

Abstract
Background:
Epigenetic dysregulation has been proposed as a key mechanism for arsenic-related cardiovascular disease (CVD). We evaluated differentially methylated positions (DMPs) as potential mediators on the association between arsenic and CVD.
Methods:
Blood DNA methylation was measured in 2321 participants (mean age 56.2, 58.6 % women) of the Strong Heart Study, a prospective cohort of American Indians. Urinary arsenic species were measured using high-performance liquid chromatography coupled to inductively coupled plasma mass spectrometry. We identified DMPs that are potential mediators between arsenic and CVD. In a cross-species analysis, we compared those DMPs with differential liver DNA methylation following early life arsenic exposure in the apolipoprotein E knock-out (apoE−/−) mouse model of atherosclerosis.
Results:
A total of 20 and 13 DMPs were potential mediators for CVD incidence and mortality, respectively, several of them annotated to genes related to diabetes. Eleven of these DMPs were similarly associated with incident CVD in three diverse prospective cohorts (Framingham Heart Study, Women’s Health Initiative and Multi-Ethnic Study of Atherosclerosis). In the mouse model, differentially methylated regions (DMRs) in 20 of those genes and DMPs in 10 genes were associated with arsenic.
Conclusions:
Differential DNA methylation might be part of the biological link between arsenic and CVD. The gene functions suggest that diabetes might represent a relevant mechanism for arsenic-related cardiovascular risk in populations with a high burden of diabetes.
Keywords: cardiovascular disease, arsenic, DNA methylation, prospective cohort, animal model
Subject terms: epigenetics, biomarkers
Graphical Abstract
Introduction
Inorganic arsenic exposure is a global health problem.1 Even at low exposure levels in water and food, arsenic has been related to multiple health outcomes including atherosclerotic cardiovascular disease (CVD).2–4 CVD outcomes associated with arsenic in Bangladesh, Chile, Taiwan, Denmark, Spain and the United States include coronary heart disease,5–9 stroke,7 peripheral arterial disease10 and overall CVD mortality.7,11,12 Arsenic has also been prospectively associated with changes in blood pressure levels9,13 and carotid atherosclerosis.9,14,15 These epidemiological findings are consistent with data from animal models showing that arsenic can induce atherosclerosis at relatively low exposure levels.16,17
The recognition of arsenic as a CVD risk factor, however, remains hindered by limited understanding of the specific mechanisms involved. Growing evidence points to the importance of epigenetic dysregulation and its influence on gene transcription pathways as a potential mechanism for arsenic-related CVD. Indeed, arsenic has been associated with changes in DNA methylation in epigenome-wide association studies in human populations from Bangladesh,18–22 South America,23,24 Taiwan,25 China,26 and the US.27–30 Increasing evidence also supports the notion that changes in DNA methylation are prospectively associated with incident CVD31,32 and coronary heart disease, the most common clinical form of heart disease.33,34
We hypothesized that epigenetics, measured based on differentially methylated CpG positions (DMPs) in blood, can partially explain arsenic-related CVD. We tested this hypothesis in the Strong Heart Study (SHS), the largest and longest study of CVD in American Indian communities, ongoing since 1989–1991. Urinary arsenic species were measured using high-performance liquid chromatography coupled to inductively coupled plasma mass spectrometry. Prior evidence in the SHS showed that baseline arsenic exposure, which was stable for decades, was associated with increased CVD risk7 and with differentially methylated blood DNA in an epigenome-wide association study (EWAS).27 We also used data from the Framingham Heart Study (FHS), Women’s Health Initiative (WHI) and Multi-Ethnic Study of Atherosclerosis (MESA) to assess if DMPs associated with arsenic-mediated CVD in the SHS were associated with incident CVD in those populations. Since MESA is, to our knowledge, the only other US cohort that has data on arsenic, DNA methylation and CVD, we also used data from MESA to assess if the same DMPs were associated with arsenic exposure. Further, we conducted an inter-species comparison in a mouse model of arsenic-enhanced atherosclerosis and measured DNA methylation in livers of adult mice that were exposed to arsenic from mating through weaning of offspring.
Methods
Data Availability.
The data underlying this article can be shared to external investigators following the procedures established by the Strong Heart Study, available at https://strongheartstudy.org/. Data from the Framingham Heart Study and from the Women’s Health Initiative are available at dbGaP (accession numbers phs000724.v8.p12 and phs000200.v11.p3). Data from the Multi-Ethnic Study of Atherosclerosis are available upon request at TopMed (https://topmed.nhlbi.nih.gov/).
Ethics.
The experimental protocol was approved by the McGill Animal Care Committee and animals were handled in accordance with institutional guidelines. McGill Animal Care Committee is certified by the Canadian Council on Animal Care.
Main study population
The SHS is an ongoing prospective cohort study of CVD and its risk factors in American Indian communities since 1989.35 At the baseline visit (1989–1991) a total of 4549 men and women aged 45–75 years members of 13 tribes based in Arizona, Oklahoma, North Dakota and South Dakota enrolled in the study (participation rate 62%). In 2016, a Tribal Nation from Arizona declined further participation, leaving 3,517 potential participants for this study. DNA methylation was measured in blood samples from 2,351 participants collected at the baseline visit (1989–1991) who were free of CVD, had community agreement, were not missing data on relevant variables, and had sufficient blood left for epigenetic analyses. Details regarding inclusion criteria for blood DNA methylation measurements have been published.36 For the main analyses, we restricted the follow-up through 2009 as water arsenic exposure, which was stable in the communities for decades,37 changed a few years after the enactment of the US EPA Final Arsenic rule in 2006.38,39 Strong Heart Study tribal review boards approved procedures for this study, and participants gave written informed consent.
Participant characteristics and urinary arsenic measurements
Trained and certified nurses and medical examiners collected information on sociodemographic factors (age, sex, study region), medical history and smoking status (never, former, current) in a personal interview. Participants who had smoked ≥ 100 cigarettes in their lifetime and were smoking at the time of the interview were considered current smokers. Participants who had smoked > 100 cigarettes in their lifetime and were not smoking at the time of the interview were considered former smokers. The examiners measured height and weight (to estimate body mass index (BMI)) and blood pressure, and collected fasting blood and urine samples.
Arsenic measurements in spot urine samples have been described in detail.40 Briefly, arsenic species (inorganic arsenic, monomethylarsonate (MMA), dimethylarsinate (DMA), and arsenobetaine) were measured using high-performance liquid chromatography coupled to inductively coupled plasma mass spectrometry (Agilent 1100 HPLC and Agilent 7700x ICP-MS; Agilent Technologies).41 Urinary creatinine was measured in the same urine sample used for arsenic measurement using an automated alkaline picrate methodology run on a rapid flow analyzer.35 As the biomarker of inorganic arsenic exposure (referred to as urinary arsenic in the manuscript for simplicity), we calculated the sum of inorganic and methylated arsenic species (MMA and DMA) concentrations (μg/L). This biomarker was divided by urinary creatinine (g/L) to account for urine dilution. In a random sample stratified by study region of 380 participants with three repeated arsenic measures over 10 years, the intraclass correlation coefficient for the log-transformed sum of inorganic and methylated arsenic species was 0.64 (95% CI, 0.60 to 0.69).
Cardiovascular disease follow-up
The endpoints are incident fatal and non-fatal CVD assessed during the follow-up by annual mortality and morbidity surveillance of medical records, which included evaluation of medical history and physical examinations, emergency room visits, medical consultations, electrocardiograms, laboratory assays, medical imaging, discharge summaries, operations, and other procedures from the Indian Health Service and other facilities. Mortality surveillance examined death certificates from state health departments, records from the Indian Health Service, autopsy and coroner’s reports, and interviews with physicians or family members. Potential CVD‐related deaths and events were reviewed by two independent physicians. In case of disagreement, they were adjudicated by a third independent physician. Detailed definitions of fatal and nonfatal events42 and definitions of the criteria used by the review committees43 have been reported. Incident CVD was defined as the first occurrence of fatal or non-fatal coronary heart disease, stroke or congestive heart failure, or other non-fatal CVD. CVD mortality was defined as any fatal CVD. Follow-up time was calculated as the time from blood drawn for DNA methylation measurement (1989–1991) to the time of CVD events (through 2009). For participants who did not develop CVD, follow-up was censored at the time of occurrence of non-CVD death, loss to follow-up, or December 31, 2009. Follow‐up rates for mortality and morbidity were at 99%.44
Microarray DNA methylation measurements
Details of microarray DNA methylation measurements have been published.36 Briefly, buffy coat was extracted from fasting blood samples and used to obtain bisulfite converted DNA methylation from white blood cells. DNA methylation was measured using Illumina’s MethylationEPIC BeadChip (850K). Individuals with low detection p-values, cross-hybridizing probes, probes located in sex chromosomes and single nucleotide polymorphisms (SNPs) with minor allele frequency > 0.05 were excluded.45 Single sample noob normalization and regression on correlated probes normalization were conducted following Illumina’s recommendations for preprocessing.46 Blood cell proportions (CD8T, CD4T, NK cells, B cells, monocytes and neutrophils) were estimated using the R package FlowSorted.Blood.EPIC, which uses the Houseman projection method.47 The preprocessing resulted in data for 2324 individuals and 788,368 CpG sites.
Replication populations
We used data from the FHS, WHI, and MESA to replicate the DMPs associated with arsenic-mediated CVD in the SHS. All of them used follow-up procedures for CVD events and analysis of blood DNA methylation similar to those used by the SHS (details reported in Supplementary Methods, DNA methylation was also measured using the 850K Illumina microarray in MESA, while the 450K Illumina microarray was used in FHS and WHI).
FHS recruited White adults of European descent from Framingham, Massachusetts starting in 1948 (original cohort). The children of the original cohort and their spouses were recruited into the Framingham Offspring study in 1971.48 The participants of exam 8 (2005–2008) of FHS offspring cohort were followed through 2014 (average follow-up of 7.7 years; range: 0.04 years – 9.8 years). This study was approved under Boston University Medical Center protocols H-27984 and H-32132. Written informed consent was obtained from each participant. Among 2,631 FHS participants with blood DNA methylation data available in the FHS Offspring, we excluded those with prior CVD (N=316) and those missing information on CVD risk factors (N=325), leaving 1,990 participants with 408,254 CpG sites available. DNA methylation measurements in the FHS were conducted in two separate batches including 1879 and 111 participants, respectively. We conducted a sensitivity analysis excluding the 111 individuals in the second batch from the analysis.
WHI enrolled 161,808 women of diverse ethnicities (including White, African American, Native American, Hispanic, Asian and pacific Islanders) starting in 1993 as part of randomized control trials that were continued as a prospective cohort study. The participants of WHI were followed from baseline (1993–1998) to 2016 with an average follow-up time of 12.18 years (range: 0.003 – 21.3 years). The WHI was approved by the institutional review boards of participating institutions from all 40 clinical centers and the coordinating center. Among 2,096 WHI participants with blood DNA methylation for 434,113 CpG sites, we excluded those with missing information on traditional risk factors of CVD, leaving 1,487 participants.
MESA followed participants of diverse ethnicities (White, African-American, Hispanic and Asian) through 2017 with an average follow-up time of 15.56 years (range: 7.76 – 17.42 years). MESA was approved by the institutional review boards of the participating institutions with the six clinical field centers and the MESA data coordinating center. Written, informed, and signed consent was obtained from each participant. From 916 participants that had DNA methylation data and prospective CVD data, 20 were excluded due to missing covariates. The final sample size for DNA methylation and CVD analyses was 896. From 214 participants that had DNA methylation and urinary arsenic data, 8 were excluded due to missing covariates. The final sample size for DNA methylation and arsenic analyses was 206.
Statistical methods
DMPs associated with CVD: To identify DMPs associated with CVD incidence and mortality, we used Iterative Sure Independence Screening coupled with Adaptive Elastic-Net (ISIS-Aenet). Adaptive elastic-net is a modified version of traditional elastic-net models that uses data-driven weights to achieve better consistency in effect estimation while preserving the advantages of elastic-net models for prediction.49 In ultra-high dimensional settings such as in epigenomics data, computational cost and algorithm instability might worsen the performance of these estimators.50 The Sure Independence Screening (SIS) method and its iterative variant (ISIS) can overcome these limitations.51 ISIS-Aenet has shown to outperform other variable selection methods in ultra-high dimensional settings.49,50,52
To account for the time to event, we used Cox ISIS – Aenet entering all the 788,368 CpG sites simultaneously to select DMPs associated with CVD incidence and mortality (dependent variables, in separate models). Confidence intervals were calculated using the quantile bootstrap method. The bootstrap tool randomly selects individuals from the database with resampling in each iteration, and fits the algorithm in those sets. We set the number of iterations to 2000. The 2.5th and the 97.5th percentiles of the effect estimates of all iterations were then selected as the lower and upper bounds of the 95 % confidence interval. Models were adjusted for baseline covariates including age, sex, smoking status (never, former, current), BMI, LDL cholesterol, HDL cholesterol, diabetes status (yes/no), hypertension medication (yes/no), systolic blood pressure and albuminuria (micro, macro, normal), which are established CVD risk factors in the SHS.53 Given the different characteristics of the three study centers (Arizona, Oklahoma, and North Dakota and South Dakota), models were also adjusted for center. DNA methylation levels are known to differ by cell type, therefore, we adjusted the models for estimated cell proportions (CD8T, CD4T, NK, B cells, and monocytes).54 To account for population stratification, models were additionally adjusted for five genetic principal components (PCs).55 Of 2,562 genotyped SHS participants as part of the CALiCo/PAGE Study, we identified 644 unrelated individuals (either founders of pedigrees or unrelated spouses of their descendants). Of 162,718 autosomal SNPs that passed quality control, we selected 15,158 based on the following criteria: minor allele frequency ≥ 0.05 (i.e., not rare variants), minimum physical separation of 1kb and pairwise correlation of genotype scores ≤ 0.1 within a 100 kb sliding window. We performed PC analysis on the genotype scores (i.e. dosages) within unrelated individuals using the R function prcomp. Non-founders doses were projected onto PC axes using the R function predict. The first five PCs were kept as they explained most of the variance. Code for implementing Cox ISIS - Aenet based on the R packages SIS and msaenet is available upon request.
Mediation analysis: To identify DMPs that may explain arsenic-related CVD, we used the Aalen additive hazards models for causal mediation analysis with survival outcomes, similar to other studies with time to event data.56–58 The DMPs tested as possible mediators included the DMPs identified as relevant for CVD by ISIS – Aenet as well as 315 DMPs previously identified as associated with arsenic exposure using an elastic-net model in the SHS in a previous study.27 The Aalen additive hazards model included time to incident CVD (or CVD mortality, in a separate model) as the outcome, baseline urine arsenic (modeled as log2) as the exposure, and DNA methylation as mediator (each DMP in a separate model). Our mediator model was a linear model with logit2-transformed methylation values (M values) as the outcome (each DMP in a separate model) and urine arsenic (modeled as log2) as the exposure. Both the outcome and mediator models included adjustment for the same covariates (age, sex, smoking status, BMI, LDL cholesterol, study center, cell counts and genetic PCs). Mediated effects (natural indirect effects) were reported as the number of CVD cases per 100,000 person-years associated with a 2-fold increase in urinary arsenic that are attributable to DNA methylation changes in that CpG site. Confidence intervals were calculated using a resampling method that takes random values from multivariate normal distribution of the estimates.56 Total effects, direct effects and indirect effects with confidence intervals not including 0 were considered significant. To account for the withdrawal of one of the Tribal Nations (see the Study Population section and 36), the primary mediation analysis used inverse probability weighting to reduce bias.59 We weighted the participants remaining in the study with approximately 1/3 of weight for each center based on the baseline SHS cohort enrollment (33.0% AZ, 33.6% OK, 33.4% ND/SD). We also present the unweighted analyses as a side by side comparison.
Protein-protein interaction network to evaluate biological plausibility of identified DMPs.
Arsenic-associated and CVD-associated DMPs were annotated to the nearest protein coding gene and included in a protein-protein interaction network. The interactions between nodes were obtained using STRING database v11.0,60 selecting all active interaction sources with a confidence score of 0.4. The confidence score (from 0 to 1) provided by STRING database estimates the likelihood that an annotated interaction between a pair of proteins is biologically meaningful, specific and reproducible.60 The network was analyzed and displayed using edge weighted spring embedded layout with Cytoscape v3.8.2.61
Gene Ontology enrichment and KEGG analyses.
We used the missmethyl R package to conduct gene ontology enrichment and KEGG analyses. We tested whether any Gene Ontology terms or pathways were enriched for the set of DMPs that were significant in the mediation analysis for both CVD incidence and mortality, as compared to the total number of CpG sites that were tested in mediation (329 for CVD incidence and 338 for CVD mortality).
Cross-reference with the EWAS catalog to evaluate biologic plausibility.
For DMPs showing significant mediated effects for arsenic-related CVD incidence and/or mortality, we looked for previously known trait associations in the EWAS Catalog.62 This catalog contains information on EWAS conducted across the literature and is regularly updated (we used the February 4, 2021 version). For DMPs with several traits in the EWAS catalog, either the most relevant trait or the study with the largest sample size were selected.
Sensitivity Analyses.
Because diabetes and hypertension might be in the arsenic-CVD causal pathway, the main models were not adjusted for those variables. We repeated the mediation analyses for CVD incidence and CVD mortality adjusting for diabetes status and for hypertension treatment and systolic blood pressure. Mediation models were also repeated considering the full follow-up (through 2017) rather than truncating it in 2009.
Differentially Methylated Genomic Regions and Positions in Livers of Arsenic-Exposed Mice
Apolipoprotein E knockout (apoE−/−) mice are a well-established animal model of atherosclerosis, where genetic manipulation results in hyperlipidemia. Importantly, the model increases disease burden in response to dietary changes (i.e. high fat)63 and environmental exposures (i.e. arsenic).16 This model is relevant for many human populations which diets are also lipid-rich, such as the typical diet of many participants in the SHS.64,65 B6.129P2-ApoEtm1Unc/J (ApoE−/−) mice were obtained from the Jackson Laboratory (see the Major Resources Table in the Supplementary Material). ApoE−/− mice were fed a purified AIN-76 diet (Harlan Laboratories Inc, WI, USA) and allowed to mate a week later. The male and female apoE−/− mice were assigned randomly into mating pairs prior to arsenic exposure. Arsenic exposure was then provided through drinking water or not to the female during the duration of pregnancy based on the random assignment of the mating pair. The mating pairs were started on either 200-ppb sodium arsenite (treated mating pair) or maintained on tap water from mating to until 3-weeks post-birth. The offspring, once weaned, were maintained on tap water and purified diet until 18 weeks of age, a time point at which enhanced atherosclerotic plaque is observed.66 At endpoint, livers were harvested from the offspring of control and treated mating pairs, and whole genome bisulfite sequencing was performed (N=3 per sex, per treatment group). A total of 12 liver samples from randomly chosen offspring of each unique litter were sequenced. DNA was isolated from liver tissues, and bisulfite conversion and whole-genome bisulfite sequencing (WGBS) were performed at McGill University and Genome Quebec Innovation Centre.
The data was processed using the GemBS pipeline from Merkel et al. 2017,67 using the MM9 mouse reference genome. A chromosome-wise matrix of methylation counts and read counts (after quality control filter) was created for all samples. The BSmooth function68 was applied in the bioconductor package bsseq to smooth the data and the t-statistics were calculated. Finally, the dmrfinder function was used to identify genomic regions that were differentially methylated in the tissue samples from the offspring of exposed dams compared to the offspring of control dams. For differentially methylated CpG sites in the genes of interest, the Bioconductor package limma was used separately for male and female. Statistical significance was determined by calculating the effective number of independent tests, separately for each site and adjusting for multiple testing as per Li and Ji 2005.69 The DMRs were annotated with the MM9 annotations using CHIPseeker70 and Annotatr.71
Results
A total of 847 participants developed incident CVD in the SHS (36.4 %), 208 in the FHS (10.4 %), 754 in the WHI (50.7 %) and 87 in MESA (9.7 %). In the SHS, individuals with incident CVD were older and more likely to have diabetes, higher LDL cholesterol, hypertension, higher systolic blood pressure and micro and macro albuminuria. Individuals who died of CVD had higher levels of urinary arsenic at baseline (Table 1). Participants’ characteristics for the replication cohorts are shown in Table S1.
Table 1.
Baseline participants’ characteristics by cardiovascular disease incidence and mortality status.
| Non-incident CVD (N=1474) |
Incident CVD (N=847) |
CVD death (N=316) |
|
|---|---|---|---|
| Age (years), median (IQR) | 53.1 (48.0, 60.0) | 57.3 (51.0, 64.4) | 58.4 (52.6, 66.2) |
| Sex, % Men | 60.0 | 58.3 | 56.8 |
| Smoking status, % | |||
| Former | 33.3 | 33.4 | 29.6 |
| Current | 32.3 | 36.4 | 34.3 |
| BMI, median (IQR) | 29.8 (26.3, 34.2) | 30.4 (27.1, 34.5) | 30.4 (27.1, 34.3) |
| LDL cholesterol (mg/dL), median (IQR) | 114 (92, 135) | 121 (99, 142) | 121 (100, 144) |
| HDL cholesterol (mg/dL), median (IQR) | 44 (38, 53) | 42 (36, 50) | 41 (36, 49) |
| Systolic blood pressure, median (IQR) | 122 (111, 135) | 129 (118, 141) | 133 (120, 144) |
| Hypertension, % | 15.3 | 30.1 | 34.5 |
| Diabetes, % | 40.3 | 61.9 | 69.2 |
| Albuminuria, % | |||
| Microalbuminuria | 15.1 | 24.5 | 24.2 |
| Macroalbuminuria | 6.4 | 15.8 | 24.4 |
| Urinary arsenic (μg/g creatinine)* | 10.2 (5.9, 16.7) | 10.3 (6.0, 17.3) | 11.2 (6.6, 18.2) |
CVD: Cardiovascular disease, IQR: interquartile range.
Urinary arsenic corresponds to the sum of inorganic and methylated species (methylarsonic acid and dimethylarsinic acid) in the urine.
The Cox ISIS-Aenet model selected 70 and 72 DMPs as relevant for CVD incidence and mortality, respectively (Excel Tables S1 and S2). Nine DMPs were common for both CVD incidence and mortality: cg13251119 (annotated to EPS8L3), cg00841849 (ID2), cg14066163 (intergenic), cg25371036 (AMOTL1), cg03362418 (TYMP), cg25452273 (PPCDC), cg18130370 (NCF4), cg00451635 (EMP2) and cg06970472 (APBB2) (Table 2).
Table 2.
Hazard ratios (95 % CIs) of the common differentially methylated positions for cardiovascular disease incidence and mortality comparing the 90th vs the 10th percentile of methylation obtained from the Cox Iterative Sure Independence Screening model coupled with adaptive elastic-net.
| CpG | Chr | Gene | Function | Location | CVD incidence |
CVD mortality |
|---|---|---|---|---|---|---|
| HR (95 % CI) | HR (95 % CI) | |||||
| cg13251119 | 1 | EPS8L3 | Unknown function | Body | 0.51 (0.29, 1.00) | 0.18 (0.06, 0.63) |
| cg00841849 | 2 | ID2 | Cellular growth, senescence, differentiation, apoptosis, angiogenesis, neoplastic transformation |
Intergenic | 0.57 (0.40, 0.84) | 0.63 (0.32, 1.01) |
| cg14066163 | 17 | Unknown | - | Intergenic | 0.63 (0.39, 1.00) | 0.67 (0.31, 1.17) |
| cg25371036 | 11 | AMOTL1 | Endothelial cell migration, capillary formation | TSS1500 | 0.71 (0.54, 0.92) | 0.42 (0.27, 0.73) |
| cg03362418 | 22 | TYMP | Angiogenesis and endothelial cell growth. Proposed as therapeutic target for CVD | Body | 0.73 (0.50, 1.02) | 0.51 (0.29, 0.94) |
| cg25452273 | 15 | PPCDC | Biosynthesis of coenzyme A. Metabolism of water-soluble vitamins | Body | 1.25 (0.96, 1.81) | 1.80 (1.00, 3.42) |
| cg18130370 | 22 | NCF4 | Arterial remodeling and advanced atherosclerosis | Body | 0.79 (0.48, 1.12) | 0.44 (0.19, 0.99) |
| cg00451635 | 16 | EMP2 | Blood vessel endothelial cell migration and angiogenesis | TSS1500 | 1.11 (0.86, 1.33) | 0.68 (0.46, 1.00) |
| cg06970472 | 4 | APBB2 | Beta cell function, insulin secretion impairment in mice | Body | 1.22 (0.93, 1.61) | 0.69 (0.43, 1.05) |
Hazard ratios are from an adaptive elastic-net model fitted on the selected CpG sites by ISIS – Aenet. 95 % confidence intervals were calculated using quantile bootstrap.
Models were adjusted for age, sex, smoking status (never, former, current), BMI, LDL cholesterol, HDL cholesterol, hypertension (yes/no), diabetes status (yes/no), systolic blood pressure, albuminuria (micro, macro, normal), study center (Arizona, Oklahoma, North Dakota and South Dakota), cell counts (CD8T, CD4T, NK, B cells and monocytes) and five genetic PCs.
In the mediation analysis for CVD incidence, which included the 70 DMPs associated with CVD incidence and 315 DMPs associated with urinary arsenic in our previous study,27 we found statistically significant mediated effects for 21 DMPs (seven from the Cox ISIS – Aenet model, and 14 among those previously associated with arsenic) (Table 3). For CVD mortality, which included 72 DMPs associated with CVD mortality and 315 DMPs associated with urinary arsenic in our previous study, we found statistically significant mediated effects for 15 CpG sites (five from the ISIS – Aenet model and 10 previously associated with arsenic) (Table 4). The DMPs cg05779585 (LOC286083), cg19693031 (TXNIP), cg06716655 (ADAR), cg17608381 (HLA-A), cg22294740 (LINGO3), cg11946459 (HLA-A), cg03362418 (TYMP) and cg06970472 (APBB2) were common significant mediators for arsenic-related CVD incidence and mortality (two from the Cox ISIS – Aenet model and four from those previously associated with arsenic). Mediated effects from unweighted models (Tables S2 and S3) were consistent with those from weighted models.
Table 3.
Incident CVD cases per 100,000 person-years for the doubling of urinary arsenic levels not attributable (direct effect) and attributable (indirect effect) to changes in DNA methylation (90th vs. 10th percentile) for each CpG site in separate models. The sum of the direct and indirect effect represents the total effect for a doubling of urinary arsenic in CVD incidence.
| Mediated effects |
|||||||
|---|---|---|---|---|---|---|---|
| CpG | Chr | Gene | Function | Location | Cases attributable to a doubling of urinary As (95% CI) (direct effect) | Cases attributable to a doubling of urinary As through DNAm (95% CI) (indirect effect) | % cases attributable to a doubling of urinary As explained by DNAm (95% CI) |
| cg19693031 | 1 | TXNIP | Binding partner for redox signaling protein thioredoxin | 3’UTR | 137.6 (−61.2, 335.9) | 95.7 (43.8, 158.8) | 41.0 (14.5, 183.0) |
| cg05779585 | 8 | LOC286083 | Unknown function | Intergenic | 200.2 (5.8, 394.2) | 69.2 (5.8, 161.2) | 25.7 (1.8, 83.6) |
| cg03497652 | 16 | ANKS3 | Vasopressin signaling in the kidney | Body | 181.7 (−14.4, 377.5) | 46.1 (12.9, 86.5) | 20.2 (3.8, 97.4) |
| cg01270753 | 9 | TGFBR1 * | Aortic disease and altered cardiovascular development | Intergenic | 200.3 (8.7, 391.4) | 43.9 (13.6, 82.9) | 18.0 (4.7, 70.6) |
| cg22294740 | 19 | LINGO3 | Unknown function | 5’UTR | 185.3 (−11.5, 381.9) | 43.3 (7.0, 8.4) | 18.9 (1.3, 92.4) |
| cg03362418 | 22 | TYMP * | Angiogenesis in vivo. Possible therapeutic target for CVD | Body | 190.3 (−3.8, 383.8) | 40.1 (9.1, 78.6) | 17.4 (2.8, 78.0) |
| cg23027596 | 9 | UBAC1 * | Glucose-induced insulin synthesis and secretion | TSS1500 | 186.3 (−6.0, 378.1) | 39.9 (11.1, 74.6) | 17.6 (3.5, 80.4) |
| cg17608381 | 6 | HLA-A | Central role in the immune system | Body | 196.3 (−0.4, 392.4) | 35.9 (5.5, 72.9) | 15.5 (1.1, 74.9) |
| cg09956442 | 19 | ARRDC2 | Unknown function | Intergenic | 195.2 (1.6, 388.4) | 35.3 (10.3, 67.9) | 15.3 (3.4, 68.2) |
| cg06668829 | 8 | EPPK1 * | Cytoskeletal linker protein involved in response to stress | TSS1500 | 203.4 (10.9, 395.5) | 33.2 (10.1, 63.8) | 14.0 (3.4, 60.5) |
| cg14827056 | 8 | EIF2C2 | RNA-mediated gene silencing | Body | 193.8 (−0.3, 387.5) | 31.0 (5.5, 63.8) | 13.8 (1.2, 67) |
| cg18032342 | 3 | NISCH | Cell growth and death in cardiac tissue | Body | 197.2 (3.3, 390.8) | 30.1 (2.2, 63.9) | 13.2 (−0.4, 61.5) |
| cg13092901 | 22 | TYMP * | Angiogenesis in vivo. Possible therapeutic target for CVD | TSS1500 | 200.1 (6.4, 393.3) | 30.3 (3.2, 62.7) | 13.1 (0.2, 59.4) |
| cg11946459 | 6 | HLA-A | Central role in the immune system | Body | 206.4 (11.6, 400.7) | 27.2 (1.9, 58.8) | 11.7 (−0.1, 55.5) |
| cg06970472 | 4 | APBB2 * | Beta cell function, insulin secretion | Body | 205.7 (13.7, 397.3) | 27.8 (7.7, 54.8) | 11.9 (2.6, 52.3) |
| cg06716655 | 1 | ADAR2 | RNA editing enzyme involved in innate immunity | Body | 203.3 (7.0, 399.2) | 25.7 (3.9, 56.5) | 11.2 (0.9, 55.7) |
| cg18618815 | 17 | COL1A1 * | Extracellular matrix. As-induced remodeling mice model | Body | 198.5 (3.1, 393.4) | 23.7 (4.8, 49.8) | 10.7 (1.2, 54.9) |
| cg01178924 | 13 | LMO7 | Development of muscle and heart tissues. Pancreatic cancer | Body | 208.7 (13.6, 403.4) | 23.7 (0.4, 54.7) | 10.2 (−0.8, 48.8) |
| cg01542019 | 19 | TECR | Sphingolipid synthesis and oxidoreductase activity | Body | 202.1 (7.7, 396.1) | 21.4 (2.3, 48.4) | 9.6 (0.2, 48.8) |
| cg02047803 | 5 | RELL2 | Apoptosis | Body | 206.3 (13.3, 398.8) | 18.7 (0.7, 45.6) | 8.3 (−0.3, 43.5) |
| cg16335098 | 6 | SMOC2 | Angiogenesis in tumor growth and myocardial ischemia | Intergenic | 219.2 (25.7, 412.2) | 13.1 (2.7, 26.9) | 5.7 (0.8, 25.4) |
Models adjusted for age, sex, smoking status, BMI, LDL cholesterol, study center (Arizona, Oklahoma or North and South Dakota), cell counts (CD8T, CD4T, NK, B cells and monocytes) and genetic PCs.
CpG sites selected by ISIS – Aenet as predictive of CVD incidence. Other CpG sites were originally identified as associated with arsenic exposure in Bozack et al. 2020.
To account for the withdrawal of one of the Tribal Nations, models were weighted with approximately 1/3 of weight for each center (33.0% AZ, 33.6% OK, 33.4% ND/SD) using inverse probability weighting.
Table 4.
CVD deaths per 100,000 person-years for the doubling of urinary arsenic levels not attributable (direct effect) and attributable (indirect effect) to changes in DNA methylation for each CpG site in separate models. The sum of the direct and indirect effect represents the total effect for a doubling of urinary arsenic in CVD deaths.
| Mediated effects |
|||||||
|---|---|---|---|---|---|---|---|
| CpG | Chr | Gene | Function | Location | Deaths attributable to a doubling of urinary As (95 % CI) (direct effect) | Deaths attributable to a doubling of urinary As through DNAm (95%CI) (indirect effect) | % deaths attributable to a doubling of urinary As explained by DNAm (95%CI) |
| cg05779585 | 8 | LOC286083 | Unknown function | Intergenic | 91.9 (−9.7, 193.3) | 52.9 (6.1, 120.4) | 36.5 (4.3, 109.0) |
| cg19693031 | 1 | TXNIP | Binding partner for redox signaling protein thioredoxin | 3’UTR | 70.7 (−35.4, 176.4) | 43.5 (18.1, 75.4) | 38.1 (9.9, 198.5) |
| cg06716655 | 1 | ADAR | RNA editing enzyme involved in innate immunity | Body | 88.9 (−16.1, 193.6) | 25.1 (7.2, 47.5) | 22 (3.2, 114.5) |
| cg17608381 | 6 | HLA-A | Central role in the immune system | Body | 91.9 (−14.2, 197.8) | 24.1 (6.5, 45.8) | 20.8 (2.8, 112.3) |
| cg22294740 | 19 | LINGO3 | Unknown function | 5’UTR | 89.9 (−14.6, 194.2) | 22.7 (1.4, 47.7) | 20.2 (−3.4, 108.3) |
| cg03362418 | 22 | TYMP * | Angiogenesis in vivo. Possible therapeutic target for CVD | Body | 93.4 (−11.1, 197.6) | 21.3 (4.6, 43.0) | 18.5 (1.6, 94.7) |
| cg11946459 | 6 | HLA-A | Central role in the immune system | Body | 98.2 (−6.2, 202.3) | 18.4 (3.6, 37.1) | 15.8 (1.2, 81.3) |
| cg21990700 | 12 | C1RL * | Complement protein in the endoplasmic reticulum | TSS200 | 92.3 (−11.9, 196.3) | 18.3 (5.7, 34.9) | 16.6 (2.6, 91.3) |
| cg06970472 | 4 | APBB2 * | Beta cell function and insulin secretion | Body | 99.2 (−4.6, 202.8) | 16.4 (5.0, 31.4) | 14.2 (2.8, 71.3) |
| cg03026982 | 11 | NAV2 * | Blood pressure regulation | Body | 101.4 (−3.2, 205.8) | 15.5 (1.9, 34.8) | 13.2 (0.3, 66.1) |
| cg05527044 | 2 | EGR4 | Transcription regulation | Intergenic | 101.3 (−2.2, 204.7) | 13.5 (0.6, 30.7) | 11.7 (−1.6, 60.6) |
| cg00451635 | 16 | EMP2 * | Endothelial cell migration and angiogenesis | TSS1500 | 106.8 (2.9, 210.4) | 9.9 (0.2, 24.2) | 8.5 (−1.0, 43.1) |
| cg27523527 | 1 | BARHL2 | Potential regulator of neural basic helix-loop-helix genes | Intergenic | 104.8 (0.9, 208.4) | 7.7 (0.1, 19.5) | 6.9 (−1.3, 37.8) |
| cg19301366 | 6 | HLA-DQB1 | Type 1 diabetes susceptibility | 3’UTR | 106.8 (3.2, 210.2) | 3.5 (0.04, 8.8) | 3.2 (−0.7, 18.6) |
Models adjusted for age, sex, smoking status, BMI, LDL cholesterol, study center (Arizona, Oklahoma or North and South Dakota), cell counts (CD8T, CD4T, NK, B cells and monocytes) and genetic PCs.
CpG sites selected by ISIS – Aenet as predictive of CVD mortality
To account for the withdrawal of one of the Tribal Nations, models were weighted with approximately 1/3 of weight for each center (33.0% AZ, 33.6% OK, 33.4% ND/SD) using inverse probability weighting.
The adjustment for diabetes in the mediation models attenuated the indirect effects for arsenic-related CVD incidence and mortality for all DMPs, although most of them remained statistically significant for both CVD incidence and mortality (data not shown). Two CpG sites that were not significant in non-diabetes-adjusted models had significant indirect effects when adjusting for diabetes; cg25371036 (annotated to AMOTL1) had a total effect of 71.1 (−35.8, 177.9) and an indirect effect of 13.5 (0.1, 31.4) CVD incidence cases per 100,000 person-years (i.e., of 71 CVD cases per 100,000 person-years associated with a doubling of arsenic exposure, 13 cases were attributed to DNA methylation). In addition, cg22130008 (annotated to FGG), showed an indirect effect of 18.8 (0.53, 46.35) for CVD incidence. The adjustment for hypertension and systolic blood pressure in the mediation models lead to similar results as the primary analysis (data not shown).
All DMPs with statistically significant mediated effects in the main analyses were also significant when considering the full follow-up (through 2017) for CVD incidence, except cg01542019 (TECR). For CVD mortality, all were significant except cg05527044 (EGR4), cg00451635 (EMP2), cg27523527 (BARHL2) and cg19301366 (HLA-DQB1) (data not shown).
Among the 21 DMPs associated with arsenic-mediated incident CVD in the SHS, all of the CpG sites were available in MESA and 14 were available in FHS and WHI. Among the 14 common CpG sites, six had hazard ratios in the same direction for the four populations (annotated to LINGO3, TXNIP, HLA-A, EIF2C2, ANKS3 and TECR), and five more had hazard ratios in the same direction for all populations except one (Table 5). Results for FHS were similar when excluding the 111 individuals from the second batch (data not shown).
Table 5.
Replication: hazard ratios (95 % CI) of the differentially methylated positions associated with arsenic-mediated CVD incidence in the Strong Heart Study in three diverse US populations (Framingham Heart Study, Women’s Health Initiative, and Multi-Ethnic Study of Atherosclerosis).
| CpG | Gene | Strong Heart Study | Framingham Heart Study | Women’s Health Initiative | Multi-Ethnic Study of Atherosclerosis |
|---|---|---|---|---|---|
| cg01178924 | LMO7 | 0.86 (0.73, 1.02) | 0.83 (0.60, 1.14) | 1.15 (0.94, 1.40) | 1.03 (0.57, 1.85) |
| cg01270753 | TGFBR1 | 0.60 (0.50, 0.73) | - | - | 1.03 (0.52, 2.03) |
| cg01542019 | TECR | 1.14 (0.96, 1.36) | 1.06 (0.74, 1.52) | 1.26 (1.03, 1.54) | 1.59 (0.78, 3.25) |
| cg02047803 | RELL2 | 0.77 (0.65, 0.92) | 0.76 (0.54, 1.07) | 1.02 (0.82, 1.26) | 1.77 (0.91, 3.42) |
| cg03362418 | TYMP | 0.60 (0.48, 0.74) | - | - | 3.36 (1.44, 7.83) |
| cg03497652 | ANKS3 | 1.50 (1.24, 1.82) | 2.32 (1.58, 3.40) | 1.15 (0.91, 1.44) | 2.36 (1.10, 5.06) |
| cg05779585 | LOC286083 | 0.89 (0.84, 0.95) | 0.87 (0.69, 1.09) | 1.18 (0.99, 1.40) | 4.02 (1.89, 8.57) |
| cg06668829 | EPPK1 | 1.44 (1.21, 1.72) | 0.77 (0.53, 1.11) | 1.15 (0.92, 1.44) | 1.96 (0.92, 4.20) |
| cg06716655 | ADAR2 | 0.76 (0.64, 0.9) | - | - | 0.57 (0.27, 1.17) |
| cg06970472 | APBB2 | 0.72 (0.59, 0.88) | 0.64 (0.41, 0.99) | 0.93 (0.73, 1.18) | 3.97 (1.93, 8.19) |
| cg09956442 | ARRDC2 | 0.71 (0.59, 0.85) | - | - | 0.89 (0.45, 1.76) |
| cg11946459 | HLA-A | 0.76 (0.63, 0.92) | 0.65 (0.46, 0.92) | 0.86 (0.70, 1.06) | 1.41 (0.71, 2.83) |
| cg13092901 | TYMP | 0.59 (0.48, 0.72) | 0.54 (0.34, 0.87) | 0.80 (0.63, 1.00) | 1.19 (0.53, 2.67) |
| cg14827056 | EIF2C2 | 1.41 (1.17, 1.69) | 1.47 (1.01, 2.13) | 1.21 (0.95, 1.54) | 1.41 (0.68, 2.89) |
| cg16335098 | SMOC2 | 0.89 (0.80, 0.99) | - | 1.08 (0.94, 1.25) | 0.89 (0.62, 1.28) |
| cg17608381 | HLA-A | 0.77 (0.64, 0.92) | 0.62 (0.45, 0.87) | 0.88 (0.72, 1.07) | 0.93 (0.50, 1.73) |
| cg18032342 | NISCH | 1.27 (1.07, 1.50) | - | - | 1.99 (1.06, 3.75) |
| cg18618815 | COL1A1 | 0.63 (0.52, 0.76) | 0.52 (0.35, 0.78) | 1.05 (0.85, 1.30) | 0.85 (0.41, 1.79) |
| cg19693031 | TXNIP | 0.51 (0.43, 0.59) | 0.72 (0.50, 1.02) | 0.76 (0.62, 0.92) | 0.93 (0.50, 1.70) |
| cg22294740 | LINGO3 | 1.42 (1.19, 1.69) | 1.84 (1.31, 2.59) | 1.21 (0.97, 1.50) | 3.87 (2.03, 7.38) |
| cg23027596 | UBAC1 | 0.65 (0.54, 0.79) | - | - | 0.90 (0.42, 1.95) |
Models adjusted for age, sex, smoking status, BMI and cell counts (CD8T, CD4T, NK, B cells [eosinophils for MESA] and monocytes) for all populations. Additionally adjusted for total cholesterol in the FHS, for LDL cholesterol, study center (Arizona, Oklahoma or North and South Dakota) and genetic PCs in the SHS, for LDL cholesterol, technical covariates (plate number and pull ID) and race in the WHI, and for race, site and LDL cholesterol in MESA.
In the SHS and MESA, DNA methylation was measured using EPIC array. In FHS and WHI, the 450K array was used.In MESA, the only cohort with urine arsenic data available (N=206), one DMP was associated with arsenic at 0.05 p-value cut-off, and two more were associated with arsenic at 0.1 p-value cut-off. These DMPs were annotated to EPPK1 (mean difference [SE] in methylation M values −0.018 [0.008] for one log-unit change in arsenic), ANKS3 (mean difference [SE]: −0.018 [0.01]) and ARRDC2 (mean difference [SE]: 0.013 [0.007]) (Excel Table S3). A DMP annotated to TXNIP associated with arsenic before adjustment for cell counts (mean difference [SE] 0.027 [0.008]), was no longer significantly associated after adjustment for cell counts (mean difference [SE] −0.014 [0.02]).
In the protein-protein interaction network, we analyzed a list of 405 unique genes (from 315 genes tagged to DMPs associated with arsenic and 70 and 72 genes tagged to DMPs associated respectively with CVD incidence and mortality). Of these, 168 ncRNA genes or unconnected nodes were discarded, obtaining a network with 237 nodes and 460 interactions (Figure 1). MAPK8, ITPKB and SMAD3 were the most connected nodes in the network with 28, 17 and 17 interactions, respectively, and all nodes associated with arsenic and SMAD3 were also associated with CVD. Other highly connected nodes associated with CVD were TGFBR1 or PKM, with more than 10 interactions. TGFBR1, LMO7, UBAC1 and COL1A1, with 11, 10, 8 and 8 interactions respectively, were significant in the mediation analysis.
Figure 1.
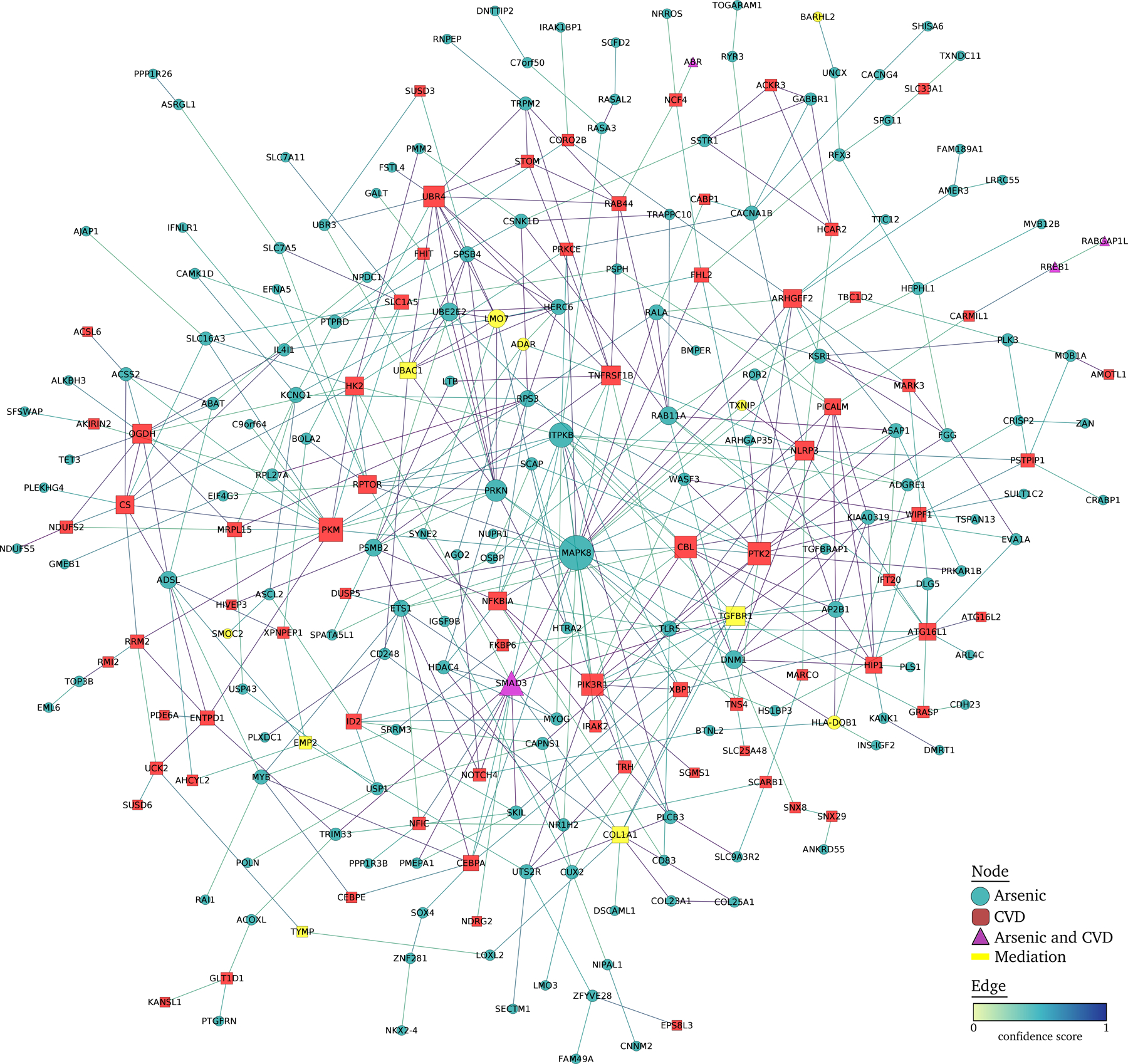
Protein-protein interaction network of differentially methylated positions associated with CVD and with arsenic in the Strong Heart Study. Arsenic-associated and CVD-associated DMPs were annotated to the nearest protein coding gene and included in a protein-protein interaction network. The interactions between nodes were obtained using STRING database v11.0,60 selecting all active interaction sources with a confidence score of 0.4. The network was analyzed and displayed using edge weighted spring embedded layout with Cytoscape v3.8.2.61.
In the Gene Ontology analysis, we found 110 enriched terms for CVD incidence (Excel Table S4), and 86 enriched terms for CVD mortality (Excel Table S5), at a cut-off of nominal p-value 0.05, none of them significant when adjusting for multiple comparisons using the FDR approach. Most of the top Gene Ontology terms were related to immune function for CVD mortality and to gene silencing for CVD incidence. In the KEGG analysis, no pathways were enriched for CVD incidence (data not shown), while 12 pathways were enriched for CVD mortality at a 0.05 nominal p-value significance threshold, including a diabetes mellitus pathway (Excel Table S6).
Cross referencing with the EWAS Catalog, 17 of the 29 DMPs that were significant in the mediation analysis for either CVD incidence or mortality showed previous associations with other traits (Table S4). The most frequently found traits were type II diabetes, smoking, and alcohol consumption.
We next investigated whether DNA methylation marks were conserved in a mouse model of early-life arsenic exposure. ApoE−/− mice exposed to arsenic during early-life (mating to weaning) exhibit increased atherosclerosis later in life and sex-specific changes to the components of the atherosclerotic plaque.66 We first interrogated differentially methylated regions (DMRs) within the 29 genes that showed significant indirect effects in the mediation analysis and were present in the animal model. We observed most (20 out of 29 DMRs) were related to arsenic-induced atherosclerosis in the animal model (Table 6, Figure 2). Further, we assessed whether individual DMPs within the 29 genes were significantly different between controls and arsenic-exposed mice. In this more stringent analysis, 43 (42 in males and one in females) DMPs mapped to 10 of 26 genes. Of note, six DMPs were annotated to Lmo7 in males, but not females, correlating with more profound arsenic-induced changes in atherosclerotic plaques found in males. The gene Nav2, significant in the mediation analysis for CVD mortality, had eight and one differentially methylated positions for male and female, respectively.
Table 6.
Significant genes in mediation analysis in the Strong Heart Study that were differentially methylated in liver samples from the mouse model of in utero arsenic exposure compared to controls.
| Mouse gene | Outcome in mediation analysis in the Strong Heart Study | Number of DMRs (male / female) annotated to the gene in the mouse model | Number of DMPs (male / female) annotated to the gene in the mouse model | Genomic position of the DMPs |
|---|---|---|---|---|
| Tgfbr1 | CVD incidence | 5 / 4 | 1 / 0 | 47429393 |
| Arrdc2 | CVD incidence | 5 / 2 | 1 / 0 | 73359785 |
| Ago2 | CVD incidence | 8 / 2 | 2 / 0 | 72999018, 72977447 |
| Nisch | CVD incidence | 2 / 0 | 1 / 0 | 32008471 |
| Lmo7 | CVD incidence | 23 / 7 | 6 / 0 | 102168435, 102232355, 102232332, 102232208, 102296394, 102136457 |
| Adar | CVD mortality | 4 / 5 | 1 / 0 | 89534367 |
| Apbb2 | CVD mortality | 3 / 16 | 4 / 0 | 66999334, 66978308, 66724458, 66733745 |
| Nav2 | CVD mortality | 31 / 15 | 8 / 1 | 56849475, 56830246, 56621107, 56724015, 56583581, 56804011, 56665515, 56605002, 56747173 |
| Egr4 | CVD mortality | 2 / 2 | 1 / 0 | 85463274 |
| Lingo3 | CVD incidence and mortality | 0 / 1 | 2 / 0 | 80308751, 80306748 |
| Ubac1 | CVD incidence | 1 / 0 | 0 / 0 | - |
| Eppk1 | CVD incidence | 1 / 2 | 0 / 0 | - |
| Tecr | CVD incidence | 3 / 1 | 0 / 0 | - |
| Smoc2 | CVD incidence | 4 / 11 | 0 / 0 | - |
| Klf9 | CVD mortality | 4 / 4 | 0 / 0 | - |
| C1rl | CVD mortality | 4 / 0 | 0 / 0 | - |
| Emp2 | CVD mortality | 4 / 9 | 0 / 0 | - |
| Barhl2 | CVD mortality | 8 / 10 | 0 / 0 | - |
| Txnip | CVD incidence and mortality | 4 / 4 | 0 / 0 | - |
| Tymp | CVD incidence and mortality | 1 / 0 | 0 / 0 | - |
DMPs: Differentially Methylated Positions
DMRs: Differentially Methylated Regions
Figure 2.
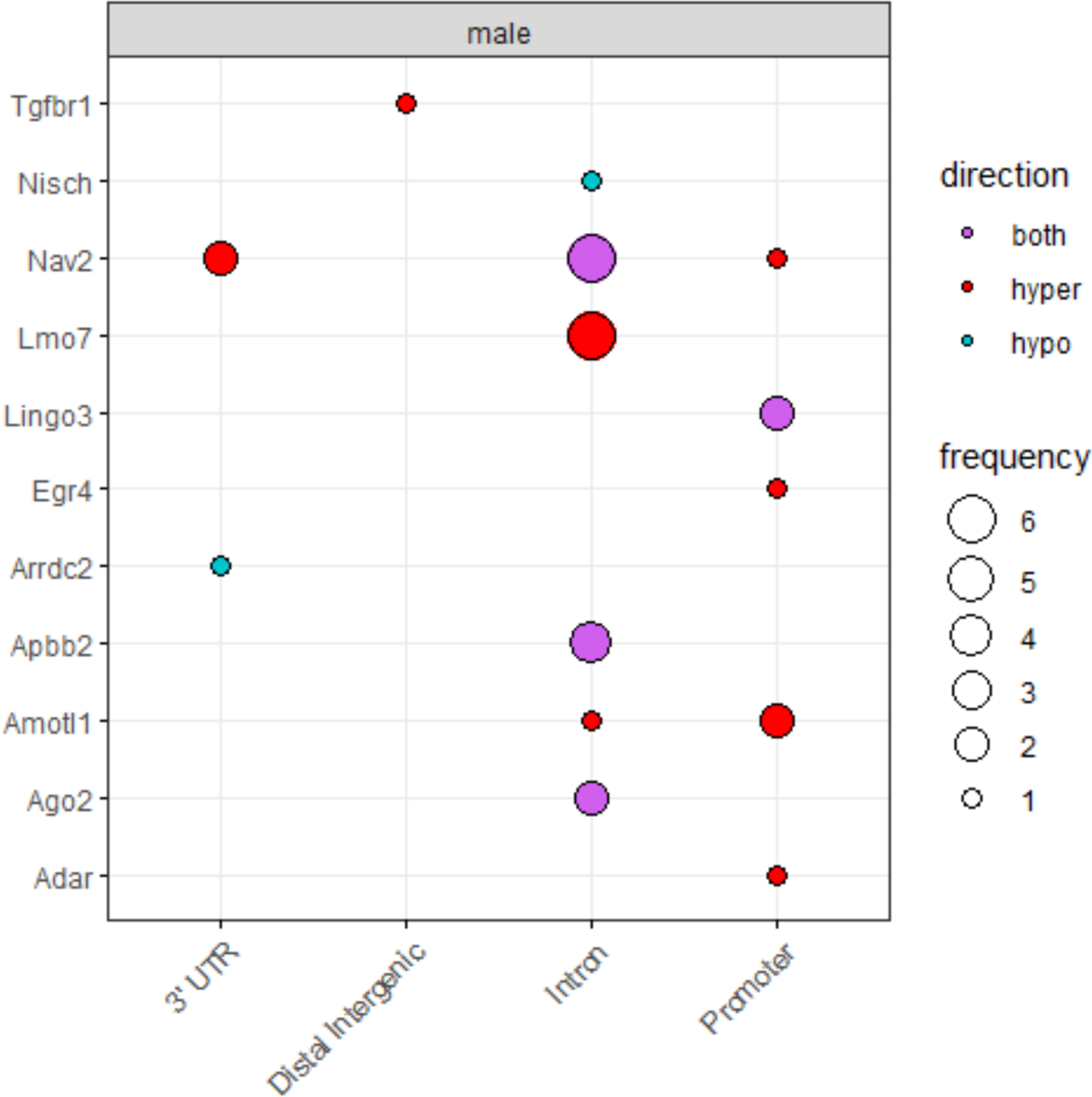
Summary of significant DMPs in mouse model of in utero arsenic exposure by gene element and the direction of differential methylation.
Discussion
In this population-based study of American Indian adults chronically exposed to arsenic in drinking water across the Southwest and the Great Plains in the US, differential methylation of several CpG sites explained part of the association of inorganic arsenic exposure, as measured in urine, with CVD incidence and mortality. Among 70 and 72 DMPs associated with CVD incidence and mortality, respectively, and 315 previously associated with arsenic in the SHS,27 we found significant mediated effects for 21 and 15 DMPs for CVD incidence and mortality, with up to 41% of mediated effects for individual DMPs (without accounting for multiple mediation). Among the 21 DMPs associated with arsenic-mediated incident CVD, six of them were associated with incident CVD in the same direction in three independent cohorts. In MESA, the only cohort with arsenic measured in a subset, despite the small sample size, the direction of association between arsenic and CVD was replicated in 13 of the 21 DMPs (N=896), and three DMPs were associated with urinary arsenic levels (N=206).
Most of the DMPs were inversely associated with CVD incidence and mortality, which would mean that hypermethylation in those CpG sites would be associated with lower risk of CVD. Only one CpG (cg25371036, annotated to AMOTL1) was located in a promoter region. As DNA methylation in promoter regions affects gene expression generally leading to gene silencing,72 our results may suggest that silencing of AMOTL1 is related to a lower risk of CVD. Several DMPs associated with arsenic-related CVD (annotated to LINGO3, UBAC1, EPPK1 and TYMP for CVD incidence, and to LINGO3, C1RL and EMP2 for CVD mortality) were located in promoter regions. Of those, TYMP, UBAC1, C1RL and EMP2 had inverse associations with CVD, potentially reflecting that silencing of those genes could be related to lower risk of CVD. EPPK1 and LINGO3, on the other hand, had positive associations with CVD incidence, potentially reflecting that overexpression of those genes could be related to higher cardiovascular risk. Functional studies exploring how DNA methylation changes in these CpG sites influence gene expression should be conducted.
The biological functions of genes annotated to the significant DMPs in the mediation analysis are relevant for CVD development and provide additional supportive evidence on the potential role of inorganic arsenic exposure on CVD through DNA methylation. Arsenic exposure has been associated with diabetes,73,74 one of the main CVD risk factors, in particular in American Indian communities,75,76 a population who has recently observed major changes in lifestyle including changes in traditional diets towards a high-fat diet in part related to limited resources and challenges of access to healthy foods in the communities.64,65 Arsenic causes impairment of pathways of glucose catabolism,77 can disrupt glucose metabolism through its reactivity toward thiol groups78 and has been related to diabetes in multiple populations including the SHS.79,80 Other mechanisms including oxidative stress, inflammation or apoptosis might also be involved in arsenic-induced diabetes.73 Several diabetes-related genes were significant in our mediation analysis. UBAC1 is a Ubiquitin-Associated Domain-Containing Protein that can influence glucose-induced insulin synthesis and secretion.81,82 Deletion of APBB2 (Amyloid Beta Precursor Protein Binding Family B) has been related to dysfunction of beta cell function in mice.83 Arsenic-induced expression changes of APBB2 were reported in primary neuronal cells in vitro.84 The EWAS Catalog has shown previous associations of RELL1 and EGR4 with diabetes or fasting glucose. In addition, methylation in FGG has been proposed as a biomarker of type 2 diabetes, while some alleles of HLA-DQB1 have been related to type 1 diabetes.85 Diabetes might be part of the biological mechanism underlying arsenic-induced CVD, at least in populations where high-fat diets have become common, as this is also the context of the animal model used in our cross-species comparison. Another possible explanation is that arsenic and diabetes share common mechanisms linking them to cardiovascular disease.
The TXNIP gene (thioredoxin interacting protein) shows one of the strongest mediated effects in our study (41%). Interestingly, cg19693031, annotated to this gene, was consistently inversely associated with CVD in all cohorts. Four DMRs annotated to this gene were also associated with arsenic in the mouse model for both males and females. TXNIP is an important binding partner for the redox signaling protein thioredoxin. Arsenic is known to directly bind thioredoxin.86 Thioredoxin plays a central role in redox control of cell functions and regulates the activity of transcription factors, such as nuclear factor kappa B (NF-KB), activating protein 1 (AP-1, an heterodimer that can include C-JUN, which is phosphorylated by MAPK8), and p53 (an important tumor suppressor protein), all of which have been involved in arsenic-toxicity, as well as in the regulation of apoptosis, a major proposed mechanism for arsenic-induced damage in multiple organs and systems.86,87 Arsenic promotes down-regulation of TXNIP in multiple myeloma cells compared to untreated cells, which could explain arsenic-induced apoptosis.88 TXNIP has also been related to prevalent diabetes,89,90 glucose homeostasis,91,92 systolic blood pressure93,94 and triglycerides.95,96 However, deletion of TXNIP is beneficial in high fat diet fed mice and streptozotocin mouse diabetes models, so the interpretation of this finding remains unclear.97,98
In addition to diabetes, the EWAS catalog linked some DMPs with smoking and alcohol intake. Smoking is a known source of arsenic,99 although it is generally not the main source. Some alcoholic beverages are known to contain arsenic, however, the estimated amount of arsenic exposure via those beverages is low.100 The EWAS catalog did not identify DMPs associated to other traits. However, this catalog is not balanced as no blood DNA methylation epigenome-wide studies have been conducted for variables that might be important for arsenic-induced CVD, such as hypertension. Hypertension is one of the most important risk factors for CVD, and it has been associated with arsenic.101 In our mediation analysis, the results did not change when adjusting for hypertension treatment and systolic blood pressure. Other EWAS are needed to evaluate the potential role of hypertension in arsenic-induced CVD.
Some of the genes in our mediation analysis have been evaluated as therapeutic targets for CVD. Mutations in the gene TGFBR1 have been associated with aortic diseases102,103 and perturbations in cardiovascular development.104 This gene has also been proposed as a prognostic biomarker after myocardial infarction.105 The DMP annotated to TYMP was consistently inversely associated with CVD in the four populations. TYMP encodes an angiogenic factor which promotes angiogenesis in vivo and contributes to endothelial cells growth in vitro. Platelets are a major source of TYMP and platelet-mediated clot formation is a key process for several types of CVD.106 The ADAR2 gene, from the ADAR gene family, has been suggested to play a vital role in preventing cardiovascular defects.107
Other significant genes have also been associated with CVD risk factors or atherosclerosis. The C1RL gene mediates the proteolytic cleavage of HP/haptoglobin in the endoplasmic reticulum. Differential expression in C1RL has been associated to CVD risk factors (hypertension, atherosclerosis) in several studies.108,109 The COL1A1 gene encodes the major component of type I collagen. Expression changes in this gene have been associated to in utero and post-natal As exposure in mice with disruptive effects in blood vessels in the heart and lungs.110 The AMOTL1 gene is related to angiomotin (an angiostatin-binding protein). This gene has been reported to be an important part of a biological mechanism by which Fat4 mutants restrict heart growth.111 Also, arsenic has been reported to be associated with dysregulations of Yap, a protein with an important role on prevention of AMOTL1 degradation.111,112 The fact that many genes with significant mediated effects in our analysis are involved in CVD-related biological pathways supports that arsenic-induced epigenetic dysregulations in those genes could be part of the biological link between arsenic and CVD, and that numerous mechanistic pathways are involved.
A recent study conducted in the same mouse model used for replication in this work showed that an in utero and early-life arsenic exposure can enhance atherosclerosis later in life in apoE−/− mice.113 Comparing the DNA methylation data from the livers harvested in that study to the top hits from our population-based study, we observed differential DNA methylation in the genes of interest. The fact that these DMPs and DMRs are validated in a different tissue (blood vs. liver) that is equally important to CVD, in particular in the context of cardiometabolic disease, provides supporting evidence of a potential causal relationship between arsenic-induced DNA methylation changes and atherosclerosis.
One of the methodological strengths of this work is the implementation of the innovative statistical tool ISIS – Aenet to evaluate the association of DNA methylation with CVD. ISIS has proven to be very efficient for variable selection, reducing the false discovery rate. It has been used in other studies paired with other shrinkage methods such as LASSO or elastic-net, however, to our knowledge, this is the first study that has incorporated Aenet, an improvement of elastic-net, to the ISIS algorithm for a survival problem. Other strengths include replication in three independent cohorts and in an animal model, having methylation data in one of the largest microarrays available (850K), the prospective study design, and the high quality of the study protocol and CVD ascertainment.
This work has some limitations. First, water arsenic levels changed a few years after the implementation of the US EPA Final Arsenic Rule in 2006.38 However, the SHS does not have updated information on urinary arsenic levels in recent years, and data from Chile support that CVD incidence changes a few years after exposure changes.114 Longitudinal studies with repeated measurements of arsenic and DNA methylation are needed to assess the reduction of CVD risk after arsenic exposure decreases. Second, DNA methylation is highly cell-type specific and results from blood cells might not be comparable to DNA methylation in other tissues. Blood DNA methylation, however, is emerging as a relevant tissue for CVD, probably because many of the immune cells in blood are involved in CVD pathogenesis. Also, it is unknown if CpG sites in human blood are comparable to mouse liver cells; indeed, there is limited homology between human and murine CpG sites. A genetically-modified mouse that induces hyperlipidemia had to be used, as wild-type mice do not develop atherosclerosis, even on a high-fat diet. Thus, the arsenic exposure cannot be studied in the absence of hyperlipidemia. Our mice were exposed to arsenic only during early-life and were all hyperlipidemic through genetic modification, although they were not on high-fat diet. This model might be well suited for the populations we studied such as SHS and MESA, but may not be representative for populations exposed to arsenic in Bangladesh and other parts of the world where high-fat diets are less common. These results lay the groundwork for developing mouse models to test specific questions regarding the epigenetic contribution to arsenic-related CVD and potential interventional strategies.
In conclusion, differential methylation of CpG sites annotated to genes relevant for arsenic-related health effects might be part of the biological link between inorganic arsenic exposure and CVD. Diabetes might be a relevant mechanism for arsenic-induced cardiovascular risk in populations with a high diabetes burden, or alternatively arsenic and diabetes might share common pathways for CVD. Replication was observed for several DMPs across diverse US populations. The inter-species comparison supports that arsenic exposure modifies methylation of the same genes in the liver of an animal model of atherosclerosis compared to unexposed animals. Additional experimental studies are needed to assess whether changes in these epigenetic signatures depending on arsenic exposure influence CVD development.
Supplementary Material
Novelty and significance.
What is known?
Arsenic, a risk factor for cardiovascular disease (CVD), induces epigenetic modifications in experimental models.
DNA methylation has been proposed as an intermediate mechanism between environmental exposures and disease.
What new information does this article contribute?
Mediation analysis in the Strong Heart Study supports that blood DNA methylation influences arsenic-related CVD.
Differential DNA methylation in several sites were replicated in three independent cohorts and in a mouse model of arsenic-induced atherosclerosis.
Gene functions support that diabetes and redox signaling are involved in arsenic-induced CVD.
This is the first study that conducts a mediation analysis to assess the potential role of DNA methylation on arsenic-related CVD. Differential methylation of DNA sites in blood were identified as potential mediators in the Strong Heart Study, and some of them were replicated as associated with CVD in three independent cohorts. Differential methylation of similar genes in the liver was observed in a mouse model of arsenic-induced atherosclerosis. The characterization of gene function related to these DNA methylation sites can help identify the biological link between arsenic exposure and CVD. Gene function analysis supported that diabetes and redox signaling are relevant pathways for arsenic-induced CVD in populations with a high diabetes burden. Alternatively, arsenic and diabetes might share common pathways for CVD.
Sources of funding
The Strong Heart Study was supported by grants from the National Heart, Lung, and Blood Institute (NHLBI) (contract numbers 75N92019D00027, 75N92019D00028, 75N92019D00029 and 75N92019D00030) and previous grants (R01HL090863, R01HL109315, R01HL109301, R01HL109284, R01HL109282, and R01HL109319 and cooperative agreements: U01HL41642, U01HL41652, U01HL41654, U01HL65520 and U01HL65521) and by the National Institute of Environmental Health Sciences (grant numbers R01ES021367, R01ES025216, P42ES010349, P30ES009089).
The Framingham Heart Study is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with Boston University (Contract No. N01-HC-25195, HHSN268201500001I and 75N92019D00031). The laboratory work for this investigation was funded by the Division of Intramural Research, National Heart, Lung, and Blood Institutes, National Institutes of Health and an NIH Director’s Challenge Award (D. Levy, Principal Investigator).
The Women’s Health Initiative is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts, HHSN268201600018C, HHSN268201600001C, HHSN268201600002C, HHSN268201600003C, and HHSN268201600004C. A full list of the WHI investigators can be found at: https://www.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Long%20List.pdf.
The datasets used for the analyses described in this manuscript in FHS and WHI were obtained from the NIH dbGaP at http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap through accession phs000007.v30.p11 and phs000200.WHI.v11.p3, respectively.
Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). Multi-Ethnic Study of Atherosclerosis (MESA)” (phs001416.v1.p1) was performed at the Broad Institute of MIT and Harvard (3U54HG003067–13S1). Centralized read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626–02S1, contract HHSN268201800002I) (Broad RNA Seq, Proteomics HHSN268201600034I, UW RNA Seq HHSN268201600032I, USC DNA Methylation HHSN268201600034I, Broad Metabolomics HHSN268201600038I). Phenotype harmonization, data management, sample-identity QC, and general study coordination, were provided by the TOPMed Data Coordinating Center (3R01HL-120393; U01HL-120393; contract HHSN268180001I). MESA and the MESA SHARe projects are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, UL1-TR-001420. Funding for SHARe genotyping was provided by NHLBI Contract N02-HL-64278. Genotyping was performed at Affymetrix (Santa Clara, California, USA) and the Broad Institute of Harvard and MIT (Boston, Massachusetts, USA) using the Affymetrix Genome-Wide Human SNP Array 6.0. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center.
Infrastructure for the CHARGE Consortium is supported in part by the National Heart, Lung, and Blood Institute (NHLBI) grant R01HL105756. Also supported in part by the National Institutes for Diabetes and Digestive and Kidney Diseases contract R01-HL151855–01 and contract R01HL146860.
ADR was supported by a fellowship from “la Caixa” Foundation (ID 100010434) (fellowship code “LCF/BQ/DR19/11740016”).
ALRC was supported by Maria Zambrano grant for the requalification of the Spanish university system - NextGeneration EU; and by ANID - Millennium Science Initiative Program - Nº NCS2021_013 – SocioMed.
KKM and KM were supported by a fellowship from the Fonds de recherche du Québec – Santé; by a grant from the Canadian Institutes of Health Research (#PJT-166142) and by the Shuk Tak Liang Fellowship.
The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health (United States) or the National Health Institute Carlos III (Spain). The funders had no role in the planning, conducting, analysis, interpretation, or writing of this study.
Non-standard Abbreviations and Acronyms
- CpG
Cytosine – Guanine dinucleotide
- CVD
Cardiovascular disease
- DMPs
Differentially methylated positions
- SHS
Strong Heart Study
- FHS
Framingham Heart Study
- WHI
Women’s Health Initiative
- MESA
Multi-Ethnic Study of Atherosclerosis
- MMA
Monomethylarsonate
- DMA
Dimethylarsinate
- SNPs
Single Nucleotide Polymorphisms
- ISIS-Aenet
Iterative Sure Independence Screening coupled with Adaptive Elastic-Net
- SIS
Sure Independence Screening
- PCs
Principal components
- DMRs
Differentially methylated regions
Footnotes
REFERENCES
- 1.Podgorski J, Berg M. Global threat of arsenic in groundwater. Science 2020;368(6493):845–850. [DOI] [PubMed] [Google Scholar]
- 2.IARC Working Group. 2009. Arsenic, metals, fibres, and dusts. IARC Monogr Eval Carcinog Risks Hum 100:11–465. [PMC free article] [PubMed] [Google Scholar]
- 3.Moon KA, Navas-Acien A, Grau-Pérez M, Francesconi KA, Goessler W, Guallar E, Umans JG, Best LG, Newman JD. Low-moderate urine arsenic and biomarkers of thrombosis and inflammation in the Strong Heart Study. Rahman M, ed. PLOS ONE 2017;12(8):e0182435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.National Research Council. Critical aspects of EPA’s IRIS assessment of inorganic arsenic: Interim report. National Academies Press; 2013. [Google Scholar]
- 5.Chen C-J, Chiou H-Y, Chiang M-H, Lin L-J, Tai T-Y. Dose-Response Relationship Between Ischemic Heart Disease Mortality and Long-term Arsenic Exposure. Arteriosclerosis, Thrombosis, and Vascular Biology 1996;16(4):504–510. [DOI] [PubMed] [Google Scholar]
- 6.Hsueh YM, Wu WL, Huang YL, Chiou HY, Tseng CH, Chen CJ. Low serum carotene level and increased risk of ischemic heart disease related to long-term arsenic exposure. Atherosclerosis 1998;141(2):249–257. [DOI] [PubMed] [Google Scholar]
- 7.Moon KA, Guallar E, Umans JG, Devereux RB, Best LG, Francesconi KA, Goessler W, Pollak J, Silbergeld EK, Howard BV., Navas-Acien A. Association Between Exposure to Low to Moderate Arsenic Levels and Incident Cardiovascular Disease. Annals of Internal Medicine 2013;159(10):649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monrad M, Ersbøll AK, Sørensen M, Baastrup R, Hansen B, Gammelmark A, Tjønneland A, Overvad K, Raaschou-Nielsen O. Low-level arsenic in drinking water and risk of incident myocardial infarction: A cohort study. Environmental Research 2017;154:318–324. [DOI] [PubMed] [Google Scholar]
- 9.Xu L, Mondal D, Polya DA. Positive association of cardiovascular disease (CVD) with chronic exposure to drinking water arsenic (As) at concentrations below the WHO provisional guideline value: A systematic review and meta-analysis. International Journal of Environmental Research and Public Health 2020;17(7). [DOI] [PMC free article] [PubMed]
- 10.Newman JD, Navas-Acien A, Kuo CC, Guallar E, Howard BV., Fabsitz, Devereux RB, Umans JG, Francesconi KA, Goessler W, Best LT, Tellez-Plaza M. Peripheral arterial disease and its association with arsenic exposure and metabolism in the strong heart study. American Journal of Epidemiology 2016;184(11):806–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Graziano JH, Parvez F, Liu M, Slavkovich V, Kalra T, Argos M, Islam T, Ahmed A, Rakibuz-Zaman M, Hasan R, Sarwar G, Levy D, Van Geen A, Ahsan H. Arsenic exposure from drinking water and mortality from cardiovascular disease in Bangladesh: Prospective cohort study. BMJ 2011;342(7806). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Medrano MJ, Boix R, Pastor-Barriuso R, Palau M, Damián J, Ramis R, del Barrio JL, Navas-Acien A. Arsenic in public water supplies and cardiovascular mortality in Spain. Environmental Research 2010;110(5):448–454. [DOI] [PubMed] [Google Scholar]
- 13.Abhyankar LN, Jones MR, Guallar E, Navas-Acien A. Arsenic exposure and hypertension: A systematic review. Environmental Health Perspectives 2012;120(4):494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mateen FJ, Grau-Perez M, Pollak JS, Moon KA, Howard BV., Umans JG, Best LG, Francesconi KA, Goessler W, Crainiceanu C, Guallar E, Devereux RB, Roman MJ, Navas-Acien A . Chronic arsenic exposure and risk of carotid artery disease: The Strong Heart Study. Environmental Research 2017;157:127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang CH, Jeng JS, Yip PK, Chen CL, Hsu LI, Hsueh YM, Chiou HY, Wu MM, Chen CJ. Biological gradient between long-term arsenic exposure and carotid atherosclerosis. Circulation 2002;105(15):1804–1809. [DOI] [PubMed] [Google Scholar]
- 16.Makhani K, Chiavatti C, Plourde D, Negro Silva LF, Lemaire M, Lemarié CA, Lehoux S, Mann KK. Using the apolipoprotein e knock-out mouse model to define atherosclerotic plaque changes induced by low dose arsenic. Toxicological Sciences 2018;166(1):213–218. [DOI] [PubMed] [Google Scholar]
- 17.Negro Silva LF, Lemaire M, Lemarié CA, Plourde D, Bolt AM, Chiavatti C, Bohle DS, Slavkovich V, Graziano JH, Lehoux S, Mann KK. Effects of inorganic arsenic, methylated arsenicals, and arsenobetaine on atherosclerosis in the apoE−/− mouse model and the role of as3mt-mediated methylation. Environmental Health Perspectives 2017;125(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demanelis K, Argos M, Tong L, Shinkle J, Sabarinathan M, Rakibuz-Zaman M, Sarwar G, Shahriar H, Islam T, Rahman M, Yunus M, Graziano JH, Broberg K, Engström K, Jasmine F, et al. Association of arsenic exposure with whole blood DNA methylation: An epigenome-wide study of Bangladeshi adults. Environmental Health Perspectives 2019;127(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Argos M, Chen L, Jasmine F, Tong L, Pierce BL, Roy S, Paul-Brutus R, Gamble MV., Harper KN, Parvez F, Rahman M, Rakibuz-Zaman M, Slavkovich, Baron JA, Graziano JH, et al. Gene-Specific Differential DNA Methylation and Chronic Arsenic Exposure in an Epigenome-Wide Association Study of Adults in Bangladesh. Environmental Health Perspectives 2015;123(1):64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Broberg K, Ahmed S, Engström K, Hossain MB, Jurkovic Mlakar S, Bottai M, Grandér M, Raqib R, Vahter M. Arsenic exposure in early pregnancy alters genome-wide DNA methylation in cord blood, particularly in boys. Journal of Developmental Origins of Health and Disease 2014;5(4):288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gliga AR, Engström K, Kippler M, Skröder H, Ahmed S, Vahter M, Raqib R, Broberg K. Prenatal arsenic exposure is associated with increased plasma IGFBP3 concentrations in 9-year-old children partly via changes in DNA methylation. Archives of Toxicology 2018;92(8):2487–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kile ML, Andres Houseman E, Baccarelli AA, Quamruzzaman Q, Rahman M, Mostofa G, Cardenas A, Wright RO, Christiani DC. Effect of prenatal arsenic exposure on DNA methylation and leukocyte subpopulations in cord blood. Epigenetics 2014;9(5):774–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ameer SS, Engström K, Hossain MB, Concha G, Vahter M, Broberg K. Arsenic exposure from drinking water is associated with decreased gene expression and increased DNA methylation in peripheral blood. Toxicology and Applied Pharmacology 2017;321:57–66. [DOI] [PubMed] [Google Scholar]
- 24.Rojas D, Rager JE, Smeester L, Bailey KA, Drobná Z, Rubio-Andrade M, Stýblo M, Stýblo S, García-Vargas G, Fry RC. Prenatal Arsenic Exposure and the Epigenome: Identifying Sites of 5-methylcytosine Alterations that Predict Functional Changes in Gene Expression in Newborn Cord Blood and Subsequent Birth Outcomes [DOI] [PMC free article] [PubMed]
- 25.Kaushal A, Zhang H, Karmaus WJJ, Everson TM, Marsit CJ, Karagas MR, Tsai SF, Wen HJ, Wang SL. Genome-wide DNA methylation at birth in relation to in utero arsenic exposure and the associated health in later life. Environmental Health: A Global Access Science Source 2017;16(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo X, Chen X, Wang J, Liu Z, Gaile D, Wu H, Yu G, Mao G, Yang Z, Di Z, Guo X, Cao L, Chang P, Kang B, Chen J, et al. Multi-generational impacts of arsenic exposure on genome-wide DNA methylation and the implications for arsenic-induced skin lesions. Environment International 2018;119:250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bozack AK, Domingo-Relloso A, Haack K, Gamble MV., Tellez-Plaza M, Umans JG, Best, Yracheta J, Gribble MO, Cardenas A, Francesconi KA, Goessler W, Tang W-Y, Fallin MD, Cole SA, et al. Locus-Specific Differential DNA Methylation and Urinary Arsenic: An Epigenome-Wide Association Study in Blood among Adults with Low-to-Moderate Arsenic Exposure. Environmental Health Perspectives 2020;128(6):067015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cardenas A, Koestler DC, Houseman EA, Jackson BP, Kile ML, Karagas MR, Marsit CJ. Differential DNA methylation in umbilical cord blood of infants exposed to mercury and arsenic in utero. Epigenetics 2015;10(6):508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green BB, Karagas MR, Punshon T, Jackson BP, Robbins DJ, Houseman EA, Marsit CJ. Epigenome-wide assessment of DNA methylation in the placenta and arsenic exposure in the New Hampshire Birth Cohort Study (USA). Environmental Health Perspectives 2016;124(8):1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koestler DC, Avissar-Whiting M, Andres Houseman E, Karagas MR, Marsit CJ. Differential DNA methylation in umbilical cord blood of infants exposed to low levels of arsenic in utero. Environmental Health Perspectives 2013;121(8):971–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Harst P, de Windt LJ, Chambers JC. Translational Perspective on Epigenetics in Cardiovascular Disease. Journal of the American College of Cardiology 2017;70(5):590–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Westerman K, Sebastiani P, Jacques P, Liu S, Demeo D, Ordovás JM. DNA methylation modules associate with incident cardiovascular disease and cumulative risk factor exposure. Clinical Epigenetics 2019;11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agha G, Mendelson MM, Ward-Caviness CK, Joehanes R, Huan TX, Gondalia R, Salfati E, Brody JA, Fiorito G, Bressler J, Chen BH, Ligthart S, Guarrera S, Colicino E, Just AC, et al. Blood Leukocyte DNA Methylation Predicts Risk of Future Myocardial Infarction and Coronary Heart Disease. Circulation 2019;140(8):645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernández-Sanlés A, Sayols-Baixeras S, Subirana I, Degano IR, Elosua R. Association between DNA methylation and coronary heart disease or other atherosclerotic events: A systematic review. Atherosclerosis 2017;263:325–333. [DOI] [PubMed] [Google Scholar]
- 35.Lee ET, Welty TK, Fabsitz R, Cowan LD, Le NA, Oopik AJ, Cucchiara AJ, Savage PJ, Howard BV. The Strong Heart Study. A study of cardiovascular disease in American Indians: design and methods. American journal of epidemiology 1990;132(6):1141–55. [DOI] [PubMed] [Google Scholar]
- 36.Domingo-Relloso A, Riffo-Campos AL, Haack K, Rentero-Garrido P, Ladd-Acosta C, Fallin DM, Tang WY, Herreros-Martinez M, Gonzalez JR, Bozack AK, Cole SA, Navas-Acien A, Tellez-Plaza M. Cadmium, Smoking, and Human Blood DNA Methylation Profiles in Adults from the Strong Heart Study. Environmental Health Perspectives 2020;128(6):067005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Navas-Acien A, Umans JG, Howard BV., Goessler W, Francesconi KA, Crainiceanu CM, Silbergeld EK, Guallar E. Urine arsenic concentrations and species excretion patterns in American Indian communities over a 10-year period: The strong heart study. Environmental Health Perspectives 2009;117(9):1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nigra AE, Sanchez TR, Nachman KE, Harvey DE, Chillrud SN, Graziano JH, Navas-Acien A. The effect of the Environmental Protection Agency maximum contaminant level on arsenic exposure in the USA from 2003 to 2014: an analysis of the National Health and Nutrition Examination Survey (NHANES). The Lancet Public Health 2017;2(11):e513–e521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nigra AE, Chen Q, Chillrud SN, Wang L, Harvey D, Mailloux B, Factor-Litvak P, Navas-Acien A. Inequalities in public water arsenic concentrations in counties and community water systems across the united states, 2006–2011. Environmental Health Perspectives 2020;128(12):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scheer J, Findenig S, Goessler W, Francesconi KA, Howard B, Umans JG, Pollak J, Tellez-Plaza M, Silbergeld EK, Guallar E, Navas-Acien A. Arsenic species and selected metals in human urine: validation of HPLC/ICPMS and ICPMS procedures for a long-term population-based epidemiological study. Analytical methods : advancing methods and applications 2012;4(2):406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scheer J, Findenig S, Goessler W, Francesconi KA, Howard B, Umans JG, Pollak J, Tellez-Plaza M, Silbergeld EK, Guallar E, Navas-Acien A. Arsenic species and selected metals in human urine: validation of HPLC/ICPMS and ICPMS procedures for a long-term population-based epidemiological study. Analytical methods : advancing methods and applications 2012;4(2):406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee ET, Welty TK, Fabsitz R, Cowan LD, Le NA, Oopik AJ, Cucchiara AJ, Savage PJ, Howard BV. The Strong Heart Study. A study of cardiovascular disease in American Indians: design and methods. American journal of epidemiology 1990;132(6):1141–55. [DOI] [PubMed] [Google Scholar]
- 43.Anon. Strongheart Study - Center for American Indian Health Research - College of Public Health.
- 44.Moon KA, Guallar E Dr., Umans JG Dr., Devereux RB Dr., Best LG Dr., Francesconi KA Dr., Goessler W Dr., Pollak J, Silbergeld EK Dr., Howard BV Dr., Navas-Acien A Dr.. Association between Low to Moderate Arsenic Exposure and Incident Cardiovascular Disease. A Prospective Cohort Study. Annals of internal medicine 2013;159(10):649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCartney DL, Walker RM, Morris SW, McIntosh AM, Porteous DJ, Evans KL. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genomics data 2016;9:22–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fortin J-P, Triche TJ, Hansen KD, Hansen KD. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics (Oxford, England) 2017;33(4):558–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012;13(1):86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsao CW, Vasan RS. Cohort Profile: The Framingham Heart Study (FHS): overview of milestones in cardiovascular epidemiology. International journal of epidemiology 2015;44(6):1800–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zou H, Hao A, Zhang H. ON THE ADAPTIVE ELASTIC-NET WITH A DIVERGING NUMBER OF PARAMETERS. The Annals of Statistics 2009;37(4):1733–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang T, Zhu L. Consistent tuning parameter selection in high dimensional sparse linear regression. Journal of Multivariate Analysis 2011;102(7):1141–1151. [Google Scholar]
- 51.Fan J, Lv J. Sure independence screening for ultrahigh dimensional feature space. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 2008;70(5):849–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hasinur M, Khan R, Ewart J, Shaw H. VARIABLE SELECTION FOR SURVIVAL DATA WITH A CLASS OF ADAPTIVE ELASTIC NET TECHNIQUES; 2013.
- 53.Lee ET, Howard BV., Wang W, Welty TK, Galloway JM, Best LG, Fabsitz RR, Zhang Y, Yeh J, Devereux RB. Prediction of Coronary Heart Disease in a Population With High Prevalence of Diabetes and Albuminuria. Circulation 2006;113(25):2897–2905. [DOI] [PubMed] [Google Scholar]
- 54.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012 13:1 2012;13(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barfield RT, Almli LM, Kilaru V, Smith AK, Mercer KB, Duncan R, Klengel T, Mehta D, Binder EB, Epstein MP, Ressler KJ, Conneely KN. Accounting for Population Stratification in DNA Methylation Studies. Genetic epidemiology 2014;38(3):231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huanga YT, Yangc HI. Causal mediation analysis of survival outcome with multiple mediators. Epidemiology 2017;28(3):370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lange T, Hansen JV. Direct and Indirect Effects in a Survival Context. Epidemiology 2011;22(4):575–581. [DOI] [PubMed] [Google Scholar]
- 58.García-Esquinas E, Pollan M, Tellez-Plaza M, Francesconi KA, Goessler W, Guallar E, Umans JG, Yeh J, Best LG, Navas-Acien A. Cadmium Exposure and Cancer Mortality in a Prospective Cohort: The Strong Heart Study. Environmental Health Perspectives 2014;122(4):363–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Höfler M, Pfister H, Lieb R, Wittchen HU. The use of weights to account for non-response and drop-out. Social Psychiatry and Psychiatric Epidemiology 2005;40(4):291–299. [DOI] [PubMed] [Google Scholar]
- 60.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, von Mering C. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Research 2019;47(D1):D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Research 2003;13(11):2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Battram T, Yousefi P, Crawford G, Prince C, Babei MS, Sharp G, Hatcher C, Vega-Salas MJ, Khodabakhsh S, Whitehurst O, Langdon R, Mahoney L, Elliott HR, Mancano G, Lee M, et al. The EWAS Catalog: a database of epigenome-wide association studies [DOI] [PMC free article] [PubMed]
- 63.Getz GS, Reardon CA. Diet and murine atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology 2006;26(2):242–249. [DOI] [PubMed] [Google Scholar]
- 64.Fretts AM Howard BV, Siscovick DS, Best LG, Beresford SA, Mete M, S Eilat-Adar N Sotoodehnia, Zhao J. Processed Meat, but Not Unprocessed Red Meat, Is Inversely Associated with Leukocyte Telomere Length in the Strong Heart Family Study. The Journal of nutrition 2016;146(10):2013–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fretts AM, Howard BV, McKnight B, Duncan GE, Beresford SA, Mete M, Eilat-Adar S, Zhang Y, Siscovick DS. Associations of processed meat and unprocessed red meat intake with incident diabetes: the Strong Heart Family Study. The American journal of clinical nutrition 2012;95(3):752–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Negro Silva LF, Lemaire M, Lemarié CA, Plourde D, Bolt AM, Chiavatti C, Bohle DS, Slavkovich V, Graziano JH, Lehoux S, Mann KK. Effects of inorganic arsenic, methylated arsenicals, and arsenobetaine on atherosclerosis in the apoE−/− mouse model and the role of as3mt-mediated methylation. Environmental Health Perspectives 2017;125(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Merkel A Fernandez-Callejo M, Casals E, Marco-Sola S, Schuyler R, Gut IG, Heath SC. gemBS: high throughput processing for DNA methylation data from bisulfite sequencing. Bioinformatics (Oxford, England) 2019;35(5):737–742. [DOI] [PubMed] [Google Scholar]
- 68.Hansen KD, Langmead B, Irizarry RA. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biology 2012 13:10 2012;13(10):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.J L L J. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity 2005;95(3):221–227. [DOI] [PubMed] [Google Scholar]
- 70.G Yu LG Wang QY He. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics (Oxford, England) 2015;31(14):2382–2383. [DOI] [PubMed] [Google Scholar]
- 71.Anon. Bioconductor - annotatr.
- 72.Luo R, Bai C, Yang L, Zheng Z, Su G, Gao G, Wei Z, Zuo Y, Li G. DNA methylation subpatterns at distinct regulatory regions in human early embryos. Open Biology 2018;8(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Navas-Acien A, Silbergeld EK, Streeter RA, Clark JM, Burke TA, Guallar E. Arsenic exposure and type 2 diabetes: A systematic review of the experimental and epidemiologic evidence. Environmental Health Perspectives 2006;114(5):641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pérez MG, Acien AN, Chilet IG, Izquierdo RL, Manzano IG, Escudero JCM, Mas JR i, Martínez FJC, Plaza MT. Arsenic, diabetes-related genes and diabetes prevalence in a general population from Spain: The Hortega Study. ? 2016;2016(1).
- 75.Howard B V, Cowan LD, Go O, Welty TK, Robbins DC, Lee ET. Adverse effects of diabetes on multiple cardiovascular disease risk factors in women: The strong heart study. Diabetes Care 1998;21(8):1258–1265. [DOI] [PubMed] [Google Scholar]
- 76.Breathett K, Sims M, Gross M, Jackson EA, Jones EJ, Navas-Acien A, Taylor H, Thomas KL, Howard BV. Cardiovascular Health in American Indians and Alaska Natives: A Scientific Statement from the American Heart Association. Circulation 2020;141(25):E948–E959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kulshrestha A Arsenic-induced abnormalities in glucose metabolism: Biochemical basis and potential therapeutic and nutritional interventions. World Journal of Translational Medicine 2014;3(2):96. [Google Scholar]
- 78.Assessment UENC for E. Biochemical toxicology of arsenic 2009.
- 79.Maull EA, Ahsan H, Edwards J, Longnecker MP, Navas-Acien A, Pi J, Silbergeld EK, Styblo M, Tseng CH, Thayer KA, Loomis D. Evaluation of the association between arsenic and diabetes: A National Toxicology Program workshop review. Environmental Health Perspectives 2012;120(12):1658–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grau-Perez M, Kuo CC, Gribble MO, Balakrishnan P, Spratlen MJ, Vaidya D, Francesconi KA, Goessler W, Guallar E, Silbergeld EK, Umans JG, Best LG, Lee ET, Howard BV., Cole SA, et al. Association of low-moderate arsenic exposure and arsenic metabolism with incident diabetes and insulin resistance in the strong heart family study. Environmental Health Perspectives 2017;125(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hofmeister-Brix A, Kollmann K, Langer S, Schultz J, Lenzen S, Baltrusch S. Identification of the ubiquitin-like domain of midnolin as a new glucokinase interaction partner. Journal of Biological Chemistry 2013;288(50):35824–35839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sternisha SM, Miller BG. Molecular and cellular regulation of human glucokinase. Archives of Biochemistry and Biophysics 2019;663:199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ye R, Gordillo R, Shao M, Onodera T, Chen Z, Chen S, Lin X, SoRelle JA, Li X, Tang M, Keller MP, Kuliawat R, Attie AD, Gupta RK, Holland WL, et al. Intracellular lipid metabolism impairs β cell compensation during diet-induced obesity. Journal of Clinical Investigation 2018;128(3):1178–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zarazúa S, Bürger S, Delgado JM, Jiménez-Capdeville ME, Schliebs R. Arsenic affects expression and processing of amyloid precursor protein (APP) in primary neuronal cells overexpressing the Swedish mutation of human APP. International Journal of Developmental Neuroscience 2011;29(4):389–396. [DOI] [PubMed] [Google Scholar]
- 85.Mosaad YM. HLA-DQB1* alleles and genetic susceptibility to type 1 diabetes mellitus. World Journal of Diabetes 2012;3(8):149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shen S, Li XF, Cullen WR, Weinfeld M, Le XC. Arsenic binding to proteins. Chemical Reviews 2013;113(10):7769–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hu Y, Jin X, Snow ET. Effect of arsenic on transcription factor AP-1 and NF-κB DNA binding activity and related gene expression. Toxicology Letters 2002;133(1):33–45. [DOI] [PubMed] [Google Scholar]
- 88.Wang M, Liu S, Liu P. Gene expression profile of multiple myeloma cell line treated by arsenic trioxide. Journal of Huazhong University of Science and Technology - Medical Science 2007;27(6):646–649. [DOI] [PubMed] [Google Scholar]
- 89.Albao DS, Cutiongco-De La Paz EM, Mercado ME, Lirio A, Mariano M, Kim S, Yangco A, Melegrito J, Wad-Asen K, Gauran II, Francisco MA, Santos-Acuin C, David-Padilla C, Murphy EJ, Paz-Pacheco E, et al. Methylation changes in the peripheral blood of Filipinos with type 2 diabetes suggest spurious transcription initiation at TXNIP. Human Molecular Genetics 2019;28(24):4208–4218. [DOI] [PubMed] [Google Scholar]
- 90.Florath I, Butterbach K, Heiss J, Bewerunge-Hudler M, Zhang Y, Schöttker B, Brenner H. Type 2 diabetes and leucocyte DNA methylation: an epigenome-wide association study in over 1,500 older adults. Diabetologia 2016;59(1):130–138. [DOI] [PubMed] [Google Scholar]
- 91.Liu J, Carnero-Montoro E, van Dongen J, Lent S, Nedeljkovic I, Ligthart S, Tsai PC, Martin TC, Mandaviya PR, Jansen R, Peters MJ, Duijts L, Jaddoe VWV, Tiemeier H, Felix JF, et al. An integrative cross-omics analysis of DNA methylation sites of glucose and insulin homeostasis. Nature Communications 2019;10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yoshihara E Txnip/tbp-2: A master regulator for glucose homeostasis. Antioxidants 2020;9(8):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang Y, Ollikainen M, Muniandy M, Zhang T, Van Dongen J, Hao G, Van Der Most PJ, Pan Y, Pervjakova N, Sun YV., Hui Q, Lahti J, Fraszczyk E, Lu X, Sun D, et al. Identification, Heritability, and Relation with Gene Expression of Novel DNA Methylation Loci for Blood Pressure. Hypertension 2020;76(1):195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Richard MA, Huan T, Ligthart S, Gondalia R, Jhun MA, Brody JA, Irvin MR, Marioni R, Shen J, Tsai PC, Montasser ME, Jia Y, Syme C, Salfati EL, Boerwinkle E, et al. DNA Methylation Analysis Identifies Loci for Blood Pressure Regulation. American Journal of Human Genetics 2017;101(6):888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Toinét Cronjé H, Elliott HR, Nienaber-Rousseau C, Pieters M. Replication and expansion of epigenome-wide association literature in a black South African population. Clinical Epigenetics 2020;12(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mamtani M, Kulkarni H, Dyer TD, Göring HHH, Neary JL, Cole SA, Kent JW, Kumar S, Glahn DC, Mahaney MC, Comuzzie AG, Almasy L, Curran JE, Duggirala R, Blangero J, et al. Genome- and epigenome-wide association study of hypertriglyceridemic waist in Mexican American families. Clinical Epigenetics 2016;8(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Elshaer SL, Mohamed IN, Coucha M, Altantawi S, Eldahshan W, Bartasi ML, Shanab AY, Lorys R, El-Remessy AB. Deletion of TXNIP Mitigates High-Fat Diet-Impaired Angiogenesis and Prevents Inflammation in a Mouse Model of Critical Limb Ischemia. Antioxidants 2017;6(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen J, Hui ST, Couto FM, Mungrue IN, Davis DB, Attie AD, Lusis AJ, Davis RA, Shalev A. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta-cell mass and protects against diabetes. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2008;22(10):3581–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lazarević K, Nikolić D, Stosić L, Milutinović S, Videnović J, Bogdanović D. Determination of lead and arsenic in tobacco and cigarettes: an important issue of public health. Central European journal of public health 2012;20(1):62–66. [DOI] [PubMed] [Google Scholar]
- 100.Huang JH, Hu KN, Ilgen J, Ilgen G. Occurrence and stability of inorganic and organic arsenic species in wines, rice wines and beers from Central European market. Food additives & contaminants. Part A, Chemistry, analysis, control, exposure & risk assessment 2012;29(1):85–93. [DOI] [PubMed] [Google Scholar]
- 101.Abhyankar LN, Jones MR, Guallar E, Navas-Acien A. Arsenic exposure and hypertension: a systematic review. Environmental health perspectives 2012;120(4):494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.De Cario R, Sticchi E, Lucarini L, Attanasio M, Nistri S, Marcucci R, Pepe G, Giusti B. Role of TGFBR1 and TGFBR2 genetic variants in Marfan syndrome. Journal of Vascular Surgery 2018;68(1):225–233.e5. [DOI] [PubMed] [Google Scholar]
- 103.Jondeau G, Ropers J, Regalado E, Braverman A, Evangelista A, Teixedo G, De Backer J, Muiño-Mosquera L, Naudion S, Zordan C, Morisaki T, Morisaki H, Von Kodolitsch Y, Dupuis-Girod S, Morris SA, et al. International Registry of Patients Carrying TGFBR1 or TGFBR2 Mutations: Results of the MAC (Montalcino Aortic Consortium). Circulation: Cardiovascular Genetics 2016;9(6):548–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nature Genetics 2005;37(3):275–281. [DOI] [PubMed] [Google Scholar]
- 105.Devaux Y, Bousquenaud M, Rodius S, Marie PY, Maskali F, Zhang L, Azuaje F, Wagner DR. Transforming growth factor β receptor 1 is a new candidate prognostic biomarker after acute myocardial infarction. BMC Medical Genomics 2011;4:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li W, Yue H. Thymidine phosphorylase: A potential new target for treating cardiovascular disease. Trends in Cardiovascular Medicine 2018;28(3):157–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Altaf F, Vesely C, Sheikh AM, Munir R, Shah STA, Tariq A. Modulation of ADAR mRNA expression in patients with congenital heart defects. PLoS ONE 2019;14(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ong WY, Ng MPE, Loke SY, Jin S, Wu YJ, Tanaka K, Wong PTH. Comprehensive Gene Expression Profiling Reveals Synergistic Functional Networks in Cerebral Vessels after Hypertension or Hypercholesterolemia. PLoS ONE 2013;8(7):68335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zeller T, Wild P, Szymczak S, Rotival M, Schillert A, Castagne R, Maouche S, Germain M, Lackner K, Rossmann H, Eleftheriadis M, Sinning CR, Schnabe RB, Lubos E, Mennerich D, et al. Genetics and beyond - the transcriptome of human monocytes and disease susceptibility. PLoS ONE 2010;5(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hays AM, Lantz RC, Rodgers LS, Sollome JJ, Vaillancourt RR, Andrew AS, Hamilton JW, Camenisch TD. Arsenic-induced decreases in the vascular matrix. Toxicologic Pathology 2008;36(6):805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ragni C V, Diguet N, Le Garrec JF, Novotova M, Resende TP, Pop S, Charon N, Guillemot L, Kitasato L, Badouel C, Dufour A, Olivo-Marin JC, Trouvé A, McNeill H, Meilhac SM. Amotl1 mediates sequestration of the Hippo effector Yap1 downstream of Fat4 to restrict heart growth. Nature Communications 2017;8(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li C, Srivastava RK, Elmets CA, Afaq F, Athar M. Arsenic-induced cutaneous hyperplastic lesions are associated with the dysregulation of Yap, a Hippo signaling-related protein. Biochemical and Biophysical Research Communications 2013;438(4):607–612. [DOI] [PubMed] [Google Scholar]
- 113.Negro-Silva LF Makhani K, Lemaire M, Lemarié CA, Plourde D, Bolt AM, Chiavatti C, Bohle DS, Lehoux, Goldberg MS, Mann KK. Sex-Specific Effects of Prenatal and Early Life Inorganic and Methylated Arsenic Exposure on Atherosclerotic Plaque Development and Composition in Adult [Formula: see text] Mice. Environmental health perspectives 2021;129(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yuan Y, Marshall G, Ferreccio C, Steinmaus C, Selvin S, Liaw J, Bates MN, Smith AH. Acute myocardial infarction mortality in comparison with lung and bladder cancer mortality in arsenic-exposed region II of Chile from 1950 to 2000. American Journal of Epidemiology 2007;166(12):1381–1391. [DOI] [PubMed] [Google Scholar]
- 115.D’Agostino RB, Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro JM, Kannel WB. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation 2008;117(6):743–753. [DOI] [PubMed] [Google Scholar]
- 116.Curb JD, Mctiernan A, Heckbert SR, Kooperberg C, Stanford J, Nevitt M, Johnson KC, Proulx-Burns L, Pastore L, Criqui M, Daugherty S. Outcomes ascertainment and adjudication methods in the Women’s Health Initiative. Annals of epidemiology 2003;13(9 Suppl). [DOI] [PubMed] [Google Scholar]
- 117.Levine ME, Hosgood HD, Chen B, Absher D, Assimes T, Horvath S. DNA methylation age of blood predicts future onset of lung cancer in the women’s health initiative. Aging 2015;7(9):690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fortin J-P, Triche TJ, Hansen KD, Hansen KD. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics (Oxford, England) 2017;33(4):558–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Triche TJ, Weisenberger DJ, Van Den Berg D, Laird PW, Siegmund KD. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Research 2013;41(7):e90–e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhou W, Laird PW, Shen H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic acids research 2017;45(4):e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Needhamsen M, Ewing E, Lund H, Gomez-Cabrero D, Harris RA, Kular L, Jagodic M. Usability of human Infinium MethylationEPIC BeadChip for mouse DNA methylation studies. BMC Bioinformatics 2017;18(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article can be shared to external investigators following the procedures established by the Strong Heart Study, available at https://strongheartstudy.org/. Data from the Framingham Heart Study and from the Women’s Health Initiative are available at dbGaP (accession numbers phs000724.v8.p12 and phs000200.v11.p3). Data from the Multi-Ethnic Study of Atherosclerosis are available upon request at TopMed (https://topmed.nhlbi.nih.gov/).