

Abstract
A series of peptidomimetic compounds containing benzothiazolyl ketone and [2.2.1] azabicyclic ring was designed, synthesized and evaluated in the hope of obtaining potent oral 3CLpro inhibitors with improved pharmacokinetic properties. Among the target compounds, 11b had the best enzymatic potency (IC50 = 0.110 μM) and 11e had the best microsomal stability (t1/2 > 120 min) and good enzyme activity (IC50 = 0.868 μM). Therefore, compounds 11b and 11e were chosen for further evaluation of pharmacokinetics in ICR mice. The results exhibited that the AUC(0-t) of 11e was 5143 h*ng/mL following single-dose oral administration of 20 mg/kg, and the F was 67.98%. Further structural modification was made to obtain compounds 11g-11j based on 11e. Among them, 11j exhibited the best enzyme inhibition activity against SARS-CoV-2 3CLpro (IC50 = 1.646 μM), the AUC(0-t) was 32473 h*ng/mL (20 mg/kg, po), and the F was 48.1%. In addition, 11j displayed significant anti-SARS-CoV-2 activity (EC50 = 0.18 μM) and low cytotoxicity (CC50 > 50 μM) in Vero E6 cells. All of the above results suggested that compound 11j was a promising lead compound in the development of oral 3CLpro inhibitors and deserved further research.
Keywords: Peptidomimetics, Benzothiazolyl ketone, 3CLpro inhibitor, Pharmacokinetic properties, SARS-CoV-2
Graphical abstract

1. Introduction
COVID-19 is an acute respiratory infectious disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) which has engendered a huge threat to the global economy and public health [1,2]. The ORF1a and ORF1b genes account for about 2/3 of the total length of the SARS-CoV-2 genome and encode two polyproteins [3,4]. The two polyproteins can be cleaved by 3C-like protease (3CLpro) and Papain-like protease (PLpro) to form sixteen functional proteins [5,6]. It is worth mentioning that 3CLpro is responsible for the cleavage of 11 sites on polyproteins and plays an essential role in viral replication and propagation [7,8]. Besides, it has been proven that the catalytic domains of different coronaviruses 3CLpro are highly conservative, thus 3CLpro inhibitors may have a broad spectrum of anti-coronaviral activities [9,10]. In addition, no human protease has high structural homology with the 3CLpro of SARS-CoV-2 [11,12]. Therefore, given the indispensable role of 3CLpro in the viral life cycle [13], the highly conserved structure [14], and the less related homologous protein in humans [15], 3CLpro is an important target for COVID-19 drugs development.
Many covalent peptidomimetics have been reported as 3CLpro inhibitors against COVID-19 [[16], [17], [18], [19], [20], [21]]. Among the reported covalent 3CLpro inhibitors, Paxlovid (co-packaged of nirmatrelvir tablets and ritonavir tablets) has been approved by the FDA for the treatment of COVID-19 on December 22nd, 2021. The 3CLpro inhibitor nirmatrelvir (Fig. 1 ) has a potent antiviral effect on the SARS-CoV-2 original strain and variants, but it is easily metabolized by CYP3A4 [22]. Ritonavir, as an inhibitor of CYP3A4, could effectively increase the blood concentration of nirmatrelvir [23]. Because of complex drug-drug interactions, Paxlovid should not be used by many people who are taking other drugs at the same time [24,25]. Therefore, oral peptidomimetic 3CLpro inhibitors with high activity and good pharmacokinetic properties deserve further investigation.
Fig. 1.

nirmatrelvir.
The catalytic pocket of SARS-CoV-2 3CLpro contains cysteine residues (Cys145), which can be covalently bound with electrophilic groups (warheads) [[26], [27], [28], [29]]. In addition to nitriles, the reported warheads of 3CLpro inhibitors were various as shown in Fig. 2 , including sulfonates, aldehydes, α-ketoamides, vinyl esters, hydroxymethyl ketones, acyloxymethyl ketones, and benzothiazolyl ketones and so on [16,[30], [31], [32], [33], [34], [35], [36]].
Fig. 2.

Structures of some reported peptidomimetic 3CLpro inhibitors.
Benzothiazolyl ketone has been reported as a promising covalent warhead bound to Cys145 of 3CLpro [[37], [38], [39]]. Konno. et al. discovered that YH-53, a peptidomimetic benzothiazolyl ketone compound, exhibited conspicuous activity against SARS-CoV-2 3CLpro [40]. Kneller. et al. reported the joint X-ray/neutron structure of the 3CLpro/BBH-1 complex, and the result showed that BBH-1's benzothiazolyl ketone-warhead reacted with Cys145-SH of 3CLpro [41]. Pfizer's researchers reported that a series of compounds with benzothiazolyl ketone as a covalent warhead exhibited various 3CLpro activities and pharmacokinetic properties, such as PF-1. It is worth mentioning that PF-1 and nirmatrelvir showed similar enzyme activity and pharmacokinetic properties [22].
The methyl substituent on the 6, 6-dimethyl-3-azabicyclo [3.1.0] hexane of nirmatrelvir was one of the major metabolic sites [22]. In order to improve pharmacokinetic properties, we carried out structural modifications of metabolic sites, as shown in Fig. 3 . First of all, we removed the methyl group of the ternary loop on nirmatrelvir. Due to the instability and high reactivity, the ternary loop was also removed. The NS5A inhibitor ledipasvir formed an [2.2.1] azabicyclic ring instead of a pyrrolidine, which could contribute to improving its pharmacokinetic properties [42]. According to the co-crystal structure of nirmatrelvir with SARS-CoV-2 3CLpro [22], the corresponding cavity in the position of pyrrolidine is large. Inspired by ledipasvir, we adopted the same transformation to convert pyrrolidine into [2.2.1] azabicyclic ring. In addition, benzothiazolyl ketone was chosen as the warhead.
Fig. 3.

Design strategy of target compounds.
In order to understand how the compound A binds to SARS-CoV-2 3CLpro, molecular docking studies of compound A were performed in the active sites of 3CLpro structure (PDB ID:7VH8) using Schrodinger, and the results were summarized in Fig. 4 . As shown in Fig. 4A, the binding pattern of compound A (light blue) with 3CLpro was similar to that of nirmatrelvir (green), and the [2.2.1] azabicyclic ring of compound A could be accommodated well in the binding pocket of the 6, 6-dimethyl-3-azabicyclo [3.1.0] hexane of nirmatrelvir. In the binding model (Fig. 4B), the benzothiazoyl ketone warhead of compound A was covalently bound to Cys145, the benzothiazolyl group formed a π-π interaction with His-41, and amide groups interacted with His-164, Glu-166, and Leu-167 to form hydrogen bond interactions.
Fig. 4.

Binding model of compound A into the SARS-CoV-2 3CLpro (PDB ID:7VH8). 4A. Comparing the binding patterns of compound A (blue) and nirmatrelvir (green); 4B. Interaction of compound A (blue) with the residues of SARS-CoV-2 3CLpro, the green dotted lines represented the π-π interaction and the yellow dotted lines represented the hydrogen bond interaction.
In summary, we designed and synthesized a series of peptidomimetic compounds containing benzothiazolyl ketone and an [2.2.1] azabicyclic ring in the hope of obtaining 3CLpro inhibitors with high potency and good pharmacokinetic properties.
2. Results and discussion
2.1. Chemistry
The synthesis of the target compounds 11a-11j was described in Scheme 1 . The reaction of commercially available Methyl(S)-2-(Boc-amino)-3-[(S)-2-oxo-3-pyrrolidinyl]propanoate with N, O-dimethyl hydroxylamine (HN(OMe)Me·HCl) in the presence of the Grignard reagent i-PrMgCl afforded Weinreb amide 2. The Weinreb amide 2 was reacted with benzothiazole in the presence of n-Butyllithium (n-BuLi) via a nucleophilic substitution reaction to give key intermediate 3. Compound 4 was esterified to obtain the intermediate 5, which was then deprotected and coupled with different carboxylic acids 6a-6e to obtain the corresponding N-protected amino acid esters 7a-7e. The intermediates 7a-7e were deprotected and reacted with trifluoromethanesulfonic anhydride or dimethylsulfamoyl chloride to afford compounds 8a-8e. Then 8a-8e were hydrolyzed with lithium hydroxide monohydrate (LiOH·H2O) to furnish the corresponding carboxylic acid fragments 9a-9e. Using the same conditions, compound 10a was obtained by hydrolyzing compound 7a. Compound 3 was deprotected and subsequently coupled with 9a-9e and 10a in the presence of the coupling agent O-(7-Azabenzotriazol-1-yl)-N, N, N′, N′-tetramethyl uronium (HATU) and N, N-diisopropylethylamine (DIEA) to afford the target compounds 11f-11j and 11a. Compound 11a was deprotected and reacted with different anhydrides and organic acids to obtain the target compounds 11b-11e.
Scheme 1.

Synthesis of the target compounds 11a-11j. Reagents and conditions: (a) HN(OMe)Me·HCl, i-PrMgCl (2 M in THF), THF, 0 °C, 3 h; (b) benzothiazole, n-BuLi (1.6 M in THF), THF, −78 °C, 3 h; (c) 4 M HCl in 1, 4-dioxane, DCM, 20–25 °C, 40 min; (d) dimethyl sulfate, NaOH, THF, 65 °C, 2 h; (e) 4 M HCl in 1, 4-dioxane, DCM, 20–25 °C, 40 min; (f) HATU, DIEA, DCM, 20–25 °C, 3–5 h; (g) LiOH·H2O, THF, H2O, MeOH, 25 °C, 3 h; (h) HATU, DIEA, DCM/DMF, 20–25 °C, 3–5 h; (i) 4 M HCl in 1, 4-dioxane, DCM, 20–25 °C, 40 min; (j) trifluoroacetic anhydride/2-fluoroisobutyric acid/acetic hydride-d6/trifluoromethanesulfonic anhydride, Et3N, DCM, 20–25 °C, overnight; (k) 4 M HCl in 1, 4-dioxane, DCM, 20–25 °C, 40 min; (l) dimethylsulfamoyl chloride/trifluoromethanesulfonic anhydride, Et3N, DCM, 0 °C, 1 h, and then 25 °C, 1 h; (m) LiOH·H2O, THF, H2O, MeOH, 25 °C, 3 h; (n) HATU, DIEA, DCM/DMF, 20–25 °C, 3–5 h.
2.2. Biological activity evaluation
2.2.1. SARS-CoV-2 3CLpro inhibitory activities of compounds 11a-11f
The crystal structures of 3CLpro with different compounds have been reported [[43], [44], [45]]. As shown in Fig. 4A, the P4 pocket has a polar gap and a hydrophobic cavity. Therefore, acyl and sulfonyl groups connected with different hydrophobic groups are selected at the R1 position, hoping to obtain compounds with high activity and good pharmacokinetic properties. A series of compounds 11a-11f were designed, synthesized, and evaluated the inhibitory activities against SARS-CoV-2 3CLpro and nirmatrelvir was used as a positive control. The results were summarized in Table 1 . This result showed that compound 11b, which introduced a trifluoroacetyl group at the R1 position, displayed the most excellent activity of all the designed compounds (IC50 = 0.110 μM), but was lower than that of nirmatrelvir (IC50 = 0.035 μM). When the two fluorine atoms of 11b were replaced with slightly larger methyl groups, the activity of 11c decreased 11 folds (IC50 = 1.268 μM). When replacing the three fluorine atoms of 11b with the smaller deuterium atoms, the activity of 11d decreased 5 folds (IC50 = 0.527 μM). In addition, the activity of 11a with the t-butyloxy carbonyl group in the R1 position decreased 45 times (IC50 = 5.003 μM) compared to that of 11b. The above results indicate that the R1 position is sensitive to the occupied space of the substituent group. Besides, compound 11e, which replaced the trifluoroacetyl group of 11b with a trifluoromethosulfonyl group, showed 8 times lower inhibitory activity against 3CLpro (IC50 = 0.868 μM) than that of compound 11b. When replacing the trifluoromethyl group with the dimethylamino group, the activity of compound 11f (IC50 = 0.584 μM) was still lower than that of compound 11b, although slightly higher than that of compound 11e.
Table 1.
Inhibition activities of compounds 11a-11f against SARS-CoV-2 3CLpro.
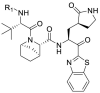 .
.
| Entry no. | R1 | IC50 (μM) |
|---|---|---|
| 11a | 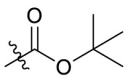 |
5.003 |
| 11b | 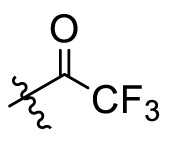 |
0.110 |
| 11c | 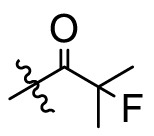 |
1.268 |
| 11d | 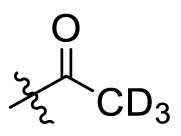 |
0.527 |
| 11e | 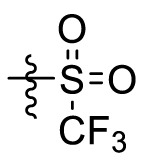 |
0.868 |
| 11f | 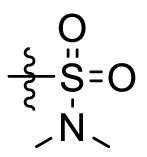 |
0.584 |
| nirmatrelvira | 0.035 |
Used as a positive control.
2.2.2. Microsomal stability of compounds 11b-11f
Based on the good enzyme activities and the concern about the metabolic properties of compounds, the microsomal stability tests of compounds 11b-11f were performed. Nirmatrelvir was used as a positive control. The data on microsomal stability in the species of human and mouse were shown in Table 2 . The microsomal stability varied widely between different compounds. Deuteration is a common strategy to improve metabolic stability in medicinal chemistry. By replacing the trifluoroacetyl group of 11b with the trideuterium acetyl group, the microsomal stability of 11d was improved compared with 11b in both human and mouse species, and was similar to that of nirmatrelvir in human species. The t1/2 of 11b and 11d were 11.60 min and 42.20 min in the human species, respectively, and 7.74 min and 33.75 min in the mouse species. When the two fluorine atoms of 11b were replaced with slightly larger methyl groups, the t1/2 of 11c was similar to that of 11b in both human and mouse species. Compared with other tested compounds, the microsomal stability of compound 11e, which replaced the trifluoroacetyl group of 11b with the trifluoromethosulfonyl group, was significantly improved. The t1/2 of 11e in both human and mouse species were more than 120 min. But when replacing the trifluoromethyl group with the dimethylamino group, the t1/2 of 11f decreased significantly compared to compound 11e.
Table 2.
Microsomal stability of compounds 11b-11f on species of human and mouse.
| Entry no. | t1/2 (minute)b |
|
|---|---|---|
| human | mouse | |
| 11b | 11.60 | 7.74 |
| 11c | 18.03 | 4.82 |
| 11d | 42.20 | 33.75 |
| 11e | >120 | >120 |
| 11f | 3.25 | 0.85 |
| nirmatrelvira | 35.5 | 17.7 |
Used as a positive control.
The tested concentration of compounds was 1 μM.
2.2.3. Pharmacokinetic properties of compounds 11b and 11e
Among the tested compounds, 11b had the best enzymatic potency, and 11e had the best metabolic stability in vitro and good enzyme activity. Therefore, compounds 11b and 11e were chosen for further evaluation of their pharmacokinetic properties. The single-dose pharmacokinetics of 11b and 11e in ICR mice were given in Table 3 . Nirmatrelvir was used as a positive control. This result exhibited that both 11b and 11e showed desirable pharmacokinetic properties. Following single-dose oral administration, the AUC(0-t) of 11e was 2.4 times higher than that of 11b and 5 times higher than that of nirmatrelvir. And the Cmax of 11e was 2.1 times greater than that of 11b and 1.8 times greater than that of nirmatrelvir. In addition, the oral bioavailability of compounds 11b and 11e was greater than that of nirmatrelvir. And 11e displayed better oral bioavailability (F = 67.98%) than that of 11b (F = 47.25%).
Table 3.
Single dose PK of compounds 11b and 11e.
| Entry no. | Administrationa | T1/2 (h) | Cmax (ng/mL) | AUC(0-t) (h*ng/mL) |
AUC(0-∞) (h*ng/mL) |
F (%) |
|---|---|---|---|---|---|---|
| 11b | IV | 0.47 ± 0.09 | -b | 2281 ± 325 | 2284 ± 324 | – |
| PO | 0.91 ± 0.09 | 1597 ± 135 | 2155 ± 168 | 2161 ± 171 | 47.25 ± 3.69 | |
| 11e | IV | 0.74 ± 0.04 | – | 3783 ± 389 | 3857 ± 388 | – |
| PO | 0.76 ± 0.03 | 3342 ± 1243 | 5143 ± 303 | 5153 ± 306 | 67.98 ± 4.01 | |
| nirmatrelvirc | IV | 0.42 ± 0.10 | – | 2240 ± 101 | 2241 ± 101 | – |
| PO | 0.51 ± 0.10 | 1819 ± 695 | 1023 ± 195 | 1029 ± 202 | 22.85 ± 4.35 |
Single IV dose was 10 mg/kg and PO dose was 20 mg/kg.
Not tested.
Used as a positive control.
2.2.4. SARS-CoV-2 3CLpro inhibitory activities of compounds 11g-11j
The compound 11e was chosen for further modification according to its inhibitory activity against 3CLpro and pharmacokinetic properties. Because the groups in R2 position may correspond to a hydrophobic pocket in the structure of 3CLpro, the hydrophobic groups are preferred at R2 position. Compounds 11g-11j were designed, synthesized and evaluated for their inhibitory activities against SARS-CoV-2 3CLpro, in which the R2 position groups were different hydrophobic groups including ethers, p-methoxybenzyl, cyclohexyl methyl and adamantyl. As shown in Table 4 , the results showed that the inhibitory activities of these compounds against 3CLpro ranged from 1.5 μM to 5.5 μM. When replacing the tert-butyl group of 11e with the tert-butoxymethyl group, the enzyme activity of 11g decreased 3 times (IC50 = 2.741 μM) compared to that of 11e (IC50 = 0.868 μM). By replacing the tert-butyl group of 11e with the p-methoxybenzyl group, the enzyme activity of 11h decreased 6 times (IC50 = 5.335 μM) compared to that of 11e. When replacing the p-methoxybenzyl group of 11h with the aliphatic cyclohexyl methyl group, the enzyme activity of 11i (IC50 = 5.140 μM) was slightly decreased compared to 11h. Among compounds 11g-11j, compound 11j, which introduced an adamantyl group at the R2 position, showed the best enzyme inhibition activity. But the 3CLpro inhibitory activity of 11j (IC50 = 1.646 μM) was slightly lower than that of 11e (IC50 = 0.868 μM).
Table 4.
Inhibition activities of compounds 11g-11j against SARS-CoV-2 3CLpro.
 .
.
| Entry no. | R2 | IC50 (μM) |
|---|---|---|
| 11g | 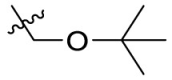 |
2.741 |
| 11h | 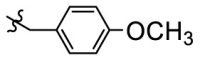 |
5.335 |
| 11i | 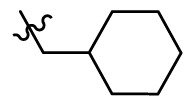 |
5.140 |
| 11j | 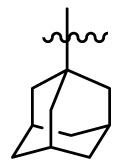 |
1.646 |
| nirmatrelvira | 0.035 |
Used as a positive control.
2.2.5. Pharmacokinetic properties of compound 11j
Although the enzyme inhibitory activity of 11e, which replaces the trifluoroacetyl group of 11b with the trifluoromethosulfonyl group, was slightly lower than that of 11b, the AUC(0-t), Cmax and oral bioavailability of 11e in ICR mice were significantly improved. Therefore, the pharmacokinetic properties of 11j whose R1 position was trifluoromethosulfonyl group, were evaluated. The single-dose pharmacokinetics of 11j in ICR mice were given in Table 5 . The oral bioavailability of 11j was 48.1%, which was similar to that of 11b (F = 47.25%). After oral administration, the AUC(0-t) of 11j (32473 h*ng/mL) was about 6.3 times higher than that of 11e (5143 h*ng/mL), and the T1/2 of 11j (2.1 h) was longer than that of 11e (0.76 h). This result exhibited that compound 11j showed high plasma exposure after oral administration.
Table 5.
Single dose PK of compound 11j.
| Administrationa | T1/2 (h) | Cmax (ng/mL) | AUC(0-t) (h*ng/mL) |
AUC(0-∞) (h*ng/mL) |
F (%) |
|---|---|---|---|---|---|
| IV | 1.38 ± 0.2 | -b | 33780 ± 4258.5 | 34308 ± 4670 | – |
| PO | 2.1 ± 0.8 | 9327 ± 3316 | 32473 ± 16572.8 | 33207 ± 16862 | 48.1 |
Single IV dose was 10 mg/kg and PO dose was 20 mg/kg.
Not tested.
2.2.6. SARS-CoV-2 antiviral activities and cytotoxicities of 11e and 11j
Finally, the antiviral activity tests against SARS-CoV-2 WIV04 and the cytotoxicity tests in Vero E6 cells of compounds 11e and 11j were performed using nirmatrelvir as the positive control. As shown in Table 6 , the antiviral activity of compound 11j (EC50 = 0.18 μM) was better than that of nirmatrelvir (EC50 = 0.24 μM) and compound 11e (EC50 = 0.32 μM). Besides, the CC50 values of 11e and 11j were all more than 50 μM. It was worth mentioning that the EC50 value (0.18 μM) of the antiviral activity of compound 11j in Vero E6 cells was lower than the IC50 value (1.646 μM) of the 3CLpro inhibitory activity. We suspected that the mismatch may be related to many factors, such as the different incubation time of the compound with SARS-CoV-2 3CLpro (10 min) and Vero E6 cells (24 h), and the cell penetration propensity of the compound. The strong hydrophobicity of the benzothiazole unit and the adamantane unit may be conducive to increasing cell penetration [40], but the real reason needs to be deeply explored in the future.
Table 6.
Antiviral activities against SARS-CoV-2 WIV04 and cytotoxicities in Vero E6 cells of compounds 11e and 11j.
| Entry no. | EC50 (μM) | CC50 (μM) | SIa |
|---|---|---|---|
| 11e | 0.32 | 137.4 | 429 |
| 11j | 0.18 | 53.99 | 300 |
| nirmatrelvirb | 0.24 | >500 | >2083 |
SI = CC50/EC50.
Used as a positive control.
In summary, in addition to high plasma exposure after oral administration in ICR mice, 11j exhibited excellent anti-SARS-CoV-2 activity in Vero E6 cells and a large security window (SI = 300). Besides, the Cmax of 11j was 70 times greater than its EC50 in Vero E6 cells. The mouse plasma concentrations at different time after oral administration of 20 mg/kg 11j and the ratio of plasma concentration to EC50 were shown in Table S1.
2.2.7. Protease inhibition selectivity of compound 11j
To evaluate the protease inhibition selectivity of 11j between 3CLpro and the other cysteine proteases, the inhibitory activities of 11j against chymotrypsin, cathepsin B and cathepsin L were tested. As shown in Table 7 , the inhibition of 11j against chymotrypsin, cathepsin B and cathepsin L at 20 μM was 55.79%, −32.93% and 26.96%, respectively. Based on the inhibitory activity of 11j against SARS-CoV-2 3CLpro (IC50 = 1.646 μM), 11j displayed high inhibition selectivity between 3CLpro and other tested cysteine proteases.
Table 7.
Protease inhibition by compound 11j.
| Protease | Inhibition (%)a, 20 μM |
|---|---|
| Chymotrypsin | 55.79 |
| Cathepsin B | −32.93 |
| Cathepsin L | 26.96 |
Data presented is the mean value of two independent determinations.
3. Conclusion
In an effort to develop high efficiency, low toxicity, and oral peptidomimetic 3CLpro inhibitors, a series of peptidomimetic compounds were designed, synthesized, and evaluated. The SARS-CoV-2 3CLpro inhibition activities of compounds 11a-11f, in which the substituent of R1 included different acyl and sulfonyl groups, were evaluated. The compounds displayed varied enzyme inhibition activities and the structure-activity relationship (SAR) of 11a-11f were discussed. The microsomal stability tests showed the half-life of 11e in both species of human and mouse were more than 120 min. Next, compound 11e showed a high AUC(0-t) (po, 20 mg/kg, 5143 h*ng/mL) and better oral bioavailability (F = 67.98%) in ICR mice. The compounds 11g-11j, in which the R2 position groups were different hydrophobic groups, were derivatives of 11e. Among them, the AUC(0-t) of 11j (po, 20 mg/kg, 32473 h*ng/mL) was 6.3 times greater than that of 11e (po, 20 mg/kg, 5143 h*ng/mL) in ICR mice. Furthermore, both compounds 11e and 11j showed good antiviral activities and low cytotoxicities (CC50 > 50 μM) in Vero E6 cells. The EC50 value of 11e was 0.32 μM and the EC50 value of 11j was 0.18 μM. Besides, 11j displayed high inhibition selectivity between 3CLpro and other tested cysteine proteases. In summary, 11j was recognized as a promising lead compound in the development of oral 3CLpro inhibitors for the treatment of a more comprehensive population of COVID-19.
4. Experimental section
4.1. Materials and methods
Reagents and solvents were commercial and were used without further purification. 1H NMR and 13C NMR spectra were recorded using a Bruker 400 MHz, 500 MHz, 600 MHz, or 800 MHz spectrometer with tetramethylsilane as an internal standard. High-resolution mass spectra (HRMS) were measured on a Micromass Ultra Q-TOF spectrometer. All target compounds possessed a purity of ≥95% as determined by HPLC. HPLC analysis was performed using an Agilent 1260 instrument or a Thermo Scientific UltiMate 3000 instrument. The HPLC methods for the target compounds were shown in Table S2.
4.2. Synthesis of 3 and 5
4.2.1. Synthetic procedure for the preparation of tert-butyl((S)-1-(benzo[d]thiazol-2-yl) -3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl) carbamate (3)
In a 500 mL round bottom flask, starting materials Methyl (S)-2-(Boc-amino)-3-[(S)-2-oxo-3-pyrrolidinyl] propanoate (8 g, 28 mmol) and N, O-dimethylhydroxylamine hydrochloride (6.817 g, 70 mmol) were added, followed by the addition of dry THF (80 mL) under nitrogen protection. And the solution was added dropwise to isopropyl magnesium chloride (i-PrMgCl) (98 mL, 2 M in THF) over 30 min using a constant pressure drip funnel at 0 °C, and the solution was stirred for 3 h. The reaction was quenched with a saturated ammonium chloride solution. The mixture was extracted with ethyl acetate, and then dried over Na2SO4. The organic layer was concentrated under reduced pressure, and the resulting residue was purified by eluting through a silica gel column with a 2:1 PE/acetone solvent system to give the pure compound 2 (8.2 g). Yield 90% from 1; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 7.62 (s, 1H), 7.00 (dd, J = 179.1, 7.0 Hz, 1H), 4.39 (dd, J = 34.4, 26.8 Hz, 1H), 3.72 (s, 3H), 3.34–3.33 (m, 1H), 3.17–3.12 (m, 2H), 3.10 (s, 3H), 2.34–2.22 (m, 1H), 2.19–2.12 (m, 1H), 1.94–1.84 (m, 1H), 1.66–1.57 (m, 1H), 1.36 (s, 9H). 13C NMR (126 MHz, DMSO‑d 6): δ 178.13, 155.60, 77.99, 61.18, 49.15, 39.44, 37.74, 32.36, 28.20, 27.26. ESI-MS: m/z 316.4 [M + H]+.
To a solution of benzothiazole (6.9 mL, 63.49 mmol) in THF (40 mL) at −78 °C was added n-BuLi (1.6 M in THF, 28 mL) dropwise over 30 min under nitrogen protection. After 1 h of stirring, the Weinreb amide 2 (4 g, 12.7 mmol) in THF (25 mL) was slowly added over 15 min, and the solution was stirred for 3 h. The reaction was quenched with a saturated ammonium chloride solution. The mixture was extracted with ethyl acetate, and then dried over Na2SO4. The organic layer was concentrated under reduced pressure, and the resulting residue was purified by eluting through a silica gel column with a 3:1 PE/acetone solvent system to give the pure compound 3 (3.9 g). Yield 79% from 2; yellow solid; 1H NMR (400 MHz, DMSO‑d 6): δ 8.25 (t, J = 8.6 Hz, 2H), 7.74 (d, J = 7.0 Hz, 1H), 7.70–7.61 (m, 3H), 5.27 (s, 1H), 3.23–3.16 (m, 2H), 2.27 (s, 1H), 2.07–1.99 (m, 1H), 1.83 (dt, J = 21.5, 10.8 Hz, 2H), 1.45–1.26 (m, 9H). 13C NMR (101 MHz, DMSO‑d 6): δ 193.75, 178.09, 164.49, 155.66, 152.92, 136.31, 128.14, 127.50, 125.18, 123.17, 78.40, 54.95, 38.08, 31.89, 28.11, 27.37. ESI-MS: m/z 390.2 [M + H]+.
4.2.2. Synthesis of 2-(tert-butyl) 3-methyl (1R,3S,4S)-2-azabicyclo[2.2.1]heptane-2,3- dicarboxylate (5)
To a solution of (3S)-N-Boc-2-azabicyclo [2.2.1] heptane-3-carboxylic acid (10 g, 41.45 mmol) in THF (150 mL) at 60 °C was added an aqueous sodium hydroxide solution (2.5 g, 62.17 mmol) and dimethyl sulfate (6 mL, 62.17 mmol) respectively and slowly at the same time. Then the solution was stirred for 2 h. The reaction was monitored by TLC. To the cooled reaction was added 100 mL of water, and the mixture was extracted with ethyl acetate, and then dried over Na2SO4. The organic layer was concentrated under reduced pressure, and the resulting residue was purified by eluting through a silica gel column with a 10:1 PE/EA solvent system to give the pure compound 5 (9 g). Yield 87% from 4; colorless oil; 1H NMR (400 MHz, DMSO‑d 6): δ 4.12 (d, J = 29.4 Hz, 1H), 3.71 (d, J = 6.2 Hz, 1H), 3.63 (d, J = 7.7 Hz, 3H), 2.59 (s, 1H), 1.71 (dd, J = 24.9, 9.9 Hz, 2H), 1.64–1.56 (m, 1H), 1.49 (d, J = 6.3 Hz, 2H), 1.35 (d, J = 33.5 Hz, 9H), 1.28–1.23 (m, 1H). 13C NMR (126 MHz, DMSO‑d 6): δ 171.10, 170.89, 153.22, 152.02, 78.79, 78.60, 63.57, 63.44, 56.92, 55.54, 51.68, 41.99, 41.37, 34.71, 34.05, 30.09, 29.87, 28.06, 27.85, 27.15, 27.06. ESI-MS: m/z 256.0 [M + H]+.
4.3. Synthesis of 7a-7e
The carboxylic acids 6a-6e were commercially available.
4.3.1. Synthesis of methyl (1R,3S,4S)-2-((S)-2-((tert-butoxycarbonyl)amino) -3,3- dimethylbutanoyl)-2-azabicyclo[2.2.1]heptane-3-carboxylate (7a)
To a solution of the N-Boc protected amine 5 (4 g, 15.69 mmol) in DCM (30 mL) was added 4 M HCl in 1, 4-dioxane (40 mL, 156.9 mmol) under nitrogen protection at 20–25 °C. After stirring for 40 min, the mixture was concentrated under reduced pressure to obtain the intermediate 3’. Then, to a solution of 3’ in DCM (40 mL), N-tert-Butylcarbamoyl-L-tert-leucine 6a (3.628 g, 15.69 mmol), and HATU (6.259 g, 15.69 mmol) were added. The resulting solution was cooled to 0 °C under ice bath conditions, and DIEA (8.2 mL, 47.07 mmol) was then added dropwise under nitrogen protection. After adding, the ice bath was removed and the mixture was allowed to stir for 3–5 h at 20–25 °C. Then, the solvent was evaporated under reduced pressure, and the residue was dissolved in ethyl acetate. The organic layer was washed with a saturated ammonium chloride solution and brine. This solution was dried over Na2SO4, filtered, and evaporated under reduced pressure to give the compound 7a (5.2 g) without further purification. Yield 90% from 5; faint yellow solid; 1H NMR (400 MHz, DMSO‑d 6): δ 6.42 (d, J = 9.2 Hz, 1H), 4.55 (s, 1H), 4.21 (d, J = 9.4 Hz, 1H), 3.86 (s, 1H), 3.60 (s, 3H), 2.61 (s, 1H), 1.81 (d, J = 9.8 Hz, 1H), 1.66 (t, J = 11.2 Hz, 4H), 1.48 (dd, J = 18.4, 13.6 Hz, 2H), 1.37 (s, 9H), 0.95 (s, 9H). 13C NMR (126 MHz, DMSO‑d 6): δ 170.29, 168.41, 155.46, 78.14, 63.21, 58.33, 58.08, 51.71, 40.71, 35.12, 34.66, 30.78, 28.17, 27.11, 26.21. ESI-MS: m/z 369.3 [M + H]+.
The compounds 7b-7e were prepared from 6b-6e with 5, using a method similar to that described for the synthesis of 7a.
4.3.2. methyl(1R,3S,4S)-2-(N-(tert-butoxycarbonyl)-O-(tert-butyl)-L-seryl)-2-azabi cyclo[2.2.1]heptane-3-carboxylate (7b)
Yield 88% from 5; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 6.84 (d, J = 8.3 Hz, 1H), 4.48 (s, 1H), 4.28 (dd, J = 14.2, 6.9 Hz, 1H), 3.80 (s, 1H), 3.59 (s, 3H), 3.42–3.36 (m, 2H), 2.60 (s, 1H), 1.88 (d, J = 9.2 Hz, 1H), 1.69 (d, J = 13.6 Hz, 4H), 1.45 (s, 1H), 1.36 (s, 9H), 1.13 (s, 9H). 13C NMR (126 MHz, DMSO‑d 6): δ 170.13, 167.91, 155.08, 78.11, 72.67, 63.28, 61.95, 57.31, 52.33, 51.64, 42.42, 40.72, 38.25, 35.08, 33.30, 30.43, 28.19, 28.09, 27.11, 27.03. ESI-MS: m/z 421.5 [M + Na]+.
4.3.3. methyl(1R,3S,4S)-2-((S)-2-((tert-butoxycarbonyl)amino)-3-(4-methoxyphenyl) propanoyl) -2-azabicyclo[2.2.1]heptane-3-carboxylate (7c)
Yield 87% from 5; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 7.23 (d, J = 8.5 Hz, 2H), 7.03 (d, J = 8.5 Hz, 1H), 6.83 (d, J = 8.6 Hz, 2H), 4.28 (td, J = 8.8, 5.6 Hz, 1H), 4.18 (s, 1H), 3.82 (s, 1H), 3.71 (s, 3H), 3.62 (s, 3H), 2.81 (dd, J = 13.9, 5.5 Hz, 1H), 2.71 (dd, J = 14.6, 9.6 Hz, 1H), 2.59 (s, 1H), 1.77 (d, J = 9.7 Hz, 1H), 1.70–1.61 (m, 3H), 1.43 (dd, J = 12.5, 7.0 Hz, 1H), 1.33 (s, 1H), 1.30 (s, 9H). 13C NMR (126 MHz, DMSO‑d 6): δ 170.26, 169.04, 157.85, 155.14, 130.43, 129.57, 113.46, 77.97, 63.25, 57.31, 54.99, 53.60, 51.74, 40.63, 36.37, 35.18, 30.51, 28.16, 27.08. ESI-MS: m/z 455.4 [M + Na]+.
4.3.4. methyl(1R,3S,4S)-2-((S)-2-((tert-butoxycarbonyl)amino)-3-cyclohexylpropanoyl) -2- azabicyclo[2.2.1]heptane-3-carboxylate (7d)
Yield 89% from 5; yellow solid; 1H NMR (500 MHz, DMSO‑d 6): δ 6.99 (d, J = 8.5 Hz, 1H), 4.28–4.19 (m, 2H), 3.81 (s, 1H), 3.59 (s, 3H), 2.69 (s, 2H), 2.60 (s, 1H), 1.81 (dd, J = 18.2, 11.5 Hz, 2H), 1.69–1.60 (m, 6H), 1.46 (dd, J = 18.0, 8.1 Hz, 2H), 1.36 (s, 9H), 1.32 (s, 1H), 1.18 (ddd, J = 28.1, 12.2, 6.9 Hz, 4H), 0.95–0.84 (m, 2H). 13C NMR (126 MHz, DMSO‑d 6): δ 170.28, 169.80, 155.39, 77.90, 62.98, 57.26, 51.73, 49.23, 40.65, 38.86, 38.25, 35.12, 33.43, 33.39, 31.88, 30.74, 28.22, 27.05, 26.06, 25.98, 25.78. ESI-MS: m/z 409.3 [M + H]+.
4.3.5. methyl(1R,3S,4S)-2-((S)-2-((3S,5S,7S)-adamantan-1-yl)-2-((tert-butoxycarbonyl) amino) acetyl)-2-azabicyclo[2.2.1]heptane-3-carboxylate (7e)
Yield 92% from 5; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 6.32 (d, J = 9.2 Hz, 1H), 4.54 (s, 1H), 4.07 (d, J = 9.4 Hz, 1H), 3.87 (s, 1H), 3.61 (s, 3H), 2.61 (s, 1H), 1.92 (s, 4H), 1.80 (d, J = 9.6 Hz, 1H), 1.66 (d, J = 18.2 Hz, 12H), 1.58 (d, J = 11.6 Hz, 4H), 1.37 (s, 9H). 13C NMR (126 MHz, DMSO‑d 6): δ 170.22, 167.79, 155.46, 78.08, 63.25, 59.14, 58.33, 51.71, 40.68, 37.51, 36.42, 36.25, 35.18, 30.73, 28.15, 27.78, 27.08. ESI-MS: m/z 447.4 [M + H]+.
4.4. Synthesis of methyl(1R,3S,4S)-2-((S)-2-((N,N-dimethylsulfamoyl)amino)-3,3- dimethylbutanoyl)-2-azabicyclo[2.2.1]heptane-3-carboxylate (8a)
To a solution of the compound 7a (368 mg, 1 mmol) in DCM (2 mL) was added 4 M HCl in 1, 4-dioxane (2.5 mL, 10 mmol) under nitrogen protection at 20–25 °C. After stirring for 40 min, the mixture was concentrated under reduced pressure to obtain the intermediate 7a’. Then, to a solution of 7a’ and DCM (2 mL), DIEA (0.52 mL, 3 mmol) was added dropwise, along with dimethylsulfamoyl chloride (93 μL, 0.9 mmol) at 0 °C under nitrogen protection. After adding, the mixture was allowed to stir at 0 °C for 1 h. Then, the ice bath was removed, and the solution was allowed to stir for 1 h at 25 °C. The organic layer was washed with a 1 M HCl solution and brine. This solution was dried over Na2SO4, filtered and evaporated under reduced pressure. The resulting residue was purified by eluting through a silica gel column with a 200:1 DCM/MeOH solvent system to give the pure compound 8a (300 mg). Yield 80% from 7a; white solid; 1H NMR (400 MHz, DMSO‑d 6): δ 7.12 (d, J = 10.1 Hz, 1H), 4.53 (s, 1H), 3.85 (s, 1H), 3.76 (d, J = 10.1 Hz, 1H), 3.62 (s, 3H), 2.63 (s, 1H), 2.58 (s, 6H), 1.82 (d, J = 9.8 Hz, 1H), 1.75–1.63 (m, 3H), 1.46 (t, J = 9.9 Hz, 1H), 1.38 (d, J = 9.7 Hz, 1H), 1.00 (s, 9H). 13C NMR (126 MHz, DMSO‑d 6): δ 170.24, 167.60, 63.47, 61.10, 58.16, 51.68, 40.59, 37.62, 35.17, 35.08, 29.70, 27.24, 26.31. ESI-MS: m/z 376.4 [M + H]+.
4.5. Synthesis of 8b-8e
4.5.1. Synthesis of methyl (1R,3S,4S)-2-(O-(tert-butyl)-N-((trifluoromethyl)sulfonyl) -L-seryl)-2-azabicyclo[2.2.1]heptane-3-carboxylate (8b)
To a solution of the compound 7b (1 g, 2.42 mmol) in DCM (5 mL) was added 4 M HCl in 1, 4-dioxane (6.1 mL, 24.2 mmol) under nitrogen protection at 20–25 °C. After 40 min of stirring, the mixture was concentrated under reduced pressure. Then, to a solution of the corresponding deprotected mixture and DCM (15 mL), Et3N (1 mL, 7.25 mmol) was added dropwise, and trifluoromethanesulfonic anhydride (406 μL, 2.42 mmol) at 0 °C under nitrogen protection. After adding, the mixture was allowed to stir for 1 h at 0 °C. Then, the ice bath was removed, and the solution was allowed to stir for 1 h at 25 °C. The organic layer was washed with a 1 M HCl solution and brine. This solution was dried over Na2SO4, filtered, and evaporated under reduced pressure. The resulting residue was purified by eluting through a silica gel column with a 150:1 DCM/MeOH solvent system to give the pure compound 8b (530 mg). Yield 51% from 7b; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 10.10 (d, J = 8.6 Hz, 1H), 4.44 (s, 1H), 4.21 (dd, J = 8.9, 4.4 Hz, 1H), 3.88 (s, 1H), 3.61 (d, J = 3.3 Hz, 3H), 3.50–3.43 (m, 2H), 2.64 (d, J = 1.7 Hz, 1H), 1.88 (d, J = 9.9 Hz, 1H), 1.75–1.68 (m, 2H), 1.54–1.46 (m, 2H), 1.40 (d, J = 9.9 Hz, 1H), 1.16 (d, J = 3.2 Hz, 9H). 13C NMR (126 MHz, DMSO‑d 6): δ 169.76, 165.89, 73.29, 63.46, 61.95, 57.62, 56.14, 51.78, 40.62, 35.14, 30.32, 26.95. ESI-MS: m/z 431.2 [M + H]+.
The compounds 8c-8e were prepared from 7c-7e with trifluoromethanesulfonic anhydride using a method similar to that described for the synthesis of 8b.
4.5.2. Synthesis of methyl (1R,3S,4S)-2-((S)-3-(4-methoxyphenyl)-2-((trifluoromethyl) sulfonamido)propanoyl)-2-azabicyclo [2.2.1] heptane-3-carboxylate (8c)
Yield 52% from 7c; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 10.03 (d, J = 8.6 Hz, 1H), 7.23 (d, J = 8.6 Hz, 2H), 6.80 (d, J = 8.6 Hz, 2H), 4.13 (s, 1H), 4.09 (td, J = 9.1, 5.1 Hz, 1H), 3.83 (s, 1H), 3.66 (s, 3H), 3.57 (s, 3H), 2.88 (dd, J = 14.0, 5.0 Hz, 1H), 2.68 (dd, J = 14.0, 9.7 Hz, 1H), 2.56 (s, 1H), 1.70 (d, J = 9.9 Hz, 1H), 1.66–1.58 (m, 2H), 1.44–1.36 (m, 2H), 1.28 (d, J = 9.8 Hz, 1H). 13C NMR (126 MHz, DMSO‑d 6): δ 169.86, 167.33, 158.25, 130.72, 127.82, 113.63, 63.40, 57.90, 57.56, 55.03, 51.87, 40.52, 37.26, 35.29, 30.45, 26.90. ESI-MS: m/z 463.2 [M − H]-.
4.5.3. Synthesis of methyl(1R,3S,4S)-2-((S)-3-cyclohexyl-2-((trifluoromethyl) sulfonamido)propanoyl)-2-azabicyclo [2.2.1] heptane-3-carboxylate (8d)
Yield 54% from 7d; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 9.96 (d, J = 8.4 Hz, 1H), 4.19–4.12 (m, 2H), 3.89 (s, 1H), 3.61 (s, 3H), 2.65 (s, 1H), 1.83 (t, J = 11.4 Hz, 2H), 1.74–1.68 (m, 4H), 1.65–1.59 (m, 2H), 1.57–1.40 (m, 6H), 1.18 (dt, J = 18.0, 5.6 Hz, 3H), 1.02–0.92 (m, 2H). 13C NMR (126 MHz, DMSO‑d 6): δ 169.88, 167.98, 63.13, 57.42, 53.46, 51.85, 40.48, 35.17, 33.21, 33.05, 31.19, 30.55, 26.87, 25.90, 25.58. ESI-MS: m/z 441.5 [M + H]+.
4.5.4. Synthesis of methyl (1R,3S,4S)-2-((S)-2-((3S,5S,7S)-adamantan-1-yl)-2-((trifluoromethyl)sulfonamido)acetyl)-2-azabicyclo [2.2.1] heptane-3-carboxylate (8e)
Yield 50% from 7e; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 9.50 (d, J = 9.3 Hz, 1H), 4.58 (s, 1H), 3.92 (d, J = 9.3 Hz, 1H), 3.87 (s, 1H), 3.63 (s, 3H), 2.63 (d, J = 2.9 Hz, 1H), 1.96 (s, 3H), 1.80 (d, J = 9.9 Hz, 1H), 1.72 (dd, J = 19.1, 8.2 Hz, 4H), 1.69–1.64 (m, 6H), 1.56 (dd, J = 23.4, 11.9 Hz, 5H), 1.45 (t, J = 9.4 Hz, 1H), 1.38 (d, J = 9.8 Hz, 1H). 13C NMR (126 MHz, DMSO‑d 6): δ 170.03, 165.40, 63.70, 58.38, 51.80, 40.57, 37.27, 36.78, 36.13, 35.25, 29.89, 27.74, 27.08. ESI-MS: m/z 479.5 [M + H]+.
4.6. Synthesis of 9a-9e, 10a
4.6.1. Synthesis of (1R, 3S, 4S)-2-((S)-2-((N, N-dimethylsulfamoyl) amino)-3,3-dimethylbutanoyl)-2-azabicyclo [2.2.1] heptane-3-carboxylic acid (9a)
To a solution of the compound 8a (300 mg, 0.8 mmol) in THF (2 mL) at room temperature, added LiOH·H2O (50 mg, 1.2 mmol) in water (1 mL) and methanol (1 mL). After stirring for 3 h at 25 °C, the solvent was evaporated under reduced pressure, and the resulting residue was dissolved in water. The pH of the mixture was adjusted to 3–4 by adding 1 M HCl (aq) dropwise. Then, the precipitated white solid was filtered and dried to obtain the compound 9a (270 mg). Yield 93% from 8a; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 12.45 (s, 1H), 7.12 (d, J = 10.1 Hz, 1H), 4.48 (s, 1H), 3.74 (d, J = 11.4 Hz, 2H), 2.61 (d, J = 2.7 Hz, 1H), 2.57 (s, 6H), 1.85 (d, J = 9.7 Hz, 1H), 1.68 (ddd, J = 30.3, 19.4, 6.5 Hz, 3H), 1.42 (dd, J = 13.7, 6.7 Hz, 1H), 1.35 (d, J = 9.6 Hz, 1H), 0.99 (s, 9H). 13C NMR (126 MHz, DMSO‑d 6): δ 171.20, 167.51, 63.81, 61.13, 58.16, 40.57, 37.61, 35.12, 35.08, 29.77, 27.41, 26.35. ESI-MS: m/z 362.4 [M + H]+.
The compounds 9b-9e and 10a were prepared from 8b-8e and 7a, using a method similar to that described for the synthesis of 9a.
4.6.2. Synthesis of (1R, 3S, 4S)-2-(O-(tert-butyl)-N-((trifluoromethyl) sulfonyl)-L-seryl)-2-azabicyclo [2.2.1] heptane-3-carboxylic acid (9b)
Yield 96% from 8b; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 12.49 (s, 1H), 10.09 (s, 1H), 4.39 (s, 1H), 4.20 (t, J = 6.7 Hz, 1H), 3.78 (s, 1H), 3.51–3.48 (m, 1H), 3.43 (d, J = 6.3 Hz, 1H), 2.64 (s, 1H), 1.90 (d, J = 9.8 Hz, 1H), 1.71 (t, J = 8.2 Hz, 2H), 1.48 (dd, J = 29.3, 9.0 Hz, 2H), 1.38 (d, J = 9.7 Hz, 1H), 1.15 (s, 9H). 13C NMR (126 MHz, DMSO‑d 6): δ 170.65, 165.81, 73.26, 63.82, 61.95, 57.61, 56.40, 40.57, 35.07, 30.43, 26.97. ESI-MS: m/z 417.3 [M + H]+.
4.6.3. Synthesis of (1R, 3S, 4S)-2-((S)-3-(4-methoxyphenyl)-2-((trifluoromethyl) sulfonamido)propanoyl)-2-azabicyclo [2.2.1] heptane-3-carboxylic acid (9c)
Yield 92% from 8c; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 12.59 (s, 1H), 10.09 (s, 1H), 7.30 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.6 Hz, 2H), 4.23 (s, 1H), 4.14 (d, J = 5.1 Hz, 1H), 3.81 (s, 1H), 3.73 (s, 3H), 2.96 (dd, J = 14.1, 4.6 Hz, 1H), 2.75 (dd, J = 14.0, 10.0 Hz, 1H), 2.64 (s, 1H), 1.83 (d, J = 9.8 Hz, 1H), 1.74–1.67 (m, 2H), 1.50 (t, J = 9.3 Hz, 1H), 1.43 (t, J = 9.1 Hz, 1H), 1.36 (d, J = 9.7 Hz, 1H). 13C NMR (126 MHz, DMSO‑d 6): δ 170.76, 167.33, 158.21, 130.72, 113.63, 63.75, 58.08, 57.57, 55.03, 40.50, 37.29, 35.24, 30.57, 27.09. ESI-MS: m/z 451.3 [M + H]+.
4.6.4. Synthesis of (1R, 3S, 4S)-2-((S)-3-cyclohexyl-2-((trifluoromethyl)sulfonamido) propanoyl)-2-azabicyclo [2.2.1] heptane-3-carboxylic acid (9d)
Yield 93% from 8d; white solid; 1H NMR (800 MHz, DMSO‑d 6): δ 12.53 (s, 1H), 9.94 (d, J = 8.1 Hz, 1H), 4.15 - 4.12 (m, 2H), 3.79 (s, 1H), 2.64 (s, 1H), 1.86 (d, J = 10.0 Hz, 1H), 1.81 (d, J = 12.2 Hz, 1H), 1.70 (ddd, J = 14.0, 9.7, 6.0 Hz, 4H), 1.64–1.60 (m, 2H), 1.58–1.55 (m, 1H), 1.50 (d, J = 11.0 Hz, 1H), 1.48–1.45 (m, 1H), 1.44–1.41 (m, 2H), 1.39 (d, J = 9.9 Hz, 1H), 1.21 (ddd, J = 12.1, 7.6, 2.6 Hz, 1H), 1.17–1.13 (m, 2H), 0.98 (ddd, J = 26.1, 17.2, 7.3 Hz, 2H). 13C NMR (201 MHz, DMSO‑d 6): δ 170.77, 167.92, 63.51, 57.38, 53.52, 40.46, 35.10, 33.24, 33.05, 31.19, 30.60, 27.05, 25.91, 25.58. ESI-MS: m/z 427.5 [M + H]+.
4.6.5. Synthesis of (1R, 3S, 4S)-2-((S)-2-((3S, 5S, 7S)-adamantan-1-yl)-2-((trifluoro methyl)sulfonamido)acetyl)-2-azabicyclo [2.2.1] heptane-3-carboxylic acid (9e)
Yield 96% from 8e; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 12.50 (s, 1H), 9.49 (d, J = 9.3 Hz, 1H), 4.54 (s, 1H), 3.91 (d, J = 9.3 Hz, 1H), 3.77 (s, 1H), 2.62 (d, J = 3.5 Hz, 1H), 1.94 (s, 3H), 1.86 (d, J = 9.8 Hz, 1H), 1.80–1.70 (m, 4H), 1.65 (dd, J = 17.7, 12.2 Hz, 6H), 1.55 (dd, J = 25.6, 11.6 Hz, 5H), 1.42 (t, J = 10.4 Hz, 1H), 1.36 (d, J = 9.6 Hz, 1H). 13C NMR (126 MHz, DMSO‑d 6): δ 171.11, 165.26, 63.75, 58.32, 40.47, 37.23, 36.81, 36.11, 35.11, 29.99, 27.78, 27.26. ESI-MS: m/z 465.4 [M + H]+.
4.6.6. Synthesis of (1R, 3S, 4S)-2-((S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethyl butanoyl)-2-azabicyclo [2.2.1] heptane-3-carboxylic acid (10a)
Yield 96% from 7a; white solid; 1H NMR (400 MHz, DMSO‑d 6): δ 12.39 (s, 1H), 6.36 (d, J = 9.3 Hz, 1H), 4.52 (s, 1H), 4.20 (d, J = 9.5 Hz, 1H), 3.77 (s, 1H), 2.60 (s, 1H), 1.85 (d, J = 11.4 Hz, 1H), 1.78–1.50 (m, 4H), 1.45–1.41 (m, 1H), 1.37 (s, 9H), 0.95 (s, 9H). 13C NMR (126 MHz, DMSO‑d 6): δ 171.20, 168.30, 155.40, 78.08, 63.53, 58.30, 58.05, 40.66, 35.02, 34.71, 30.85, 28.14, 27.26, 26.22. ESI-MS: m/z 355.3 [M + H]+.
4.7. Synthesis of 11a-11j
4.7.1. Synthesis of tert-butyl ((S)-1-((1R,3S,4S)-3-(((S)-1-(benzo[d]thiazol-2-yl)-1-oxo -3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamoyl)-2-azabicyclo[2.2.1]heptan-2-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (11a)
To a solution of the intermediate 3 (660 mg, 1.69 mmol) in DCM (4 mL) was added 4 M HCl in 1, 4-dioxane (4.3 mL, 16.9 mmol) under nitrogen protection at 20–25 °C. After stirring for 40 min, the mixture was concentrated under reduced pressure to obtain the intermediate 3’. Then, to a solution of 3’ in DCM (5 mL) and DMF (5 mL), the compound 10a (600 mg, 1.69 mmol), and HATU (677 mg, 1.69 mmol) were added. The resulting solution was cooled to 0 °C under ice bath conditions, and DIEA (884 μL, 5.07 mmol) was then added dropwise under nitrogen protection. After adding, the ice bath was removed and the mixture was allowed to stir for 3–5 h at 20–25 °C. Then, the dichloromethane was evaporated under reduced pressure, and the residue was dissolved in ethyl acetate. The organic layer was washed with a saturated ammonium chloride solution and brine. This solution was dried over Na2SO4, filtered, and evaporated under reduced pressure. The resulting crude compound was purified by silica gel column chromatography using 200:1 DCM/MeOH as eluents to obtain compound 11a (602 mg). Yield 57% from 10a; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 8.67 (d, J = 7.9 Hz, 1H), 8.27 (dd, J = 7.2, 1.8 Hz, 1H), 8.24–8.21 (m, 1H), 7.66 (td, J = 7.0, 1.4 Hz, 2H), 7.63 (s, 1H), 6.47 (d, J = 9.2 Hz, 1H), 5.59 (ddd, J = 11.4, 8.1, 3.0 Hz, 1H), 4.48 (d, J = 10.3 Hz, 1H), 4.18 (d, J = 9.3 Hz, 1H), 3.91 (s, 1H), 3.19 (t, J = 9.1 Hz, 1H), 3.10 (dd, J = 16.5, 9.1 Hz, 1H), 2.58 (td, J = 11.3, 3.5 Hz, 1H), 2.46 (s, 1H), 2.33–2.26 (m, 1H), 2.09–2.03 (m, 1H), 1.98 (d, J = 9.4 Hz, 1H), 1.82–1.76 (m, 2H), 1.65 (s, 3H), 1.37 (s, 9H), 1.28–1.19 (m, 2H), 0.94 (s, 9H). 13C NMR (126 MHz, DMSO‑d 6): δ 193.15, 178.15, 169.95, 168.04, 164.31, 155.51, 152.90, 136.38, 128.19, 127.56, 125.18, 123.22, 77.97, 64.04, 58.19, 58.09, 52.80, 41.16, 37.58, 34.83, 34.54, 32.83, 30.87, 28.16, 27.48, 27.35, 26.30. ESI-MS: m/z 626.3 [M + H]+. HRMS (ESI): m/z calcd for C32H44N5O6S [M + H]+ 626.3007, found 626.3007. HPLC purity: 95.15% (Rt: 17.464 min).
The compounds 11f-11j were prepared from 9a-9e with 3, using a method similar to that described for the synthesis of 11a.
4.7.2. Synthesis of (1R, 3S, 4S)-N-((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxo pyrrolidin-3-yl)propan-2-yl)-2-((S)-2-((N,N-dimethylsulfamoyl)amino)-3,3-dimethyl butanoyl)-2-azabicyclo [2.2.1] heptane-3-carboxamide (11f)
Yield 57% from 9a; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 8.69 (d, J = 8.2 Hz, 1H), 8.29–8.26 (m, 1H), 8.24–8.20 (m, 1H), 7.67 (dq, J = 6.9, 5.6 Hz, 2H), 7.63 (s, 1H), 7.12 (d, J = 10.0 Hz, 1H), 5.64 (ddd, J = 11.5, 8.3, 3.0 Hz, 1H), 4.44 (s, 1H), 3.93 (s, 1H), 3.75 (d, J = 10.0 Hz, 1H), 3.20–3.09 (m, 2H), 2.59 (s, 6H), 2.48 (s, 1H), 2.34–2.28 (m, 1H), 2.08–1.99 (m, 2H), 1.85–1.76 (m, 2H), 1.72–1.60 (m, 3H), 1.33 (d, J = 10.8 Hz, 1H), 1.23 (s, 2H), 0.98 (s, 9H). 13C NMR (126 MHz, DMSO‑d 6): δ 193.11, 178.22, 169.87, 167.30, 164.29, 152.91, 136.39, 128.22, 127.58, 125.19, 123.24, 64.22, 61.14, 58.04, 52.64, 41.13, 37.64, 37.57, 35.13, 34.84, 32.99, 29.93, 27.47, 26.37. ESI-MS: m/z 633.4 [M + H]+. HRMS (ESI): m/z calcd for C29H41N6O6S2 [M + H]+ 633.2524, found 633.2529. HPLC purity: 96.96% (Rt: 7.952 min).
4.7.3. Synthesis of (1R, 3S, 4S)-N-((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxo pyrrolidin-3-yl)propan-2-yl)-2-(O-(tert-butyl)-N-((trifluoromethyl)sulfonyl)-L-seryl)-2-azabicyclo [2.2.1] heptane-3-carboxamide (11g)
Yield 43% from 9b; white solid; 1H NMR (800 MHz, DMSO‑d 6): δ 10.11 (s, 1H), 8.64 (s, 1H), 8.27 (d, J = 8.0 Hz, 1H), 8.21 (d, J = 7.9 Hz, 1H), 7.67–7.65 (m, 3H), 5.42 (t, J = 7.6 Hz, 1H), 4.33 (s, 1H), 4.20 (s, 1H), 3.90 (s, 1H), 3.59–3.56 (m, 1H), 3.41 (d, J = 8.0 Hz, 1H), 3.20 (d, J = 8.9 Hz, 1H), 3.13 (d, J = 7.3 Hz, 1H), 2.47 (dd, J = 10.5, 3.6 Hz, 1H), 2.43 (s, 1H), 2.30–2.27 (m, 1H), 2.09 (d, J = 10.3 Hz, 1H), 2.05 (d, J = 8.9 Hz, 1H), 1.81 (ddd, J = 12.6, 9.2, 5.4 Hz, 2H), 1.68 (d, J = 7.7 Hz, 2H), 1.53 (s, 1H), 1.36–1.33 (m, 1H), 1.22 (d, J = 4.1 Hz, 1H), 1.15 (s, 9H). 13C NMR (201 MHz, DMSO‑d 6): δ 193.26, 178.65, 169.78, 164.90, 153.32, 136.78, 128.61, 128.01, 125.53, 123.68, 73.80, 65.17, 58.14, 53.70, 41.47, 38.13, 35.49, 32.82, 30.98, 27.76, 27.56, 27.49, 27.35. 19F NMR (753 MHz, DMSO‑d 6): δ −77.33. ESI-MS: m/z 688.3 [M + H]+. HRMS (ESI): m/z calcd for C29H37F3N5O7S2 [M + H]+ 688.2081, found 688.20. HPLC purity: 95.02% (Rt: 17.845 min).
4.7.4. Synthesis of (1R, 3S, 4S)-N-((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxo pyrrolidin-3-yl)propan-2-yl)-2-((S)-3-(4-methoxyphenyl)-2-((trifluoromethyl)sulfonamido)propanoyl)-2-azabicyclo [2.2.1] heptane-3-carboxamide (11h)
Yield 45% from 9c; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 10.11 (d, J = 8.7 Hz, 1H), 8.74 (d, J = 7.3 Hz, 1H), 8.28 (d, J = 5.2 Hz, 1H), 8.23 (dd, J = 7.2, 1.7 Hz, 1H), 7.69–7.65 (m, 3H), 7.30 (d, J = 8.6 Hz, 2H), 6.86 (d, J = 8.6 Hz, 2H), 5.45–5.42 (m, 1H), 4.24 (s, 1H), 4.12 (dd, J = 9.6, 3.6 Hz, 1H), 3.92 (s, 1H), 3.72 (s, 3H), 3.22–3.18 (m, 1H), 3.11 (d, J = 10.2 Hz, 1H), 2.73–2.68 (m, 1H), 2.45 (s, 1H), 2.32 (dd, J = 11.8, 5.9 Hz, 1H), 2.11 (d, J = 11.0 Hz, 1H), 2.02–1.99 (m, 1H), 1.86–1.80 (m, 2H), 1.69 (s, 2H), 1.51 (d, J = 10.5 Hz, 1H), 1.35 (d, J = 11.7 Hz, 1H), 1.28 (d, J = 9.5 Hz, 1H), 1.23 (d, J = 5.0 Hz, 2H). 13C NMR (201 MHz, DMSO‑d 6): δ 192.85, 178.18, 169.45, 167.82, 164.46, 158.16, 152.86, 136.32, 130.64, 128.21, 128.15, 127.54, 125.10, 123.22, 113.63, 64.68, 58.21, 57.58, 55.00, 53.39, 40.94, 37.79, 37.14, 35.20, 32.25, 30.59, 27.34, 27.13. ESI-MS: m/z 722.4 [M + H]+. HRMS (ESI): m/z calcd for C32H35F3N5O7S2 [M + H]+ 722.1925, found 722.1922. HPLC purity: 96.44% (Rt: 17.868 min).
4.7.5. Synthesis of (1R, 3S, 4S)-N-((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxo pyrrolidin-3-yl)propan-2-yl)-2-((S)-3-cyclohexyl-2-((trifluoromethyl)sulfonamido)propanoyl)-2-azabicyclo [2.2.1] heptane-3-carboxamide (11i)
Yield 48% from 9d; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 9.98 (d, J = 8.5 Hz, 1H), 8.73 (d, J = 7.3 Hz, 1H), 8.27 (d, J = 1.7 Hz, 1H), 8.23–8.21 (m, 1H), 7.66 (dd, J = 11.1, 4.4 Hz, 3H), 5.41–5.37 (m, 1H), 4.13 (d, J = 8.2 Hz, 1H), 4.10 (s, 1H), 3.89 (s, 1H), 3.20 (d, J = 8.7 Hz, 1H), 3.14 (d, J = 7.2 Hz, 1H), 2.65 (dd, J = 18.2, 16.3 Hz, 1H), 2.44 (s, 1H), 2.40–2.28 (m, 1H), 2.30–2.24 (m, 1H), 2.12–2.06 (m, 1H), 1.98 (s, 1H), 1.83–1.77 (m, 3H), 1.69 (d, J = 6.3 Hz, 3H), 1.59–1.49 (m, 4H), 1.34–1.28 (m, 2H), 1.23 (d, J = 5.0 Hz, 2H), 1.14 (dd, J = 22.9, 11.2 Hz, 3H), 0.95 (dd, J = 19.9, 10.3 Hz, 2H). 13C NMR (126 MHz, DMSO‑d 6): δ 192.87, 178.10, 169.46, 168.30, 164.44, 152.86, 136.31, 128.15, 127.54, 125.09, 123.22, 64.32, 57.35, 53.58, 53.35, 40.92, 37.72, 35.03, 33.25, 33.03, 32.19, 31.14, 30.62, 27.27, 27.09, 25.94, 25.84, 25.54. 19F NMR (753 MHz, DMSO‑d 6): δ −77.69. ESI-MS: m/z 698.5 [M + H]+. HRMS (ESI): m/z calcd for C31H39F3N5O6S2 [M + H]+ 698.2288, found 698.2296. HPLC purity: 95.47% (Rt: 3.570 min).
4.7.6. Synthesis of (S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)pro pan-2-yl(1R,3S,4S)-2-((S)-2-((3S,5S,7S)-adamantan-1-yl)-2-((trifluoromethyl)sulfonamido)acetyl)-2-azabicyclo[2.2.1]heptane-3-carboxylate (11j)
Yield 47% from 9e; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 9.65 (d, J = 9.3 Hz, 1H), 8.69 (d, J = 9.1 Hz, 1H), 8.28 (dd, J = 7.0, 2.1 Hz, 1H), 8.22 (dd, J = 7.1, 2.0 Hz, 1H), 7.70–7.65 (m, 2H), 7.62 (s, 1H), 5.74 (ddd, J = 11.9, 9.2, 2.5 Hz, 1H), 4.53 (s, 1H), 3.92 (d, J = 7.8 Hz, 2H), 3.18 (d, J = 9.1 Hz, 1H), 3.03 (d, J = 7.4 Hz, 1H), 2.60 (d, J = 10.6 Hz, 1H), 2.43 (s, 1H), 2.40–2.35 (m, 1H), 2.11 (d, J = 9.2 Hz, 1H), 2.02 (d, J = 12.1 Hz, 1H), 1.94 (s, 3H), 1.79 (t, J = 14.7 Hz, 5H), 1.69–1.62 (m, 8H), 1.59 (d, J = 11.8 Hz, 3H), 1.49 (d, J = 11.2 Hz, 1H), 1.31–1.25 (m, 2H). 13C NMR (126 MHz, DMSO‑d 6): δ 192.89, 178.41, 169.64, 164.81, 164.32, 152.93, 136.40, 128.22, 127.60, 125.16, 123.25, 64.29, 63.67, 58.17, 52.18, 40.99, 37.36, 37.18, 36.82, 36.16, 34.86, 33.60, 30.09, 27.86, 27.38. 19F NMR (471 MHz, DMSO‑d 6): δ −77.08. ESI-MS: m/z 736.4 [M + H]+. HRMS (ESI): m/z calcd for C34H41F3N5O6S2 [M + H]+ 736.2445, found 736.2447. HPLC purity: 97.20% (Rt: 3.887 min).
4.8. Synthesis of 11b-11e
4.8.1. Synthesis of (1R,3S,4S)-N-((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrr olidin-3-yl)propan-2-yl)-2-((S)-3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanoyl)-2- azabicyclo[2.2.1]heptane-3-carboxamide (11b)
To a solution of the compound 11a (300 mg, 0.48 mmol) in DCM (1 mL) was added 4 M HCl in 1, 4-dioxane (1.2 mL, 4.8 mmol) under nitrogen protection at 20–25 °C. After stirring for 40 min, the mixture was concentrated under reduced pressure to obtain the intermediate 11a’. Then, to a solution of 11a’ and DCM (5 mL), Et3N (2 mL, 1.44 mmol) and trifluoroacetic anhydride (67 μL, 0.48 mmol) were added dropwise at 0 °C under nitrogen protection. After adding, the ice bath was removed and the solution was allowed to stir all night at 20–25 °C. The organic layer was washed with a 1 M HCl solution and brine. This solution was dried over Na2SO4, filtered, and evaporated under reduced pressure. The resulting residue was purified by eluting through a silica gel column with a 150:1 DCM/MeOH solvent system to give the pure compound 11b (100 mg). Yield 32% from 11a; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 8.67 (d, J = 7.9 Hz, 1H), 8.27 (dd, J = 7.2, 1.8 Hz, 1H), 8.24–8.21 (m, 1H), 7.66 (td, J = 7.0, 1.4 Hz, 2H), 7.63 (s, 1H), 6.47 (d, J = 9.2 Hz, 1H), 5.59 (ddd, J = 11.4, 8.1, 3.0 Hz, 1H), 4.48 (d, J = 10.3 Hz, 1H), 4.18 (d, J = 9.3 Hz, 1H), 3.91 (s, 1H), 3.19 (t, J = 9.1 Hz, 1H), 3.10 (dd, J = 16.5, 9.1 Hz, 1H), 2.58 (td, J = 11.3, 3.5 Hz, 1H), 2.46 (s, 1H), 2.33–2.26 (m, 1H), 2.09–2.03 (m, 1H), 1.98 (d, J = 9.4 Hz, 1H), 1.82–1.76 (m, 2H), 1.65 (s, 3H), 1.37 (s, 9H), 1.28–1.19 (m, 2H), 0.94 (s, 9H). 13C NMR (126 MHz, DMSO‑d 6): δ 193.15, 178.15, 169.95, 168.04, 164.31, 155.51, 152.90, 136.38, 128.19, 127.56, 125.18, 123.22, 77.97, 64.04, 58.19, 58.09, 52.80, 41.16, 37.58, 34.83, 34.54, 32.83, 30.87, 28.16, 27.48, 27.35, 26.30. ESI-MS: m/z 626.3 [M + H]+. HRMS (ESI): m/z calcd for C32H44N5O6S [M + H]+ 626.3007, found 626.3007. HPLC purity: 95.15% (Rt: 9.760 min).
4.8.2. Synthesis of (1R,3S,4S)-N-((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopy rrolidin-3-yl)propan-2-yl)-2-((S)-2-(2-fluoro-2-methylpropanamido)-3,3-dimethylbutanoyl)-2-azabicyclo[2.2.1]heptane-3-carboxamide (11c)
To a solution of the compound 11a (300 mg, 0.48 mmol) in DCM (4 mL) was added 4 M HCl in 1, 4-dioxane (1.2 mL, 4.8 mmol) under nitrogen protection at 20–25 °C. After stirring for 40 min, the mixture was concentrated under reduced pressure to obtain the intermediate 11a’. Then, to a solution of 11a’ and DCM (5 mL), the 2-fluoroisobutyric acid (46 μL, 0.48 mmol), and HATU (192 mg, 0.48 mmol) were added. The resulting solution was cooled to 0 °C under ice bath conditions, and DIEA (250 μL, 1.44 mmol) was then added dropwise under nitrogen protection. After adding, the ice bath was removed, and the mixture was allowed to stir all night at 20–25 °C. Then, the dichloromethane was evaporated under reduced pressure, and the residue was dissolved in ethyl acetate. The organic layer was washed with a saturated ammonium chloride solution and brine. This solution was dried over Na2SO4, filtered, and evaporated under reduced pressure. The resulting crude compound was purified by silica gel column chromatography using 200:1 DCM/MeOH as eluents to obtain the compound 11c (170 mg). Yield 58% from 11a; white solid; 1H NMR (800 MHz, DMSO‑d 6): δ 8.71 (d, J = 7.7 Hz, 1H), 8.27 (t, J = 5.9 Hz, 1H), 8.23 (d, J = 7.8 Hz, 1H), 7.68–7.64 (m, 3H), 7.06 (dd, J = 17.8, 13.2 Hz, 1H), 5.59–5.56 (m, 1H), 4.53 (d, J = 9.1 Hz, 2H), 3.94 (s, 1H), 3.20 (t, J = 9.2 Hz, 1H), 3.11 (d, J = 9.0 Hz, 1H), 2.56–2.52 (m, 1H), 2.32–2.28 (m, 1H), 2.07 (dd, J = 26.9, 13.1 Hz, 1H), 1.96 (d, J = 9.0 Hz, 1H), 1.81 (dd, J = 21.4, 10.5 Hz, 2H), 1.65 (dd, J = 28.0, 11.9 Hz, 3H), 1.52–1.49 (m, 3H), 1.45 (d, J = 22.2 Hz, 3H), 1.39–1.37 (m, 1H), 1.27–1.18 (m, 2H), 0.94 (d, J = 24.6 Hz, 9H). 13C NMR (201 MHz, DMSO‑d 6): δ 193.10, 178.07, 171.48, 171.37, 169.73, 167.01, 164.27, 152.90, 136.38, 128.20, 127.56, 125.19, 123.22, 96.26, 95.37, 64.09, 58.15, 55.80, 54.91, 52.95, 41.19, 37.65, 35.37, 34.74, 32.71, 30.98, 27.41, 27.25, 26.07, 25.29, 25.17, 24.47, 24.35. 19F NMR (753 MHz, DMSO‑d 6): δ −145.33. ESI-MS: m/z 614.4 [M + H]+. HRMS (ESI): m/z calcd for C31H41FN5O5S [M + H]+ 614.2807, found 614.281. HPLC purity: 97.42% (Rt: 12.635 min).
4.8.3. Synthesis of (1R, 3S, 4S)-2-((S)-2-(acetamido-2,2,2-d3)-3,3-dimethylbutanoyl) -N-((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-2- azabicyclo [2.2.1] heptane-3-carboxamide (11d)
To a solution of the compound 11a (300 mg, 0.48 mmol) in DCM (1 mL) was added 4 M HCl in 1, 4-dioxane (1.2 mL, 4.8 mmol) under nitrogen protection at 20–25 °C. After 40 min of stirring, the mixture was concentrated under reduced pressure. Then, to a solution of the corresponding deprotected mixture and DCM (5 mL), Et3N (2 mL, 1.44 mmol) and acetic anhydride-d 6 (46 μL, 0.48 mmol) were added dropwise at 0 °C under nitrogen protection. After adding, the ice bath was removed and the solution was allowed to stir all night at 20–25 °C. The organic layer was washed with a 1 M HCl solution and brine. This solution was dried over Na2SO4, filtered, and evaporated under reduced pressure. The resulting residue was purified by eluting through a silica gel column with a 150:1 DCM/MeOH solvent system to give the pure compound 11d (150 mg). Yield 55% from 11a; yellow solid; 1H NMR (500 MHz, DMSO‑d 6): δ 8.66 (d, J = 8.2 Hz, 1H), 8.27 (dd, J = 7.2, 1.8 Hz, 1H), 8.22 (dd, J = 7.2, 1.6 Hz, 1H), 7.86 (d, J = 9.2 Hz, 1H), 7.66 (td, J = 7.1, 1.4 Hz, 2H), 7.63 (s, 1H), 5.63 (ddd, J = 11.6, 8.3, 3.1 Hz, 1H), 4.54 (d, J = 9.2 Hz, 1H), 4.48 (s, 1H), 3.89 (s, 1H), 3.19 (t, J = 9.1 Hz, 1H), 3.12–3.06 (m, 1H), 2.59 (dt, J = 11.5, 5.8 Hz, 1H), 2.45 (s, 1H), 2.34–2.28 (m, 1H), 2.08–1.99 (m, 2H), 1.83–1.76 (m, 2H), 1.61 (d, J = 11.9 Hz, 3H), 1.34 (d, J = 9.5 Hz, 1H), 1.24 (d, J = 9.4 Hz, 1H), 0.96 (s, 9H). 13C NMR (126 MHz, DMSO‑d 6): δ 193.15, 178.26, 169.92, 169.15, 167.73, 164.32, 152.91, 136.40, 128.20, 127.57, 125.18, 123.23, 64.09, 58.12, 56.24, 52.63, 41.12, 37.53, 34.89, 34.57, 33.01, 30.80, 27.47, 27.31, 26.38. ESI-MS: m/z 571.4 [M + H]+. HRMS (ESI): m/z calcd for C29H35D3N5O5S [M + H]+ 571.2776, found 571.2779. HPLC purity: 95.72% (Rt: 9.556 min).
4.8.4. Synthesis of (1R, 3S, 4S)-N-((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopy rrolidin-3-yl)propan-2-yl)-2-((S)-3,3-dimethyl-2-((trifluoromethyl)sulfonamido)butanoyl)-2-azabicyclo [2.2.1] heptane-3-carboxamide (11e)
To a solution of the compound 11a (300 mg, 0.48 mmol) in DCM (1 mL) was added 4 M HCl in 1, 4-dioxane (1.2 mL, 4.8 mmol) under nitrogen protection at 20–25 °C. After stirring for 40 min, the mixture was concentrated under reduced pressure. Then, to a solution of the corresponding deprotected mixture 11a’ and DCM (5 mL), Et3N (2 mL, 1.44 mmol) was added dropwise, along with trifluoromethanesulfonic anhydride (81 μL, 0.48 mmol) at 0 °C under nitrogen protection. After adding, the ice bath was removed, and the solution was allowed to stir all night at 20–25 °C. The organic layer was washed with a 1 M HCl solution and brine. This solution was dried over Na2SO4, filtered, and evaporated under reduced pressure. The resulting residue was purified by eluting through a silica gel column with a 150:1 DCM/MeOH solvent system to give the pure compound 11e (100 mg). Yield 32% from 11a; white solid; 1H NMR (500 MHz, DMSO‑d 6): δ 9.63 (d, J = 9.4 Hz, 1H), 8.72 (d, J = 8.6 Hz, 1H), 8.28 (dd, J = 7.0, 2.1 Hz, 1H), 8.22 (dd, J = 7.2, 2.0 Hz, 1H), 7.70–7.65 (m, 2H), 7.64 (s, 1H), 5.68 (ddd, J = 11.7, 8.6, 2.8 Hz, 1H), 4.54 (s, 1H), 4.07 (d, J = 9.5 Hz, 1H), 3.94 (s, 1H), 3.18 (d, J = 9.1 Hz, 1H), 3.11–3.06 (m, 1H), 2.60 (dt, J = 18.6, 5.8 Hz, 1H), 2.46 (s, 1H), 2.32 (dd, J = 12.7, 7.1 Hz, 1H), 2.02 (dd, J = 9.3, 4.4 Hz, 2H), 1.81 (dt, J = 22.3, 10.7 Hz, 2H), 1.70–1.61 (m, 2H), 1.52 (t, J = 9.3 Hz, 1H), 1.33 (d, J = 6.0 Hz, 1H), 1.24 (d, J = 7.5 Hz, 1H), 1.03 (s, 9H). 13C NMR (126 MHz, DMSO‑d 6): δ 193.02, 178.25, 169.65, 165.57, 164.27, 152.91, 136.41, 128.24, 127.60, 125.19, 123.25, 64.18, 62.76, 58.16, 52.49, 41.10, 37.48, 35.41, 34.79, 33.19, 30.10, 27.45, 27.36, 26.15. ESI-MS: m/z 658.4 [M + H]+. HRMS (ESI): m/z calcd for C28H35F3N5O6S2 [M + H]+ 658.1975, found 658.1979. HPLC purity: 96.47% (Rt: 4.101 min).
4.9. Molecular docking procedure
The X-ray structure of SARS-CoV-2 3CLpro (PDB ID: 7VH8) was downloaded from the RCSB protein data bank. The molecular docking was performed using the Glide Covalent Docking module in the software package Schrödinger Suite 2020 to perform protein-ligand interaction studies.
4.10. Biological experimental methods
4.10.1. SARS-CoV-2 3CLpro inhibition assay for 11a-11j
A fluorescence resonance energy transfer (FRET) protease assay was applied to measure the inhibitory activity of compounds against SARS-CoV-2 3CLpro. The recombinant SARS-CoV-2 3CLpro at a concentration of 40 nM was mixed with serial dilutions of each compound in 80 μL of assay buffer (50 mM Tris-HCl, pH 7.3, 1 mM EDTA) and incubated for 10 min. The reaction was initiated by adding 40 μL of a fluorogenic substrate (Dacyl-KTSAVLQSGFRKME-Edans) at a final concentration of 5 μM. After that, the fluorescence signal at 340 nm (excitation) and 490 nm (emission) was measured immediately every 1 min for 5 min with a Bio-Tek SynergyH1 plate reader. The velocities of reactions with compounds added at various concentrations compared to the reaction added with DMSO were calculated and used to generate inhibition profiles. For each compound, at least three biological replicates were performed for the determination of IC50 values.
4.10.2. Microsomal stability assay for 11b-11f and nirmatrelvir
Preheat 100 mM K-buffer with 5 mM MgCl2 to pH 7.41. Solutions of test and reference compounds were spiking. The tested concentration of compounds was 1 μM. NADPH stock solution (6 mM, 5 mg/mL) is prepared by dissolving NADPH in K/Mg-buffer. Dispense 30 μL of 1.5 μM spiking solution containing 0.75 mg/mL microsomes solution to the assay plates designated for different time points (0, 5, 15, 30, and 45 min). Pre-incubate the other plate at 37 °C for 5 min. For 0 min, add 150 μL of ACN containing IS to the wells before adding 15 μL of NADPH stock solution (6 mM). For other time points, add 15 μL of NADPH stock solution (6 mM) to the wells to start the reaction and timing. At 5 min, 15 min, 30 min, 45 min add 150 μL of ACN containing IS to the wells of corresponding plates, respectively, to stop the reaction. After quenching, shake the plates for 10 min (600 rpm) and then centrifuge at 6000 rpm for 15 min. Transfer 80 μL of the supernatant from each well into a 96-well sample plate containing 140 μL of pure water for LC/MS analysis.
4.10.3. Pharmacokinetics of 11b, 11e, 11j and nirmatrelvir
All experiment procedures involving animals were in accordance with the guidelines of the Institutional Animal Care and Use Committee. The mice were randomly assigned into IV and PO groups, three mice per group. The mice received an intravenous (10 mg/kg) or oral (20 mg/kg) dose. After that, the whole blood samples were collected at 0.083 h, 0.25 h, 0.5 h, 1 h, 2 h, 4 h, 8 h and 24 h post-dose by intravenous administration. And the whole blood samples were collected at 0.25 h, 0.5 h, 1 h, 2 h, 4 h, 6 h, 8 h and 24 h post-dose by oral administration. An aliquot of a 20 μL plasma sample was protein precipitated with 400 μL MeOH, which contains 100 ng/mL IS. The mixture was vortexed for 1 min and centrifuged at 18000g for 10 min 400 μL of the supernatant should be transferred to 96-well plates. An aliquot of 8 μL supernatant was injected for LC-MS/MS analysis. The data analyzed and treated by the non-atrioventricular model, data acquisition and control system software were Phoenix WinNonlin 7.0 (Pharsight, USA).
4.10.4. Antiviral assay for 11e, 11j and nirmatrelvir
Vero E6 cells (50000 cells/well) were plated into 48-well plates, and 200 μL/well medium containing 1 μM compound was added and incubated at 37 °C for 1 h. Then, SARS-CoV-2 WIV04 was added at a multiplicity of infection (MOI) of 0.01. After 24 h, the supernatant was collected, and the viral RNA was extracted from the supernatant. The viral copy number of the supernatant was detected by real-time fluorescence quantitative PCR. The inhibition rate of the compound was calculated based on the viral copy number.
4.10.5. Cytotoxic assay for 11e, 11j and nirmatrelvir
Vero E6 cells were plated in the 384-well plates at a density of 2.5 × 103 cells per well for 48 h. Then the cells were incubated with the test articles at different concentrations (0.5–200 μM) for another 48 h (n = 3). A Luminescent Cell Viability Assay Kit purchased from Meilun Biotech Co., Ltd. (Dalian, China) was used for the cytotoxicity assay, with 10 μL of the Luminescent Cell Viability Assay Kit added to each well for 10 min. The absorbance was measured by an automatic microplate reader (Biotek, Winooski, VT, USA) at Luminescent. The half inhibitory concentration (IC50) values for each compound were calculated by GraphPad Prism 8.3.0 software (GraphPad Software Inc., La Jolla, CA, USA).
4.10.6. Protease selectivity assay for 11j
The chymotrypsin assay was performed as follows: Chymotrypsin (Sigma, catalog # SLCH1926) at a final concentration of 5 nM was mixed with 20 μM compound 11j in an assay buffer (phosphate buffer saline, pH 7.4) and incubated for 10 min. Then, the fluorogenic substrate N-succinyl-AAPF-AMC at a final concentration of 10 μM was added to initiate the reaction. After that, the fluorescence signal was immediately measured at 380 nm (excitation) and 460 nm (emission) every 1 min for 10 min using a Bio-Tek Synergy-H1 plate reader.
The cathepsin B assay was performed in a reaction buffer containing 20 mM sodium acetate (pH 5.5), 1 mM EDTA, and 2 mM DTT. 0.25 units of cathepsin B (Sigma, catalog # SLCJ4379) and the testing compound 11j were added to each well and incubated for 30 min at ambient temperature. The enzymatic reaction was started by adding 40 μL substrate (Z-RR-AMC) at a final concentration of 1 μM. After that, the fluorescence signal was immediately measured at 340 nm (excitation) and 440 nm (emission) every 1 min for 10 min with a Bio-Tek Synergy-H1 plate reader.
The cathepsin L (2 nM at a final concentration) was mixed with compound 11j in 80 μL buffer (100 mM potassium phosphate, pH 6.8, 5 mM EDTA-Na, 0.001% Triton X-100, and 2 mM DDT). The reaction was initiated by adding 40 μL of the fluorogenic substrate Z-FR-AMC (20 μM). After that, the fluorescence signal was immediately measured at 360 nm (excitation) and 450 nm (emission) every 1 min for 10 min with a Bio-Tek Synergy-H1 plate reader.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
This research was supported by National Key Research and Development Plan of China (2021YFC2300700 to L.K.Z., 2022YFC2303300 to L.K.Z.) and the Strategic Priority Research Program of Chinese Academy of Sciences (SIMM010120).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2023.115512.
Abbreviations
- COVID-19
Coronavirus disease 2019
- FDA
Food and Drug Administration
- THF
tetrahydrofuran
- Et3N
triethylamine
- DMF
N, N-dimethylformamide
- AUC
area under the curve
- EC50
half-maximal effective concentration
- DCM
dichloromethane
- MeOH
methanol
- PE
petroleum ether
- TLC
thin layer chromatography
- DMSO
dimethyl sulfoxide
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Tang D.L., Comish P., Kang R. The hallmarks of COVID-19 disease. PLoS Pathog. 2020;16:1–24. doi: 10.1371/journal.ppat.1008536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hao Y.J., Wang Y.L., Wang M.Y., Zhou L., Shi J.Y., Cao J.M., Wang D.P. The origins of COVID-19 pandemic: a brief overview. Transbound Emerg Dis. 2022;69:3181–3197. doi: 10.1111/tbed.14732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao K.F., Wang R., Chen J.H., Tepe J., Huang F.Q., Guo W. Perspectives on SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2021;64:16922–16955. doi: 10.1021/acs.jmedchem.1c00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiong M.Y., Su H.X., Zhao W.F., Xie H., Shao Q., Xu Y.C. What coronavirus 3C-like protease tells us: from structure, substrate selectivity, to inhibitor design. Med. Res. Rev. 2021;41:1965–1998. doi: 10.1002/med.21783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dampalla C.S., Rathnayake A.D., Perera K.D., Jesri A.R.M., Nguyen H.N., Miller M.J., Thurman H.A., Zheng J., Kashipathy M.M., Battaile K.P., Lovell S., Perlman S., Kim Y., Groutas W.C., Chang K.O. Structure-guided design of potent inhibitors of SARS-CoV-2 3CL protease: structural, biochemical, and cell-based studies. J. Med. Chem. 2021;64:17846–17865. doi: 10.1021/acs.jmedchem.1c01037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dampalla C.S., Kim Y., Bickmeier N., Rathnayake A.D., Nguyen H.N., Zheng J., Kashipathy M.M., Baird M.A., Battaile K.P., Lovell S., Perlman S., Chang K.O., Groutas W.C. Structure-guided design of conformationally constrained cyclohexane inhibitors of severe acute respiratory syndrome Coronavirus-2 3CL protease. J. Med. Chem. 2021;64:10047–10058. doi: 10.1021/acs.jmedchem.1c00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Su H.X., Yao S., Zhao, Zhao W.F., Zhang Y.M., Liu J., Shao Q., Wang Q.X., Li M.J., Xie H., Shang W.J., Ke C.Q., Feng L., Jiang X.R., Shen J.S., Xiao G.F., Jiang H.L., Zhang L.K., Ye Y., Xu Y.C. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nat. Commun. 2021;12:3623. doi: 10.1038/s41467-021-23751-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qiao J.X., Li Y.S., Zeng R., Liu F.L., Luo R.H., Huang C., Wang Y.F., Zhang J., Quan B., Shen C.J., Mao X., Liu X.L., Sun W.N., Yang W., Ni X.C., Wang K., Xu L., Duan Z.L., Zou Q.C., Zhang H.L., Qu W., Long Y.H.P., Li M.H., Yang R.C., Liu X.L., You J., Zhou Y.L., Yao R., Li W.P., Liu J.M., Chen P., Liu Y., Lin G.F., Yang X., Zou J., Li L.L., Hu Y.G., Lu G.W., M Li W., Wei Y.Q., Zheng Y.T., Lei J., Yang S.Y. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371:1374–1378. doi: 10.1126/science.abf1611. https://www.science.org/doi/10.1126/science.abf1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang K.S., Leeuwon S.Z., Xu S.Q., Liu W.S.R. Evolutionary and structural insights about potential SARS-CoV-2 evasion of nirmatrelvir. J. Med. Chem. 2022;65:8686–8698. doi: 10.1021/acs.jmedchem.2c00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cannalire R., Cerchia C., Beccari A.R., Leva F.S.D., Summa V. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: state of the art and future opportunities. J. Med. Chem. 2022;65:2716–2746. doi: 10.1021/acs.jmedchem.0c01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Y., Fang C., Zhang Q., Zhang R.X., Zhao X.B., Duan Y.K., Wang H.F., Zhu Y., Feng L., Zhao J.Y., Shao M.L., Yang X.N., Zhang L.K., Peng C., Yang K.L., Ma D.W., Rao Z.H., Yang H.T. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein & Cell. 2022;13:689–693. doi: 10.1007/s13238-021-00883-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao S.H., Song L.T., Claff T., Woodson M., Sylvester K., Jing L.L., Weiße R.H., Cheng Y.S., Sträter N., Schäkel L., Gütschow M., Ye B., Yang M.L., Zhang T., Kang D.W., Toth K., Tavis J., Tollefson A.E., Müller C.E., Zhan P., Liu X.Y. Discovery and crystallographic studies of nonpeptidic piperazine derivatives as covalent SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2022;65:16902–16917. doi: 10.1021/acs.jmedchem.2c01716. [DOI] [PubMed] [Google Scholar]
- 13.Hirose Y., Shindo N., Mori M., Onitsuka S., Isogai H., Hamada R., Hiramoto T., Ochi J., Takahashi D., Ueda T., Caaveiro J.M.M., Yoshida Y., Ohdo S., Matsunaga N., Toba S., Sasaki M., Orba Y., Sawa H., Sato A., Kawanishi E., Ojida A. Discovery of chlorofluoroacetamide-based covalent inhibitors for severe acute respiratory syndrome coronavirus 2 3CL protease. J. Med. Chem. 2022;65:13852–13865. doi: 10.1021/acs.jmedchem.2c01716. [DOI] [PubMed] [Google Scholar]
- 14.Ma X.Y.R., Alugubelli Y.R., Ma Y.Y., Vatansever E.C., Scott D.A., Qiao Y.C., Yu G., Xu S.Q., Liu W.S.R. MPI8 is potent against SARS-CoV-2 by inhibiting dually and selectively the SARS-CoV-2 main protease and the host cathepsinl. ChemMedChem. 2021;16:1–9. doi: 10.1002/cmdc.202100456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang K.S., Ma X.Y.R., Ma Y.Y., Alugubelli Y.R., Scott D.A., Vatansever E.C., Drelich A.K., Sankaran B., Geng Z.Z., Blankenship L.R., Ward H.E., Sheng Y.J., Hsu J.C., Kratch K.C., Zhao B.Y., Hayatshahi H.S., Liu J., Li P.W., Fierke C.A., Tseng C.T.K., Xu S.Q., Liu W.S.R. A Quick route to multiple highly potent SARS-CoV-2 main protease inhibitors. ChemMedChem. 2021;16:942–948. doi: 10.1002/cmdc.202000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bai B., Belovodskiy A., Hena M., Kandadai A.S., Joyce M.A., Saffffran H.A., Shields J.A., Khan M.B., Arutyunova E., Lu J., Bajwa S.K., Hockman D., Fischer C., Lamer T., Vuong W., Belkum M.J., Gu Z.X., Lin F., Du Y.H., Xu J., Rahim M., Young H.S., Vederas J.C., Tyrrell D.L., Lemieux M.J., Nieman J.A. Peptidomimetic α-acyloxymethylketone warheads with six membered lactam P1 glutamine mimic: SARS-CoV-2 3CL protease inhibition, coronavirus antiviral activity, and in vitro biological stability. J. Med. Chem. 2022;65:2905–2925. doi: 10.1021/acs.jmedchem.1c00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banerjee R., Perera L., Tillekeratne L.M.V. Potential SARS-CoV-2 main protease inhibitors. Drug Discov. Today. 2021;26:804–816. doi: 10.1016/j.drudis.2020.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vuong W., Khan M.B., Fischer C., Arutyunova E., Lamer T., Shields J., Saffran H.A., McKay R.T., Belkum M.J., Joyce M.A., Young H.S., Tyrrell D.L., Vederas J.C., Lemieux M.J. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020;11:4282. doi: 10.1038/s41467-020-18096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu L.F., Ye F., Feng Y., Yu F., Wang Q.S., Wu Y., Zhao C., Sun H., Huang B.Y., Niu P.H., Song H., Shi Y., Li X.B., Tan W.J., Qi J.X., Gao G.F. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020;11:4417. doi: 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boras B., Jones R.M., Anson B.J., Arenson D., Aschenbrenner L., Bakowski M.A., Beutler N., Binder J., Chen E., Eng H., Hammond J., Hoffman R., Kadar E.P., Kania R., Kimoto E., Kirkpatrick M.G., Lanyon L., Lendy E.K., Lillis J.R., Luthra S.A., Ma C., Noell S., Obach R.S., O'Brien M.N., O'Connor R., Ogilvie K., Owen D., Pettersson M., Reese M.R., Rogers T.F., Rossulek M.I., Sathish J.G., Steppan C., Ticehurst M., Updyke L.W., Zhu Y., Wang J., Chatterjee A.K., Mesecar A.D., Anderson A.S., Allerton C. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID-19. Nat. Commun. 2021;12:6055. doi: 10.1038/s41467-021-26239-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020;30 doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Owen D.R., Allerton C.M.N., Anderson A.S., Aschenbrenner L., Avery M., Berritt S., Boras B., Cardin R.D., Carlo A., Coffman K.J., Dantonio A., Di L., Eng H., Ferre R.A., Gajiwala K.S., Gibson S.A., Greasley S.E., Hurst B.L., Kadar E.P., Kalgutkar A.S., Lee J.C., Lee J., Liu W., Mason S.W., Noell S., Novak J.J., Obach R.S., Ogilvie K., Patel N.C., Pettersson M., Rai D.K., Reese M.R., Sammons M.F., Sathish J.G., Singh R.S.P., Steppan C.M., Stewart A.E., Tuttle J.B., Updyke L., Verhoest P.R., Wei L.Q., Yang Q.Y., Zhu Y. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374:1586–1593. doi: 10.1126/science.abl4784. https://www.science.org/doi/10.1126/science.abl4784 [DOI] [PubMed] [Google Scholar]
- 23.Lamb Y.N. Nirmatrelvir plus ritonavir: first approval. Drugs. 2022;82:585–591. doi: 10.1007/s40265-022-01692-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sindelar M., McCabe D., Carroll E. Tacrolimus drug-drug interaction with nirmatrelvir/ritonavir (Paxlovid) managed with phenytoin. J. Med. Toxicol. 2023;19:45–48. doi: 10.1007/s13181-022-00922-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marzolini C., Kuritzkes D.R., Marra F., Boyle A., Gibbons S., Flexner C., Pozniak A., Boffito M., Waters L., Burger D., Back D.J., Khoo S. Recommendations for the management of drug-drug interactions between the COVID-19 antiviral nirmatrelvir/ritonavir (Paxlovid) and comedications. Clin. Pharmacol. Ther. 2022;112:1191–1200. doi: 10.1002/cpt.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang H.H., Yang J.F. A review of the latest research on Mpro targeting SARS-COV inhibitors. RSC Med Chem. 2021;12:1026–1036. doi: 10.1039/D1MD00066G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stille J.K., Tjutrins J., Wang G.Y., Venegas F.A., Hennecker C., Rueda A.M., Sharon I., Blaine N., Miron C.E., S P., Labarre A., Plescia J., Patrascu M.B., Zhang X.C., Wahba A.S., Vlaho D., Huot M.J., Schmeing T.M., Mittermaier A.K., Moitessier N. Design, synthesis and in vitro evaluation of novel SARS-CoV-2 3CLpro covalent inhibitors. Eur. J. Med. Chem. 2022;229 doi: 10.1016/j.ejmech.2021.114046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao S.H., Sylvester K., Song L.T., Claff T., Jing L.L., Woodson M., Weiße R.H., Cheng Y.S., Schäkel L., Petry M., Gütschow M., Schiedel A.C., Sträter N., Kang D.W., Xu S.J., Toth K., Tavis J., Tollefson A.E., Müller C.E., Liu X.Y., Zhan P. Discovery and crystallographic studies of trisubstituted piperazine derivatives as non-covalent SARS-CoV-2 main protease inhibitors with high target specificity and low toxicity. J. Med. Chem. 2022;65:13343–13364. doi: 10.1021/acs.jmedchem.2c01146. [DOI] [PubMed] [Google Scholar]
- 29.Breidenbach J., Lemke C., Pillaiyar T., Schäkel L., Hamwi G.A., Diett M., Gedschold R., Geiger N., Lopez V., Mirza S., Namasivayam V., Schiedel A.C., Sylvester K., Thimm D., Vielmuth C., Vu L.P., Zyulina M., Bodem J., Gütschow M., Müller C.E. Targeting the main protease of SARS-CoV-2: from the establishment of high throughput screening to the design of tailored inhibitors. Angew. Chem., Int. Ed. Engl. 2021;60:10423–10429. doi: 10.1002/anie.202016961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen R.F., Gao Y.L., Liu H., Li H., Chen W.F., Ma J.J. Advances in research on 3C-like protease (3CLpro) inhibitors against SARS-CoV-2 since 2020. RSC Med. Chem. 2023;14:9–21. doi: 10.1039/D2MD00344A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dai W.H., Zhang B., Jiang X.M., Su H.X., Li J., Zhao Y., Xie X., Jin Z.M., Peng J.J., Liu F.J., Li C., Li Y., Bai F., Wang H.F., Cheng X., Cen X.B., Hu S.L., Yang X.N., Wang J., Liu X., Xiao G.F., Jiang H.L., Rao Z.H., Zhang L.K., Xu Y.C., Yang H.T., Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. https://www.science.org/doi/10.1126/science.abb4489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vuong W., Fischer C., Khan M.B., van Belkum M.J., Lamer T., Willoughby K.D., Lu J., Arutyunova E., Joyce M.A., Saffran H.A., Shields J.A., Young H.S., Nieman J.A., Tyrrell D.L., Lemieux M.J., Vederas J.C. Improved SARS-CoV-2 Mpro inhibitors based on feline antiviral drug GC376: structural enhancements, increased solubility, and micellar studies. Eur. J. Med. Chem. 2021;222 doi: 10.1016/j.ejmech.2021.113584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Citarella A., Scala A., Piperno A., Micale N. SARS-CoV-2 Mpro: a potential target for peptidomimetics and small-molecule inhibitors. Biomolecules. 2021;11:607. doi: 10.3390/biom11040607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang L.L., Lin D.Z., Sun X.Y.Y., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeid R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. https://www.science.org/doi/10.1126/science.abb3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin Z.M., Du X.Y., Xu Y.C., Deng Y.Q., Liu M.Q., Zhao Y., Zhang B., Li X.F., Zhang L.K., Peng C., Duan Y.K., Yu J., Wang L., Yang K.L., Liu F.J., Jiang R.D., Yang X.L., You T., Liu X.C., Yang X.N., Bai F., Liu H., Liu X., Guddat L.K.W., Xu W.Q., Xiao G.F., Qin C.F., Shi Z.L., Jiang H.L., Rao Z.H., Yang H.T. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 36.Hoffman R.L., Kania R.S., Brothers M.A., Davies J.F., Ferre R.A., Gajiwala K.S., He M.Y., Hogan R.J., Kozminski K., Li L.L.Y., Lockner J.W., Lou J.H., Marra M.T., Mitchell L.J., Murray B.W., Nieman J.A., Noell S., Planken S.P., Rowe T., Ryan K., Smith G.J., Solowiej J.E., Steppan C.M., Taggart B. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020;63:12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Konno S., Thanigaimalai P., Yamamoto T., Nakada K., Kakiuchi R., Takayama K., Yamazaki Y., Yakushiji F., Akaji K., Kiso Y., Kawasaki Y., Chen S.E., Freire E., Hayashi Y. Design and synthesis of new tripeptide-type SARS-CoV 3CL protease inhibitors containing an electrophilic arylketone moiety. Bioorg. Med. Chem. 2013;21:412–424. doi: 10.1016/j.bmc.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thanigaimalai P., Konno S., Yamamoto T. Design, synthesis, and biological evaluation of novel dipeptide-type SARS-CoV 3CL protease inhibitors: structure-activity relationship. Eur. J. Med. Chem. 2013;65:436–447. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thanigaimalai P., Konno S., Yamamoto T., Koiwai Y., Taguchi A., Takayama K., Yakushiji F., Akaji K., Chen S.E., Tavakolian A.N., Schön A., Freire E., Hayashi Y. Development of potent dipeptide-type SARS-CoV 3CL protease inhibitors with novel P3 scaffolds: design, synthesis, biological evaluation, and docking studies. Eur. J. Med. Chem. 2013;68:372–384. doi: 10.1016/j.ejmech.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Konno S., Kobayashi K., Senda M., Funai Y., Seki Y., Tamai I., Schäkel L., Sakata K., Pillaiya T., Taguchi A., Taniguchi A., Gütschow M., Müller C.E., Takeuchi K., Hirohama M., Kawaguchi A., Kojima M., Senda T., Shirasaka Y., Kamitani W., Hayashi Y. 3CL protease inhibitors with an electrophilic arylketone moiety as anti-SARS-CoV-2 agents. J. Med. Chem. 2022;65:2926–2939. doi: 10.1021/acs.jmedchem.1c00665. [DOI] [PubMed] [Google Scholar]
- 41.Kneller D.W., Li H., Phillips G., Weiss K.L., Zhang Q., Arnould M.A., Jonsson C.B., Surendranathan S., Parvathareddy J., Blakeley M.P., Coates L., M Louis J., Bonnesen P.V., Kovalevsky A. Covalent narlaprevir- and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nat. Commun. 2022;13:2268. doi: 10.1038/s41467-022-29915-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Link J.O., Taylor J.G., Xu L.H., Mitchell M., Guo H.Y., Liu H.T., Kato D., Kirschberg T., Sun J.Y., Squires N., Parrish J., Kellar T., Yang Z.Y., Yang C., Matles M., Wang Y.J., Wang K., Cheng G.F., Tian Y., Mogalian E., Mondou E., Cornpropst M., Perry J., Desai M.C. Discovery of Ledipasvir (GS-5885): a potent, once-daily oral NS5A inhibitor for the treatment of hepatitis C virus infection. J. Med. Chem. 2014;57:2033–2046. doi: 10.1021/jm401499g. [DOI] [PubMed] [Google Scholar]
- 43.Iketani S., Forouhar F., Liu H.R., Hong S.J., Lin F.Y., Nair M.S., Zask A., Huang Y.X., Xing L., Stockwell B.R., Chavez A., Ho D.D. Lead compounds for the development of SARS-CoV-2 3CL protease inhibitors. Nat. Commun. 2021;12:2016. doi: 10.1038/s41467-021-22362-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu H.R., Iketani S., Zask A., Khanizeman N., Bednarova E., Forouhar F., Fowler B., Hong S.J., Mohri H., Nair M.S., Huang Y.X., Tay N.E.S., Lee S., Karan C., Resnick S.J., Quinn C., Li W.J., Shion H., Xia X., Daniels J.D., Cruz M.B., Farina M., Rajbhandari P., Jurtschenko C., Lauber M.A., Donald T.M., Stokes M.E., Hurst B.L., Rovis T., Chavez A., Ho D.D., Stockwell B.R. Development of optimized drug-like small molecule inhibitors of the SARS-CoV-2 3CL protease for treatment of COVID-19. Nat. Commun. 2022;13:1891. doi: 10.1038/s41467-022-29413-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281:4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.