

Abstract
Patients with metastatic melanoma have historically had dismal outcomes. The last several years has seen the emergence of effective immune and targeted therapies for metastatic melanoma. Targeted therapies have primarily impacted the 40–50 % of patients with BRAFV600 mutated melanoma. The remainder of patients with advanced melanoma harbor a wide spectrum of mutations other than BRAFV600 that are associated with unique pathophysiological, prognostic, and therapeutic implications. The treatment of this subset of patients is a challenging problem. In recent years, preclinical and early clinical studies have suggested that inhibitors of mitogen activated protein kinase (MAPK) pathway and parallel signaling networks may have activity in treatment of BRAFV600 wild-type (WT) melanoma. In this review, we will discuss available and developing therapies for BRAF WT patients with metastatic melanoma, particularly focusing on molecular targeted options for various genetically defined melanoma subsets.
Keywords: Melanoma, BRAF wild-type, Mutation, NRAS, MAPK, MEK, Inhibitor, GNAQ, GNA11, Angiogenesis, CDK4, CKIT, NF, Immunotherapy, Ipilimumab, Atypical, Trametinib, Binimetinib, Selumetinib, Bevacizumab
Introduction
Melanoma of the skin constitutes the sixth most common cancer among American men and women, and its incidence continues to rise [1]. While many patients with localized disease have a favorable outcome following complete surgical resection, others present later with either advanced regional or metastatic disease and have historically had dismal outcomes. Standard chemotherapy was associated with a median survival of 6–8 months with objective responses only in the 10–15 % range in metastatic disease [2]. A growing understanding of the biology and pathogenesis of melanoma has led to the discovery of crucial signaling pathways and the developmentofimmuneand targeted therapies,which havedramatically improved the outcomes of patients with metastatic disease. Approximately 40–50 % of advanced melanomas harbor the BRAFV600 mutation and can be successfully treated with selective BRAF and MEK inhibitors, leading to significant prolongation of progression free survival (PFS) and overall survival (OS) [3–5]. However, the remaining 50–60 % of patients with advanced melanoma are BRAF wild-type (BRAF WT) and do not benefit from treatment with BRAF inhibitors. A number of other mutations are present in these BRAF WT tumors, such as NRAS, NF1, MEK1/2, CKIT, and GNAQ/GNA11, and atypical (non-V600) BRAF mutations or BRAF fusions. These other genetically defined cohorts are associated with unique risk factors, pathophysiologic, clinical, and prognostic features, that differ from those related to BRAF tumors. The treatment of this subset of patients without a BRAFV600 mutation is a challenging problem. In this review, we will discuss available targeted and immune therapies for BRAF WT patients with metastatic melanoma, particularly focusing on molecular targeted options for various genetically defined melanoma subsets.
Pathogenesis
Nearly all melanomas, including BRAF WT melanomas, harbor mutations that activate the mitogen activated protein kinase (MAPK) signaling pathway. NRAS, the most frequent non-BRAF oncogene activated in melanoma, belongs to a family of GTP-binding proteins connected with the plasma membrane. RAS proteins exert their influence on a multitude ofcellularfunctions including promotion ofcellgrowth, transformation, and survival. They achieve this by activating an array of downstream signaling pathways, including the RAF/MEK/ERK, MAPK, and the PI3K/AKT pathways.These signaling pathways are crucial to the development and progression of melanoma that cause [6] uncontrolled cell growth, defective apoptosis, angiogenesis, cell migration, invasion, and metastasis [7]. Loss of the tumor suppressor gene neurofibromatosis 1 (NF1) is another frequent event in melanoma and functionally activates MAPK and PI3K/AKT pathway signaling. Other less common events in melanoma causing dysregulated MAPK signaling include mutations in GNAQ, GNA11, BRAF (non-V600), MEK1/2, KRAS, HRAS, and RAF1. Stepwise acquisition of alterations in genes regulating key cellular functions cooperates with these MAPK-activating mutations, including loss of CDKN2A, TP53 mutations, RAC1 mutations, and CCND1 amplifications. Overexpression and/or mutations of various growth factor receptors (MET, EGFR, VEGFR, KIT) also play key roles in melanoma growth and progression. Each of these genetic alterations contributes to melanoma pathogenesis but also may present therapeutic targets. These targets and their frequencies are noted in Fig. 1.
Fig. 1.
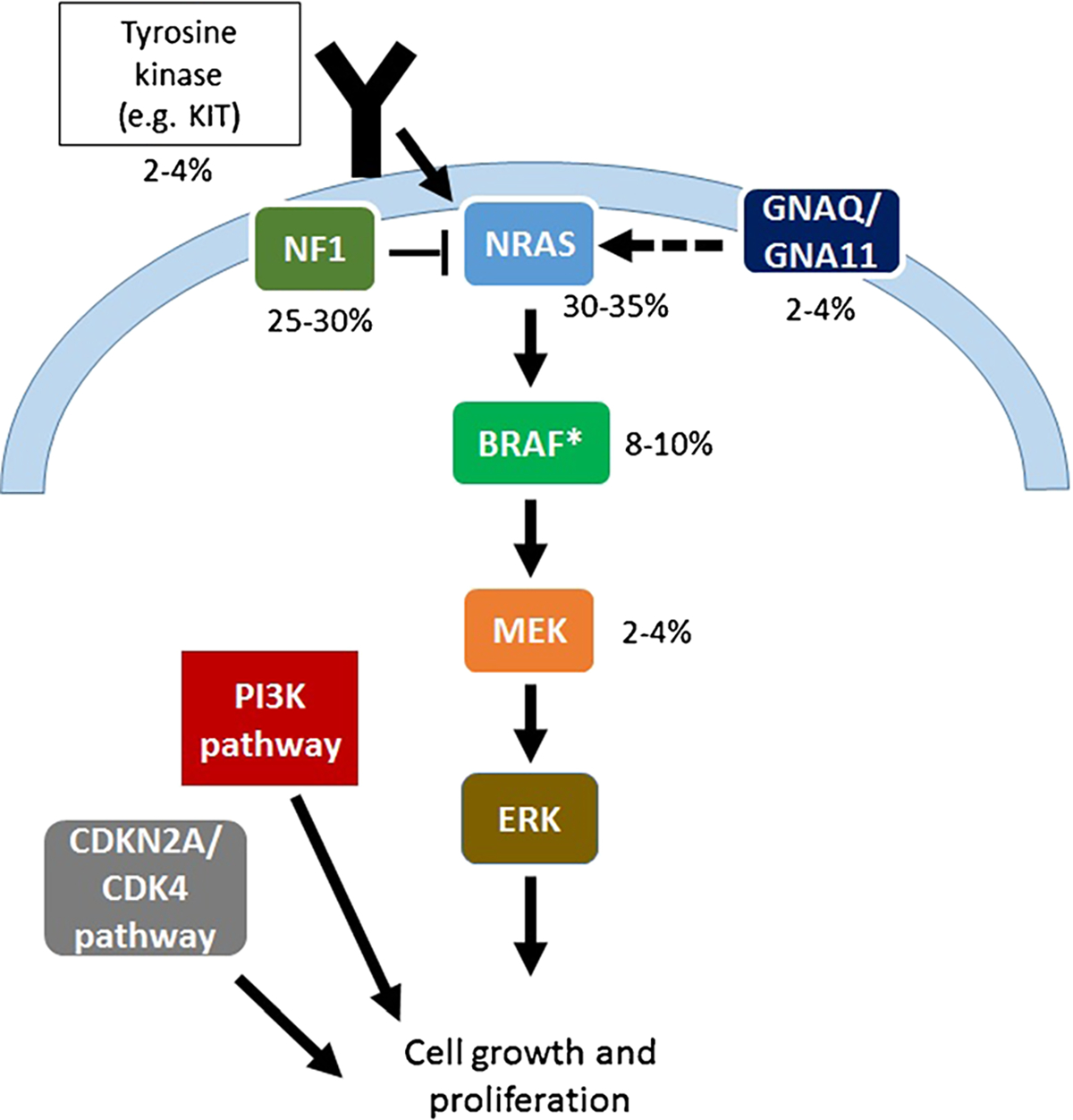
Genetic alterations and their frequency in BRAFV600 wild-type melanoma. The asterisk symbol indicates atypical (non-V600) BRAF mutations and BRAF fusions
Treatment
Immune Therapy
At this time, the current standard of care for patients with BRAF WT melanoma is various immune therapy strategies. In fact, other than cytotoxic chemotherapy, immune therapies are the only approved agents for BRAF WT melanoma. A full review of these incredibly promising treatments is beyond the scope of this review, but we will briefly discuss available agents.
A number of clinical trials have demonstrated the activity of immune therapy in BRAF mutant and BRAF wild-type melanoma. High-dose interleukin-2 (IL-2) has shown durable responses in 5–8 % of patients, albeit with significant toxicities, thus limiting therapy to young and otherwise healthy individuals [8–10]. Ipilimumab is a monoclonal antibody against cytotoxic T lymphocyte antigen-4 (CTLA4), which abrogates T cell inhibition, thereby activating a T cell antitumor response. In a phase III clinical trial, ipilimumab with or without a peptide vaccine versus a peptide vaccine showed improved OS in pretreated patients with metastatic melanoma [11]. Similar results were seen with ipilimumab in combination with dacarbazine versus chemotherapy alone as front-line therapy for metastatic melanoma [12]. Toxicities, including skin rash, colitis, endocrinopathies, and hepatitis, are a reflection of aberrant immune activation by therapy and in some cases can be formidable.
The advent of programmed cell death-1 receptor (PD-1) and ligand (PD-L1) inhibitors has further broadened the therapeutic armamentarium for metastatic melanoma. Treatment with nivolumab and pembrolizumab are accompanied by response rates of 25–45 %, many of which are durable [13–16]. Furthermore, immune-related toxicities occur at a lower rate than with ipilimumab. Pembrolizumab and nivolumab are each FDA-approved for metastatic melanoma and are now commonly used as first-line and salvage therapy for patients failing other treatments. Of note, a pooled analysis of several trials suggests that clinical outcomes are similar in patients with and without BRAF mutations [17]. More recently, ipilimumab and nivolumab in combination demonstrated an impressive 59 % response rate. In September 2015, the FDA-approved nivolumab and ipilimumab for previously untreated metastatic BRAF WT melanoma based on significantly better overall response rate (ORR), duration of response, and PFS compared to ipilimumab alone.
Finally, talimogene laherparepvec, a distinct immunotherapeutic, was approved by the FDA in October 2015. This agent [18] is a modified herpes simplex virus that replicates selectively within tumors and produces granulocyte macrophage colony stimulating factor (GM-CSF) within the tumor microenvironment. This directly injectable agent improved the durable response rate (defined as 6 months of objective response) compared to GM-CSF alone (16.3 vs. 2.1 %, p<0.001). A trend toward improved OS was also observed (median 23.3 vs. 18.9 months, p=0.051).
NRAS Mutation
NRAS mutations are seen in approximately 15–20 % of cutaneous, mucosal, and acral melanoma and are largely mutually exclusive with BRAFV600 mutations [19, 20]. Patients with these mutations are more often older individuals with a longer duration of sun exposure. Their tumors also tend to be thicker with increased mitotic activity and located on the extremities [21]. NRAS mutations portend a worse overall survival [21–23]. Currently, direct targeting of NRAS has not been successful. Single agents and combination treatments targeting downstream effectors of NRAS (MEK, PI3K/mTOR, and cell-cycle-related targets) may hold promise, however.
NRAS-mutant disease may be particularly responsive to immune therapy. In one retrospective study, high-dose IL-2 therapy resulted in a 47 % response rate among a small number of patients [24, 25]. In a more recent study, 229 patients with metastatic melanoma treated with a variety of immune therapies (IL-2, ipilimumab, or anti-PD-1/PD-L1) were compared for clinical outcomes based on their NRAS status. Patients with NRAS mutation had superior response rates with a trend toward improvedPFSandOS.Ofinterest,inthesamestudy,NRAS-mutant tumors seemed to have higher expression of PD-L1, which may contribute to superior responses [25]. Ultimately, prospective validation is needed to confirm these findings.
Direct targeting of NRAS (or other activated RAS proteins) has been challenging. Through downstream activation of MAPK signaling, mutant NRAS drives tumorigenesis and unrestrained tumor growth. Hence, targeted therapy against NRAS has been focused on blocking key downstream molecules, particularly inhibitors of MEK.
Binimetinib (MEK162), a dual MEK1/2 inhibitor, is the first targeted agent to show robust efficacy against NRAS-mutant melanoma. In a phase II trial of patients with NRAS mutation treated with binimetinib, partial responses (PR) were seen in 20 % and stable disease (SD) in 43 %. Median PFS was 3.7 months and was comparable to the patients with BRAF-mutant melanoma [26••]. Toxicities were manageable and comparable to other MEK inhibitors, including acneiform dermatitis, peripheraledema,and diarrhea. Elevated creatinine kinase also occurred in approximately one third of treated patients but was not associated with any clinical morbidity. Based on these promising results, a randomized, phase III, open-label, multicenter, study comparing the efficacy of binimetinib versus dacarbazine in patients with advanced NRAS mutation-positive melanoma is ongoing (NCT01763164).
Other MEK inhibitors (trametinib and RO4987655) have also shown modest clinical efficacy in early studies with NRAS-mutant melanoma [27•, 28]. In two multicenter, phase I trials, patients with NRAS-mutant and WT melanoma (as well as other mutations and malignancies) were treated with MEK inhibitors. These trials demonstrated single agent clinical activity with anobjective response rate of10–20 % and SD in ~20 % of patients in NRAS-mutant and uveal melanoma. Other novel MEK inhibitors with preclinical activity against NRAS-mutant melanoma include GDC-0623, which disrupts feedback activation of MEK by RAF [29•]. Prior MEK inhibitors, by contrast, directly inhibit activated MEK and are more active in BRAF-mutant cancers.MEK inhibitors with differing mechanisms of action may be more effective in RAS-mutant tumors compared to those with BRAF mutations. Currently, GDC-0623 is being tested in a phase I clinical trial.
Results from preclinical and clinical studies of combination therapy incorporating MEK inhibitors are encouraging for further development. CDK pathway dysregulation occurs frequently in melanoma through loss of CDKN2A, CCND1 amplification, and CDK4 mutations [30, 31]. These alterations predict response to CDK4 inhibition in vitro [32, 33]. Inhibition of MEK and CDK4 induces both cell apoptosis and cell cycle arrest, producing superior efficacy compared to MEK inhibition alone [34•]. Two phase I/II trials targeting NRAS mutation with this approach are ongoing with binimetinib+ribociclib (LEE011) (NCT01781572) and trametinib+palbociclib (NCT02065063). Early results from the binimetinib+ribociclib trial demonstrated encouraging clinical efficacy with 33 % ORR and 52 % experiencing SD [35].
Inhibition of PI3K/AKT may also be important in NRAS-mutant melanoma. Preclinical studies have suggested that MEK+PI3K inhibition is superior to MEK+mTOR inhibition, and a combination of MEK and PI3K/mTOR synergistically induces extreme tumor suppression [36, 37]. There are ongoing early phase trials of dual pathway inhibition including the combination of trametinib and an AKT inhibitor (uprosertib; GSK2141795) in BRAF wild-type melanoma (NCT01941927) and binimetinib and various PI3K/AKT pathway inhibitors (NCT01363232, NCT01337765, NCT01449058).
Several other ongoing phases I/II trials with preclinical support in NRAS-mutant and other subsets include studies utilizing inhibitors of Aurora kinase A (MLN8237/alisertib, GSK1070916A), CDK (PD0332991, dinaciclib, LY2835219, BAY1000394, LEE011), and the Notch pathway (RO4929097). XL888 (HSP90 inhibitor), BI-69-A11 (inhibitor of NF-KB and AKT), inhibitors of ERK, RAF-265 (inhibitor of BRAF and VEGFR2), interruption of IQGAP1 (a scaffolding protein in the MAPK pathway), are all potential therapeutic targets for NRAS-mutant melanoma [38, 39].
KIT Mutations
KIT amplifications or mutations are seen in up to 30 % of mucosal, 20 % of acral, and 2 % of melanomas occurring in patients with chronic sun-damaged skin (CSD). BRAF and NRAS mutations, by contrast, are found less frequently in these populations. KIT mutations are heterogeneously distributed across the gene and are most commonly encountered in exon 11 (L576P) and exon 13 (K642E) [40–42]. Although KIT mutations portend an overall poor prognosis compared to KIT wild-type melanomas, specific mutations, particularly in exon 11, confer sensitivity to KIT inhibitors [43]. Clinical efficacy of KIT inhibitors in chronic myelogenous leukemia (CML) [44] and gastrointestinal stromal tumors (GIST) [45] has already been reported.
Several studies in unselected melanoma populations using the KIT inhibitor imatinib as a single agent demonstrated minimal clinical benefit [46]. More recently, three phase II clinical trials of imatinib limited to melanoma patients with KIT alterations (mutations and amplifications) have been conducted [43, 47, 48•]. In one study [47] that enrolled 43 patients, 23 % had a PR and 42 % exhibited tumor regression. The 1-year OS rate was 51 %. Of note, nine of the 10 PRs were recorded in patients with mutations in exons 11 or 13. In another series [48], 24 patients with metastatic mucosal, acral, or CSD melanoma with KIT mutations (33 %), amplifications (46 %), or both (21 %) were treated with imatinib. Best overall response rate was seen in 54 % of patients with KIT mutations versus 0 % in KIT amplified patients. Furthermore, overall disease control (defined as PR or SD) also favored tumors with KIT mutation (77 % versus 18 %). Imatinib therefore can be considered a treatment option for patients with KIT mutations (but not KIT amplifications), particularly in exons 11 and 13. There have been other case reports and studies utilizing various KIT inhibitors, including dasatinib [49], sorafenib [50], sunitinib [51], and nilotinib [52], that reported responses in patients with KIT exon 11 or 13 mutations. Studies also suggest that preexisting NRAS mutations and KIT amplifications may confer intrinsic resistance to KIT inhibition [48•, 51]. There are also ongoing trials exploring the efficacy of combination therapy such as ipilimumab and imatinib.
Atypical BRAF Mutations
As mentioned, discovery of the activating BRAF V600E mutation [19, 53] in approximately 40–50 % of melanomas has revolutionized treatment of metastatic disease by the use of targeted therapy with BRAF inhibitors, leading to improved outcomes [3, 4, 54]. However, identification of less common variant BRAF mutations is crucial, as they confer sensitivity to BRAF and/or MEK inhibitors in many cases. The standard BRAF V600E assay may not detect these alternative mutations, including V600K/R/M/D mutations. The activity of BRAF inhibitors in patients with BRAF V600K and R mutations has now been well-described [55, 56]. In less common V600 variants, activity has also been demonstrated. A 66-year-old female with metastatic melanoma harboring a rare and complex mutation V600DK601del, was treated with vemurafenib with symptomatic improvement and stable disease on imaging [57]. In another case report, a 54-year-old man with a BRAF V600M mutation was treated with combination therapy of the BRAF inhibitor dabrafenib and MEK inhibitor trametinib, resulting in a clinical and radiological response [58].
Other non-V600 BRAF alterations may have clinical relevance,including those at the L597,K601, and G466positions. BRAF fusions have also been recently described. Together, these atypical, non-V-600 BRAF alterations comprise up to 5 % of all melanomas [59]. Critically, preclinical and early clinical data suggest that these alterations confer sensitivity to MEK inhibitors [60••, 61•, 62]. Several case reports have described responses to various MEK inhibitors in patients with BRAF L597 and K601 mutations [63]. Moreover, in vitro data and case reports suggest that BRAF fusions are sensitive to trametinib or sorafenib [64]. Thus, identification of atypical BRAF mutations is important,as more patients may be eligible for targeted therapy and improved clinical outcomes. There have been several case reports demonstrating responses with MEK inhibitors in patients with rare BRAF mutations. A prospective phase II study is ongoing to characterize the activity of trametinib across the spectrum of BRAF non-V600 mutations and fusions (NCT02296112).
NF1 Mutations
Mutations and loss of NF1 also activate MAPK signaling. NF1 mutations are identified in approximately 14 % of all melanomas and up to 70 % of BRAF/NRAS WT [65]. Several preclinical studies suggest that NF1 loss of function mutations or loss confer sensitivity to MEK inhibition [66, 67]. The clinical experience is not quite so clear, however, since the majority of patients with BRAF/NRAS WT melanoma failed to respond to MEK inhibitors [27•]. Still, MEK inhibition in combination with other agents is an attractive strategy for this group. NF1 mutations are also associated with a high overall mutational burden, which may correlate with high response rates to immune therapy as well.
GNAQ and GNA11 Mutations
Uveal melanoma comprises tumors arising from the iris, ciliary body, and choroid. Based on large population studies [68, 69], the 5-year disease-specific survival is approximately 70 %. It is an extremely aggressive tumor, and outcomes have been poor in the metastatic setting secondary to limited therapeutic options [70]. According to the Collaborative Ocular Melanoma Study (COMS), the 10-year overall metastasis rate is 34 %, and among these patients, 80 % will die within 1 year ofdiagnosis ofadvanceddisease [71].Historically, the median OS for patients with metastatic uveal melanoma has ranged from 2 to 12 months [72, 73]. When compared to patients with advanced cutaneous melanoma, those with uveal melanoma are characterized by more systemic symptoms, hepatic dysfunction, and oligometastatic disease [74, 75].
Similar to cutaneous melanoma, uveal melanoma is driven by activation of the MAPK pathway. However, uveal melanoma has a unique mutational profile that is characterized by the absence of BRAF and NRAS mutations, and the presence of activating mutations in the G-protein α-subunits q (GNAQ) and 11 (GNA11) that are identified >80 % of primary uveal melanomas. These mutations are found most frequently at the Q209 position of exon 5, or less often, at the R183 position of exon 4 [76, 77]. However, to date, no approved targeted therapy against these genes is available.
Although the advent of immunotherapy has changed the treatment landscape for metastatic cutaneous melanoma, there is paucity of data for patients with advanced uveal melanoma. In landmark studies of ipilimumab or anti-PD-1, uveal melanoma patients were largely excluded [12]. A multicenter, retrospective analysis of 39 patients with advanced uveal melanoma treated with ipilimumab suggested uncommon durable responses with acceptable side effects [78]. These findings were supported by a phase II clinical trial enrolling patients with advanced uveal melanoma, who were treated with ipilimumab. Results demonstrated favorable outcomes in some patients, with 34 % immune-related disease control, 1-year OS of 31 %, and tolerable sides effects similar to those seen in patients with cutaneous melanoma [79]. Studies evaluating anti-PD-1 with or without ipilimumab are ongoing.
Another area of great interest is targeted therapy, particularly focusing on MEK inhibition. In a randomized, open-label, multicenter phase II clinical trial, 101 patients with either untreated or previously treated metastatic uveal melanoma were randomized to receive either selumetinib, a selective MEK1/2 inhibitor, versus chemotherapy (investigator’s choice of dacarbazine or temozolomide). This study demonstrated improved median PFS in the selumetinib arm of 15.9 versus 7 weeks in the chemotherapy arm (P < 0.001). Objective responses were observed in 14 vs. 0 % in the selumetinib and chemotherapy arms, respectively, and 49 % of patients treated with selumetinib had some tumor regression. Only a trend toward improved overall survival favoring selumetinib was seen (P = 0.09), although crossover from chemotherapy to selumetinib was allowed after progression. Although treatment-related toxicity was noted in 97 % of patients on selumetinib that included rash, visual symptoms, edema, and abnormal creatine kinase, the majority of patients were managed successfully with supportive therapy, and only 6 % discontinued therapy secondary to intolerable side effects [80••]. Preliminary results from the randomized phase III SUMIT trial (NCT01974752) of selumetinib in combination with dacarbazine versus chemotherapy alone for the treatment of patients with metastatic uveal melanoma failed to meet its primary endpoint of PFS. In addition, greater toxicity was seen in the combination arm.
GNAQ/GNA11 mutations activate both MAPK and PI3K/AKT signaling. [81]. Inhibition of both pathways by various combinations has shown synergy in preclinical studies. However, currently, there are no clinical trials testing these combinations. A trial of the mTOR inhibitor everolimus (Rad001) and the somatostatin- receptor-activating peptide pasireotide/SOM232 in advanced uveal melanoma is ongoing (NCT01252251). Protein kinase C (PKC) is also involved in signal transduction from GNAQ to MEK. PKC inhibitors, Enzastaurin and AEB071, have demonstrated activity against uveal melanoma in vitro [82, 83]. Phase I and II clinical trials are studying these findings further [82], (NCT01430416, NCT01801358).
GNAQ/GNA11 mutants found in uveal melanoma promote tumorigenesis by activating YAP [84, 85]. Mutant GNAQ/GNA11, but not wild-type GNAQ/GNA11, trigger dephosphorylation and nuclear localization of YAP, associated with YAP-dependent transcription. Pharmacologic targeting of this novel YAP-dependent pathway may be critical for effective therapy against GNAQ/GNA11 mutant cancers.
Angiogenesis Inhibitors
Bevacizumab is a monoclonal antibody targeted at the vascular endothelial growth factor (VEGF) receptor. In many different tumor types, including advanced colorectal, breast, and non-small-cell lung cancer, the combination of standard chemotherapy plus bevacizumab conferred favorable outcomes [86–88]. Overexpression of VEGF receptor has been identified in melanoma [89]. Preclinical studies have demonstrated that VEGF results in the growth and maintenance of melanoma, while anti-VEGF therapy leads to the inhibition of melanoma cell growth [90, 91]. Other data have shown a direct correlation between tumor VEGF concentration and tumor survival [92].
The clinical utility of adding anti-VEGF therapy to chemotherapy was studied in a single-arm multicenter phase II trial in which 62 patients with previously untreated melanoma were treated with temozolomide and bevacizumab until disease progression or intolerable side effects. The primary end point of disease stabilization rate at 12 weeks (DSR12), defined as complete response (CR), PR, or SD, was found in 52 % of patients. A statistically significant improvement in both median PFS and OS was found, favoring the BRAF WT group. Grade 3–4 toxicity was seen in 32 % of all patients and included thrombocytopenia, neutropenia, hypertension, fatigue, and hemorrhage [93]. The BEAM trial, a phase II, randomized, multicenter trial, evaluated PFS and OS in 214 patients with previously untreated metastatic melanoma treated with carboplatin and paclitaxel with or without bevacizumab. Although this trial did not achieve its primary endpoint, in the absence of any phase III data, the study suggests modest efficacy of VEGF inhibitors and chemotherapy in advanced melanoma. The BEAM trial demonstrated numerically greater but generally not statistically significant improvements in ORR, PFS, and OS at most analysis time points [94].
Given equivocal results of concurrent chemotherapy and VEGF inhibition, a prospective, single-arm, phase II trial enrolled 38 previously treated patients with advanced melanoma to receive sequential therapy with axitinib, a potent VEGF inhibitor, followed by carboplatin plus paclitaxel every 3 weeks. The median PFS was 8.7 months, the median OS was 14.0 months, 22 % had a PR, and 55 % had SD. Therapy was well tolerated. BRAF WT patients did significantly better than those with BRAF mutation [95]. Most current development approaches for angiogenesis inhibitors is proceeding in combination with immune therapy.
Other Agents
Glembatumumab vedotin is an antibody-drug conjugate that has shown efficacy in advanced melanoma and triple negative breast cancer. This agent is a monoclonal antibody to glycoprotein NMB (gpNMB) linked to a microtubule inhibitor (monomethyl auristatin E). Many melanomas (including uveal melanomas) overexpress gpNMB, a molecule that has been linked to poor prognosis in several cancers [96, 97]. Glembatumumab [98•] was evaluated in a phase I/II study and produced a 15 % ORR with an additional 24 % stable disease rate at the recommended phase II dose level. Although treatment-related toxicities (including rash, neutropenia, and fatigue) were manageable overall, there were three treatment-related deaths from sepsis, renal failure, and toxic epidermal necrolysis at doses above the maximum-tolerated dose (MTD). A phase II study is currently ongoing to determine efficacy and whether gpNMB overexpression is a biomarker of response.
Conclusion
The landscape of melanoma therapy is rapidly advancing to include numerouseffective immuneand targeted therapies.As 50–60 % of patients harbor mutations other than BRAFV600 and are associated with various unique pathophysiological, prognostic, and therapeutic implications, it remains critical to capture this cohort of patients in clinical trials to determine effective treatment options. Elucidating biomarkers predictive of response to therapy and determining the best combination and/or sequence of therapies will be crucial in further improvement of treatment and outcomes in these patients.
Footnotes
Compliance with Ethical Standards
Conflict of Interest Romany A. N. Johnpulle declares that she has no conflict of interest.
Douglas B. Johnson has received compensation from Genoptix and Bristol-Myers Squibb for service on advisory boards.
Jeffrey A. Sosman has received compensation from Merck for service as a consultant, and has received research funding from Bristol-Myers Squibb and Novartis.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Howlader N SEER cancer statistics review, 1975–2012. Bethesda: National Cancer Institute; 2015. Available from: http://seer.cancer.gov/csr/1975_2012/. [Google Scholar]
- 2.Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. N Engl J Med. 2004;351(10):998–1012. [DOI] [PubMed] [Google Scholar]
- 3.Chapman PB et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hauschild A et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65. [DOI] [PubMed] [Google Scholar]
- 5.Flaherty KT et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–14. [DOI] [PubMed] [Google Scholar]
- 6.Exploring the pathway: the RAS/RAF/MEK/ERK pathway in cancer: combination therapies and overcoming feedback in ASCO Daily News. 2015.
- 7.Platz A et al. Human cutaneous melanoma; a review of NRAS and BRAF mutation frequencies in relation to histogenetic subclass and body site. Mol Oncol. 2008;1(4):395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenberg SA et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271(12):907–13. [PubMed] [Google Scholar]
- 9.Atkins MB et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105–16. [DOI] [PubMed] [Google Scholar]
- 10.Schwartzentruber DJ. Guidelines for the safe administration of high-dose interleukin-2. J Immunother. 2001;24(4):287–93. [DOI] [PubMed] [Google Scholar]
- 11.Hodi FS et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robert C et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–26. [DOI] [PubMed] [Google Scholar]
- 13.Topalian SL et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamid O et al. Safety and tumor responses with lambrolizumab (Anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robert C et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384(9948):1109–17. [DOI] [PubMed] [Google Scholar]
- 16.Weber J Phase I/II trial of PD-1 antibody nivolumab with peptide vaccine in patients naive to or that failed ipilimumab, in ASCO Annual Meeting. 2013. [Google Scholar]
- 17.Larkin J et al. Efficacy and safety of nivolumab in patients with BRAF V600 mutant and BRAF wild-type advanced melanoma: a pooled analysis of 4 clinical trials. JAMA Oncol. 2015;1(4):433–40. [DOI] [PubMed] [Google Scholar]
- 18.Andtbacka RH et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–8. [DOI] [PubMed] [Google Scholar]
- 19.Curtin JA et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353(20):2135–47. [DOI] [PubMed] [Google Scholar]
- 20.Lovly CM et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS One. 2012;7(4):e35309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Devitt B et al. Clinical outcome and pathological featuresassociated with NRAS mutation in cutaneous melanoma. Pigment Cell Melanoma Res. 2011;24(4):666–72. [DOI] [PubMed] [Google Scholar]
- 22.Jakob JA et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118(16):4014–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carlino MS et al. Correlation of BRAF and NRAS mutation status with outcome, site of distant metastasis and response to chemotherapy in metastatic melanoma. Br J Cancer. 2014;111(2):292–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joseph RW et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother. 2012;35(1):66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson DB et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res. 2015;3(3):288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ascierto PA et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14(3):249–56. •• This isthe first study to identifyanactive targeted therapy for NRAS mutant melanoma.
- 27. Falchook GS et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):782–9. • This study describes the activity of trametinib in a small number of other melanomas outside of the BRAF V600 mutant cohort.
- 28.Zimmer L et al. Phase I expansion and pharmacodynamic study of the oral MEK inhibitor RO4987655 (CH4987655) in selected patients with advanced cancer with RAS-RAF mutations. Clin Cancer Res. 2014;20(16):4251–61. [DOI] [PubMed] [Google Scholar]
- 29. Hatzivassiliou G et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013;501(7466):232–6. • This pre-clinical study supports the use of MEK inhibitors in various RAS mutant cancers.
- 30.Hodis E et al. A landscape of driver mutations in melanoma. Cell. 2012;150(2):251–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krauthammer M et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44(9):1006–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sheppard KE, McArthur GA. The cell-cycle regulator CDK4: an emerging therapeutic target in melanoma. Clin Cancer Res. 2013;19(19):5320–8. [DOI] [PubMed] [Google Scholar]
- 33.Young RJ et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991inmelanoma cell lines. PigmentCell Melanoma Res. 2014;27(4):590–600. [DOI] [PubMed] [Google Scholar]
- 34. Kwong LN et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18(10):1503–10. • This preclinical study provides the rationale for combining MEK and CDK4/6 inhibition in NRAS mutant melanoma.
- 35.Sosman J A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: early encouraging clinical activity, in ASCO Annual Meeting. 2014. [Google Scholar]
- 36.Roberts PJ et al. Combined PI3K/mTOR and MEK inhibition provides broad antitumor activity in faithful murine cancer models. Clin Cancer Res. 2012;18(19):5290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Posch C et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc Natl Acad Sci U S A. 2013;110(10):4015–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fedorenko IV et al. Beyond BRAF: where next for melanoma therapy? Br J Cancer. 2015;112(2):217–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson DB, Puzanov I. Treatment of NRAS-mutant melanoma. Curr Treat Options Oncol. 2015;16(4):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Curtin JA et al. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24(26):4340–6. [DOI] [PubMed] [Google Scholar]
- 41.Beadling C et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008;14(21):6821–8. [DOI] [PubMed] [Google Scholar]
- 42.Slipicevic A, Herlyn M. KIT in melanoma: many shades of gray. J Investig Dermatol. 2015;135(2):337–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carvajal RD et al. KITas a therapeutic target in metastatic melanoma. JAMA. 2011;305(22):2327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Druker BJ et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–7. [DOI] [PubMed] [Google Scholar]
- 45.Demetri GD et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–80. [DOI] [PubMed] [Google Scholar]
- 46.Wyman K et al. Multicenter phase II trial of high-dose imatinib mesylate in metastatic melanoma: significant toxicity with no clinical efficacy. Cancer. 2006;106(9):2005–11. [DOI] [PubMed] [Google Scholar]
- 47.Guo J et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29(21):2904–9. [DOI] [PubMed] [Google Scholar]
- 48.•. Hodi FS et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin.J ClinOncol.2013;31(26):3182–90. • Thisphase II study demonstrates the activity of imatinib in KIT-altered subsets of melanoma.
- 49.Woodman SE et al. Activity of dasatinib against L576P KIT mutant melanoma: molecular, cellular, and clinical correlates. Mol Cancer Ther. 2009;8(8):2079–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quintas-Cardama A et al. Complete response of stage IVanal mucosal melanoma expressing KIT Val560Asp to the multikinase inhibitor sorafenib. Nat Clin Pract Oncol. 2008;5(12):737–40. [DOI] [PubMed] [Google Scholar]
- 51.Minor DR et al. Sunitinib therapy for melanoma patients with KIT mutations. Clin Cancer Res. 2012;18(5):1457–63. [DOI] [PubMed] [Google Scholar]
- 52.Carvajal RD et al. PhaseII study ofnilotinibinmelanomaharboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res. 2015;21(10):2289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Houben R et al. Constitutive activation of the Ras-Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J Carcinog. 2004;3(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Long GV et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239–46. [DOI] [PubMed] [Google Scholar]
- 55.Klein O et al. BRAF inhibitor activity in V600R metastatic melanoma. Eur J Cancer. 2013;49(5):1073–9. [DOI] [PubMed] [Google Scholar]
- 56.McArthur GA et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15(3):323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trudel S et al. The clinical response to vemurafenibina patient with a rare BRAFV600DK601del mutation-positive melanoma. BMC Cancer. 2014;14:727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parakh S et al. Response to MAPK pathway inhibitors in BRAF V600M-mutated metastatic melanoma. J Clin Pharm Ther. 2015;40(1):121–3. [DOI] [PubMed] [Google Scholar]
- 59.Greaves WO et al. Frequency and spectrumofBRAFmutationsina retrospective, single-institution study of 1112 cases of melanoma. J Mol Diagn. 2013;15(2):220–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dahlman KB et al. BRAF(L597) mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer Discov. 2012;2(9):791–7. •• This study showed the dramatic activity of MEK inhibition in a patient with BRAF L597 mutated melanoma.
- 61. Menzies AM et al. Clinical activity of the MEK inhibitor trametinib in metastatic melanoma containing BRAF kinase fusion. Pigment Cell Melanoma Res. 2015;28(5):607–10. • This study shows the activity of trametinib in patients with BRAF fusions.
- 62.Bowyer SE et al. Activity of trametinib in K601E and L597Q BRAF mutation-positive metastatic melanoma. Melanoma Res. 2014;24(5):504–8. [DOI] [PubMed] [Google Scholar]
- 63.Kim KB et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31(4):482–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hutchinson KE et al. BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clin Cancer Res. 2013;19(24):6696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Genomic classification of cutaneous melanoma. Cell. 2015;161(7):1681–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nissan MH et al. Loss of NF1 in Cutaneous Melanoma Is Associated with RAS Activation and MEK Dependence. Cancer Res. 2014;74(8):2340–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ranzani M et al. BRAF/NRAS wild-type melanoma, NF1 status and sensitivity to trametinib. Pigment Cell Melanoma Res. 2015;28(1):117–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bishop KD, Olszewski AJ. Epidemiology and survival outcomes of ocular and mucosal melanomas: a population-based analysis. Int J Cancer. 2014;134(12):2961–71. [DOI] [PubMed] [Google Scholar]
- 69.Chang AE, Karnell LH, Menck HR. The National Cancer Data Base report on cutaneous and noncutaneous melanoma: a summary of 84,836 cases from the past decade. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer. 1998;83(8):1664–78. [DOI] [PubMed] [Google Scholar]
- 70.Gragoudas ES et al. Survival of patients with metastases from uveal melanoma. Ophthalmology. 1991;98(3):383–9. discussion 390. [DOI] [PubMed] [Google Scholar]
- 71.Diener-West M et al. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative Ocular Melanoma Study Group Report No. 26. Arch Ophthalmol. 2005;123(12):1639–43. [DOI] [PubMed] [Google Scholar]
- 72.Rietschel P et al. Variates of survival in metastatic uveal melanoma. J Clin Oncol. 2005;23(31):8076–80. [DOI] [PubMed] [Google Scholar]
- 73.Augsburger JJ, Correa ZM, Shaikh AH. Effectiveness of treatments for metastatic uveal melanoma. Am J Ophthalmol. 2009;148(1):119–27. [DOI] [PubMed] [Google Scholar]
- 74.Albert DM, Ryan LM, Borden EC. Metastatic ocular and cutaneous melanoma: a comparison of patient characteristics and prognosis. Arch Ophthalmol. 1996;114(1):107–8. [PubMed] [Google Scholar]
- 75.Singh AD, Bergman L, Seregard S. Uveal melanoma: epidemiologic aspects. Ophthalmol Clin N Am. 2005;18(1):75–84. viii. [DOI] [PubMed] [Google Scholar]
- 76.Van Raamsdonk CD et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457(7229):599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.VanRaamsdonk CD et al. Mutations inGNA11 inuvealmelanoma. N Engl J Med. 2010;363(23):2191–9.21083380 [Google Scholar]
- 78.Luke JJ et al. Clinical activity of ipilimumab for metastatic uveal melanoma: a retrospective review of the Dana-Farber Cancer Institute, Massachusetts General Hospital, Memorial SloanKettering Cancer Center, and University Hospital of Lausanne experience. Cancer. 2013;119(20):3687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maio M et al. Efficacy and safety of ipilimumab in patients with pre-treated, uveal melanoma. Ann Oncol. 2013;24(11):2911–5. [DOI] [PubMed] [Google Scholar]
- 80. Carvajal RD et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA. 2014;311(23):2397–405. •• This is the first study to show improved clinical outcomes in uveal melanoma.
- 81.Ho AL et al. Impact of combined mTOR and MEK inhibition in uveal melanoma is driven by tumor genotype. PLoS One. 2012;7(7):e40439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shtivelman E et al. Pathways and therapeutic targets in melanoma. Oncotarget. 2014;5(7):1701–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu X et al. The protein kinase C inhibitor enzastaurin exhibits antitumor activity against uveal melanoma. PLoS One. 2012;7(1):e29622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yu FX et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell. 2014;25(6):822–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Feng X et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell. 2014;25(6):831–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hurwitz H et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–42. [DOI] [PubMed] [Google Scholar]
- 87.Miller KD. E2100: a phase III trial of paclitaxel versus paclitaxel/ bevacizumab for metastatic breast cancer. Clin Breast Cancer. 2003;3(6):421–2. [DOI] [PubMed] [Google Scholar]
- 88.Sandler A et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355(24):2542–50. [DOI] [PubMed] [Google Scholar]
- 89.Brychtova S et al. The role of vascular endothelial growth factors and their receptors in malignant melanomas. Neoplasma. 2008;55(4):273–9. [PubMed] [Google Scholar]
- 90.Liu B et al. Melanoma cell lines express VEGF receptor KDR and respond to exogenously added VEGF. Biochem Biophys Res Commun. 1995;217(3):721–7. [DOI] [PubMed] [Google Scholar]
- 91.Danielsen T, Rofstad EK. VEGF, bFGF and EGF in the angiogenesis of human melanoma xenografts. Int J Cancer. 1998;76(6):836–41. [DOI] [PubMed] [Google Scholar]
- 92.Ugurel S et al. Increased serum concentration of angiogenic factors in malignant melanoma patients correlates with tumor progression and survival. J Clin Oncol. 2001;19(2):577–83. [DOI] [PubMed] [Google Scholar]
- 93.von Moos R et al. First-line temozolomide combined with bevacizumab in metastatic melanoma: a multicentre phase II trial (SAKK 50/07). Ann Oncol. 2012;23(2):531–6. [DOI] [PubMed] [Google Scholar]
- 94.Kim KB et al. BEAM: a randomized phase II study evaluating the activity of bevacizumab in combination with carboplatin plus paclitaxel in patients with previously untreated advanced melanoma. J Clin Oncol. 2012;30(1):34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Algazi AP et al. The combination of axitinib followed bypaclitaxel/carboplatin yields extended survival in advanced BRAF wild-type melanoma: results of a clinical/correlative prospective phase II clinical trial. Br J Cancer. 2015;112(8):1326–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rose AA et al. Glycoprotein nonmetastatic B is an independent prognostic indicator of recurrence and a novel therapeutic target in breast cancer. Clin Cancer Res. 2010;16(7):2147–56. [DOI] [PubMed] [Google Scholar]
- 97.Williams MD et al. GPNMB expression in uveal melanoma: a potential for targeted therapy. Melanoma Res. 2010;20(3):184–90. [DOI] [PubMed] [Google Scholar]
- 98. Ott PA et al. Phase I/II study of the antibody-drug conjugate glembatumumab vedotin in patients with advanced melanoma. J Clin Oncol. 2014;32(32):3659–66. • This study shows activity of an antibody-drug conjugate in melanoma.