

Abstract
Resistance to EGFR tyrosine kinase inhibitors (TKIs) in non-small cell lung cancers (NSCLCs) with activating EGFR mutations generally involve development of acquired secondary or tertiary EGFR mutations, such as T790M or C797S. However, case reports have demonstrated that actionable receptor tyrosine kinase fusions such as EML4-ALK, CCDC6-RET, and FGFR3-TACC3 can potentially confer resistance to EGFR TKIs. We seeked to identify the prevalence of FGFR3-TACC3 fusion transcripts as resistance mechanism to EGFR TKIs. Hybrid-capture based genomic profiling was performed on FFPE tissue samples and circulating tumor DNA isolated from peripheral whole blood in the course of clinical care. We performed a comprehensive survey of 17,319 clinical NSCLC samples (14,170 adenocarcinomas and 3149 NSCLC not otherwise specified (NOS)) and identified 5 cases of FGFR3-TACC3 containing the intact kinase domain of FGFR3 and the coiled-coil domain of TACC3 emerging after treatment with EGFR TKIs, including one previously reported index case. Of the 4 novel cases of FGFR3-TACC3, one emerged after erlotinib, one after afatinib, one after osimertinib, and one after ASP8273. These 5 cases of FGFR3-TACC3 fusions acquired post-EGFR TKI, while rare, indicate that FGFR3-TACC3 is a recurrent resistance mechanism, which can bypass EGFR blockade by all generations of EGFR TKIs in NSCLC. Routine rebiopsy and genomic profiling using platforms capable of detecting kinase fusions has the potential to inform new therapeutic strategies for patients with EGFR-mutant NSCLC progressing on TKIs.
1. Introduction
The majority of reported acquired resistance mechanisms to EGFR tyrosine kinase inhibitors (TKIs) generally involve mutations in the kinase domain which alter drug binding and/or ATP affinity, including T790M, which mediates resistance to first- and second-generation EGFR TKIs [1,2], and C797S, which mediates resistance to the third-generation EGFR TKI such as osimertinib [3,4]. MET amplification is another recognized resistance mechanism to all generations of EGFR TKIs. Other common resistance mechanisms include bypass pathway activation, via mechanisms such as MET amplification, and small cell transformation, both of which have been observed as resistance mechanism to all generations of EGFR TKIs [1,2,5,6]. Recently, reports have identified known oncogenic receptor tyrosine kinase (RTK) fusions (CCDC6-RET, FGFR3-TACC3, EML4-ALK) emerging after disease progression on EGFR TKIs [7–9]. Hence we performed a survey of the genomic database of Foundation Medicine, Inc for cases with FGFR3-TACC3 fusions that also had co-existing activating EGFR mutations.
2. Materials and methods
Comprehensive genomic profiling was performed on tissue samples using the FoundationOne® assay as previously described [10]. Briefly, DNA was extracted from 40 μM of FFPE sections (generally 10 unstained 4 μM slides), and CGP (Comprehensive Genomic Profiling) was performed on hybridization-captured, adaptor ligation based libraries to a mean coverage depth of >550X for 236 or 315 cancer-related genes plus select introns from 19 or 28 genes frequently rearranged in cancer. Genomic profiling of circulating tumor DNA (ctDNA) using the assay FoundationACT™ assay was performed as described previously [11] where two 10 mL aliquots of peripheral, whole blood were collected in proprietary, stabilizing collection tubes. A double spin protocol was used to isolate plasma and 50–100 ng of ctDNA was extracted to create adapted sequencing libraries prior to hybrid capture and sample-multiplexed sequencing on an Illumina HSQ2500. This ctDNA test covers 62 genes to >3000× unique coverage and employs propriety algorithms to call alterations at low allele frequencies (0.1% for substitutions, 1% for indels and rearrangements, and 20% for copynumber amplifications). Mutant allele frequency (MAF) for the tissue-based assay represents the percentage of DNA obtained from the biopsied tumor which contains the mutation. In the blood, MAF represents the percentage of ctDNA in the blood stream harboring the mutation on the given day and time of specimen procurement. MAF for the tissue assay and ctDNA assays cannot be directly compared. Both assays are validated and performed on similar Clinical Laboratory Improvement Amendments (CLIA)-certified genomic profiling platforms (Foundation Medicine, Inc., Cambridge, MA). Approval for this study, including a waiver of informed consent and a HIPAA waiver of authorization, was obtained from the Western Institutional Review Board (Protocol No. 20152817).
3. Results
We surveyed 17,319 clinical lung cancer samples (14,170 adenocarcinomas and 3149 NSCLC not otherwise specified (NOS)) submitted between August 2012 and March 2017. We identified a total of 5 cases of FGFR3-TACC3 fusions and activating EGFR mutations in the setting of known resistance to EGFR TKIs. No co-occurring FGFR3 fusions and EGFR activating mutations were identified in 2609 cases of lung squamous cell carcinoma. One of the five cases has been reported [8], and here we report the 4 additional cases identified. All fusions involved the complete FGFR3 kinase domain and were predicted to be in frame.
Case 1.
Patient is a 49-old Asian female never-smoker who presented with stage IV adenocarcinoma of the lung. Her tumor was submitted for CGP, which revealed only EGFR exon 19 deletion (ex19del), TP53 Y163C, and RB1 loss (Table 1). She received afatinib and achieved a partial response (PR) by RECIST for 16 months in the primary tumor before appearance of new metastatic nodules (Fig. 1) and eventual progression of her primary tumor. Because of the location and size of the primary tumor and pulmonary nodules (Fig. 1A), a plasma-based circulating tumor DNA (ctDNA) assay was performed as previously described [10] and revealed the original EGFR ex19del as well as an acquired FGFR3-TACC3 fusion (F18; T10) (Fig. 1B). Details of the detected genomic changes pre- and post-EGFR TKI are listed in Table 1. The patient is currently receiving carboplatin/pemetrexed chemotherapy with response after two cycles of chemotherapy (Fig. 1A).
Table 1.
Clinical characteristics and genomic alterations identified in 5 patients with EGFR-driven lung adenocarcinomas with acquired FGFR fusions. (For interpretation of the references to color in this table, the reader is referred to the web version of this article.)
| Case | Age | Gender | Genomic alterations identified pre-most immediate EGFR TKI | Genomic alterations identified post-most immediate EGFR TKI | Interim EGFR targeted therapy (response) |
|---|---|---|---|---|---|
| 1 | 49 | F | EGFR E746_A750del TP53 T163C RB1 loss^ |
EGFR E746_A750del TP53 T163C FGFR3-TACC3 (F18; T10) |
afatinib (PR, 18 months) |
| 2 | 45 | M | EGFR E745_A750del EGFR T790M* CDKN2A/B^loss TP53 C176Y |
EGFR E745_A750del CDKN2A/B loss TP53 C176Y APC truncation exon 15, E1229fs*93 RB1 S618 FGFR3-TACC3 (F18; T7) |
ASP8273 (PR, 8 months) |
| 3 | 66 | F | EGFR exon 19 deletion# | EGFR L747_A750>P FGFR3 amplification FGFR3-TACC3 (F17; T11) |
erlotinib (PR, 11 months) |
| 4 | 74 | M | EGFR E746_A750del EGFR T790M* TP53 V172F |
EGFR E746_A750del TP53 V172F, A86fs*55 FGFR3-TACC3 (F18; T13) |
osimertinib (PR, 4 months) |
| 5** | 65 | F | EGFR L858R, E709K NFKB1A amplification^ NKX2–1 amplification^ ZNF217 amplification^ TP53 V272L |
EGFR L858R TP53 V272L CDKN2A p16 D108IM p14 R122Q FGFR3-TACC3 (F18; T8) |
afatinib+cetuximab (SD, 10 months) |
The presence of T790M in patient 2 and 4 is due to earlier treatment with erlotinib.
Previously published index case [8].
Detected on local testing that did not cover detection of FGFR3 alterations.
Not covered on ctDNA assay. Boxes shaded in green indicate testing was performed using the FoundationOne® tissue-based assay. Boxes shaded in blue indicated testing was performed using the FoundationACT™ ctDNA-based assay.
Fig. 1.
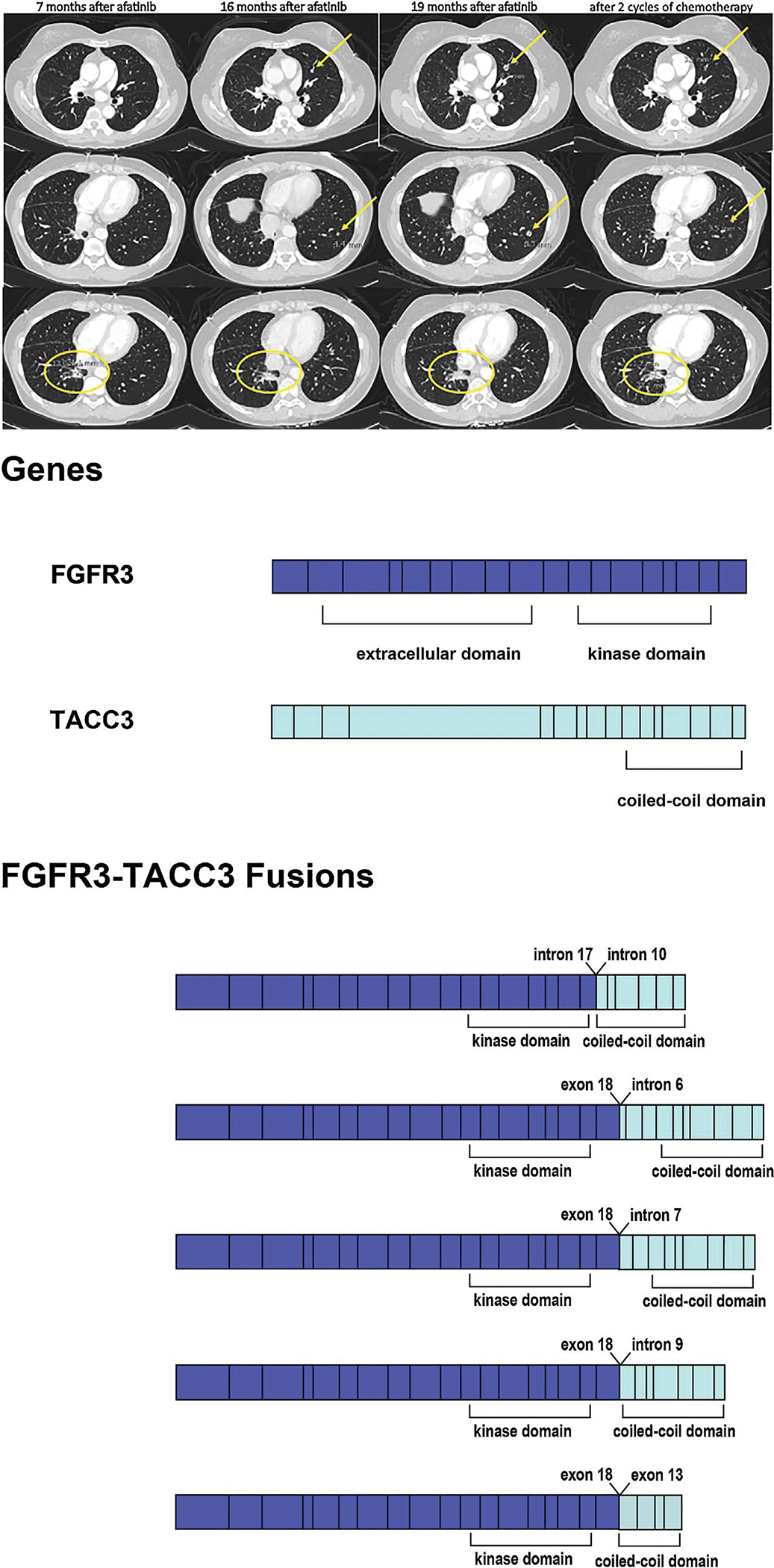
(A) Ememrgence of the resistance pulmonary nodules during afatinib treatment and subsequence response to chemotherapy of patient as described in Case 1. Primary tumor response (yellow circle) and no metastatic pulmonary nodules after 7 months of afatinib treatment. Stable pulmonary primary response (yellow circle) but appearance of new pulmonary nodules (yellow arrows) after 16 months of afatinib treatment. Increase in sizes of both primary tumor (yellow circle) and pulmonary nodules (yellow arrows) after 19 months of afatinib treatment.Primary tumor (yellow circle) and metastatic pulmonary nodules (yellow arrows) response to two cycles of chemotherapy. (B) Schematic of the full length FGFR3 and TACC3 genes and the FGFR3-TACC3 fusion variants identified.
Case 2.
Patient was a 45-year-old male with EGFR ex19del positive stage IV NSCLC who achieved a PR on erlotinib for 8 months. On progression, CGP revealed the original EGFR ex19del and EGFR T790M resistance mutation. The patient was treated on a clinical trial with a third-generation EGFR TKI, ASP8273. After achieving a PR for another 8 months, the patient’s disease again progressed and repeat tumor biopsy was performed. CGP of this sample revealed loss of EGFR T790M mutation, but the emergence of FGFR3-TACC3 (F18; T7) (Fig. 1B) and retention of the original activating EGFR ex19del mutation. Patient received palliative radiation and received osimertinib but passed away 2 months after discontinuation of ASP8273.
Case 3.
Patient was a 66-year-old female diagnosed with NSCLC positive for EGFR ex19del mutation identified by local testing. She was treated with erlotinib and achieved a PR (> 80% tumor shrinkage) for 11 months prior to disease progression. She then received 4 cycles of paclitaxel/carboplatin/bevacizumab followed by maintenance erlotinib with bevacizumab for 7 cycles. Upon subsequent disease progression she received 7 cycles of afatinib with cetuximab and whole brain radiation therapy for asymptomatic brain metastasis. At this time, CGP of a lung tumor biopsied at progression revealed FGFR3 amplification and FGFR3-TACC3 (F17; T11) (Fig. 1B) as well as the primary EGFR ex19del mutation. The patient received gemcitabine and vinorelbine and then docetaxel with some clinical benefit.
Case 4.
Patient is a 74-year-old male diagnosed with lung adenocarcinoma positive for EGFR ex19del by local testing. He was treated with erlotinib with a PR; however, after 5 months of therapy he developed pneumonitis and therapy was discontinued. Post-erlotinib, a blood sample was sent for analysis using a plasma-based ctDNA assay, which revealed EGFR T790M mutation in addition to the EGFR ex19del. Osimertinib was begun and the patient had a PR lasting for approximately 4 months, when he presented with increasing symptoms and worsening disease on imaging. Upon progression on osimertinb a second blood sample was sent for ctDNA testing, and the T790M mutation was not detected; however, an FGFR3-TACC3 (F18; T13) fusion (Fig. 1B) and retention of the original EGFR ex19del mutation was observed. Subsequently, the patient received one dose of paclitaxel, but was hospitalized for symptom management and has now received two doses of nivolumab.
4. Discussion
In addition to the first case report of an FGFR3-TACC3 fusion as potential resistance mechanism to EGFR TKI [8], herein, we report four additional cases of EGFR-mutated NSCLC with FGFR3-TACC3 fusions acquired post EGFR targeted therapy, implicating FGFR3-TACC3 fusion as a rare but recurrent mechanism of acquired resistance to all three generations of EGFR TKIs (erlotinib, afatinib, osimeritnib, ASP8273). In each case, the acquired fusion occurred in the absence of other known mechanisms of resistance, and in 3 of 4 cases a prior sample was assayed on the same or similar platform and was negative for any FGFR alterations including kinase fusion. In this series, each FGFR3-TACC3 fusion results in C-terminal truncation of FGFR3 (breakpoint in intron 17 or exon 18) with a retained kinase domain, fused to TACC3 (breakpoints ranging between intron 6 and exon 13), which retains its coiled-coil dimerization domain (Fig. 1B). In two cases presented in this series T790M was no longer detected post-osimertinib progression and instead the FGFR3-TACC3 fusion emerged as a potential resistance mechanism. FGFR3-TACC3 fusions have been reported in both squamous cell carcinoma [12,13] and adenocarcinoma of the lung [13,14]. Notably, the FGFR3-TACC3 fusion transcript was shown to activate Erk signaling to escape EGFR/ERBB3 blockade in a head and neck squamous cell carcinoma (HNSCC) xenograft model. Daly and colleagues demonstrated during combination antibody blockade of EGFR and ERBB3 in the HNSCC xenograft model that EGFR blockade preferentially inhibited Erk activation while ERBB3 blockade inhibited AKT activation. Upon progression of the xenograft, there was emergence of the FGFR3-TACC3 fusion transcripts detected. Furthermore, introduction of FGFR3-TACC3 conferred resistance to osimertinib inhibition of the NCI-H1975 (EGFR L858R + T790M) cell line, but did not confer resistance to PI3K inhibition of PI3K mutated cell lines [15].
The observation that RTK fusions especially FGFR3-TACC3 can emerge as mechanisms of acquired resistance to all current generations of EGFR TKIs in EGFR-driven NSCLC is relatively new. For patients with FGFR3-TACC3 fusion-positive disease, there are no approved US FDA approved TKIs targeting FGFR, thus severely limiting options of combining FGFR inhibitors and EGFR TKIs. Although patient in case 1 responded to chemotherapy, given the multiple individual cases of acquired resistance presented herein, as well as the potentially prominent role of FGFR3-TACC3 fusions as driver alterations in a diverse group of malignancies, and the recent in vitro experiments suggesting that FGFR3-TACC3 can activate bypass signaling to circumvent EGFR blockade, development and approval of either pan-FGFR or specific FGFR3 inhibitors is urgently needed. This reports highlights the important role of analysis of repeat biopsies and ctDNA using well validated methods capable of sensitive detection of all classes of genomic alterations, including kinase fusions, to identify rare mechanisms of acquired resistance and guide future therapy selection.
Footnotes
Conflict of interest
Sai-Hong Ignatius Ou has received speaking and adviser honorarium from Astra Zeneca and Foundation Medicine Inc. Christine M. Lovy has received honorarium from Novartis, Pfizer, ARIAD, Sequenom, Genoptix. Leora Horn has received honorarium from Abbive, X-covery, BMS, Lilly, Merck, and Roche.
Alexa B. Schrock, Adrienne Johnson, Vincent A. Miller, and Siraj M. Ali are employees of Foundation Medicine, Inc. and own stocks in Foundation Medicine, Inc.
Marcelo Cruz, Davood Vafai, Allison Spradlin, Michael J. Williamson, Ibiayi Dagogo-Jack, and Shirish Gadgeel have nothing to declare.
References
- [1].Sequist LV, Waltman BA, Dias-Santagata D, et al. , Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors, Sci. Transl. Med. 3 (March (75)) (2011) 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yu HA, Arcila ME, Rekhtman N, et al. , Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers, Clin. Cancer Res. 19 (2013) 2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Thress KS, Paweletz CP, Felip E, et al. , Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M, Nat.Med. 21 (2015) 560–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yu HA, Tian SK, Drilon AE, et al. , Acquired resistance of EGFR-mutant lung cancer to a T790M-specific EGFR inhibitor: emergence of a third mutation (C797S) in the EGFR tyrosine kinase domain, JAMA Oncol 1 (2015) 982–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ou SH, Agarwal N, Ali SM, High MET amplification level as a resistance mechanism to osimertinib (AZD9291) in a patient that symptomatically responded to crizotinib treatment post-osimertinib progression, Lung Cancer 98 (2016) 59–61. [DOI] [PubMed] [Google Scholar]
- [6].Chabon JJ, Simmons AD, Lovejoy AF, et al. , Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients, Nat. Commun. 7 (June) (2016) 11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Klempner SJ, Bazhenova LA, Braiteh FS, et al. , Emergence of RET rearrangement co- existing with activated EGFR mutation in EGFR-mutated NSCLC patients who had progressed on first- or second-generation EGFR TKI, Lung Cancer 89 (2015) 357–359. [DOI] [PubMed] [Google Scholar]
- [8].Allen JM, Schrock AB, Erlich RL, et al. , Genomic profiling of circulating tumor DNA in relapsed EGFR-mutated lung adenocarcinoma reveals an acquired FGFR3-TACC3 fusion, Clin. Lung Cancer 22 (December (16)) (2016) 30382–30385 pii: S1525–7304. [DOI] [PubMed] [Google Scholar]
- [9].Liang W, He Q, Chen Y, et al. , Metastatic EML4-ALK fusion detected by circulating DNA genotyping in an EGFR-mutated NSCLC patient and successful management by adding ALK inhibitors: a case report, BMC Cancer 16 (February (62)) (2016) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Frampton GM, Fichtenholtz A, Otto GA, et al. , Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing, Nat. Biotechnol. 31 (2013) 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ou SI, Young L, Schrock AB, et al. , Emergence of preexisting MET Y1230C mutation as a resistance mechanism to crizotinib in NSCLC with MET exon 14 skipping, J. Thorac. Oncol. 12 (2017) 137–140. [DOI] [PubMed] [Google Scholar]
- [12].Kim Y, Hammerman PS, Kim J, et al. , Integrative and comparative genomic analysis of lung squamous cell carcinomas in East Asian patients, J. Clin. Oncol. 32 (2014) 121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang R, Wang L, Li Y, et al. , FGFR1/3 tyrosine kinase fusions define a unique molecular subtype of non-small cell lung cancer, Clin. Cancer Res. 20 (2014) 4107–4114. [DOI] [PubMed] [Google Scholar]
- [14].Capelletti M, Dodge ME, Ercan D, et al. , Identification of recurrent FGFR3-TACC3 fusion oncogenes from lung adenocarcinoma, Clin. Cancer Res. 20 (2014) 6551–6558. [DOI] [PubMed] [Google Scholar]
- [15].Daly C, Castanaro C, Zhang W, et al. , FGFR3-TACC3 fusion proteins act as naturally occurring drivers of tumor resistance by functionally substituting for EGFR/ERK signaling, Oncogene 36 (2017) 471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]