

Abstract
The COVID-19 pandemic caused by SARS-CoV-2 continues to pose a great threat to public health while various vaccines are available worldwide. Main protease (Mpro) has been validated as an effective anti-COVID-19 drug target. Using medicinal chemistry and rational drug design strategies, we identified a quinazolin-4-one series of nonpeptidic, noncovalent SARS-CoV-2 Mpro inhibitors based on baicalein, 5,6,7-trihydroxy-2-phenyl-4H-chromen-4-one. In particular, compound C7 exhibits superior inhibitory activity against SARS-CoV-2 Mpro relative to baicalein (IC50 = 0.085 ± 0.006 and 0.966 ± 0.065 μM, respectively), as well as improved physicochemical and drug metabolism and pharmacokinetics (DMPK) properties. In addition, C7 inhibits viral replication in SARS-CoV-2-infected Vero E6 cells more effectively than baicalein (EC50 = 1.10 ± 0.12 and 5.15 ± 1.64 μM, respectively) with low cytotoxicity (CC50 > 50 μM). An X-ray co-crystal structure reveals a non-covalent mechanism of action, and a noncanonical binding mode not observed by baicalein. These results suggest that C7 represents a promising lead for development of more effective SARS-CoV-2 Mpro inhibitors and anti-COVID-19 drugs.
Keywords: SARS-CoV-2 Mpro, Noncovalent Mpro inhibitors, Antiviral drugs, Antiviral activity
Graphical abstract

1. Introduction
The Coronavirus Disease 2019 (COVID-19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has heavily impacted the global economy and threatened public health. According to the World Health Organization (WHO) report, there have been more than 645 million confirmed cases of COVID-19 worldwide, including more than 6.6 million deaths, as of 13 December 2022 [1]. Since the outbreak of COVID-19, enormous global efforts from academic and pharmaceutical companies have been made to discover preventive and therapeutic strategies to fight against this life-threatening disease [2,3]. To date, more than 20 effective SARS-CoV-2 vaccines have been made available globally, and play an important preventive role in controlling the COVID-19 pandemic. Nevertheless, a large number of people have not yet been vaccinated due to personal unwillingness or limited medicinal conditions; on the other hand, increasing numbers of breakthrough infections have been observed in convalescent and/or vaccinated populations, mainly due to decline in protective efficacy of vaccines with time since vaccination, or compromised effectiveness against the emerging omicron variants and subvariants [[4], [5], [6], [7], [8]]. Accordingly, the public are still at high risk of being infected, and effective antiviral drugs against SARS-CoV-2 and its emerging variants are still highly needed.
A large number of anti-COVID-19 drug targets have been reported, with SARS-CoV-2 main protease [Mpro; also known as 3C-like (3CL) protease or nonstructural protein 5 (nsp5)] capturing much attention. Mpro has been validated as an effective target for development of orally available small-molecule antiviral drugs [9,10]. Mpro cleaves viral polyproteins at 11 distinct sites to release functional non-structural proteins (nsps) that are essential for viral replication. Inhibition of Mpro is therefore able to block the viral replication and shut down the viral life cycle. Moreover, Mpro features a unique substrate specificity of glutamine (Gln) at the P1 position and no homologous human proteases have been known, and hence Mpro inhibitors are likely to cause no side-effects by interfering with host proteases. In contrast, Mpro are highly conserved among various coronaviruses, such as SARS-CoV-2, SARS-CoV and MERS, making it a promising target for development of broad-spectrum coronavirus antivirals. Notably, Mpro inhibitors should be effective against all the emerging variants of SARS-CoV-2, since the mutations occurring in spike proteins of variant strains cannot affect Mpro, and Mpro itself was shown to have an extremely low mutation rate in the emerging SARS-CoV-2 variants [[10], [11], [12]]. To date, a large number of SARS-CoV-2 Mpro inhibitors have been reported, and are mainly divided into two types: peptide-like covalent inhibitors represented by nirmatrelvir (PF-07321332, 1) [13], PF-00835231 (2) [14], PF-07304814 (3) [15], GC-376 (4) [16], MI-09 (5), MI-30 (6) [17] and compounds (7–9) [18,19], as well as nonpeptidic, noncovalent inhibitors represented by Ensitrelvir (S-217622, 10) [11], ML-188 (11), compound 12 [20], HL-3-68 (13) [21], baicalein (14) [22] and compounds 15–17 [[23], [24], [25]] (Fig. 1 ). Potential off-target effects, low membrane permeability and poor metabolic stability pose challenges for the extensive clinical application of peptidelike covalent inhibitors, notwithstanding the approval of Paxlovid (a combination of PF-07321332 and ritonavir) by the US Food and Drug Administration (FDA). The nonpeptidic, noncovalent Mpro inhibitor seems to be a more attractive modality. Ensitrelvir (S-217622) developed by Shionogi & Co., Ltd. showed robust antiviral potency against SARS-CoV-2 and its variants, excellent selectivity for Mpro over host proteases, as well as an outstanding DMPK profile in preclinical models [11]. Ensitrelvir (S-217622) exhibited promising single-agent antiviral potency and safety profiles in phase II/III clinical trials [26]. More recently, it was approved for clinical application by the Pharmaceuticals and Medical Device Agency (PMDA). But effective nonpeptidic, non-covalent Mpro inhibitors are very limited, and it is still necessary to search for more non-covalent Mpro inhibitors with diverse chemical scaffolds and improved properties.
Fig. 1.

Chemical structures of representative peptide-like covalent, and nonpeptidic, noncovalent SARS-CoV-2 Mpro inhibitors.
Here, we describe our discovery of quinazolin-4-one-based nonpeptidic, non-covalent inhibitors of SARS-CoV-2 Mpro that are derived from baicalein (14). Baicalein was the first reported nonpeptidic, noncovalent inhibitor of SARS-CoV-2 Mpro discovered by Shanghai Institute of Materia Medica, Chinese Academy of Sciences [22], and its good biochemical and cellular antiviral activity captured our interest. Nevertheless, the potency of baicalein requires further structural optimization and extensive structure activity relationship (SAR) studies. Moreover, the drug metabolism and pharmacokinetics (DMPK) properties and target specificity were largely unknown. In order to fill in gaps in this area and explore the potential of this series as a lead for the development of anti-COVID-19 drugs, we firstly used a scaffold hopping strategy turning baicalein's chromen-4-one core to the alternative privileged scaffold, quinazolin-4-one. In this way, we discovered the first quinazolin-4-one-based SARS-CoV-2 Mpro inhibitor, and further structural optimizations allowed us to produce quinazolin-4-one-based SARS-CoV-2 Mpro inhibitors that are superior to baicalein in terms of biochemical potency, cellular antiviral activity and DMPK profile. An X-ray cocrystal structure of D8 in SARS-CoV-2 Mpro revealed a ligand-induced conformation change, which allows the sec-butyl moiety of D8 to occupy a newly formed binding site between the canonical S1′ and S2 subpockets, suggesting a binding mode to Mpro of quinazolin-4-one-based SARS-CoV-2 Mpro inhibitors that is different from that of baicalein.
2. Results and discussion
2.1. Design of the quinazolin-4-one class of SARS-CoV-2 Mpro inhibitors
The active site of Mpro is composed of five subpockets, S4–S1′, which can accommodate substrate and inhibitor groups at positions P4–P1′, with a Cys145-His41 catalytic dyad. As summarized by researchers from Shionogi Pharmaceutical in a recent publication, the pharmacophore based on the known Mpro inhibitors generally includes: (i) a hydrogen acceptor that interacts with the side-chain NH of His163 in the S1 subpocket, (ii) a hydrogen acceptor that forms a hydrogen bond with the main-chain NH of Glu166, and (iii) a lipophilic group in the S2 subpocket [11]. As shown in Fig. 2 A, the cocrystal structure of baicalein (14) in SARS-CoV-2 Mpro (PDB code: 6M2N [22]) showed that baicalein follows this pharmacophore model. Specially, three phenolic hydroxyl groups in baicalein form a hydrogen-bond network with the side chains of Ser144/His163 and main chains of Leu141/Gly143 directly or indirectly through water molecules. The carbonyl group at C4 position forms a critical hydrogen bond with the Glu166. Accordingly, three phenolic hydroxyl groups and C4 carbonyl group are necessary for the potency, and therefore were retained in our design. The free phenyl ring (C ring) occupies the S2 subpocket with extensive hydrophobic interactions. The core chromen-4-one forms many key interactions contributing to baicalein's binding affinity with protein: (i) the A ring forming S–π and NH2–π with the catalytic Cys145 and Asn142, respectively; (ii) the B ring π–π stacking with catalytic His41, and forming hydrophobic interactions with Met165. In addition, the chromen-4-one motif acts as a scaffold to assemble these necessary components. We envisioned that other aromatic heterocycles with similar structure could replace the chromen-4-one structure in baicalein. Because quinazolin-4-one and quinolin-4-one have been often seen in many natural products, biologically active compounds and clinically used drugs, together with well-established efficient synthesis and functionalization methodologies of these heterocycles, we firstly used a scaffold hopping strategy to replace the core chromen-4-one in baicalein with quinazolin-4-one or quinolin-4-one, respectively, in order to discover structurally novel non-covalent Mpro inhibitors. The resulting quinolin-4-one-based compound 18 had a complete loss of Mpro enzymatic inhibitory activity, while quinazolin-4-one-based compound 19 exhibited comparable inhibitory activity with that of baicalein. Therefore, we proceeded with 19 as a lead compound. A step-by-step optimization strategy and SAR studies of substituent groups at the C2 and N3 positions of quinazolin-4-one were conducted (Fig. 2B). Finally, the optimal substituents at the C2 and N3 positions were integrated, leading to a series of potent SARS-CoV-2 Mpro inhibitors (Fig. 2B).
Fig. 2.

Rational design of non-covalent SARS-CoV-2 Mpro inhibitors. (A) Binding pocket of baicalein in SARS-CoV-2 Mpro (PDB code 6M2N). (B) Detailed binding mode of baicalein complexed with Mpro. The protein is shown in gray cartoon, baicalein in blue sticks, and the selected residues in yellow sticks. Hydrogen bonds are indicated as gray dashes, and water molecule as red sphere. S–π, NH2–π and π–π stacks are indicated as green dashes. (C) Step-by-step optimization strategy of non-covalent SARS-CoV-2 Mpro inhibitors starting from baicalein.
2.2. Chemistry
As shown in Scheme 1 , the quinolin-4-one derivative 18 was obtained by the base-mediated cyclization of N-(ketoaryl)amide (the Camps cyclization) according to the published literature [27]. Condensation of compound 20 and benzoyl chloride afforded benzamide (21). The Friedel-Crafts acylation of 21 using acetyl chloride in the presence of SnCl4 gave the cyclization precursor (22) [28], which was then cyclized at 110 °C in the presence of KOH to generate compound 23 [27], and this was followed by BBr3-mediated demethylation to yield the desired product (18).
Scheme 1.

Synthesis of compound 18. Reagents and conditions: (a) benzoyl chloride, TEA, THF, 0 °C to rt, 2 h, 91%; (b) acetyl chloride, SnCl4, anhydrous DCM, 0 °C to rt, 5 h, 45%; (c) KOH, anhydrous 1,4-dioxane, reflux, 4 h, 35%; (d) BBr3/DCM, DCM, −10 °C to rt, 24 h, 75%.
There are many efficient ways to construct quinazolin-4-one derivatives. We selected the appropriate synthetic method for each target compound based on the availability of raw materials. Compounds A1–A6, A8–A12, A14, A16, A18, A19, A21–A27 and 19 were obtained in two steps: (i) the reaction of key intermediate 24 [29] with various amidine hydrochlorides 25a–25w that are commercially available or easily synthesized in efficient mild copper-catalyzed conditions to yield compounds 26a–26w [30]; (ii) BBr3-mediated demethylation of compounds 26a–26w (Scheme 2 ). A one-pot I2-mediated oxidative cyclization of o-anthranilamide 27 [31] and various commercially available aldehydes 28a–28v [32] and subsequent demethylation were used to synthesize compounds A7, A13, A15, A17, A20 and compounds B1–B17 (Scheme 3 ). As depicted in Scheme 4 , the N 3 substituted quinazolin-4-one derivatives C1–C15 were prepared by condensation of the prepared o-anthranilamides 30a–30o with benzaldehyde under similar reaction conditions with B series compounds followed by demethylation. Compounds D6–D9 were obtained by the oxidative cyclization approach that was used to access compounds in the B and C series in a good yield (Scheme 7), but compounds D1–D5 and D10–D12 could not be synthesized under the same conditions. D1–D4, D10 and D11 were successfully achieved by cyclization of N-acylanthranilic acids and amines in the presence of PCl3 in a good yield (Scheme 5 ) [33]. However, N 3-alkyl substituted compounds D5 and D12 could not be prepared with the synthetic method shown in Scheme 5, and these were obtained by a one-pot, two step cyclization: (i) the reaction of compound 36 with the prepared 2-phenylpropanoyl chloride or commercially available phenylacetyl chloride in the presence of triphenyl phosphite (TPP) and pyridine, and (ii) the substitution with isobutylamine [34], followed by demethylation (Scheme 6 ).
Scheme 2.

Synthesis of compounds A1–A6, A8–A12, A14, A16, A18, A19, A21–A27 and 19. Reagents and conditions: (a) CuI, CsCO3, DMF, rt, overnight, 55%–90%; (b) BBr3/DCM, DCM, −10 °C to rt, 24–36 h, 35%–64%.
Scheme 3.

Synthesis of compounds A7, A13, A15, A17, A20, B1–B17. Reagents and conditions: (a) I2, EtOH, reflux, 3–5 h, 40%–80%; (b) BBr3/DCM, DCM, −10 °C to rt, 24–36 h, 40%–63%.
Scheme 4.

Synthesis of compounds C1–C15. Reagents and conditions: (a) I2, EtOH, reflux, 3–5 h, 45%–87%; (b) BBr3/DCM, DCM, −10 °C to rt, 24–36 h, 35%–57%.
Scheme 7.

Synthesis of compounds D6–D9. Reagents and conditions: (a) I2, EtOH, reflux, 5 h, 72%–88%; (b) BBr3/DCM, DCM, −10 °C to rt, 24 h, 55%–56%.
Scheme 5.

Synthesis of compounds D1–D4, D10 and D11. Reagents and conditions: (a) PCl3, 50 °C, 4–12 h, 70%–82%; (b) BBr3/DCM, DCM, −10 °C to rt, 24–36 h, 45%–63%.
Scheme 6.

Synthesis of compounds D5 and D12. Reagents and conditions: (a) For 37a: (i) 2-phenylpropanoic acid (38), (COCl)2, DCM, cat. DMF, 0 °C to rt, 2 h; (ii) triphenyl phosphite (TPP), Py, 0 °C–70 °C, 2 h; iii) isobutylamine (40), 70 °C, overnight, 40%; (b) For 37b: (i) phenylacetyl chloride (39), TPP, Py, 0 °C–70 °C, 2 h; (ii) isobutylamine (40), 70 °C, overnight, 45%; (c) BBr3/DCM, DCM, −10 °C, 24 h, 50%–60%.
2.3. SAR studies of the quinazolin-4-one class of SARS-CoV-2 Mpro inhibitors
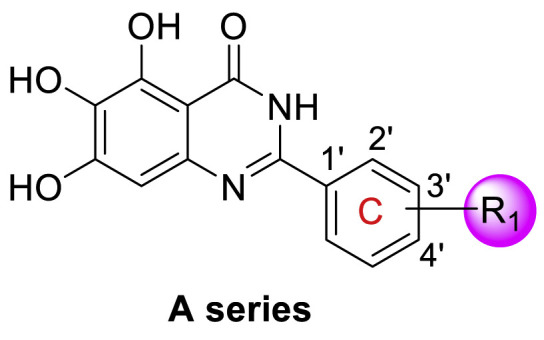
All target compounds were preliminarily evaluated for inhibitory activity against SARS-CoV-2 Mpro in fluorescence resonance energy transfer (FRET)-based enzymatic assays. As shown in Table 1 , quinolin-4-one-based compound 18 had a complete loss of enzymatic inhibitory activity against SARS-CoV-2 Mpro. To our delight, quinazolin-4-one-based compound 19 was only slightly less potent than baicalein (IC50 = 1.372 ± 0.047 μM and 0.966 ± 0.065 μM, respectively). With the promising potency for compound 19 in hand, we firstly performed the SAR analysis of the substituents on the free phenyl group (C ring) in 19. Generally, the positions on the phenyl ring led to this order of potency: 2′-position > 3′-position > 4′-position (Table 1). In particular, compounds with fluorine (F), trifluoromethyl (CF3), trifluoromethoxy (OCF3) or tert-butyl at the C2′ position exhibited much more potency than the corresponding compounds with 3′ and/or 4′ substituents (A1 vs A2 and A3, A13 vs A14, A15 vs A16, A21 vs A22). The significant influence of the substituent position on activity can also be concluded from the difluorine-substituted compounds: 2′,3′-difluoro (A25, IC50 = 1.130 ± 0.031 μM) > 2′,4′-difluoro (A26, IC50 = 1.541 ± 0.042 μM) > 3′,4′-difluoro (A27, IC50 = 2.716 ± 0.051 μM).
Table 1.
SAR exploration of the substituent groups on the C ring of quinazolin-4-one a.
| Compd. | R1 | IC50 (μM) |
|---|---|---|
| A1 | 2′-F | 1.443 ± 0.060 |
| A2 | 3′-F | 5.132 ± 0.094 |
| A3 | 4′-F | 12.760 ± 0.067 |
| A4 | 2′-Cl | 0.435 ± 0.041 |
| A5 | 3′-Cl | 6.960 ± 0.076 |
| A6 | 4′-Cl | 7.706 ± 0.055 |
| A7 | 2′-Br | 0.554 ± 0.041 |
| A8 | 3′-Br | 8.017 ± 0.033 |
| A9 | 4′-Br | 9.797 ± 0.051 |
| A10 | 2′-methyl | 0.365 ± 0.033 |
| A11 | 3′-methyl | 2.277 ± 0.029 |
| A12 | 4′-methyl | 1.867 ± 0.034 |
| A13 | 2′-CF3 | 1.481 ± 0.047 |
| A14 | 3′-CF3 | 10.53 ± 0.050 |
| A15 | 2′-OCF3 | 1.798 ± 0.061 |
| A16 | 4′-OCF3 | >20 |
| A17 | 2′-NO2 | 10.588 ± 0.040 |
| A18 | 3′-NO2 | 11.169 ± 0.034 |
| A19 | 4′-NO2 | >20 |
| A20 | 3′-isopropyl | 7.635 ± 0.058 |
| A21 | 3′-tert-butyl | 8.423 ± 0.031 |
| A22 | 4′-tert-butyl | >20 |
| A23 | 2′-OH | 18.285 ± 0.040 |
| A24 | 4′-OH | >20 |
| A25 | 2′,3′-di-F | 1.130 ± 0.031 |
| A26 | 2′,4′-di-F | 1.541 ± 0.042 |
| A27 | 3,4′-di-F | 2.716 ± 0.051 |
| 19 | – | 1.372 ± 0.047 |
| baicalein (14) | – | 0.966 ± 0.065 |
Baicalein was used as the positive control. Inhibitory activity against SARS-CoV-2 Mprowas determined with the FRET protease activity assay. Values represent a mean ± SD of at least three independent experiments.
The volume, polarity and electronegativity of the substituent groups also affect the potency significantly. Introduction of a bulky isopropyl (A20) and tert-butyl (A21 and A22), or a polar hydroxyl (A23 and A24) and a nitro group (A17–A19) resulted in a sharp decline in potency. It seemed that less bulky substituents with moderate-to-low electronegativity benefit the inhibitory activity against Mpro. A10 with 2′-methyl and A4 with 2′-chlorine were the two most active compounds among the A series, with IC50 values of 0.365 ± 0.033 μM and 0.435 ± 0.04 μM, respectively. Adding a bromine (Br) atom at the C2′ position, whose van der Waals radius is slightly larger than that of chlorine (Cl) atom and whose electronegativity is lower than that of Cl, led to compound A7, which showed a slightly decreased potency relative to A4 (A7, IC50 = 0.554 ± 0.041 μM), but was more active than 19 and baicalein. The introduction of more electronegative substituents at the C2′ position led to a poorer potency against Mpro, for example, F (IC50 = 1.443 ± 0.060 μM) > CF3 (IC50 = 1.481 ± 0.047 μM) > nitro group (IC50 = 10.588 ± 0.040 μM). Compound A7 containing an electron-donating trifluoromethoxy (OCF3) group was slightly less potent than compound A13 with an electron-withdrawing CF3 (IC50 = 1.798 ± 0.061 μM for A7 vs 1.481 ± 0.047 μM for A13), which might be attributed to the larger volume of OCF3 than that of CF3, and the volume effect affected the potency more significantly. Together, these results suggested that the position, steric size, polarity and electronic property of substituents on the C ring integratedly affect the inhibitory activity against SARS-CoV-2 Mpro.
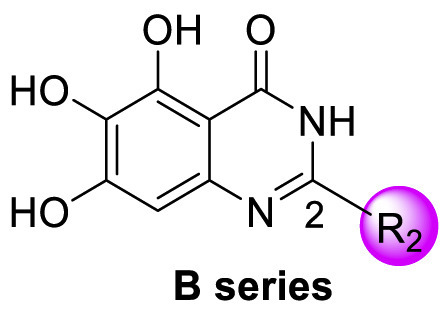
Next, we replaced the C ring with different types of groups in order to diversify the structures of quinazolin-4-one based Mpro inhibitors and search for more desirable substituents at the C2 position. As shown in the Table 2 , the replacement of phenyl (the C ring) with naphthyl and heteroaromatic rings, including thienyl, 3-pyridyl, 4-pyridyl and pyrazolyl, led to compounds B1–B5, and resulted in a remarkable decrease in potency against Mpro. Different cycloalkyl replacements afforded compounds B6–B8. Substituting phenyl with cyclopentyl (B7, IC50 = 0.539 ± 0.061 μM) led to improved potency against Mpro over that of 19 (IC50 = 1.372 ± 0.047 μM) and baicalein (14) (IC50 = 0.966 ± 0.065 μM). B8 with a cyclohexyl (IC50 = 1.370 ± 0.140 μM) was equipotent with 19, while B6 with cyclopropyl (IC50 = 5.485 ± 0.791 μM) remarkably decreased the potency. The phenyl was replaced by different alkyl groups leading to compounds B9–B12. The introduction of isopropyl (B9) and tert-butyl (B10) resulted in a sharp drop of the potency. In contrast, B11 with sec-butyl (IC50 = 0.385 ± 0.024 μM) and B12 with a bulkier tert-amyl (IC50 = 0.970 ± 0.075 μM) were more potent than or comparable to 19, respectively. Subsequently, we explored the effects of styryl and benzyl groups at the C2 position on the inhibitory activity against Mpro. Compound B13 with a rigid styryl (IC50 = 2.874 ± 0.030 μM) led to some loss of potency against Mpro compared to 19. The relatively flexible benzyl group in compound 14 improved the IC50 value to 0.327 ± 0.052 μM. Small groups, such as methyl or ethyl, when introduced to the benzyl in 14 could further increase the activity, and the resulting compounds 15 and 16 exhibited the most potent inhibitory activity against Mpro among B series, with IC50 values of 0.174 ± 0.038 μM and 0.210 ± 0.028 μM, respectively. Addition of a larger isopropyl group (compound 17) led to a slight loss of the potency of 14, with an IC50 value of 0.390 ± 0.048 μM.
Table 2.
SAR exploration of substituent groups at the C2 position of quinazolin-4-onea.
| Compd | R2 | IC50 (μM) |
|---|---|---|
| B1 | 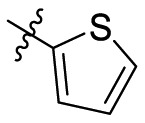 |
>20 |
| B2 | 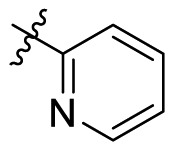 |
>20 |
| B3 | 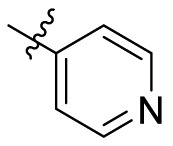 |
15.19 ± 0.0405 |
| B4 | 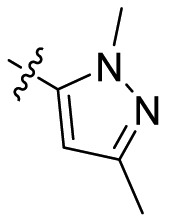 |
>20 |
| B5 | 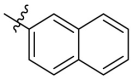 |
3.565 ± 0.295 |
| B6 | 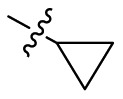 |
5.485 ± 0.791 |
| B7 | 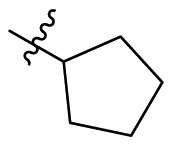 |
0.539 ± 0.061 |
| B8 | 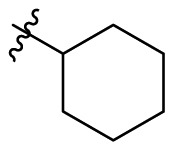 |
1.370 ± 0.140 |
| B9 | 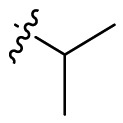 |
4.943 ± 0.504 |
| B10 | 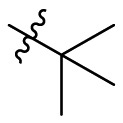 |
4.086 ± 0.647 |
| B11 | 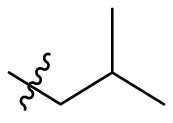 |
0.385 ± 0.024 |
| B12 | 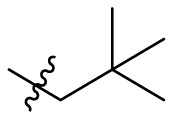 |
0.970 ± 0.075 |
| B13 | 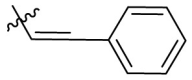 |
2.874 ± 0.030 |
| B14 | 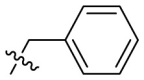 |
0.327 ± 0.052 |
| B15 | 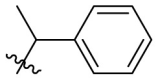 |
0.174 ± 0.038 |
| B16 | 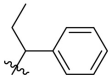 |
0.210 ± 0.028 |
| B17 | 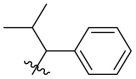 |
0.390 ± 0.048 |
| 19 | 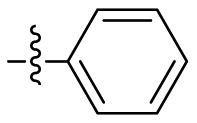 |
1.372 ± 0.047 |
| baicalein (14) | – | 0.966 ± 0.065 |
Baicalein was used as the positive control. Inhibitory activity against SARS-CoV-2 Mprowas determined by using the FRET protease activity assay. Values represent a mean ± SD of at least three independent experiments.
We tentatively kept R2 as phenyl and explored the SAR at the N3 position (Table 3 ). Generally, C series compounds with different substituents at the N-3 position of quinazolin-4-one exhibited improved inhibitory potency against Mpro compared to compound 19 and baicalein (14), except for compounds C2 with the polar hydroxyethyl (IC50 = 1.365 ± 0.062 μM), C3 with a bulky tert-butyl (0.949 ± 0.077 μM) and C14 with a biphenylyl (IC50 = 1.476 ± 0.117 μM). For cycloalkyl replacements, cyclopentyl was the optimal substituent group, and the corresponding compound C5 showed significantly increased potency relative to 19, with an IC50 value of 0.124 ± 0.014 μM (Table 3, Fig. S1). C1 with a less bulky sec-butyl substituent showed potency as good as C5 (Table 3, Fig. S1). The introduction of phenyl leading to compound C7 improved the IC50 value to 0.085 ± 0.006 μM (Table 3, Fig. S1). Inspired by this result, different substituted phenyl groups were introduced to the N3 position. F, Br and hydroxyl substituents affording compounds C8–C11 retained the most of the potency of C7, showing IC50 values of about 0.2 μM. Compound C12 containing a 3′-methyl-4′-fluorophenyl substituent was equipotent with C7 (IC50 = 0.117 ± 0.016 μM, Table 3, Fig. S1). In addition, adding a 3-pyridyl or 4-pyrazolyl at the N3 position resulted in compounds C13 and C14, which had good inhibitory activity against Mpro (IC50 = 0.212 ± 0.024 and 0.390 ± 0.003 μM, respectively).
Table 3.
SAR exploration of substituent groups at the N3 position of quinazolin-4-onea.
| Compd | R3 | IC50 (μM) |
|---|---|---|
| C1 | 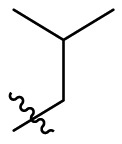 |
0.124 ± 0.018 |
| C2 | 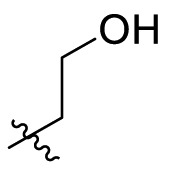 |
1.365 ± 0.062 |
| C3 | 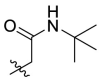 |
0.949 ± 0.077 |
| C4 |  |
0.290 ± 0.028 |
| C5 | 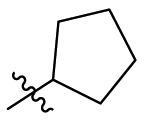 |
0.124 ± 0.016 |
| C6 | 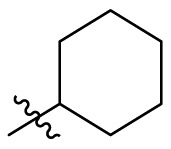 |
0.274 ± 0.022 |
| C7 | 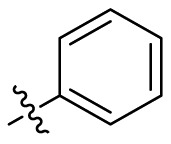 |
0.083 ± 0.006 |
| C8 | 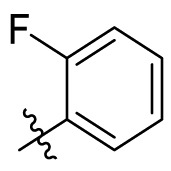 |
0.205 ± 0.033 |
| C9 | 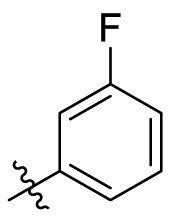 |
0.236 ± 0.018 |
| C10 | 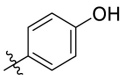 |
0.271 ± 0.018 |
| C11 | 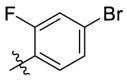 |
0.207 ± 0.024 |
| C12 | 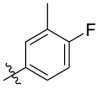 |
0.117 ± 0.016 |
| C13 | 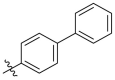 |
1.476 ± 0.117 |
| C14 | 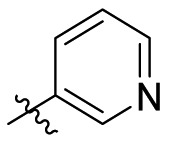 |
0.212 ± 0.024 |
| C15 | 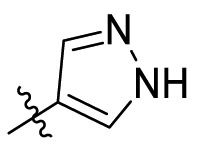 |
0.390 ± 0.003 |
| 19 | 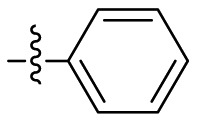 |
1.372 ± 0.047 |
| baicalein (14) | – | 0.966 ± 0.065 |
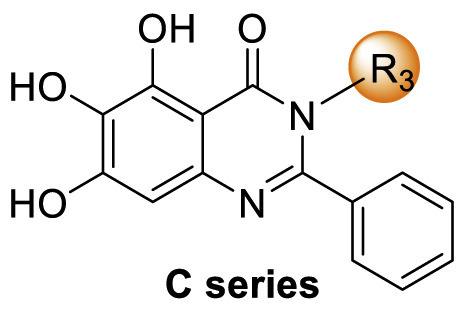
Baicalein was used as the positive control. Inhibitory activity against SARS-CoV-2 Mprowas determined by using the FRET protease activity assay. Values represent a mean ± SD of at least three independent experiments.
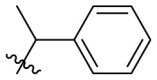
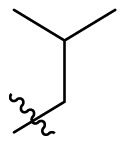
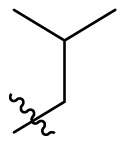
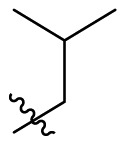
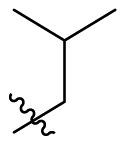
D series compounds were derived from the combination of the desirable substitutions at the C2 and N3 positions that we have identified (Table 4 ). Unexpectedly, the combination of 2-methyl-benzyl that is the optimal substitution at the C2 position with phenyl and sec-butyl groups that were the favorable substituents at the N3 position resulted in decreased activity compared to the corresponding C series compounds. In particular, compounds D2 (IC50 = 1.005 ± 0.136 μM) and D5 (IC50 = 0.692 ± 0.119 μM) showed a sharp decrease in potency relative to the corresponding compounds C12 (IC50 = 0.117 ± 0.016 μM) and C1 (IC50 = 0.124 ± 0.018 μM). The possible reason for the decreased inhibitory activity was that C2-2-methyl-benzyl clashed with N3-phenyl or N3-sec-butyl, leading one or both of them to be unable to smoothly enter into the corresponding Mpro protein subpockets. N3-sec-butyl series compounds D6–D9 did not improve the potency compared to C7, with IC50 values ranging from 0.100 to 0.284 μM. The combination of C2-benzyl with N3-phenyl resulted in compound D10 being equipotent with C7 (IC50 = 0.103 ± 0.014 μM), while the combination of C2-benzyl with 3-methyl-4-fluorobenzyl and sec-butyl at the N3 position led to some loss of the potency relative to C7.
Table 4.
SAR exploration of substituent groups at C-2 and N-3 positions of quinazolin-4-onea.
| Compd | R4 | R5 | IC50 (μM) |
|---|---|---|---|
| D1 |  |
 |
0.477 ± 0.078 |
| D2 |  |
 |
1.005 ± 0.136 |
| D3 |  |
 |
0.290 ± 0.030 |
| D4 |  |
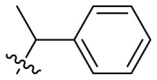 |
0.386 ± 0.017 |
| D5 | 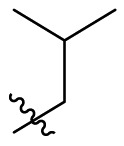 |
 |
0.692 ± 0.119 |
| D6 |  |
 |
0.107 ± 0.023 |
| D7 | 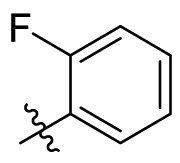 |
 |
0.239 ± 0.028 |
| D8 | 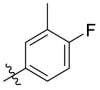 |
 |
0.100 ± 0.012 |
| D9 | 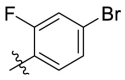 |
 |
0.284 ± 0.028 |
| D10 | 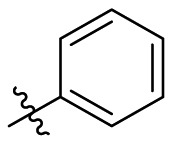 |
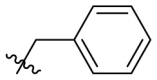 |
0.131 ± 0.022 |
| D11 | 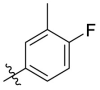 |
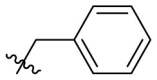 |
0.502 ± 0.038 |
| D12 | 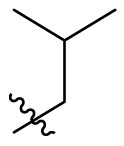 |
 |
0.466 ± 0.040 |
| C7 |  |
 |
0.085 ± 0.006 |
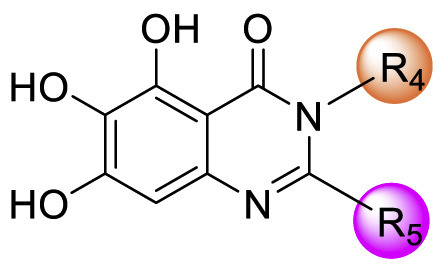
C7 was used as the positive control. Inhibitory activity against SARS-CoV-2 Mprowas determined with the FRET protease activity assay. Values represent a mean ± SD of at least three independent experiments.
2.4. Target validation and selectivity
To examine if the inhibitory activity against Mpro is associated with covalent modification of cysteine residues, we compared the inhibition of C5, C7, D6, D8 and D10 in the presence or absence of the reducing agent dithiothreitol (DTT) (Table S1). Addition of DTT guarantees accessible cysteine residues of a protein at their reduced state and will indicate whether an inhibitor acts by a false mechanism of redox-cycling [24,35]. There were no significant differences in the IC50 values in the presence of DTT and in the absence of DTT.
One of the major limitations encountered by cysteine protease inhibitors is the target selectivity. Compound C7 was selected as an example of our target scaffold to be profiled with the selectivity over several common human proteases, including caspase 2, cathepsin L, thrombin, cathepsin B and cathepsin D. Compound C7 showed no apparent inhibitory activity against these host proteins at a concentration of 10 μM, suggesting that C7 had a good target specificity against Mpro over host proteases (Table S2).
2.5. In vitro DMPK profiling
As shown in Table S3, compounds C7 (Papp AB = 9.67 × 10−6 cm/s, Efflux radio (ER) = 0.44), D6 (Papp AB = 7.45 × 10−6 cm/s, ER = 0.40), and D8 (Papp AB = 7.83 × 10−6 cm/s, ER = 0.41) exhibited improved Madin-Darby canine kidney (MDCK) membrane permeability relative to baicalein (Papp AB = 6.07 × 10−6 cm/s, ER = 0.34). The kinetic solubility in phosphate buffer solution (PBS, pH = 7.4) of C7 and D6 was measured as 179.35 and 155.98 μM, respectively, which were superior to baicalein (112.18 μM). C7, D6, D8 and baicalein exhibited reasonable plasma protein binding (PPB) in human plasma (fraction unbound fu = 0.70%, 2.61%, 3.17% and 0.93%, respectively). Baicalein exhibited high intrinsic clearance in human liver microsome (HLM), with clearance rate (CLint) of 333.05 μL/min/mg protein and half-time (T1/2) of 4.16 min. C7 and D6 showed improved HLM stability relative to baicalein (CLint = 108.68 and 68.34 μL/min/mg protein, respectively; T1/2 = 12.75 and 20.28 min). The two compounds showed relatively high HLM metabolic stability in the presence of the cofactor NADPH alone, indicative of mainly undergoing phase II metabolism. With the presence of three phenolic hydroxyl groups in the structures, the above results were not unexpected. Despite the improvement relative to baicalein, the metabolic stability of this quinazolin-4-one series inhibitors requires further optimization.
2.6. Cellular cytotoxicity and antiviral activity
The antiviral activity of compounds C7, D6 and D8 was evaluated in SARS-CoV-2-infected Vero E6 cells. Prior to antiviral assays, the cytotoxicity of selected Mpro inhibitors against Vero E6 cells was evaluated by using cell counting kit-8 (CCK-8) assays. The test compounds exhibited no significant cytotoxicity at the highest tested concentration (50% cytotoxicity concentration, CC50 > 50 μM, Table 5 and Fig. S2). The antiviral activity was determined in a yield reduction assay that assesses the inhibitory activity of the test compounds on the viral replication using quantitative real-time polymerase chain reaction (qRT-PCR), with baicalein as the positive control. As shown in Table 5 and Fig. S2, compounds C7 (EC50 = 1.10 ± 0.12 μM), D6 (EC50 = 2.87 ± 1.43 μM) and D8 (EC50 = 2.11 ± 1.12 μM) showed better antiviral activity in Vero E6 cells than baicalein (EC50 = 5.15 ± 1.64 μM).
Table 5.
Antiviral activity and cytotoxicity of selected compounds in Vero E6 cells.
| Compd. | EC50 (μM)a | CC50 (μM) |
|---|---|---|
| C7 | 1.10 ± 0.12 | >50 |
| D6 | 2.87 ± 1.43 | >50 |
| D8 | 2.11 ± 1.16 | >50 |
| baicalein | 5.15 ± 2.46 | >50 |
Inhibitory effect on viral replication induced by SARS-CoV-2 infection in Vero E6 cells (RT-qPCR assay). Values are expressed as mean ± SD from three independent experiments.
2.7. X-ray crystal structure of D8 in complex with SARS-CoV-2 Mpro
To further examine the binding mode of quinazolin-4-one inhibitors in SARS-CoV-2 Mpro, we resolved an X-ray co-crystal structure of D8 complexed with Mpro at a resolution of 2.2 Å (PDB code 8I4S, Fig. 3 A–3C). The X-ray data and refinement statistics are depicted in Table S4. The trihydroxyphenyl moiety of D8 sits in a similar position as in the baicalein/Mpro complex (Fig. 3B and E). Three phenolic hydroxyl groups of D8 form critical hydrogen bond interactions with the main chains of Gly143/Ser144/Gly145, and also form hydrogen bond interactions with the side chain of His163 via a buried water molecule (Fig. 3B). Distinct from the case of baicalein, the C4 carbonyl oxygen atom of D8 was a little aside from Glu166, and forms a hydrogen bond with the backbone NH of Glu166 with the aid of a buried water molecule (Fig. 3B). Unexpectedly, the 3′-methyl-4′-fluorophenyl at the N3 position occupies the S2 pocket, forming hydrophobic interactions with Gln189 and Met49, while the sec-butyl at the C2 position projects into a newly formed binding site that was observed in CCF0058981 analogue (12) complexed with Mpro (PDB code 7LMF), termed the S2c pocket by Han et al. (Fig. 3D).20 When D8 bound to Mpro, the flexible side chains of some amino acid residues of the protein, particularly Met49 and Gln189, exhibited a ligand-induced conformation change relative to most other inhibitor/Mpro structures (e.g., baicalein, nirmatrelvir and ensitrelvir), further rearranging the binding surface of subpockets S4 and S2 (Fig. 3D and S3). This allows the sec-butyl to occupy the S2c pocket, forming favorable hydrophobic interactions with Cys44, Thr45 and Ser46. As with baicalein, the phenyl ring of D8 with three hydroxyl groups is sandwiched between Cys145 and Asn142 by forming S–π and NH2-π with Cys145 and Asn142, respectively (Fig. 3B). In addition, the middle ring π–π stacks with catalytic His41, also contributing to the binding affinity of D8 with protein (Fig. 3B).
Fig. 3.

(A) Overview of the co-crystal structure of D8 bound to SARS-CoV-2 Mpro (PDB code 8I4S). The protein is shown in cartoon, and domains I, II and III are colored light orange, magenta and violet, respectively. D8 is shown as spheres with carbons in cyan. (B) Detailed binding mode of compound D8 in complex with SARS-CoV-2 Mpro. The protein is shown in gray cartoon, D8 in cyan sticks, and the selected residues in yellow sticks. Hydrogen bonds are indicated as gray dashes, and the water molecule as a red sphere. S–π, NH2–π and π–π stacks are indicated as green dashes. (C) Fo−Fc density map (contoured at 3.00σ) around D8 (cyan mesh). (D) Overlay of D8/SARS-CoV-2 Mpro (PDB code 8I4S, cyan) with 12/SARS-CoV-2 Mpro (PDB code 7LMF20, pink) complexes, highlighting the residues Gln189 and Met49. (E) Overlay of D8/SARS-CoV-2 Mpro (cyan) with baicalein/SARS-CoV-2 Mpro (PDB code 6M2N22, blue) complexes, highlighting the residues Gln189 and Met49.
Although highly conserved among variants of SARS-CoV-2, some mutated amino acid residues in Mpro have been observed, such as K90R in Beta strain B.1.351, K90R and A193V in Beta B.1.351.2, L205V in Zeta P.2, and P132H in Omicron B.1.529 [36]. However, it is worth noting that these amino residues are more than 10 Å away from the binding site of D8 (Fig. S4), suggesting that D8 should be have inhibitory effects against Mpro from SARS-CoV-2 variants. The assays assessing the inhibitory activity of the inhibitors against Mpro from SARS-CoV-2 Omicron variant are currently in progress.
3. Conclusions
Mpro has been validated as an effective target for development of orally available small molecule anti-COVID19 drugs. In this study, we sought to use medicinal chemistry and rational drug design approaches to structurally modify the first reported nonpeptidic, noncovalent SARS-CoV-2 Mpro inhibitor, baicalein. These efforts led to a series of quinazolin-4-one-derived noncovalent inhibitors with nanomolar potencies against SARS-CoV-2 Mpro. In particular, an optimized compound, C7 exhibited superior inhibitory potency against Mpro relative to baicalein and is endowed with improved physicochemical and DMPK properties. Significantly, C7 also showed more potent antiviral activity than baicalein (EC50 = 1.1 and 5.15 μM, respectively) in SARS-CoV-2-infected Vero E6 cells. Moreover, C7 exhibited relatively high selectivity over a panel of human proteases (IC50 > 10 μM) and low cytotoxicity against Vero E6 cells (CC50 > 50 μM). The co-crystal structure of another potent inhibitor D8 complexed with Mpro showed that the inhibitor noncovalently binds to the active site of Mpro, and occupies a newly formed S2c pocket that is not observed with baicalein and most other Mpro inhibitors, and can be further exploited for inhibitor design. Meanwhile, the S1 and S4 subpocket remain largely unoccupied by D8 and its analogues, leaving room for further improvement. While having improvements, the inhibitors still lack sufficient properties necessary to be profiled for in vivo antiviral efficacy in SARS-CoV-2-infected animal models. Further structural optimizations of this series of inhibitors are ongoing, and are focused on improving the DMPK properties, as well as further improving biochemical and cellular potencies. In addition, profiling the potency against Mpro from SARS-CoV-2 Omicron variant and other coronavirus is underway in our laboratory. Collectively, compound C7 represents a promising lead for further development of more effective Mpro inhibitors and antiviral drugs against SARS-CoV-2 infection.
4. Experimental
4.1. Chemistry
Reagents and solvents from commercial sources were used without further purification. The progress of all reactions was monitored by TLC using EtOAc/petroleum ether (PE) or dichloromethane (DCM)/MeOH as the solvent system, and spots were visualized by irradiation with UV light (254 nm) or by staining with phosphomolybdic acid. Flash chromatography was performed using silica gel (200−300 mesh). 1H NMR and 13C NMR spectra were recorded on a Bruker Avance ARX-400, a Bruker Avance ARX-500 or a Bruker Avance ARX-600. Chemical shifts δ are reported in ppm, and multiplicity of signals are denoted as: br = broad, s = singlet, d = doublet, t = triplet, q = quartet and m = multiplet. The low resolution ESI-MS was recorded on Shimadzu GCMS-2010 instruments and the high resolution mass spectra (HRMS) on a Water Q-Tofmicro mass spectrometer. Anhydrous DCM and N,N-dimethylformamide (DMF) were freshly distilled from calcium hydride. Anhydrous tetrahydrofuran (THF) was freshly distilled over sodium using benzophenone as the indicator. All other solvents were reagent grade. All moisture sensitive reactions were carried out in flame dried flasks under an argon atmosphere. The chemical purity of the target compounds was analyzed by high performance liquid chromatography (HPLC) on an InertSustain C18 column (4.6 mm × 250 mm, 5 μm) under gradient 60–100% MeOH in water (with 0.1% TFA in each mobile phase), with a flow rate of 1.0 mL/min and peak detection at 254 nm. All the target compounds showed purity greater than 95%. The synthesis and compound information of the intermediates can be found in the Supporting Information.
4.1.1. 5,6,7-Trihydroxy-2-phenylquinolin-4(1H)-one(18)
Step1: To the stirred solution of compound 20 (1.83 g, 10 mmol) in dry THF (25 mL) was added TEA (2 mL, 15 mmol) and benzoyl chloride (1.8 mL, 15 mmol) at 0 °C. Then the reaction mixture was stirred at room temperature (rt) for 2 h. The resulting mixture was concentrated and partitioned between water and DCM. The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under the reduced pressure. The resulting residue triturated with n-hexane, filtered, washed with n-hexane, dried in vacuum to afford 21 as a white solid (2.61g, 91%), which was used directly in the next step without further purification.
Step2: To the stirred solution of compound 21 in anhydrous DCM (30 mL) was successively dropwise added acetyl chloride (0.78 mL, 11 mmol) and SnCl4 (2 mL, 20 mmol) at 0 °C. The resulting reaction mixture was stirred at 0 °C for 4 h and then moved to room temperature for 1 h, after which the reaction mixture was poured into ice water. The aqueous portion was extracted with DCM (3 × 20 mL), and the combined organics were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica column chromatography and eluted with petroleum ether: EtOAc (20:1) to give compound 22 as a white solid (1.48 g, 45%). 1H NMR (400 MHz, CDCl3) δ 12.37 (s, 1H), 8.42 (s, 1H), 8.05–7.99 (m, 2H), 7.59–7.47 (m, 3H), 4.00 (s, 6H), 3.85 (s, 3H), 2.67 (s, 3H). m/z (ESI-MS): 330.2 [M + H ]+.
Step3: To the stirred solution of compound 22 (1.32 g, 4.01 mmol) in anhydrous 1,4-dioxane (40 mL) was added NaOH (480 mg, 12 mmol), and the reaction mixture was heated to reflux under a nitrogen atmosphere for 4 h. Then, the reaction mixture was cooled to room temperature, and the solvent was removed under reduced pressure. Next, the residue was treated with water and n-hexane and the resulting mixture was sonicated for approximately 2 min. The resulting suspension was adjusted to pH∼7 with 1 M HCl and filtered. The precipitate obtained was washed with n-hexane and dried under vacuum to afford compound 23 as a white solid (435 mg, 35%). 1H NMR (400 MHz, CDCl3) δ 8.07–8.01 (m, 2H), 7.96 (s, 1H), 7.54–7.45 (m, 3H), 7.02 (s, 1H), 4.21 (s, 3H), 4.05 (s, 3H), 3.95 (s, 3H). m/z (ESI-MS): 312.2 [M + H ]+.
Step4: To the stirred suspension of compound 23 (200 mg, 0.64 mmol) in anhydrous DCM (1.5 mL) was dropwise added BBr3/DCM (9.6 mL, 9.6 mmol, 1 M) at −10 °C under a nitrogen atmosphere. The resulting reaction mixture was stirred at −10 °C overnight and then moved to room temperature for 12 h. Then, the mixture was moved to −10 °C again, and was quenched by slowly adding ice methanol, after which the solvent was removed under reduced pressure. The resulting residue was triturated with water, filtered, washed with water and DCM, dried in vacuum to afford 18 as a pale yellow solid (130 mg, 75%). 1H NMR (400 MHz, DMSO‑d 6) δ 14.02 (s, 1H), 11.26 (s, 1H), 9.31 (s, 1H), 8.04–7.95 (m, 2H), 7.67–7.57 (m, 3H), 7.32 (s, 1H), 7.03 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.7, 156.1, 152.6, 139.2, 134.4, 132.2, 131.8, 131.5, 129.1, 128.5, 105.3, 101.1, 94.6. HRMS (ESI): m/z calcd for C15H10NO4 [M − H]–: 268.0615, found 268.0610. HPLC analysis: tR = 10.216 min, 96.3%.
4.1.2. General procedure a for the preparation of A1–A6, A8 – A12, A14, A16, A18, A19, A21 – A27 and 19
Compound 24 (337 mg, 1 mmol) was dissolved in anhydrous DMF (6 mL), and compounds 25a–25w (1.5 mmol), CsCO3 (652 mg, 2 mmol) and CuI (39 mg, 0.2 mmol) were added. The resulting mixture was stirred at room temperature under nitrogen atmosphere overnight. After completion of the reaction, excess saturated aqueous NH4Cl was added and filtered. The precipitate obtained was washed with a large amount of water, dried in vacuum to afford trimethoxyquinazolinones 26a–26w.
To the stirred suspension of the obtained trimethoxyquinazolinone (0.5 mmol) in anhydrous DCM (1.5 mL) was dropwise added BBr3/DCM (5 mL, 6 mmol, 1 M) at −10 °C under a nitrogen atmosphere. The resulting reaction mixture was stirred at −10 °C overnight and then moved to room temperature for 12–24 h. Then, the mixture was moved to −10 °C again, and was quenched by slowly adding ice methanol, after which the solvent was removed under the reduced pressure. The resulting residue was triturated with water, filtered, washed with water and DCM, dried in vacuum to afford target compounds A1–A6, A8–A12, A14, A16, A18, A19, A21–A27 and 19.
4.1.3. 5,6,7-Trihydroxy-2-phenylquinazolin-4(3H)-one (19)
The product was obtained as a offwhite solid (91 mg), yield 67%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.46 (s, 1H), 11.76 (s, 1H), 10.33 (s, 1H), 8.91 (s, 1H), 8.10 (d, J = 6.7 Hz, 3H), 7.59–7.49 (m, 3H), 6.65 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.4, 154.0, 149.4, 146.8, 142.1, 132.8, 131.1, 131.1, 128.6, 127.5, 103.2, 100.6. HRMS (ESI): m/z calcd for C14H11N2O4 [M + H]+: 271.0713, found 271.0717. HPLC analysis: tR = 6.765min, 96.9%.
4.1.4. 2-(2-Fluorophenyl)-5,6,7-trihydroxyquinazolin-4(3H)-one (A1)
According to general procedure A, the product was obtained as a white solid (65 mg), yield 45%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.48 (s, 1H), 11.71 (s, 1H), 10.33 (s, 1H), 8.93 (s, 1H), 7.72 (t, J = 7.4 Hz, 1H), 7.63–7.56 (m, 1H), 7.41–7.31 (m, 2H), 6.63 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.6, 159.5 (d, J = 250.2 Hz), 153.9, 146.8 (d, J = 6.8 Hz), 142.0, 132.6 (d, J = 8.5 Hz), 131.3, 131.0, 124.6 (d, J = 3.7 Hz), 122.2 (d, J = 13.1 Hz), 116.1 (d, J = 21.3 Hz), 103.2, 100.6. HRMS (ESI): m/z calcd for C14H10FN2O4 [M + H]+: 289.0619, found 289.0623. HPLC analysis: tR = 6.825 min, 95.3%.
4.1.5. 2-(3-Fluorophenyl)-5,6,7-trihydroxyquinazolin-4(3H)-one (A2)
According to general procedure A, the product was obtained as a white solid (68 mg), yield 47%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.51 (br, 1H), 11.77 (s, 1H), 10.29 (s, 1H), 8.97 (s, 1H), 8.04–7.87 (m, 1H), 7.63–7.52 (m, 1H), 7.41 (td, J = 8.5, 2.6 Hz, 1H), 6.67 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.6, 162.1 (d, J = 243.5 Hz), 154.0, 148.4, 146.8, 141.4, 135.0 (d, J = 8.1 Hz), 131.4, 130.7 (d, J = 8.4 Hz), 123.7 (d, J = 2.7 Hz), 118.0 (d, J = 21.0 Hz), 114.3 (d, J = 23.8 Hz), 103.0, 101.0. HRMS (ESI): m/z calcd for C14H10FN2O4 [M + H]+: 289.0619, found 289.0622. HPLC analysis: tR = 7.435 min, 95.5%.
4.1.6. 2-(4-Fluorophenyl)-5,6,7-trihydroxyquinazolin-4(3H)-one (A3)
According to general procedure A, the product was obtained as a white solid (56 mg), yield 39%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.80 (s, 1H), 10.33 (s, 1H), 8.89 (br, 1H), 8.25–8.08 (m, 2H), 7.44–7.29 (m, 2H), 6.64 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.7, 163.8 (d, J = 247.5 Hz), 153.9, 148.9, 146.8, 141.5, 131.0, 130.1 (d, J = 8.9 Hz), 129.2 (d, J = 2.9 Hz), 115.6 (d, J = 21.9 Hz), 102.7, 100.4. HRMS (ESI): m/z calcd for C14H10FN2O4 [M + H]+: 289.0619, found 289.0617. HPLC analysis: tR = 8.683 min, 98.9%.
4.1.7. 2-(2-Chlorophenyl)-5,6,7-trihydroxyquinazolin-4(3H)-one(A4)
According to general procedure A, the product was obtained as a white solid (90 mg), yield 59%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.56 (s, 1H), 11.73 (s, 1H), 10.35 (s, 1H), 8.93 (s, 1H), 7.65–7.52 (m, 3H), 7.48 (td, J = 7.4, 1.5 Hz, 1H), 6.60 (s, 1H). 13C NMR (150 MHz, DMSO‑d 6) δ 165.5, 153.8, 149.1, 146.8, 141.7, 133.6, 131.6, 131.5, 131.2, 131.0, 129. 6, 127.2, 102.9, 100.7. HRMS (ESI): m/z calcd for C14H10ClN2O4 [M + H]+: 305.0324, found 305.0323. HPLC analysis: tR = 6.920 min, 95.2%.
4.1.8. 2-(3-Chlorophenyl)-5,6,7-trihydroxyquinazolin-4(3H)-one (A5)
According to general procedure A, the product was obtained as a white solid (94 mg), yield 62%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.50 (br, 1H), 11.73 (s, 1H), 10.35 (br, 1H), 8.98 (br, 1H), 8.16 (t, J = 1.9 Hz, 1H), 8.07 (d, J = 7.9 Hz, 1H), 7.69–7.48 (m, 2H), 6.67 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.5, 153.9, 148.2, 146.8, 141.7, 134.8, 133.4, 131.4, 130.8, 130.5, 127.3, 126.2, 103.4, 100.7. HRMS (ESI): m/z calcd for C14H10ClN2O4 [M + H]+: 305.0324, found 305.0323. HPLC analysis: tR = 12.106 min, 96.2%.
4.1.9. 2-(4-Chlorophenyl)-5,6,7-trihydroxyquinazolin-4(3H)-one (A6)
According to general procedure A, the product was obtained as a white solid (97 mg), yield 64%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.51 (s, 1H), 11.72 (s, 1H), 10.36 (s, 1H), 8.94 (s, 1H), 8.15–8.09 (m, 2H), 7.62–7.56 (m, 2H), 6.64 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.3, 154.0, 148.4, 146.8, 141.9, 135.9, 131.6, 131.3, 129.3, 128.7, 103.3, 100.6. HRMS (ESI): m/z calcd for C14H10ClN2O4 [M + H]+: 305.0324, found 305.0317. HPLC analysis: tR = 9.457 min, 98.0%.
4.1.10. 2-(3-Bromophenyl)-5,6,7-trihydroxyquinazolin-4(3H)-one (A8)
According to general procedure A, the product was obtained as a white solid (104 mg), yield 60%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.54 (br, 1H), 11.78 (s, 1H), 10.37 (s, 1H), 8.96 (s, 1H), 8.29 (s, 1H), 8.11 (d, J = 7.9 Hz, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.49 (t, J = 7.9 Hz, 1H), 6.66 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.6, 153.9, 148.3, 146.8, 141.3, 134.9, 133.7, 131.3, 130.7, 130.1, 126.5, 121.9, 102.9, 100.6. HRMS (ESI): m/z calcd for C14H10 79BrN2O4 [M + H]+: 348.9819, found 348.9823. HPLC analysis: tR = 10.440 min, 95.4%.
4.1.11. 2-(4-Bromophenyl)-5,6,7-trihydroxyquinazolin-4(3H)-one (A9)
According to general procedure A, the product was obtained as a white solid (108 mg), yield 62%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.51 (s, 1H), 11.77 (s, 1H), 10.35 (s, 1H), 8.94 (s, 1H), 8.09–8.02 (m, 2H), 7.780–7.71 (m, 2H), 6.65 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.6, 153.9, 148.8, 146. 8, 141.5, 131.9, 131.6, 131.2, 129.5, 124.8, 102.8, 100.6. HRMS (ESI): m/z calcd for C14H8 79BrN2O4 [M − H]–: 346.9673, found 346.9666. HPLC analysis: tR = 11.866 min, 96.6%.
4.1.12. 5,6,7-Trihydroxy-2-(o-tolyl)quinazolin-4(3H)-one (A10)
According to general procedure A, the product was obtained as a white solid (50 mg), yield 35%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.37 (s, 1H), 11.78 (s, 1H), 10.28 (s, 1H), 8.87 (s, 1H), 7.47–7.38 (m, 2H), 7.35–7.28 (m, 2H), 6.58 (s, 1H), 2.35 (s, 3H). 13C NMR (150 MHz, DMSO‑d 6) δ 166.3, 153.8, 151.7, 146.8, 141.3, 136.2, 134.0, 131.0, 130.5, 129.8, 129.2, 125.7, 102.8, 100.5, 19.6. HRMS (ESI): m/z calcd for C15H11N2O4 [M − H] –: 283.0724, found 283.0718. HPLC analysis: tR = 9.143 min, 99.2%.
4.1.13. 5,6,7-Trihydroxy-2-(m-tolyl)quinazolin-4(3H)-one (A11)
According to general procedure A, the product was obtained as a white solid (57 mg), yield 40%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.84 (br, 1H), 10.36 (s, 1H), 8.90 (br, 1H), 7.93 (s, 1H), 7.88 (d, J = 7.5 Hz, 1H), 7.45–7.34 (m, 2H), 6.65 (s, 1H), 2.39 (s, 3H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.8, 153.9, 150.1, 146.8, 141.5, 137.9, 132.5, 131.8, 131.0, 128.5, 128.1, 124.7, 102.6, 100.5, 21.0. HRMS (ESI): m/z calcd for C15H11N2O4 [M − H] –: 283.0724, found 283.0722. HPLC analysis: tR = 7.412 min, 95.1%.
4.1.14. 5,6,7-Trihydroxy-2-(p-tolyl)quinazolin-4(3H)-one (A12)
According to general procedure A, the product was obtained as a white solid (61 mg), yield 43%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.38 (br, 1H), 11.82 (s, 1H), 10.31 (s, 1H), 8.87 (s, 1H), 8.05–7.98 (m, 2H), 7.36–7.30 (m, 2H), 6.62 (s, 1H), 2.38 (s, 3H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.8, 154.4, 150.1, 147.3, 141.6, 131.3, 130.3, 129.6, 127.9, 102.7, 100.9, 21.42. HRMS (ESI): m/z calcd for C15H11N2O4 [M − H] –: 283.0724, found 283.0717. HPLC analysis: tR = 5.061 min, 97.0%.
4.1.15. 5,6,7-Trihydroxy-2-(3-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (A14)
According to general procedure A, the product was obtained as a white solid (68 mg), yield 40%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.68 (s, 1H), 11.72 (s, 1H), 10.38 (s, 1H), 8.99 (s, 1H), 8.46 (s, 1H), 8.41 (d, J = 8.1 Hz, 1H), 7.93 (d, J = 7.7 Hz, 1H), 7.77 (t, J = 7.8 Hz, 1H), 6.68 (s, 1H). 13C NMR (150 MHz, DMSO‑d 6) δ 166.3, 154.0, 148.0, 146.7, 141.8, 133.7, 131.53, 131.45, 129.8, 129.4 (q, J = 32.0 Hz), 127.5, 124.2, 124.0 (q, J = 272.3 Hz), 103.4, 100.7. HRMS (ESI): m/z calcd for C15H8F3N2O4 [M − H] –: 337.0442, found 337.0436. HPLC analysis: tR = 8.083 min, 97.2%.
4.1.16. 5,6,7-Trihydroxy-2-(4-(trifluoromethoxy)phenyl)quinazolin-4(3H)-one (A16)
According to general procedure A, the product was obtained as a yellow solid (76 mg), yield 43%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.78 (br, 1H), 10.35 (br, 1H), 8.27–8.18 (m, 2H), 7.56–7.49 (m, 2H), 6.66 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.5, 158.2, 153.9, 150.2, 148.6, 146.8, 141.4, 131.8, 131.2, 129.8, 128.3, 120.8, 120.0 (q, J = 257.4 Hz), 102.7, 100.5. HRMS (ESI): m/z calcd for C15H8F3N2O5 [M − H] –: 353.0391, found 353.0381. HPLC analysis: tR = 8.742 min, 96.3%.
4.1.17. 5,6,7-Trihydroxy-2-(3-nitrophenyl)quinazolin-4(3H)-one (A18)
According to general procedure A, the product was obtained as a yellow solid (71 mg), yield 45%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.76 (s, 1H), 11.71 (s, 1H), 10.42 (s, 1H), 9.02 (s, 1H), 8.94 (s, 1H), 8.53 (d, J = 7.8 Hz, 1H), 8.38 (dd, J = 8.2, 2.2 Hz, 1H), 7.81 (t, J = 8.0 Hz, 1H), 6.69 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 167.0, 154.4, 148.4, 147.8, 147.2, 141.9, 134.7, 134.2, 132.0, 130.7, 125.9, 122.8, 103.9, 101.2. HRMS (ESI): m/z calcd for C14H8N3O6 [M − H] –: 314.0419, found 314.0412. HPLC analysis: tR = 11.254 min, 97.5%.
4.1.18. 5,6,7-Trihydroxy-2-(4-nitrophenyl)quinazolin-4(3H)-one (A19)
According to general procedure A, the product was obtained as a yellow solid (61 mg), yield 39%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.74 (s, 1H), 11.70 (s, 1H), 10.44 (s, 1H), 9.07 (s, 1H), 8.41–8.30 (s, 4H), 6.70 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.1, 153.9, 148.7, 147.6, 146.7, 141.6, 138.5, 131.8, 128.9, 123.6, 103.6, 100.7. HRMS (ESI): m/z calcd for C14H8N3O6 [M − H] –: 314.0419, found 314.0412. HPLC analysis: tR = 8.392 min, 97.7%.
4.1.19. 2-(3-(tert-Butyl)phenyl)-5,6,7-trihydroxyquinazolin-4(3H)-one (A21)
According to general procedure A, the product was obtained as a white solid (68 mg), yield 42%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.53 (br, 1H), 11.84 (s, 1H), 10.37 (s, 1H), 8.89 (s, 1H), 8.10 (s, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.58 (d, J = 7.9 Hz, 1H), 7.45 (t, J = 7.8 Hz, 1H), 6.66 (s, 1H), 1.34 (s, 9H). 13C NMR (150 MHz, DMSO‑d 6) δ 166.9, 153.9, 151.1, 150.0, 146.8, 141.4, 132.4, 131.0, 128.4, 128.1, 125.0, 124.3, 102.5, 100.5, 34.8, 31.1.
HRMS (ESI): m/z calcd for C18H17N2O4 [M − H] –: 325.1194, found 325.1189. HPLC analysis: tR = 12.850 min, 95.2%.
4.1.20. 2-(4-(tert-Butyl)phenyl)-5,6,7-trihydroxyquinazolin-4(3H)-one (A22)
According to general procedure A, the product was obtained as a white solid (67 mg), yield 41%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.38 (s, 1H), 11.80 (s, 1H), 10.29 (s, 1H), 8.85 (s, 1H), 8.10–8.01 (m, 2H), 7.56–7.51 (m, 2H), 6.63 (s, 1H), 1.32 (s, 9H). 13C NMR (150 MHz, DMSO‑d 6) δ 166.3, 153.9, 149.6, 146.8, 142.2, 130. 9, 130.0, 128.6, 127.3, 125.4, 102.9, 100.5, 34.7, 30.9. HRMS (ESI): m/z calcd for C18H17N2O4 [M − H] –: 325.1194, found 325.1185. HPLC analysis: tR = 10.744 min, 97.0%.
4.1.21. 5,6,7-Trihydroxy-2-(2-hydroxyphenyl)quinazolin-4(3H)-one (A23)
According to general procedure A, the product was obtained as a white solid (50 mg), yield 35%. 1H NMR (400 MHz, DMSO‑d 6) δ 13.71 (br, 1H), 12.42 (br, 1H), 11.60 (s, 1H), 10.48 (s, 1H), 9.03 (s, 1H), 8.15 (dd, J = 8.1, 1.6 Hz, 1H), 7.47–7.37 (m, 1H), 7.01–6.92 (m, 2H), 6.63 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.4, 159.8, 154.2, 151.1, 147.0, 139.0, 133.4, 131.4, 127.4, 118.8, 117.8, 113.8, 101.8, 100.4. HRMS (ESI): m/z calcd for C14H9N2O5 [M − H] –: 285.0517, found 285.0509. HPLC analysis: tR = 9.255 min, 95.4%.
4.1.22. 5,6,7-Trihydroxy-2-(4-hydroxyphenyl)quinazolin-4(3H)-one (A24)
According to general procedure A, the product was obtained as a white solid (54 mg), yield 38%. 1H NMR (300 MHz, DMSO‑d 6) δ 12.27 (br, 1H), 11.85 (s, 1H), 10.27 (s, 1H), 10.13 (s, 1H), 8.81 (s, 1H), 8.06–7.93 (m, 2H), 6.93–6.82 (m, 2H), 6.58 (s, 1H). 13C NMR (150 MHz, DMSO‑d 6) δ 166.4, 160.3, 154.0, 149.2, 146.8, 142.4, 130.5, 129.3, 123.3, 115.3, 102.5, 100.2. HRMS (ESI): m/z calcd for C14H9N2O5 [M − H] –: 285.0517, found 285.0514. HPLC analysis: tR = 10.262 min, 96.1%.
4.1.23. 2-(2,3-Difluorophenyl)-5,6,7-trihydroxyquinazolin-4(3H)-one (A25)
According to general procedure A, the product was obtained as a white solid (77 mg), yield 50%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.60 (s, 1H), 11.67 (s, 1H), 10.40 (s, 1H), 9.01 (s, 1H), 7.69–7.60 (m, 1H), 7.58–7.51 (m, 1H), 7.40–7.32 (m, 1H), 6.64 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.0, 154.35, 151.17 (d, J = 12.5 Hz), 149.17 (dd, J = 13.2, 9.5 Hz), 147.20, 146.07 (d, J = 3.5 Hz), 142.26, 132.04, 126.60 (d, J = 3.5 Hz), 125.88–125.24 (m), 124.68 (d, J = 9.6 Hz), 119.93 (d, J = 17.3 Hz), 103.9, 101.1. HRMS (ESI): m/z calcd for C14H7F2N2O4 [M − H] –: 305.0379, found 305.0369. HPLC analysis: tR = 8.571 min, 96.1%.
4.1.24. 2-(2,4-Difluorophenyl)-5,6,7-trihydroxyquinazolin-4(3H)-one (A26)
According to general procedure A, the product was obtained as a white solid (84 mg), yield 55%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.52 (s, 1H), 11.69 (s, 1H), 10.36 (s, 1H), 8.97 (s, 1H), 7.83–7.76 (m, 1H), 7.50–7.42 (m, 1H), 7.25 (td, J = 8.5, 2.5 Hz, 1H), 6.62 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.6, 163.5 (dd, J = 248.8, 11.3Hz), 160.0 (J = 253.2, 12.6 Hz), 153.9, 146.7, 146.0, 141.9, 132.6 (dd, J = 10.6, 3.7 Hz), 131.4, 119.0 (dd, J = 13.1, 3.5 Hz), 111.9 (dd, J = 22.1, 3.1 Hz), 104.6 (t, J = 26.0 Hz), 103.2, 100.6. HRMS (ESI): m/z calcd for C14H7F2N2O4 [M − H] –: 305.0379, found 305.0369. HPLC analysis: tR = 7.082 min, 95.4%.
4.1.25. 2-(3,4-Difluorophenyl)-5,6,7-trihydroxyquinazolin-4(3H)-one (A27)
According to general procedure A, the product was obtained as a white solid (77 mg), yield 50%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.53 (s, 1H), 11.69 (s, 1H), 10.38 (s, 1H), 8.97 (s, 1H), 8.20–8.13 (m, 1H), 8.04–7.97 (m, 1H), 7.67–7.57 (m, 1H), 6.65 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.6 (d, J = 141.3 Hz), 153.9, 152.2 (d, J = 13.4 Hz), 150.2 (t, J = 12.7 Hz), 148.3 (d, J = 12.9 Hz), 147.3, 146.7, 141.7, 131.32, 130.2 (dd, J = 6.2, 3.4 Hz), 124.84, 117.79 (d, J = 17.7 Hz), 116.76 (d, J = 19.3 Hz), 103.3, 100.5. HRMS (ESI): m/z calcd for C14H7F2N2O4 [M − H] –: 305.0379, found 305.0371. HPLC analysis: tR = 7.863 min, 95.4%.
4.1.26. General procedure B for the preparation of compounds A7, A13, A15, A17, A20, B1–B17, C1–C15 and D6–D9
To the stirred solution of 27 or 30a–30p (1 mmol) in anhydrous EtOH (10 mL) was added I2 (1.2 mmol) and the corresponding aldehyde (1.2 mmol). The resulting reaction mixture was heated to reflux and stirred for 3–5 h. After completion of the reaction, the reaction mixture was cooled to room temperature and was quenched with excess 5% aqueous Na2S2O3. The aqueous portion was extracted with DCM (3 × 10 mL), and the combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica column chromatography (DCM/MeOH = 300:1 to 150:1) to give the corresponding quinazolinones 29a–29v, 32a–32o, and 41a–41d.
According to general procedure A, the demethylation of the above quinazolinones afforded the target compounds A7, A13, A15, A17, A20, B1–B17, C1–C15 and D6–D9.
4.1.27. 2-(2-Bromophenyl)-5,6,7-trihydroxyquinazolin-4(3H)-one (A7)
According to general procedure B, the product was obtained as a white solid (99 mg), yield 57%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.55 (s, 1H), 11.76 (s, 1H), 10.36 (s, 1H), 8.94 (s, 1H), 7.76 (dd, J = 7.7, 1.5 Hz, 1H), 7.60 (dd, J = 7.4, 2.1 Hz, 1H), 7.56–7.42 (m, 2H), 6.60 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.5, 153.8, 150.2, 146.8, 141.8, 135.7, 132.6, 131.6, 131.3, 130.9, 127.6, 121.1, 103.2, 100.7. HRMS (ESI): m/z calcd for C14H10 79BrN2O4 [M + H]+: 348.9819, found 348.9822. HPLC analysis: tR = 9.626 min, 98.2%.
4.1.28. 5,6,7-Trihydroxy-2-(2-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (A13)
According to general procedure B, the product was obtained as a yellow solid (71 mg), yield 42%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.62 (s, 1H), 11.70 (s, 1H), 10.35 (s, 1H), 8.94 (s, 1H), 7.94–7.85 (m, 1H), 7.84–7.69 (m, 3H), 6.57 (s, 1H). 13C NMR (150 MHz, DMSO‑d 6) δ 165.5, 153.9, 149.3, 147.0, 141.6, 132.9, 132.4, 131.3, 130.8, 130.4, 127.09 (q, J = 30.8 Hz), 126.41 (q, J = 4.7 Hz), 123.76 (q, J = 273 Hz), 103.0, 100.6. HRMS (ESI): m/z calcd for C15H8F3N2O4 [M − H] –: 337.0442, found 337.0431. HPLC analysis: tR = 7.223 min, 96.4%.
4.1.29. 5,6,7-Trihydroxy-2-(2-(trifluoromethoxy)phenyl)quinazolin-4(3H)-one (A15)
According to general procedure B, the product was obtained as a white solid (71 mg), yield 40%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.58 (s, 1H), 11.70 (s, 1H), 10.36 (s, 1H), 8.97 (s, 1H), 7.76 (dd, J = 7.6, 1.8 Hz, 1H), 7.68 (td, J = 7.8, 1.8 Hz, 1H), 7.58–7.50 (m, 2H), 6.62 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.6, 153.9, 147.2, 146.7, 145.7, 141.9, 132.1, 131.4, 128.0, 127.6, 121.5, 120.0 (q, J = 257.4 Hz), 103.3, 100.5. HRMS (ESI): m/z calcd for C15H8F3N2O5 [M − H] –: 353.0391, found 353.0377. HPLC analysis: tR = 9.658 min, 95.0%.
4.1.30. 5,6,7-Trihydroxy-2-(2-nitrophenyl)quinazolin-4(3H)-one (A17)
According to general procedure B, the product was obtained as an orange solid (68 mg), yield 43%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.76 (s, 1H), 11.68 (s, 1H), 10.37 (s, 1H), 8.97 (s, 1H), 8.20–8.16 (m, 1H), 7.94–7.76 (m, 3H), 6.54 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.7, 153.9, 148.4, 147.6, 146.8, 141.9, 133.8, 131.6, 131.5, 131.4, 129.0, 124.5, 103.2, 100.6. HRMS (ESI): m/z calcd for C14H8N3O6 [M − H] –: 314.0419, found 314.0411. HPLC analysis: tR = 6.699 min, 95.1%.
4.1.31. 5,6,7-Trihydroxy-2-(3-isopropylphenyl)quinazolin-4(3H)-one (A20)
According to general procedure B, the product was obtained as a white solid (72 mg), yield 46%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.46 (s, 1H), 11.83 (s, 1H), 10.30 (s, 1H), 8.87 (s, 1H), 7.98 (s, 1H), 7.94–7.89 (m, 1H), 7.44 (d, J = 4.8 Hz, 2H), 6.64 (s, 1H), 2.99 (hept, J = 6.8 Hz, 1H), 1.27 (s, 3H), 1.26 (s, 3H). 13C NMR (150 MHz, DMSO‑d 6) δ 166.6, 153.9, 150.0, 148.8, 146.8, 141.8, 132.6, 131.0, 129.2, 128.6, 125.4, 125.2, 102.8, 100.5, 33.5, 23.8. HRMS (ESI): m/z calcd for C17H15N2O4 [M − H] –: 311.1037, found 311.1033. HPLC analysis: tR = 13.162 min, 97.7%.
4.1.32. 5,6,7-Trihydroxy-2-(thiophen-2-yl)quinazolin-4(3H)-one (B1)
According to general procedure B, the product was obtained as a offwhite solid (83 mg), yield 60%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.59 (br, 1H), 11.75 (s, 1H), 10.33 (s, 1H), 8.90 (br, 1H), 8.16–8.11 (m, 1H), 7.85–7.78 (m, 1H), 7.24–7.17 (m, 1H), 6.54 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.3, 154.0, 147.00, 145.40, 141.5, 137.5, 131.5, 131.0, 128.7, 128.5, 102.4, 100.4. HRMS (ESI): m/z calcd for C12H7N2O4S [M − H] –: 275.0123, found 275.0132. HPLC analysis: tR = 7.024 min, 96.7%.
4.1.33. 5,6,7-Trihydroxy-2-(pyridin-2-yl)quinazolin-4(3H)-one (B2)
According to general procedure B, the product was obtained as an orange solid (61 mg), yield 45%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.92 (s, 1H), 10.45 (s, 1H), 9.04 (s, 1H), 8.73 (d, J = 4.6 Hz, 1H), 8.36 (d, J = 7.8 Hz, 1H), 8.04 (t, J = 7.8 Hz, 1H), 7.62 (t, J = 6.1 Hz, 1H), 6.74 (s, 1H). 13C NMR (150 MHz, DMSO‑d 6) δ 166.1, 153.8, 148.9, 148.8, 148.0, 147.0, 140.7, 137.9, 131.6, 126.3, 122.0, 102.8, 101.3. HRMS (ESI): m/z calcd for C13H8N3O4 [M − H] –: 270.0520, found 270.0520. HPLC analysis: tR = 5.840 min, 95.0%.
4.1.34. 5,6,7-Trihydroxy-2-(pyridin-4-yl)quinazolin-4(3H)-one (B3)
According to general procedure B, the product was obtained as an yellow solid (68 mg), yield 50%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.68 (s, 1H), 11.73 (s, 1H), 10.44 (s, 1H), 9.07 (s, 1H), 8.82–8.73 (m, 2H), 8.12–8.05 (m, 2H), 6.70 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.9, 153.9, 149.9, 149.8, 147.5, 146.7, 146.0, 141.3, 140.2, 131.8, 121.5, 103.6, 101.0. HRMS (ESI): m/z calcd for C13H8N3O4 [M − H] –: 270.0520, found 270.0519. HPLC analysis: tR = 5.971 min, 96.7%.
4.1.35. 2-(1,3-Dimethyl-1H-pyrazol-5-yl)-5,6,7-trihydroxyquinazolin-4(3H)-one (B4)
According to general procedure B, the product was obtained as a yellow solid (75 mg), yield 52%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.34 (br, 1H), 11.73 (s, 1H), 10.38 (s, 1H), 8.97 (br, 1H), 6.89 (s, 1H), 6.62 (s, 1H), 4.10 (s, 3H), 2.18 (s, 3H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.2, 153.9, 146.9, 145.7, 142.4, 141.1, 134.7, 131.4, 107.8, 103.0, 100.4, 39.6, 13.2. HRMS (ESI): m/z calcd for C13H11N4O4 [M − H] –: 287.0786, found 287.0777. HPLC analysis: tR = 7.618 min, 96.5%.
4.1.36. 5,6,7-Trihydroxy-2-(naphthalen-2-yl)quinazolin-4(3H)-one (B5)
According to general procedure B, the product was obtained as a white solid (99 mg), yield 62%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.84 (s, 1H), 10.37 (s, 1H), 8.73 (s, 1H), 8.23 (dd, J = 8.6, 1.9 Hz, 1H), 8.08–7.97 (m, 3H), 7.67–7.56 (m, 2H), 6.70 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.6, 154.0, 150.0, 146.9, 141.2, 134.0, 132.3, 131.3, 129.7, 128.9, 128.2, 127.9, 127.8, 127.7, 127.0, 124.4, 102.5, 100.6. HRMS (ESI): m/z calcd for C18H11N2O4 [M − H] –: 319.0724, found 319.0721. HPLC analysis: tR = 8.708 min, 97.4%.
4.1.37. 2-Cyclopropyl-5,6,7-trihydroxyquinazolin-4(3H)-one (B6)
According to general procedure B, the product was obtained as a white solid (49 mg), yield 42%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.34 (s, 1H), 11.71 (s, 1H), 10.15 (s, 1H), 8.65 (s, 1H), 6.35 (s, 1H), 1.92–1.84 (m, 1H), 1.06–0.93 (m, 4H). 13C NMR (150 MHz, DMSO‑d 6) δ 166.0, 156.3, 153.8, 146.9, 141.8, 129.8, 102.0, 100.2, 13.4, 9.2. HRMS (ESI): m/z calcd for C11H9N2O4 [M − H] –: 233.0568, found 233.0560. HPLC analysis: tR = 5.988 min, 95.3%.
4.1.38. 2-Cyclopentyl-5,6,7-trihydroxyquinazolin-4(3H)-one (B7)
According to general procedure B, the product was obtained as a white solid (49 mg), yield 48%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.06 (s, 1H), 11.75 (s, 1H), 10.19 (s, 1H), 8.71 (s, 1H), 6.46 (s, 1H), 3.00–2.90 (m, 1H), 1.99–1.89 (m, 2H), 1.88–1.77 (m, 2H), 1.76–1.65 (m, 2H), 1.63–1.52 (m, 2H). 13C NMR (150 MHz, DMSO‑d 6) δ 165.9, 157.4, 153.7, 146.8, 142.2, 130.2, 102.6, 100.4, 43.7, 30.8, 25.3. HRMS (ESI): m/z calcd for C13H13N2O4 [M − H] –: 261.0881, found 261.0875. HPLC analysis: tR = 8.081min, 95.7%.
4.1.39. 2-Cyclohexyl-5,6,7-trihydroxyquinazolin-4(3H)-one (B8)
According to general procedure B, the product was obtained as a white solid (49 mg), yield 53%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.01 (s, 1H), 11.75 (s, 1H), 10.19 (s, 1H), 8.71 (s, 1H), 6.47 (s, 1H), 2.53–2.45 (m, 1H), 1.91–1.82 (m, 2H), 1.80–1.72 (m, 2H), 1.66 (d, J = 10.6 Hz, 1H), 1.58–1.45 (m, 2H), 1.35–1.13 (m, 3H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.8, 157.6, 153.7, 146.8, 142.3, 130.3, 102.1, 100.4, 42.6, 30.2, 25.5, 25.3. HRMS (ESI): m/z calcd for C14H15N2O4 [M − H] –: 275.1037, found 275.1034. HPLC analysis: tR = 9.657 min, 96.3%.
4.1.40. 5,6,7-Trihydroxy-2-isopropylquinazolin-4(3H)-one (B9)
According to general procedure B, the product was obtained as a white solid (71 mg), yield 60%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.03 (s, 1H), 11.73 (s, 1H), 10.16 (s, 1H), 8.69 (s, 1H), 6.48 (s, 1H), 2.81 (hept, J = 6.8 Hz, 1H), 1.21 (d, J = 6.9 Hz, 6H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.1, 158.7, 153.7, 146.9, 142.0, 130.3, 102.4, 100.4, 33.1, 20.4. HRMS (ESI): m/z calcd for C11H11N2O4 [M − H] –: 235. 0724, found 235.0721. HPLC analysis: tR = 5.456 min, 96.1%.
4.1.41. 2-(tert-Butyl)-5,6,7-trihydroxyquinazolin-4(3H)-one (B10)
According to general procedure B, the product was obtained as a white solid (70 mg), yield 56%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.79 (s, 1H), 10.22 (s, 1H), 8.75 (s, 1H), 6.51 (s, 1H), 1.29 (s, 9H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.5, 160.1, 153.8, 146.7, 141.4, 130.5, 102.5, 100.1, 37.1, 27.8. HRMS (ESI): m/z calcd for C12H13N2O4 [M − H] –: 249.0881, found 249.0873. HPLC analysis: tR = 7.203 min, 95.2%.
4.1.42. 5,6,7-Trihydroxy-2-isobutylquinazolin-4(3H)-one (B11)
According to general procedure B, the product was obtained as a white solid (58 mg), yield 56%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.08 (s, 1H), 11.70 (s, 1H), 10.21 (s, 1H), 8.74 (s, 1H), 6.48 (s, 1H), 2.39 (d, J = 7.3 Hz, 2H), 2.11 (hept, J = 6.7 Hz, 1H), 0.90 (d, J = 6.6 Hz, 6H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.1, 153.7, 146.8, 142.1, 130.3, 102.4, 100.4, 43.1, 27.0, 22.1. HRMS (ESI): m/z calcd for C12H13N2O4 [M − H] –: 249.0881, found 249.0879. HPLC analysis: tR = 9.580 min, 98.8%.
4.1.43. 5,6,7-Trihydroxy-2-neopentylquinazolin-4(3H)-one (B12)
According to general procedure B, the product was obtained as a white solid (79 mg), yield 60%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.99 (s, 1H), 11.71 (s, 1H), 10.21 (s, 1H), 8.74 (s, 1H), 6.49 (s, 1H), 2.41 (s, 2H), 0.97 (s, 9H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.7, 153.7, 152.4, 146.8, 142.1, 130.3, 102.2, 100.1, 47.0, 31.9, 29.4. HRMS (ESI): m/z calcd for C13H15N2O4 [M − H] –: 263.1037, found 263.1030. HPLC analysis: tR = 7.704 min, 95.1%.
4.1.44. (E)-5,6,7-Trihydroxy-2-styrylquinazolin-4(3H)-one (B13)
According to general procedure B, the product was obtained as a white solid (93 mg), yield 63%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.27 (s, 1H), 11.78 (s, 1H), 10.28 (s, 1H), 8.89 (s, 1H), 7.84 (d, J = 16.2 Hz, 1H), 7.69–7.60 (m, 2H), 7.51–7.37 (m, 3H), 6.93 (d, J = 16.2 Hz, 1H), 6.58 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.6, 153.8, 148.4, 146.8, 142.1, 137.2, 135.1, 130.9, 129.5, 129.0, 127.5, 121.0, 102.8, 100.6. HRMS (ESI): m/z calcd for C16H11N2O4 [M − H] –: 295.0724, found 295.0720. HPLC analysis: tR = 9.694 min, 95.3%.
4.1.45. 2-Benzyl-5,6,7-trihydroxyquinazolin-4(3H)-one (B14)
According to general procedure B, the product was obtained as a white solid (85 mg), yield 60%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.34 (s, 1H), 11.68 (s, 1H), 10.23 (s, 1H), 8.77 (s, 1H), 7.38–7.28 (m, 4H), 7.27–7.21 (m, 1H), 6.48 (s, 1H), 3.86 (s, 2H). 13C NMR (150 MHz, DMSO‑d 6) δ 166.1, 153.9, 153.6, 146.8, 141.1, 136.4, 130.7, 128.8, 128.5, 126.9, 101.6, 100.2, 40.3. HRMS (ESI): m/z calcd for C15H11N2O4 [M − H] –: 283.0724, found 283.0716. HPLC analysis: tR = 8.088 min, 95.0%.
4.1.46. 5,6,7-Trihydroxy-2-(1-phenylethyl)quinazolin-4(3H)-one (B15)
According to general procedure B, the product was obtained as a white solid (85 mg), yield 57%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.16 (s, 1H), 11.65 (s, 1H), 10.25 (s, 1H), 8.76 (s, 1H), 7.40–7.27 (m, 4H), 7.26–7.18 (m, 1H), 6.54 (s, 1H), 4.03 (q, J = 7.0 Hz, 1H), 1.55 (d, J = 7.0 Hz, 3H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.8, 155.9, 153.8, 146.8, 142.4, 142.0, 130.5, 128.5, 127.4, 126.8, 102.8, 100.4, 43.6, 19.3. HRMS (ESI): m/z calcd for C16H13N2O4 [M − H] –: 297. 0881, found 297. 0871. HPLC analysis: tR = 7.318 min, 97.7%.
4.1.47. 5,6,7-Trihydroxy-2-(1-phenylpropyl)quinazolin-4(3H)-one (B16)
According to general procedure B, the product was obtained as a white solid (92 mg), yield 59%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.69 (br, 1H), 10.27 (br, 1H), 7.42–7.35 (m, 2H), 7.34–7.29 (m, 2H), 7.26–7.20 (m, 1H), 6.55 (s, 1H), 3.74 (t, J = 7.7 Hz, 1H), 2.27–2.15 (m, 1H), 1.96–1.84 (m, 1H), 0.83 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.1, 156.2, 154.5, 153.9, 146.8, 140.7, 130.7, 128.5, 127.9, 127.1, 101.8100.4, 51.2, 26.3, 12.2. HRMS (ESI): m/z calcd for C17H15N2O4 [M − H] –: 311.1037, found 311.1028. HPLC analysis: tR = 7.867 min, 95.1%.
4.1.48. 5,6,7-Trihydroxy-2-(2-methyl-1-phenylpropyl)quinazolin-4(3H)-one (B17)
According to general procedure B, the product was obtained as a white solid (103 mg), yield 63%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.23 (s, 1H), 11.61 (s, 1H), 10.24 (s, 1H), 8.76 (s, 1H), 7.44 (d, J = 7.1 Hz, 2H), 7.31 (t, J = 7.5 Hz, 2H), 7.25–7.18 (m, 1H), 6.55 (s, 1H), 3.40 (d, J = 11.3 Hz, 1H), 2.67–2.53 (m, 1H), 0.93 (d, J = 6.4 Hz, 3H), 0.69 (d, J = 6.6 Hz, 3H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.7, 155.4, 153.8, 146.7, 142.3, 140.3, 130.4, 128.33, 128.28, 127.0, 102.7, 100.2, 57.8, 30.9, 21.2, 20.5. HRMS (ESI): m/z calcd for C18H18N2O4Na [M + Na]+: 349. 1159, found 349. 1158. HPLC analysis: tR = 9.494 min, 96.0%.
4.1.49. 5,6,7-Trihydroxy-3-isobutyl-2-phenylquinazolin-4(3H)-one (C1)
According to general procedure B, the product was obtained as a light yellow solid (77 mg), yield 47%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.83 (s, 1H), 10.37 (s, 1H), 8.95 (s, 1H), 7.61–7.56 (m, 2H), 7.55–7.47 (m, 3H), 6.57 (s, 1H), 3.83 (d, J = 7.4 Hz, 2H), 1.73 (hept, J = 6.9 Hz, 1H), 0.61 (d, J = 6.7 Hz, 6H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.2, 153.9, 153.0, 146.4, 140.3, 135.2, 131.4, 129.5, 128.5, 128.3, 102.8, 100.4, 50.6, 27.3, 19.7. HRMS (ESI): m/z calcd for C18H19N2O4 [M + H] +: 327.1339, found 327.1342. HPLC analysis: tR = 14.369 min, 96.7%.
4.1.50. 5,6,7-Trihydroxy-3-(2-hydroxyethyl)-2-phenylquinazolin-4(3H)-one (C2)
According to general procedure B, the product was obtained as a white solid (71 mg), yield 45%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.80 (s, 1H), 10.33 (s, 1H), 8.93 (s, 1H), 7.62–7.55 (m, 2H), 7.54–7.47 (m, 3H), 6.55 (s, 1H), 4.83 (t, J = 5.7 Hz, 1H), 3.94 (t, J = 6.2 Hz, 2H), 3.48 (q, J = 5.9 Hz, 2H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.1, 153.9, 153.4, 146.4, 140.5, 135.4, 131.3, 129.3, 128.6, 128.3, 102.6, 100.7, 57.8, 46.9. HRMS (ESI): m/z calcd for C16H15N2O5 [M + H] +: 315. 0976, found 315. 0973. HPLC analysis: tR = 11.691 min, 98.1%.
4.1.51. N-(tert-butyl)-2-(5,6,7-trihydroxy-4-oxo-2-phenylquinazolin-3(4H)-yl)acetamide (C3)
According to general procedure B, the product was obtained as a white solid (82 mg), yield 43%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.57 (s, 1H), 10.39 (s, 1H), 8.96 (br, 1H), 7.67 (s, 1H), 7.58–7.43 (m, 5H), 6.58 (s, 1H), 4.36 (s, 2H), 1.17 (s, 9H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.4, 164.7, 154.0, 153.1, 146.3, 140.4, 134.8, 131.4, 129.6, 128.3, 128.1, 102.8, 100.2, 50.3, 47.1, 28.3. HRMS (ESI): m/z calcd for C20H21N3O5Na [M + Na] +: 406. 1373, found 406.1370. HPLC analysis: tR = 12.294 min, 96.8%.
4.1.52. 3-Cyclopropyl-5,6,7-trihydroxy-2-phenylquinazolin-4(3H)-one (C4)
According to general procedure B, the product was obtained as a white solid (71 mg), yield 46%.1H NMR (400 MHz, DMSO‑d 6) δ 11.88 (s, 1H), 10.26 (s, 1H), 8.88 (s, 1H), 7.84–7.66 (m, 2H), 7.56–7.41 (m, 3H), 6.55 (s, 1H), 3.17 (tt, J = 7.3, 4.1 Hz, 1H), 0.87–0.65 (m, 2H), 0.57–0.30 (m, 2H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.7, 153.9, 153.6, 146.4, 140.1, 136.0, 131.2, 129.3, 128.4, 127.9, 102.7, 100.4, 29.0, 10.7. HRMS (ESI): m/z calcd for C17H15N2O4 [M + H] +: 311. 1026, found 311. 1023. HPLC analysis: tR = 12.671 min, 98.4%.
4.1.53. 3-Cyclopentyl-5,6,7-trihydroxy-2-phenylquinazolin-4(3H)-one (C5)
According to general procedure B, the product was obtained as a white solid (85 mg), yield 50%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.90 (s, 1H), 10.30 (s, 1H), 8.91 (s, 1H), 7.70–7.38 (m, 5H), 6.53 (s, 1H), 4.35–4.25 (m, 1H), 2.34–2.21 (m, 2H), 1.96–1.82 (m, 2H), 1.79–1.65 (m, 2H), 1.46–1.34 (m, 2H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.4, 153.8, 153.7, 146.5, 140.1, 135.9, 131.3, 129.5, 128.6, 127.8, 102.4, 101.4, 61.0, 28.7, 25.4. HRMS (ESI): m/z calcd for C19H19N2O4 [M + H] +: 339.1339, found 339.1338. HPLC analysis: tR = 11.188 min, 96.5%.
4.1.54. 3-Cyclohexyl-5,6,7-trihydroxy-2-phenylquinazolin-4(3H)-one (C6)
According to general procedure B, the product was obtained as a white solid (97 mg), yield 55%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.98 (s, 1H), 10.30 (s, 1H), 8.91 (s, 1H), 7.60–7.48 (m, 5H), 6.52 (s, 1H), 3.71 (t, J = 12.1 Hz, 1H), 2.58–2.43 (m, 2H), 1.81–1.62 (m, 4H), 1.54–1.42 (m, 1H), 1.16–0.98 (m, 1H), 0.91–0.73 (m, 2H). 13C NMR (125 MHz, DMSO‑d 6) δ 166.4, 154.2, 153.9, 147.0, 140.5, 136.4, 131.7, 129.9, 129.0, 127.9, 102.9, 101.9, 62.0, 28.8, 26.3, 25.1. HRMS (ESI): m/z calcd for C20H21N2O4 [M + H] +: 353.1496, found 353.1503. HPLC analysis: tR = 8.000 min, 98.4%.
4.1.55. 5,6,7-Trihydroxy-2,3-diphenylquinazolin-4(3H)-one (C7)
According to general procedure B, the product was obtained as a white solid (99 mg), yield 57%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.60 (s, 1H), 10.42 (s, 1H), 9.00 (s, 1H), 7.37–7.15 (m, 10H), 6.67 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.0, 154.1, 152.1, 146.8, 140.7, 136.9, 135.3, 131.5, 129.6, 129.0, 128.8, 128.6, 128.3, 127.5, 103.2, 100.4. HRMS (ESI): m/z calcd for C20H15N2O4 [M + H] +: 347.1026, found 347.1025. HPLC analysis: tR = 8.939 min, 98.9%.
4.1.56. 3-(2-Fluorophenyl)-5,6,7-trihydroxy-2-phenylquinazolin-4(3H)-one (C8)
According to general procedure B, the product was obtained as a white solid (73 mg), yield 40%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.32 (s, 1H), 10.50 (s, 1H), 9.07 (s, 1H), 7.51 (td, J = 7.8, 1.7 Hz, 1H), 7.43–7.20 (m, 7H), 7.16 (td, J = 7.7, 1.4 Hz, 1H), 6.70 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 164.3, 157.0 (d, J = 248.1 Hz), 154.3, 151.7, 146.8, 140.4, 134.5, 131.7 (d, J = 24.6 Hz) 131.2 (d, J = 8.2 Hz), 129.3, 128.3, 127.7, 124.7 (d, J = 3.4 Hz), 124.5, 124.4, 115.8 (d, J = 19.6 Hz), 103.6, 99.9. HRMS (ESI): m/z calcd for C20H14FN2O4 [M + H] +: 365.0932, found 365.0931. HPLC analysis: tR = 7.934 min, 98.3%.
4.1.57. 3-(3-Fluorophenyl)-5,6,7-trihydroxy-2-phenylquinazolin-4(3H)-one (C9)
According to general procedure B, the product was obtained as a white solid (73 mg), yield 40%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.50 (s, 1H), 10.44 (s, 1H), 9.03 (s, 1H), 7.45–7.07 (m, 9H), 6.67 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 164.8, 161.6 (d, J = 244.4 Hz), 154.1, 151.8, 146.8, 140.5, 138.4 (d, J = 10.6 Hz), 135.1, 131.6, 130.1 (d, J = 9.0 Hz), 128.9, 127.6, 126.1 (d, J = 3.1 Hz), 117.2 (d, J = 23.7 Hz), 115.4 (d, J = 20.8 Hz), 103.3, 100.3. HRMS (ESI): m/z calcd for C20H14FN2O4 [M + H] +: 365.0932, found 365.0930. HPLC analysis: tR = 7.986 min, 96.7%.
4.1.58. 5,6,7-Trihydroxy-3-(4-hydroxyphenyl)-2-phenylquinazolin-4(3H)-one (C10)
According to general procedure B, the product was obtained as a white solid (94 mg), yield 52%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.69 (s, 1H), 10.37 (s, 1H), 9.62 (s, 1H), 8.97 (s, 1H), 7.34–7.29 (m, 2H), 7.27–7.19 (m, 3H), 7.10–7.05 (m, 2H), 6.65–6.60 (m, 3H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.3, 157.0, 154.0, 152.6, 146.8, 140.7, 135.6, 131.4, 130.5, 129.0, 128.7, 127.9, 127.5, 115.1, 103.1, 100.5. HRMS (ESI): m/z calcd for C20H15N2O5 [M + H] +: 363.0976, found 363.0984. HPLC analysis: tR = 7.195 min, 98.2%.
4.1.59. 3-(4-Bromo-2-fluorophenyl)-5,6,7-trihydroxy-2-phenylquinazolin-4(3H)-one(C11)
According to general procedure B, the product was obtained as a white solid (93 mg), yield 42%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.23 (s, 1H), 10.57 (s, 1H), 9.13 (br, 1H), 7.66 (dd, J = 9.4, 2.1 Hz, 1H), 7.53 (t, J = 8.2 Hz, 1H), 7.42 (dd, J = 8.6, 2.1 Hz, 1H), 7.39–7.25 (m, 5H), 6.70 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 164.1, 156.9 (d, J = 253.0 Hz), 154.4, 151.4, 146.8, 140.3, 134.3, 133.2, 131.9, 129.5, 128.3, 128.0 (d, J = 3.4 Hz), 127.9, 124.2 (d, J = 13.1 Hz), 122.7 (d, J = 9.0 Hz), 119.4 (d, J = 23.2 Hz), 103.7, 99.8. HRMS (ESI): m/z calcd for C20H11 79BrFN2O4 [M − H]–: 440.9892, found 440.9878. HPLC analysis: tR = 8.670 min, 97.1%.
4.1.60. 3-(4-Fluoro-3-methylphenyl)-5,6,7-trihydroxy-2-phenylquinazolin-4(3H)-one (C12)
According to general procedure B, the product was obtained as a white solid (108 mg), yield 57%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.57 (s, 1H), 10.42 (br, 1H), 9.01 (br, 1H), 7.38–7.29 (m, 3H), 7.31–7.13 (m, 4H), 7.06 (t, J = 9.1 Hz, 1H), 6.66 (s, 1H), 2.12 (d, J = 1.9 Hz, 3H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.0, 159.8 (d, J = 244.8 Hz), 154.1, 152.0, 146.8, 140.5, 135.2, 132.69 (d, J = 3.4 Hz), 132.66, 132.61, 131.5, 128.9, 128.8 (d, J = 9.2 Hz), 127.5, 124.5 (d, J = 18.8 Hz), 115.0 (d, J = 23.7 Hz), 103.2, 100.3, 14.0 (d, J = 3.1 Hz). HRMS (ESI): m/z calcd for C21H16FN2O4 [M + H] +: 379. 1089, found 379.1089. HPLC analysis: tR = 8.177 min, 96.6%.
4.1.61. 3-([1,1′-biphenyl]-4-yl)-5,6,7-trihydroxy-2-phenylquinazolin-4(3H)-one (C13)
According to general procedure B, the product was obtained as a white solid (74 mg), yield 35%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.61 (s, 1H), 10.44 (s, 1H), 9.02 (s, 1H), 7.67–7.60 (m, 4H), 7.50–7.33 (m, 7H), 7.28–7.19 (m, 3H), 6.68 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.1, 154.1, 152.0, 146.8, 140.7, 139.7, 138.8, 136.2, 135.3, 131.6, 130.1, 129.1, 129.0, 128.9, 127.8, 127.6, 126.7, 126.7, 126.6, 103.3, 100.4. HRMS (ESI): m/z calcd for C26H19N2O4 [M + H] +: 423. 1340, found 423. 1345. HPLC analysis: tR = 10.271 min, 97.1%.
4.1.62. 5,6,7-Trihydroxy-2-phenyl-3-(pyridin-3-yl)quinazolin-4(3H)-one (C14)
According to general procedure B, the product was obtained as a white solid (74 mg), yield 50%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.42 (s, 1H), 10.49 (s, 1H), 9.06 (s, 1H), 8.52 (d, J = 2.4 Hz, 1H), 8.43 (dd, J = 4.8, 1.5 Hz, 1H), 7.88–7.83 (m, 1H), 7.44–7.30 (m, 3H), 7.29–7.19 (m, 3H), 6.69 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 164.9, 154.2, 151.7, 150.0, 149.0, 146.8, 140.6, 137.2, 134.9, 133.9, 131.7, 129.1, 129.0, 127.7, 123.5, 103.4, 100.2. HRMS (ESI): m/z calcd for C19H14N3O4 [M + H] +: 348.0979, found 348.0974. HPLC analysis: tR = 6.881min, 97.9%.
4.1.63. 5,6,7-Trihydroxy-2-phenyl-3-(1H-pyrazol-4-yl)quinazolin-4(3H)-one (C15)
According to general procedure B, the product was obtained as a white solid (82 mg), yield 49%. 1H NMR (400 MHz, DMSO‑d 6) δ 12.86 (br, 1H), 11.62 (s, 1H), 10.44 (s, 1H), 9.00 (s, 1H), 7.69 (br, 1H), 7.42–7.35 (m, 2H), 7.32–7.23 (m, 3H), 6.64 (s, 1H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.1, 154.1, 152.9, 146.8, 140.4, 135.4, 131.5, 128.9, 128.9, 127.6, 117.8, 103.2, 100.2. HRMS (ESI): m/z calcd for C17H11N4O4 [M − H] –: 335.0786, found 335.0779. HPLC analysis: tR = 8.686 min, 95.4%.
4.1.64. 5,6,7-Trihydroxy-2-isobutyl-3-phenylquinazolin-4(3H)-one (D6)
According to general procedure B, the product was obtained as a white solid (98 mg), yield 60%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.53 (s, 1H), 10.27 (s, 1H), 8.82 (s, 1H), 7.62–7.48 (m, 3H), 7.47–7.39 (m, 2H), 6.57 (s, 1H), 2.16 (d, J = 6.9 Hz, 2H), 2.08–1.96 (m, 1H), 0.78 (d, J = 6.6 Hz, 6H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.1, 153.9, 152.7, 146.7, 140.6, 136.4, 130.8, 129.4, 129.0, 128.9, 102.6, 100.1, 43.2, 25.8, 22.2. HRMS (ESI): m/z calcd for C18H19N2O4 [M + H]+: 327.1339, found 327.1345. HPLC analysis: tR = 8.174 min, 99.8%.
4.1.65. 3-(2-Fluorophenyl)-5,6,7-trihydroxy-2-isobutylquinazolin-4(3H)-one (D7)
According to general procedure B, the product was obtained as a white solid (107 mg), yield 62%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.26 (s, 1H), 10.43 (br, 1H), 7.71–7.58 (m, 2H), 7.54–7.46 (m, 1H), 7.42 (t, J = 7.4 Hz, 1H), 6.60 (s, 1H), 2.29–2.12 (m, 2H), 1.99 (hept, J = 6.7 Hz, 1H), 0.80 (dd, J = 8.2, 6.6 Hz, 6H). 13C NMR (125 MHz, DMSO‑d 6) δ 163.9, 157.3 (d, J = 248.5 Hz), 154.4, 153.1, 146.7, 145.9, 139.3, 132.0 (d, J = 7.9 Hz), 131.3 (d, J = 43.4 Hz), 125.5 (d, J = 3.6 Hz), 123.4 (d, J = 13.4 Hz), 116.5 (d, J = 19.4 Hz), 102.3, 99.6, 42.7, 25.9, 22.1 (d, J = 7.7 Hz). HRMS (ESI): m/z calcd for C18H17FN2O4 [M − H]–: 343.1100, found 343.1090. HPLC analysis: tR = 9.123 min, 96.3%.
4.1.66. 3-(4-Fluoro-3-methylphenyl)-5,6,7-trihydroxy-2-isobutylquinazolin-4(3H)-one (D8)
According to general procedure B, the product was obtained as a white solid (107 mg), yield 60%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.51 (s, 1H), 10.31 (s, 1H), 8.84 (s, 1H), 7.42–7.37 (m, 1H), 7.34–7.29 (m, 2H), 6.56 (s, 1H), 2.28 (d, J = 2.1 Hz, 3H), 2.17 (d, J = 6.3 Hz, 2H), 2.11–1.98 (m, 1H), 0.81 (dd, J = 6.6, 1.9 Hz, 6H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.1, 160.5 (d, J = 245.1 Hz), 154.0, 152.7, 146.7, 140.6, 132.2 (d, J = 3.2 Hz), 132.0 (d, J = 5.8 Hz), 130.9, 128.3 (d, J = 8.8 Hz), 125.7 (d, J = 18.7 Hz), 115.9 (d, J = 23.7 Hz), 102.6, 100.1, 43.1, 25.8, 22.3 (d, J = 5.4 Hz), 14.1 (d, J = 2.9 Hz). HRMS (ESI): m/z calcd for C19H18FN2O4 [M − H]–: 357.1256, found 357.1252. HPLC analysis: tR = 10.674 min, 95.2%.
4.1.67. 3-(4-Bromo-2-fluorophenyl)-5,6,7-trihydroxy-2-isobutylquinazolin-4(3H)-one (D9)
According to general procedure B, the product was obtained as a white solid (116 mg), yield 55%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.17 (s, 1H), 10.43 (br, 1H), 7.94–7.87 (m, 1H), 7.70–7.61 (m, 2H), 6.60 (s, 1H), 2.29–2.11 (m, 2H), 2.01 (hept, J = 6.6 Hz, 1H), 0.82 (dd, J = 7.8, 6.6 Hz, 6H). 13C NMR (125 MHz, DMSO‑d 6) δ 164.1, 157.2 (d, J = 253.0 Hz), 154.3, 152.0, 146.7, 140.2, 132.6, 131.3, 128.8 (d, J = 3.5 Hz), 123.3, 123.2 (d, J = 5.4 Hz), 120.1(d, J = 23.2 Hz), 102.9, 99.5, 42.8, 25.7, 22.1 (d, J = 2.7 Hz). HRMS (ESI): m/z calcd for C18H15 79BrFN2O4 [M − H]–: 421.0205, found 421.0200. HPLC analysis: tR = 10.166 min, 96.6%.
4.1.68. General procedure C for the preparation of D1–D4, D10 and D11
To a solution of compound 33a or 33b (1 mmol) in anhydrous acetonitrile (8 mL) was added the corresponding anilines (1.2 mmol) and PCl3 (0.18 mL, 2 mmol) at 0 °C, and the resulting suspension was heated to 50 °C and stirred for 4–12 h. Then, the reaction was moved to 0 °C and was quenched by adding 1 N aqueous HCl solution. The aqueous portion was extracted with DCM (3 × 20 mL), and the combined organics were washed with 10% aqueous NaHCO3 solution and brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica column chromatography (petroleum ether/EtOAc = 8:1 to 6:1) to give the corresponding quinazolinones 35a–35f.
According to general procedure A, the demethylation of the above quinazolinones afforded the target compounds D1–D4, D10 and D11.
4.1.69. 5,6,7-Trihydroxy-3-phenyl-2-(1-phenylethyl)quinazolin-4(3H)-one (D1)
According to general procedure C, the product was obtained as a white solid (118 mg), yield 63%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.49 (s, 1H), 10.39 (s, 1H), 8.92 (s, 1H), 7.57 (d, J = 4.5 Hz, 2H), 7.49–7.40 (m, 1H), 7.23–7.12 (m, 4H), 6.80 (dd, J = 6.6, 2.9 Hz, 2H), 6.68 (s, 1H), 6.54–6.48 (m, 1H), 3.81 (q, J = 6.8 Hz, 1H), 1.46 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.2, 155.0, 154.0, 146.7, 142.3, 140.3, 135.8, 131.2, 129.6, 129.4, 128.9, 128.7, 128.4, 127.1, 126.6, 103.1, 100.3, 43.2, 21.8.
HRMS (ESI): m/z calcd for C22H19N2O4 [M + H]+: 375. 1339, found 375. 1345. HPLC analysis: tR = 9.099 min, 99.1%.
4.1.70. 3-(4-Fluoro-3-methylphenyl)-5,6,7-trihydroxy-2-(1-phenylethyl)quinazolin-4(3H)-one (D2)
According to general procedure C, the product was obtained as a white solid (106 mg), yield 52%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.46 (s, 1H), 10.40 (s, 1H), 8.92 (s, 1H), 7.54–7.48 (m, 0.4 H), 7.49–7.39 (m, 0.6 H), 7.32 (t, J = 9.0 Hz, 0.6 H), 7.23–7.11 (m, 3H), 6.94 (t, J = 9.1 Hz, 0.4H), 6.88–6.81 (m, 1H), 6.80–6.72 (m, 1H), 6.68 (d, J = 4.5 Hz, 1H), 6.36–6.28 (m, 0.4 H), 6.20–6.14 (m, 0.6H), 3.89–3.74 (m, 1H), 2.31 (s, 1.2 H), 1.91 (s, 1.9 H), 1.45 (dd, J = 6.9, 3.1 Hz, 3H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.2, 161.6 (d, J = 5.0 Hz), 159.2 (d, J = 5.0 Hz), 154.9 (d, J = 8.7 Hz), 154.1 (d, J = 1.9 Hz), 146.7, 142.6, 142.4, 140.3 (d, J = 1.2 Hz), 133.1 (d, J = 6.0 Hz), 131.7 (d, J = 5.7 Hz), 131.5 (d, J = 3.3 Hz), 131.4 (d, J = 3.2 Hz), 131.2, 128.9 (d, J = 8.9 Hz), 128.4 (d, J = 4.7 Hz), 128.0 (d, J = 8.8 Hz), 127.1 (d, J = 6.9 Hz), 126.6 (d, J = 7.8 Hz), 125.5 (d, J = 18.7 Hz), 124.7 (d, J = 18.9 Hz), 115.7 (d, J = 23.9 Hz), 115.0 (d, J = 23.6 Hz), 103.2, 103.1, 100.2, 43.7, 43.3, 22.0, 21.9, 14.2 (d, J = 2.9 Hz), 13.8 (d, J = 3.1 Hz). HRMS (ESI): m/z calcd for C23H20FN2O4 [M + H]+: 407.1402, found 407.1395. HPLC analysis: tR = 13.337 min, 96.5%.
4.1.71. 3-(2-Fluorophenyl)-5,6,7-trihydroxy-2-(1-phenylethyl)quinazolin-4(3H)-one (D3)
According to general procedure C, the product was obtained as a white solid (110 mg), yield 56%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.28 (s, 1H), 10.45 (s, 1H), 8.96 (s, 1H), 7.81–7.71 (m, 1H), 7.58–7.44 (m, 1H), 7.43–7.35 (m, 1H), 7.26–7.03 (m, 3H), 7.03–6.93 (m, 1H), 6.72 (s, 1H), 6.71–6.60 (m, 2H), 3.95 (q, J = 6.7 Hz, 1H), 1.47 (d, J = 6.8 Hz, 3H). 13C NMR (125 MHz, DMSO‑d 6) δ 164.4, 157.7 (d, J = 250.6 Hz), 154.5, 154.3, 146.7, 141.3, 140.2, 131.6 (d, J = 8.0 Hz), 131.5, 130.6, 128.5, 128.3, 126.8, 126.7, 125.0 (d, J = 3.5 Hz), 123.3 (d, J = 13.4 Hz), 116.1, 115.9, 103.4, 99.7, 43.5, 21.9. HRMS (ESI): m/z calcd for C22H18FN2O4 [M + H]+: 393. 1245, found 393. 1251. HPLC analysis: tR = 8.657 min, 96.6%.
4.1.72. 3-(4-Bromo-2-fluorophenyl)-5,6,7-trihydroxy-2-(1-phenylethyl)quinazolin-4(3H)-one (D4)
According to general procedure C, the product was obtained as a white solid (141 mg), yield 60%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.18 (s, 1H), 10.48 (s, 1H), 8.98 (s, 1H), 7.77 (t, J = 8.2 Hz, 1H), 7.63 (dd, J = 8.5, 2.1 Hz, 1H), 7.32 (dd, J = 9.3, 2.1 Hz, 1H), 7.17–7.09 (m, 3H), 6.74–6.68 (m, 3H), 3.95 (q, J = 6.7 Hz, 1H), 1.47 (d, J = 6.8 Hz, 3H). 13C NMR (125 MHz, DMSO‑d 6) δ 164.1, 158.6, 156.6, 154.3, 154.1, 146.7, 141.2, 140.0, 132.1, 131.5, 128.4, 128.3 (d, J = 3.5 Hz), 126.8, 126.7, 123.1–122.9 (m), 119.3 (d, J = 23.2 Hz), 103.5, 99.6, 43.6, 21.9. HRMS (ESI): m/z calcd for C22H17 79BrFN2O4 [M + H]+: 471.0350, found 471.0352. HPLC analysis: tR = 13.244 min, 99.4%.
4.1.73. 2-Benzyl-5,6,7-trihydroxy-3-phenylquinazolin-4(3H)-one (D10)
According to general procedure C, the product was obtained as a white solid (112 mg), yield 62%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.48 (s, 1H), 10.33 (s, 1H), 8.88 (s, 1H), 7.49–7.38 (m, 3H), 7.23–7.14 (m, 5H), 6.90–6.82 (m, 2H), 6.59 (s, 1H), 3.75 (s, 2H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.1, 154.0, 152.2, 146.7, 140.5, 136.0, 135.8, 131.2, 129.1, 129.0, 128.5, 128.2, 126.6, 102.8, 100.3, 41.3. HRMS (ESI): m/z calcd for C21H17N2O4 [M + H]+: 361.1183, found 361.1190. HPLC analysis: tR = 8.737 min, 95.8%.
4.1.74. 2-Benzyl-3-(4-fluoro-3-methylphenyl)-5,6,7-trihydroxyquinazolin-4(3H)-one (D11)
According to general procedure C, the product was obtained as a white solid (88 mg), yield 45%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.44 (s, 1H), 10.35 (s, 1H), 8.89 (s, 1H), 7.23–7.16 (m, 4H), 7.14–7.08 (m, 1H), 6.94 (dd, J = 7.0, 2.6 Hz, 1H), 6.88–6.83 (m, 2H), 6.60 (s, 1H), 3.77 (q, J = 15.5 Hz, 2H), 2.12 (d, J = 1.8 Hz, 3H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.6, 160.9 (d, J = 245.1 Hz), 154.5, 152.8, 147.2, 140.9, 136.2, 132.8 (d, J = 5.8 Hz), 132.1 (d, J = 3.2 Hz), 131.7, 128.9, 128.7 (d, J = 9.0 Hz), 128.6, 127.0, 125.6 (d, J = 18.8 Hz), 116.0 (d, J = 23.9 Hz), 103.3, 100.7, 42.0, 14.4 (d, J = 2.9 Hz). HRMS (ESI): m/z calcd for C25H18FN2O4 [M + H]+: 393.1245, found 393.1249. HPLC analysis: tR = 12.068 min, 96.2%.
4.1.75. General procedure D for the preparation of D5 and D12
To the stirred solution of the prepared 2-phenylpropanoyl chloride or commercially available phenylacetyl chloride (1 mmol) in pyridine (5 mL) was added TPP (816 mg, 2.5 mmol) under the N2 atmosphere at 0 °C, and the reaction mixture was heated to 70 °C and stirred for 2 h. Then the reaction mixture was cooled to room temperature, and was treated with isobutylamine (40) (0.12 mL, 1.2 mmol). Then the resulting mixture was heated to 70 °C and stirred overnight. After completion, the reaction mixture was cooled to room temperature, and the solvent was removed under reduced pressure to obtain the residue. The residue was then diluted with DCM (50 mL), washed with 3 N aqueous HCl solution (1 × 20 mL) and brine (3 × 20 mL), and dried anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica column chromatography (petroleum ether: EtOAc = 6:1) to give the corresponding quinazolinones 37a, 37b.
According to general procedure A, the demethylation of the above quinazolinones afforded the target compounds D5, D12.
4.1.76. 5,6,7-Trihydroxy-3-isobutyl-2-(1-phenylethyl)quinazolin-4(3H)-one (D5)
According to general procedure D, the product was obtained as a white solid (97 mg), yield 55%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.75 (s, 1H), 10.30 (s, 1H), 8.83 (s, 1H), 7.36–7.20 (m, 5H), 6.61 (s, 1H), 3.80 (d, J = 15.5 Hz, 1H), 3.73 (d, J = 15.5 Hz, 1H), 4.02–3.93 (m, 1H), 3.50 (dd, J = 14.0, 7.1 Hz, 1H), 2.04 (hept, J = 6.9 Hz, 1H), 1.56 (d, J = 6.7 Hz, 3H), 0.90 (d, J = 6.7 Hz, 3H), 0.82 (d, J = 6.6 Hz, 3H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.3, 155.1, 153.9, 146.3, 142.7, 139.9, 131.1, 128.9, 127.1, 126.9, 102.6, 100.0, 48.0, 42.3, 28.1, 22.5, 19.7, 19.6. HRMS (ESI): m/z calcd for C20H23N2O4 [M + H]+: 355.1652, found 355.1655. HPLC analysis: tR = 11.131 min, 96.2%.
4.1.77. 2-Benzyl-5,6,7-trihydroxy-3-isobutylquinazolin-4(3H)-one (D12)
According to general procedure D, the product was obtained as a white solid (95 mg), yield 56%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.74 (s, 1H), 10.30 (s, 1H), 8.84 (s, 1H), 7.36–7.30 (m, 2H), 7.28–7.22 (m, 3H), 6.52 (s, 1H), 4.18 (s, 2H), 3.77 (d, J = 7.5 Hz, 2H), 2.08 (hept, J = 7.0 Hz, 1H), 0.86 (d, J = 6.6 Hz, 6H). 13C NMR (125 MHz, DMSO‑d 6) δ 165.3, 153.9, 152.6, 146.3, 140.3, 136.2, 131.0, 128.7, 128.4, 126.8, 102.3, 100.1, 49.0, 40.6, 27.8, 19.7. HRMS (ESI): m/z calcd for C19H21N2O4 [M + H]+: 341.1496, found 341.1495. HPLC analysis: tR = 11.644 min, 97.8%.
4.2. SARS-CoV-2 Mpro protein expression and purification
The cDNA of SARS-CoV-2 Mpro (GeneBank: YP_009725301.1) was synthesized and cloned into pET-28a (+) vector with codon optimization by Sangon (Sangon Biotech, Shanghai, China). The SARS-CoV-2 Mpro construct contains protease autoprocessing site of Mpro (SAVLQ⬇SGFRK, arrow indicating the cleavage site) and PreScission site (SGVTFQ⬇GP, arrow indicating the cleavage site) in the N terminus and C terminus, respectively. The pET-28a(+)-SARS-CoV-2 M pro plasmid was then transformed into competent E.coli BL21 (DE3) cells and a single colony was picked, which was inoculated in 5 ml LB medium supplemented with 50 g/mL kanamycin and grown at 37 °C to an optical density at 600 nm of 0.8, and then induced using 0.5 mM isopropyl-β-d-thiogalactoside (IPTG). Induced cultures were grown at 25 °C for an additional 16 h. Cells were harvested via centrifugation at 10,000 rpm for 10 min and the precipitate were resuspended in a buffer (20 mM HEPES, pH 7.5, 500 mM NaCl, 20 mM imidazole) and homogenized with an ultrahigh-pressure cell disrupter at 4 °C. The insoluble material was removed by centrifugation at 18,000 rpm for 60 min. The protein was eluted by elution buffer (20 mM HEPES, pH 7.5, 500 mM NaCl, 300 mM imidazole) after incubation with Ni-resin and washed by buffer (20 mM HEPES, pH 7.5, 500 mM NaCl, 20 mM imidazole). After auto-cleaved and cleaved by PreScission protease at 4 °C overnight, native Mpro was generated. In order to obtain homogeneous and pure protein suitable for crystallization and enzymatic inhibition assays, the further purification using Q column was performed with A buffer (20 mM HEPES, pH 7.5, 50 mM NaCl) and B buffer (20 mM HEPES, pH 7.5, 1 M NaCl). Protein sample was concentrated into 10 mg/mL and stored at −80 °C.
4.3. SARS-CoV-2 Mpro enzymatic assays
The enzyme activity of SARS-CoV-2 Mpro was determined using a FRET assay, which was performed as previously reported [37]. The FRET assay uses fluorogenic peptide Dabcyl-KLSAVLQSGFRKM-Edans-NH2 as the substrate, which was custom synthesized and obtained from GL Biochem Ltd (Shanghai, China). In brief, protease SARSCov-2 Mpro (600 nM at a final concentration) was mixed with compounds at the indicated concentration in 50 μL of assay buffer (20 mM Tris-HCl, pH 7.5, 10 mM EDTA, 150 mM NaCl) and incubated at room temperature for 30 min. Then, the reaction was initiated by adding 50 μL of peptide substrate to the mixture (20 μM at a final concentration) and monitored at 340 nM (excitation)/490 nM (emission) with a Tecan's Spark multimode reader. The Vmax of reactions with compounds added at the indicated concentrations compared to the solvent control was calculated and used to generate IC50 curves. For each compound, the half-maximal inhibitory concentration (IC50) values against SARS-CoV-2 Mpro were measured at 10 different concentrations, and three independent experiments were performed. The data were analyzed using GraphPad Prism 8.4.3 software.
4.4. Cellular antiviral assays
A clinical isolate of SARS-CoV-2 (GenBank: MT121215.1) was propagated in Vero E6 cells and the viral titer was determined as 50% tissue culture infectious dose (TCID50) per milliliter (mL) by CPE quantification. All the infection experiments were performed at biosafety level-3 (BLS-3) as previously reported [38]. Pre-seeded Vero E6 cells (5 × 104 cells/well) were incubated with test compounds at the indicated concentrations at 37 °C in 5% CO2 for 1 h. Then, 100 μL of the cell supernatant was removed, followed by addition of the prepared virus-drug mixture (100 μL) to the final virus titer of 100 TCID50/well. After infection for 1 h, the supernatant medium was discarded, and the cells were washed twice with PBS and further cultured with maintenance medium at 37 °C in 5% CO2 for 48 h. At 48 h postinfection, the cell supernatant was collected and lysed using TRIzol LS Reagent (Invitrogen, Carlsbad, USA). Subsequently, RNA was extracted using a PureLink™ RNA Mini Kit (Thermo Fisher, Waltham, USA). The viral RNA copy number was quantified by RT-qPCR using Verso SYBR Green 1-Step qRT-PCR Kit (Thermo Fisher, Waltham, USA) on CFX96™ Real-Time PCR System (Bio-Rad, Hercules, CA) according to the manufacturer's instructions. The PCR primers targeting the N gene (nt608-706) of SARS-CoV-2 are as follows:
5′-GGGGAACTTCTCCTGCTAGAAT-3’;
5′-CAGACATTTTGCTCTCAAGCTG-3’.
4.5. SARS-CoV-2 Mpro protein crystallization and structure determination
8 mg/mL and 12 mg/mL SARS-CoV-2 Mpro in buffer (20 mM Tris-HCl, pH 7.5, 10 mM EDTA, 150 mM NaCl) were incubated with 2 mM compound D8 at 4 °C for 18 h. Then, the mixture was centrifuged at 12,500 rpm for 10 min to remove the precipitate, and the supernatant was used for crystallization. All crystallization experiments were set up at 293 K using the hanging drop approach where 1 μL of each precipitant solution was mixed with 1 μL of protein solution. Crystals were observed after a few days of incubation in 12% PEG 20,000, 0.1 M MES monohydrate (pH 6.5), which were next immersed in the crystallization solution containing 15% glycerol, and were frozen in liquid nitrogen before diffraction. The diffraction data sets were collected at the Shanghai Synchrotron Radiation Facility (SSRF, Shanghai, China) on beamline BL18U1. Using the molecular-replacement method, the structure was resolved with the Phenix program Phaser-MR [39,40], and the electron density could be clearly observed in the protein catalytic site. The eLBOW and LigandFit program in Phenix was used to fit the ligand D8 [41]. Next, adjustment of the structural model was performed in Coot, and structure refinement in Phenix was conducted with NCS restrains [42,43]. Finally, the MolProbity program in Phenix was used to check the quality of the resulting structure [44]. Data collection and structural refinement statistics are depicted in Table S3.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We thank Biosafety Level 3 Laboratory, Fudan University for providing the experiment platform of cellular antiviral assay. This work was supported by grants from the National Natural Science Foundation of China (No. 82161138005, 22107117 and 22001267), China Postdoctoral Science Foundation (No. 2021M693516), and Postdoctoral Research Program of Jiangsu Province (No. 2021K218B).
Handling Editor: Dr. Z Liu
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2023.115487.
Abbreviations
- CCK-8
cell counting kit-8
- COVID-19
Coronavirus Disease 2019
- DTT
dithiothreitol
- DMPK
drug metabolism and pharmacokinetics
- FRET
fluorescence resonance energy transfer
- HLM
human liver microsome
- IPTG
isopropyl-β-d-thiogalactoside
- Mpro
main protease
- nsp
non-structural protein
- PBS
phosphate buffer solution
- PPB
plasma protein binding
- SAR
structure activity relationship
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- TPP
triphenyl phosphite
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Supporting data to this article can be found online.
Data availability
Data will be made available on request.
References
- 1.WHO, WHO Coronavirus Disease (COVID-19) Dashboard. https://covid19.who.int/(accessed date: 2022.December.14).
- 2.Ledford H. Hundreds of COVID trials could provide a deluge of new drugs. Nature. 2022;603:25–27. doi: 10.1038/d41586-022-00562-0. [DOI] [PubMed] [Google Scholar]
- 3.Li Y., Tenchov R., Smoot J., Liu C., Watkins S., Zhou Q.A. A comprehensive review of the global efforts on COVID-19 vaccine development. ACS Cent. Sci. 2021;7:512–533. doi: 10.1021/acscentsci.1c00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lipsitch M., Krammer F., Regev-Yochay G., Lustig Y., Balicer R.D. SARS-CoV-2 breakthrough infections in vaccinated individuals: measurement, causes and impact. Nat. Rev. Immunol. 2022;22:57–65. doi: 10.1038/s41577-021-00662-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feikin D.R., Higdon M.M., Abu-Raddad L.J., Andrews N., Araos R., Goldberg Y., Groome M.J., Huppert A., O'Brien K.L., Smith P.G., Wilder-Smith A., Zeger S., Deloria Knoll M., Patel M.K. Duration of effectiveness of vaccines against SARS-CoV-2 infection and COVID-19 disease: results of a systematic review and meta-regression. Lancet (London, England) 2022;399:924–944. doi: 10.1016/S0140-6736(22)00152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ren S.Y., Wang W.B., Gao R.D., Zhou A.M. Omicron variant (B.1.1.529): infectivity, vaccine breakthrough, and antibody resistance. J. Chem. Inf. Model. 2022;10:1–11. doi: 10.1021/acs.jcim.1c01451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cele S., Jackson L., Khoury D.S., Khan K., Moyo-Gwete T., Tegally H., San J.E., Cromer D., Scheepers C., Amoako D.G., Karim F., Bernstein M., Lustig G., Archary D., Smith M., Ganga Y., Jule Z., Reedoy K., Hwa S.H., Giandhari J., Blackburn J.M., Gosnell B.I., Abdool Karim S.S., Hanekom W., von Gottberg A., Bhiman J.N., Lessells R.J., Moosa M.S., Davenport M.P., de Oliveira T., Moore P.L., Sigal A. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature. 2022;602:654–656.8. doi: 10.1038/s41586-021-04387-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8..
- 9.Cannalire R., Cerchia C., Beccari A.R., Di Leva F.S., Summa V. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: state of the art and future opportunities. J. Med. Chem. 2022;65:2716–2746. doi: 10.1021/acs.jmedchem.0c01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao K., Wang R., Chen J., Tepe J.J., Huang F., Wei G.W. Perspectives on SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2021;64:16922–16955. doi: 10.1021/acs.jmedchem.1c00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Unoh Y., Uehara S., Nakahara K., Nobori H., Yamatsu Y., Yamamoto S., Maruyama Y., Taoda Y., Kasamatsu K., Suto T., Kouki K., Nakahashi A., Kawashima S., Sanaki T., Toba S., Uemura K., Mizutare T., Ando S., Sasaki M., Orba Y., Sawa H., Sato A., Sato T., Kato T., Tachibana Y. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J. Med. Chem. 2022;65:6499–6512. doi: 10.1021/acs.jmedchem.2c00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drayman N., DeMarco J.K., Jones K.A., Azizi S.A., Froggatt H.M., Tan K., Maltseva N.I., Chen S., Nicolaescu V., Dvorkin S., Furlong K., Kathayat R.S., Firpo M.R., Mastrodomenico V., Bruce E.A., Schmidt M.M., Jedrzejczak R., Muñoz-Alía M., Schuster B., Nair V., Han K.Y., O'Brien A., Tomatsidou A., Meyer B., Vignuzzi M., Missiakas D., Botten J.W., Brooke C.B., Lee H., Baker S.C., Mounce B.C., Heaton N.S., Severson W.E., Palmer K.E., Dickinson B.C., Joachimiak A., Randall G., Tay S. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS-CoV-2. Science (New York, N.Y.) 2021;373:931–936. doi: 10.1126/science.abg5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Owen D.R., Allerton C.M.N., Anderson A.S., Aschenbrenner L., Avery M., Berritt S., Boras B., Cardin R.D., Carlo A., Coffman K.J., Dantonio A., Di L., Eng H., Ferre R., Gajiwala K.S., Gibson S.A., Greasley S.E., Hurst B.L., Kadar E.P., Kalgutkar A.S., Lee J.C., Lee J., Liu W., Mason S.W., Noell S., Novak J.J., Obach R.S., Ogilvie K., Patel N.C., Pettersson M., Rai D.K., Reese M.R., Sammons M.F., Sathish J.G., Singh R.S.P., Steppan C.M., Stewart A.E., Tuttle J.B., Updyke L., Verhoest P.R., Wei L., Yang Q., Zhu Y. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science (New York, N.Y.) 2021;374:1586–1593. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- 14.Hoffman R.L., Kania R.S., Brothers M.A., Davies J.F., Ferre R.A., Gajiwala K.S., He M., Hogan R.J., Kozminski K., Li L.Y., Lockner J.W., Lou J., Marra M.T., Mitchell L.J., Jr., Murray B.W., Nieman J.A., Noell S., Planken S.P., Rowe T., Ryan K., Smith G.J., 3rd, Solowiej J.E., Steppan C.M., Taggart B. Discovery of Ketone-based bovalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020;63:12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boras B., Jones R.M., Anson B.J., Arenson D., Aschenbrenner L., Bakowski M.A., Beutler N., Binder J., Chen E., Eng H., Hammond H., Hammond J., Haupt R.E., Hoffman R., Kadar E.P., Kania R., Kimoto E., Kirkpatrick M.G., Lanyon L., Lendy E.K., Lillis J.R., Logue J., Luthra S.A., Ma C., Mason S.W., McGrath M.E., Noell S., Obach R.S., Mn O.B., O'Connor R., Ogilvie K., Owen D., Pettersson M., Reese M.R., Rogers T.F., Rosales R., Rossulek M.I., Sathish J.G., Shirai N., Steppan C., Ticehurst M., Updyke L.W., Weston S., Zhu Y., White K.M., García-Sastre A., Wang J., Chatterjee A.K., Mesecar A.D., Frieman M.B., Anderson A.S., Allerton C. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun. 2021;12:6055. doi: 10.1038/s41467-021-26239-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma C., Sacco M.D., Hurst B., Townsend J.A., Hu Y., Szeto T., Zhang X., Tarbet B., Marty M.T., Chen Y., Wang J., Boceprevir G.C.- 376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qiao J., Li Y.S., Zeng R., Liu F.L., Luo R.H., Huang C., Wang Y.F., Zhang J., Quan B., Shen C., Mao X., Liu X., Sun W., Yang W., Ni X., Wang K., Xu L., Duan Z.L., Zou Q.C., Zhang H.L., Qu W., Long Y.H., Li M.H., Yang R.C., Liu X., You J., Zhou Y., Yao R., Li W.P., Liu J.M., Chen P., Liu Y., Lin G.F., Yang X., Zou J., Li L., Hu Y., Lu G.W., Li W.M., Wei Y.Q., Zheng Y.T., Lei J., Yang S. SARS-CoV-2 M(pro) inhibitors with antiviral activity in a transgenic mouse model. Science (New York, N.Y.) 2021;371:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai W., Zhang B., Jiang X.M., Su H., Li J., Zhao Y., Xie X., Jin Z., Peng J., Liu F., Li C., Li Y., Bai F., Wang H., Cheng X., Cen X., Hu S., Yang X., Wang J., Liu X., Xiao G., Jiang H., Rao Z., Zhang L.K., Xu Y., Yang H., Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science (New York, N.Y.) 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science (New York, N.Y.) 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han S.H., Goins C.M., Arya T., Shin W.J., Maw J., Hooper A., Sonawane D.P., Porter M.R., Bannister B.E., Crouch R.D., Lindsey A.A., Lakatos G., Martinez S.R., Alvarado J., Akers W.S., Wang N.S., Jung J.U., Macdonald J.D., Stauffer S.R. Structure-based optimization of ML300-derived, noncovalent inhibitors targeting the severe acute respiratory syndrome coronavirus 3CL protease (SARS-CoV-2 3CL(pro)) J. Med. Chem. 2022;65:2880–2904. doi: 10.1021/acs.jmedchem.1c00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kneller D.W., Li H., Galanie S., Phillips G., Labbé A., Weiss K.L., Zhang Q., Arnould M.A., Clyde A., Ma H., Ramanathan A., Jonsson C.B., Head M.S., Coates L., Louis J.M., Bonnesen P.V., Kovalevsky A. Structural, electronic, and electrostatic determinants for inhibitor binding to subsites S1 and S2 in SARS-CoV-2 main protease. J. Med. Chem. 2021;64:17366–17383. doi: 10.1021/acs.jmedchem.1c01475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Su H.X., Yao S., Zhao W.F., Li M.J., Liu J., Shang W.J., Xie H., Ke C.Q., Hu H.C., Gao M.N., Yu K.Q., Liu H., Shen J.S., Tang W., Zhang L.K., Xiao G.F., Ni L., Wang D.W., Zuo J.P., Jiang H.L., Bai F., Wu Y., Ye Y., Xu Y.C. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 2020;41:1167–1177. doi: 10.1038/s41401-020-0483-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitamura N., Sacco M.D., Ma C., Hu Y., Townsend J.A., Meng X., Zhang F., Zhang X., Ba M., Szeto T., Kukuljac A., Marty M.T., Schultz D., Cherry S., Xiang Y., Chen Y., Wang J. Expedited approach toward the rational design of noncovalent SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2022;65:2848–2865. doi: 10.1021/acs.jmedchem.1c00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luttens A., Gullberg H., Abdurakhmanov E., Vo D.D., Akaberi D., Talibov V.O., Nekhotiaeva N., Vangeel L., De Jonghe S., Jochmans D., Krambrich J., Tas A., Lundgren B., Gravenfors Y., Craig A.J., Atilaw Y., Sandström A., Moodie L.W.K., Lundkvist Å., van Hemert M.J., Neyts J., Lennerstrand J., Kihlberg J., Sandberg K., Danielson U.H., Carlsson J. Ultralarge virtual screening identifies SARS-CoV-2 main protease inhibitors with broad-spectrum activity against coronaviruses. J. Am. Chem. Soc. 2022;144:2905–2920. doi: 10.1021/jacs.1c08402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang C.H., Stone E.A., Deshmukh M., Ippolito J.A., Ghahremanpour M.M., Tirado-Rives J., Spasov K.A., Zhang S., Takeo Y., Kudalkar S.N., Liang Z., Isaacs F., Lindenbach B., Miller S.J., Anderson K.S., Jorgensen W.L. Potent noncovalent inhibitors of the main protease of SARS-CoV-2 from molecular sculpting of the drug perampanel guided by free energy perturbation calculations. ACS Cent. Sci. 2021;7:467–475. doi: 10.1021/acscentsci.1c00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukae H., Yotsuyanagi H., Ohmagari N., Doi Y., Imamura T., Sonoyama T., Fukuhara T., Ichihashi G., Sanaki T., Baba K., Takeda Y., Tsuge Y., Uehara T. A randomized phase 2/3 study of Ensitrelvir, a novel oral SARS-CoV-2 3C-Like protease inhibitor, in Japanese patients with mild-to-moderate COVID-19 or asymptomatic SARS-CoV-2 infection: results of the phase 2a part. Antimicrob. Agents Chemother. 2022;66 doi: 10.1128/aac.00697-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones C.P., Anderson K.W., Buchwald S.L. Sequential Cu-catalyzed amidation-base-mediated camps cyclization: a two-step synthesis of 2-aryl-4-quinolones from o-halophenones. J. Org. Chem. 2007;72:7968–7973. doi: 10.1021/jo701384n. [DOI] [PubMed] [Google Scholar]
- 28.Hadjeri M., Peiller E.L., Beney C., Deka N., Lawson M.A., Dumontet C., Boumendjel A. Antimitotic activity of 5-hydroxy-7-methoxy-2-phenyl-4-quinolones. J. Med. Chem. 2004;47:4964–4970. doi: 10.1021/jm049876x. [DOI] [PubMed] [Google Scholar]
- 29.Carreras J., Gopakumar G., Gu L., Gimeno A., Linowski P., Petuškova J., Thiel W., Alcarazo M. Polycationic ligands in gold catalysis: synthesis and applications of extremely π-acidic catalysts. J. Am. Chem. Soc. 2013;135:18815–18823. doi: 10.1021/ja411146x. [DOI] [PubMed] [Google Scholar]
- 30.Liu X., Fu H., Jiang Y., Zhao Y. A simple and efficient approach to quinazolinones under mild copper-catalyzed conditions. Angew. Chem., Int. Ed. Engl. 2009;48:348–351. doi: 10.1002/anie.200804675. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y., Jin L., Xiang H., Wu J., Wang P., Hu D., Xue W., Yang S. Synthesis and anticancer activities of 5,6,7-trimethoxy-N-phenyl(ethyl)-4-aminoquinazoline derivatives. Eur. J. Med. Chem. 2013;66:335–344. doi: 10.1016/j.ejmech.2013.05.043. [DOI] [PubMed] [Google Scholar]
- 32.Tian X., Song L., Li E., Wang Q., Yu W., Chang J. Metal-free one-pot synthesis of 1,3-diazaheterocyclic compounds via I2-mediated oxidative C–N bond formation. RSC Adv. 2015;5:62194–62201. [Google Scholar]
- 33.Xue S., McKenna J., Shieh W.C., Repic O. A facile synthesis of C2,N3-disubstituted-4-quinazolone. J. Org. Chem. 2004;69:6474–6477. doi: 10.1021/jo049118e. [DOI] [PubMed] [Google Scholar]
- 34.Liu K., Li D., Zheng W., Shi M., Chen Y., Tang M., Yang T., Zhao M., Deng D., Zhang C., Liu J., Yuan X., Yang Z., Chen L. Discovery, optimization, and evaluation of quinazolinone derivatives with novel linkers as orally efficacious phosphoinositide-3-kinase delta inhibitors for treatment of inflammatory diseases. J. Med. Chem. 2021;64:8951–8970. doi: 10.1021/acs.jmedchem.1c00004. [DOI] [PubMed] [Google Scholar]
- 35.Dahlin J.L., Nissink J.W., Strasser J.M., Francis S., Higgins L., Zhou H., Zhang Z., Walters M.A. PAINS in the assay: chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J. Med. Chem. 2015;58:2091–2113. doi: 10.1021/jm5019093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quan B.X., Shuai H., Xia A.J., Hou Y., Zeng R., Liu X.L., Lin G.F., Qiao J.X., Li W.P., Wang F.L., Wang K., Zhou R.J., Yuen T.T., Chen M.X., Yoon C., Wu M., Zhang S.Y., Huang C., Wang Y.F., Yang W., Tian C., Li W.M., Wei Y.Q., Yuen K.Y., Chan J.F., Lei J., Chu H., Yang S. An orally available M(pro) inhibitor is effective against wild-type SARS-CoV-2 and variants including Omicron. Nat. Microbiol. 2022;7:716–725. doi: 10.1038/s41564-022-01119-7. [DOI] [PubMed] [Google Scholar]
- 37.Yu W., Zhao Y., Ye H., Wu N., Liao Y., Chen N., Li Z., Wan N., Hao H., Yan H., Xiao Y., Lai M. Structure-based design of a dual-targeted covalent inhibitor against papain-like and main proteases of SARS-CoV-2. J. Med. Chem. 2022;65:16252–16267. doi: 10.1021/acs.jmedchem.2c00954. [DOI] [PubMed] [Google Scholar]
- 38.Ni Y., Liao J., Qian Z., Wu C., Zhang X., Zhang J., Xie Y., Jiang S. Synthesis and evaluation of enantiomers of hydroxychloroquine against SARS-CoV-2 in vitro. Bioorg. Med. Chem. 2022;53 doi: 10.1016/j.bmc.2021.116523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., McCoy A.J., Moriarty N.W., Oeffner R., Read R.J., Richardson D.C., Richardson J.S., Terwilliger T.C., Zwart P.H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moriarty N.W., Grosse-Kunstleve R.W., Adams P.D. Electronic ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D. Towards automated crystallographic structure refinement with phenix. refine. Acta Crystallogr. D Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B., Jain S., Lewis S.M., Arendall W.B., 3rd, Snoeyink J., Adams P.D., Lovell S.C., Richardson J.S., Richardson D.C. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 2018;27:293–315. doi: 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.