

Abstract
Exome sequencing is a powerful tool in prenatal and postnatal genetics and can help identify novel candidate genes critical to human development. We describe seven unpublished probands with rare likely pathogenic variants or variants of uncertain significance that segregate with recessive disease in TBC1D32, including four fetal probands in three unrelated pedigrees and three pediatric probands in unrelated pedigrees. We also report clinical comparisons with seven previously published patients. Index probands were identified through an ongoing prenatal exome sequencing study and through an online data sharing platform (Gene Matcher™). A literature review was also completed. TBC1D32 is involved in the development and function of cilia and is expressed in the developing hypothalamus and pituitary gland. We provide additional data to expand the phenotype correlated with TBC1D32 variants, including a severe prenatal phenotype associated with life-limiting congenital anomalies.
Keywords: ciliopathy, exome sequencing, prenatal phenotype, TBC1D32
1 |. INTRODUCTION
Ciliopathies describe a heterogeneous group of disorders resulting from abnormal ciliary function or structure. oro-facial-digital (OFD) syndromes are defined by abnormalities of the face, oral cavity, and extremities. Multiple OFD subtypes have been defined based on additional features, such as kidney disease, brain malformations and dysfunction, and retinal abnormalities (Bruel et al., 2017). Adly et al. (2014) reported TBC1D32, encoding TBC1 domain family member 32, as a candidate causal gene in a simplex patient associated with a severe ciliopathy phenotype. Based on the observation of facial cleft, microphthalmia and coloboma, and polydactyly, the authors characterized the phenotype within the OFD spectrum (type IX).
Exome sequencing can be a powerful tool to uncover novel candidate genes. However, one of the challenges encountered with whole exome and genome sequencing is the limited ability to define genotype–phenotype correlations for rare genetic conditions (Gray et al., 2019). This is especially true in prenatal diagnosis as the fetal phenotype is often not well described. Absent functional information, imperfect ultrasound and MRI resolution and absent detailed fetal autopsy contribute to the challenge of interpreting rare variants identified through fetal exome sequencing (Vora et al., 2020). Clinicians often rely on the literature reviews and collaboration with other genetics professionals to classify variants. Additionally, there is currently no publicly available prenatal database with genotype–phenotype information.
To date, variants in TBC1D32 have been reported in seven individuals (Adly et al., 2014; Alsahan & Alkuraya, 2020; Hietamäki et al., 2020; Monies et al., 2019); however, TBC1D32 has no associated phenotype cataloged in Online Mendelian Inheritance in Man (OMIM). We present seven additional probands, including four with severe prenatal phenotypes, associated with TBC1D32 gene variants to provide additional data on the spectrum of phenotypes associated with this complex ciliopathy.
2 |. MATERIALS AND METHODS
Index probands (Probands 5 and 6) were identified through the Fetal Exome Sequencing (FES) Study at the University of North Carolina at Chapel Hill. Anomalous fetuses with normal microarray were identified prospectively and retrospectively and exome sequencing and variant filtering was completed on the fetus-mother–father trios, as previously described (Vora et al., 2020). The UNC Hospitals Clinical Molecular Genetics Laboratory performed Sanger DNA sequencing analysis to confirm the presence of the genetic variant and appropriate segregation with the disease in the trio. This study was approved from the University of North Carolina at Chapel Hill Institutional Review Board (IRB) (13–4084) before patient consent and enrollment. Participants had pretest counseling about exome sequencing and possible results and were consented to participate in the research study.
Gene Matcher™ (Sobreira et al., 2015) was queried to identify unpublished affected individuals with TBC1D32 gene variants. Researchers were contacted via email and asked to contribute patient information for inclusion in this series. A literature review was completed using PubMed to identify all previously published patients.
3 |. RESULTS
3.1 |. Proband 1
A 3-month-old female was referred for evaluation for microcephaly (Table 1). She was the product of a full-term uncomplicated pregnancy with no known maternal exposures. Consanguinity was denied and both parents were reportedly healthy and of European descent. Significant physical exam findings included microcephaly, brachycephaly, deep set eyes, mild prominent nose, long philtrum, and thin upper lip, and mild 2–3 toe syndactyly. Increased muscle tone, mild restriction of joints, and developmental delay were reported.
TABLE 1.
Summary of clinical and genetic findings in pedigrees with recessive TBC1D32 variants.
| Hietamäki et al. (2020) | Hietamäki et al. (2020) | Hietamâki et al. (2020) | Monies et al. (2019) | Adly et al. (2014) | Hietamâki et al. (2020) | Proband 1 | Proband 2 | Proband 3 | Proband 4 | Proband 5 | Proband 6 | Proband 7 | Alsahan and Alkuraya (2020) | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TBC1D32 variants | c.ll65_1166dup and c.2151del | c.ll65_1166dup and c.2151del | c.1372 + 1 G > T homozygous | c.1372 + 1 G > T homozygous | c.1372 + 1 G > T homozygous | c.1372 + 1 G > T homozygous | c.2134 A > G and C3182G > C | c.2481 + 4 A > T and C.3572A > G | c.2650 C > T homozygous | c.2650 C > T homozygous | c.3325_3326delAG homozygous | c.3325_3326delAG homozygous | c.3527 A > G homozygous | C3724C > T homozygous | – |
| Oral/Facial | |||||||||||||||
| Lip/Palate | – | – | High arched palate | Cleft lip | Midline facial cleft | Midline facial cleft | – | – | – | – | – | Midline facial cleft | – | – | 5/14 |
| Nose | Upturned nose | Upturned nose | Upturned nose, choanal atresia | – | Choanal atresia | – | Prominent | Choanal atresia | Single fused nostril | High position, partially fused nares | Absent | – | – | Single nostril | 10/14 |
| Ears | Low set | Low set | Low set | – | – | – | – | – | – | Low set | Low set | – | – | – | 5/14 |
| Tongue | – | – | Bifid | – | – | – | – | – | – | – | – | – | – | – | 1/14 |
| Dentition | – | Abnormal | – | – | – | – | – | Median central incisor | – | – | – | – | – | – | 2/14 |
| Facial | Hyper-telorism, prominent forehead | Hyper-telorism, prominent forehead | Hyper-telorism, prominent forehead | – | Midface hypoplasia | – | Deep set eyes, long philtrum, thin upper lip | Midface hypoplsia | Long philtrum, micrognathia | Midface hypo-plasia | Midface hypoplasia | – | Hyper-telorism, deep set eyes, coarse facies | – | 10/14 |
| Eye | |||||||||||||||
| Anophthalmia | – | – | – | – | – | – | – | – | – | – | Bilateral | – | – | Bilateral | 2/14 |
| Microphthalmia | – | – | – | – | Bilateral | – | – | – | Bilateral | Bilateral | – | – | – | – | 3/14 |
| Cyclopia | – | – | – | + | – | – | – | – | – | – | – | – | – | – | 1/14 |
| Coloboma | – | – | – | – | Bilateral | – | – | – | – | – | – | – | – | – | 1/14 |
| Vision | Astig-matism | Retinal dystrophy | Reduced visual acuity | – | – | – | – | Strabismus, hetero chromia iridis | Retinal dysplasia | Retinal dysplasia | – | – | – | – | 6/14 |
| CNS | |||||||||||||||
| Size | – | – | – | – | Macro-cephaly | – | Micro-cephaly | Macro-cephaly | Enlarged cranium | – | Enlarged cranium | Anen-cephaly | – | Enlarged cranium | 7/14 |
| Holoprosencephaly | – | – | – | + | – | – | – | – | Lobar | Lobar | – | – | – | – | 3/14 |
| Ventriculomegaly | + | – | – | – | – | – | + | + | + | + | – | – | – | + | 6/14 |
| Pituitary gland | Absent | Absent | Hypoplastic | – | Absent | – | – | Ectopic | Absent sella turcica | Dysplastic sella turcica | Absent sella turcica | – | – | – | 8/14 |
| Optic nerve | – | – | Septo-optic dysplasia | – | – | – | – | – | Fused | Fused | Absent | – | – | – | 4/14 |
| Thalamus | – | – | – | – | – | – | – | – | Fused thalami, dysplastic hypo-thalami | Fused thalami, dysplastic hypo-thalami | – | – | – | – | 2/14 |
| Cerebellum | – | – | – | – | Vermis hypoplasia | – | Dandy Walker | – | Abnormal vermis | Abnormal vermis | – | – | – | – | 4/14 |
| Corpus callosum | – | – | Agenesis | Agenesis | Agenesis | – | – | – | – | – | – | – | – | – | 3/14 |
| Limb | |||||||||||||||
| Polydactyly | – | – | Postaxial | – | + | – | – | – | – | – | – | Unilateral postaxial | – | – | 3/14 |
| Syndactyly | – | 2,3 toes | – | – | – | – | 2, 3 toes | – | – | – | 2–5 toes | – | – | – | 3/14 |
| Digits | – | Sandal gap | – | Sandal gap | – | – | – | Brachy-dactyly | – | Short digits | Sandal gap | Sandal gap | Absent 5th toe phal-ange | – | 7/14 |
| Other | – | Hyper-mobile joints | Rhizomelic shortening | Club foot | – | Short long bones | Mild restriction of joints | – | Flexed wrists, rocker bottom feet | Short, curved ulnea | – | – | Short long bones | Short long bones | 9/14 |
| Cardiac | |||||||||||||||
| Congenital heart defect | – | – | – | VSD | PDA, ASD | – | – | – | AVSD | – | Dextro-cardia | – | – | – | 4/14 |
| Gastrointestinal | |||||||||||||||
| Omphalocele | – | – | – | – | – | – | – | – | – | – | + | + | – | – | 2/14 |
| Malrotation | – | – | – | – | – | + | – | – | – | – | – | – | – | – | 1/14 |
| Situs | – | – | – | – | – | – | – | – | – | – | Situs inversus | – | – | – | 1/14 |
| Genitourinary | |||||||||||||||
| Renal | – | – | – | – | – | – | – | – | – | Hypo-plasia | – | – | – | – | 1/14 |
| Adrenal | – | – | – | – | – | – | – | – | Hypoplasia | Hypo-plasia | – | – | – | – | 2/14 |
| Genitalia | Micro-penis, Crypt orchidism | – | – | – | Ambiguous | – | – | – | – | Micro-penis | – | – | – | – | 3/14 |
| Developmental | |||||||||||||||
| Delays | Global | Motor | Global | – | – | – | Global | Global | – | – | – | – | ID | – | 6/14 |
| Seizures | – | – | – | – | + | – | – | – | – | – | – | – | – | – | 1/14 |
| Prenatal presentation | |||||||||||||||
| – | – | Short long bones, midline cystic brain changes, poly- hydramnios | Multiple anomalies | Cleft lip, abnormal hands, small head | Midline facial cleft, short femur length | – | Short long bones, ventriculo- megaly | Multiple anomalies | Multiple anomalies fetal growth restriction | Multiple anomalies | Multiple anomalies, fetal growth restriction | – | Multiple anomalies | 10/14 | |
Abbreviations: ASD, atrial septal defect; AVSD, atrioventricular septal defect; ID, intellectual disability; PDA, patent ductus arteriosus; VSD, ventricular septal defect.
Head CT imaging showed ventriculomegaly involving the lateral ventricles bilaterally as well as the third ventricle and sulcal prominence. Increased extra-axial space overlying the convexities of the frontal lobes bilaterally was also present. A small cerebellum was noted, and multiple skull deformities were identified, including asymmetric flattening of the right posterior parietal bone and hypoplastic frontal bones bilaterally. There was no evidence of craniosynostosis. Brain and spinal cord MRI confirmed the head CT scan findings and also showed enlargement of the posterior fossa with hypoplasia of the inferior cerebellar vermis.
3.1.1 |. Genetics
Karyotype and microarray were normal. Trio exome sequencing revealed compound heterozygous missense variants in TBC1D32 GenBank ID: NM_152730.6:c.3182G > C p.(Gly1061Ala) and c.2134A > G p.(Asn712Asp). Both changes are rare (gnomAD frequency: 0.0006684 and 0.000004047, respectively), and neither is present in homozygosity in gnomAD (www.gnomad.broadinstitute.org). They are both considered variants of uncertain significance. (Figures 1 and 2; Table S1).
FIGURE 1.
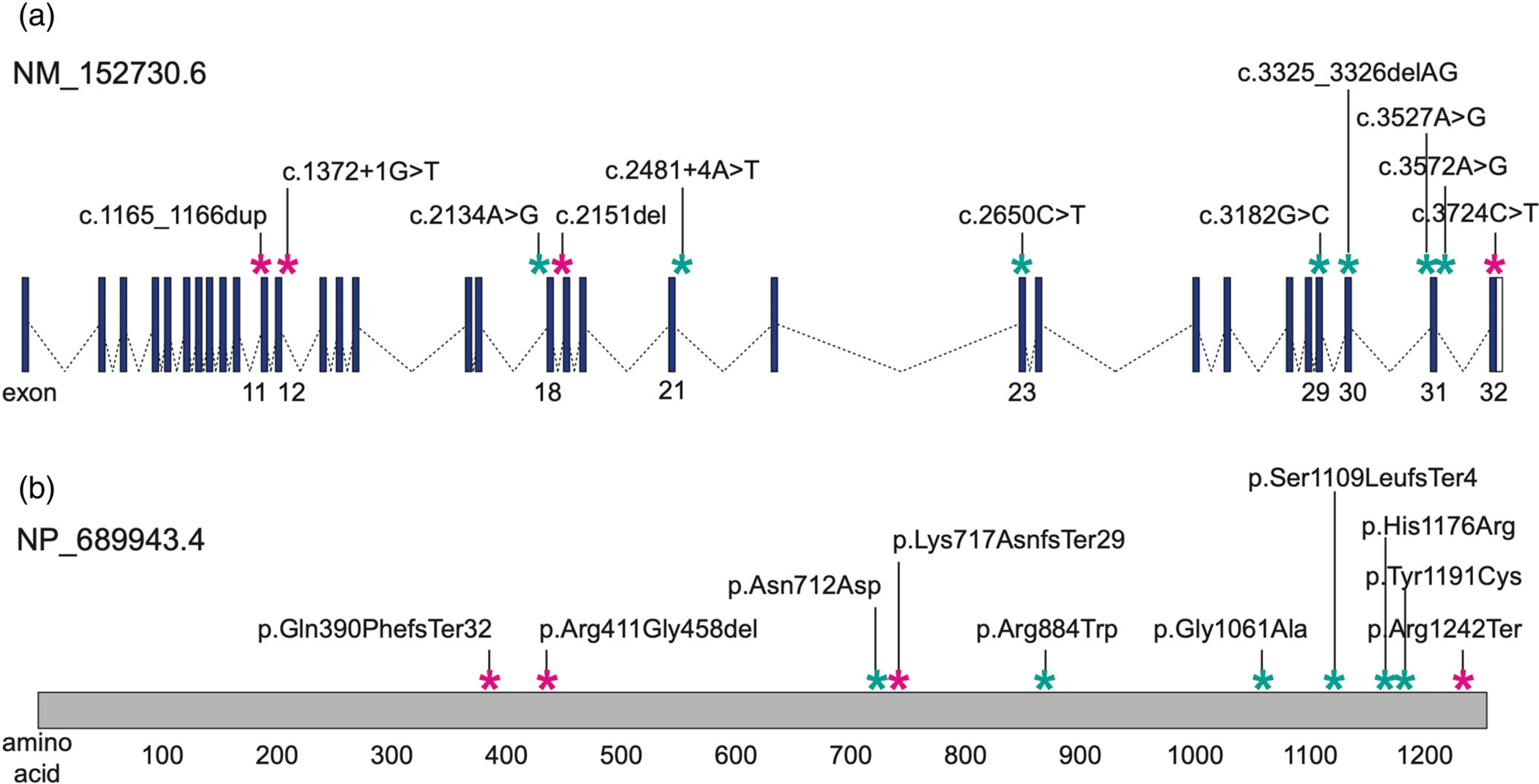
TBC1D32 Schematic. (a) Genomic locus on chr6:121,079,494–121,334,480 (GRCh38, reverse strand). Blue boxes, exons; white boxes, untranslated regions; dashed lines, introns; exons harboring case-associated variants are labeled at the bottom. (b) Location of variants on a linear protein schematic; amino acids are labeled at the bottom. For both panels: teal asterisks, hitherto unreported case-associated variants; magenta asterisks, previously published pathogenic variants.
FIGURE 2.
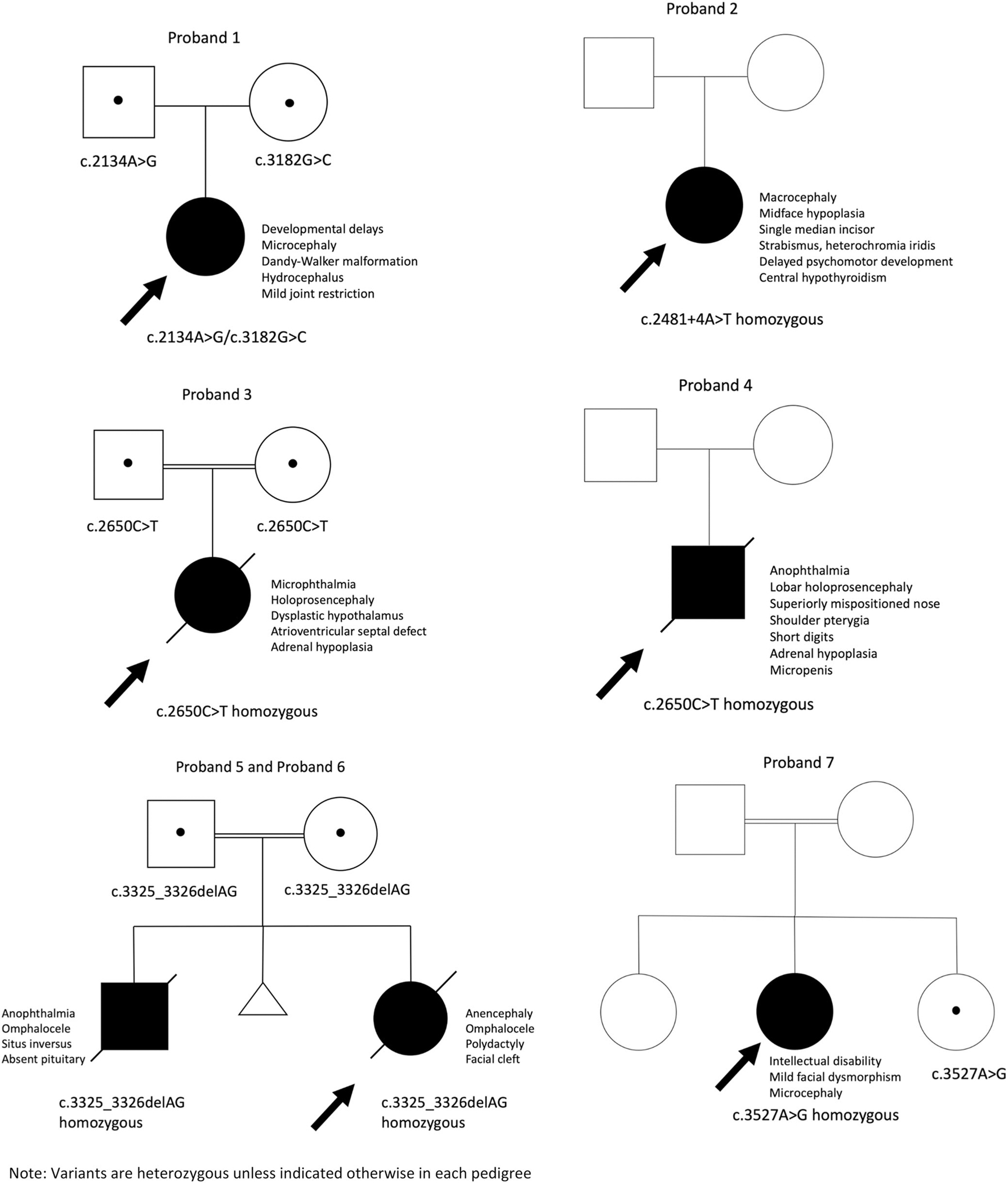
Pedigrees of previously unpublished TBC1D32 variants that segregate with disease (variants are heterozygous unless indicated otherwise in each pedigree).
3.2 |. Proband 2
A 3-month-old female was referred for clinical genetics assessment due to micromelia and brachydactyly (Table 1). She was the first child of unrelated and healthy parents. During pregnancy, mild short long bones and borderline ventriculomegaly were identified. On examination, the patient was noted to have strabismus, a single median incisor, choanal stenosis, short limbs, and relative macrocephaly. She was noted to have a large forehead, prominent forehead, epicanthus inversus, narrow palpebral fissures, ptosis, and midface hypoplasia. Obstructive sleep apnea and global developmental delay were also diagnosed.
MRI of the brain suspected an ectopic pituitary gland in relation to growth hormone (GH) and thyroid stimulating hormone (TSH) deficiency.
3.2.1 |. Genetics
Karyotype was not performed. Sequencing of recurrent FGFR3 mutations was normal. Microarray revealed 17p12 microdeletion (including PMP22) unrelated to the patient phenotype. Exome sequencing identified compound heterozygous variants in TBC1D32 GenBank ID: NM_152730.6: c.2481 + 4A > T, considered to be a pathogenic splicing variant by performing a splicing reporter minigene assay (exon 21 skipping) and c.3572A > G (p.Tyr1191Cys), considered to be a missense likely pathogenic variant. Both changes are rare (gnomAD frequency: 0.00002685 and 0.000008025, respectively), and neither is present in homozygosity in gnomAD. (Figures 1 and 2; Table S1).
3.3 |. Proband 3
A 26-year-old primigravid of Sri Lankan descent was referred for the targeted ultrasound at 20 weeks gestation for multiple fetal anomalies. The patient and her partner were healthy and consanguineous. No significant family history was reported. Ultrasound findings were significant for hydrocephalus, possible lobar holoprosencephaly, bilateral microphthalmia/anophthalmia, proboscis and severe midface hyperplasia, congenital heart defect, possible esophageal atresia, wrists in a flexed position, rocker bottom feet and dorsiflexed large toes (Table 1).
The patient underwent induction of labor for pregnancy termination at 20 weeks gestation and autopsy was completed. Findings included abnormal facies with an enlarged cranium, microphthalmia, patent partially fused nares superior to orbits, long philtrum, and micrognathia (Figure 3). The sella turcica was absent and there was no identifiable pituitary gland. The adrenal glands were hypoplastic. Lobar holoprosencephaly with fused a fused optic nerve containing dysplastic retinal elements, severely dysplastic hypothalamus, fused thalami, aqueductal atresia, brainstem disorganization with absent corticospinal tracts, and a poorly demarcated cerebellar vermis were observed (Figures 4 and 5). The heart demonstrated an incomplete, balanced atrioventricular septal defect.
FIGURE 3.
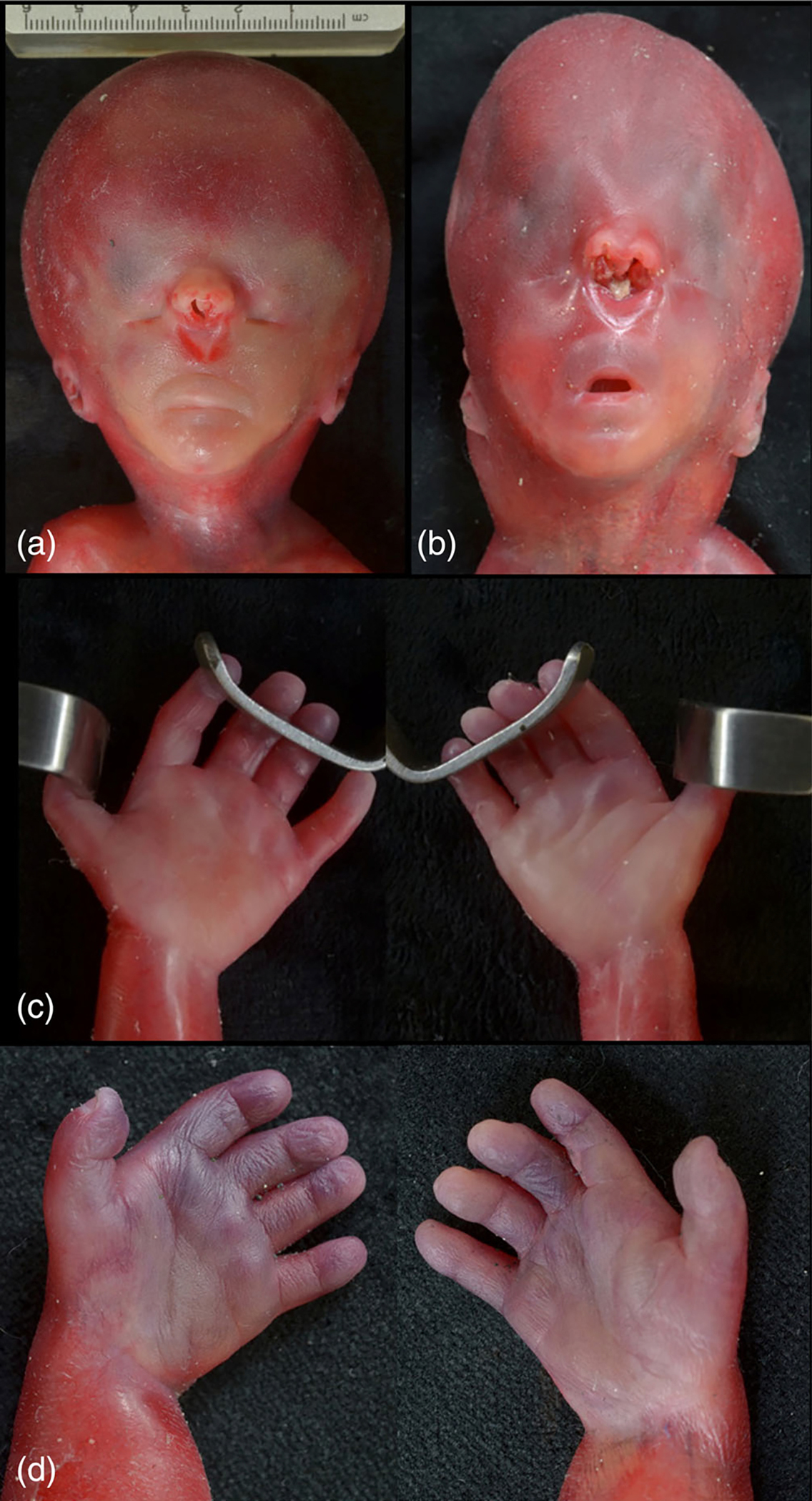
Facies comparison of similar facies of Proband 3 (a) and Proband 4 (b). Proband 3 (a) with an enlarged head with microphthalmia and hypotelorism. The columella is absent, resulting in a fused single nostril although the nasal septum is present internally. The philtrum is long with a wedge-shaped depression below the fused nostril, and there is micrognathia. Comparison of hands of Proband 3 (C) and Proband 4 (D). Proband 4 (D) with hypoplastic, poorly demarcated middle phalanges on digits 2–5, with short, thick fingers.
FIGURE 4.
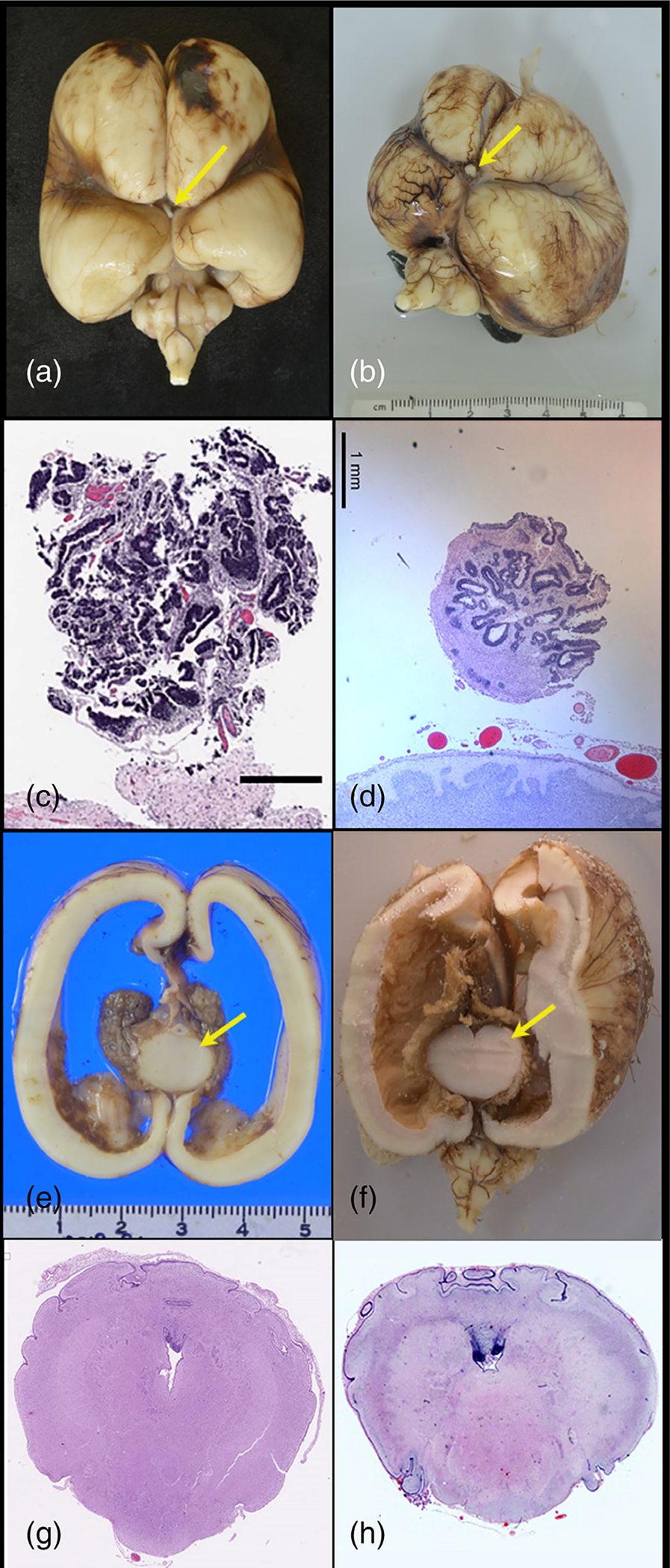
Central nervous system. The inferior surface of the brain in Proband 3 (a) and Proband 4 (b) demonstrates well-marked interhemispheric fissure with absent olfactory bulbs and tracts, absent infundibulum, and single, thick fused optic nerve (arrow). Section through optic nerve in Proband 3 (c) and Proband 4 (d) demonstrating rosettes of ciliated neuroepithelial elements resembling fetal retina. Coronal sections in Proband 3 (e) and Proband 4 (f) demonstrate small fused thalamus (arrow) with massive ventriculomegaly and thinning of the cortical mantle. In both cases, the midbrain was small and the aqueduct stenotic: Cross sections of the cerebellum and pons in Proband 3 (g) and Proband 4 (h) demonstrate a minute fourth ventricle, a poorly demarcated cerebellar vermis, and absent corticospinal tracts.
FIGURE 5.
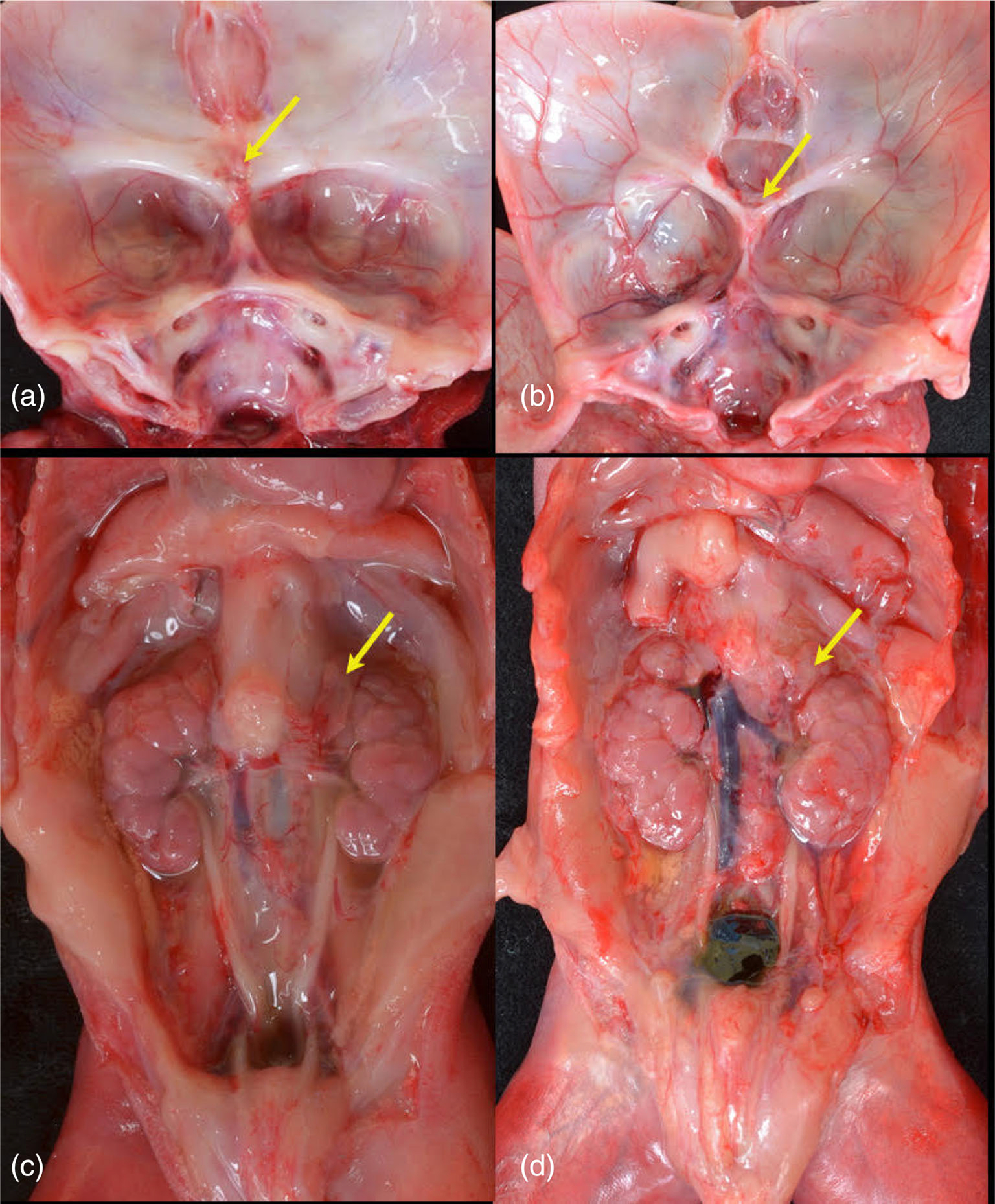
Internal structures. Comparison of skull bases of Proband 3 (a) and Proband 4 (b). Proband 3 (a) with absent sella turcica. Arrow indicates fused optic nerve. No pituitary was present, even on subserial sectioning of the sphenoid. Proband 4 (b) demonstrates wide, shallow sella anterior to (arrow) fused optic nerve. A small pituitary was present. In both fetuses, the hypothalamus was disorganized and no infundibulum was present. Retroperitoneum demonstrating small adrenal glands (arrows) in Proband 3 (c) and Proband 4 (d).
3.3.1 |. Genetics
Karyotype and microarray were normal. A homozygous variant of uncertain significance was identified in TBC1D32, GenBank ID: NM_152730.6:c.2650C > T p.(Arg884Trp). Sequence analysis confirmed that both parents were carriers of the variant. This variant is rare in gnomAD (minor allele frequency 0.0000441) and is not present in homozygosity in healthy populations. In silico analysis supports that this missense variant has a deleterious effect on protein structure and function. (Figures 1 and 2; Table S1).
3.4 |. Proband 4
A 33-year-old primigravid woman of Sri Lankan descent was referred for the targeted ultrasound at 19 weeks following abnormal maternal serum screening. Maternal history was significant for poorly controlled epilepsy treated with levetiracetam and perampanel. Consanguinity was reported. There was no significant family history. Ultrasound showed fetal growth restriction, severe ventriculomegaly, severe midface hypoplasia, possible proboscis, and abnormal posturing of the upper extremities (Table 1).
The patient underwent induction of labor for pregnancy termination at 22 weeks gestation and an autopsy was completed. On autopsy, there was severe midface hypoplasia with high positioned nose, low set and posteriorly rotated ear, microphthalmia, patent partially fused nares superior to the orbits, long philtrum, and micrognathia (Figure 3). The sella turcica was shallow and dysplastic and the adrenal glands hypoplastic. The brain demonstrated lobar holoprosencephaly with fused a fused optic nerve containing dysplastic retinal elements, severely dysplastic hypothalamus, fused thalami, aqueductal atresia, brainstem disorganization with absent corticospinal tracts, and a poorly demarcated cerebellar vermis (Figures 4 and 5). Additionally, axillary pterygia, short digits, renal hypoplasia, and a micropenis were noted. Radiology demonstrated short, curved ulnae bilaterally.
3.4.1 |. Genetics
Microarray reported a normal male. Holoprosencephaly Panel at Gene Dx was negative for TGIF, SHH, SIX3, and ZIC2. A homozygous variant of uncertain significance was identified in TBC1D32, GenBank ID: NM_152730.6:c.2650C > T p.(Arg884Trp). Sequence analysis confirmed that both parents were heterozygous carriers of the variant. This variant is rare in gnomAD (minor allele frequency 0.0000441) and is not present in homozygosity in healthy populations. In silico analysis supports that this missense variant has a deleterious effect on protein structure and function. (Figures 1 and 2; Table S1).
3.5 |. Probands 5 and 6
A consanguineous Pakistani couple with two affected pregnancies was identified through an ongoing fetal exome sequencing study. In the couple’s first affected pregnancy (Proband 5; Table 1), an abnormal maternal serum screen prompted a detailed anatomy ultrasound. Multiple anomalies were identified, including suspected holoprosencephaly, proboscis, suspected anophthalmia, situs inversus, bilateral echogenic kidneys, omphalocele, ambiguous genitalia, and shortened long bones.
The patient underwent induction of labor for pregnancy termination at 16 weeks gestation and autopsy was completed. Significant findings included, hypoplastic midface, anophthalmia, low-set posteriorly rotated ears, absent nose, bilateral submental pterygia, a small omphalocele, partial syndactyly of toes 2–5 on the right, bilateral sandal gap, complete situs inversus with dextrocardia absent sella turcica, no pituitary tissue identified grossly, possible absent corpus callosum, absence of olfactory tracts and bulbs, rectus gyri, and optic nerves. Small/hypoplastic adrenal glands were also noted.
In the couple’s second affected pregnancy (Proband 6; Table 1), ultrasound showed multiple fetal anomalies including, suspected anencephaly, midline facial cleft, small omphalocele, polydactyly, clubbed feet, and intrauterine growth restriction. Intrauterine fetal death was diagnosed at 21 weeks gestation followed by a spontaneous vaginal delivery. A fetal autopsy was not completed. As per the delivery note, the fetus was female with anencephaly, facial cleft, small omphalocele, clubbed feet, left foot postaxial polydactyly, and left-hand postaxial polydactyly.
Karyotype and microarray were normal in both affected fetuses. Exome sequencing data identified homozygous loss of function variants in TBC1D32, GenBank ID: NM_152730.6 c.3325_3326del (p.-Ser1109Leufs*4) (Figure 1). Sanger sequence analysis confirmed that both the mother and the father are heterozygous carriers of the frameshifting variant. This change is absent from the genome aggregation database and considered likely pathogenic according to ACMG variant classification criteria (Figures 1 and 2; Table S1).
3.6 |. Proband 7
A 10-year-old Brazilian female with short stature and intellectual disability was referred for clinical genetics assessment (Table 1). Consanguinity was reported. The patient was noted to have proportional postnatal onset short stature. Mild facial dysmorphism including coarse face, deep set eyes, pectus excavatum, mild ptosis, and full lips. She had moderate-to-severe intellectual disability. Additional findings included microcephaly and blue sclera.
A skeletal survey showed nonspecific shortening of the long bones and the absence of the distal phalanges of the fifth toe. She had a negative screen for mucopolysaccharidosis type 1 and type IV and normal IGF-1 and GH peak. She had a normal brain MRI.
3.6.1 |. Genetics
Karyotype and microarray were normal. Exome sequencing identified a homozygous missense variant in TBC1D32 GenBank ID: NM_152730.6:c.3527A > G p.(His1176Arg), considered to be a variant of uncertain significance (Figure 1). This change is rare in gnomAD (MAF 0.00000803) and is not present in homozygosity in healthy populations (Table S1). The change is also rare in ABraOM (https://abraom.ib.usp.br), a Brazilian public genomic database, (MAF 0.001708) and only present in the heterozygous state. (Figures 1 and 2; Table S1).
4 |. DISCUSSION
In this study, we report seven new probands in six unrelated pedigrees with biallelic TBC1D32 variants and seven previously reported patients associated with a complex ciliopathy phenotype (Tables 1; Table S1). Common findings include ocular differences in the majority of individuals (10/14), including anophthalmia/microphthalmia (5/14), absent or abnormal nose formation (10/14), and facial clefting (5/14). Digital differences, including polydactyly and syndactyly, were noted in 10 individuals. CNS abnormalities were identified in all probands including, anencephaly, hydrocephalus, holoprosencephaly, microcephaly, and a poorly demarcated vermis with cerebellar hypoplasia. Five of 14 reported probands had absence/hypoplasia of the pituitary gland. Additional findings include congenital heart disease, intestinal malrotation, omphalocele, and situs inversus (Table 1).
Ciliopathies comprise a heterogeneous group of disorders caused by dysfunction of the primary cilium, a microtubule-based organelle found in almost every mammalian cell that is critical for integrating and transducing extracellular signals (Satir & Christensen, 2007; Shamseldin et al., 2020). TBC1D32 was first associated with a severe ciliopathy phenotype by Adly et al. (2014). In their report, an affected male infant born to consanguineous parents was noted to have multiple facial anomalies, including left anophthalmia, right microphthalmia, midline facial cleft, and choanal stenosis. Polydactyly and congenital heart defects were also noted. MRI was significant for the absent pituitary gland and agenesis of the corpus callosum. Exome sequencing identified a homozygous splice junction variant in TBC1D32I, c.1372 + 1G > T p.(Arg411_Gly458del). The authors concluded that this phenotypic constellation most closely matched OFD Type IX, given the prominent eye involvement (Adly et al., 2014). Tongue differences, including bifid and hamartomatous tongues, have also been reported with OFD Type IX (Gurrieri et al., 2007). Notably, there is a lack of classic tongue findings in individuals with TBC1D32 variants (1/14; Table 1).
TCB1D32 has been identified as a primary ciliary protein through cilia proteomic studies (Ishikawa et al., 2012). In mice and zebrafish models, TCB1D32 has been shown to be involved in the development and function of cilia. It has been hypothesized that TCB1D32 interacts with cell cycle-related kinase (CCRK) to allow for the correct assembly of the cilia axoneme and membrane, which is required for the sonic hedgehog (Shh) signaling pathway (Ishikawa et al., 2012; Ko et al., 2010). In the bromi mouse model, mutants harbor a deletion of the TBC1D32 homologue that prevents the correct development of cilia and interrupts Shh signaling. Bromi mutants have exencephaly, poorly developed eyes, and preaxial polydactyly (Ko et al., 2010). Similar findings are noted in the individuals with a severe fetal presentation in this series, including anencephaly and holoprosencephaly, anophthalmia, and polydactyly (Table 1).
Human gene expression analyses have identified TCB1D32 in the developing hypothalamus and pituitary gland and in adult cDNA libraries (Hietamäki et al., 2020). It has been suggested that TCB1D32 is activated during the early stages of hypothalamo-pituitary formation and may play a role in other parts of the developing brain (Hietamäki et al., 2020). Differences in the CNS were observed in all of the reported probands harboring TBC1D32 variants (Table 1).
The probands reported in this study further define the phenotype associated with TBC1D32 variants. However, consistent with the notorious phenotypic variability of the ciliopathies, clearly defined genotype–phenotype are difficult to establish. This is exemplified by the spectrum of phenotypes displayed among the four individuals harboring the recurrent c.1372 + 1G > T, p.(Arg411Gly458del) variant (Tables 1). Second-site modifiers, potentially impacting other ciliary proteins, may explain the heterogeneity of clinical presentation in this small cohort as described for other ciliopathies (Kousi et al., 2020).
Fetal phenotypes often vary significantly from the well-defined pediatric or adult phenotype and accurate and thorough documentation is imperative to assist with the diagnosis and classification of variants (Gray et al., 2019). Prenatal ultrasound findings were noted in 10 of the 14 reported probands, including a severe phenotype with multiple life-limiting anomalies in seven individuals. Two of the pediatric patients had minor ultrasound findings reported, including short longs bones, ventriculomegaly, and polyhydramnios. We also present an affected individual diagnosed with an absent pituitary gland by fetal autopsy, a finding reported in four of the pediatric patients. Although prenatal diagnosis continues to improve with advances in the field of ultrasound, a fetal autopsy can aid in the diagnosis of additional anomalies and improve diagnostic yield (Nayak et al., 2015). Deep phenotyping with additional postnatal evaluation is critical for defining the fetal phenotype for rare genetic conditions.
5 |. CONCLUSION
We describe a total of 14 affected individuals with TBC1D32 variants, expanding the phenotype of this complex ciliopathy. Prior functional studies and the probands presented provide the support that TBC1D32 plays a critical role in the development of the pituitary gland, eyes, and brain.
This report also highlights the importance of data sharing for rare genetic diseases. It is essential for clinicians and researchers to collaborate through online platforms to accurately define rare variants. Additionally, detailed documentation of prenatal ultrasound findings and autopsy results is essential for defining the prenatal phenotype of rare genetic conditions.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by US National Institutes of Health grants R21TR002770 (Neeta L. Vora and Erica E. Davis), R01HD105868 (Neeta L. Vora and Erica E. Davis), R01DK072301 (Erica E. Davis), and R01HD042601 (Erica E. Davis). Sao Paulo Research Foundation—FAPESP (grants 2013/03236-5 to Alexander A. L. Jorge and 2018/10893-6 to Bruna L. Freire); and the National Council for Scientific and Technological Development—CNPq (grant 303294/2020-5 to Alexander A. L. Jorge). Erica E. Davis is the Ann Marie and Francis Klocke, MD research scholar.
FUNDING INFORMATION
US National Institutes of Health grants R21TR002770 (Neeta L. Vora and Erica E. Davis), R01HD105868 (Neeta L. Vora and Erica E. Davis), R01DK072301 (Erica E. Davis), and R01HD042601 (Erica E. Davis). Sao Paulo Research Foundation—FAPESP (grants 2013/03236-5 to Alexander A. L. Jorge and 2018/10893-6 to Bruna L. Freire) and the National Council for Scientific and Technological Development—CNPq (grant 303294/2020-5 to Alexander A. L. Jorge). Erica E. Davis is the Ann Marie and Francis Klocke, MD research scholar.
Footnotes
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Adly N, Alhashem A, Ammari A, & Alkuraya FS (2014). Ciliary genes TBC1D32/C6orf170 and SCLT1 are mutated in patients with OFD type IX. Human Mutation, 35(1), 36–40. 10.1002/humu.22477 [DOI] [PubMed] [Google Scholar]
- Alsahan N, & Alkuraya FS (2020). Confirming TBC1D32-related ciliopathy in humans. American Journal of Medical Genetics. Part A, 182(8), 1985–1987. 10.1002/ajmg.a.61717 [DOI] [PubMed] [Google Scholar]
- Bruel AL, Franco B, Duffourd Y, Thevenon J, Jego L, Lopez E, Deleuze JF, Doummar D, Giles RH, Johnson CA, Huynen MA, Chevrier V, Burglen L, Morleo M, Desguerres I, Pierquin G, Doray B, Gilbert-Dussardier B, Reversade B, … Thauvin-Robinet C (2017). Fifteen years of research on oral-facial-digital syndromes: From 1 to 16 causal genes. Journal of Medical Genetics, 54(6), 371–380. 10.1136/jmedgenet-2016-104436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray KJ, Wilkins-Haug LE, Herrig NJ, & Vora NL (2019). Fetal phenotypes emerge as genetic technologies become robust. Prenatal Diagnostica, 39(9), 811–817. 10.1002/pd.5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurrieri F, Franco B, Toriello H, & Neri G (2007). Oral-facial-digital syndromes: Review and diagnostic guidelines. American Journal of Medical Genetics. Part A, 143A(24), 3314–3323. 10.1002/ajmg.a.32032 [DOI] [PubMed] [Google Scholar]
- Hietamäki J, Gregory LC, Ayoub S, Iivonen AP, Vaaralahti K, Liu X, Brandstack N, Buckton AJ, Laine T, Känsäkoski J, Hero M, Miettinen PJ, Varjosalo M, Wakeling E, Dattani MT, & Raivio T (2020). Loss-of-function variants in TBC1D32 underlie syndromic hypopituitarism. The Journal of Clinical Endocrinology and Metabolism, 105(6), 1748–1758. 10.1210/clinem/dgaa078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Thompson J, Yates JR 3rd, & Marshall WF (2012). Proteomic analysis of mammalian primary cilia. Current Biology, 22(5), 414–419. 10.1016/j.cub.2012.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko HW, Norman RX, Tran J, Fuller KP, Fukuda M, & Eggenschwiler JT (2010). Broad-minded links cell cycle-related kinase to cilia assembly and hedgehog signal transduction. Developmental Cell, 18(2), 237–247. 10.1016/j.devcel.2009.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousi M, Söylemez O, Ozanturk A, Mourtzi N, Akle S, Jungreis I, Muller J, Cassa CA, Brand H, Mokry JA, Wolf MY, Sadeghpour A, McFadden K, Lewis RA, Talkowski ME, Dollfus H, Kellis M, Davis EE, Sunyaev SR, & Katsanis N (2020). Evidence for secondary-variant genetic burden and non-random distribution across biological modules in a recessive ciliopathy. Nature Genetics, 52(11), 1145–1150. 10.1038/s41588-020-0707-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monies D, Abouelhoda M, Assoum M, Moghrabi N, Rafiullah R, Almontashiri N, Alowain M, Alzaidan H, Alsayed M, Subhani S, Cupler E, Faden M, Alhashem A, Qari A, Chedrawi A, Aldhalaan H, Kurdi W, Khan S, Rahbeeni Z, … Alkuraya FS (2019). Lessons learned from large-scale, first-tier clinical exome sequencing in a highly consanguineous population. American Journal of Human Genetics, 104, 1182–1201. 10.1016/j.ajhg.2019.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak SS, Shukla A, Lewis L, Kadavigere R, Mathew M, Adiga PK, Vasudeva A, Kumar P, Shetty J, Shah H, & Girisha KM (2015). Clinical utility of fetal autopsy and its impact on genetic counseling. Prenatal Diagnosis, 35(7), 685–691. 10.1002/pd.4592 [DOI] [PubMed] [Google Scholar]
- Satir P, & Christensen ST (2007). Overview of structure and function of mammalian cilia. Annual Review of Physiology, 69, 377–400. 10.1146/annurev.physiol.69.040705.141236 [DOI] [PubMed] [Google Scholar]
- Shamseldin HE, Shaheen R, Ewida N, Bubshait DK, Alkuraya H, Almardawi E, Howaidi A, Sabr Y, Abdalla EM, Alfaifi AY, Alghamdi JM, Alsagheir A, Alfares A, Morsy H, Hussein MH, Al-Muhaizea MA, Shagrani M, Al Sabban E, Salih MA, … Alkuraya FS (2020). The morbid genome of ciliopathies: An update. Genetics in Medicine, 22(6), 1051–1060. 10.1038/s41436-020-0761-1 [DOI] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, & Hamosh A (2015). Gene-Matcher: A matching tool for connecting investigators with an interest in the same gene. Human Mutation, 36(10), 928–930. 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vora NL, Gilmore K, Brandt A, Gustafson C, Strande N, Ramkissoon L, Hardisty E, Foreman AKM, Wilhelmsen K, Owen P, Weck KE, Berg JS, Powell CM, & Powell BC (2020). An approach to integrating exome sequencing for fetal structural anomalies into clinical practice. Genetics in Medicine, 22(5), 954–961. 10.1038/s41436-020-0750-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.