

Abstract
Stroke is a devastating neurological disease and the leading cause of long-term disability, particularly in the elderly. CaMKK β is a major kinase activated by elevated levels of intracellular calcium. Our previous findings in young mice have suggested that CaMKK β is neuroprotective as KO mice had worse stroke outcomes. Because age is an important determinant of stroke outcome, we evaluated the functional role of CaMKK β in stroke in aged mice. We used middle cerebral artery occlusion to induce stroke in aged wild type (WT) and CaMKK β KO male mice. Lentiviral vectors carrying CaMKK β (LV-CaMKK β) were used to overexpress CaMKK β in the mouse brain. Baseline levels of CaMKK β in the aged brain were significantly lower than those in young mice. LV-CaMKK β treatment reduced infarcts and neurological deficits assessed 3 days after stroke. In chronic survival experiments, CaMKK β KO mice showed increased tissue loss in the ipsilateral hemisphere 3 weeks after stroke. Additionally, KO mice showed poorer functional recovery during the 3-week survival period, as measured by the rotarod test, corner test, locomotor activity assay, and novel object recognition test, compared with WT controls. The loss of blood- brain barrier proteins, inactivation of survival gene expression such as B-cell lymphoma 2 (Bcl-2) and an increase in inflammatory cytokines in the serum were observed after stroke with CaMKK β inhibition. We demonstrate that CaMKK β is neuroprotective in stroke in aged mice. Therefore, our data suggest that CaMKK β may be a potential target for reducing long-term disability after stroke.
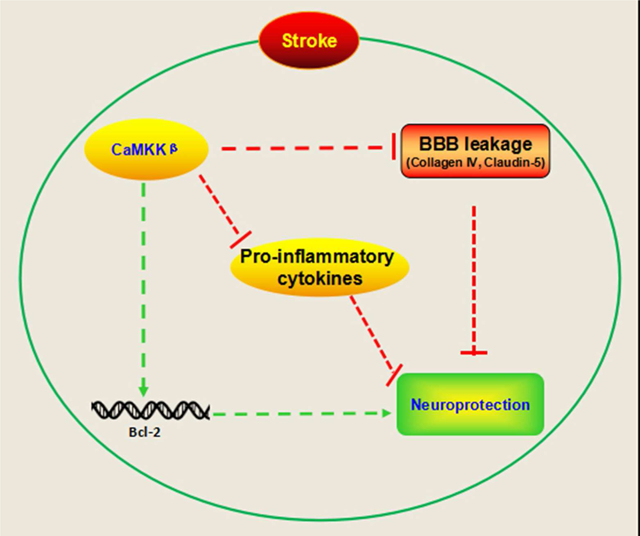
Graphical abstract
This study demonstrates that CaMKK β is neuroprotective following stroke in aged mice. Inhibition of CaMKK β worsened stroke outcome by reducing expression of pro survival factors, as well as the levels of proteins important for the integrity of blood brain barrier, and increasing pro inflammatory cytokines in the serum after stroke. Conversely, overexpression of CaMKK β reduced infarcts and behavioral deficits in aged mice.
Introduction
Cerebral ischemia induces an elevation of intracellular calcium levels. Calcium has traditionally been believed to play detrimental roles in the pathology of stroke (McCullough et al., 2013). However, clinical trials testing treatments that block calcium have failed to show neuroprotection in ischemic stroke (McCullough et al., 2013). Recently, it has been recognized that the pathways activated by calcium signaling may contribute to neuronal survival after stroke. Therefore, the failure of these trials may be due to nonspecific blockage of calcium activated pathways. Calcium/calmodulin dependent protein kinase kinase (CaMKK) is a major kinase that is activated by elevated intracellular calcium and is highly abundant in the brain (Liu et al., 2014). Two isoforms of CaMKK (α and β) exist, with β having longer lasting activity. Upon activation, CaMKK phosphorylates its downstream substrates, CaMK I/IV. Together, these three kinases act in what is termed the CaMK cascade.
The functional role of CaMKK signaling in stroke is currently unestablished, although earlier studies have implicated CaMKK in neuroprotection (McCullough et al., 2013, Liu et al., 2014). CaMKK has been implicated in protecting the integrity of the blood- brain barrier (BBB), the disruption of which is a major cause of neuronal death after stroke. Indeed, our previous work has shown that inhibiting the CaMKK pathway exacerbates infarcts and behavioral deficits after stroke (McCullough et al., 2013, Liu et al., 2014), further suggesting the importance of CaMKK in neuronal survival after stroke. However, it is unclear whether activating CaMKK can be therapeutically beneficial in stroke. Furthermore, downstream mediators of CaMKK have not been identified in the context of stroke.
Age is the most important independent risk factor for stroke (Liu et al., 2009). Nearly 90 % of all strokes occur in humans aged > 65 years (Bix et al., 2013), and elderly stroke patients have a high in-hospital mortality and poor functional outcome after stroke. The aged brain undergoes numerous neurochemical and physiological changes compared with the young brain (Liu et al., 2012). However, the vast majority of stroke studies have been performed exclusively in young animals. Interestingly, studies have provided considerable evidence that CaMK cascade signaling declines with age. Age-related reductions in cortical CaMK IV levels have been observed in the brains of humans, rhesus macaques and mice. Mounting evidence has suggested an age related decline in this important endogenous pro survival mechanism. Therefore, we sought to define the neuroprotective role of CaMKK in stroke using a clinically relevant model- a stroke model in aged mice.
Methods
Animals
The study was performed following the National Institutes of Health Guide for the Care and Use of Laboratory Animals and was approved by the Center for Laboratory Animal Care of the University of Connecticut Health Center. CaMKK β KO mice were a gift from Dr. Anthony Means at Duke University and were backcrossed into a C57BL/6J background for over 9 generations. The C57BL/6J WT mice used in the current study were purchased from Charles River. Age- and weight- matched mice were used in all experiments (16 to 20 months of age). Researchers who performed all analyses and surgeries were blinded to the treatment (drug and mouse phenotype).
Middle cerebral artery occlusion
Sixty or 90 minutes of middle cerebral artery occlusion (MCAO) was employed in the stroke models. We used 90 minutes MCAO for acute survival studies as previously published (Liu et al., 2014). In chronic survival (21 day) experiments, we reduced the MCAO duration to 60 minutes because of the higher mortality in aged mice during long-term survival after stroke. Briefly, the right common and external carotid arteries (CCA and ECA) were exposed. An occluder (6 0 monofilament, silicone coated, 0.23 mm) was advanced into the internal carotid artery through ECA to occlude the middle cerebral artery (MCA). Laser Doppler flowmetry was used to confirm vascular occlusion in each animal. After 60 or 90 minutes of occlusion, mice were reperfused by suture withdrawal. During the ischemic period, body temperature was kept at a physiological level by a heating pad with a feedback thermo control system (FST). The mice were randomly assigned to a stroke or surgical sham cohort. Mice in the sham cohort underwent the same procedure as those in the stroke cohort, except for the MCA occlusion. Animals without neurological deficits at reperfusion time were excluded (see below paragraph for behavior measurement). Totally, 3 mice were excluded and 13 mice died prematurely. Animals were observed over a period of 1 day to 21 days depending on the treatment group. Food pellets were placed directly in the cage so the animals did not have to rear to obtain food. For chronic survival experiments, we placed a wet mash (standard lab chow that was moistened and ground into a mash) into the cage.
Behavior measurements
For acute survival after stroke, neurological deficit scoring was performed at reperfusion and at 72 hours after stroke using a previously described four point scale. The scores were assigned according to the following criteria: 0, no deficit; 1, forelimb weakness and torso turning to the ipsilateral side when animals were held by the tail; 2, circling to the affected side; 3, inability to bear weight on the affected side; and 4, no spontaneous locomotor activity or barrel rolling (Liu et al., 2014, Liu et al., 2012, Jin et al., 2014,). Animals without neurological deficits at reperfusion time were excluded.
In chronic survival experiments, the open field test, corner test, rotarod test, and novel object recognition test (NORT) were performed as previously described (White et al., 2012). Open field testing was performed by placing each mouse in the open field chamber and recording the total number of locomotor activities within a 20-min period by using a computer-operated PAS Open Field System (San Diego Instruments, San Diego, CA). The NORT was performed according to previously published procedures (White et al., 2012). The corner test was performed by placing the mice between two pieces of cardboard, which were gradually moved to encourage the mouse to enter a 300 corner. Once the vibrissae were stimulated, the mice reared forward and upward and then turned to the face the open end. Only turns involving full rearing along either board were recorded. Turn percentage was calculated with previously published procedures (White et al., 2012). For rotarod test performance, the rotarod was rotated at a speed of 2 to 20 revolutions per minute. The time at which each mouse fell from the drum was recorded. Mice were trained before the stroke onset, and they were tested weekly after stroke onset (White et al., 2012).
Histological assessment
For the acute survival study, the stroke mice were killed with isoflurane overdose followed by cervical dislocation and then brains were removed 72 hours after stroke and sliced into 5 2 mm sections. Brain slices were then stained by 1.5% 2, 3, 5-triphenyltetrazolium (TTC) at 37 °C for 8 minutes. Images were digitized, and the infarction was analyzed with Sigmascan Pro5 software, as previously described (Liu et al., 2014). For the chronic survival study, mice were perfused with saline and 4% paraformaldehyde, then the brains were harvested 21 days after stroke for cresyl violet staining following previously published procedures (Liu et al., 2012, Jin et al., 2014). The brain tissue loss was determined according to the following calculation: (contralateral hemisphere ipsilateral hemisphere)/contralateral hemisphere × 100%.
Western blots
The mice were killed by overdose of isoflurane anesthesia followed by cervical dislocation and then brain samples were harvested 6 hours after stroke and then homogenized by RIPA lysis buffer (Boston Bioproducts). The concentration of each sample was measured with a BCA assay (Thermo Scientific). Precast gels were used to separate the proteins, and polyvinylidene difluoride membranes (Bio-Rad) were used for the transfer. After blocking with 5% fat-free milk, the blots were incubated overnight at 4 °C with primary antibodies (anti-CaMKK β antibody, 1:1000, sc-50341, Santa Cruz; anti-Collagen IV antibody, 1:500, ab-19808, Abcam; Claudin-5 antibody, 1:500, ab-15106, Abcam, anti-p-JNK antibody, 1:500, #4671, Cell Signaling Technology; or anti Bcl-2 antibody, 1:1000, sc-7382, Santa Cruz). The appropriate secondary antibodies (anti rabbit IgG, 1:5000, 7074s, Cell Signaling Technology; or anti-mouse IgG, 1:5000, PI-2000, Vector) were then incubated for one hour at room temperature. Signals were detected using an electrochemiluminescence detection kit (Thermo Scientific). β-actin (primary antibody 1:5000, a5316, Sigma) was used as a loading control.
Enzyme-linked immunosorbent assay (ELISA)
Blood samples were harvested transcardially 21 days after stroke and were spun at 6000 rpm for 10 minutes at 4 °C. IL-6 and TNF-alpha were detected with ELISA kits (eBioscience, US) and following the manufacturer’s instructions.
Immunohistochemistry
Samples were processed following our previously published methods (Liu et al., 2014). Twenty-micrometer coronal slices were blocked for 1 hour at room temperature, incubated with anti-collagen IV antibody (1:100, ab-19808, Abcam) overnight at 4 °C and then labeled with an Alexa Flour 488 labeled secondary antibody (1:500, Invitrogen). Images were digitized and analyzed with a Zeiss Axiovert 200 M microscope (Carl Zeiss, Oberkochen, Germany) with Zeiss image acquisition software (Zeiss LSM 510) and an X-Cite 120Q fluorescence illumination system (Lumen Dynamics Group Inc., Mississauga, ON, Canada). The collagen IV level was semi-quantified as the integrated optical density (IOD) of green-positive signals in the penumbra of the ischemic hemisphere. The unit of the IOD results was adjusted to 1 by dividing it by the mean IOD of the WT controls.
Production and titration of lentiviral vector
The GFP-CaMKK β (pRRLsinPPThPGK-CamKK2-IRESeGFP, Lot#G0756) plasmid was purchased from the University of Iowa, whereas the packaging plasmids of pHDM-tat1b, pRC/CMV-rev1B, pHDM-Hgpm2 and pHDM-G were kindly provided by Dr. Alexander Lichtler (University of Connecticut Health Center, Farmington, CT). A 4-plasmid system was employed for lentiviral packaging. Briefly, 293T cells were cultured in Dulbecco’s modified Eagle’s/F12 medium (DMEM/F12; Gibco, Grand Island, NY, USA) with 1% penicillin or streptomycin (Gibco) and 10% fetal bovine serum (Gibco). Following the calcium phosphate precipitation method (Tiscornia et al., 2006), 18 separate T175 flasks including 1.5-2 × 107 HEK-293T cells were transiently infected by a mixture of 360 μg of transfer vectors, 54 μg of pHDM-tat1b, 27 μg of pRC/CMV-rev1B, 27 μg of pHDM-Hgpm2 and 54 μg of pHDM-G. Supernatants were collected 48 and 72 hours after transfection, and the viral particles were concentrated with ultracentrifugation. The viruses were further concentrated by sucrose gradient ultracentrifugation. Viruses were stored in Hank’s balanced salt solution (HBSS) at -80 0C before injection. The viral particles were used for titration by transducing 293FT cells and counting eGFP-positive colonies (Tiscornia et al., 2006).
Lentivirus and drug administration in vivo
Mice were anesthetized and placed in a stereotaxic frame (Stoetling, IL, USA). A 4-point injection was performed at the following coordinates: 0.5 mm anterior to the bregma, 2.0 mm or 3.0 mm lateral (right) to the sagittal suture, and 1.0 mm or 2.8 mm from the surface of the skull (Zeng et al., 2014). At each position, 1 μL of concentrated lentivirus (109 transducing units/mL) was injected at a speed of 0.5 μL/min with a 30 gauge needle on a 10-μL Hamilton syringe (Cat# 80010). After the lentivirus was injected, the needle remained in position for 10 min before it was withdrawn. The stroke surgery was performed 7 days after the lentivirus injection. STO-609, a CaMKK inhibitor, was intracerebroventricularly injected to mice as previously described (McCullough et al., 2013).
Statistical analysis
The neurological deficit scores are presented as the median (IQR); all other data are shown as the mean ± SEM. One way or two way ANOVA was performed for mean comparisons whenever appropriate. Posthoc test used for multiple comparisons was Tukey test. Neurological deficit scores were compared with the Mann-Whitney U test. A value of p<0.05 was considered statistically significant. Statistical power for each variable measured in our study was over 80%.
Results
CaMKK β levels were reduced in the brains of aged mice
Western blotting was performed to measure the baseline levels of CaMKK β protein in both young and aged brains. We found that CaMKK β levels were reduced in the aged brains compared with those in the young brains (n=3 p/g; Figure 1A and 1B, p<0.05).
Fig. 1. The CaMKK β level is reduced in the brains of aged mice.

A, Representative western blots of CaMKK β in young and aged mice. B, Quantification of CaMKK β levels, n = 3 p/g. Data were normalized to young mice, # P= 0.01 compared with young mice; data are presented as the mean ± SEM.
Overexpression of CaMKK β reduced injury, assessed 3 days after stroke
For the acute survival experiments, we used a lentivirus to overexpress CaMKK β in WT aged mice. We found that the vector efficiently increased CaMKK β levels in aged brains 7 days after induction (n=3 p/g; Figure 2A and 2C). Furthermore, our results demonstrated that the overexpression of CaMKK β reduced infarct volume assessed 3 days after stroke (GFP control 26.43 ± 4.30% vs. CaMKK vector 16.76 ±3.16%, p<0.05; n=7 p/g; Figure 2B and 2D). Consistently with the reduced infarcts, neurological deficit scores were also decreased by the overexpression of CaMKK β [Median (IQR): LV GFP 2(1) vs. LV CaMKK β 1(1), p<0.05].
Fig. 2. The overexpression of CaMKK β reduced injury assessed 3 days after stroke.

A and B, Representative western blots of CaMKK β and brains with cerebral infarcts in LV-GFP and LV-CaMKK β mice. C, Quantification of CaMKK β levels in mouse brains 7 days after induction, n = 3 p/g. D, infarct volumes 3 days after stroke, n = 7 p/g. P= 0.008 in C while P= 0.047 in D, # P < 0.05 compared with LV-GFP; data are presented as the mean ± SEM.
Deletion of CaMKK β increased injury in aged mice, assessed 3 days after stroke
We next compared the outcomes in CaMKK β KO and WT aged mice 3 days after stroke. Consistently with the kinase overexpression data, the deletion of CaMKK β increased both the infarct volume (WT 23.02 ±3.67% vs. KO 35.22 ±4.15%, p<0.05; n=7 p/g; Figure 3A and 3B) and neurological deficit scores [Median (IQR): WT 2(1) vs. CaMKK β KO 3(1), p<0.05].
Fig. 3. Deletion of CaMKK β increased injury in aged mice assessed 3 days after stroke.

A, Representative brains with cerebral infarcts in WT and CaMKK β KO mice. B, Quantification of infarct volumes 3 days after stroke, n=7 p/g. P= 0.048, # P<0.05 compared with WT stroke; data are presented as the mean±SEM.
Inhibition of CaMKK β reduced Bcl-2, collagen IV and claudin-5 but increased p-JNK 6 hours after stroke
Next, we examined the mechanisms of CaMKK β during acute survival after stroke in aged mice using STO-609, an inhibitor of CaMKK. We found that the inhibition of CaMKK β reduced the levels of Bcl-2 (n=4 p/g; Figure 4A and 4B). Bcl-2 is an anti apoptotic protein, and its transcription is suggested to be regulated by CaMKK signaling. Our data suggested that CaMKK KO reduced transcriptional activation after stroke. We also observed an increase in the levels of the proapoptotic protein p-JNK in KO mice (n=4 p/g; Figure 4C and 4D, p<0.05). Furthermore, CaMKK β genetic deletion downregulated the levels of collagen IV and claudin-5, two important components of the BBB (n=4 p/g; Figure 4E, 4F, 4G and 4H, p<0.05), implying exacerbated BBB leakage.
Fig. 4. The inhibition of CaMKK β reduced Bcl-2, collagen IV and claudin-5 but increased p-JNK after stroke.

A, C, E and G, Representative western blots of Bcl-2, p-JNK, collagen IV and claudin-5 in vehicle and inhibitor groups. B, D, F and H, Quantification of Bcl-2, p-JNK, collagen IV and Claudin-5 expression, n=4 p/g. Data were normalized to corresponding vehicle sham. B, [ANOVA, F(3, 12)= 6.147, P= 0.01]; D, [ANOVA, F(3, 12)= 5.064, P= 0.02]; F, [ANOVA, F(3, 12)= 7.780, P< 0.01]; H, [ANOVA, F(3, 12)= 12.82, P< 0.01]. # P<0.05 compared with vehicle stroke; data are presented as the mean±SEM.
Deletion of CaMKK β suppressed functional recovery after stroke in chronic survival
Long-term disability is a major consequence of stroke. Therefore, we examined the roles of CaMKK β during chronic survival using CaMKK β KO and aged control mice. We tested memory, as well as sensory, motor, and cognitive functions 21 days after the onset of stroke by performing a battery of tests including the rotarod test (Day 21: WT control 0.72 ± 0.05 vs. CaMKK β KO 0.57 ± 0.04, n=10-11 p/g; Figure 5A), corner test (Day 21: WT control 0.63 ± 0.02 vs. CaMKK β KO 0.76 ± 0.06, n=10-11 p/g; Figure 5B), open field test (Day 21: WT control 12562 ± 1206 vs. CaMKK β KO 4652 ± 652, n=6-7 p/g; Figure 5C) and NORT (Day 21: WT control 0.65 ± 0.02 vs. CaMKK β KO 0.52 ± 0.04, n=6-7 p/g; Figure 5D) (p<0.05). We found that the deletion of CaMKK β significantly worsened behavioral recovery compared with that in the WT controls (Figure 5 A–D, p<0.05).
Fig. 5. The deletion of CaMKK β suppressed functional recovery after stroke in chronic survival.

A and B, rotarod and corner tests conducted within 21 days of stroke onset. The pretest was conducted one day before stroke onset, n=10 or 11. C and D, Open field test on days 14 and 21, and Novel object recognition test on day 21, n=7 in WT and n=6 in KO. A, [F(1, 78) = 18.64, P < 0.01], B, [F(1, 78) = 12.40, P< 0.01], C, [F(1, 22)= 28.76, P < 0.01], D, (P= 0.007), * or # P<0.05 compared with WT stroke; data are presented as the mean±SEM.
Loss of CaMKK β increased stroke-induced tissue loss after chronic survival
By CV staining, we found that the deletion of CaMKK β increased the amount of brain tissue loss 21 days after stroke onset (WT 11.89 ± 1.00% vs. KO 20.59 ± 1.90%; n=7 for the WT group; n=6 for the KO group; Figure 6A and 6B, p<0.05). Together, the results of the histological analysis (Figure 6A and 6B) and functional tests (Figure 5 A–D) showed that CaMKK β is neuroprotective in aged mice, even after long-term survival.
Fig. 6. The loss of CaMKK β increased stroke-induced tissue loss after chronic survival.

A, Representative brains with tissue loss in WT and CaMKK β KO mice 21 days after stroke. B, Quantification of brain tissue loss resulting from stroke, n=7 in WT and n=6 in KO. P= 0.001, # P<0.05 compared with WT controls; data are presented as the mean±SEM.
CaMKK β deletion reduced collagen IV and increased inflammation in chronic survival
To explore the underlying mechanisms of CaMKK β in chronic survival, we next examined collagen IV using IHC staining 21 days after stroke. CaMKK modulates neutrophils, and neutrophils are the major source of MMPs, the enzymes that degrade collagen IV (McCullough et al., 2013). We found that the loss of CaMKK β resulted in a reduction of collagen IV (n=5 p/g; Figure 7A and 7B). Furthermore, we found that inflammation was increased by deleting CaMKK β (n=3 p/g), because serum levels of IL-6 and TNF-α in CaMKK β KO and WT aged mice were significantly elevated 21 days after stroke (IL-6: WT control 146.6 ± 16.86 vs. CaMKK β KO 311.8 ±12.13; TNF-α: WT control 123.2 ± 17.94 vs. CaMKK β KO 281.0 ±11.55. Figure 7C and 7D, p<0.05).
Fig. 7. CaMKK β deletion reduced collagen IV and increased inflammation during chronic survival.

A, Representative collagen IV in WT and CaMKK β KO mice 21 days after stroke, scale bar= 50 um. B, Quantification of collagen IV, n=5 p/g. C and D, IL-6 and TNF α in serum 21 days after stroke onset. IL-6 and TNF α were analyzed with ELISA kits. n=3 p/g. B, (P<0.01); C, [ANOVA, F (3, 8) = 67.83, P< 0.01]; D, [ANOVA, F(3, 8) = 61.27, P< 0.01]; # P<0.05 compared with WT stroke; data are presented as the mean±SEM.
Discussion
We reveal several significant findings in the present study. First, the baseline levels of CaMKK β in aged mice were significantly lower than those in young mice. Second, in the acute survival experiments, the overexpression of CaMKK β improved stroke outcomes by reducing the infarcts and neurological deficits, whereas KO mice showed poorer outcomes during acute survival. Third, pharmacological inhibition of CaMKK reduced the levels of Bcl-2 and the BBB protein collagen IV in the acute survival studies. Fourth, in the chronic survival experiments, genetic deletion of CaMKK β worsened sensory, motor and memory functions and increased brain tissue loss. A loss of BBB proteins and an increase in inflammatory cytokines in the serum were observed in the KO stroke mice after chronic survival.
Our present study is the first to report that CaMKK is neuroprotective in stroke. We demonstrated that augmenting CaMKK signaling improved stroke outcomes in mice. CaMKK resides in both the cytosol and nucleus, where it responds to changes in Ca2+ levels and signals CaM-kinase. CaMKK is held in an inactive state by its autoinhibitory domain, which interacts with the catalytic domain to prevent kinase activity. The binding of Ca2+/CaM releases this autoinhibitory domain, thus activating the kinase (McCullough et al 2013, Liu et al., 2014). It has been suggested that CaMKK provides neuroprotection after stroke by protecting the integrity of the BBB, improving transcriptional activation and reducing inflammation. There are other calcium/calmodulin dependent protein kinases, for instance, death-associated protein kinase (DAPK). Inhibition of DAPK showed neuroprotective effects in both in vitro and in vivo stroke models (Glover et al., 2012). The interaction of CaMKK and DAPK remains an interesting topic for future research.
The inhibition of CaMKK reduced Bcl-2 after stroke in aged mice, thus indicating that endogenous CaMKK activates transcriptional machinery. In young mice, the deletion of CaMKK β or CaMK IV in males has been suggested to exacerbate the nuclear accumulation of histone deacetylase 4 (HDAC4) (McCullough et al 2013), which may subsequently reduce levels of the neuronal survival factors p-CREB and Bcl-2 (Li et al., 2012, Paroni et al., 2007, McCullough et al 2013). Nevertheless, whether and how CaMKK signaling regulates HDAC4 translocation in aged mice in the setting of cerebral ischemia warrants future investigation.
The inhibition of CaMKK by STO-609 or genetic deletion reduced the BBB protein collagen IV and claudin-5. It is known that CaMKK inhibits maturation of neutrophils, and that neutrophils are important sources of MMPs, which could mediate the degradation of collagen IV, a basal lamina protein, and claudin-5, a tight junction protein. STO-609 was injected directly into the brain, suggesting the involvement of other mechanisms besides peripheral neutrophils. It is known that endothelial cells express CaMKK (McCullough et al 2013), but whether endothelial CaMKK directly regulates collagen IV expression under ischemic condition remains to be investigated. Macroscopic hemorrhagic transformation in the ischemic brain was observed in young KO mice following stroke; however, it was not observed in aged mice. Additionally, levels of both collagen IV and claudin-5 showed no difference between sham and vehicle stroke in our study. In young animals, some studies reported reductions in collagen IV levels after stroke (Lu et al., 2013), although others suggested that its levels in stroke mice were similar to sham-operated mice (McColl et al., 2008). The opening of BBB after stroke is biphasic, so changes in collagen IV could be time-dependent. We only studied one time point after stroke as our focus was to see the effect of CaMKK inhibition. More importantly, in our model, aging could be a confounding factor contributing to the discrepancies. The brain undergoes numerous neurochemical and physiological changes with aging, highlighting the importance of using an aged stroke model. For instance, we previously found that aged mice have less BBB leakage after stroke than young (Liu et al., 2009), although the underlying mechanisms are not known. In the chronic survival experiments, we also observed increased levels of serum inflammatory cytokines in KO mice after stroke. A similar observation has been made in young female mice after stroke (Liu et al., 2014), further highlighting the importance of the CaMKK pathway for immune regulation in stroke. The inflammatory response is a key contributor to stroke related injury. Because it occurs in a delayed manner after stroke, it is an attractive target for neuroprotective therapies. It has been suggested that activator protein-1, myocyte enhancer factor-2, and members of the retinoid orphan receptor family may be mediators of the CaMKK pathway-induced effects in the immune regulation in stroke (Liu et al., 2014). Inflammation is important in stroke (Jin et al., 2013). We are aware that assessments of the changes in cytokines at 21 days only provided limited mechanistic insights for the anti-inflammatory action of CaMKK. More time points will need to be assessed. Our focus of the chronic survival study was to see if there is a sustained/long-lasting effect in histological and behavioral outcomes with the manipulation of CaMKK. The potential regulatory role of CaMKK in serum proinflammatory cytokines will be a very interesting future research direction.
Performing chronic survival studies and functional assessment in an aged-animal model is key to moving pre-clinical stroke research into the clinic. The vast majority of stroke occurs in humans aged over 65 years, and aged patients have less capacity for recovery (Bix et al., 2013). We demonstrated a reduction of CaMKK protein in aged mice compared with young mice, a result consistent with previous studies showing a decline in other molecules in this cascade (Fukushima et al., 2008). Stroke is the leading cause of long-term adult disability, especially in the elderly population, which has poorer functional recovery. Therefore, we performed 21-day survival experiments to determine whether manipulation of CaMKK affects the functional outcomes after prolonged survival. Assessments of sensory and motor functions were performed with well-established methods. Cognitive and memory functions were also tested, as recommended by the Stroke Therapy Academic Industry Roundtable (STAIR) and because CaMKK has been implicated in memory formation. We demonstrated that augmenting this cascade in the aged brain could potentially reduce injury or disability resulting from stroke in the elderly. However, the functional recovery data should be interpreted with caution. The recovery might reflect the role that CaMKK plays not only in cell death and survival but also in neurogenesis, because neurogenesis is influenced by CaMKK because of its effects on CREB and BDNF. Both of these proteins enhance neuronal proliferation.
In the near future, pharmacological agents capable of specifically activating this cascade should be designed and tested in stroke models and hopefully moved into clinical trials. With significant effects seen in global deletion (KO) of CaMKK β in our stroke model, it can be speculated that systemic treatments that are clinically relevant (intravenous, oral, et al.) could be effective in providing neuroprotection. Of course, treatments’ capability of penetration of blood brain barrier and their interaction with peripheral environments are to be considered. Mechanistically, CaMKK targets both inflammatory responses as well as apoptotic cell death pathways (Bcl2 and p-JNK signaling) after stroke. As both inflammation and apoptosis take place after stroke in a delayed manner, we envision that a delayed pharmacological treatment (hours after the onset of stroke) that activate CaMK cascade would provide neuroprotection.
In summary, we established a neuroprotective role of CaMKK in stroke by using an aged mice model. Because the elderly population has poorer functional recovery after stroke, our data suggest that this protein may be a potential target for reducing long-term disability in stroke patients.
Acknowledgments
This work was supported by NIH grants R01 NS078446 (J. L.) and R21 NS079137 (J.L.).
Abbreviations
- LV-CaMKK β
Lentiviral vectors carrying CaMKK β
- WT
wild type
- CaMKK
Calcium/Calmodulin-Dependent Protein Kinase Kinase
- BBB
blood- brain barrier
- MCAO
middle cerebral artery occlusion
- FST
feedback thermo-control system
- NORT
novel object recognition test
- TTC
2, 3, 5-triphenyltetrazolium
- HBSS
Hank’s balanced salt solution
- IQR
Interquartile range
References
- Bix GJ, Gowing EK, & Clarkson AN (2013). Perlecan Domain V Is Neuroprotective and Affords Functional Improvement in a Photothrombotic Stroke Model in Young and Aged Mice. Translational Stroke Research, 4(5), 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima H, Maeda R, Suzuki R, Suzuki A, Nomoto M, Toyoda H, Wu LJ, Xu H, Zhao MG, Ueda K, Kitamoto A, Mamiya N, Yoshida T, Homma S, Masushige S, Zhuo M, Kida S(2008). Upregulation of calcium/calmodulin-dependent protein kinase IV improves memory formation and rescues memory loss with aging. J Neurosci. 28(40), 9910–9919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover LE, Tajiri N, Weinbren NL, Ishikawa H, Shinozuka K, Kaneko Y, Watterson DM, Borlongan CV (2012). A Step-up Approach for Cell Therapy in Stroke: Translational Hurdles of Bone Marrow Derived Stem Cells. Transl Stroke Res, 3(1), 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R, Zhu X, Liu L, Nanda A, Granger DN, & Li G (2013). Simvastatin attenuates stroke induced splenic atrophy and lung susceptibility to spontaneous bacterial infection in mice. Stroke; a Journal of Cerebral Circulation, 44(4), 1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R, Liu L, Zhang S, Nanda A, Li G (2013). Role of inflammation and its mediators in acute ischemic stroke.J Cardiovasc Transl Res, 6(5), 834–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Chen J, Ricupero CL, Hart RP, Schwartz MS, Kusnecov A, & Herrup K (2012). Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia-telangiectasia. Nature Medicine, 18(5), 783–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Doran S , Xu Y , Manwani B , Ritzel R , Benashski S, McCullough L, Li J.(2014). Inhibition of mitogen activated protein kinase phosphatase-1 (MKP-1) increases experimental stroke injury. Exp. Neurol, 261, 404–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Fang YQ, Xue ZF, He YP, Fang RM, Li L (2012). Beta asarone attenuates ischemia reperfusion induced autophagy in rat brains via modulating JNK, p-JNK, Bcl-2 and Beclin 1. Eur. J Pharmacol, 680 (1#3), 34–40. [DOI] [PubMed] [Google Scholar]
- Liu F, Benashski SE, Persky R, Xu Y, Li J, & McCullough LD (2012). Age-related changes in AMP-activated protein kinase after stroke. Age, 34(1), 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Yuan R, Benashski SE, & McCullough LD (2009). Changes in experimental stroke outcome across the lifespan. Journal of Cerebral Blood Flow and Metabolism : Official Journal of the International Society of Cerebral Blood Flow and Metabolism, 29(4), 792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, McCullough L, Li J (2014). Genetic deletion of calcium/calmodulin dependent protein kinase kinase β (CaMKK β) or CaMK IV exacerbates stroke outcomes in ovariectomized (OVXed) female mice. BMC Neuroscience, 15, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A, Suofu Y, Guan F, Broderick JP, Wagner KR, Clark JF (2013). Matrix metalloproteinase-2 deletions protect against hemorrhagic transformation after 1 hour of cerebral ischemia and 23 hours of reperfusion. Neuroscience, 253, 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl BW, Rothwell NJ, Allan SM (2008). Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci, 28(38), 9451–9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough LD, Tarabishy S, Benashski S, Liu L, Xu Y, Ribar T, Means A, & Li J (2013). Inhibition of CaMKK β and CaMK IV is detrimental in cerebral ischemia. Stroke; a Journal of Cerebral Circulation, 44(9), 10.1161/STROKEAHA.113.001030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paroni G, Fontanini A, Cernotta N, Foti C, Gupta MP, Yang X-J, … Brancolini C (2007). Dephosphorylation and Caspase Processing Generate Distinct Nuclear Pools of Histone Deacetylase 4. Molecular and Cellular Biology, 27(19), 6718–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiscornia G, Singer O, and Verma IM (2006) Production and purification of lentiviral vectors. Nat Protoc. 1, 241 245. [DOI] [PubMed] [Google Scholar]
- White BJ, Tarabishy S, Venna VR, Manwani B, Benashski S, McCullough LD, & Li J (2012). Protection from Cerebral Ischemia by Inhibition of TGFβ-activated kinase. Experimental Neurology, 237(1), 238–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, He X, Wang Y, Tang Y, Zheng C, Cai H Liu J, Wang Y, Fu Y, Yang GY (2014) . MicroRNA-210 overexpression induces angiogenesis and neurogenesis in the normal adult mouse brain. Gene Ther, 21, 37–43. [DOI] [PubMed] [Google Scholar]