

Abstract
De novo protein design enhances our understanding of the principles that govern protein folding and interactions, and has the potential to revolutionize biotechnology through the engineering of novel protein functionalities. Despite recent progress in computational design strategies, de novo design of protein structures remains challenging, given the vast size of the sequence‐structure space. AlphaFold2 (AF2), a state‐of‐the‐art neural network architecture, achieved remarkable accuracy in predicting protein structures from amino acid sequences. This raises the question whether AF2 has learned the principles of protein folding sufficiently for de novo design. Here, we sought to answer this question by inverting the AF2 network, using the prediction weight set and a loss function to bias the generated sequences to adopt a target fold. Initial design trials resulted in de novo designs with an overrepresentation of hydrophobic residues on the protein surface compared to their natural protein family, requiring additional surface optimization. In silico validation of the designs showed protein structures with the correct fold, a hydrophilic surface and a densely packed hydrophobic core. In vitro validation showed that 7 out of 39 designs were folded and stable in solution with high melting temperatures. In summary, our design workflow solely based on AF2 does not seem to fully capture basic principles of de novo protein design, as observed in the protein surface's hydrophobic vs. hydrophilic patterning. However, with minimal post‐design intervention, these pipelines generated viable sequences as assessed experimental characterization. Thus, such pipelines show the potential to contribute to solving outstanding challenges in de novo protein design.
Keywords: AlphaFold2, computational structural biology, De novo protein design, machine learning, structure prediction network inversion
1. INTRODUCTION
De novo protein design aims to create stable, well‐folded proteins with sequences distant from those found in nature and potentially new functions. To date, de novo proteins remain challenging to design because the solution space expands exponentially with each additional amino acid in the sequence. Therefore, it is crucial to develop new computational methods that capture the underlying principles that govern protein structure, allowing for efficient exploration of the structure‐sequence space and the design of more complex protein folds and functions.
To date, computational protein design has seen significant advances in creating proteins with novel folds and functionalities such as enzymes (Jiang et al., 2008; Röthlisberger et al., 2008), protein–protein interactions (Gainza et al., 2022; Koday et al., 2016; Marchand et al., 2022), protein switches (Giordano‐Attianese et al., 2020; Langan et al., 2019), and vaccines (Correia et al., 2014; Sesterhenn et al., 2020; Yang et al., 2021).
A classical approach in computational design is fixed backbone design, where a novel sequence is fitted to an existing protein topology from the Protein Data Bank (Berman et al., 2000). Backbone and side‐chain rotamer conformations of the residues are sampled and scored with various scoring functions allowing the creation of new protein structures and functions such as zinc finger domains (Dahiyat and Mayo, 1997), protein sensors (Feng et al., 2015; Glasgow et al., 2019), enzymes (Jiang et al., 2008; Röthlisberger et al., 2008; Siegel et al., 2010), and small molecule binders (Tinberg et al., 2013).
Fragment assembly design methods have been extensively used to generate diverse backbones from scratch. This method assembles structural protein fragments into the desired fold which has proven successful in the design of de novo beta barrels (Dou et al., 2018), TIM barrels (Huang et al., 2016), jellyroll structures (Marcos et al., 2018), and various alpha‐beta proteins (Correia et al., 2014; Koga et al., 2012). Fragment assembly has also successfully designed novel protein folds that were not found in nature (Kuhlman et al., 2003).
De novo proteins generated with fragment assembly have also been functionalized by constructing topologies that stabilize a functional motif. This method has resulted in the design of biosensors and vaccine candidates (Sesterhenn et al., 2020; Yang et al., 2021). However, this method is completely dependent on fragments extracted from native proteins that are included in limited libraries and are also notoriously inefficient in generating good quality backbones if structural constraints are not applied.
Recently, de novo protein design has taken an exciting turn with the emergence of deep learning tools for protein modeling, allowing the generation of proteins without relying on fragment libraries that explore diverse solutions of the sequence‐structure space. With an increase in sequence and structural data combined with significant progress in deep learning, these techniques have transformed protein structure prediction and design (Ovchinnikov and Huang, 2021). For example, deep learning has proven to be an excellent tool for sampling the backbone conformational space through, for example, Generative Adversarial Networks (Anand et al., 2022) and Variational Auto‐Encoders (Eguchi et al., 2022; Guo et al., 2021). Deep learning methods can also be used for sequence generation given a target backbone; through the interpolation between sequence and structural data, sequences that fit a target topology can be found (Anand et al., 2022; Dauparas et al., 2022; Ingraham et al., 2019).
The trRosetta structure prediction network (Yang et al., 2020) was recently applied for sequence generation given a target structure. This was achieved by using the error gradient between the sequence and the target contact map to optimize a Position Specific Scoring Matrix (PSSM; Henikoff and Henikoff, 1996; Norn et al., 2020). Protein folding network sequence generation was followed by removal of the structure constraints and only optimizing for protein stability. This method allows the network to “hallucinate” a sequence and structure different from those in nature (Anishchenko et al., 2021). Hallucination involves optimizing a randomly initialized amino acid sequence to a loss function. Experimental data confirmed that several hallucinated proteins adopted the target fold. Additionally, it was shown that a scaffold could be hallucinated to stabilize a functional site, supporting the desired structure (Tischer et al., 2020). The latest version of this hallucination pipeline uses a more accurate prediction pipeline using RoseTTAfold (RF; Baek et al., 2021). Additionally, an inpainting step was used to optimize designs by randomly masking and predicting the most likely amino acids. This method successfully designed de novo proteins with active sites, epitopes, and protein binding sites (Wang et al., 2022).
Recently, AlphaFold2 (AF2), an end‐to‐end structure prediction network, reached unprecedented accuracy levels close to experimental methods for structure determination (Jumper et al., 2021). AF2 predicts 3D atom coordinates of the protein structure given a Multiple Sequence Alignment (MSA) and structures of homologs (templates). Although the network was trained on MSAs containing co‐evolutionary signals and templates as inputs, AF2 can accurately predict protein structures of de novo proteins from a single sequence alone (Pereira et al., 2021). This indicates that AF2 can generalize to de novo designed protein sequences, potentially providing a new tool for protein design.
New ideas for harnessing the power of AF2 followed shortly after its release. The first attempts started with an initialized sequence from a generative model and applied a greedy algorithm to optimize the designs for various loss functions (Jendrusch et al., 2021; Moffat et al., 2021). These AF2‐based design pipelines were used to design monomers, oligomers and protein switches which were validated using various in silico metrics. In recent research, it was also demonstrated how symmetric protein homo‐oligomers could be designed through the help of deep network hallucination using AF2. A backbone was hallucinated using AF2 and the sequence is redesigned using protein MPNN, a message‐passing graph neural network that improves design success rates (Dauparas et al., 2022; Ingraham et al., 2019; Wicky et al., 2022). However, the conformational search of these pipelines is solely based on stochastic Markov Chain Monte Carlo (MCMC) methods and is, therefore, computationally expensive.
We hypothesized that we could devise an efficient design strategy by inverting the AF2 structure prediction network. Hence, a structural loss is backpropagated through AF2 to generate amino acid sequences compatible with a target fold. Through error gradient backpropagation combined with MCMC optimization, we explored several protocols to generate sequences given a target structure using AF2‐based pipelines we call AF2‐design. We analyzed the generated sequences both in silico and in vitro, showing that our AF2‐based protocol can be leveraged for de novo protein design.
2. RESULTS
2.1. AF2‐design methodological approach
The general goal of protein structure prediction networks such as AF2 is to predict the tertiary structure given the sequence. Here, we propose using AF2 for the inverse problem, generating protein sequences given a target backbone. One inversion strategy is error backpropagation (Simonyan et al., 2014), where given a target protein structure, the input sequence is optimized to a loss function. Inspired by the trDesign method (Norn et al., 2020), we developed an AF2‐design pipeline as illustrated in Figure 1a. The first step was to initialize an input sequence and predict its structure. Since we started with non‐natural sequences, natural homologs are unavailable and, therefore, MSAs and structural templates are disabled in the AF2 network (also referred to as single sequence mode).
FIGURE 1.
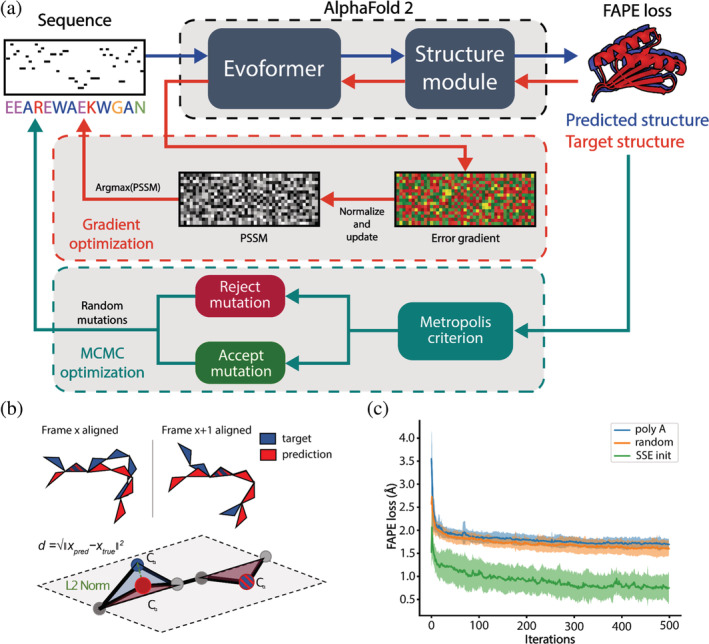
AlphaFold‐based pipeline for sequence design. (a) The design pipeline consists of two sequence optimization methods, gradient descent (GD) and Markov Chain Monte Carlo (MCMC)‐based optimization. In the GD optimization step, the design process is initialized with a secondary‐structure biased sequence, where the structure is predicted upon each sequence change (blue arrows). Next, sequence information from the AlphaFold2 (AF2) network is extracted by backpropagating the error gradient between the predicted and desired target structures (red arrows). This error gradient is then used to optimize a Position Specific Scoring Matrix (PSSM) where the amino acids with the highest probability are used as the input to the next optimization round. After several rounds of gradient‐based optimization, the sequence with the lowest error (Frame Aligned Point Error [FAPE] loss) is selected and used for further MCMC‐based optimization to decrease the distance between the target and the predicted protein structure (green arrows). Random mutations are introduced to the sequence in the refinement step and accepted based on the Metropolis criterion. (b) The FAPE loss is computed by taking the Euclidean distance between the C‐alpha coordinates after alignment to each of the residue frames. (c) Example of design trajectories of the top7 fold using different initialization sequences. SSE sequence initialization was the only approach that converged to the correct folds within 500 iterations.
After predicting the structures of these sequences, the structural error is computed using the Frame Aligned Point Error (FAPE; Jumper et al., 2021). The FAPE loss is similar to the root mean square deviation, however, there is no alignment of the complete structure, making it independent of the orientation and rotation of the structure. The backbone atoms of each residue, which are represented as frames, are aligned and the C‐alpha distances between the predicted and target structure are computed (Figure 1b). Since the FAPE loss was the primary loss component during training, we reasoned that backpropagating this loss would result in the most accurate generation of protein sequences.
AF2 consists of five neural networks, each of which has been trained with different parameters, resulting in the generation of five structural models per input sequence. The advantage of using multiple networks is that it reduces the risk of overfitting to a single model, which means that the designed sequences will be in agreement with all the structural models generated by the five networks. The error gradients in the input sequence are calculated and combined to create an N × 20 matrix that contains the average error gradient for each residue. The size of the matrix depends on the length of the sequence (N), and the error gradient indicates how much each residue contributes to the overall loss. This allows us to apply a gradient descent (GD) algorithm to find a sequence optimized to our loss function. The GD algorithm is used to update a PSSM by updating the entries in the direction of the negative gradient, which means it moves them in a way that reduces the value of the loss function. It can be thought of as the optimization of a probability distribution of the amino acids to a structure. After each round, the most probable amino acids are selected from the PSSM and used in the next iteration as the input. To test the performance of AF2‐design, we sought to de novo design a collection of protein folds. Specifically, we attempted to generate new sequences that would adopt the folds of top7, protein A, protein G, ubiquitin, and a four‐helix bundle (Figure S1 and Table S1). Throughout our design trajectories with AF2‐design, we observed that the initial input sequence significantly impacts the rate of structural convergence of AF2‐design. When initializing the design protocol with a poly‐alanine or a random sequence, AF2‐design did not converge within 500 iterations for any of the attempted folds (Figure 1c). One possible explanation is that when predicting the structure of a poly‐alanine, the resulting structure may be disordered, meaning that the predicted positions of the atoms are highly uncertain. This results in an error gradient that is very noisy, which means that the direction and magnitude of the error changes rapidly and unpredictably. As a result, the optimization algorithm may have difficulty finding a smooth, continuous trajectory toward a folded conformation, which is the desired end state. We hypothesized that this problem could be solved by introducing sequences that favor the local secondary structure propensity; hence we proposed a Secondary Structural Element (SSE) initialization strategy (Figure S2). In the starting sequence, the amino acid identities are assigned according to which SSE they are expected to form: helical residues get assigned alanines, beta‐sheets valines, and loops with glycines (Costantini et al., 2006). This strategy led to convergence to the correct fold within 500 iterations for all the tested design targets. However, since AF2 is deterministic in inference mode, in AF2‐design the same designed sequence will be obtained for each run with the same input. To address this issue, we randomly mutate 10% of the amino acids in the starting sequence for each design trajectory, resulting in the generation of diverse sequences. Additionally, we do not allow the use of cysteines in the designed sequences to avoid an overrepresentation of these residues and disulfide bonds.
In addition to GD optimization, we also implemented a MCMC search algorithm. The MCMC search algorithm randomly samples mutations each step and accepts or rejects these mutations based on the loss function. When we compare MCMC with GD we find that the design performance differs per fold. GD has the advantage of speed, for example, it is 1.5 times faster in each mutation step for a 92 residue protein and is not dependent on random sampling of mutations as is the case for MCMC. We observed that GD allows for a quick convergence within the sequence space, but further improvement in TM scores could be achieved (Zhang and Skolnick, 2004, 2005) by adding an MCMC optimization step (Figures 1a and 2a). We hypothesized that this extra MCMC step allows the escape from local minima where GD converges. During MCMC optimization, four random positions in the sequence are chosen and mutated every iteration. We sample amino acids from a probability distribution of natural protein compositions for the mutations, identical to the trDesign approach (Anishchenko et al., 2021). Finally, the designs with the lowest FAPE loss are selected and relaxed in an AMBER force field to improve structure quality by removing steric clashes (Hornak et al., 2006).
FIGURE 2.
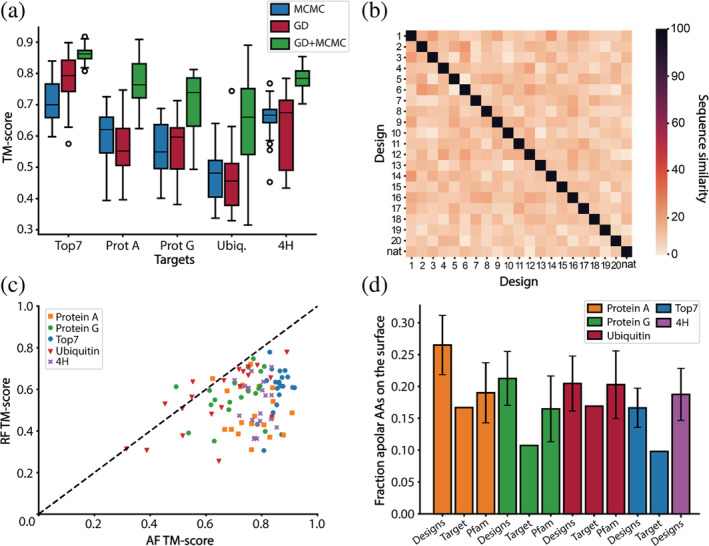
Features of AlphaFold2 (AF2) designed sequences. (a) TM‐scores of designed sequences using Markov Chain Monte Carlo (MCMC), gradient descent (GD) optimization and a combination of both (GD + MCMC). Blue–TM‐scores after MCMC design; red–TM‐scores after GD design; green–TM‐scores after GD design followed by MCMC optimization. For each fold, 20 rounds of GD and MCMC optimization were performed. A combination of GD and MCMC optimization significantly improves the TM scores compared to MCMC and GD only designs. (b) Evaluation of the sequence diversity obtained within the top7 designs. The designed sequences have a low sequence similarity (between 10% and 30%) when compared to one another and to the native sequence. (c) Structure prediction of the AF2 designed sequences using AF2 and RF. In most instances, RF predicts lower TM‐scores than AF2. (d) Fraction of hydrophobics on the surface before the surface redesign step. All the designs have more hydrophobic residues on their surface than their target fold. When comparing the designs to their protein family, we find that the designs of protein A and protein G have slightly more hydrophobics on their surface than similar folds found in nature. Top7 and 4H are de novo proteins hence do not have a protein family, additionally, 4H is a backbone model designed with the TopoBuilder and as such there is no sequence to be compared to.
An additional surface design step was performed with Rosetta to increase solubility and prevent aggregation by removing solvent‐exposed hydrophobic amino acids. Accordingly, surface residues are randomly mutated to hydrophilic amino acids and optimized using the Rosetta energy function (Alford et al., 2017). The refined sequences are then predicted using both AF2 and RF. The final designs are selected based on both TM‐score (>0.6 for AF2 and 0.5 for RF‐generated models) and AF2 confidence in the predicted structure (AF2 pLDDT >60). The pLDDT threshold of 60 represents a balance between having a sufficient number of designs for in vitro testing while enabling an exploration of the utility of this metric for design selection.
2.2. Computational evaluation of designed sequences
To test the performance of AF2‐design, five distinct folds were selected as design targets. The attempted folds were: top7, one of the first de novo proteins (Kuhlman et al., 2003); protein A, ubiquitin, and protein G, small globular folds; 4H, a 4‐helix bundle designed by TopoBuilder (Yang et al., 2021; Figure S1). For each target fold, 20 designs were generated using 500 iteration trajectories of MCMC and GD or both, after which the structural similarity between designs and target is evaluated using TM‐score (Figure 2a).
By combining the GD and MCMC design stages, a good balance between speed and performance was achieved, resulting in improved TM‐scores for all designs (Figure 2a, green boxes). To assess the sequence diversity of the generated designs, we performed an all against all comparison of the designed and native sequences and observed a low sequence similarity between 20% and 30% (Figures 2b and S3). Additionally, we sought to evaluate the designed sequences for structural accuracy using structure prediction tools. We used RF, a structure prediction network developed and trained independently from AF2 (Baek et al., 2021) and compared the results to AF2 predictions. Generally, RF predictions of the designed sequences showed lower TM scores than AF2 (Figure 2c), this is likely related to the fact that the sequences were generated with AF2. Therefore, orthogonal prediction tools may be valuable help to identify the best designs. In further analysis of the designs, we noticed a slight overrepresentation of hydrophobic amino acids on the surface in three out of the four folds where comparisons could be established (Figure 2d). In general, the presence of hydrophobic amino acids at the surface of the protein is seen as unfavorable for the solubility of the designs. Hence, a Rosetta surface redesign step was performed on the AF2‐designed sequences to correct the exposed surface hydrophobics. The resulting sequences were validated by AF2 and RF predictions and the best designs were selected for experimental characterization. When comparing the final models to the presurface redesign models, we noticed that the BLAST e‐values increased, indicating that the proteins became even less similar to those found in nature (Figure S4).
The designs characterized experimentally were generated both by GD‐MCMC optimization as well as MCMC‐only design pipelines; trajectories are shown in Figures S5 and S6. Next, the designs were filtered on AF2 TM scores >0.6, RF TM scores >0.5, and AF2 confidence (pLDDT) > 60 of the predicted structures resulting in 39 designs (Table S1–3). The predicted structures of the designs closely resemble their target folds as shown by structural superpositions (Figures 3a, S7, S8) and the TM scores (Figure 3b). At the sequence level, the designs explore novel sequence space as observed by the high e‐values derived from sequence alignments performed with BLAST on nonredundant protein sequences (Figure 3c) (Gish and States, 1993). In summary, our AF2‐design pipelines can efficiently explore the non‐natural sequence space given a predefined fold and can be validated using orthogonal structure prediction networks.
FIGURE 3.
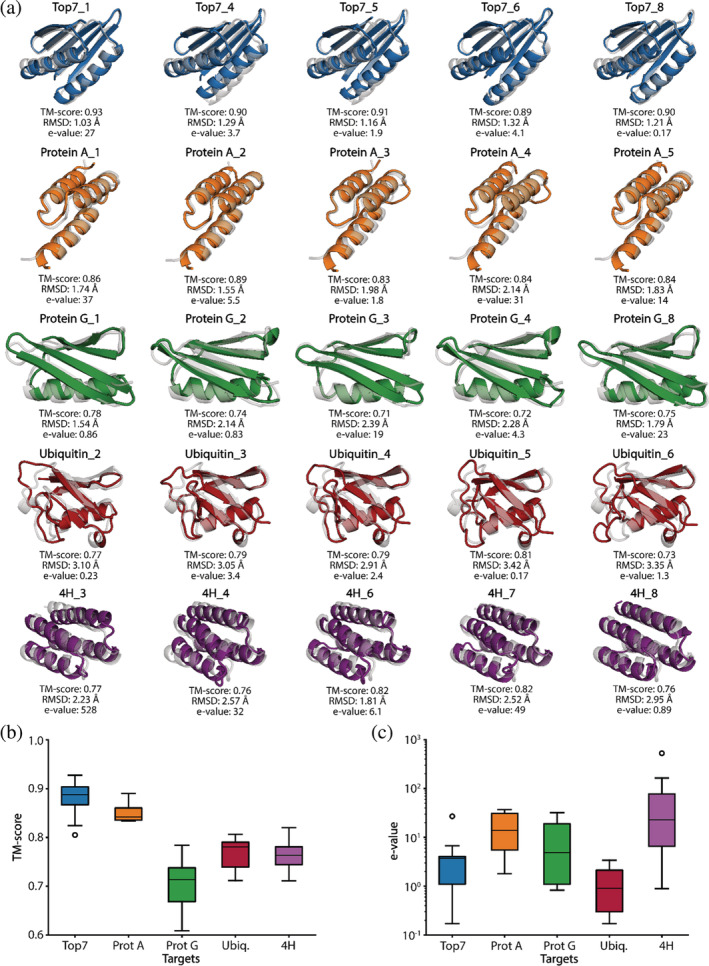
Overview of the structural and sequence properties of the AlphaFold2 generated designs. (a) Alignment of the best design prediction vs. the reference structure (gray) according to the TM‐score. top7–blue, Protein A–orange, Protein G–green, Ubiquitin–red and 4H–purple. (b) TM‐scores of the final designs with a minimum above 0.60. (c) The distribution of the nearest e‐value of all designs.
2.3. In vitro characterization of designed sequences
We next sought to biochemically characterize the AF2‐designed sequences. Thirty‐nine designs were cloned and expressed in E. coli (11 top7, 5 protein A, 9 protein G, 6 ubiquitin, and 8 4H designs). Twenty‐five designs were expressed solubly and purified by affinity and size exclusion chromatography (SEC; 3 top7, 5 protein A, 7 protein G, 6 ubiquitin, and 4 4H designs). Ultimately, only three of the five target folds yielded proteins with acceptable biochemical behavior (protein G, top7, and 4H), meaning that the designs were soluble, folded and had defined oligomeric species in solution. Nine designs from these folds displayed monodisperse peaks indicating monomeric or dimeric states in solution as shown by SEC coupled to multiangle light scattering (Figures 4 and S9). The oligomeric states observed in solution were single‐species monomeric (4H_1) and mixed species between monomer/dimer (top7_1, protein G_1). Several designs showed similar secondary structure content to the original proteins, as assessed by CD spectroscopy. The designs based on de novo backbones (top7 and 4H) showed very high thermal stability with all but one design having melting temperatures (Tm) over 90°C (Figures 4 and S9). The original top7 was also a hyper‐stable protein (Tm > 90°C; Kuhlman et al., 2003); this is an interesting observation given that the designs have only 12.4% of shared sequence identity, suggesting that such backbone may be prone to sequences conferring high thermal stability. The protein G designs showed cooperative unfolding with Tms of 49°C and 46°C, respectively, which are considerably lower than the wildtype protein G (Tm = 89°C; Ross et al., 2001).
FIGURE 4.
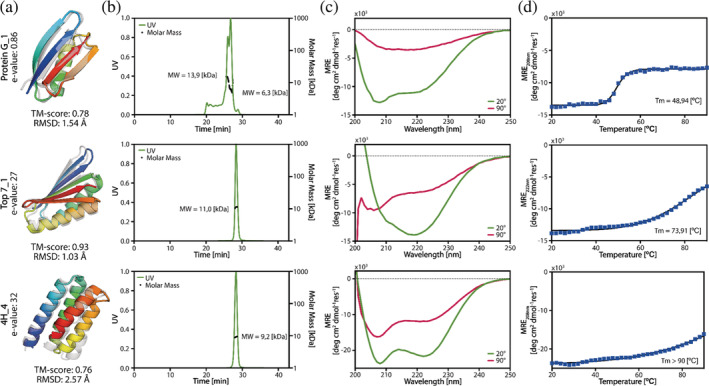
Experimental characterization of the designs. (a) The superimposed structures of the design predicted by AlphaFold2 (colored) versus the original (gray) structure. (b) The SEC‐MALS measurements indicated that protein G_1 and top7_10 appear in monomeric and dimeric forms, whereas 4H_1 appears purely as a monomer. The expected Molecular Weight (MW) of the designs was 7.7 kDa for protein G_1, 11.7 kDa for protein top7_10, and 11.2 kDa for protein 4H_1. (c) The CD spectra of the designs showed that the designs were folded. (d) Temperature melting curves per design. Full experimental characterization of all folded designs can be found in Figure S9.
In light of the experimental results obtained, we sought to learn which metrics could be the best predictors for folded designs. We trained a simple logistic regression model for folded and nonfolded classification based on TM‐score, pLDDT, Rosetta energy score (ddG), and packing score. It is important to highlight that our dataset is extremely small and this analysis should be taken as merely indicative. We tested our model using stratified k‐fold cross‐validation in which the test set of each fold contained one positive and one negative example (for each fold, the test set contained a unique positive sample and a random negative sample). After training and validating our model, we obtained a mean training AUC of 0.86 (±0.02) on the training set and 0.88 (±0.33) on the testing set. Analyzing the logistic regression coefficients, we concluded that the most important features are AF2 and RF TM‐score and AF2 pLDDT (Figure 5a–c). The RF TM‐score showed the most significant impact on the folded classification. Unexpectedly, a high AF2 TM‐score showed to correlate with the unfolded designs. This correlation is likely an effect of filtering on AF2 TM‐score in combination with our small sample size. These results indicate that increasing the RF TM‐scores and AF2 pLDDT filters might increase success rates in obtaining folded designs in vitro. However, and as mentioned above, the number of designs is small and more computational and experimental data is necessary for more robust conclusions. When inspecting the per residue pLDDT of protein G and ubiquitin‐like fold designs, we found regions of the designs with low confidence (ubiquitin) or noticeable lower on the unfolded designs compared to the folded ones (protein G, Figure 5d). This observation is consistent with the findings reported by Listov et al., where per residue pLDDT scores were also utilized as an indicator of problematic areas in designed proteins (Listov et al. 2022). In conclusion, we obtained seven soluble and well‐folded proteins out of 39 designs. Our analysis points to some quality indicators derived from structure prediction networks for selecting folded designed sequences.
FIGURE 5.
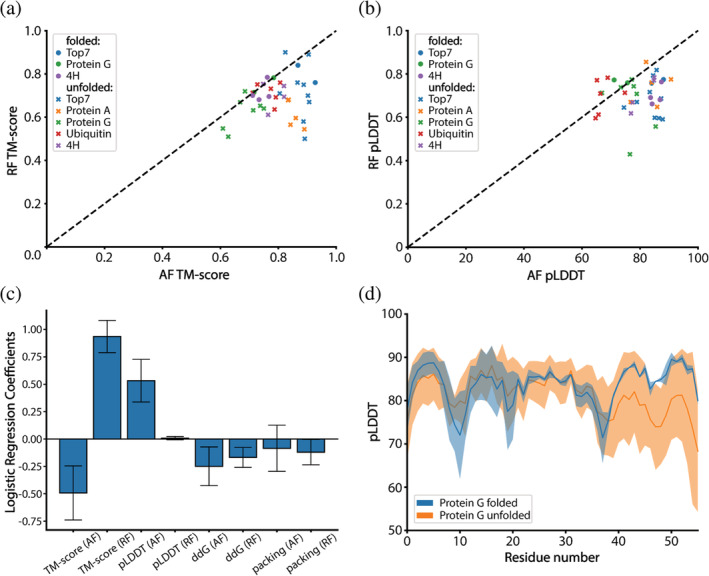
In silico analysis of folded and nonfolded designs. (a) TM‐scores of AlphaFold2 (AF2) predicted models versus RF predicted models. (b) Confidence in the models as predicted by AF2 and RF. (c) Logistic regression coefficients for a model trained on features of all folded and nonfolded designs. The AF2 and RF TM‐score, and AF2 pLDDT have the strongest impact on the classification. (d) Per residue confidence of protein G designs as predicted by AF2. For two of the non‐folded designs, AF2 predicted the two beta sheets at the C‐terminal end with lower confidence than the other beta strands in the structure. The per residue pLDDT for all designs can be found in Figures S10 and S11.
3. CONCLUSION
Machine learning approaches have had a transformative impact on protein structure prediction. Historically, predicting structure from sequence has been considered an essential requirement for protein design endeavors. However, design exercises pose a much more complex challenge due to the vast sequence space and the restricted subset that encodes particular folds. With this challenge in mind, we tested if the state‐of‐the‐art structure prediction network AF2 could be repurposed for design using several sequence‐structure generation and selection workflows. Several important observations arose from our studies for the generated designs: (I) the networks converged relatively fast (small number of steps) into the target folds and showed dependency on the starting sequence; (II) a substantial amount of designs generated lacked the observed/expected hydrophobicity patterns in soluble proteins and displayed a high proportion of hydrophobic residues in solvent‐exposed areas, despite this lack of agreement with the fundamental physicochemical properties of proteins the prediction network still produced structures very similar to those of the target folds; (III) upon refinement of the sequences with Rosetta, specifically of surface positions, 7 out of 39 designs were soluble, folded and stable in solution. Our results demonstrate that a combination of AF2 and RosettaDesign can be used for de novo protein design. However, despite our efforts, we were unable to generate successful designs for protein A and ubiquitin fold in vitro. Several factors may have contributed to this, including the suitability of AF2's training procedure for sequence generation. For instance, during training, 15% of the input sequences in the multiple sequence alignment (MSA) are masked, which may force AF2 to be insensitive to several unfavorable residues in the designed sequence (Jumper et al. 2021). Designing the small core of Ubiquitin and protein A requires a meticulous approach, which our design protocol was likely unable to accomplish. This may also explain the observed strong correlation between RF structures and successful designs, as RF recognizes some of these sequences as incorrect. We speculate that a different training protocol that recognizes and mispredicts some of the incorrect sequences may lead to improved design success. In a recent study, Wicky et al. demonstrated that the use of proteinMPNN in combination with AF2‐based design pipelines led to the generation of proteins with high success rates. Specifically, the sequence was redesigned using proteinMPNN followed by structure prediction with AF2, resulting in a significant increase in the number of expressed and folded designs (Wicky et al. 2022). These findings highlight the potential benefits of incorporating multiple neural networks to validate each other, ultimately enhancing the chances of successful protein design. Hence, in the next iteration of this pipeline, it would likely be beneficial to use the error gradient‐based AF2‐design protocol to generate high‐quality backbones followed by a proteinMPNN sequence generation step. Followed by structure prediction by AF2 and RF, having three independent networks involved in the design process should increase the success rate of our designs and allow the fast design of complex protein folds.
The exploration of the non‐natural sequence space given a protein topology is useful for the design of new functional proteins. Such functional design could be achieved within our pipeline by adding sidechain losses allowing control the configuration of binding sites, as has also been shown by Wang et al. (2022). This framework has potential uses for designing small molecule binding sites, catalytic sites and protein–protein interactions (PPIs). Additionally, for PPI design, the ability of AF2 to predict multimers can be used to sample binder sequences that can be optimized given a positional loss and confidence through error backpropagation. Additionally, a network capable of predicting PPIs, like the MaSIF framework (Gainza et al. 2020), could be used as an extra filter or replace the loss function entirely. This should allow for the design of sequence to function in an end‐to‐end fashion. To conclude, this work demonstrates that inversion of structure prediction networks allows for the design of de novo proteins and holds promise for tackling complex problems such as protein binder and functional site design.
4. METHODS
4.1. Loss function
AF2's loss function was built of several components of which the FAPE loss was the major contributor for the training of the structural module. The FAPE loss measures the L2 norm (Euclidean distance) of all predicted C‐alpha atoms compared to the ground truth, making the loss invariant to rotations and translations. Hence, we chose the FAPE loss to calculate the error gradients guiding our sequence search (Jumper et al. 2021). The FAPE loss can be clamped by an L1 norm, which sets a cap of 10 Å between two C‐alpha positions, allowing for more precise local positioning and reducing the impact of long‐range errors on the overall loss.
4.2. Gradient descent optimization
We initialized the target amino acid sequences based on the secondary structure of the residue derived from the target fold. The secondary structure assignments were then encoded in sequences using alanines for helix, valines for beta sheet, and glycines for loop residues. This introduces a bias toward the correct local structure, aiding faster convergence of the design trajectories. To ensure sequence diversity of the generated designs we randomly mutate 10% of the amino acids in the initial sequence of each design trajectory.
The starting sequence is then one‐hot‐encoded and passed through the AF2 network. AF2 consists of five differently tuned networks to generate five structural models, in our design pipeline we make use of all the five generated models. Next, we compute the mean FAPE of the models and calculate the mean error gradient of the input by taking the average value for each entry in the matrix, resulting in a matrix of shape N × 20, N being the number of residues in the sequence. An empty PSSM of shape N × 20 is initialized with a starting value of 0.01 assigned to the input residues. We used the ADAM optimizer (Kingma and Ba 2017) to update the PSSM with the normalized error gradient. After the update the PSSM is put through a softmax function, converting the matrix into a probability distribution of the amino acids per position. Next, the most probable amino acid identities per position are selected using the argmax function and used to construct the new input sequence for the next iteration. Additionally, we set the values for cysteine in the PSSM to negative infinity resulting in designed sequences without cysteines.
4.3. Markov chain Monte Carlo optimization
As starting inputs we use SSE‐initialized sequences or the designed sequences from GD runs. In each iteration, we mutate four random residues in the sequence to an amino acid sampled from a natural amino acid distribution (Anishchenko et al. 2021). We predict the structure using AF2 with three recycles, enabling more accurate predictions (Mirdita et al. 2022). Recycling entails utilizing the network's outputs as inputs, allowing for iterative refinement of the prediction. Next, we calculate the mean FAPE loss between the predicted structures and target fold and use a Metropolis‐Hastings algorithm to accept or reject the mutated sequence (Hastings 1970). If the introduced mutations result in a reduction of the FAPE loss, the new sequence is accepted. If not, the sequence can be accepted based on the following metropolis criterion:
Where is sampled from a uniform distribution between 0 and 1 and is set to an initial value of 80 and doubled every 250 steps, making it unlikely that structures with a much higher loss are accepted. The Metropolis‐Hastings search allows us to escape from local minima while exploring the sequence space.
4.4. Hardware settings
For the design pipeline one Nvidia Tesla V100 (32GB) was used. The prediction of a protein with a sequence length of 92 (top7) and calculation of the gradients takes ~6 s for one iteration without recycling. For MCMC optimization one iteration took ~9 s using 3 recycles.
4.5. Model settings
We initialized our AF2 network configurations using the settings of “model_5_ptm.” Additionally, since we run AF2 in single sequence mode, we disabled MSAs and template processing in the settings, reducing runtime and memory usage. We run all five components of the AF2 network in parallel, speeding up the prediction time fivefold compared to the original AF2 pipeline.
4.6. Computational design protocol
The five protein folds chosen as design targets were: top7 (1QYS, 2.5 Å x‐ray)–a 92 residue de novo protein with a fold unknown to nature; protein A (1DEE, 2.7 Å x‐ray)–a small three helix bundle with 54 residues; protein G (1FCL, NMR)–a mixed alpha/beta protein with 56 residues; ubiquitin (1UBQ, 1.8 Å x‐ray)–a mainly beta protein with 76 residues; 4H (Rosetta model)–a de novo design with 84 residues constructed using TopoBuilder (Yang et al. 2021) and Rosetta FunFolDes (Bonet et al. 2018).
We employed our AF2‐design pipeline using 500 rounds of GD and 1000–6000 steps of MCMC optimization. Of the experimentally validated and folded designs 1QYS_1 and 1QYS_10 were obtained purely through MCMC optimization.
After AF2 sequence generation a Rosetta fixed backbone design protocol was used to redesign the surface residues. The surface residues were defined as residues with Solvent Accessible Surface Area (SASA) > 40 Å2, and were allowed to mutate to hydrophilic or charged amino acids common in their respective secondary structural element: “DEHKPQR” for helices, “EKNQRST” for beta sheets and “DEGHKNPQRST” for loops. For relaxation and scoring of the designs the REF15 energy function was used (Alford et al. 2017). The TM‐scores and C‐alpha RMSDs between design and target structure were determined using TM‐align (Zhang and Skolnick 2005). The sequence similarity is defined as the number of positions at which the corresponding residue is different. The validation using RF was performed without MSAs or structural templates.
4.7. Protein expression and purification
The designs were ordered as synthetic gene fragments from Twist Bioscience with the addition of a C‐terminal 6‐His‐Tag and cloned into a pET11b vector using NdeI and BlpI restriction sites. The designs were transformed into XL‐10‐Gold cells and the DNA was extracted and validated by Sanger sequencing. The validated DNA sequences were transformed into BL21 DE3 cells and put in 20 mL of LB medium with 100 μg/mL Ampicillin overnight at 37°C as starting cultures. The next day, 500 mL of Auto‐Induction medium with 100 μg/mL Ampicillin was inoculated with 10 mL overnight culture and grown at 37°C to OD of 0.6 then the cultures were grown for ~20 h at 20°C. Bacteria were pelleted by centrifugation and resuspended in lysis buffer (100 mM Tris‐Cl pH 7.5, 500 mM NaCl, 5% Glycerol, 1 mM Phenylmethanesulfonyl fluoride, 1 μg/mL lysozyme and 1:20 of CelLyticTm B Cell Lysis Reagent). The resuspensions were put at room temperature on a shaker at 40 rpm for 2 h and then centrifuged at 48′300 g for 20 min. We filtered the supernatant with a 0.2 μm filter and loaded the mixture on a 5 mL HisTrapTm FF column using an AKTApure system and a predefined method regarding Cytiva's recommendations with that column. We used 50 mM Tris–HCl pH 7.5, 500 mM NaCl, 10 mM Imidazole as was buffer and processed the elution with 50 mM Tris–HCl pH 7.5, 500 mM NaCl, 500 mM Imidazole. We collected the main fraction released through the elution step and injected it on a Gel Filtration column Superdex 16/600 75 pg filled with PBS. The peaks corresponding to the size of the design were collected and concentrated to a concentration of approximately 1 mg/mL for further analysis. Folding and secondary structure content was assessed using circular dichroism in a Chirascan V100 instrument from Applied Photophysics. Melting temperature determination was performed by ranging the temperature from 20°C to 90°C with measurements every 2°C using a wavelength of 208 nm and 222 nm.
AUTHOR CONTRIBUTIONS
Casper A. Goverde: Conceptualization (equal); data curation (equal); formal analysis (equal); investigation (equal); methodology (equal); resources (equal); software (equal); writing – original draft (equal); writing – review and editing (equal). Benedict Wolf: Data curation (equal); formal analysis (equal); investigation (equal); methodology (equal). Hamed Khakzad: Supervision (equal); writing – review and editing (equal). Stephane Rosset: Investigation (equal). Bruno Correia: Funding acquisition (equal); project administration (equal); supervision (equal); writing – original draft (equal); writing – review and editing (equal).
Supporting information
Data S1: Supporting Information
ACKNOWLEDGMENTS
The authors are funded by the European Research Council (Starting grant—716058), the Swiss National Science Foundation, the National Center of Competence in Research in Molecular Systems Engineering, the high‐performance computing facility at EFPL–SCITAS, the support from the Swiss Data Science Center and the CSCS Swiss National Supercomputing Centre. Open access funding provided by Ecole Polytechnique Federale de Lausanne.
Goverde CA, Wolf B, Khakzad H, Rosset S, Correia BE. De novo protein design by inversion of the AlphaFold structure prediction network. Protein Science. 2023;32(6):e4653. 10.1002/pro.4653
Casper A. Goverde and Benedict Wolf contributed equally.
Review Editor: Nir Ben‐Tal
DATA AVAILABILITY STATEMENT
The code used for this study is available in the following repository https://github.com/bene837/af2seq.
REFERENCES
- Alford RF, Leaver‐Fay A, Jeliazkov JR, O'Meara MJ, DiMaio FP, Park H, et al. The Rosetta all‐atom energy function for macromolecular modeling and design. J Chem Theory Comput. 2017;13(6):3031–48. 10.1021/acs.jctc.7b00125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand N, Eguchi R, Huang P‐S. Fully differentiable full‐atom protein backbone generation. 2022. https://openreview.net/forum?id=SJxnVL8YOV
- Anishchenko I, Pellock SJ, Chidyausiku TM, Ramelot TA, Ovchinnikov S, Hao J, et al. De novo protein design by deep network hallucination. Nature. 2021;600(7889):552. 10.1038/s41586-021-04184-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, et al. Accurate prediction of protein structures and interactions using a three‐track neural network. Science. 2021;373(6557):871–6. 10.1126/science.abj8754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–42. 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonet J, Wehrle S, Schriever K, Yang C, Billet A, Sesterhenn F, et al. Rosetta FunFolDes––a general framework for the computational design of functional proteins. PLoS Comput Biol. 2018;14(11):e1006623. 10.1371/journal.pcbi.1006623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia BE, Bates JT, Loomis RJ, Baneyx G, Carrico C, Jardine JG, et al. Proof of principle for epitope‐focused vaccine design. Nature. 2014;507(7491):201–6. 10.1038/nature12966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini S, Colonna G, Facchiano AM. Amino acid propensities for secondary structures are influenced by the protein structural class. Biochem Biophys Res Commun. 2006;342(2):441–51. 10.1016/j.bbrc.2006.01.159 [DOI] [PubMed] [Google Scholar]
- Dahiyat BI, Mayo SL. De novo protein design: fully automated sequence selection. Science. 1997;278(5335):82–7. 10.1126/science.278.5335.82 [DOI] [PubMed] [Google Scholar]
- Dauparas J, Anishchenko I, Bennett N, Bai H, Ragotte RJ, Milles LF, et al. Robust deep learning–based protein sequence design using ProteinMPNN. Science. 2022;378(6615):49–56. 10.1126/science.add2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou J, Vorobieva AA, Sheffler W, Doyle LA, Park H, Bick MJ, et al. De novo design of a fluorescence‐activating β‐barrel. Nature. 2018;561(7724):491. 10.1038/s41586-018-0509-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi RR, Choe CA, Huang P‐S. Ig‐VAE: generative modeling of protein structure by direct 3D coordinate generation. PLoS Comput Biol. 2022;18(6):e1010271. 10.1371/journal.pcbi.1010271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Jester BW, Tinberg CE, Mandell DJ, Antunes MS, Chari R, et al. A general strategy to construct small molecule biosensors in eukaryotes. eLife. 2015;4:e10606. 10.7554/eLife.10606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainza P, Sverrisson F, Monti F, Rodolà E, Boscaini D, Bronstein MM, et al. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat Methods. 2020;17(2):192. 10.1038/s41592-019-0666-6 [DOI] [PubMed] [Google Scholar]
- Gainza P, Wehrle S, Hall‐Beauvais AV, Marchand A, Scheck A, Harteveld Z, et al. De novo design of site‐specific protein interactions with learned surface fingerprints. (p. 2022.06.16.496402) bioRxiv 2022. 10.1101/2022.06.16.496402 [DOI]
- Giordano‐Attianese G, Gainza P, Gray‐Gaillard E, Cribioli E, Shui S, Kim S, et al. A computationally designed chimeric antigen receptor provides a small‐molecule safety switch for T‐cell therapy. Nat Biotechnol. 2020;38(4):432. 10.1038/s41587-019-0403-9 [DOI] [PubMed] [Google Scholar]
- Gish W, States DJ. Identification of protein coding regions by database similarity search. Nat Genet. 1993;3(3):266–72. 10.1038/ng0393-266 [DOI] [PubMed] [Google Scholar]
- Glasgow AA, Huang Y‐M, Mandell DJ, Thompson M, Ritterson R, Loshbaugh AL, et al. Computational design of a modular protein sense‐response system. Science. 2019;366(6468):1024–8. 10.1126/science.aax8780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Du Y, Tadepalli S, Zhao L, Shehu A. Generating tertiary protein structures via an interpretative variational autoencoder. (arXiv:2004.07119). arXiv 2021. 10.48550/arXiv.2004.07119 [DOI] [PMC free article] [PubMed]
- Hastings WK. Monte Carlo sampling methods using Markov chains and their applications. Biometrika. 1970;57(1):13–109. [Google Scholar]
- Henikoff JG, Henikoff S. Using substitution probabilities to improve position‐specific scoring matrices. Bioinformatics. 1996;12(2):135–43. 10.1093/bioinformatics/12.2.135 [DOI] [PubMed] [Google Scholar]
- Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65(3):712–25. 10.1002/prot.21123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P‐S, Feldmeier K, Parmeggiani F, Fernandez Velasco DA, Höcker B, Baker D. De novo design of a four‐fold symmetric TIM‐barrel protein with atomic‐level accuracy. Nat Chem Biol. 2016;12(1):29–34. 10.1038/nchembio.1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingraham J, Garg V, Barzilay R, Jaakkola T. Generative models for graph‐based protein design. Adv Neural Inf Process Syst. 2019;32 https://proceedings.neurips.cc/paper/2019/hash/f3a4ff4839c56a5f460c88cce3666a2b‐Abstract.html [Google Scholar]
- Jendrusch M, Korbel JO, Sadiq SK. AlphaDesign: a de novo protein design framework based on AlphaFold. (p. 2021.10.11.463937). bioRxiv 2021. 10.1101/2021.10.11.463937 [DOI]
- Jiang L, Althoff EA, Clemente FR, Doyle L, Röthlisberger D, Zanghellini A, et al. De novo computational design of Retro‐Aldol enzymes. Science. 2008;319(5868):1387–91. 10.1126/science.1152692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):589. 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers. 1983;22(12):2577–637. 10.1002/bip.360221211 [DOI] [PubMed] [Google Scholar]
- Kingma DP, Ba J. Adam: a method for stochastic optimization. (arXiv:1412.6980). arXiv 2017. 10.48550/arXiv.1412.6980 [DOI]
- Koday MT, Nelson J, Chevalier A, Koday M, Kalinoski H, Stewart L, et al. A computationally designed hemagglutinin stem‐binding protein provides In vivo protection from influenza independent of a host immune response. PLoS Pathog. 2016;12(2):e1005409. 10.1371/journal.ppat.1005409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga N, Tatsumi‐Koga R, Liu G, Xiao R, Acton TB, Montelione GT, et al. Principles for designing ideal protein structures. Nature. 2012;491(7423):222–7. 10.1038/nature11600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a novel globular protein fold with atomic‐level accuracy. Science. 2003;302(5649):1364–8. 10.1126/science.1089427 [DOI] [PubMed] [Google Scholar]
- Langan RA, Boyken SE, Ng AH, Samson JA, Dods G, Westbrook AM, et al. De novo design of bioactive protein switches. Nature. 2019;572(7768):210. 10.1038/s41586-019-1432-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listov D, Lipsh‐Sokolik R, Rosset S, Yang C, Correia BE, Fleishman SJ. Assessing and enhancing foldability in designed proteins. Protein Sci. 2022;31(9):e4400. 10.1002/pro.4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand A, Van Hall‐Beauvais AK, Correia BE. Computational design of novel protein–protein interactions––an overview on methodological approaches and applications. Curr Opin Struct Biol. 2022;74:102370. 10.1016/j.sbi.2022.102370 [DOI] [PubMed] [Google Scholar]
- Marcos E, Chidyausiku TM, McShan AC, Evangelidis T, Nerli S, Carter L, et al. De novo design of a non‐local β‐sheet protein with high stability and accuracy. Nat Struct Mol Biol. 2018;25(11):1034. 10.1038/s41594-018-0141-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold: making protein folding accessible to all. Nat Methods. 2022;19(6):679–82. 10.1038/s41592-022-01488-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat L, Greener JG, Jones DT. Using AlphaFold for rapid and accurate fixed backbone protein design. (p. 2021.08.24.457549). bioRxiv 2021. 10.1101/2021.08.24.457549 [DOI]
- Norn C, Wicky BIM, Juergens D, Liu S, Kim D, Koepnick B, et al. Protein sequence design by explicit energy landscape optimization. (p. 2020.07.23.218917). bioRxiv 2020. 10.1101/2020.07.23.218917 [DOI]
- Ovchinnikov S, Huang P‐S. Structure‐based protein design with deep learning. Curr Opin Chem Biol. 2021;65:136–44. 10.1016/j.cbpa.2021.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira J, Simpkin AJ, Hartmann MD, Rigden DJ, Keegan RM, Lupas AN. High‐accuracy protein structure prediction in CASP14. Proteins. 2021;89(12):1687–99. 10.1002/prot.26171 [DOI] [PubMed] [Google Scholar]
- Ross SA, Sarisky CA, Su A, Mayo SL. Designed protein G core variants fold to native‐like structures: sequence selection by ORBIT tolerates variation in backbone specification. Protein Sci. 2001;10(2):450–4. 10.1110/ps.32501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röthlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453(7192):195. 10.1038/nature06879 [DOI] [PubMed] [Google Scholar]
- Sesterhenn F, Yang C, Bonet J, Cramer JT, Wen X, Wang Y, et al. De novo protein design enables the precise induction of RSV‐neutralizing antibodies. Science. 2020;368(6492):eaay5051. 10.1126/science.aay5051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JB, Zanghellini A, Lovick HM, Kiss G, Lambert AR, St. Clair JL, et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels‐Alder reaction. Science. 2010;329(5989):309–13. 10.1126/science.1190239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonyan K, Vedaldi A, Zisserman A. Deep inside convolutional networks: visualising image classification models and saliency maps. (arXiv:1312.6034). arXiv 2014. 10.48550/arXiv.1312.6034 [DOI]
- Tinberg CE, Khare SD, Dou J, Doyle L, Nelson JW, Schena A, et al. Computational design of ligand‐binding proteins with high affinity and selectivity. Nature. 2013;501(7466):Article 7466. 10.1038/nature12443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischer D, Lisanza S, Wang J, Dong R, Anishchenko I, Milles LF, et al. Design of proteins presenting discontinuous functional sites using deep learning. (p. 2020.11.29.402743) bioRxiv 2020. 10.1101/2020.11.29.402743 [DOI]
- Wang J, Lisanza S, Juergens D, Tischer D, Watson JL, Castro KM, et al. Scaffolding protein functional sites using deep learning. Science. 2022;377(6604):387–94. 10.1126/science.abn2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicky BIM, Milles LF, Courbet A, Ragotte RJ, Dauparas J, Kinfu E, et al. Hallucinating symmetric protein assemblies. Science. 2022;378(6615):56–61. 10.1126/science.add1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Sesterhenn F, Bonet J, van Aalen EA, Scheller L, Abriata LA, et al. Bottom‐up de novo design of functional proteins with complex structural features. Nat Chem Biol. 2021;17(4):Article 4. 10.1038/s41589-020-00699-x [DOI] [PubMed] [Google Scholar]
- Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D. Improved protein structure prediction using predicted interresidue orientations. Proc Natl Acad Sci. 2020;117(3):1496–503. 10.1073/pnas.1914677117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004;57(4):702–10. 10.1002/prot.20264 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Skolnick J. TM‐align: a protein structure alignment algorithm based on the TM‐score. Nucleic Acids Res. 2005;33(7):2302–9. 10.1093/nar/gki524 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Supporting Information
Data Availability Statement
The code used for this study is available in the following repository https://github.com/bene837/af2seq.