

Abstract

Here, we present an intriguing photoinduced chloroamination cyclization of allenes bearing a tethered sulfonylamido group to afford 2-(1-chlorovinyl)pyrrolidines and related heterocycles in the presence of N-chlorosuccinimide (NCS) as the chlorine source. An in depth experimental and computational mechanistic study revealed the existence of multiple reaction pathways leading to a common nitrogen centered radical (NCR). This key NCR can be, in fact, originated from (a) the oxidation of the deprotonated allene by the photoexcited state of the Ru-catalyst and (b) the photodissociation of the in situ formed N-chloroallene. The NCR formation triggers an intramolecular cyclization to a highly reactive pyrrolidine vinyl radical, which upon chlorination delivers the final product. Thus, NCS plays a dual role, serving both as an activator of the sulfonamido functionality and as the chlorinating agent.
Introduction
Alkenyl chlorides are ubiquitous across natural products,1,2 active pharmaceutical ingredients,3 and agrochemicals.4 In addition, they are relevant intermediates in the preparation of complex molecules and plastics through transition-metal catalyzed and radical reactions. Compared to heavier halides, only a limited number of known reactions deliver alkenyl chlorides in high yields and stereoselectivity. Such methods usually rely on the functionalization of benchmark chlorinated substrates (e.g., 1,1- and 1,2-dichloroethene) by Suzuki coupling5 and olefin metathesis,6 or on the copper-catalyzed retro-Finkelstein reaction which requires high temperatures and long reaction times.7−9 Other methods, like Hunsdiecker-type transformations, are restricted to α,β-unsaturated compounds.10−13 An alternative approach is the chlorination of alkynes and allenes. The strategy presents higher generality and, most importantly, allows the hetero 1,2-difunctionalization, which rapidly increases the molecular complexity of the product and opens to downstream transformations. Alkynes have extensively been used in hydrochlorination,14−16 chlorosulfonylation,17−19 and chloroamination20−22 (Scheme 1A), while the addition of chlorine to allenes is still underexplored (Scheme 1B). Dichlorination was observed using different chlorine donors (TMSCl, oxalyl chloride) and strong oxidizers, like KMnO4,23 and Selectfluor.24 The dichlorination of propadiene was accomplished using Cl2 in molten NaAlCl4–KAlCl4 eutectic at 140 °C.25 In 2018, Murphy reported the radical 1,2-dichlorination of aryllallenes in refluxing acetonitrile (CH3CN) with a chlorinated hypervalent iodine reagent, producing E/Z mixtures of vinyl chlorides. Even fewer examples of vicinal hetero-difunctionalizations are reported. Ma developed a regio- and stereoselective chlorohydroxylation of 1,2-allenyl phenylsulfoxides with stoichiometric CuCl2 and silica gel under ball milling.26 In 2020 Schomaker studied the addition of amidyl radicals to allenes producing N-heterocycles (Scheme 1C, middle).27 In this report, only in one example was N-chlorosuccinimide (NCS) utilized to quench the vinyl radical intermediate, affording the corresponding alkenyl chloride. The sole previous report of allene chloroamination is from 1967, by Neale, who published a study on the radical addition of dialkyl N-chloramines to 1,3-dienes, olefins, acetylenes, and allenes (Scheme 1C, top). A mixture of N-chloramine and allene in sulfuric and acetic acids generated the corresponding product via a free-radical chain mechanism, by the use of a Fe(II) catalyst or UV irradiation.28
Scheme 1. (A,B) General Strategies for the 1,2-Functionalization of Alkynes and Allenes; (C) Chloroamination of Allenes, Previous and Current Approaches.

Despite being essentially unexplored, the radical intramolecular chloroamination of allenes is an intriguing approach to provide chlorovinyl N-heterocycles. Here, we describe a photochemical chloroamino cyclization of allenes bearing a tethered sulfonylamido group to access 2-(1-chlorovinyl)pyrrolidines and related heterocycles (Scheme 1C, bottom). Such an approach allows both access to small saturated heterocycles, the presence of which is widespread in drugs, natural alkaloids, and organocatalysts, and the concomitant installation of a chloroalkenyl moiety, which can be further exploited for the lateral functionalization of the obtained molecules.
Results and Discussion
Since our previous studies on the reactivity of neutral nitrogen-centered radicals generated from sulfonylhydrazones through photoredox catalysis,29,30 we became interested in other precursors of open-shell intermediates suitable for domino cyclization processes. Recently, we investigated the beneficial effect of blue-light irradiation on a Pd(0) catalyzed reaction between aryl bromides and allenyltosyl amides, thus developing a room temperature Heck reaction to access 2-(1-arylvinyl)pyrrolidines and piperidines.31 Similar substrates have already been subjected to haloamination with a brominating reagent (LiBr, NBS or 1-bromopyrrolidin-2-one) in combination with transition metal catalysis,32−34 or with iodine alone.35 As demonstrated by the recent blooming of chlorination protocols exploiting photoredox catalysis,36 we envisioned that a light mediated process could have been an ideal approach to tackle this chloroamino cyclization. Therefore, we selected a tosyl protected allenylamine (1a) as our model substrate. The feasibility of the reaction was tested reacting N-tosylhexa-4,5-dien-1-ylamine 1a with 1 equiv of NCS in the presence of 5% mol of [Ru(bpy)3]Cl2 as the photocatalyst, 1 equiv of K2CO3 as the base in CH3CN under irradiation with a 40W blue LED. As shown in Table 1 entry 2, the desired chlorovinylpyrrolidine 2a was obtained in 33% yield. The design of the reaction condition was inspired by a paper of Leonori and co-workers, which described the NCS-promoted chlorination of primary and secondary amines providing the corresponding aminium NCR.37 This latter was generated exploiting the combination of both acidic and photoredox conditions. In our opinion, the substitution of the amine, with a sulfonamide as the starting material and the acid with a base, would unveil a dual role for NCS. Indeed, it could serve both as an initiator of the sulfonamidic nitrogen reactivity and as a chlorinating agent of the double bond. To optimize the reaction conditions, the influence of the solvent, base, and photocatalyst was investigated (Table 1). It should be underlined that, according to the reaction conditions, a certain amount of N-chloro-N-tosylhexa-4,5-dien-1-ylamine 3a was formed. Moreover, the tricyclic compound 2a′ was identified as byproduct and never produced with yields higher than 8% (see SI for full screening and characterization). The role of this species will be clarified in the reaction mechanism study. An extensive screening of the photocatalyst was performed, testing both metal complexes and fully organic compounds (see SI). [Acr-Mes]+(BF4)− was revealed to be less efficient in CH3CN than [Ru(bpy)3]Cl2 affording pyrrolidine 2a in 29% (Table 1, entry 3). It must be noticed that in this case the N-chlorinated allene 3a was obtained in 14% yield. As shown in Table 1, entry 4, the usage of [Ru(bpy)3](PF6)2 was beneficial for the reaction outcome, causing an increase in yield up to 43%. Probably, this can be ascribed to the counterion PF6–, which increases the catalyst solubility.
Table 1. Screening and Optimization of the Reaction Conditionsa.

| entry | deviation | 2a [%]b | 3a [%]c | byproduct [%]c |
|---|---|---|---|---|
| 1 | none | 56 | 0 | 4 |
| 2 | [Ru(bpy)3]Cl2 (5% mol), K2CO3 (1.0 equiv) in CH3CN | 33 | 0 | 2 |
| 3 | [Acr-Mes]+(BF4)− (5% mol), K2CO3 (1.0 equiv) in CH3CN | 29 | 14 | <1 |
| 4 | [Ru(bpy)3](PF6)2 (5% mol), K2CO3 (1.0 equiv) in CH3CN | 43 | 0 | 4 |
| 5 | KOH (1.0 equiv) in CH3CN | 29 | 0 | 1 |
| 6 | Cs2CO3 (1.0 equiv) in CH3CN | 27 | 0 | 3 |
| 7 | 2,6-lutidine (1.0 equiv) in CH3CN | 10 | 3 | <1 |
| 8 | K2CO3 (1.0 equiv) in PhCH3 | 47 | 0 | 5 |
| 9 | K2CO3 (1.0 equiv) in CHCl3 or DMF | 28 | 0 | 2/not obs |
| 10 | K2CO3 (1.0 equiv) in acetone | 43 | 0 | 2 |
| 11 | K2CO3 (1.0 equiv) in HCO2Me | 31 | 0 | 4 |
| 12 | K2CO3 (1.0 equiv) in PhCH3:CH3CN 3:1 | 46 | 0 | 4 |
| 13 | K2CO3 (0.5 equiv) | 52 | 0 | 4 |
| 14 | NCS (2 equiv) | 45 | 6 | 6 |
| 15 | NCS (0.5 equiv) | 10 | 0 | 4 |
| 16 | no base or NCS | 0 | 0 | 0 |
| 17 | no irradiation | 0 | 82 | 0 |
| 18 | no catalyst | 11 | 66 | <1 |
Reaction conditions: 1a (0.20 mmol), base, and catalyst as indicated, anhydrous solvent(s) (4 mL total) under irradiation with 456 nm Kessil blue LED. See SI for the complete screening.
Yield determined on isolated products.
Yield determined after 2 repetitions with 1H NMR using dichloroethane and nitromethane as internal standards.
Subsequently, the effect of the base was studied. Neither a stronger base such KOH nor organic bases such 2,6-lutidine resulted beneficial for the reaction outcome. In all cases yields below 30% were observed (Table 1, entries 5 and 7, see also SI). Also the change of the cation was not advantageous; for example, Cs2CO3 produced the chlorivinylpirrolidine 2a in only 27% (Table 1, entry 6). When the solvent was studied, only the apolar toluene demonstrated to be slightly more efficient than CH3CN raising the yield to 47% (Table 1, entry 8). Acetone performed as CH3CN (43%, Table 1, entry 10), whereas CHCl3 (28%, entry 9), DMF (28%, entry 9), and methyl formate (31%, entry 11) afforded the vinylpirrolidine 2a in lower yields. Anyway, the apolar nature of toluene did not allow the catalyst to be completely solubilized, so different solvent mixtures were tested to increase the polarity of the reaction medium and, at the same time, to exploit the advantages offered by the use of toluene. So, a mixture of toluene/CH3CN 3:1 was tested at first and a 46% yield of 2a was observed (Table 1, entry 12). On the contrary, the use of toluene/methylformate in ratio 3:1 ensured the recovery of product 2a in 56% yield (Table 1, entry 15). Interestingly, when the equivalents of the base were reduced to a substoichiometric amount, we observed comparable yields; in fact, pyrrolidine 2a was obtained in 52% when 0.5 equiv of K2CO3 were utilized (Table 1, entry 13) and 56% in the case of 0.2 equiv (Table 1, entry 1). The amount of NCS had a central role in the reaction performance, and an excess was always required. A decrease in yield was, in fact, observed reducing the equivalents of NCS from 3 to 2 (56% versus 45% yield; Table 1, entries 1 and 14). An additional loss in yield up to 10% was obtained with 0.5 equiv of NCS (Table 1, entry 15). Also, control experiments were accomplished. As reported in entry 16 of Table 1, the reaction needs both the base and NCS to afford the product. Interestingly, the absence of NCS also impeded the formation of any not-halogenated cyclization product, thus confirming the crucial role of such a chlorinating agent as the initiator of the process. In the absence of the photocatalyst, the chlorinated amide 3a was recovered as the major product in 66% yield, whereas the desired pyrrolidine 2a was obtained in only 11% yield (Table 1, entry 18). Finally, when the reaction was conducted without irradiation, the desired product was not observed. Surprisingly, a total conversion of the starting material to the chlorinated derivative 3a as the unique product occurred in remarkable yield (82%, Table 1, entry 17). Therefore, the standard reaction conditions, described in entry 1, were considered as the best and were applied for the following studies. These reaction conditions included 5 mol % of [Ru(bpy)3](PF6)2, 0.2 equiv of K2CO3, 3 equiv of NCS in dry toluene/methylformate 3:1 under irradiation at 456 nm.
Scope of the Reaction
Once the best conditions were determined, the scope of the reaction was explored, and the results are reported in Table 2. First, the influence of variations of the sulfonyl moiety were investigated. Both aryl- and alkylsulfonyl allenes were prepared following three different strategies (Mitsunobu reaction followed by a Crabbè–Ma homologation,38,39 Johnson–Claisen rearrangement,38 or the isomerization of propargyl(thio)ether).40 Those substrates afforded chlorovinyl pyrrolidines 2a–i from moderate to good yields without significant differences between aromatic and aliphatic sulfonyl substitution. Indeed, a similar outcome was observed with mesyl compound 2h (50% yield) and for the aromatic derivatives 2a, 2b, and 2c, which were obtained in 56, 52, and 51%, respectively. Electron-poor arylsulfonyl allenes seemed to be slightly favored when compared to electron-rich derivatives. See, for example, p-methoxyphenyl (2f) and 2,3-dihydrobenzo[b][1,4]dioxane (2g) derivatives, which were recovered in 40% yield, whereas when the p-acetyl group was introduced on the phenyl ring, a yield of 50% for product 2e was observed. Also, the presence of a chlorine in para position (2d) resulted in 40% yield. It must be noticed that both chloro and acetyl substituents were tolerated in reaction conditions. Finally, a decrease in yield was observed, when sterically hindered aliphatic (+)-camphor sulfonyl allene 1i was utilized as the starting material. The resulting diastereoisomeric mixture was recovered in 32% yield and diastereomeric ratio of 1:1. Next, internal allenes were explored as starting materials. A decrease in yield was observed with the increase of the number of substituents on the double bond in the corresponding chlorovinyl pyrrolidines. Actually, trisubstituted olefins 2k, 2l, and 2m were obtained in 48, 44, and 54% yield, respectively, superior to those of tetrasubstituted 2n–q (from 28 to 32% yield, see Scheme 2). Notably, the nature of the substituents, whether aromatic or aliphatic, seemed not to influence the reaction outcome.
Table 2. Scope of the Reactiona.

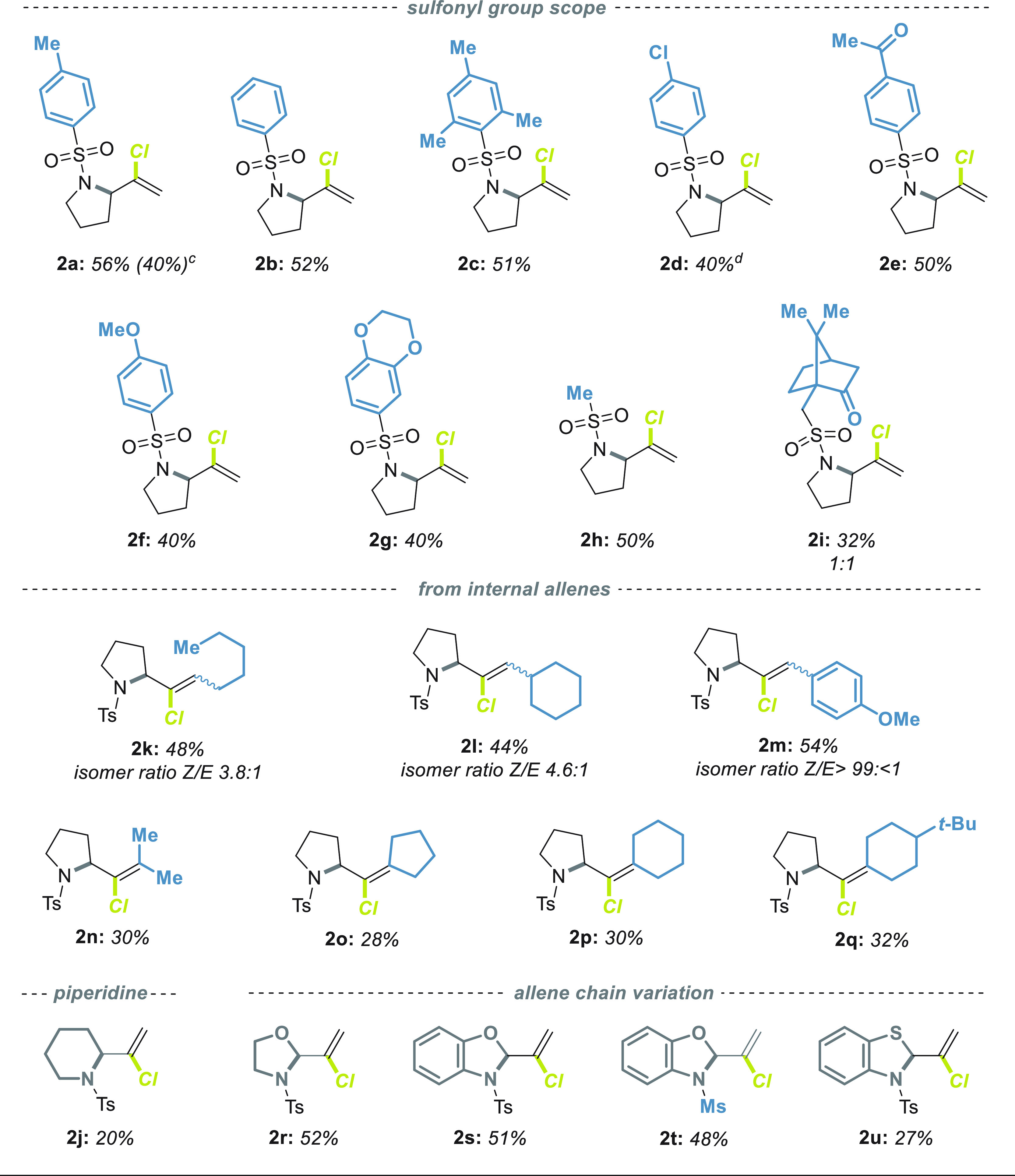
Standard reaction conditions: (a) 1 (0.20 mmol), K2CO3 (0.20 equiv, 0.04 mmol), NCS (3.0 equiv, 0.60 mmol), [Ru(bpy)3](PF6)2 (5 mol %, 0.01 mmol), anhydrous PhCH3 (3 mL), anhydrous HCO2CH3 (1 mL), N2 atmosphere irradiation with 456 nm light source for 21 h, blue light. (b) Yields determined on isolated products. (c) Reaction scaled up to 1 mmol, 36 h reaction time. (d) 36 h reaction time.
Scheme 2. Summary of the Reaction Mechanism.
Finally, the variations in the chain moiety bearing the allene substituents were contemplated, aiming for a change in the nature of the produced heterocycle. With a seven carbon chain, we observed a drop in yield and the resulting piperidine 2j was produced in only 20% yield, probably because the 1,5-HAT by the NCR might become a competitive process.41 Heteroatoms could also be incorporated, and the introduction of an oxygen to afford vinyl oxazoles and hydrogenated vinyl oxazolidines did not result in any significant change in the reaction outcome. Products 2r, 2s, and 2t were obtained in yields comparable to that of analogue vinyl pyrrolidines regardless the substituent of the nitrogen atom (tosyl versus mesyl) and the rigidity of the system (52, 51, and 48%, respectively). The replacement of the oxygen with a sulfur atom to obtain the hydrogenated benzothiazole scaffold resulted instead in a drop in the yield (27% for product 2u).
Mechanistic Studies
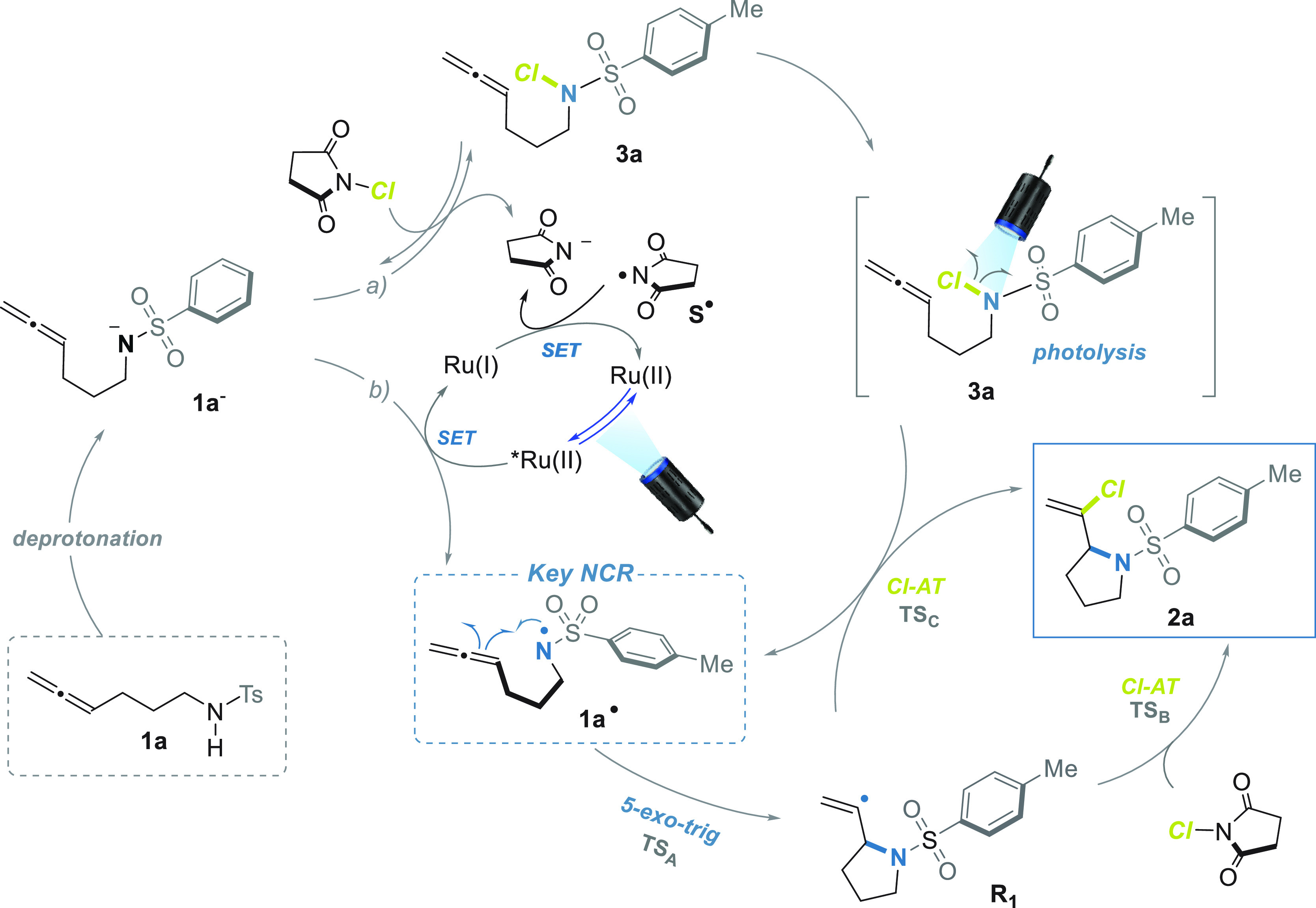
To attempt an elucidation of the reaction mechanism, several tests were carried out both experimental and computational (see Section S1, Supporting Information—Computational Data (SI-CD), for the details). First, to further confirm the essential role played by the light and to exclude a possible thermal reaction due to the heating provided by the operating light source, the same transformation was realized under thermal conditions (see SI for the complete study). Only when the reaction was carried out at 90 °C, the vinylpyrrolidine 2a was obtained in traces. Since the highest temperature recorded inside the reaction mixture during irradiation was 45 °C, we excluded the possibility of a thermal mechanism, as proved by the lack of pyrrolidine 2a formation observed without irradiation at this temperature. In all cases the main product was the chlorinated sulfonamide 3a. This compound is also the main product recovered in all the control experiments (Table 1, entries 17–18) in the presence of the base. Therefore, in order to clarify the fate of the allene starting material 1a in the presence of K2CO3 and NCS, prior to the irradiation, an NMR investigation was conducted adding a component at a time to the allene 1a in the NMR tube (Figure 1). The studied solutions were prepared in CDCl3, given the wide availability of CDCl3 as deuterated solvent and considering that a complete conversion of the starting material was obtained also in CHCl3 albeit the lower yield in product 2a (28%, Table 1, entry 9). The evolution of the starting material was studied using both NBS (N-bromosuccinimide) and NCS as reactants. Since in our hypothesis, other two main species could be involved in the process together with allenyltosyl amine 1a, the corresponding Na+ salt of allene 1a (Na+1a–) and N-chloro allene 3a were synthesized to be used as references (for their synthesis and complete characterization, see the SI).
Figure 1.

1H NMR monitoring of the reaction mixture components prior to irradiation. Top: Spectra recorded after the preparation of the solutions in CDCl3 (t = 5 min). Bottom: Spectra of the same solutions above recorded after 60 min from preparation. The same solutions were monitored every hour for 6 h.
As depicted in the stacked NMR spectra reported in Figure 1, no changes were observed when allene 1a and NCS were mixed, whereas the concurrent presence of the base and NCS immediately triggered the formation of 3a. Monitoring the same solution every 60 min for 6 h showed that the chlorinated species 3a was not able to evolve to the desired product 2a in the absence of light and catalyst. The same experiment, accomplished using NBS, afforded the corresponding bromo vinylpyrrolidine 4a in 1 h, and N-brominated allene was completely consumed. This confirmed that, in the case of NBS, no irradiation was required, and a thermal pathway was followed. So, a first hypothesis is that in the presence of a base and NCS an equilibrium between the starting allene 1a and chloro allene 3a may be established. This has been confirmed by the DFT study (Scheme S1 in the Section S2 SI-CD). The deprotonation by K2CO3 is thermodynamically favored (ΔG = −5.1 kcal mol–1, Table S1a in the SI-CD) and yields the potassium salt allene K+1a–. This intermediate can rapidly react with NCS (ΔG‡ = 3.7 kcal mol–1, Figure S1, left) to generate the N-chloroallene 3a and potassium succinimmide K+S– (Table S1b). This reaction is isoergonic (ΔG = 0.04 kcal mol–1). On the contrary, the reaction of allene 1a with NCS to form 3a and the K+S, although thermodynamically feasible (ΔG = +0.7 kcal mol–1), is kinetically forbidden because its activation free energy barrier is very large (79.3 kcal mol–1, Figure S1, right, Table S1c).
In order to confirm the role of the visible light, ON/OFF experiments were performed (Figure 2a). As already suggested by previous investigations, we observed that a mixture of allene 1a and N-chlorinated allene 3a was found in 30:70 ratio at time = 5 min. After 300 min, the chlorovinyl pyrrolidine 2a was recovered in 36%; 10% of starting allene 1a together with a 35% of allene 3a were still present. Anyway, the formation of product 2a was observed only under irradiation, confirming the photocatalyzed nature of this process. Moreover, when the lamp was off, neither the consumption of allenes 1a and 3a nor the formation of 2a took place. Moreover, quantum yield measurement (Φ = 0.54) was coherent with a photochemical mechanism excluding a radical chain.
Figure 2.

(a) The ON/OFF experiments: blue line, product 2a; gray line, starting reagent allene 1a; light blue line, chlorinated sulfonamide 3a. (b) Fluorescence quenching experiments for [Ru(bpy)3](PF6)2. Left: Fluorescence emission with allene sodium salt 1a–Na+ as the quencher. Right: Stern–Volmer plot for the quenching of [Ru(bpy)3](PF6)2 by 1a–Na+.
Fluorescence quenching and Stern–Volmer experiments were also carried out (Figure 2b). Allenes 1a and 3a, the sodium salt of allene Na+1a– and NCS were tested as the possible fluorescence quenchers of the excited state of [Ru(bpy)3](PF6)2. Despite the lower yield observed in CH3CN (Table 1, entry 4), this solvent was chosen for these measurements because it provided the complete dissolution of all the species involved in the fluorescence quenching experiments, thus ensuring the reproducibility of the measurements. NCS was not able to act as a quencher, even when the reaction conditions were reproduced in the presence of K2CO3 and of the allene, confirming the results reported by König and Lamar.42,43 Thus, an electrophilic amplification of NCS promoted by the oxidative quenching of the excited photocatalyst (PC*) had to be excluded. Furthermore, both allene 1a and N-chloroallene 3a were not able to provide fluorescence quenching. These findings suggested that a mechanism different from a typical photoredox process could be considered for the conversion of the chlorinated sulfonylamide 3a into the final product 2a. However, as evidenced in Figure 2b, the sodium salt of allene Na+1a– was able to quench the excited species of [Ru(bpy)3](PF6)2. Thus, a plausible oxidation of this anion to the corresponding NCR 1a• is a process that cannot be excluded.
The radical nature of the process was then investigated using TEMPO as the radical scavenger. A drop in yield was noticed and the pyrrolidine 2a was recovered in 15% yield in the presence of 35% of the starting allene 1a. Anyway, neither adducts with TEMPO nor allene 3a were detected. The same results were obtained using the chloroallene 3a and the sodium salt of allene 1a–Na+ as the starting materials.
To elucidate the role and the evolution of the putative reactive intermediates involved, the reactivity of the allene derivatives 3a and 1a–Na+ was explored. A selection of the results is reported in Table 3 (refer to the SI for the complete report). Given the fluorescence quenching experiments results, the anionic species 1a–Na+ was first analyzed and reacted in the optimized reaction conditions. No loss in yield was observed (56% of 2a) when compared to the reaction in which the allene 1a is the starting material (Table 3a, entry 1). Instead, in the absence of the photocatalyst, only 13% conversion to product 2a was observed with the formation of chlorinated 3a as the major outcome (Table 3a, entry 2), whereas in the absence of the NCS and of the base no reaction was observed and allene 1a was quantitatively recovered (Table 3a, entry 3). We deduced that 1a–Na+ could be involved in two reaction pathways: chlorination by NCS to deliver the N-chloroallene 3a and oxidation (upon reductive quenching of the photocatalyst) to the corresponding NCR that triggers an intramolecular aminochlorination of the allene moiety yielding the product 2a. A similar investigation was then accomplished using N-chlorinated allene 3a as the starting reagent. Also in this case, no deviation in yield was observed when compared to the results obtained in the case of allene 1a, when 3a was subjected to standard reaction conditions (Table 3b, entry 1).
Table 3. Screening of Sodium Salt of Allene 1a–Na+ and N-Chloroallene 3a Reactivitya.
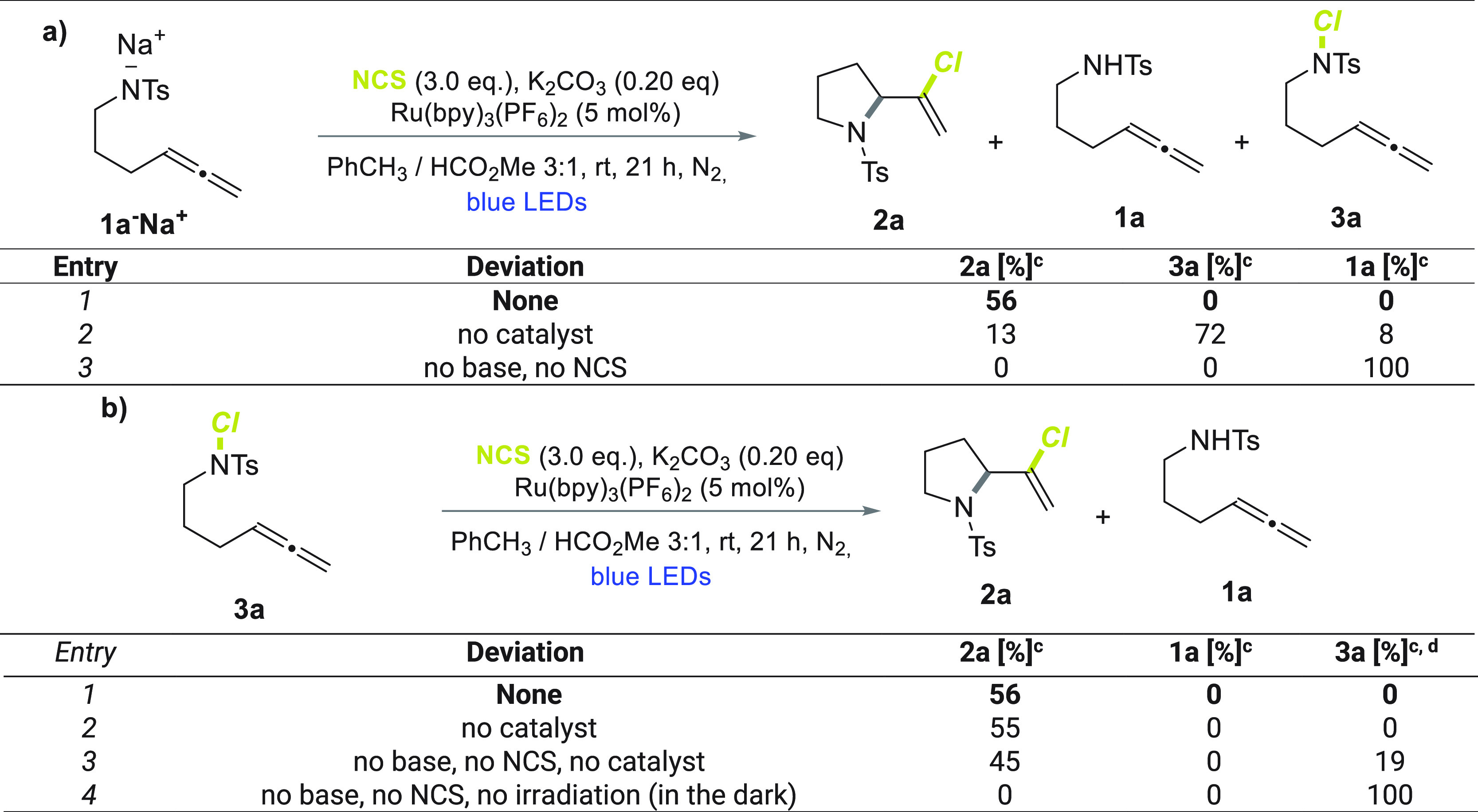
Standard reaction conditions: (a) 1–Na+ (0.20 mmol), K2CO3 (0.20 equiv, 0.04 mmol), NCS (3.0 equiv, 0.60 mmol), [Ru(bpy)3](PF6)2 (5 mol %, 0.01 mmol), anhydrous PhCH3 (3 mL), anhydrous HCO2CH3 (1 mL) under irradiation with 456 nm light source, blue light. (b) 3a (0.20 mmol), K2CO3 (0.20 equiv, 0.04 mmol), NCS (3.0 equiv, 0.60 mmol), [Ru(bpy)3](PF6)2 (5 mol %, 0.01 mmol), anhydrous PhCH3 (3 mL), anhydrous HCO2CH3 (1 mL) under irradiation with 456 nm light source, blue light. (c) Yields were determined after 2 repetitions with 1H NMR using dichloroethane and nitromethane as internal standards. (d) Amount of unreacted 3a.
Surprisingly, no erosion in 2a yield was observed under irradiation in the absence of [Ru(bpy)3](PF6)2 (Table 3b, entry 2). Furthermore, vinyl pyrrolidine 2a could be obtained in 45% yield starting from N-chlorinated allene 3a also in the absence of both base, NCS, and photocatalyst under blue light (Table 3b, entry 3). In this case, unreacted chloroallene 3a was recovered in 19% yield. On the contrary, no reaction was observed in the dark employing also 3a as the starting material (Table 3b, entry 4). Moreover, the unreacted chloroallene 3a was recovered when the reaction was performed without irradiation. These findings suggested that the chloroamide 3a could act as a source of the corresponding sulfonamidyl NCR 1a•. However, the fluorescence quenching experiments excluded that 3a could participate in the quenching of the excited state of the photocatalyst to yield such NCR 1•. Therefore, we hypothesized that a photolytic cleavage of the N–Cl bond of 3a occurs upon the interaction of this chloroamide species 3a with blue light. Such homolysis would generate both the NCR 1a• species and a chlorine radical. This hypothesis was supported by the results of Alexanian and co-workers concerning the development of N-chloroamides as reagents for the site selective C–H chlorination provided by the direct visible light cleavage of the N-Cl bond.44,45 Also the computational study is in favor of this hypothesis (for a full discussion, see Section S3, SI-CD). While the homolysis of the N–Cl bond is thermodynamically prohibitive (ΔG = 43.7 kcal mol–1, Table S2a), the photodissociation from the first triplet state T1 (used as a model for the singlet first excited state S1) shows a free energy barrier of only 7.1 kcal mol–1 and leads to the energetically favored separation of the radicals 1a• and Cl• (ΔG = −3.0 kcal mol–1, Table S2b). The dissociation to the separated radicals is energetically even more favored (ΔE = −22.0 kcal mol–1) when starting from the S1 state. All the experimental data collected agreed with a radical mechanism. The key radical is the NCR 1a•, which can be generated by the oxidation of the deprotonated allene 1a–, the photoexcited 3RuII(bpy)3, and the photodissociation of 3a (Scheme 2). Once the NCR 1a• has been generated (Scheme 2, for a full discussion of the reaction mechanism see Section S4, SI-CD), the formation of the pyrrolidine ring occurs through an intramolecular radical addition (TSA, Figure 3) of the N-centered radical to the allene moiety yielding the highly reactive vinyl radical R1. The step is fast (ΔG‡ = 9.5 kcal mol–1, kA = 6.9 × 105 s–1) and thermodynamically favored (ΔG = −11.1 kcal mol–1). As shown in Scheme 2, from the radical intermediate R1 two main pathways open: the radical Chlorine-Atom-Transfer (Cl-AT) from the NCS (TSB, Figure 3, ΔG‡ = 10.0 kcal mol–1, kB′ = 7.2 × 106 × [NCS] s–1, ΔG = −15.3 kcal mol–1) yielding the main product 2a and the succinimi-N-yl S• (the latter will be reduced to the anion by 2RuI(bpy)3 regenerating the photocatalyst); the Cl-AT from the N-chloro allene 3a (TSC, Figure 3, ΔG‡ = 8.3 kcal mol–1, kC′ = 1.3 × 108 × [ACl] s–1, ΔG = −40.3 kcal mol–1) also yielded the main product 2a and a new NCR 1a•. Both Cl-AT are bimolecular processes requiring the presence of the chlorine donor. Apart from the obvious choice of NCS, the experimental and computational study suggested that the same role can also be assumed by 3a easily formed in the reaction environment (see above).
Figure 3.

Transition structures: TSA for the intramolecular cyclization of 1a• yielding the 1-(N-tosyl-pyrrol-1-yl)vinyl radical R1; TSB for the Cl-AT from NCS to R1; TSC for the Cl-AT from 3a to R1.
Conclusions
In conclusion, in this article we report a cyclization chlorination domino process as the synthetic strategy to obtain chlorovinyl pyrrolidines by means of blue light and NCS on N-substituted sulfonyl allenes in good yields. In our hypothesis, two different pathways contribute to the formation of a common NCR triggering the chloroamination of the allene moiety. Thus, both the homolytic cleavage of the N–Cl bond from the in situ formed N-chlorinated sulfonamide and a photoredox event involving the excited state of the photocatalyst might be implicated. The use of visible light was fundamental to provide access to the heterocyclic scaffold and to the synthetically valuable alkenyl chloride moiety.
Experimental Section
Materials and Methods
Flasks and all equipment used for the generation and reaction of moisture-sensitive compounds were dried by an electric heat gun under a vacuum and backfilled with N2, then used under a N2 atmosphere. All commercially available reagents and solvents were used as received. Anhydrous solvents were purchased by Sigma-Aldrich or distilled as indicated by Armarego.46 Products were purified by preparative column chromatography on Macherey-Nagel silica-gel for flash chromatography, 0.04–0.063 mm/230–400 mesh. Reactions were monitored by TLC using silica-gel on TLC-PET foils Sigma-Aldrich, 2–25 μm, layer thickness 0.2 mm, medium pore diameter 60 Å. NMR spectra were recorded employing a Jeol ECZR instrument. 1H NMR spectra were recorded in CDCl3 or DMSO-d6 at 600 MHz. 13C{1H} NMR spectra were recorded in CDCl3 at 150 MHz. Chemical shifts were reported in ppm relative to the resonance of CHCl3 (δ = 7.26) for 1H NMR, or referred to the central peak of CDCl3 (δ = 77.0) for 13C NMR. 13C NMR spectra were measured with complete proton decoupling; thus, 13C NMR implies 13C{1H} NMR in the NMR characterization of new products. DEPT experiments were carried out with a DEPT-135 sequence. 1H NMR coupling constants (J) were reported in Hertz (Hz), and multiplicities are indicated as follows: s (singlet), bs (broad singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublets), ddd (doublet of doublets of doublets), dm (doublets of multiplet), td (triplet of doublets), tm (triplet of multiplets). Structural assignments were made with additional information from gCOSY, gNOESY experiments. Complete characterization of the light source and the instruments employed is reported in the SI.
Typical Procedure for the Synthesis of 2-Chlorovinyl Saturated Nitrogen Heterocycles (2a–u)
A 10 mL Schlenk tube containing a magnetic stirring bar was dried with a heat gun under a vacuum, and then the tube was backfilled with N2. Three mL of PhCH3 and 1 mL of HCO2CH3 were added via syringe and degassed with N2 for 20 min. Then NCS (3.0 equiv, 0.60 mmol, 80 mg), [Ru(bpy)3](PF6)2 (0.05 equiv, 0.01 mmol, 8 mg), and K2CO3 (0.20 equiv, 0.04 mmol, 5 mg) were added in one portion. The resulting mixture was stirred and degassed for 2 min. Then, the allene 1 (1.0 equiv, 0.20 mmol) was added via syringe under N2 and the mixture was stirred and degassed for additional 2 min. Finally, the tube was sealed and placed under irradiation with a Kessil A160PR Blue LED (456 nm) placed at 3 cm distance for 21 h under continuous stirring. In order to analyze the crude reaction mixture by NMR, the reaction was filtered over a short pad of silica and eluted with 25 mL of AcOEt. Then, the solvent was evaporated. The crude mixture purified by flash chromatography to afford product 2.
2-(1-Chlorovinyl)-1-tosylpyrrolidine (2a)
Following the described procedure, allene 1a (0.2 mmol, 50 mg) was reacted to obtain 32 mg of 2-(1-chlorovinyl)-1-tosylpyrrolidine 2a as a colorless oil (eluent: EP 92/8 Acetone, yield: 56%). The reaction was scaled up to 1 mmol of allene 1a (250 mg) to obtain 114 mg of 2-(1-chlorovinyl)-1-tosylpyrrolidine 2a (eluent: EP 92/8 Acetone, yield: 40%). 1H NMR (600 MHz, CDCl3, Me4Si) δ 7.73 (d, J = 8.1 Hz, 2H, Ar-H), 7.32 (d, J = 7.8 Hz, 2H, Ar-H), 5.57 (s, 1H, CCl=CHaHb), 5.33 (s, 1H, CCl=CHaHb), 4.32 (m, 1H, N–CH–CCl), 3.49 (m, 1H, N–C(H)H–CH2), 3.28 (m, 1H, N–C(H)H–CH2), 2.43 (s, 3H, CH3), 1.97 (m, 1H, N–CH2–C(H)H), 1.89 (m, 1H, N–CH2–C(H)H), 1.73 (m, 1H, N–CH–C(H)H), 1.66 (m, 1H, N–CH–C(H)H). 13C{1H} NMR (151 MHz, CDCl3, Me4Si) δ 143.7 (Cq), 141.8 (Cq), 135.0 (Cq), 129.8 (2 × CH), 127.6 (2 × CH), 113.7 (CH2), 64.3 (CH), 49.3 (CH2), 31.1 (CH2), 23.9 (CH2), 21.6 (CH3). The data matches the one reported in the literature.47 HRMS (ESI) m/z [M + H]+ calcd for C13H17ClNO2S 286.0663, found 286.0664. IR ν max (neat)/cm–1 2921, 2873, 1634, 1597, 1334, 1155, 812, 662.
2-(1-Chlorovinyl)-1-(phenylsulfonyl)pyrrolidine (2b)
Following the described procedure, allene 1b (0.2 mmol, 47 mg) was reacted to obtain 28 mg of 2-(1-chlorovinyl)-1-(phenylsulfonyl)pyrrolidine 2b as a colorless oil (EP/Acetone 90/10, 52% yield). 1H NMR (600 MHz, CDCl3, Me4Si) δ 7.84 (m, 2H, Ar-H), 7.59 (m, 1H, Ar-H), 7.52 (m, 2H, Ar-H), 5.55 (t, J = 1.5 Hz, 1H, CCl=CHaHb), 5.32 (d, J = 1.5 Hz, 1H, CCl=CHaHb), 4.35 (m, 1H, m, 1H, N–CH–CCl), 3.50 (m, 1H, N–C(H)H–CH2), 3.30 (m, 1H, N–C(H)H–CH2), 1.98 (m, 1H, N–CH2–(CH)H), 1.89 (m, 1H, N–CH–C(H)H), 1.74 (m, 1H, N–CH–C(H)H), 1.67 (m, 1H, N–CH2–(CH)H). 13C{1H} NMR (151 MHz, CDCl3, Me4Si) δ 141.7 (Cq), 138.0 (Cq), 132.9 (CH), 129.2 (2 × CH), 127.5 (2 × CH), 113.9 (CH2), 64.4 (CH), 49.3 (CH2), 31.1 (CH2), 23.9 (CH2). HRMS (ESI) m/z [M + H]+ calcd for C12H15ClNO2S 272.0507, found 272.0506. IR ν max (neat)/cm–1 2956, 1633, 1445, 1347,1159, 1092, 892, 723, 587.
2-(1-Chlorovinyl)-1-(mesitylsulfonyl)pyrrolidine (2c)
Following the described procedure, allene 1c (0.2 mmol, 56 mg) was reacted to obtain 32 mg of 2-(1-chlorovinyl)-1-(mesitylsulfonyl)pyrrolidine 2c as a colorless oil (EP/Acetone 95/5, yield 51%). 1H NMR (600 MHz, CDCl3, Me4Si) δ 6.91 (m, 2H, Ar-H), 5.19 (t, J = 1.6 Hz, 1H, CCl=CHaHb), 4.98 (d, J = 1.6 Hz, 1H, CCl=CHaHb), 4.52 (m, 1H, N–CH–CCl), 3.58 (m, 1H, N–C(H)H–CH2), 3.34 (m, 1H, N–C(H)H–CH2), 2.61 (s, 6H, o-CH3-Ar), 2.27 (s, 3H, p-CH3-Ar), 2.18–2.10 (m, 1H, N–CH–C(H)H), 2.09–1.94 (m, 2H, N–CH–C(H)H), 1.93–1.85 (m, 1H, N–CH2–CH2). 13C{1H} NMR (151 MHz, CDCl3, Me4Si) δ 142.7 (Cq), 141.8 (Cq), 140.3 (2 × Cq), 133.2 (Cq), 131.9 (2 × CH), 113.6 (CH2), 63.7 (CH), 48.8 (CH2), 31.9 (CH2), 24.5 (CH2), 23.0 (2 × CH3), 21.1 (CH3). HRMS (ESI) m/z [M + H]+ calcd for C15H21ClNO2S 314.0976, found 314.0974. IR ν max (neat)/cm–1 2930, 2886, 1602, 1321, 1311, 1147, 674.
1-((4-Chlorophenyl)sulfonyl)-2-(1-chlorovinyl)pyrrolidine (2d)
Following the described procedure, allene 1d (0.2 mmol, 54 mg) was reacted to obtain, after 32 h, 24.5 mg of 1-((4-chlorophenyl)sulfonyl)-2-(1-chlorovinyl)pyrrolidine 2d as a colorless oil (EP/Acetone 92/8, 40% yield). 1H NMR (600 MHz, CDCl3, Me4Si): 7.78 (dm, J = 8.1 Hz, 2H, Ar-H), 7.50 (dm, J = 8.1 Hz, 2H, Ar-H), 5.53 (d, J = 1.7 Hz, 1H, CCl=CHaHb), 5.32 (d, J = 1.7 Hz, 1H, CCl=CHaHb), 4.37 (m, 1H, N–CH–CCl), 3.48 (m, 1H, N–C(H)H–CH2), 3.33 (m, 1H, N–C(H)H–CH2), 2.05–1.98 (m, 1H, N–CH–C(H)H), 1.98–1.88 (m, 1H, N–CH2–(CH)H), 1.86–1.77 (m, 1H, N–CH–C(H)H), 1.77–1.69 (m, 1H, N–CH2–(CH)H). 13C{1H} NMR (151 MHz, CDCl3) δ 141.6 (Cq), 139.5 (Cq), 136.9 (Cq), 129.5 (2 × CH), 128.9 (2 × CH), 114.2 (CH2), 64.5 (CH), 49.3 (CH2), 31.2 (CH2), 24.0 (CH2). HRMS (ESI) m/z [M + H]+ calcd for C12H14Cl2NO2S 306.0117, found 306.0117. IR ν max (neat)/cm–1 3095, 2975, 1634, 1346, 1181, 1086, 843, 755.
1-(4-((2-(1-Chlorovinyl)pyrrolidin-1-yl)sulfonyl)phenyl)ethan-1-one (2e)
Following the described procedure, allene 1e (0.2 mmol, 56 mg) was reacted to obtain 31 mg of 2-(1-chloro-2-cyclohexylvinyl)-1-tosylpyrrolidine 2e as a colorless oil (EP/Acetone 85/15, yield 50%). 1H NMR (600 MHz, CDCl3, Me4Si) δ 8.07 (d, J = 8.5 Hz, 2H, Ar-H), 7.93 (d, J = 8.5 Hz, 2H, Ar-H), 5.52 (t, J = 1.7 Hz, 1H, CCl=CHaHb)), 5.31 (d, J = 1.7 Hz, 1H, CCl=CHaHb), 4.40 (m, 1H, N–CH–CCl), 3.52–3.47 (m, 1H, N–C(H)H–CH2), 3.39–3.35 (m, 1H, N–C(H)H–CH2), 2.65 (s, 3H, CO–CH3), 2.04–1.96 (m, 1H, N–CH–C(H)H), 1.97–1.89 (m, 1H, N–CH2–(CH)H), 1.88–1.76 (m, 1H, N–CH–C(H)H), 1.76–1.68 (m, 1H, N–CH2–(CH)H). 13C{1H} NMR (151 MHz, CDCl3, Me4Si) δ 196.9 (Cq), 142.3 (Cq), 141.5 (Cq), 140.2 (Cq), 129.0 (2 × CH), 127.8 (2 × CH), 114.3 (CH2), 64.5 (CH), 49.4 (CH2), 31.3 (CH2), 27.0 (CH3), 24.1 (CH2). HRMS (ESI) m/z [M + H]+ calcd for C14H17ClNO3S 314.0612, found 314.0612. IR ν max (neat)/cm–1 1689, 1635, 1348, 903, 835.
2-(1-Chlorovinyl)-1-((4-methoxyphenyl)sulfonyl)pyrrolidine (2f)
Following the described procedure, allene 1f (0.2 mmol, 54 mg) was reacted to obtain 24 mg of 2-(1-chlorovinyl)-1-((4-methoxyphenyl)sulfonyl)pyrrolidine 2f as a light yellow oil (eluent: from EP to EP/Acetone 80/20, yield: 40%). 1H NMR (600 MHz, CDCl3, Me4Si) δ 7.78 (d, J = 8.9 Hz, 2H, Ar-H), 6.99 (d, J = 8.9 Hz, 2H, Ar-H), 5.57 (t, J = 1.2 Hz, 1H, CCl=CHaHb), 5.32 (d broad, J = 1.2 Hz, 1H, CCl=CHaHb), 4.32 (dd, J = 8.5, 3.1 Hz, 1H, N–CH), 3.88 (s, 3H, OMe), 3.48 (ddd, J = 9.8, 7.2, 4.1 Hz, 1H, N–C(H)H–CH2), 3.28 (m, 1H, N–C(H)H–CH2), 1.98 (m, 1H, N–CH2–C(H)H), 1.88 (m, 1H, N–CH–C(H)H), 1.75 (m, 1H, CH–C(H)H–CH2), 1.67 (m, 1H, N–CH2–C(H)H–CH2). 13C{1H} NMR (151 MHz, CDCl3, Me4Si) δ 163.0 (Cq), 141.8 (Cq), 129.7 (Cq), 129.5 (2 × CH), 114.2 (2 × CH), 113.7 (CH2), 64.2 (CH), 55.6 (OCH3), 49.2 (CH2), 31.0 (CH2), 23.8 (CH2). HRMS (ESI) m/z [M + H]+ calcd for C13H17ClNO3S 302.0612, found 302.0615. IR ν max (neat)/cm–1 1496, 1344, 1092, 833, 667.
2-(1-Chlorovinyl)-1-((2,3-dihydrobenzo[b][1,4]dioxin-6-yl)sulfonyl)pyrrolidine (2g)
Following the described procedure, allene 1g (0.2 mmol, 59 mg) was reacted to obtain 26 mg of 2-(1-chlorovinyl)-1-((2,3-dihydrobenzo[b][1,4]dioxin-6-yl)sulfonyl)pyrrolidine 2g as a light yellow oil (eluent: from EP to EP/Et2O 50/50, yield: 40%). 1H NMR (600 MHz, CDCl3, Me4Si) δ 7.37 (d, J = 2.1 Hz, 1H, Ar-H), 7.33 (dd, J = 8.4, 2.2 Hz, 1H, Ar-H), 6.96 (d, J = 8.5 Hz, 1H, Ar-H), 5.58 (t, J = 1.0 Hz, 1H, CCl=CHaHb), 5.33 (d broad, J = 1.8 Hz, 1H, CCl=CHaHb), 4.29–4.33 (m, 5H, O–(CH2)2–O and N–CH–CH2), 3.48 (ddd, J = 10.3, 6.8, 4.2 Hz, 1H, N–C(H)H–CH2), 3.28 (m, 1H, N–C(H)H–CH2), 1.98 (m, 1H, CH–C(H)H), 1.88 (m, 1H, N–CH2–C(H)H), 1.75 (m, 1H, N–CH2–C(H)H), 1.68 (m, 1H, N–CH–C(H)H–CH2). 13C{1H} NMR (151 MHz, CDCl3, Me4Si) δ 147.5 (Cq), 143.5 (Cq), 141.8 (Cq), 130.4 (Cq), 121.2 (CH), 117.7 (CH), 117.0 (CH), 113.6 (CH2), 64.5 (CH2), 64.2 (CH), 64.2 (CH2), 49.2 (CH2), 31.0 (CH2), 23.7 (CH2). HRMS (ESI) m/z [M + H]+ calcd for C14H17ClNO4S 330.0561, found 330.0565. IR ν max (neat)/cm–1 1633, 1344, 1285, 877, 699.
2-(1-Chlorovinyl)-1-(methylsulfonyl)pyrrolidine (2h)
Following the described procedure, allene 1h (0.2 mmol, 40 mg) was reacted to obtain 21 mg of 2-(1-chlorovinyl)-1-(methylsulfonyl)pyrrolidine 2h as a colorless oil (eluent: EP 92/8 Acetone, yield: 50%). 1H NMR (600 MHz, CDCl3, Me4Si) δ 5.51 (d, J = 1.7, 1H, CCl=CHaHb), 5.34 (d, J = 1.7 Hz, 1H, CCl=CHaHb), 4.48 (m, 1H, N–CH–CH2), 3.55–3.52 (m, 1H, N–C(H)H–CH2), 3.45–3.42 (m, 1H, N–C(H)H–CH2), 2.87 (s, 3H, CH3), 2.18–1.99 (m, 3H, N–CH–CH2), 1.97–1.87 (m, 1H, N–CH2–CH2). 13C{1H} NMR (151 MHz, CDCl3, Me4Si) δ 141.9 (Cq), 114.6 (CH2), 64.1 (CH), 49.0 (CH2), 38.5 (CH3), 31.5 (CH2), 24.5 (CH2). HRMS (ESI) m/z [M + H]+ calcd for C7H13ClNO2S 210.0350, found 210.0354. IR ν max (neat)/cm–1 2931, 1630, 1324, 1141, 1060, 1008, 970, 889.
1-(((2-(1-Chlorovinyl)pyrrolidin-1-yl)sulfonyl)methyl)-7,7-dimethylbicyclo[2.2.1]heptan-2-one (2i)
Following the described procedure, allene 1i (0.2 mmol, 69 mg) was reacted to obtain 22 mg of 2-(1-chlorovinyl)-1-tosylpyrrolidine 2i as a colorless oil (eluent: EP 92/8 Acetone, yield: 32%, mixture of 2 isomers ratio 1:1). 1H NMR (600 MHz, CDCl3, Me4Si) δ 5.56 (s, 1H, CCl=CHaHbisomer A), 5.53 (s, 1H, CCl=CHaHbisomer B), 5.36 (s, 1H, CCl=CHaHbisomer A), 5.34 (s, 1H, CCl=CHaHbisomer B), 4.56–4.49 (m, 1H, N–CH–CCl, both isomers), 3.66–3.56 (m, 1H, N–CHaHb–CH2, both isomers), 3.50–3.44 (m, 1H, SO2–C(H)H, both isomers), 3.48–3.41 (m, 1H, N–CHaHb–CH2, both isomers), 2.92–2.82 (m, 1H, SO2–C(H)H, both isomers), 2–55–2.48 (m, 1H, C(=O)—C(H)H, both isomers), 2.40–2.30 (m, 1H, CH2–C(C)H–CH2, both isomers), 2.20–2.12 (m, 2H, N–CH2–(CH2)2, both isomers), 2.13–1.99(m, 3H, C—(C(H)H)2—CH—C(=O), N–CH2–(CH2)2, both isomers), 1.96–1.89 (m, 2 H, C(=O)-C(H)H, CH2–C(C)H-CH2, both isomers), 1.70–1.60 (m, 1H, C—(C(H)H)2—CH—C(=O), both isomers), 1.48–1.40 (m, 1H, C—(C(H)H)2—CH—C(=O), both isomers), 1.13 (s, 3H, C(CH3)CH3, isomer A), 1.11 (s, 3H, C(CH3)CH3, isomer A), 0.87 (s, 3H, C(CH3)CH3, isomer A), 0.86 (s, 3H, C(CH3)CH3, isomer B). 13C{1H} NMR (151 MHz, CDCl3, Me4Si): mixture of isomers δ 215.4 (Cq), 215.3 (Cq), 142.4 (Cq), 142.0 (Cq), 114.5 (CH2), 114.5 (CH2), 64.2 (CH), 64.0 (CH), 58.5 (Cq), 58.4 (Cq), 49.2 (CH2), 48.0 (Cq), 47.9 (CH2), 47.9 (Cq), 43.2 (CH), 42.8 (CH), 42.7 (CH2), 42.7 (CH2), 31.5 (CH2), 31.3 (CH2), 27.1 (CH2), 27.0 (CH2), 38.5 (CH3), 31.5 (CH2), 25.2 (CH2), 25.1 (CH2), 24.6 (CH2), 24.5 (CH2), 20.2 (CH3), 20.2 (CH3), 19.9 (CH3), 19.9 (CH3). HRMS (ESI) m/z [M + H]+ calcd for C16H25ClNO3S 346.1238, found 346.1235. IR ν max (neat)/cm–1 2960, 2900, 1742, 1336, 1160, 573.
2-(1-Chlorovinyl)-1-tosylpiperidine (2j)
Following the described procedure, allene 1j (0.2 mmol, 53 mg) was reacted to obtain, 12 mg of 2-(1-chlorovinyl)-1-tosylpiperidine 2j as a colorless oil (eluent EP 95/5 Acetone, yield: 20%). 1H NMR (600 MHz, CDCl3, Me4Si) δ 7.72 (dm, J = 8.1 Hz, 2H, Ar-H), 7.29 (dm, J = 8.1 Hz, 2H, Ar-H), 5.44 (t, J = 2.0 Hz, 1H, CCl=CHaHb), 5.38 (t, J = 2.0 Hz, 1H, CCl=CHaHb), 4.77 (m, 1H, N–CH–CH2), 3.74 (dm, J = 13.7, 1H, N–C(H)H–CH2), 3.09 (tm, J = 13.7, 1H, N–C(H)H–CH2), 2.43 (s, 3H, CH3), 2.22 (m, 1H, N–C(CCl)H–(C(H)H), 1.58–1.39 (m, 3H, N–CH2–(CH2), N–C(CCl)H–CH2–(C(H)H), 1.29 (m, 2H, m, 1H, N–C(CCl)H–(C(H)H), N–C(CCl)H–CH2–(C(H)H)). 13C{1H} NMR (151 MHz, CDCl3) δ 143.3 (Cq), 139.6 (Cq), 138.1 (Cq), 129.7 (2 × CH), 127.1 (2 × CH), 115.2 (CH2), 57.2 (CH), 41.8 (CH2), 26.6 (CH2), 24.4 (CH2), 21.6 (CH3), 18.8 (CH2). HRMS (ESI): Chemical Formula C14H18ClNO2S; m/z [M + K]+ calcd for C14H18ClKNO2S 338.0378, found 338.0378. IR ν max (neat)/cm–1 2924, 2855, 1596, 1446, 1337, 1156, 1092, 956, 813, 653.
2-(1-Chlorohept-1-en-1-yl)-1-tosylpyrrolidine (2k)
Following the described procedure, allene 1k (0.2 mmol, 64 mg) was reacted to obtain 34 mg of 2-(1-chlorohept-1-en-1-yl)-1-tosylpyrrolidine 2k as a colorless oil (EP/Acetone 90/10, yield 48%). Mixture of E/Z isomers, isomer A/B ratio 1:3.8.1H NMR (600 MHz, CDCl3, Me4Si) δ 7.70 (d, J = 8.2 Hz, 2H, Ar-H isomer A), 7.69 (d, J = 8.2 Hz, 2H, Ar-H isomer B), 7.27 (d, J = 8.2 Hz, 2H, Ar-H isomer B), 7.28 (d, J = 8.2 Hz, 2H, Ar-H isomer A), 5.82 (t, J = 7.8 Hz, 1H, CCl=CH, isomer B), 5.63 (t, J = 7.8 Hz, 1H, CCl=CH, isomer A), 4.81 (dd, J = 8.2, 3.5 Hz, 1H, N–CH–CH2, isomer A), 4.35 (dd, J = 8.2, 3.5 Hz, 1 H, N–CH–CH2, isomer B), 3.60–3.53 (m, 1H, N–C(H)H–CH2, isomer A), 3.47–3.40 (m, 1H, N–C(H)H–CH2, isomer B), 3.39–3.31 (m, 2H, N–C(H)H–CH2 mixture of isomers), 2.41 (s, 3H, Ar–CH3), 2.26–2.10 (m, 2H, CH2—CH=, isomer A), 2.12 (q, J = 7.3 Hz, 2H, CH2—CH=, isomer B), 2.04–1.84 (m, 3H, N–CH2–(CH2)2), 1.80–1.71 (m, 1H, N–CH–C(H)H, isomer B), 1.70–1.61 (m, 1H, N–CH2–C(H)H, isomer B), 1.51–1.22 (m, 6H, CH3–CH2–CH2, CH2–(CH2)2–CH2isomer A), 1.36 (quin, 2H, J = 7.3 Hz, CH3–CH2–CH2, isomer B), 1.33–1.23 (m, 4H, CH2–(CH2)2–CH2isomer B), 0.89 (t, J = 7.0 Hz, 3H, CH2–CH3), 0.88 (t, J = 7.0 Hz, 3H, CH2–CH3). 13C{1H} NMR (151 MHz, CDCl3, Me4Si): Isomer A δ 143.3 (Cq), 136.4 (Cq), 134.0 (Cq), 131.1 (CH), 129.5 (2 × CH), 127.4 (2 × CH), 58.2 (CH), 49.4 (CH2), 31.6 (CH2), 31.5 (CH2), 29.1 (CH2), 28.5 (CH2), 25.3 (CH2), 24.1 (CH2), 21.6 (CH3), 14.1 (CH3); Isomer B δ 143.4 (Cq), 135.7 (Cq), 133.7 (Cq), 129.6 (2 × CH), 127.9 (CH), 127.5 (2 × CH), 64.7 (CH), 49.2 (CH2), 31.5 (CH2), 31.3 (CH2), 28.3 (CH2), 28.1 (CH2), 24.1 (CH2), 22.5 (CH2), 21.6 (CH2), 14.1 (CH3). HRMS (ESI) m/z [M + H]+ calcd for C19H29ClNO2S 370.1602, found 370.1602. IR ν max (neat)/cm–1 2954, 2857, 1348, 1157, 991, 586.
2-(1-Chloro-2-cyclohexylvinyl)-1-tosylpyrrolidine (2l)
Following the described procedure, allene 1l (0.2 mmol, 65 mg) was reacted to obtain 34 mg of 2-(1-chloro-2-cyclohexylvinyl)-1-tosylpyrrolidine 2l as a colorless oil (EP/Acetone 90/10, yield 44%). Mixture of E/Z isomers, isomer A/B ratio 1:4.6. 1H NMR (600 MHz, CDCl3, Me4Si) δ 7.70 (d, J = 8.2 Hz, 2H, Ar-H isomer A), 7.69 (d, J = 8.2 Hz, 2H, Ar-H isomer B), 7.28 (d, J = 8.2 Hz, 2H, Ar-H isomer B), 7.28 (d, J = 8.2 Hz, 2H, Ar-H isomer A), 5.63 (d, J = 8.8 Hz, 1H, CCl=CH isomer B), 5.49 (d, J = 8.8 Hz, 1H, CCl=CH isomer A), 4.80 (dd, J = 8.2, 3.5 Hz, 1 H, N–CH–CH2, isomer A), 4.33 (dd, J = 8.2, 3.5 Hz, 1 H, N–CH–CH2, isomer B), 3.62–3.52 (m, 1H, N–C(H)H–CH2, isomer A), 3.47–3.41 (m, 1H, N–C(H)H–CH2, isomer B), 3.41–3.32 (m, 2H, N–C(H)H–CH2), 2.43 (s, 3H, Ar–CH3isomer A), 2.41 (s, 3H, Ar–CH3isomer B), 2.41–2.31 (m, 1H, —CH—CH=CCl), 2.06–1.83 (m, 1H, m, 1H, N–CH2–(CH2)2), 1.81–1.72 (m, 2H, m, 1H, N–CH2–(CH2)2), 1.71–1.59 (m, 5H, N–CH2–(CH2)2, (CH2)2–CH2-(CH2)2, cyclohexyl CH2) 1.37–1.12 (m, 4H, cyclohexyl CH2), 1.12–0.98 (m, 2H, CCl—C=CH—C(CH2)H2). 13C{1H} NMR (151 MHz, CDCl3, Me4Si) Isomer A δ 143.3 (Cq), 136.5 (CH), 136.4 (Cq), 135.8 (Cq), 129.6 (2 × CH), 127.4 (2 × CH), 58.5 (CH), 49.5 (CH2), 38.1 (CH), 33.6 (CH2), 32.7 (CH2), 32.7 (CH2), 25.9 (CH2), 25.8 (2 × CH2), 25.3 (CH2), 21.7 (CH3). Isomer B δ 143.4 (Cq), 135.8 (Cq), 133.0 (CH), 132.0 (Cq), 129.6 (2 × CH), 127.5 (2 × CH), 64.7 (CH), 49.3 (CH2), 37.5 (CH), 31.9 (CH2), 31.9 (CH2), 31.3 (CH2), 26.1 (CH2), 25.7 (CH2), 24.1 (CH2), 21.7 (CH3). HRMS (ESI) m/z [M + H]+ calcd for C19H26ClNO2S 368.1446, found 368.1447. IR ν max (neat)/cm–1 2962, 1258, 1087, 1008, 788, 663.
(Z)-2-(1-Chloro-2-(4-methoxyphenyl)vinyl)-1-tosylpyrrolidine (2m)
Following the described procedure, allene 1m (0.2 mmol, 71 mg) was reacted to obtain 40 mg 2-(1-chloro-2-(4-methoxyphenyl)vinyl)-1-tosylpyrrolidine 2m as a colorless oil (EP/Acetone 90/10, 54% yield). Only one isomer was recovered. 1H NMR (600 MHz, CDCl3, Me4Si) δ 7.80 (dm, J = 8.1 Hz, 2H, Ar1-H), 7.56 (dm, J = 8.7 Hz, 2H, Ar2-H), 7.36 (dm, J = 8.1 Hz, 2H, Ar1-H), 6.88 (dm, J = 8.7 Hz, 2H, Ar2-H), 5.89 (s, 1H, CCl=CH—Ar2), 4.74 (m, 1H, N–CH–CH2), 3.86 (m, 1H, N–C(H)H–CH2) 3.82 (s, 3H, O–CH3), 3.55 (m, 1H, N–C(H)H–CH2), 2.45 (s, 3H, CH3), 2.33 (m, 1H, N–CH–C(H)H), 1.92 (m, 1H, N–CH–C(H)H), 1.78 (m, 1H, N–CH2–C(H)H), 1.10 (m, 1H, N–CH2–C(H)H). 13C{1H} NMR (151 MHz, CDCl3, Me4Si) δ 160.1 (Cq), 144.4 (Cq), 135.3 (Cq), 131.6 (2 × CH), 130.2 (2 × CH), 128.9 (Cq), 127.7 (2 × CH), 113.2 (2 × CH), 99.7 (Cq), 67.0 (CH), 66.7 (CH), 55.4 (CH3), 52.3 (CH2), 30.1 (CH2), 24.8 (CH2), 21.7 (CH3). HRMS (ESI) m/z [M + H]+ calcd for C20H23ClNO3S 392.1082, found 392.1073. IR ν max (neat)/cm–1 2924, 1610, 1152, 1348, 1250, 1159, 1087, 1030, 841, 664.
2-(1-Chloro-2-methylprop-1-en-1-yl)-1-tosylpyrrolidine (2n)
Following the described procedure, allene 1n (0.2 mmol, 53 mg) was reacted to obtain 19 mg of 2-(1-chloro-2-methylprop-1-en-1-yl)-1-tosylpyrrolidine 2n as a colorless oil (EP/Acetone 90/10, 30% yield). 1H NMR (600 MHz, CDCl3, Me4Si) δ 7.66 (dm, J = 8.1 Hz, 2H, Ar-H), 7.25 (dm, J = 8.1 Hz, 2H, Ar-H), 4.93 (m, 1H, N–CH–CH2), 3.62 (m, 1H, N–C(H)H–CH2), 3.38 (m, 1H, N–C(H)H–CH2), 2.40 (s, 3H, Ar–CH3), 2.01 (m, 2H, N–CH2–(CH2)2), 1.93 (m, 1H, m, 2H, N–CH2–CH2), 1.89 (s, 3H, ClC=C—C(H3)CH3), 1.73 (s, 3H, ClC=C—C(H3)CH3), 1.64 (m, 2H, N–CH2–(CH2)2). 13C{1H} NMR (151 MHz, CDCl3, Me4Si) δ 143.1 (Cq), 136.8 (Cq), 131.0 (Cq), 129.3 (2 × CH), 128.9 (Cq), 127.3 (2 × CH), 59.4 (CH), 49.3 (CH2), 31.4 (CH2), 25.3 (CH2), 22.4 (CH3), 21.6 (CH3), 20.6 (CH3). HRMS (ESI) m/z [M + H]+ calcd for C15H21ClNO2S 314.0976, found 314.0977. IR ν max (neat)/cm–1 2920, 2850, 1340, 1185, 1152, 586.
2-(Chloro(cyclopentylidene)methyl)-1-tosylpyrrolidine (2o)
Following the described procedure, allene 1o (0.2 mmol, 61 mg) was reacted to obtain 19 mg of 2-(chloro(cyclopentylidene)methyl)-1-tosylpyrrolidine 2o as a colorless oil (EP/Acetone 9/1, 28% yield). 1H NMR (600 MHz, CDCl3, Me4Si):δ 7.66 (d, J = 8.3 Hz, 2H, Ar-H), 7.25 (d, J = 8.3 Hz, 2H, Ar-H), 4.75 (m, 1H, N–CH–CH2), 3.62 (m, 1H, N–C(H)H–CH2), 3.38 (m, 1H, N–C(H)H–CH2), 2.67 (m, 1H, N–CH2–(CH2)2), 2.40 (s, 3H, Ar–CH3), 2.33–2.26 (m, 1H, N–CH2–(CH2)2), 2.25–2.15 (m, 2H, C=C—(C(H)H)2), 2.09–1.97 (m, 2H, N–CH2–(CH2)2), 1.97–1.89 (m, 1H, C=C—Cb(H)H), 1.79–1.69(m, 2H, N–CH2–(CH2)2) 1.65 (m, 3H, C=C—Ca(H)H, C=C—(CH2)2—(CH2)2). 13C{1H} NMR (151 MHz, CDCl3, Me4Si) δ 143.4 (Cq), 143.1 (Cq), 136.8 (Cq), 129.3 (2 × CH), 127.4 (2 × CH), 124.8 (Cq), 61.2 (CH), 49.3 (CH2), 33.4 (CH2), 31.2 (CH2), 31.2 (CH2), 27.6 (CH2), 25.8 (CH2), 25.2 (CH2), 21.6 (CH3). HRMS (ESI) m/z [M + H]+ calcd for C17H23ClNO2S 340.1133, found 340.1136. IR ν max (neat)/cm–1 2923, 2865, 1598, 1347, 1154, 1091, 1001, 809, 670, 586 cm.
2-(Chloro(cyclohexylidene)methyl)-1-tosylpyrrolidine (2p)
Following the described procedure, allene 1p (0.2 mmol, 65 mg) was reacted to obtain 21 mg of 2-(chloro(cyclohexylidene)methyl)-1-tosylpyrrolidine 2p as a white solid (EP/Acetone 9/1, 30% yield). 1H NMR (600 MHz, CDCl3, Me4Si) δ 7.68 (d, J = 8.3 Hz, 2H, Ar-H), 7.25 (d, J = 8.3 Hz, 2H, Ar-H), 5.01 (m, 1H, N–CH–CH2), 3.63 (m, 1H, N–C(H)H–CH2), 3.38 (m, 1H, N–C(H)H–CH2), 2.46–2.39 (m, 2H, C=C—Ca(H)H and C=C—Ca′(H)H), 2.40 (s, 3H, Ar–CH3,), 2.35–2.19 (m, 2H, C=C—Ca(H)H and C=C—Ca′(H)H), 2.08–1.93 (m, 3H, N–CH–(CH2)2, CH2cyclohexyl), 1.74–1.68 (m, 1H, CH2cyclohexyl), 1.67–1.60 (m, 2H, N–CH–(CH2)2), 1.59–1.48 (m, 4H, CH2cyclohexyl). 13C{1H} NMR (151 MHz, CDCl3, Me4Si) δ 143.0 (Cq), 138.2 (Cq), 136.9 (Cq), 129.4 (2 × CH), 127.4 (2 × CH), 126.5 (Cq), 58.8 (CH), 49.4 (CH2), 32.1 (CH2), 31.8 (CH2), 31.3 (CH2), 27.6 (CH2), 26.9 (CH2), 26.4 (CH2), 25.4 (CH2), 21.6(CH3). HRMS (ESI) m/z [M + H]+ calcd for C18H25ClNO2S 354.1289, found 354.1286. IR ν max (neat) cm–1 2975, 2846, 1331, 1154, 1010, 579. mp 82 °C.
2-((4-(tert-Butyl)cyclohexylidene)chloromethyl)-1-tosylpyrrolidine (2q)
Following the described procedure, allene 1q (0.2 mmol, 71 mg) was reacted to obtain 26 mg of 2-((4-(tert-butyl)cyclohexylidene)chloromethyl)-1-tosylpyrrolidine 2q as a colorless oil (EP/Acetone 9/1, 32% yield). Two conformers are observed in NMR spectra. 1H NMR (600 MHz, CDCl3, Me4Si): 7.69 (d, J = 8.3 Hz, 2H, Ar-H, conformer B), 7.64 (d, J = 8.3 Hz, 2H, Ar-H, conformer A), 7.25 (m, 4H, Ar-H, both conformers), 5.07 (m, 1H, N–CH–CH2, conformer B), 4.96 (m, 1H, N–CH–CH2, conformer A), 3.68 (m, 1H, N–C(H)H–CH2, conformer B), 3.62 (m, 1H, N–C(H)H–CH2, conformer A), 3.39 (m, 2H, N–C(H)H–CH2, both conformers), 2.96–2.90 (m, 2H, C=C-(C(H)H)2, conformer B), 2.90–2.81 (m, 2H, C=C—(C(H)H)2, conformer A), 2.41 (s, 6H, Ar–CH3, both conformers), 2.07–1.79 (m, 12H, C=C—(C(H)H)2, N–CH2–(CH2)2, both conformers), 1.75–1.57 (m, 4H, C=C(CH2)2—C(H)H), N–CH2–(CH2)2, both conformers), 1.32–1.12 (m, 2H, C(t-Bu)H–(C(H)H)2, both conformers), 1.06–0.93 (m, 4H, C(t-Bu)H–(C(H)H)2, C(t-Bu)H–(C(H)H)2, both conformers), 0.86 (s, 9H, (CH3)3, conformer A), 0.83 (s, 9H, (CH3)3, conformer B). 13C{1H} NMR (151 MHz, CDCl3, Me4Si) δ 143.1 (Cq), 143.0 (Cq), 138.4 (Cq), 137.9 (Cq), 137.3 (Cq), 136.7 (Cq), 129.3 (2 × CH), 129.3 (2 × CH), 127.4 (2 × CH), 127.2 (2 × CH), 126.3 (Cq), 126.0 (Cq), 58.9 (CH), 58.8 (CH), 49.5 (CH2), 49.4 (CH2), 48.1 (CH), 48.0 (CH), 32.6 (Cq), 32.5 (Cq), 31.9 (CH2), 31.9 (CH2), 31.8 (CH2), 31.7 (CH2), 31.1 (CH2), 31.0 (CH2), 28.7 (CH2) 27.9 (CH2), 27.7 (3 × CH3), 27.6 (3 × CH3), 27.5 (CH2), 27.4 (CH2), 25.5 (CH2), 25.3 (CH2), 21.6 (CH3), 21.6 (CH3). HRMS (ESI) m/z [M + H]+ calcd for C22H33ClNO2S 410.1915, found 410.1912. IR ν max (neat)/cm–1 2948, 2866, 1596, 1344, 1156, 1092, 664.
2-(1-Chlorovinyl)-3-tosyloxazolidine (2r)
Following the described procedure, allene 1r (0.2 mmol, 51 mg) was reacted to obtain 30 mg of 2-(1-chlorovinyl)-3-tosyloxazolidine 2r as a colorless oil (EP/Acetone 92/8, 52% yield). 1H NMR (600 MHz, CDCl3, Me4Si) δ 7.75 (d, J = 8.3 Hz, 2H, Ar-H), 7.33 (d, J = 8.0 Hz, 2H, Ar-H), 5.72 (dm, J = 1.7, 1H, CCl=CHaHb), 5.64 (s, 1H, N–CH–O), 5.49 (d, J = 1.7 Hz, 1H, CCl=CHaHb), 3.97 (m, 1H, N–(CH2)2–O), 3.58 (m, 2H, N–(CH2)2–O), 3.50 (m, 1H, N–(CH2)2–O), 2.43 (s, 3H, CH3). 13C{1H} NMR (151 MHz, CDCl3, Me4Si) δ 144.6 (Cq), 138.5 (Cq), 134.5 (Cq), 130.1 (2 × CH), 127.9 (2 × CH), 116.7 (CH2), 90.8 (CH), 66.1 (CH2), 46.5 (CH2), 21.7 (CH3). HRMS (ESI) m/z [M + H]+ calcd for C12H15ClNO3S 288.0456, found 288.0455. IR ν max (neat)/cm–1 2921, 1631, 1596, 1344, 1161, 1088, 912, 819, 661.
2-(1-Chlorovinyl)-3-tosyl-2,3-dihydrobenzo[d]oxazole (2s)
Following the described procedure, allene 1s (0.2 mmol, 60 mg) was reacted to obtain 34.2 mg of 2-(1-chlorovinyl)-3-tosyl-2,3-dihydrobenzo[d]oxazole 2s as a colorless solid (EP/acetone 95:5, 51% yield). 1H NMR (600 MHz, CDCl3, Me4Si) δ 7.49 (dd, J = 7.8, 1.4 Hz, 1H, Ar-H), 7.45–7.40 (m, 2H, Ar–H), 7.13–7.08 (m, 2H, Ar-H), 6.97 (td, J = 7.8, 1.4 Hz, 1H, Ar-H), 6.89 (td, J = 7.7, 1.2 Hz, 1H, Ar-H), 6.63 (dd, J = 7.9, 1.1 Hz, 1H, Ar-H), 6.20 (bs, 1H, O–CH–N), 5.75 (dd, J = 2.0, 1.0 Hz, CCl=C(H)H), 5.43 (d, J = 2.1 Hz, 1H, CCl=C(H)H), 2.29 (s, 3H, CH3). 13C{1H} NMR (151 MHz, CDCl3, Me4Si) δ 151.2 (Cq), 145.3 (Cq), 136.6 (Cq), 132.9 (Cq), 130.0 (2 × CH), 128.8 (Cq), 127.7 (2 × CH), 127.0 (CH), 122.2 (CH), 117.9 (CH), 117.0 (CH2), 109.9 (CH), 94.2 (CH), 21.7 (CH3). HRMS (ESI) m/z [M + H]+ calcd for C16H15ClNO3S 336.0456, found: 336.0453. IR ν max (neat)/cm–1 2924, 1635, 1595, 1477, 1165, 671. mp 142.1–143.6 °C.
2-(1-Chlorovinyl)-3-(methylsulfonyl)-2,3-dihydrobenzo[d]oxazole (2t)
Following the described procedure, allene 1t (0.2 mmol, 45 mg) was reacted to obtain 25 mg of 2-(1-chlorovinyl)-3-(methylsulfonyl)-2,3-dihydrobenzo[d]oxazole 2t as a colorless oil (EP/acetone 95:5, 48% yield). 1H NMR (600 MHz, CDCl3, Me4Si): 7.39 (dd, J = 7.8, 1.3 Hz, 1H, Ar-H), 7.13 (td, J = 7.8, 1.3 Hz, 1H, Ar-H), 6.99 (td, J = 7.8, 1.1 Hz, 1H, Ar-H), 6.95 (dd, J = 8.0, 1.1 Hz, 1H, Ar-H), 6.40 (d, J = 0.7 Hz, 1H, O–CH–N), 5.81 (dd, J = 2.1, 0.9 Hz, 1H, CCl=C(H)H), 5.53 (d, J = 2.1 Hz, 1H, CCl=C(H)H), 2.85 (s, 3H, CH3). 13C{1H} NMR (151 MHz, CDCl3, Me4Si): 150.9 (Cq), 136.4 (Cq), 128.7 (Cq), 127.0 (CH), 122.6 (CH), 117.3 (CH2), 116.9 (CH), 110.2 (CH), 94.3 (CH), 36.5 (CH3). HRMS (ESI) m/z [M + H]+ calcd for C10H11ClNO3S 260.0143, found 260.0139. IR ν max (neat)/cm–1 2933, 2888 1633, 1470, 1346, 1162, 1054, 742.
2-(1-Chlorovinyl)-3-tosyl-2,3-dihydrobenzo[d]thiazole (2u)
Following the described procedure, allene 1u (0.2 mmol, 65 mg) was reacted to obtain 19 mg of 2-(1-chlorovinyl)-3-tosyl-2,3-dihydrobenzo[d]thiazole2u as a colorless oil (EP/acetone 95:5, 27% yield). 1H NMR (600 MHz, CDCl3, Me4Si): 7.69 (s, 1H, Ar-H) 7.46 (d, J = 8.2 Hz, 2H, Ar-H), 7.15 (d, J = 8.2 Hz, 2H, Ar-H), 7.08 (s, 1H, Ar-H), 7.03 (s, 1H. Ar-H), 6.07 (s, 1H, S–CH–N), 5.68 (s, 1H, CCl=C(H)H), 5.36 (s, 1H, CCl=C(H)H), 2.36 (s, 3H, CH3). 13C{1H} NMR (151 MHz, CDCl3, Me4Si) δ 145.0 (Cq), 139.0 (Cq), 137.2 (Cq), 134.0 (Cq), 132.1 (Cq), 129.7 (2 × CH), 127.2 (2 × CH), 127.5 (CH), 125.8 (CH), 122.6 (CH), 120.5 (CH), 114.2 (CH2), 70.8 (CH), 21.7 (CH3). HRMS (ESI) m/z [M + H]+ calcd for C16H15ClNO2S2 352.0227, found 352.0224. IR ν max (neat)/cm–1 2992, 1554, 1469, 1455, 1156, 680.
Acknowledgments
We thank Dr. Francesco Pellegrino for helping us in the determination of the quantum yield. This work was supported by MIUR (Ministero dell’Istruzione, dell’Università e della Ricerca).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.2c01963.
The authors declare no competing financial interest.
Supplementary Material
References
- Nunnery J. K.; Engene N.; Byrum T.; Cao Z.; Jabba S. V.; Pereira A. R.; Matainaho T.; Murray T. F.; Gerwick W. H. Biosynthetically Intriguing Chlorinated Lipophilic Metabolites from Geographically Distant Tropical Marine Cyanobacteria. J. Org. Chem. 2012, 77 (9), 4198–4208. 10.1021/jo300160e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhukova N. V.; Gloriozova T. A.; Poroikov V. V.; Dembitsky V. M. Halogenated (Cl, Br and I) marine steroids and their biological activities: A brief review. Pharma Innov. J. 2017, 6 (11), 456–462. [Google Scholar]
- Fang W.-Y.; Ravindar L.; Rakesh K. P.; Manukumar H. M.; Shantharam C. S.; Alharbi N. S.; Qin H.-L. Synthetic approaches and pharmaceutical applications of chloro-containing molecules for drug discovery: A critical review. Eur. J. Med. Chem. 2019, 173, 117–153. 10.1016/j.ejmech.2019.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraj R.; Megha P.; Sreedev P. Review Article. Organochlorine pesticides, their toxic effects on living organisms and their fate in the environment. Interdiscip. Toxicol. 2016, 9 (3–4), 90–100. 10.1515/intox-2016-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barluenga J.; Moriel P.; Aznar F.; Valdés C. A Very Simple Synthesis of Chloroalkenes and Chlorodienes by Selective Suzuki Couplings of 1,1- and 1,2-Dichloroethylene. Adv. Synth. Catal. 2006, 348 (3), 347–353. 10.1002/adsc.200505302. [DOI] [Google Scholar]
- Koh M. J.; Nguyen T. T.; Zhang H.; Schrock R. R.; Hoveyda A. H. Direct synthesis of Z-alkenyl halides through catalytic cross-metathesis. Nature 2016, 531 (7595), 459–465. 10.1038/nature17396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitelet A.; Kairouz V.; Lebel H.; Charette A. B.; Evano G. Continuous Flow Chlorination of Alkenyl Iodides Promoted by Copper Tubing. Synthesis 2019, 51 (01), 251–257. 10.1055/s-0037-1610398. [DOI] [Google Scholar]
- Nitelet A.; Evano G. A General Copper-Catalyzed Vinylic Halogen Exchange Reaction. Org. Lett. 2016, 18 (8), 1904–1907. 10.1021/acs.orglett.6b00678. [DOI] [PubMed] [Google Scholar]
- Axelrad G.; Laosooksathit S.; Engel R. Reactions of copper(I) halide complexes of trivalent phosphorus with vinylic halides. J. Org. Chem. 1981, 46 (25), 5200–5204. 10.1021/jo00338a028. [DOI] [Google Scholar]
- Liu L.; Zhang-Negrerie D.; Du Y.; Zhao K. PhICl2 and Wet DMF: An Efficient System for Regioselective Chloroformyloxylation/α-Chlorination of Alkenes/α,β-Unsaturated Compounds. Org. Lett. 2014, 16 (2), 436–439. 10.1021/ol403321n. [DOI] [PubMed] [Google Scholar]
- Pawluć P.; Hreczycho G.; Szudkowska J.; Kubicki M.; Marciniec B. New One-Pot Synthesis of (E)-β-Aryl Vinyl Halides from Styrenes. Org. Lett. 2009, 11 (15), 3390–3393. 10.1021/ol901233j. [DOI] [PubMed] [Google Scholar]
- Yin J.; Gallis C. E.; Chisholm J. D. Tandem Oxidation/Halogenation of Aryl Allylic Alcohols under Moffatt–Swern Conditions. J. Org. Chem. 2007, 72 (18), 7054–7057. 10.1021/jo0711992. [DOI] [PubMed] [Google Scholar]
- Kim K.-M.; Park I.-H. A Convenient Halogenation of α,β-Unsaturated Carbonyl Compounds with OXONE® and Hydrohalic Acid (HBr, HCl). Synthesis 2004, 2004 (16), 2641–2644. 10.1055/s-2004-831232. [DOI] [Google Scholar]
- Weidkamp A. J.; Oestreich M. Metal-free transfer hydrochlorination of internal C–C triple bonds with a bicyclo[3.1.0]hexane-based surrogate releasing two molecules of hydrogen chloride. Chem. Commun. 2022, 58 (7), 973–976. 10.1039/D1CC06591B. [DOI] [PubMed] [Google Scholar]
- Yu P.; Bismuto A.; Morandi B. Iridium-Catalyzed Hydrochlorination and Hydrobromination of Alkynes by Shuttle Catalysis. Angew. Chem., Int. Ed. 2020, 59 (7), 2904–2910. 10.1002/anie.201912803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebule R.; Liang S.; Hammond G. B.; Xu B. Chloride-Tolerant Gold(I)-Catalyzed Regioselective Hydrochlorination of Alkynes. ACS Catal. 2017, 7 (10), 6798–6801. 10.1021/acscatal.7b02567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Shi X.; Fang M.; Xu X. Iron Halide-Mediated Regio- and Stereoselective Halosulfonylation of Terminal Alkynes with Sulfonylhydrazides: Synthesis of (E)-β-Chloro and Bromo Vinylsulfones. J. Org. Chem. 2013, 78 (18), 9499–9504. 10.1021/jo401581n. [DOI] [PubMed] [Google Scholar]
- Iwai T.; Fujihara T.; Terao J.; Tsuji Y. Iridium-Catalyzed Addition of Aroyl Chlorides and Aliphatic Acid Chlorides to Terminal Alkynes. J. Am. Chem. Soc. 2012, 134 (2), 1268–1274. 10.1021/ja209679c. [DOI] [PubMed] [Google Scholar]
- Snelders D. J. M.; Dyson P. J. Efficient Synthesis of β-Chlorovinylketones from Acetylene in Chloroaluminate Ionic Liquids. Org. Lett. 2011, 13 (15), 4048–4051. 10.1021/ol201182t. [DOI] [PubMed] [Google Scholar]
- Giofrè S.; Loro C.; Molteni L.; Castellano C.; Contini A.; Nava D.; Broggini G.; Beccalli E. M. Copper(II)-Catalyzed Aminohalogenation of Alkynyl Carbamates. Eur. J. Org. Chem. 2021, 2021 (11), 1750–1757. 10.1002/ejoc.202100202. [DOI] [Google Scholar]
- Wang W.; Liu L.; Chang W.; Li J. Copper Promoted Regio- and Stereoselective Aminochlorination of Alkynes and Alkenes with NFSI. Chem. Eur. J. 2018, 24 (34), 8542–8547. 10.1002/chem.201801262. [DOI] [PubMed] [Google Scholar]
- Danielec H.; Klügge J.; Schlummer B.; Bach T. FeCl2-Catalyzed Intramolecular Chloroamination Reactions. Synthesis 2006, 2006 (03), 551–556. 10.1055/s-2005-918500. [DOI] [Google Scholar]
- Boyes A. L.; Wild M. The manganese-mediated regioselective chlorination of allenes in synthetic approaches towards the spongistatins and halomon natural products. Tetrahedron Lett. 1998, 39 (37), 6725–6728. 10.1016/S0040-4039(98)01410-5. [DOI] [Google Scholar]
- Laali K. K.; Nandi G. C.; Bunge S. D. Selectfluor-mediated mild oxidative halogenation and thiocyanation of 1-aryl-allenes with TMSX (X = Cl, Br, I, NCS) and NH4SCN. Tetrahedron Lett. 2014, 55 (15), 2401–2405. 10.1016/j.tetlet.2014.02.110. [DOI] [Google Scholar]
- Mueller W. H.; Butler P. E.; Griesbaum K. Allene chemistry. VII. Reaction of chlorine with allene under ionic conditions. J. Org. Chem. 1967, 32 (8), 2651–2654. 10.1021/jo01283a073. [DOI] [Google Scholar]
- Ma S.; Ren H.; Wei Q. Highly Regio- and Stereoselective Halohydroxylation Reaction of 1,2-Allenyl Phenyl Sulfoxides. Reaction Scope, Mechanism, and the Corresponding Pd- or Ni-Catalyzed Selective Coupling Reactions. J. Am. Chem. Soc. 2003, 125 (16), 4817–4830. 10.1021/ja034039q. [DOI] [PubMed] [Google Scholar]
- Liu L.; Ward R. M.; Schomaker J. M. Regioselective Intramolecular Allene Amidation Enabled by an EDA Complex**. Chem. Eur. J. 2020, 26 (61), 13783–13787. 10.1002/chem.202002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale R. S. Chemistry of nitrogen radicals. V. Free radical addition of dialkyl-N-chloramines to olefinic and acetylenic hydrocarbons. J. Org. Chem. 1967, 32 (11), 3263–3273. 10.1021/jo01286a001. [DOI] [Google Scholar]
- Azzi E.; Ghigo G.; Parisotto S.; Pellegrino F.; Priola E.; Renzi P.; Deagostino A. Visible Light Mediated Photocatalytic N-Radical Cascade Reactivity of γ,δ-Unsaturated N-Arylsulfonylhydrazones: A General Approach to Structurally Diverse Tetrahydropyridazines. J. Org. Chem. 2021, 86 (4), 3300–3323. 10.1021/acs.joc.0c02605. [DOI] [PubMed] [Google Scholar]
- Parisotto S.; Garreffa G.; Canepa C.; Diana E.; Pellegrino F.; Priola E.; Prandi C.; Maurino V.; Deagostino A. Visible-Light-Driven Photocatalytic Transformation of α,β-Unsaturated-N-Tosylhydrazones: A Novel Route to Allylic Sulfones. ChemPhotoChem. 2017, 1 (2), 56–59. 10.1002/cptc.201600037. [DOI] [Google Scholar]
- Renzi P.; Azzi E.; Bessone E.; Ghigo G.; Parisotto S.; Pellegrino F.; Deagostino A. Blue light enhanced Heck arylation at room temperature applied to allenes. Org. Chem. Front. 2022, 9 (4), 906–916. 10.1039/D1QO01631H. [DOI] [Google Scholar]
- Jiang H.-J.; Liu K.; Yu J.; Zhang L.; Gong L.-Z. Switchable Stereoselectivity in Bromoaminocyclization of Olefins: Using Brønsted Acids of Anionic Chiral Cobalt(III) Complexes. Angew. Chem., Int. Ed. 2017, 56 (39), 11931–11935. 10.1002/anie.201705066. [DOI] [PubMed] [Google Scholar]
- Miles D. H.; Veguillas M.; Toste F. D. Gold(i)-catalyzed enantioselective bromocyclization reactions of allenes. Chem. Sci. 2013, 4 (9), 3427–3431. 10.1039/c3sc50811k. [DOI] [Google Scholar]
- Jonasson C.; Horváth A.; Bäckvall J.-E. Intramolecular Palladium(II)-Catalyzed 1,2-Addition to Allenes. J. Am. Chem. Soc. 2000, 122 (40), 9600–9609. 10.1021/ja001748k. [DOI] [Google Scholar]
- Davies I. W.; Shaw R. W.; Wisedale R.; Gallagher T. Synthesis of medium ring azacycles via allene-based cyclisations: evaluation of possible mechanistic pathways. J. Chem. Soc. Perk. T. 1 1994, (24), 3557–3561. 10.1039/p19940003557. [DOI] [Google Scholar]
- Parisotto S.; Azzi E.; Lanfranco A.; Renzi P.; Deagostino A. Recent Progresses in the Preparation of Chlorinated Molecules: Electrocatalysis and Photoredox Catalysis in the Spotlight. Reactions 2022, 3 (2), 233–253. 10.3390/reactions3020018. [DOI] [Google Scholar]
- Ruffoni A.; Juliá F.; Svejstrup T. D.; McMillan A. J.; Douglas J. J.; Leonori D. Practical and regioselective amination of arenes using alkyl amines. Nat. Chem. 2019, 11 (5), 426–433. 10.1038/s41557-019-0254-5. [DOI] [PubMed] [Google Scholar]
- Brummond K. M.; DeForrest J. E. Synthesizing Allenes Today (1982–2006). Synthesis 2007, 2007 (06), 795–818. 10.1055/s-2007-965963. [DOI] [Google Scholar]
- Huang X.; Ma S. Allenation of Terminal Alkynes with Aldehydes and Ketones. Acc. Chem. Res. 2019, 52 (5), 1301–1312. 10.1021/acs.accounts.9b00023. [DOI] [PubMed] [Google Scholar]
- Hashmi A. S. K. Synthesis of Allenes by Isomerization Reactions. Modern Allene Chemistry 2004, 2–50. 10.1002/9783527619573.ch1. [DOI] [Google Scholar]
- Pratley C.; Fenner S.; Murphy J. A. Nitrogen-Centered Radicals in Functionalization of sp2 Systems: Generation, Reactivity, and Applications in Synthesis. Chem. Rev. 2022, 122 (9), 8181–8260. 10.1021/acs.chemrev.1c00831. [DOI] [PubMed] [Google Scholar]
- Rogers D. A.; Gallegos J. M.; Hopkins M. D.; Lignieres A. A.; Pitzel A. K.; Lamar A. A. Visible-light photocatalytic activation of N-chlorosuccinimide by organic dyes for the chlorination of arenes and heteroarenes. Tetrahedron 2019, 75 (36), 130498. 10.1016/j.tet.2019.130498. [DOI] [Google Scholar]
- Hering T.; König B. Photocatalytic activation of N-chloro compounds for the chlorination of arenes. Tetrahedron 2016, 72 (48), 7821–7825. 10.1016/j.tet.2016.06.028. [DOI] [Google Scholar]
- Quinn R. K.; Könst Z. A.; Michalak S. E.; Schmidt Y.; Szklarski A. R.; Flores A. R.; Nam S.; Horne D. A.; Vanderwal C. D.; Alexanian E. J. Site-Selective Aliphatic C–H Chlorination Using N-Chloroamides Enables a Synthesis of Chlorolissoclimide. J. Am. Chem. Soc. 2016, 138 (2), 696–702. 10.1021/jacs.5b12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carestia A. M.; Ravelli D.; Alexanian E. J. Reagent-dictated site selectivity in intermolecular aliphatic C–H functionalizations using nitrogen-centered radicals. Chem. Sci. 2018, 9 (24), 5360–5365. 10.1039/C8SC01756E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armarego W. L. F.Purification of Laboratory Chemicals; Butterworth-Heinemann, 2017. [Google Scholar]
- Lu H.; Chen Q.; Li C. Control of the Regioselectivity of Sulfonamidyl Radical Cyclization by Vinylic Halogen Substitution. J. Org. Chem. 2007, 72 (7), 2564–2569. 10.1021/jo0625857. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.