× human) strains. With extensive structural polymorphism across mouse genomes, the use of a single linear reference may therefore be inadequate for mapping genomic data, especially from more diverged strains. For example, per genome, we find that SV insertions duplicate whole genes (20 in domesticus, 37 in castaneus, and 28 in musculus), suggesting that the effect of paralogous gene copies has been systematically underestimated by short-read approaches due to reference biases.
Long-read sequencing data reveals novel SVs
Previously, a number of studies used short-read sequencing data to interrogate SVs across large cohorts of genetically diverse inbred mice.7,8,22,23 To identify the subset of variation from our study that could be detected from short reads and to add orthogonal support for our assembly-based SV callset, we performed SV calling from Illumina whole-genome sequencing of 18 previously sequenced strains that overlapped our cohort.7,22 On average 91% short-read deletions and 84% short-read insertions were detected in our long-read SV callset (Figure 1D). Conversely, short-read SV calling was only able to detect 46% of deletions, 14% of insertions, and 39% of inversions discovered by long-read sequencing. Across the 18 strains, SV calling from long reads detected an additional distinct 213,688 insertions, 64,277 deletions, and 97 inversions. Notably, short-read sequencing is particularly underpowered to detect SVs in repeat regions12,24; the number of SVs that are supported by short reads drops considerably for both deletions (46%–24%) and insertions (14%–9%) when considering variants within tandem repeat regions. In total, 155,156 simple repeat SVs were not detected by short reads.
To compare our SV resource with previously published variant calls, we intersected our long-read callset with the most recent mouse genomes project (MGP) SVs.8,23 Overall, 54% (215,262) of SVs we call are novel to our study (Figure 1E). Interestingly, we find a large number of MGP insertions (445,538) that were not detected by long-read methods (Figure 1E). To determine the validity of these MGP specific calls, we mapped long reads generated from each sample to the GRCm39 reference genome and surveyed each SV region for changes in sequence length (mean read length difference ≥50 bp). From this we determined that 95% (117,715) of the insertions specific to long-read detection were supported by raw long-read alignments, while only 28% (122,981) of the MGP-only insertions were supported, leaving a majority (322,574, 72%) of potential false insertion calls within the MGP dataset. Calls detected only with long reads were significantly smaller (mean of 425 vs. 1,393 bp for MGP-supported calls, p < 0.0001, Student’s t test). This was also true for variants which contain simple repeat sequences (mean of 279 bp for novel variants vs. 1,631 bp for MGP-supported calls; p < 0.0001, Student’s t test). These data suggest that short-read variant calling falters in the detection of smaller SVs.
Long-read assemblies reveal extensive transposon variation at a nucleotide level
The most notable improvement from using long-read genome assemblies comes from the ability to reconstruct long repetitive regions. This is particularly imperative when characterizing mouse genetic variation as mice contain elevated retrotransposon insertion rates when compared with human.25,26 To create a more complete resource and investigate the impact of mobile element variation in diverse mice, we identified TE variants (TEVs) in each mouse genome. We find that 39% (162,787) of SVs between all samples are attributable to TEVs, with most of the TEVs being insertions (60%, 97,100). TEVs are dominated by LINE-1 polymorphisms (47%, 76,640), followed by SINEs (B1 and B2; 24%, 39,389), and various endogenous retroviruses (ERVs) (ERVK, ERVL-MaLR, ERVL, and ERV1; 28%, 45,204) (Figure 2A). We observe various modes of TEV length consistent with retrotransposition, with accumulations of TEVs at ∼200 bp (SINEs), ∼7.2 kbp (ERVK LTRs), and a ∼6.3 kbp peak for full-length LINE-1s (Figure 2B). In addition to comprising 39% of non-redundant variant sites, TEVs constitute 76% (278 Mbp) of the variable sequence content (Figure 2C). LINE-1 variants alone constitute 47% (172 Mbp) of variable base pairs, in contrast with the 24% (92 Mbp) contributed by non-TEVs. Although SINEs comprise 25% of TE variant sites, they only account for 2.1% (7.9 Mbp) of variable sequence.
Figure 2.

TEV in diverse mouse genomes
(A) Total number of TEVs discovered from long read sequencing by transposon family.
(B) Length distribution of TEVs by transposon family.
(C) Total number of variable bases due to TEV by transposon families and non-TE SVs. Each block equals one megabase of variable sequence.
(D) Count of subspecies-specific intracisternal A-particle (IAP) variants. Highlighted size ranges are solo-LTR (∼300 bp), IΔ1 variants (4.5–5.5 kbp), and full-length (FL) variants (6.5–7.5 kbp).
(E) Size distribution of species-specific IAP insertions (Mann-Whitney U test; ∗∗∗∗p ≤ 1 × 10−4).
(F) Size distribution of strain-specific IAP insertions within the domesticus lineage. Substrains that share a parental origin (C3H, C3H/HeJ and C3H/HeOuJ; BALB, BALB/cByJ and BALB/cJ) are grouped to represent each lineage.
(G) Count of strain-specific IAP insertions in closely related domesticus animals.
As expected, we observe fewer TEVs within and around genic regions (43% intergenic variants vs. 26% non-intergenic, Table S4). Coding sequence and intronic variants are also underrepresented for TEVs. At the repeat family level, we find LINE-1 variants are enriched in intergenic regions and depleted for coding sequence, intronic, UTR, and nearby gene regions. We observe an orientation bias for intronic LINE-1 variants in antisense orientation respective to nearby genes (60% antisense vs. 40% sense), similar to previous observations.27,28 Conversely, SINE elements tend to accumulate in and around genic regions with significant enrichment in introns, UTRs, and up- and downstream regions. Various ERVs are depleted in introns; however, splice donor site variants are significantly enriched for ERVL and ERV1 sequences when compared with other SVs. We find that 41% of all ERV variants that contain splice donor sites are from the MT retrotransposon family, which are known to contribute to chimeric RNA sequences and are developmentally regulated.29
When compared with previously published TE variant calls from diverse mice,27 we detected 80% (59,344) of previously identified TE polymorphisms (Figure S1). We find that 37% of the variants we detect were previously identified using short-read methods, with 99,349 TE variants unique to our study. Using our sequence-resolved SVs, we further annotate TEVs by subtype (Figure S2) and find that Mus musculus LINE-1 variants are predominantly L1MdA (26,717 SVs) followed by L1MdTf (17,010 SVs) and L1MdGf (10,649 SVs), fitting previous studies that have detailed active LINE-1 subfamilies in Mus musculus.30 Interestingly, we find that L1MdTf-I and L1MdTf-II elements, which are the most active LINE-1 subtypes in the C57BL/6J genome,25 comprise a much larger percentage of LINE-1 insertions in domesticus genomes when compared with musculus and castaneus (3% castaneus, 2% musculus, and 34% domesticus). By looking at total copy number of L1MdTf-I and L1MdTf-II elements in each assembly, we find that non-domesticus animals contain an extremely low copy number when compared with domesticus animals (average of 4,808 copies per domesticus genome compared with 679 copies in non-domesticus genomes) (Figure S3A). We find that non-domesticus genomes contain a greater proportion of L1MdTf-I/II deletions when compared with insertions, suggesting a recent expansion of these subfamilies occurring after the divergence of these subspecies (Figure S3B). These data provide a new and comprehensive catalog of mouse TE variants built from long reads and provide evidence of an expansion of L1MdTf-I and L1MdTf-II transposons in the domesticus lineage.
IAP endogenous retroviral elements have variable rates of insertion in mouse genomes
Intracisternal A-particle (IAP) elements are murine-specific retroviral elements that drive variation in mice.31 Full-length (∼7.2 kbp) IAPs are autonomous long terminal repeat (LTR) retrotransposons that can cause aberrant splicing and disease if they insert near or within genes.32,33 Proper annotation of LTR elements is especially difficult with short reads as many subtypes are differentiated by their internal structure. We utilized our TEV callset to investigate polymorphic IAP LTRs within the Mus musculus lineage. We find that strains of the domesticus lineage contain 43% (3,363 insertions) of all subspecies-specific IAP polymorphisms (Figure 2D). Strains from the domesticus subspecies contain a significant increase (p ≤ 2.41 × 10−2, Fisher’s exact test) in the proportion of IAPs that constitute all ERVK insertions (54%) when compared with castaneus (44%) and musculus (43%), which is consistent with previous findings that suggest an ongoing domesticus-specific IAP expansion.27 We further detail this expansion by providing all variable IAP sequences between these subspecies and observe a domesticus-specific increase in the proportion of variable bases due to IAP insertions in comparison with other ERVKs (67% from active IAP subtypes, total of 15 Mbp), representing an increase in the variable sequence content of domesticus when compared with castaneus (56%, 6 Mbp) and musculus (56%, 7 Mbp) IAP sequences. IAP insertions in domesticus also contain a different nucleotide length distribution, with notable accumulation of full-length elements as well as variants that are within a 4.5–5.5 kbp size range (Figure 2E). These shorter, or intermediate-sized sequences are indicative of IΔ1 IAPs, which contain an internal deletion relative to full-length elements and remain active in Mus musculus.34,35 Here, we show evidence that the expansion of IAP TEs in domesticus is driven by both full-length and variable-length IAPs.
Interestingly, even between domesticus strains we observe a difference in the distribution of IAP lengths. Mice with a parental C3H background contain a notable size discrepancy for IAP insertions that are 4.5–5.5 kbp in length when compared with other domesticus strains (Figure 2F). Previous studies have found that IAP elements are more highly expressed and active in C3H/HeJ relative to other domesticus strains.31 We find that C3H/HeJ contains a significant increase in the proportion of variants within the IΔ1 size range when compared with every non-C3H strain in our study (p < 0.05, Fisher’s exact test). Within closely related domesticus strains, C3H mice contain over one-third of strain-specific IAP insertions in the IΔ1 size range (Figure 2G). Hyperactive IAP expression and numerous gene altering polymorphisms have been well documented in C3H mice.11,31 Here, we uncover a large number of unique IAP insertions specific to C3H mice and catalog a higher number of insertion polymorphisms when compared with other domesticus lines (34% of strain-specific insertions in the IΔ1 size range).
The consequences of structural variation on mouse genomes
To assess the potential functional impact of mouse SV, we used the Ensembl Variant Effect Predictor tool to intersect SV calls with known genomic features and predict variant severity.36 We find that 55% (228,232) of SVs map within or around (5 kbp up-/downstream) genes, with 43% (179,239) intergenic and 13% (52,411) intersecting mouse regulatory regions (Figure 3A). Most genic SVs (98%) are intronic, with 2% overlapping a non-intron feature (3′ UTR, 5′ UTR, and coding regions) (Figure 3B). We report 829 coding sequence variants (512 deletions and 317 insertions), 2,647 3′ UTR variants (1,148 deletions and 1,499 insertions), and 451 5′ UTR SVs (280 deletions and 171 insertions). We then selected all SVs with potential functional consequences and performed an association analysis based on multiple criteria (Figure 3C). First, we determined the tendency of a particular SV class to be detected by short-read sequencing when compared with long reads. Among all coding sequence variants, 577 (70%) are uniquely called with long reads and 252 (30%) are detected by both long and short reads. We then compared all variant consequences with previously published mouse SVs.8,23 Of all variants that were unique to our study, we report 94,863 to be intronic, 1,469 UTR, and 510 coding sequence variants. Interestingly, across all SV calls, coding sequence variants were enriched for simple repeat sequences (Table S4). We additionally conducted 120 PCR validations (15 PCR reactions across 8 strains) for SVs that alter coding regions across the 8 CC founder strains yielding orthogonal data for SVs leading to frameshift and insertion variants (Table S3).
Figure 3.

SV consequences
(A) Count of SVs overlapping various intergenic regions, with the percentage of variants that overlap genic and non-genic regions.
(B) Count of SVs overlapping genic features, with percentage of variants that overlap a non-intronic or intronic region of a gene.
(C) Genic, regulatory, and intergenic SV consequences. Each SV consequence is shown with the proportion that is specific to long-read detection (LR, long read only; SR, short read support), novel to our study when compared with the mouse genomes project database (Novel or MGP), tandem repeat composition (Tandem Repeat and Non-Tandem Repeat), and insertion or deletion SVs (Insertion and Deletion). Each consequence was correlated (or not) with simple repeat SVs (red and blue).
(D) Mutyh expression differences between strains that contain a 5.3 kbp IAP insertion within intron 2 of Mutyh.
(E) LOD score for a significant cis-eQTL for the Mutyh gene.
(F) Effect score of each collaborative cross founder strain for the Mutyh eQTL on chromosome 4.
Recent studies have detailed a domesticus-specific C>A transversion mutator allele associated with a haplotype of the Mutyh gene37; however, a causal variant has not been identified. To investigate this allele for potentially causative SVs, we surveyed each mouse genome for large variants in the Mutyh gene locus. We uncovered a 5.3 kbp IAP insertion within intron 2 of Mutyh that is unique to long-read detection. IAPs have been shown to alter gene structure and negatively impact gene expression through chromatin silencing.32,33,38 To investigate Mutyh expression, we performed bulk RNA sequencing on mESCs derived from 10 strains (STAR Methods) and grouped each sample by insertion status to perform differential gene expression analysis of Mutyh. Mice that contained the transposon insertion contained a significant decrease in Mutyh expression compared with those with no IAP insertion (Figure 3D). Interestingly, all strains that contain the IAP insertion (DBA/2J, A/J, and WSB/EiJ) also contain the five distinct single nucleotide variants previously associated with the mutator phenotype, otherwise known as the “D-like allele.”37 We additionally searched previously published mESC eQTL data derived from a large, outbred stock of mice (DO) constructed from the CC founder strains. We found an extremely significant (LOD score = 19.46) cis-eQTL for the Mutyh gene (Figure 3E).4 Samples that contain a negative effect score for Mutyh expression (A/J and WSB/EiJ) match the samples we find to contain the IAP insertion (Figure 3F). These data show that important regions of the mouse genome can be altered by previously unannotated SVs and that these SVs can reside in important genes associated with phenotypic variation.
Polymorphic transposons alter mESC chromatin dynamics and gene expression
TEs often promote genome diversity and give rise to species-specific neofunctionalization events of biological pathways.39,40 Furthermore, numerous early embryonic tissue types exhibit cell-type-specific patterning of TE expression and chromatin accessibility in human and mouse development, such as the expression of ERVs, which is a hallmark of the two-cell stage.41,42,43,44 They can inherently harbor promoters and regulatory sequences, therefore, we hypothesized that polymorphic TEs would alter the cis-regulatory landscape of early development, resulting in altered chromatin accessibility and transcript variation. To investigate the functional impact of these polymorphisms, we utilized our long-read callset to determine the genome-wide impact of TEVs on mESC chromatin accessibility. We performed ATAC-seq on mESCs derived from 10 genetically diverse strains (STAR Methods) to detect open chromatin regions and surveyed small (5 kbp) regions surrounding SVs in each mouse to detect variant associations with chromatin accessibility (Figure 4A). From this, we found 22,123 (14%) TEVs that are associated with a significant change (p < 0.05, Mann-Whitney Wilcoxon test) in chromatin accessibility.
Figure 4.

TEV effects on chromatin accessibility and gene expression
(A) Diagram showing the experimental approach; long-read whole-genome sequencing of diverse mouse genomes allows for comprehensive identification of TEV. Open chromatin regions found with ATAC-seq were used to profile changes in chromatin accessibility at sites of TEVs.
(B) Change in chromatin accessibility (normalized ATAC coverage) for 18 transposable element subtypes whose insertion status (insertion, blue; deletion, purple) is correlated with changes in chromatin accessibility (Mann-Whitney U test; ∗1 × 10−2 < p ≤ 5 × 10−2, ∗∗1 × 10−3 < p ≤ 1 × 10−2, ∗∗∗1 × 10−4 < p ≤ 1 × 10−3, ∗∗∗∗p ≤ 1 × 10−4).
(C) Change in chromatin accessibility (normalized ATAC coverage) plotted against TEV length for L1MdTf variants. Each point represents an L1MdTf variant, categorized by insertion (blue) and deletion (purple). Two distinct clusters of length represent full-length (FL) and truncated elements.
(D) Change in chromatin accessibility (normalized ATAC coverage) plotted against TEV length for L1MdA variants.
(E) Change in chromatin accessibility (normalized ATAC coverage) plotted against TEV length for IAPLTR1/1a variants. Three distinct clusters of length represent FL, IΔ1, and solo LTR elements.
(F) Genome browser diagram showing gene Slc47a2. Strain-specific mESC RNA transcriptome assemblies for strains C57BL/6J and CAST/EiJ are shown with ATAC-seq signal (blue). Site of a de novo ETn insertion specific to strain CAST/EiJ is shown (red arrow) and is accompanied by an alternative transcript not found in C57BL/6J (red transcript).
(G) ATAC coverage at the site of CAST/EiJ ETn insertion.
(H) mESC ATAC-seq coverage profiles of 10 strains surrounding the ETn insertion site between CAST/EiJ and all other strains which do not contain the insertion.
(I) Changes in Slc47a2 gene expression between CAST/EiJ and all other strains that do not contain the insertion.
To further investigate if specific TEVs are responsible for altered chromatin accessibility, we grouped TEVs by family and subtype to determine if the insertion status of a given element is associated with a change in chromatin accessibility. We identified 18 distinct TE subtypes where there was a significant chromatin accessibility difference between insertions and deletions (Figure 4B), with the most frequent genome-wide changes facilitated by LINE-1 variants. Full-length (∼6.3 kbp) polymorphic L1MdTf elements were associated with strain-specific changes in chromatin accessibility with a cluster of elements that are ∼6 kbp long (Figure 4C), suggesting that the 5′ UTR of L1MdTf elements contains regulatory sequences that promote the formation of euchromatin in mESCs. This distribution was not seen in L1MdA elements and is most likely due to the variation in the 5′ UTR monomeric repeat (Figure 4D). Given that active mouse and human LINE-1 subtypes have highly conserved ORF2p, they presumably integrate with similar insertion preferences45,46,47 and likely lack a preference for open chromatin. Thus, our data suggest that L1MdTf elements contain mESC regulatory sequences that are lacking in L1MdA sequences, and that these changes are not due to integration preferences. Overall, 18% (2,522 of 16,304) of L1MdTf variants from the 10 strains were associated with a significant change in chromatin accessibility, compared with 8% (2,048 of 23,863) of L1MdA variants. IAP variants had the opposite effect, with insertions associated with heterochromatin formation (Figure 4E). Three distinct IAP size clusters were associated with chromatin variation; however, only two clusters (∼7.2 and ∼5.3 kbp) showed a strong trend in accessibility changes based on insertion status. The third cluster consists of short (∼300 bp) sequences that are remnants of recombination between the 5′ and 3′ LTR ends. IAP sequences are associated with heterochromatin in the mouse genome48; however, we find that IAP polymorphisms that lack their internal sequence show no association with heterochromatin, suggesting that insertion of the element, rather than integration preference, is linked to accessibility changes. This pattern reflects the effects of the internal structure of the IAP element, which contains a short heterochromatin inducing sequence known to be active in mESCs.38 We now show that these sequences are often propagated in the mouse genome, leading to genome-wide changes in chromatin accessibility.
We highlight a strain-specific (CAST/EiJ) ETn (early transposon) insertion within intron 8 of the gene Scl47a2 (Figure 4F). This insertion is accompanied by a differential ATAC-seq signal unique to CAST/EiJ closely flanking the insertion site (Figures 4G and 4H). To investigate potential transcriptomic changes due to this polymorphic TE, we used our RNA sequencing dataset generated from the same cell lines to quantify mESC Slc47a2 gene expression relative to this SV. From this, we observe elevated levels of Slc47a2 expression in CAST/EiJ compared with strains that lack the insertion (Figure 4I). Transcript assemblies revealed a CAST/EiJ-specific transcript that initiates at the site of the polymorphic ETn insertion (Figure 4F, red arrow and transcript). These data suggest that the polymorphic ETn insertion introduced a novel alternative transcriptional start site within the canonical intron 8 of Slc47a2 and align with previous studies that show that ETn sequences contain binding regions for the pluripotency factor Oct4.49 We find that polymorphic ETn sequences are strongly associated with chromatin changes (p = 1.94 × 10−27, Student’s t test) in mESCs.
Discussion
The laboratory mouse is an important model for mammalian genetics and dissecting the relationship of genotype to phenotype.50,51 Genetic reference populations such as the CC and DO mice are reproducible resources of genomic complexity that have led to numerous discoveries of disease-relevant loci, and accurate knowledge of genetic variation is imperative for finding the causative alleles at these loci. Furthermore, diverse subspecies of Mus musculus contain greater nucleotide divergence than human populations, offering unique advantages to population research.52,53 Genetic variation between diverged mice has not been captured in its entirety, stemming from technological limitations of short-read DNA sequencing, which results in poor discovery and annotation for SVs. SVs are often underrepresented in variant catalogs due to their complexity, association with repetitive regions of genomes, and lack of standardized detection methods.54,55 Recent advances in long-read whole-genome sequencing have surpassed short reads in their ability to accurately detect SVs15,16,24 and have enabled researchers to resolve regions of the mouse genome that were previously represented as gaps.17 However, the comprehensive detection of SVs between diverse mouse genomes and discerning the full potential of SVs on mouse genetic reference populations cannot be completed until their genomes are entirely sequence resolved. We have made important steps in rectifying this deficit.
We characterized genome-wide SVs present in 20 laboratory mouse strains with long-read sequencing. This cohort represents popular research models such as: (1) the parental founders of the CC and DO crosses, which are powerful selective breeding panels used for trait mapping,56 (2) six resultant CC strains that have interesting phenotypes, (3) a strain often crossed with C57BL/6J that is used for studying genotype-phenotype interactions,37,57,58 and (4) numerous other models that contain unique phenotypes due to genetic background, such as the PWD/PhJ strain, which is used to model hybrid sterility.59,60 Here, we find a 221 Mbp of insertion sequence not present in the Mus musculus reference. These sequences are important for the creation of genetically engineered mice on backgrounds other than C57BL/6J, yet targeted mutagenesis constructs are often guided by the Mus musculus reference. For example, substrains from the 129 background are regularly used for genetic engineering, including CRISPR-mediated modification.61,62 Efforts to create strain-specific assemblies have aimed to correct these biases7; however, current assemblies lack large inserted sequences, duplications, and full annotation of TEs.13,24 From human studies, we have found that long reads excel in identifying SVs and can be used to create highly contiguous genome assemblies that can also be used for SV detection.9,12,13,63 Our study creates strain-specific genome assemblies with greater continuity and comprehensive SV callsets. In total we discovered 278,062 variants that were undetected with short-read SV calling methods while also capturing 90% of the variants from short-read calls (Figure 1D). We call 215,262 SVs that are absent from public mouse SV catalogs,23,64 including 510 coding sequence SVs, indicating the importance of using long-read data.22 Newly emerging tools and techniques can further integrate our callset in the genotyping of large breeding panels, such as the CC, DO, and BXD.65
An ongoing difficulty in genome assembly is the reconstruction of repetitive element sequences, including TEs. Mice have a higher de novo TE insertion rate than that of humans (∼1 LINE-1 insertion in every 8 live births in contrast to 1 in 20–200 live births in humans)25,26 and contain retrotransposition competent ERVs66; therefore, the role of TEs in genomic change is an important contributor to murine genomic variation. We find that TEs drive extensive SVs in mice and are the dominant source of variable sequence content in mouse genomes (Figure 2C). Our resource provides classification of polymorphic elements, distinguishes closely related subfamilies, including the active L1MdA, L1MdTf, and L1MdGf LINE-1 transposons, and reveals evidence of an L1MdTf-I/II expansion within the domesticus subspecies. Interestingly, we find that non-domesticus genomes contain L1MdTf-I/II copies, although they are at a much lower copy number than domesticus. This could be due to (1) misclassification of these elements as the Mus musculus database of TEs has been constructed primarily using C57BL/6J sequences, (2) introduction of these elements into non-domesticus genomes through hybridization between subspecies, which has been observed previously,67 or (3) L1MdTf-I/II elements have existed before the estimated divergence of domesticus, musculus, and castaneus and became increasingly active within the domesticus genome, similar to the IAP insertions observed within C3H mice.11 Surprisingly, we observe no large differences in L1MdA-I copy number between each subspecies despite the estimated age of this subfamily being more proximal than divergence of domesticus, musculus, and castaneus (0.3.–0.5 Myr). We believe that our work indicates a need to investigate the expansion or regulation of these elements between these major subspecies, and our resource will enable future studies on this topic.
By resolving TEVs, we detect domesticus-specific enrichment of IΔ1 and full-length IAP element variants when compared with castaneus and musculus (Figures 2D and 2E). Previous studies suggest that post-transcriptional processing of IAP-containing sequences varies between castaneus and domesticus mice due to mutations in the Nxf1 protein.68 We observe that the level of IAP polymorphism in musculus strains is similar to castaneus and that both are lower than domesticus, suggesting that variable activity of IAP elements may have different mechanisms in different subspecies. Even within the domesticus lineage, IAP insertions unique to C3H mice contain a distinct length distribution, with a greater number of variants in the 4.5–5.5 kbp range (Figures 2F and 2G). Increased expression of IAP retrotransposons has been observed in mESCs derived from C3H/HeJ mice, with multiple accounts of aberrant gene expression from insertion polymorphisms.11,31 Here, we sequence-resolve 187 C3H-specific IAP insertions that can be used to further investigate this phenotype. Furthermore, fixed transposon sequences can be differentially methylated in the mouse genome and are regulated by trans factors such as KRAB zinc finger proteins.69 Fixed TEs can also accumulate mutations that are otherwise blind to short-read detection methods70,71; the long-read de novo assemblies we provide can be used to investigate allelic and epigenetic heterogeneity within a given repeat and allow for precise mapping of orthogonal sequencing data in a strain-specific manner.72,73
The biological effects of TEVs are not confined to structural alteration of genomic DNA.74 TEVs frequently carry with them potent regulatory sequences that alter gene expression and chromatin accessibility, sometimes in a tissue-specific manner.41,75,76 Furthermore, genomic TEs are increasingly recognized as important contributors to early development as they can be co-opted as enhancers, regions of transcription factor binding sites,77 and early expression of TEs can be used to profile early embryonic stages such as zygotic genome activation.78 Our TEV callset and assemblies will enable the examination of the effects of TE polymorphisms on early embryonic tissues and can act as a model for studies in other organisms. Our study examines polymorphic TEs, showing that many families affect nearby chromatin accessibility and that they may differentiate mouse transcriptomes in early development. Interestingly, our data suggest that recently transposed events from closely related LINE-1 subfamilies likely differ in the cis-regulatory elements they bind to. Mus musculus LINE-1 subtypes are known to contain alternative promoters constructed from monomeric repeats and are differentially methylated in male germ cell development.79,80 Here, we show that polymorphic L1MdTf elements are more often associated with increases in mESC chromatin accessibility when compared with L1MdA variants despite representing fewer polymorphisms, and this effect is more marked with full-length elements, suggesting that the L1MdTf promoter is active in mESCs (Figures 4C and 4D). In contrast, we find that IAPs are associated with chromatin closing (Figure 4E), consistent with previous studies that detail an internal short heterochromatin inducing sequence that is active in mESCs.38 It is important to note that we detail these changes at one developmental time point, and that TE activity is dynamic during mammalian development.42 Further utilization of this callset with orthogonal data to support additional tissue types will aid in characterizing the effects of TE polymorphisms throughout development.
Several important technological advances in sequencing have arisen recently, including ultralong ONT and PacBio HiFi,81 which improve accuracy, assembly contiguity, and assembly-based variant detection.6,13,14,82 While telomere-to-telomere assembly is not yet routine, complete sequence resolution of diverse mouse genomes may be possible in the near future. These technologies will enable phasing for the approximately 5% of mouse genomes that are heterozygous after inbreeding and for wild-derived individuals and would more completely resolve segmental duplication loci. Complete assemblies may also enable other studies, such as an examination of genome-wide synteny over 500,000 years of evolution and the examination of rearrangements between complex segmental duplications. The data we generate here are important to modernize mouse genetics, and with it we provide a sequence-resolved SV resource, a mESC expression resource, and mESC chromatin accessibility data, which will enable evolutionary research and phenotype-genotype correlations in mice.
Limitations of the study
There remain two outstanding limitations to our study. First, our methods exclude phasing, and therefore our analysis is confined to haploid genomic assemblies. This drawback is not as important when sequencing classical inbred laboratory mouse genomes as ∼95% of their genomes are homozygous. We estimate that a small portion (∼2.5%, 50% within regions of residual heterozygosity) of SVs are lost as these regions are represented as one haplotype. Second, the mouse genome contains many large complex repetitive loci, such as segmental duplications and centromeres. Some of these regions prove to be too large to assemble with our PacBio long reads, resulting in collapsed or discontinuous sequences. Resolution of these regions will require a combination of longer lengths to span repeats, such as Oxford Nanopore ultralong reads, and lower error to separate paralogs and homologs, such as PacBio HiFi reads.6,83 In future studies, assemblies constructed using these technologies will allow for more complete profiling of diverse mouse genomes.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Whole genome PacBio long reads | This Study | NCBI: PRJNA923323 |
| mESC ATAC-seq reads | This study | NCBI: PRJNA923323 |
| mESC RNA-seq reads | This study | NCBI: PRJNA923323 |
| Long read genome assemblies | This study | NCBI: PRJNA923323 |
| Structural variant calls | This study | https://doi.org/10.5281/zenodo.7644286 |
| RepeatMasker annotation of mouse assemblies | This study | https://doi.org/10.5281/zenodo.7644286 |
| Biological samples | ||
| Mouse strain 129S1/SvImJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:02448 |
| Mouse strain A/J Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:00646 |
| Mouse strain BALB/cByJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:01026 |
| Mouse strain BALB/cJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:00651 |
| Mouse strain C3H/HeJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:00659 |
| Mouse strain C3H/HeOuJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:00635 |
| Mouse strain C57BL/6NJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:05304 |
| Mouse strain DBA/2J Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:00671 |
| Mouse strain NOD/ShiLtJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:01976 |
| Mouse strain NZO/HlLtJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:02105 |
| Mouse strain CAST/EiJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:00928 |
| Mouse strain PWD/PhJ Male Testis | The Jackson Laboratory | RRID:IMSR_JAX:04660 |
| Mouse strain PWK/PhJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:03715 |
| Mouse strain WSB/EiJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:01145 |
| Mouse strain CC005/TauUncJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:20945 |
| Mouse strain CC015/UncJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:18859 |
| Mouse strain CC032/GeniUncJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:20946 |
| Mouse strain CC055/TauUncJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:25422 |
| Mouse strain CC060/UncJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:26427 |
| Mouse strain CC074/UncJ Female Kidney | The Jackson Laboratory | RRID:IMSR_JAX:18855 |
| Experimental models: Cell lines | ||
| 129S1/SvImJ mouse embryonic stem cells | The Jackson Laboratory | RRID:IMSR_JAX:02448 |
| A/J mouse embryonic stem cells | The Jackson Laboratory | RRID:IMSR_JAX:00646 |
| C3H/HeJ mouse embryonic stem cells | The Jackson Laboratory | RRID:IMSR_JAX:00659 |
| C57BL/6J mouse embryonic stem cells | The Jackson Laboratory | RRID:IMSR_JAX:00664 |
| DBA/2J mouse embryonic stem cells | The Jackson Laboratory | RRID:IMSR_JAX:00671 |
| NOD/ShiLtJ mouse embryonic stem cells | The Jackson Laboratory | RRID:IMSR_JAX:01976 |
| NZO/HlLtJ mouse embryonic stem cells | The Jackson Laboratory | RRID:IMSR_JAX:02105 |
| CAST/EiJ mouse embryonic stem cells | The Jackson Laboratory | RRID:IMSR_JAX:00928 |
| PWK/PhJ mouse embryonic stem cells | The Jackson Laboratory | RRID:IMSR_JAX:03715 |
| WSB/EiJ mouse embryonic stem cells | The Jackson Laboratory | RRID:IMSR_JAX:01145 |
| Software and algorithms | ||
| Flye (v2.8.3) | Kolmogorov et al.19 | https://github.com/fenderglass/Flye |
| Arrow (v2.0.2) | PacBio | https://github.com/PacificBiosciences/gcpp |
| PAV (v1.1.2) | Ebert et al.13 | https://github.com/EichlerLab/pav |
| SV-Pop (v2.0.0) | Ebert et al.13 | https://github.com/EichlerLab/svpop |
| pbsv (v2.6.2) | PacBio | https://github.com/PacificBiosciences/pbsv |
| Sniffles (v1.0.12) | Sedlazeck et al.84 | https://github.com/fritzsedlazeck/Sniffles |
| SVIM (v2.0.0) | Heller et al.85 | https://github.com/eldariont/svim |
| SVIM-asm (v1.0.2) | Heller et al.63 | https://github.com/eldariont/svim-asm |
| BWA-MEM (v0.7.17) | Li and Durbin86 | https://github.com/lh3/bwa |
| Manta (v1.6.0) | Chen et al.87 | https://github.com/Illumina/manta |
| DELLY (v0.8.7) | Rausch et al.88 | https://github.com/dellytools/delly |
| LUMPY (v0.2.13) | Layer et al.89 | https://github.com/arq5x/lumpy-sv |
| WGAC | Bailey et al.90 | https://github.com/EichlerLab/WGAC |
| ReapeatMasker (v4.1.2) | Smit, AFA, Hubley, R & Green, P | https://github.com/rmhubley/RepeatMasker |
| subseq (commit c790053) | Ebert et al.13 | https://github.com/EichlerLab/seqtools |
| quast (5.0.2) | Gurevich et al.91 | https://github.com/ablab/quast |
| nextflow (v21.04.0) | Di Tommaso et al.92 | https://www.nextflow.io |
| nf-core/atacseq (v1.2.1) | https://doi.org/10.5281/zenodo.2634132 | https://nf-co.re/atacseq |
| BEDTools (2.30.0) | Quinlan and Hall93 | https://github.com/arq5x/bedtools2 |
| Conda (4.10.3) | Anaconda Software Distribution | https://github.com/conda/conda |
| Ensembl Variant Effect Predictor (v104.3) | McLaren et al.36 | https://github.com/Ensembl/ensembl-vep |
| STAR (v2.7.8a) | Dobin et al.94 | https://github.com/alexdobin/STAR |
| Tetranscripts (v2.2.1) | Jin et al.95 | https://github.com/mhammell-laboratory/Tetranscripts |
| DESeq (v3.15) | Love et al.96 | https://github.com/mikelove/DESeq2 |
| StringTie (v2.1.7) | Kovaka et al.97 | https://github.com/skovaka/stringtie2 |
| featureCounts (v2.0.1) | Liao et al.98 | https://github.com/topics/featurecounts |
| Duphold (v0.2.3) | Pederson and Quinlan99 | https://github.com/brentp/duphold |
| bam2fastx (v1.3.0) | PacBio | https://www.pacb.com/wp-content/uploads/SMRT_Tools_Reference_Guide_v600.pdf |
| NGMLR (v0.2.7) | Sedlazeck et al.84 | https://github.com/philres/ngmlr |
| Minimap2 (v2.17) | Li100 | https://github.com/lh3/minimap2 |
| pbmm2 (v1.4.0) | PacBio | https://github.com/PacificBiosciences/pbmm2 |
| LRA (v1.3.0) | Ren et al.101 | https://github.com/ChaissonLab/LRA |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Christine R. Beck (christine.beck@jax.org).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Information of diverse inbred mouse strains and derived cell lines in this study can be found, and are accompanied by research resource identifiers, in the key resources table and method details.
Method details
Sample selection
We assembled the genomes of 20 diverse inbred laboratory mouse strains (129S1/SvImJ, A/J, CAST/EiJ, NOD/ShiLtJ, NZO/HILtJ, PWK/PhJ, WSB/EiJ, CC005, CC015, CC032, CC055, CC060, CC074, BALB/cByJ, BALB/cJ, C3H/HeJ, C3H/HeOuJ, DBA/2J, C57BL/6NJ, and PWD/PhJ) with whole-genome long-read sequencing and called SVs in each genome generated in this study. We chose to sequence the founders of the collaborative cross as they represent various diverse genetic backgrounds of Mus musculus and have been used to create numerous backcrossed offspring for complex phenotype mapping. We also sequenced six resultant strains created from breeding of these founders. Lastly, we sequenced seven additional diverse mice which are important for other recombinant crosses and other mouse genetics purposes. Because we do not have orthogonal sequencing data to support genome phasing such as Strand-seq, our assemblies and SV calls represent a single haplotype. Approximately 95% of inbred mouse genomes are homozygous, and we expect to lose 50% of SVs in residual heterozygous loci. For short-read sequencing analysis, we used data from previously published datasets of Illumina whole-genome sequencing reads.7,22
mESCs
mESC cell lines from ten diverse mice (129S1/SvImJ, A/J, C57BL/6J, CAST/EiJ, NOD/ShiLtJ, NZO/HILtJ, PWK/PhJ, WSB/EiJ, C3H/HeJ, and DBA/2J) were derived and cultured using previously published methods.102 Cells were thawed onto gelatin in ESM/FBS/2i media and grown to 60–80% confluency. All cell lines were between P11-P13 at the time of the harvest. Cells were dissociated with 0.05% trypsin-EDTA, resuspended in PBS for counting, with 1 × 106 cells reserved for RNA and 1 × 105 cells reserved for ATAC-seq. For the RNA sample, the cells were spun down, the supernatant removed, and the pellet was flash frozen and then placed on dry ice. The samples were stored at −80°C until harvest. For the ATAC samples, the cells were spun down and resuspended in 1 mL of freeze media (80% ESM/10% FBS/10%DMSO). The volume was divided in 1/2 among 2 cryovials. The ATAC samples were placed in Cool Cells in the −80°C and then transferred to LN2 24–48 h later.
PacBio long-read whole-genome sequencing
Kidney tissue from female mice were supplied from The Jackson Laboratory mouse services, other than the PWD/PhJ sample, which was derived from testis (key resources table). gDNA was first extracted using the Gentra Puregene (Qiagen) kit. Frozen kidney tissues from each mouse were first pulverized using a mortar and pestle and transferred to a 15mL tube containing Qiagen Cell Lysis Solution. Lysate was then incubated with Proteinase K for 3 h at 55°C, followed by the addition RNase A and continued incubation for 40 min at 37°C. Samples were cooled on ice and Protein Precipitation Solution was added. Samples were then vortexed and centrifuged. The supernatant was transferred to a new tube containing isopropanol for precipitation. The remaining pellet was washed with 70% ethanol, air dried, and rehydrated in PacBio Elution Buffer until dissolved. Sample preparation was performed following the continuous long read (CLR) protocol from PacBio using the PacBio SMRTbell Express Template Prep Kit 2.0. 15 μg of high molecular weight gDNA was sheared using 26G Needles for 10–20 passes. Sample showing a broad distribution of DNA 20–100 kbp on Femto Pulse (Agilent) was selected to proceed. Additional shearing was performed to achieve the targeted distribution if needed. Sheared DNA Sample was then concentrated with AMPure PB Beads (PacBio). 10μg of the sheared DNA was end repaired and A-tailed to remove single-strand overhangs and repair DNA damage. This is followed by a ligation to an overhang V3 adapter and clean up with 0.45x AMPure PB. The purified library was subjected to size-selection (>10 kbp) using the Blue Pippin system (Sage Science). The library was purified with 1⨉ AMPure PB library and sequenced on a PacBio Sequel II. Each sample was sequenced to a minimum of 30-fold coverage.
ATAC sequencing (ATAC-seq)
ATAC-seq103 data was generated from mESCs derived from 10 mouse strains (129S1/SvImJ, A/J, C57BL/6J, CAST/EiJ, NOD/ShiLtJ, NZO/HILtJ, PWK/PhJ, WSB/EiJ, C3H/HeJ, and DBA/2J). ATAC-seq libraries were prepared using 50,000 cells as previously described104 with the following modifications: pelleted cells were washed in 150 μL PBS before the addition transposase mixture; transposition reactions were agitated at 1000 rpm; transposed DNA was purified using a Genomic DNA Clean & Concentrator (Zymo); purified DNA was eluted in 21 μL elution buffer (10 mM Tris-HCl, pH 8); transposed fragments were amplified using 2⨉ KAPA HiFi HotStart ReadyMix (Roche) and Nextera DNA CD Indexes (Illumina) for 10 cycles of PCR; PCR reactions were purified using 1.7⨉ KAPAPure beads (Roche). Libraries were checked for quality and concentration using the DNA High-Sensitivity TapeStation assay (Agilent Technologies) and quantitative PCR (Roche), according to the manufacturers’ instructions. Libraries were sequenced 100 bp paired-end on an Illumina NovaSeq 6000 using the S2 Reagent Kit v1.5.
RNA sequencing (RNA-seq)
RNA-seq data was generated from mESCs derived from 10 mouse strains (129S1/SvImJ, A/J, C57BL/6J, CAST/EiJ, NOD/ShiLtJ, NZO/HILtJ, PWK/PhJ, WSB/EiJ, C3H/HeJ, and DBA/2J). Tissues were lysed and homogenized in TRIzol Reagent (Ambion), then RNA was isolated using the miRNeasy Mini kit (Qiagen), according to manufacturers’ protocols, including the optional DNase digest step. RNA concentration and quality were assessed using the Nanodrop 8000 spectrophotometer (Thermo Scientific) and the RNA ScreenTape Assay (Agilent Technologies). Libraries were constructed using the KAPA mRNA Hyper-Prep Kit (Roche Sequencing and Life Science), according to the manufacturer’s protocol. Briefly, the protocol entails isolation of polyA containing mRNA using oligo-dT magnetic beads, RNA fragmentation, first and second strand cDNA synthesis, ligation of Illumina-specific adapters containing a unique barcode sequence for each library, and PCR amplification. The quality and concentration of the libraries were assessed using the D5000 ScreenTape (Agilent Technologies) and Qubit dsDNA HS Assay (ThermoFisher), respectively, according to the manufacturers’ instructions. Libraries were sequenced 100 bp paired-end on an Illumina NovaSeq 6000 using the S2 Reagent Kit v1.5.
Genome assembly, variant calling, and QC
For each sample, raw PacBio CLR bam files were converted to FASTA files with bam2fastx (https://github.com/PacificBiosciences/bam2fastx) (v1.3.0). Strain-specific assemblies were then constructed with Flye19 (v2.8.3) and Arrow (https://github.com/PacificBiosciences/gcpp) (v2.0.2) was used for polishing. Assembly metrics were computed and summarized with quast91 (v5.02) (Table S1).
Variant discovery was performed against the GRCm39 (mm39, Mus musculus) reference genome. SVs were called with PAV13 (v1.1.2) using minimap2100 (v2.17) alignments. Orthogonal callset support was obtained with pbsv (https://github.com/PacificBiosciences/pbsv) (v2.6.2), Sniffles84 (v1.0.22), SVIM85 (v2.0.0), SVIM-asm63 (v1.0.2), and PAV run with the LRA101 (v1.3.0) aligner (PAV-LRA). For pbsv and SVIM, we used the pbmm2 (v1.4.0) alignment tool to align raw long reads to the GRCm39 (mm39, Mus musculus) genome. For Sniffles, NGMLR84 (v0.2.7) alignments were used. Additional raw-read support was annotated by extracting aligned segments of long-reads in regions around SVs with subseq13 (commit c790053) (2 + supporting reads).
For short-read SV calling, raw Illumina FASTQ files were aligned to the GRCm39 reference genome with BWA-MEM86 (v0.7.17). We then called SVs with Manta87 (v1.6.0), LUMPY89 (v0.2.13), and DELLY88 (v0.8.7). We then ran Duphold99 (v0.2.3) on all SV calls to obtain a read depth value for regions overlapping SV calls.
Variants were accepted into the final callset if they have a) either pbsv or SVIM support in addition to raw-read subseq support, b) support from two or more of Sniffles, SVIM-asm, and PAV-LRA in addition to raw-read subseq support, c) both SVIM-asm and PAV-LRA support and are greater than 250 bp, or d) they have raw-read subseq support or duphold-CLR support, have duphold-SR support, and have support from one or more of DELLY, LUMPY, or Manta. The full reference assembly was present during read and contig alignments; however, the final callset was filtered to only include chromosome scaffolds excluding chrY (chr1-19 and chrX).
Finally, a non-redundant callset across samples was achieved by merging with SV-Pop13 (v2.0.0). The per-genome average number of SVs for each subspecies was calculated from Table 1; C57BL/6NJ was excluded from the domesticus mean due to its extreme conservation with the C57BL/6J reference genome. The total percentage of the mouse genome affected by SVs was calculated by first running BEDTools merge (v2.30.0) on the SV calls to remove redundant overlap between SVs not found in the same samples. The sum all SV lengths were then divided by the length of the GRCm39 reference genome (2,728,206,152 bp, obtained from https://www.ncbi.nlm.nih.gov/grc/mouse/data).
Segmental duplication annotation and callable regions
Segmental duplications (SD) were characterized in GRCm39 using the whole-genome assembly comparison (WGAC) method as previously described.90 Briefly, WGAC identifies non-homologous pairwise alignments by performing an all-by-all comparison after removing common repeat elements (RepeatMasker v4.1.0 with engine crossmatch v1.090518 and the 'Mus musculus' and Tandem Repeat Masker v4.09 while ignoring repeats with >12 periodicity available from the UCSC browser). WGAC fragments the genome into 400 kbp repeat-masked segments and performs an all-by-all comparison between segments (blastall v2.2.11) and within 400 kbp alignments using the LASTZ tool (v1.02.00) (https://www.bx.psu.edu/∼rsharris/lastz/). Common repeats are reintroduced to construct optimal global alignments that are at least >1 kbp and >90% sequence identical to define the SD set for the mouse genome.
Intersection with mouse genomes project callset, Nellaker transposable element variant calls, and subseq validation
Variants were intersected with the Mouse Genomes Project SV release v5 (Project URL: http://www.sanger.ac.uk/resources/mouse/genomes/; FTP: ftp://ftp-mouse.sanger.ac.uk/; and lifted over to GRCm39 coordinates using UCSC LiftOver: https://genome.ucsc.edu/cgi-bin/hgLiftOver). Because callset characteristics were different than modern callsets, we adopted a customized approach. Because insertions lengths are unknown, we counted variants as supported if the insertion site was within 250 bp and ignored the size. Large insertions are called duplicated reference loci, and to allow direct comparisons, we re-mapped insertion sequences to the reference and intersected the resulting loci with duplication calls using 50% reciprocal overlap. Deletions were also matched by 50% reciprocal overlap. An SV was considered detected by both methods if the SV was called in the same strain. This same approach was used to intersect transposable element variants we detected with previously published transposable element variants.27
To validate SV calls with long-read support, we used subseq to extract read lengths (200 bp surrounding region of SV) from pbmm2 alignments. Calls were accepted as PASS if the mean read length difference was ≥50 bp.
Transposable element annotation
In order to characterize transposable element variation, we obtained a FASTA output from SV-Pop containing the full sequences of insertion and deletion SV. We then used RepeatMasker105 (v4.1.2) with the “-species mus_musculus” option to annotate repeat sequences within these variant sequences. Transposable element variants (TEVs) are accepted as insertion or deletion variants which have 60% of their content match a specific transposable element family. Full length long terminal repeat (LTR) transposable element consensus sequences are fragmented in transposable element databases as their internal sequence is separate from their flanking LTRs. We further annotated LTR TEVs by accepting the flanking LTR annotation if it were adjacent to an internal sequence. Distribution of target site duplication lengths are shown in Figure S5. TEV annotations are present in the final data table (Table S2), with annotation for both TE type and TE subtype.
Variant effects and association analysis
A VCF file containing our primary SV callset was used as input for the Ensembl Variant Effect predictor36 (v104.3) tool (command-line) to intersect SV coordinates with GRCm39 features. Frameshift_variant, inframe_insertion, inframe_deletion, frameshift_variant, stop_gained, stop_lost, protein_altering_variant, stop_retained_variant, and start_lost annotations were then merged into one “coding_sequence_variant” annotation. A VEP summary analysis was performed and is reported in the Table S2 (VEP_SUMMARY column). We performed an association analysis for each variant consequence (https://useast.ensembl.org/info/genome/variation/prediction/predicted_data.html) and four criteria: tandem repeat SVs, transposable element variants, variants which have short-read support, and variants which have support from the Mouse Genomes Project. For each criterion, we performed a Fisher’s exact test to determine contingency of each SV consequence and criteria (Table S4).
Chromatin accessibility, gene expression profiling, and transposable element association
Open chromatin peaks were detected for each sample by using the nextflow92 atacseq (nf-core/atacseq v1.2.1) pipeline. Raw read counts for each open chromatin region were extracted by using featureCounts98 (v2.0.1). For each sample, regions surrounding SV coordinates (±2500 bp flanking the breakpoint) were intersected with open chromatin regions using BEDTools93 intersect (v2.30.0). The sum of raw read counts for overlapping peaks were accepted for further analysis. Read count values for each sample was merged into a matrix and normalized with DESeq296 (v3.15). For each SV, normalized read counts for each sample were grouped into two groups depending on whether the sample contained the SV or not and a Mann-Whitney U test was performed between each group to determine a statistically significant (p < 0.05) difference. To assess TE families and changes in chromatin accessibility association, SVs which were associated with changes in chromatin accessibility were first grouped by TE subtype. Subtype-specific calls were further grouped whether they were an insertion or deletion variant, and a Student’s t test was performed between normalized read count values for insertion and deletions of a particular TE subtype, followed by Bonferroni correction.
For RNA-seq analysis, raw FASTQ files were aligned to GRCm39 reference with STAR94 (v2.7.8a) (default settings). Raw gene read counts were obtained for each sample with the TEtranscripts package (v2.2.1) and normalized with the DESeq2 package (v3.15). Association between candidate SVs and gene expression changes were calculated by a nonparametric test (Mann-Whitney U) of normalized gene read counts between samples which contained the candidate SV and those that did not. For transcript assemblies, we used the StringTie package (v2.1.7).
PCR validations and false discovery rate
For each SV, 500 bp of flanking DNA sequence was obtained from selected SV breakpoints using BEDTools getfasta. Primers were designed using Primer3web (version 4.1.0; https://primer3.ut.ee/). Ideal primer conditions were set to have a length of 25 bp, melting temperature of 65°C, and 50% GC content. Primers were designed to anneal at least 50 bp from SV breakpoints. At least one primer was required to anneal in a unique region of the genome using UCSC BLAT (https://genome.ucsc.edu/cgi-bin/hgBlat), and primer pairs were analyzed using UCSC In-Silico PCR (https://genome.ucsc.edu/cgi-bin/hgPcr) to confirm a unique amplicon for each SV. If primer conditions were not met in the first 500 bp flanking region, the design window was increased in increments of 500 bp until all primer requirements are fulfilled. Takara LA Taq Polymerase (Takara RR002M) was used for PCR validation. Each primer pair was tested across 8 founder mice strains and a negative control. Touchdown PCR was used for desirable primer annealing and amplification of each predicted amplicon. OrangeG gel loading dye was mixed with PCR product and ran in a 1% agarose gel (Bio-Rad Certified Molecular Biology Agarose 1613102) supplemented with ethidium bromide (Bio-Rad 1610433). Each primer pair was designed to show a resulting amplicon whether or not the SV was present. PCR validations and primer sequences can be found in Table S3.
In order to provide an unbiased FDR estimate, variant callset filters were informed by inspecting callset support from all sources and identifying sources of likely false SV calls, was not informed by PCR validation results, and adjustments to the filters were not made once FDR estimates were computed.
Thermocycler program for amplicon sizes less than 5 kbp:
Step 1. 95°C for 1 min.
Step 2. 95°C for 30 s.
Step 3. 68°C for 30 s (with a 1°C ramp down per cycle)
Step 4. 72°C for 5 min.
Step 5. Return to step 2 and repeat for 5 cycles.
Step 6. 95°C for 30 s.
Step 7. 63°C for 30 s.
Step 8. 72°C for 5 min.
Step 9. Return to step 6 and repeat for 25 cycles.
Step 10. 72°C for 10 min.
Step 11. 4°C infinite hold.
Thermocycler program for amplicon sizes greater than 5 kbp:
Step 1. 95°C for 1 min.
Step 2. 95°C for 30 s.
Step 3. 68°C for 30 s (with a 1°C ramp down per cycle)
Step 4. 72°C for 9 min.
Step 5. Return to step 2 and repeat for 5 cycles.
Step 6. 95°C for 30 s.
Step 7. 63°C for 30 s.
Step 8. 72°C for 9 min.
Step 9. Return to step 6 and repeat for 25 cycles.
Step 10. 72°C for 10 min.
Step 11. 4°C infinite hold.
Quantification and statistical analysis
Statistical analyses were performed with Python (v3.8.3) and the SciPy (v1.5.2) library. Details of each test can be found in the method details section and all tests performed were two-sided.
Acknowledgments
This research was supported by The Jackson Laboratory Director’s Innovation Fund to C.R.B. and L.G.R., The Jackson Laboratory Cancer Center (P30CA034196), the National Institutes of Health (R35 GM133600 to C.R.B., R24 OD021325-06 to L.G.R., R01 HG002385 to E.E.E., and 1T32HG010463 for support of A.F.). We would like to thank Denisse Tafur, Natalie Powers, and Petko Petkov for their aid in tissue sample extraction and submission for sequencing. We would like to thank Charles Lee for his review of the manuscript, Tonia Brown and Martina Miranda for their critical proofreading assistance, and the reviewers for their thoughtful input during manuscript revisions. We would also like to thank The Jackson Laboratory Genome Technologies core for help with sequencing. E.E.E. is an investigator of the Howard Hughes Medical Institute. The graphical abstract and Figure 4A were created with BioRender.
Author contributions
C.R.B. conceived of and supervised the study. C.R.B. and L.G.R. procured funding for the study. A.F. performed data preprocessing, data analysis, and data interpretation. P.A.A. performed quality control, method development, and data analysis. A.C. and L.G.R. performed cell culture work, sample preparation, and sample submission for sequencing. P.B. and I.A.W. aided in sample submission, data management, and preprocessing. P.B. performed data archiving. J.I.F., A.A.R., and V.M. designed primers and conducted PCRs for validation work. D.S.G. and E.E.E. generated and provided the GRCm39 segmental duplication track. A.F., P.A.A., and C.R.B. wrote the manuscript.
Declaration of interests
E.E.E. is a scientific advisory board (SAB) member of Variant Bio, Inc.
Published: April 5, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100291.
Supplemental information
Structural variant calls for each mouse genome were merged and annotated into a non-redundant callset. Descriptions of each field can be found within the “Fields” sheet.
PCR validations of SVs in eight strains of mice (129S1/SvImJ, A/J, CAST/EiJ, NOD/ShiLtJ, NZO/HILtJ, PWK/PhJ, and WSB/EiJ): 1 indicates an animal contains the SV and 0 denotes absence of SV. Animals computationally predicted to contain the SV are shown in the MERGE_SAMPLES column.
Summary of statistical analysis performed for VEP output (consequence_stats sheet, Fisher’s exact test), VEP consequences for transposable element variants (TE_family_consequence_stats sheet, Fisher’s exact test), and chromatin accessibility associations by transposable element type (chromatin_accessibility_stats sheet, Student’s t test). SR_SUPPORT, SVs that are supported by short-read SV calling; TR_ANNO, tandem repeat SVs; MGP_ANNO, SVs supported by the mouse genomes project dataset; TE_ANNO, SVs that are transposable elements.
Data and code availability
The genome assemblies, long-read sequencing, ATAC-seq, and RNA-seq data generated for this study can be obtained through the NCBI sequencing read archive under BioProject NCBI: PRJNA923323. Structural variant VCFs for each individual mouse strain, the merged structural variant VCF, and RepeatMasker annotations for each assembly were deposited on Zenodo at https://doi.org/10.5281/zenodo.7644286. All software used in this study can be found in the key resources table.
References
- 1.Threadgill D.W., Miller D.R., Churchill G.A., de Villena F.P.M. The collaborative cross: a recombinant inbred mouse population for the systems genetic era. ILAR J. 2011;52:24–31. doi: 10.1093/ilar.52.1.24. [DOI] [PubMed] [Google Scholar]
- 2.Churchill G.A., Airey D.C., Allayee H., Angel J.M., Attie A.D., Beatty J., Beavis W.D., Belknap J.K., Bennett B., Berrettini W., et al. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat. Genet. 2004;36:1133–1137. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- 3.Keller M.P., Rabaglia M.E., Schueler K.L., Stapleton D.S., Gatti D.M., Vincent M., Mitok K.A., Wang Z., Ishimura T., Simonett S.P., et al. Gene loci associated with insulin secretion in islets from non-diabetic mice. J. Clin. Invest. 2019;129:4419–4432. doi: 10.1172/JCI129143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skelly D.A., Czechanski A., Byers C., Aydin S., Spruce C., Olivier C., Choi K., Gatti D.M., Raghupathy N., Keele G.R., et al. Mapping the effects of genetic variation on chromatin state and gene expression reveals loci that control ground state pluripotency. Cell Stem Cell. 2020;27:459–469.e8. doi: 10.1016/j.stem.2020.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bubier J.A., Jay J.J., Baker C.L., Bergeson S.E., Ohno H., Metten P., Crabbe J.C., Chesler E.J. Identification of a QTL in Mus musculus for alcohol preference, withdrawal, and Ap3m2 expression using integrative functional genomics and precision genetics. Genetics. 2014;197:1377–1393. doi: 10.1534/genetics.114.166165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nurk S., Koren S., Rhie A., Rautiainen M., Bzikadze A.V., Mikheenko A., Vollger M.R., Altemose N., Uralsky L., Gershman A., et al. The complete sequence of a human genome. Science. 2022;376:44–53. doi: 10.1126/science.abj6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lilue J., Doran A.G., Fiddes I.T., Abrudan M., Armstrong J., Bennett R., Chow W., Collins J., Collins S., Czechanski A., et al. Sixteen diverse laboratory mouse reference genomes define strain-specific haplotypes and novel functional loci. Nat. Genet. 2018;50:1574–1583. doi: 10.1038/s41588-018-0223-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keane T.M., Goodstadt L., Danecek P., White M.A., Wong K., Yalcin B., Heger A., Agam A., Slater G., Goodson M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477:289–294. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Audano P.A., Sulovari A., Graves-Lindsay T.A., Cantsilieris S., Sorensen M., Welch A.E., Dougherty M.L., Nelson B.J., Shah A., Dutcher S.K., et al. Characterizing the major structural variant alleles of the human genome. Cell. 2019;176:663–675.e19. doi: 10.1016/j.cell.2018.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mouse Genome Sequencing Consortium. Waterston R.H., Lindblad-Toh K., Birney E., Rogers J., Abril J.F., Agarwal P., Agarwala R., Ainscough R., Alexandersson M., et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 11.Gagnier L., Belancio V.P., Mager D.L. Mouse germ line mutations due to retrotransposon insertions. Mob. DNA. 2019;10:15. doi: 10.1186/s13100-019-0157-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaisson M.J.P., Sanders A.D., Zhao X., Malhotra A., Porubsky D., Rausch T., Gardner E.J., Rodriguez O.L., Guo L., Collins R.L., et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019;10:1784. doi: 10.1038/s41467-018-08148-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ebert P., Audano P.A., Zhu Q., Rodriguez-Martin B., Porubsky D., Bonder M.J., Sulovari A., Ebler J., Zhou W., Serra Mari R., et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science. 2021;372:eabf7117. doi: 10.1126/science.abf7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nurk S., Walenz B.P., Rhie A., Vollger M.R., Logsdon G.A., Grothe R., Miga K.H., Eichler E.E., Phillippy A.M., Koren S. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 2020;30:1291–1305. doi: 10.1101/gr.263566.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou W., Emery S.B., Flasch D.A., Wang Y., Kwan K.Y., Kidd J.M., Moran J.V., Mills R.E. Identification and characterization of occult human-specific LINE-1 insertions using long-read sequencing technology. Nucleic Acids Res. 2020;48:1146–1163. doi: 10.1093/nar/gkz1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ewing A.D., Smits N., Sanchez-Luque F.J., Faivre J., Brennan P.M., Richardson S.R., Cheetham S.W., Faulkner G.J. Nanopore sequencing enables comprehensive transposable element epigenomic profiling. Mol. Cell. 2020;80:915–928.e5. doi: 10.1016/j.molcel.2020.10.024. [DOI] [PubMed] [Google Scholar]
- 17.Sarsani V.K., Raghupathy N., Fiddes I.T., Armstrong J., Thibaud-Nissen F., Zinder O., Bolisetty M., Howe K., Hinerfeld D., Ruan X., et al. The genome of C57BL/6J "eve", the mother of the laboratory mouse genome reference strain. G3. 2019;9:1795–1805. doi: 10.1534/g3.119.400071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chaisson M.J.P., Huddleston J., Dennis M.Y., Sudmant P.H., Malig M., Hormozdiari F., Antonacci F., Surti U., Sandstrom R., Boitano M., et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature. 2015;517:608–611. doi: 10.1038/nature13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolmogorov M., Yuan J., Lin Y., Pevzner P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019;37:540–546. doi: 10.1038/s41587-019-0072-8. [DOI] [PubMed] [Google Scholar]
- 20.Gan P., Patterson M., Watanabe H., Wang K., Edmonds R.A., Reinholdt L.G., Sucov H.M. Allelic variants between mouse substrains BALB/cJ and BALB/cByJ influence mononuclear cardiomyocyte composition and cardiomyocyte nuclear ploidy. Sci. Rep. 2020;10:7605. doi: 10.1038/s41598-020-64621-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chesler E.J., Miller D.R., Branstetter L.R., Galloway L.D., Jackson B.L., Philip V.M., Voy B.H., Culiat C.T., Threadgill D.W., Williams R.W., et al. The collaborative cross at oak ridge national laboratory: developing a powerful resource for systems genetics. Mamm. Genome. 2008;19:382–389. doi: 10.1007/s00335-008-9135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srivastava A., Morgan A.P., Najarian M.L., Sarsani V.K., Sigmon J.S., Shorter J.R., Kashfeen A., McMullan R.C., Williams L.H., Giusti-Rodríguez P., et al. Genomes of the mouse collaborative cross. Genetics. 2017;206:537–556. doi: 10.1534/genetics.116.198838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doran A.G., Wong K., Flint J., Adams D.J., Hunter K.W., Keane T.M. Deep genome sequencing and variation analysis of 13 inbred mouse strains defines candidate phenotypic alleles, private variation and homozygous truncating mutations. Genome Biol. 2016;17:167. doi: 10.1186/s13059-016-1024-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao X., Collins R.L., Lee W.P., Weber A.M., Jun Y., Zhu Q., Weisburd B., Huang Y., Audano P.A., Wang H., et al. Expectations and blind spots for structural variation detection from long-read assemblies and short-read genome sequencing technologies. Am. J. Hum. Genet. 2021;108:919–928. doi: 10.1016/j.ajhg.2021.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richardson S.R., Gerdes P., Gerhardt D.J., Sanchez-Luque F.J., Bodea G.O., Muñoz-Lopez M., Jesuadian J.S., Kempen M.J.H.C., Carreira P.E., Jeddeloh J.A., et al. Heritable L1 retrotransposition in the mouse primordial germline and early embryo. Genome Res. 2017;27:1395–1405. doi: 10.1101/gr.219022.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feusier J., Watkins W.S., Thomas J., Farrell A., Witherspoon D.J., Baird L., Ha H., Xing J., Jorde L.B. Pedigree-based estimation of human mobile element retrotransposition rates. Genome Res. 2019;29:1567–1577. doi: 10.1101/gr.247965.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nellåker C., Keane T.M., Yalcin B., Wong K., Agam A., Belgard T.G., Flint J., Adams D.J., Frankel W.N., Ponting C.P. The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol. 2012;13:R45. doi: 10.1186/gb-2012-13-6-r45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han J.S., Szak S.T., Boeke J.D. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature. 2004;429:268–274. doi: 10.1038/nature02536. [DOI] [PubMed] [Google Scholar]
- 29.Peaston A.E., Evsikov A.V., Graber J.H., de Vries W.N., Holbrook A.E., Solter D., Knowles B.B. Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev. Cell. 2004;7:597–606. doi: 10.1016/j.devcel.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Jachowicz J.W., Torres-Padilla M.E. LINEs in mice: features, families, and potential roles in early development. Chromosoma. 2016;125:29–39. doi: 10.1007/s00412-015-0520-2. [DOI] [PubMed] [Google Scholar]
- 31.Rebollo R., Galvao-Ferrarini M., Gagnier L., Zhang Y., Ferraj A., Beck C.R., Lorincz M.C., Mager D.L. Inter-strain epigenomic profiling reveals a candidate IAP master copy in C3H mice. Viruses. 2020;12:783. doi: 10.3390/v12070783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duhl D.M., Vrieling H., Miller K.A., Wolff G.L., Barsh G.S. Neomorphic agouti mutations in obese yellow mice. Nat. Genet. 1994;8:59–65. doi: 10.1038/ng0994-59. [DOI] [PubMed] [Google Scholar]
- 33.Morgan H.D., Sutherland H.G., Martin D.I., Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat. Genet. 1999;23:314–318. doi: 10.1038/15490. [DOI] [PubMed] [Google Scholar]
- 34.Ishihara H., Tanaka I., Wan H., Nojima K., Yoshida K. Retrotransposition of limited deletion type of intracisternal A-particle elements in the myeloid leukemia Clls of C3H/He mice. J. Radiat. Res. 2004;45:25–32. doi: 10.1269/jrr.45.25. [DOI] [PubMed] [Google Scholar]
- 35.Kuff E.L., Lueders K.K. The intracisternal A-particle gene family: structure and functional aspects. Adv. Cancer Res. 1988;51:183–276. doi: 10.1016/s0065-230x(08)60223-7. [DOI] [PubMed] [Google Scholar]
- 36.McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F. The Ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sasani T.A., Ashbrook D.G., Beichman A.C., Lu L., Palmer A.A., Williams R.W., Pritchard J.K., Harris K. A natural mutator allele shapes mutation spectrum variation in mice. Nature. 2022;605:497–502. doi: 10.1038/s41586-022-04701-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sadic D., Schmidt K., Groh S., Kondofersky I., Ellwart J., Fuchs C., Theis F.J., Schotta G. Atrx promotes heterochromatin formation at retrotransposons. EMBO Rep. 2015;16:836–850. doi: 10.15252/embr.201439937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Judd J., Sanderson H., Feschotte C. Evolution of mouse circadian enhancers from transposable elements. Genome Biol. 2021;22:193. doi: 10.1186/s13059-021-02409-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Modzelewski A.J., Shao W., Chen J., Lee A., Qi X., Noon M., Tjokro K., Sales G., Biton A., Anand A., et al. A mouse-specific retrotransposon drives a conserved Cdk2ap1 isoform essential for development. Cell. 2021;184:5541–5558.e22. doi: 10.1016/j.cell.2021.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miao B., Fu S., Lyu C., Gontarz P., Wang T., Zhang B. Tissue-specific usage of transposable element-derived promoters in mouse development. Genome Biol. 2020;21:255. doi: 10.1186/s13059-020-02164-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He J., Babarinde I.A., Sun L., Xu S., Chen R., Shi J., Wei Y., Li Y., Ma G., Zhuang Q., et al. Identifying transposable element expression dynamics and heterogeneity during development at the single-cell level with a processing pipeline scTE. Nat. Commun. 2021;12:1456. doi: 10.1038/s41467-021-21808-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Macfarlan T.S., Gifford W.D., Driscoll S., Lettieri K., Rowe H.M., Bonanomi D., Firth A., Singer O., Trono D., Pfaff S.L. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature. 2012;487:57–63. doi: 10.1038/nature11244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hendrickson P.G., Doráis J.A., Grow E.J., Whiddon J.L., Lim J.W., Wike C.L., Weaver B.D., Pflueger C., Emery B.R., Wilcox A.L., et al. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat. Genet. 2017;49:925–934. doi: 10.1038/ng.3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flasch D.A., Macia Á., Sánchez L., Ljungman M., Heras S.R., García-Pérez J.L., Wilson T.E., Moran J.V. Genome-wide de novo L1 Retrotransposition Connects Endonuclease Activity with Replication. Cell. 2019;177:837–851.e28. doi: 10.1016/j.cell.2019.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sultana T., van Essen D., Siol O., Bailly-Bechet M., Philippe C., Zine El Aabidine A., Pioger L., Nigumann P., Saccani S., Andrau J.C., et al. The landscape of L1 retrotransposons in the human genome is shaped by pre-insertion sequence biases and post-insertion selection. Mol. Cell. 2019;74:555–570.e7. doi: 10.1016/j.molcel.2019.02.036. [DOI] [PubMed] [Google Scholar]
- 47.Sookdeo A., Hepp C.M., McClure M.A., Boissinot S. Revisiting the evolution of mouse LINE-1 in the genomic era. Mob. DNA. 2013;4:3. doi: 10.1186/1759-8753-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campos-Sánchez R., Cremona M.A., Pini A., Chiaromonte F., Makova K.D. Integration and fixation preferences of human and mouse endogenous retroviruses uncovered with functional data analysis. PLoS Comput. Biol. 2016;12:e1004956. doi: 10.1371/journal.pcbi.1004956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bourque G., Leong B., Vega V.B., Chen X., Lee Y.L., Srinivasan K.G., Chew J.L., Ruan Y., Wei C.L., Ng H.H., Liu E.T. Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res. 2008;18:1752–1762. doi: 10.1101/gr.080663.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Griffin L.E., Essenmacher L., Racine K.C., Iglesias-Carres L., Tessem J.S., Smith S.M., Neilson A.P. Diet-induced obesity in genetically diverse collaborative cross mouse founder strains reveals diverse phenotype response and amelioration by quercetin treatment in 129S1/SvImJ, PWK/EiJ, CAST/PhJ, and WSB/EiJ mice. J. Nutr. Biochem. 2021;87:108521. doi: 10.1016/j.jnutbio.2020.108521. [DOI] [PubMed] [Google Scholar]
- 51.Saul M.C., Philip V.M., Reinholdt L.G., Center for Systems Neurogenetics of Addiction. Chesler E.J. High-diversity mouse populations for complex traits. Trends Genet. 2019;35:501–514. doi: 10.1016/j.tig.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fujiwara K., Kawai Y., Takada T., Shiroishi T., Saitou N., Suzuki H., Osada N. Insights into Mus musculus population structure across eurasia revealed by whole-genome analysis. Genome Biol. Evol. 2022;14:evac068. doi: 10.1093/gbe/evac068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmidt S. Capturing genetic diversity: the power of the CC and DO mouse models. Environ. Health Perspect. 2018;126:014003. doi: 10.1289/EHP2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carvalho C.M.B., Lupski J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016;17:224–238. doi: 10.1038/nrg.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Balachandran P., Beck C.R. Structural variant identification and characterization. Chromosome Res. 2020;28:31–47. doi: 10.1007/s10577-019-09623-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iraqi F.A., Churchill G., Mott R. The Collaborative Cross, developing a resource for mammalian systems genetics: a status report of the Wellcome Trust cohort. Mamm. Genome. 2008;19:379–381. doi: 10.1007/s00335-008-9113-1. [DOI] [PubMed] [Google Scholar]
- 57.Geisert E.E., Williams R.W. Using BXD mouse strains in vision research: a systems genetics approach. Mol. Vis. 2020;26:173–187. [PMC free article] [PubMed] [Google Scholar]
- 58.Martins A.C., López-Granero C., Ferrer B., Tinkov A.A., Skalny A.V., Paoliello M.M.B., Aschner M. BXD recombinant inbred mice as a model to study neurotoxicity. Biomolecules. 2021;11:1762. doi: 10.3390/biom11121762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lustyk D., Kinský S., Ullrich K.K., Yancoskie M., Kašíková L., Gergelits V., Sedlacek R., Chan Y.F., Odenthal-Hesse L., Forejt J., Jansa P. Genomic structure of Hstx2 modifier of prdm9-dependent hybrid male sterility in mice. Genetics. 2019;213:1047–1063. doi: 10.1534/genetics.119.302554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chubb C., Nolan C. Mouse hybrid sterility and testicular function. Biol. Reprod. 1987;36:1343–1348. doi: 10.1095/biolreprod36.5.1343. [DOI] [PubMed] [Google Scholar]
- 61.Simpson E.M., Linder C.C., Sargent E.E., Davisson M.T., Mobraaten L.E., Sharp J.J. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat. Genet. 1997;16:19–27. doi: 10.1038/ng0597-19. [DOI] [PubMed] [Google Scholar]
- 62.Qin W., Kutny P.M., Maser R.S., Dion S.L., Lamont J.D., Zhang Y., Perry G.A., Wang H. Generating mouse models using CRISPR-Cas9-mediated genome editing. Curr. Protoc. Mouse Biol. 2016;6:39–66. doi: 10.1002/9780470942390.mo150178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heller D., Vingron M. SVIM-asm: structural variant detection from haploid and diploid genome assemblies. Bioinformatics. 2020;36:5519–5521. doi: 10.1093/bioinformatics/btaa1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yalcin B., Wong K., Agam A., Goodson M., Keane T.M., Gan X., Nellåker C., Goodstadt L., Nicod J., Bhomra A., et al. Sequence-based characterization of structural variation in the mouse genome. Nature. 2011;477:326–329. doi: 10.1038/nature10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ebler J., Ebert P., Clarke W.E., Rausch T., Audano P.A., Houwaart T., Mao Y., Korbel J.O., Eichler E.E., Zody M.C., et al. Pangenome-based genome inference allows efficient and accurate genotyping across a wide spectrum of variant classes. Nat. Genet. 2022;54:518–525. doi: 10.1038/s41588-022-01043-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Deniz Ö., Frost J.M., Branco M.R. Regulation of transposable elements by DNA modifications. Nat. Rev. Genet. 2019;20:417–431. doi: 10.1038/s41576-019-0106-6. [DOI] [PubMed] [Google Scholar]
- 67.Vanlerberghe F., Boursot P., Catalan J., Gerasimov S., Bonhomme F., Botev B.A., Thaler L. [Genetic analysis of the hybridization zone between two subspecies Mus musculus domesticus and Mus musculus musculus in Bulgaria] Genome. 1988;30:427–437. [PubMed] [Google Scholar]
- 68.Concepcion D., Ross K.D., Hutt K.R., Yeo G.W., Hamilton B.A. Nxf1 natural variant E610G is a semi-dominant suppressor of IAP-induced RNA processing defects. PLoS Genet. 2015;11:e1005123. doi: 10.1371/journal.pgen.1005123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bertozzi T.M., Elmer J.L., Macfarlan T.S., Ferguson-Smith A.C. KRAB zinc finger protein diversification drives mammalian interindividual methylation variability. Proc. Natl. Acad. Sci. USA. 2020;117:31290–31300. doi: 10.1073/pnas.2017053117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chuang N.T., Gardner E.J., Terry D.M., Crabtree J., Mahurkar A.A., Rivell G.L., Hong C.C., Perry J.A., Devine S.E. Mutagenesis of human genomes by endogenous mobile elements on a population scale. Genome Res. 2021;31:2225–2235. doi: 10.1101/gr.275323.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sanchez-Luque F.J., Kempen M.J.H.C., Gerdes P., Vargas-Landin D.B., Richardson S.R., Troskie R.L., Jesuadian J.S., Cheetham S.W., Carreira P.E., Salvador-Palomeque C., et al. LINE-1 evasion of epigenetic repression in humans. Mol. Cell. 2019;75:590–604.e12. doi: 10.1016/j.molcel.2019.05.024. [DOI] [PubMed] [Google Scholar]
- 72.Lutz S.M., Vincent B.J., Kazazian H.H., Jr., Batzer M.A., Moran J.V. Allelic heterogeneity in LINE-1 retrotransposition activity. Am. J. Hum. Genet. 2003;73:1431–1437. doi: 10.1086/379744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Salvador-Palomeque C., Sanchez-Luque F.J., Fortuna P.R.J., Ewing A.D., Wolvetang E.J., Richardson S.R., Faulkner G.J. Dynamic methylation of an L1 transduction family during reprogramming and neurodifferentiation. Mol. Cell Biol. 2019;39:00499-18. doi: 10.1128/MCB.00499-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chiang C., Scott A.J., Davis J.R., Tsang E.K., Li X., Kim Y., Hadzic T., Damani F.N., Ganel L., et al. GTEx Consortium The impact of structural variation on human gene expression. Nat. Genet. 2017;49:692–699. doi: 10.1038/ng.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maksakova I.A., Mager D.L. Transcriptional regulation of early transposon elements, an active family of mouse long terminal repeat retrotransposons. J. Virol. 2005;79:13865–13874. doi: 10.1128/JVI.79.22.13865-13874.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maksakova I.A., Zhang Y., Mager D.L. Preferential epigenetic suppression of the autonomous MusD over the nonautonomous ETn mouse retrotransposons. Mol. Cell Biol. 2009;29:2456–2468. doi: 10.1128/MCB.01383-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Todd C.D., Deniz Ö., Taylor D., Branco M.R. Functional evaluation of transposable elements as enhancers in mouse embryonic and trophoblast stem cells. Elife. 2019;8:e44344. doi: 10.7554/eLife.44344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kigami D., Minami N., Takayama H., Imai H. MuERV-L is one of the earliest transcribed genes in mouse one-cell embryos. Biol. Reprod. 2003;68:651–654. doi: 10.1095/biolreprod.102.007906. [DOI] [PubMed] [Google Scholar]
- 79.Zhou M., Smith A.D. Subtype classification and functional annotation of L1Md retrotransposon promoters. Mob. DNA. 2019;10:14. doi: 10.1186/s13100-019-0156-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kong L., Saha K., Hu Y., Tschetter J.N., Habben C.E., Whitmore L.S., Yao C., Ge X., Ye P., Newkirk S.J., An W. Subfamily-specific differential contribution of individual monomers and the tether sequence to mouse L1 promoter activity. Mob. DNA. 2022;13:13. doi: 10.1186/s13100-022-00269-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wenger A.M., Peluso P., Rowell W.J., Chang P.C., Hall R.J., Concepcion G.T., Ebler J., Fungtammasan A., Kolesnikov A., Olson N.D., et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019;37:1155–1162. doi: 10.1038/s41587-019-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang Y., Zhao Y., Bollas A., Wang Y., Au K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021;39:1348–1365. doi: 10.1038/s41587-021-01108-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rautiainen M., Nurk S., Walenz B.P., Logsdon G.A., Porubsky D., Rhie A., Eichler E.E., Phillippy A.M., Koren S. Telomere-to-telomere assembly of diploid chromosomes with Verkko. Nat. Biotechnol. 2023 doi: 10.1038/s41587-023-01662-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sedlazeck F.J., Rescheneder P., Smolka M., Fang H., Nattestad M., von Haeseler A., Schatz M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods. 2018;15:461–468. doi: 10.1038/s41592-018-0001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Heller D., Vingron M. SVIM: structural variant identification using mapped long reads. Bioinformatics. 2019;35:2907–2915. doi: 10.1093/bioinformatics/btz041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen X., Schulz-Trieglaff O., Shaw R., Barnes B., Schlesinger F., Källberg M., Cox A.J., Kruglyak S., Saunders C.T. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–1222. doi: 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 88.Rausch T., Zichner T., Schlattl A., Stütz A.M., Benes V., Korbel J.O. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–i339. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Layer R.M., Chiang C., Quinlan A.R., Hall I.M. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15:R84. doi: 10.1186/gb-2014-15-6-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bailey J.A., Yavor A.M., Massa H.F., Trask B.J., Eichler E.E. Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 2001;11:1005–1017. doi: 10.1101/gr.gr-1871r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gurevich A., Saveliev V., Vyahhi N., Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Di Tommaso P., Chatzou M., Floden E.W., Barja P.P., Palumbo E., Notredame C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017;35:316–319. doi: 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- 93.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jin Y., Tam O.H., Paniagua E., Hammell M. TEtranscripts: a package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics. 2015;31:3593–3599. doi: 10.1093/bioinformatics/btv422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kovaka S., Zimin A.V., Pertea G.M., Razaghi R., Salzberg S.L., Pertea M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019;20:278. doi: 10.1186/s13059-019-1910-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 99.Pedersen B.S., Quinlan A.R. Duphold: scalable, depth-based annotation and curation of high-confidence structural variant calls. Gigascience. 2019;8:giz040. doi: 10.1093/gigascience/giz040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ren J., Chaisson M.J.P. lra: a long read aligner for sequences and contigs. PLoS Comput. Biol. 2021;17:e1009078. doi: 10.1371/journal.pcbi.1009078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Czechanski A., Byers C., Greenstein I., Schrode N., Donahue L.R., Hadjantonakis A.K., Reinholdt L.G. Derivation and characterization of mouse embryonic stem cells from permissive and nonpermissive strains. Nat. Protoc. 2014;9:559–574. doi: 10.1038/nprot.2014.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Buenrostro J.D., Wu B., Chang H.Y., Greenleaf W.J. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015;109:21.29.1–21.29.9. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Corces M.R., Buenrostro J.D., Wu B., Greenside P.G., Chan S.M., Koenig J.L., Snyder M.P., Pritchard J.K., Kundaje A., Greenleaf W.J., et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016;48:1193–1203. doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics. 2004;Chapter 4:Unit 4.10. doi: 10.1002/0471250953.bi0410s05. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Structural variant calls for each mouse genome were merged and annotated into a non-redundant callset. Descriptions of each field can be found within the “Fields” sheet.
PCR validations of SVs in eight strains of mice (129S1/SvImJ, A/J, CAST/EiJ, NOD/ShiLtJ, NZO/HILtJ, PWK/PhJ, and WSB/EiJ): 1 indicates an animal contains the SV and 0 denotes absence of SV. Animals computationally predicted to contain the SV are shown in the MERGE_SAMPLES column.
Summary of statistical analysis performed for VEP output (consequence_stats sheet, Fisher’s exact test), VEP consequences for transposable element variants (TE_family_consequence_stats sheet, Fisher’s exact test), and chromatin accessibility associations by transposable element type (chromatin_accessibility_stats sheet, Student’s t test). SR_SUPPORT, SVs that are supported by short-read SV calling; TR_ANNO, tandem repeat SVs; MGP_ANNO, SVs supported by the mouse genomes project dataset; TE_ANNO, SVs that are transposable elements.
Data Availability Statement
The genome assemblies, long-read sequencing, ATAC-seq, and RNA-seq data generated for this study can be obtained through the NCBI sequencing read archive under BioProject NCBI: PRJNA923323. Structural variant VCFs for each individual mouse strain, the merged structural variant VCF, and RepeatMasker annotations for each assembly were deposited on Zenodo at https://doi.org/10.5281/zenodo.7644286. All software used in this study can be found in the key resources table.