

Abstract
Cellular senescence increases with aging. While senescence is associated with an exit of the cell cycle, there is ample evidence that post-mitotic cells including neurons can undergo senescence as the brain ages, and that senescence likely contributes significantly to the progression of neurodegenerative diseases (ND) such as Alzheimer’s Disease (AD) and Amyotrophic Lateral Sclerosis (ALS). Stress granules (SGs) are stress-induced cytoplasmic biomolecular condensates of RNA and proteins, which have been linked to the development of AD and ALS. The SG seeding hypothesis of NDs proposes that chronic stress in aging neurons results in static SGs that progress into pathological aggregates Alterations in SG dynamics have also been linked to senescence, though studies that link SGs and senescence in the context of NDs and the aging brain have not yet been performed. In this Review, we summarize the literature on senescence, and explore the contribution of senescence to the aging brain. We describe senescence phenotypes in aging neurons and glia, and their links to neuroinflammation and the development of AD and ALS. We further examine the relationships of SGs to senescence and to ND. We propose a new hypothesis that neuronal senescence may contribute to the mechanism of SG seeding in ND by altering SG dynamics in aged cells, thereby providing additional aggregation opportunities within aged neurons.
Keywords: stress granules, senescence, neuronal aging, neurodegeneration, Alzheimer’s Disease, Amyotrophic Lateral Sclerosis (ALS), neuroinflammation, senescence-associated secretory phenotype (SASP)
1. Introduction
Cellular senescence is an irreversible state characterized most obviously by cell cycle suppression (Collado et al., 2007). Cellular senescence increases with aging cells, and can be triggered by multiple factors associated with aging including shortened telomeres, DNA damage, oncogenic stress, chromatin perturbation, oxidative stress, reactive oxygen species (ROS), and other stress conditions (Collado et al., 2007; Serrano et al., 2001; Sikora et al., 2021). While senescence is most often associated with an exit of the cell cycle, there is now ample evidence that post-mitotic cells – including neurons – can express other hallmarks of senescence as the brain ages (Sah et al., 2021), and that these senescence phenotypes contribute to age-related disease (Martínez-Cué et al., 2020).
Stress granules (SGs) are stress-induced cytoplasmic biomolecular condensates of RNA and proteins (Hofmann et al., 2021). SGs are generally thought to be a cytoprotective response for surviving brief acute stresses (Arimoto et al., 2008), though chronic and pathogenic SGs have been linked to the development of a variety of disease states. Notably, SGs are hypothesized to seed or exacerbate pathogenic protein aggregation in an array of neurodegenerative diseases (Advani et al., 2020). Alteration in SG dynamics has also been linked to the onset of cellular senescence (Al-Rushadi, 2022; Chatterjee et al., 2022), though studies that link SGs and senescence in neurons have not yet been performed.
In this review, we explore the current state of understanding of the contribution of senescence to the aging brain (Figure 1). We describe senescence phenotypes in aging neurons and glia, and their links to neuroinflammation and the development of neurodegenerative disease (ND), with a focus on Alzheimer’s Disease (AD) and Amyotrophic Lateral Sclerosis (ALS). We further examine the known relationships of SGs to senescence and ND, and propose a hypothesis that integrates both SG formation and senescence in the pathogenesis of ND.
Figure 1. Aging connects neuroinflammation, senescence, SGs formation, and NDs.
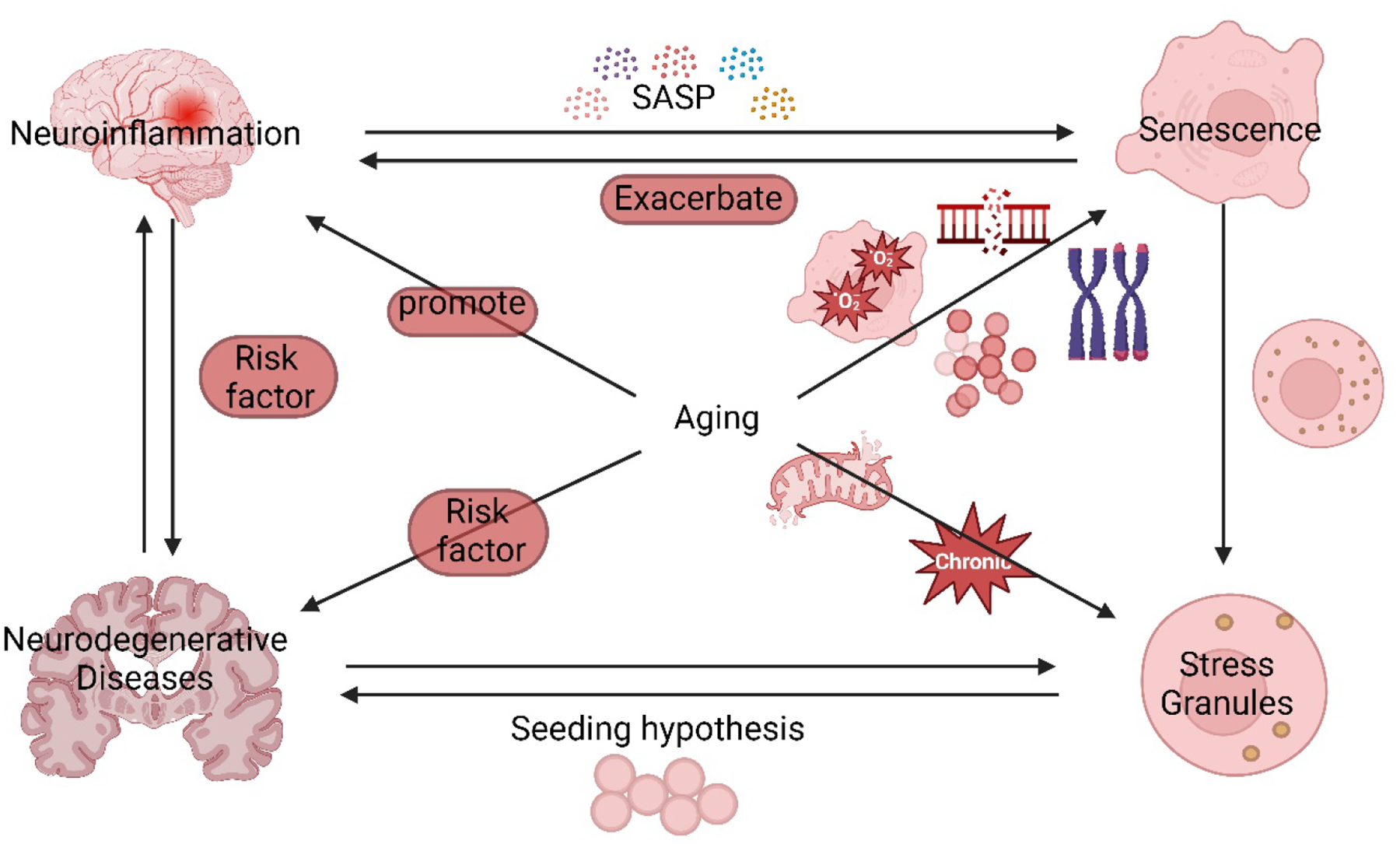
Aging is a central factor that connects neuroinflammation, senescence, SGs, and ND. Shortened telomeres, oncogenes, DNA damage accumulation, and other factors lead to senescence during aging. Both neuroinflammation and aging are major risk factors of neurodegenerative diseases. Aging promotes SGs formation by dysfunctional mitochondria, presence of chronic stress, and decline in protein homeostasis. SGs promote NDs by seeding hypothesis which causes aggregation of pathological proteins such as TDP-43 and tau. Accumulation of senescent cells during aging promotes neuroinflammation at the early stage by secreting SASP, and SASP further aggravates senescence. The senescent cells form smaller but more numerous SGs which implies impairments in the SGs formation pathway. See text for more information.
2. Senescence
Senescence in mitotic somatic cells is characterized by several key hallmarks, the details of which are described in Table 1: (1) enhanced senescence - associated β-galactosidase (SA-β-gal) activity (Al-Rushadi, 2022), (2) flat and enlarged cell phenotype, (3) blebbing and dysmorphic nuclei (Campisi et al., 2007), (4) cell cycle arrest, (5) DNA damage, (6) increased nuclear lamin A abundance (Moujaber et al., 2017) (7) elevated expression of p16INK4A and hypophosphorylated RB (Sharpless et al., 2015), (8) senescence-associated secretory phenotype (SASP), and (9) senescence-associated heterochromatic foci (SAHFs). Senescence is proposed to be an anti-cancer or tumor suppressive mechanism since it restricts cells from indefinite proliferation, which is a hallmark of cancer cells (Campisi et al., 2007). Senescence also limits tissue regenerative capacity and is increased in aging tissues (Campisi, 2001). Because it can be beneficial to young organisms as a cancer suppressor, but harmful to old organisms as a contributor to aging, senescence is said to be a mechanism of antagonistic pleiotropy (Kirkwood et al., 2000). Thus, senescence is a complicated and context-dependent process that can impact organisms throughout their lifecycles.
Table 1.
Comparison of senescence phenotypes in mitotic and post-mitotic cells.
| Senescence phenotypes in mitotic cells | Senescence phenotypes in post-mitotic neurons | Description of Senescence Phenotypes |
|---|---|---|
| enhanced senescence - associated β-galactosidase (SA-β-GAL) activity | ✓ | The enzyme β-galactosidase is a lysosomal hydrolases which cleaves off the terminal β-galactose from the compounds (Morgunova et al., 2015). β-galactosidase expression increases with aging both in vitro and in vivo (Sharpless et al., 2015). |
| flat and enlarged cell phenotype | ✕ | Increase in cell size limits cell proliferation and contributes to the permanent cell-cycle arrest (Neurohr et al., 2019). The cell cycle arrest further causes the increase in cell size and promotes senescence. |
| Cell cycle arrest | ✓* | P53-P21 pathway and P16 pathway causes RB hypophosphorylation which sequesters E2F and causes cell arrest in G1 phase (Fielder et al., 2017). |
| DNA damage | ✓ | DNA damage activates P53-P21 pathway which leads to cell cycle arrest (Fielder et al., 2017). |
| increased nuclear lamin A abundance | ✕ | The amount of lamin A increases during senescence (Moujaber et al., 2017). The aberrant form of lamin A leads to nuclear blebbing, DNA damage accumulation, and senescence aggravation (Tran et al., 2016). |
| blebbing and dysmorphic nuclei | ✕ | Senescent cells change their nuclear morphology (Freyter et al., 2022). The nuclear envelop plays a key role in regulating chromatin organization regulation and nuclear activity. |
| elevated expression of hypophosphorylated RB | ✓ | Hypophosphorylated RB causes cell arrest in the G1 phase (Sharpless et al., 2015). |
| senescence-associated secretory phenotype (SASP) | ✓ | SASP describes increased secretion of cytokines, chemokines, growth factors, and remodeling proteases (LeBrasseur et al., 2015). SASP activates an innate immune response to clear senescent cells, and failure in clearance leads to senescent cell accumulation (Saez-Atienzar et al., 2020). SASP aggravates the senescent phenotype and exacerbates senescence by spreading senescence from adjacent mitotic cells to neighboring neurons (Guerrero et al., 2021; McHugh et al., 2017) |
| senescence-associated heterochromatic foci (SAHFs). | ✕ | The SAHFs promote cell cycle arrest by inhibiting the expression of genes which promote proliferation, such as E2F target genes which plays crucial role in G1 to S phase transition (Funayama et al., 2007). |
✓ indicates there is evidence that this phenotype occurs in neurons.
✕ indicates there is no evidence that this phenotype occurs in neurons.
Note: Cell cycle arrest in G0 is a normal feature of terminally differentiated post-mitotic neurons and is not in and of itself a senescence phenotype in this context.
2.1. Mechanisms of Senescence in Mitotic Cells
Much is known about the mechanism of senescence induction in mitotic cells, which is closely related to the mechanism of cell cycle progression (Figure 2). The cell cycle consists of several phases: the G0 phase where quiescent cells reside, the G1 phase which prepares the cell for DNA replication in S phase, and then the G2 phase which primes cells for mitosis (M phase) (Fielder et al., 2017). Entry from G0 to G1 is promoted by the expression of cyclin D. Cyclin D and CDK4/6 form a complex that results in pRb-E2F phosphorylation (Fielder et al., 2017). In actively cycling mammalian cells, CDK4, CDK6, and CDK2 phosphorylate proteins from the retinoblastoma family including RB1, p107 (RBL1), and p130 (RBL2) inG1 phase (Rizzolio et al., 2012; Vassilis Gorgoulis et al., 2019). Phosphorylation restricts the capability of RB family members to repress E2F transcription factor activity, which is crucial in cell-cycle progression (Sharpless et al., 2015). During senescence, this molecular mechanism is interrupted by various molecular triggers including genome instability, telomere dysfunction, epigenetic alterations, proteostatic stress, and compromised mitochondrial function (Schumacher et al., 2021).
Figure 2. Mechanisms of senescence.
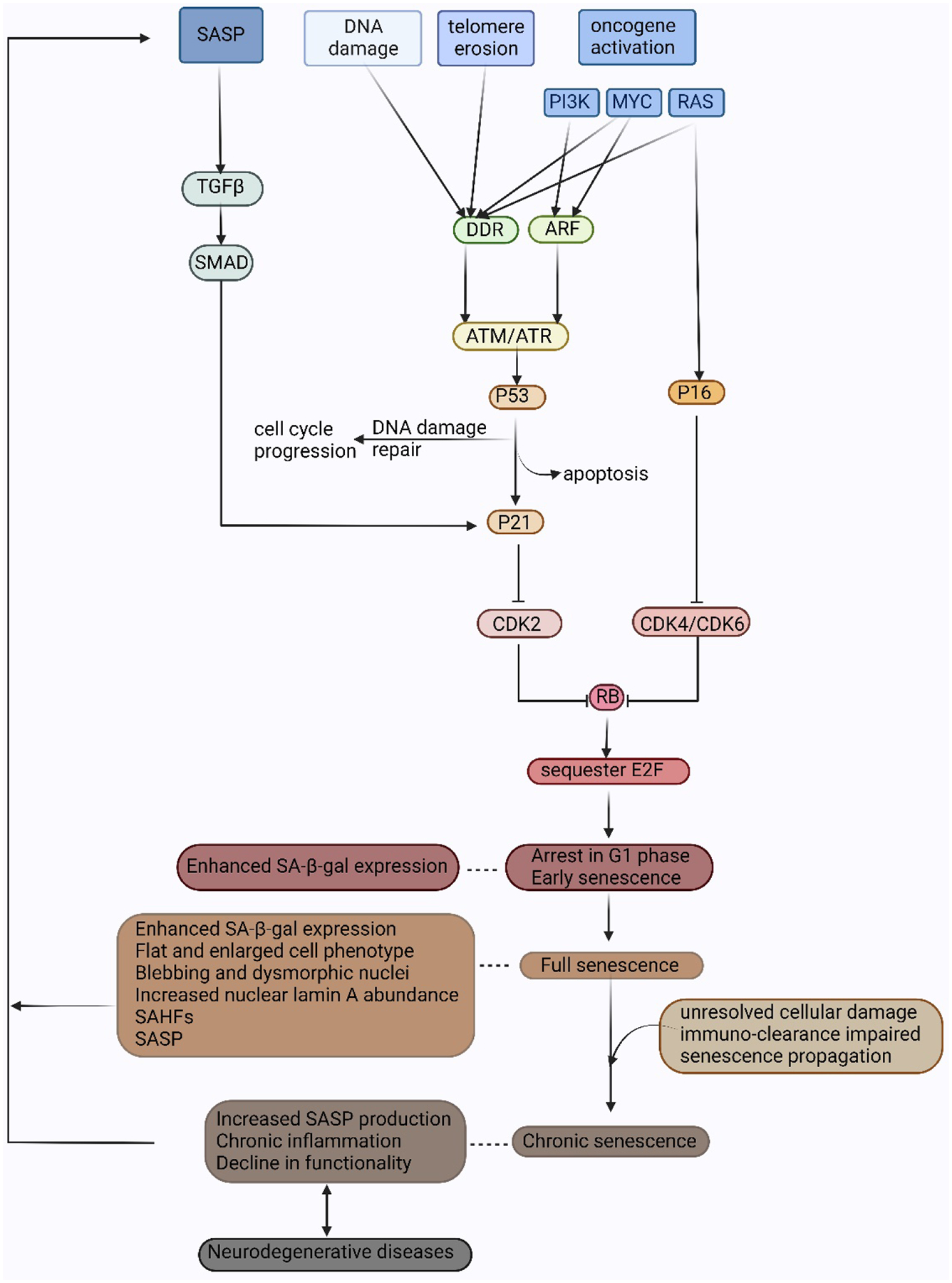
Senescence can be induced by DNA damage, telomere erosion, and oncogene activation. DNA damage, telomere erosion, and activation of oncogenes MYC and RAS lead to activation of DDR. Activation of PI3K and MYC also induces ARF transcription which leads to senescence via the p53 pathway. Activated ATM/ATR kinase results in p53 induction. Successful DNA damage repair allows cell cycle progression. Failure in DNA damage repair leads to either apoptosis or senescence by p53-induced p21 transcription. P21 inhibits the cyclinE-CDK2 complex and causes RB to be maintained in a hypophosphorylated state. Activation of RAS also mediates P16INK4A induction which inhibits cyclin D-CDK4/CDK6 complex and RB hyperphosphorylation. Hypophosphorylated RB binds and sequesters transcription factor E2F and causes cell cycle arrest in the G1 phase. This early step in senescence results in senescence-associated β-galactosidase activity. Later events include morphological effects, SAFH, and SASP. Increasing SASP in chronic senescence can trigger senescence in neighboring cells. The exact mechanism of how post-mitotic neurons undergo senescence is unknown, though increased SASP, neuroinflammation, and declining cell function attributable to senescence are believed to play a role in neurodegeneration. Adapted from Saez-Atienzar and Masliah, 2020.
While senescence can be induced by a variety of stresses, a common trigger for senescence is the activation of the DNA damage response (DDR) (Di Micco et al., 2006; Schumacher et al., 2021). In senescent cells, the DDR consists of DNA damage detection and amplification of activities of upstream sensor kinases Ataxia Telangiectasia Mutated (ATM) and Rad3-related (ATR), the signal cascade via downstream kinases CHK2 and CHK1, and effector proteins such as p53 and CDC25 (d’Adda di Fagagna, 2008). DDR-positive telomeres sequester TRF2, a telomeric protein that prevents chromosome end fusion as well as suppresses DNA repair, and further results in persistent DDR (Salama et al., 2014). Once the telomere reaches the uncapped state, TRF2 will be lost, which causes interchromosomal or intrachromosomal fusion as well as an increase in genomic instability and cell death (Salama et al., 2014). DDR activates ATM and ATR to recognize and phosphorylate H2AX histone at DNA breaks (Firsanov et al., 2011). ATM is the major response mechanism for double-strand breaks (DSBs) (Herbig et al., 2004). ATR plays a secondary role but is the major modulator of responses to ultraviolet (UV) radiation damage and replication fork stalling (Herbig et al., 2004). ATM/ATR further phosphorylates p53 which stimulates the transcription of p21 and leads to reduced activities of CDK2 and CDK4 (Herbig et al., 2004), activation of RB family proteins (Vassilis Gorgoulis, 2019), inhibition of E2F transactivation, and consequent cell-cycle arrest. The high level of p21 protein arrests cells inG1 phase (Al-Rushadi, 2022; Fielder et al., 2017). Once the p21 protein level declines, p16 protein levels become upregulated, and cell proliferation is halted (Al-Rushadi, 2022). p16 prevents p16-retinoblastoma protein (pRB) phosphorylation and inactivation (Campisi et al., 2007) which slows the G1 phase to the S phase transition (Al-Rushadi, 2022; Rayess et al., 2012). pRB blocks proliferation by inhibiting the activity of E2F. The persistence of RB protein activation is enhanced by E2F target gene heterochromatinization, cytokine secretion, and ROS production (Salama et al., 2014). Senescence is induced by the activation of the p53-p21 pathway and maintained by elevated expression of p16 which functions as an additional barrier to cell proliferation (Herbig et al., 2004; Rayess et al., 2012).
2.1.1. Classification of Senescence Mechanisms
Since senescence can be induced by multiple reagents and can activate different pathways, the classification of senescence mechanisms has remained a challenge. Currently, there are two major ways to classify senescence: first, by the nature of the damage that induces the senescence, and second, by the kinetics (chronic or acute) of the senescence inducer. Under the first method of classification, there are 3 major categories of senescence: telomere-initiated cellular senescence (also known as replicative senescence), stress-induced premature senescence (SIPS), and oncogene-induced senescence (Plygawko, 2016).
Telomere-initiated cellular senescence
Telomere-initiated cellular senescence, also known as replicative senescence, is a progressive process caused by gradual base pair loss during the S phase, and consequent exposure and destabilization of telomeric DNA ends (Beck et al., 2020; Harley et al., 1990). The DNA damage associated with telomeric shortening foci activates the DDR, which in turn activates p53 and induces cell cycle inhibitor p21, which triggers telomere-initiated cellular senescence (Beck et al., 2020; Campisi et al., 2007; Collado et al., 2007; Salama et al., 2014).
Stress-induced premature senescence (SIPS)
Stress-induced premature senescence (SIPS) requires a shorter timescale for cell cycle arrest and can be induced by a telomere-independent pathway (Al-Rushadi, 2022; Campisi et al., 2007; von Kobbe, 2018). Instead of being driven by the progressive shortening of the telomere, SIPS is a rapid senescence phenotype also induced by DDR. SIPS can be accelerated by various physiological and pathological acute stresses including exposure to ionizing radiation (Bakkenist et al., 2015; Munro et al., 2004; Wiley et al., 2016), mitochondrial dysfunction (Al-Rushadi, 2022; Saez-Atienzar et al., 2020; Wiley et al., 2016), genotoxic drugs, oncogenes, and age-associated oxidative stress (Beck et al., 2020).
Oncogene-mediated senescence
Oncogene-mediated senescence is a telomere-independent tumor suppressor mechanism in the RAS-activated pathway, Myc pathway, and PI3K pathway (Rayess et al., 2012; Salama et al., 2014; Wu et al., 2007). Oncogene-induced replication stress is an early promotor of genomic instability and is ascribed to multiple factors including reactive oxygen species (ROS), replication-transcription collision, and defective nucleotide metabolism (Kotsantis et al., 2018). Activated oncogenes contribute to genomic instability and promote tumorigenesis (Kotsantis et al., 2018). Since oncogenes that induce senescence boost cell proliferation, senescence may be a way to offset the excessive mitogenic stimulation that places cells at risk of potential oncogenic transformation (Campisi et al., 2007).
Senescent cells can alternatively be classified into acute senescence, chronic senescence, or a combination based on the kinetics of senescence induction (Mas-Bargues et al., 2021; van Deursen, 2014). Acute senescence is induced by acute extrinsic stimuli that target specific cells and produce cell-specific SASP factors with concrete functions (Mas-Bargues et al., 2021; van Deursen, 2014). Acutely senescent cells generate persistent DDR as a rapid response to excessive genomic damage (Roninson, 2003). In this case, SASP activation triggers an innate immune response to eliminate senescent cells (Saez-Atienzar et al., 2020). Chronic senescence is induced by non-cell-type-specific, gradually increasing stress or macromolecular damage, which causes stable cell cycle arrest and contributes to aging-related compromises in cellular function (Martínez-Cué et al., 2020; Mas-Bargues et al., 2021; van Deursen, 2014). Due to age-related immunodeficiency and/or decreased SASP production, immune cells may inefficiently eliminate chronically senescent cells, which allows senescent cell accumulation (van Deursen, 2014). Chronic senescence includes telomere-initiated senescence (also named replicative senescence), stress-induced premature senescence (SIPS), and mitochondrial dysfunction-associated senescence (Kuilman et al., 2010). Chemotherapy or radiation-induced senescence during cancer therapy is thought to be a combination of acute and chronic senescence (Allan et al., 2005).
2.2. Senescence in the Brain
Cellular senescence is not confined to mitotic cells only. Under stress conditions, non-dividing cells can also display many of the hallmarks of senescence (Table 1) (Jacome Burbano et al., 2020). Both senescent cells and post-mitotic cells are permanently blocked from re-entering the cell cycle (Campisi et al., 2007). The mechanisms of post-mitotic cells re-entering the cell cycle under rare circumstances are poorly understood; however, they may share a few common mechanisms with senescence. The majority of current knowledge on senescence comes from mitotic cells (Jacome Burbano et al., 2020), and the relationship between senescence in mitotic cells and post-mitotic cells is only just beginning to be elucidated. Whether neurons and other post-mitotic cells should even be called “senescent” is controversial; some articles refer to “senescent neurons”, and others refer to “senescent-like neurons” to differentiate post-mitotic neurons from mitotic cells. While we acknowledge the ongoing debate, we will refer to neurons as senescent for simplicity throughout this review. Regardless of whether or not neurons can be properly considered as senescent, clear connections have been made between hallmark phenotypes of senescence and ND. Therefore, understanding cellular senescence could provide new avenues for slowing brain aging and ND disease progression.
2.2.1. Senescence in Post-Mitotic Neurons
Neurons are classified as long-lived post-mitotic cells (LLPMCs) which depend on intracellular repair and renewal capacities, such as autophagy and mitophagy (Jacome Burbano et al., 2020; Rao, 2009). The hallmarks of aging in neurons include compromised DNA repair (van Deursen, 2014), intracellular accumulation of oxidatively damaged proteins (López-Otín et al., 2013), nucleic acids, and lipids, dysregulated energy metabolism, impaired cellular “waste disposal” mechanisms (autophagy, lysosomal degradation, and proteasome function), impaired adaptive stress response signaling, dysregulated neuronal Ca2+ homeostasis, stem cell exhaustion, inflammation, a decline in axonal transport (Jacome Burbano et al., 2020), focal accumulation of cytoplasmic and membrane proteins, mitochondrial dysfunction, and synaptic transmission alteration. The persistence of senescent neurons is believed to also alter tissue organization (Moreno-Blas et al., 2019). As indicated by proliferation markers, post-mitotic cells’ re-entry from G0 into G1 is directly related to neuronal dysfunction and disease, and such re-entry makes post-mitotic cells susceptible to apoptosis or senescence (Sah et al., 2021). In AD, about 20% of post-mitotic neurons re-enter the cell cycle via DNA content variation and cyclin B expression (Fischer et al., 2012). However, whether proliferation markers could be applied to a pre-senescence phase in post-mitotic neurons is poorly understood due to the difficulty of distinguishing quiescent cells from senescent cells (Sah et al., 2021).
While cell cycle arrest cannot be used to identify senescence in post-mitotic cells, several other common senescence hallmarks have been identified in neurons (Table 1). One difficulty in studying the role of senescent cells in brain aging and neurodegenerative disorders (NDs) is the absence of tools for identifying, isolating, and specifically targeting or eliminating senescent cells (Baker et al., 2018). Currently, senescent neurons are detected based on a mix of markers including activation of p16INK4A, p21, SA β-gal, ROS, and senescence-associated heterochromatin foci (SAHF) (Baker et al., 2018; Herdy et al., 2022). However, other processes, such as inflammation and DNA damage unrelated to senescence, can induce phenotypes similar to senescence. Thus, the absence of unique markers makes it an ongoing challenge to investigate the contribution of senescence in aging and NDs.
Senescence is implicated to contribute to brain aging in three specific ways (LeBrasseur et al., 2015). First, senescent cell accumulation alters tissue function (Mas-Bargues et al., 2021). Senescence-induced chromatin reorganization causes heterochromatin formation, cell enlargement, and changes in organelle and cell morphologies (Rodier et al., 2011). The brain is highly vulnerable to oxidative stress due to the large production of reactive oxygen species (ROS) during cellular metabolism, and the presence of a large quantity of polyunsaturated fatty acids as well as a small amount of antioxidants results in increased ROS (Chinta et al., 2013; Vicencio et al., 2008). Accumulation of stress, such as ROS, protein aggregates, and DNA damage, boosts senescence and apoptosis in post-mitotic cells, thereby decreasing the functional capacity of the tissue (Sah et al., 2021). Second, senescence limits tissue regeneration capacity (Guerrero et al., 2021; LeBrasseur et al., 2015; Mas-Bargues et al., 2021). This limited regenerative capacity includes limited replacement of mitotic glia, as well as limited internal regenerative capacity (i.e. autophagy, mitophagy) of LLPMCs. Third, excess activation of the senescence-associated secretory phenotype (SASP), which triggers secretion of cytokines, chemokines, growth factors, and matrix remodeling proteases, causes chronic inflammation and tissue dysfunction by spreading senescence from adjacent mitotic cells to neighboring neurons (Kang, 2019; LeBrasseur et al., 2015; Mas-Bargues et al., 2021). Thus, the matrix of contributions of senescence to impaired brain function is highly complex. However, the exact relationship between neuron functional decline and senescence is poorly understood (Jacome Burbano et al., 2020).
2.2.2. Senescence in Glia
Proliferating glial cells, such as astrocytes, oligodendrocytes, and microglia, are an important source of inflammation in the aging brain and provide structural and metabolic support to neurons (Allen et al., 2009; Chinta et al., 2013). The brain endures modest chronic inflammation during aging due to the dysregulation of glial cell activation, which causes the release of proinflammatory cytokines (Martínez-Cué et al., 2020). Although the mechanisms of senescence in the brain and how non-neuronal cells potentially affect brain functions remain unclear, evidence of cellular senescence has been detected in some glial cell types (Chinta et al., 2013). Senescence causes aging-related inflammation in glial cells in the brain which affects adjacent neurons by chronic production of pro-inflammatory agents, such as ROS and SASP (Chinta et al., 2013).
Astrocytes are the most predominant and proliferative glial cells. Senescent astrocytes are defined by increased SA-β-gal expression, flat cell morphology, increased p21 and p16INK4a production, senescence-associated heterochromatin foci (SAHF) development, and increased cytokine expression (Chinta et al., 2013; Ungerleider et al., 2021). Senescent astrocytes build up non-cell-autonomous neurotoxicity through SASPs when co-cultured with mature neurons, motor neurons, neural progenitor cells (NPCs), or neural stem cells (NSCs) (Ungerleider et al., 2021). Inhibiting senescence in astrocytes reduces SASPs and prevents astrocyte-mediated neurotoxicity in vitro (Ungerleider et al., 2021). SIPS in irradiated astrocytes showed an increase in DNA damage and SASPs production (Ungerleider et al., 2021). Tau induces senescence in human astrocytes, and astrocytes acquire SASPs in response to extracellular tau (Ungerleider et al., 2021). Thus, there is significant evidence from in vitro experiments of a relationship between glial senescence and neurotoxicity, though the relationship in the brain in situ is less clear.
Other glial cells have also been documented to show senescence phenotypes (Baker et al., 2018; Martínez-Cué et al., 2020). Microglia are resident macrophages in the central nervous system (CNS), and senescent microglia showed increased SA-β-gal expression (Yu et al., 2012), growth arrest, SAHF development, shortened telomeres (Flanary et al., 2007), and increased inflammatory factors (Sierra et al., 2007). Oligodendrocytes have also been shown to display increased SA-β-gal expression upon aging (Al-Mashhadi et al., 2015). Thus, the contribution of glial cells to neurotoxic phenotypes in the aging brain may arise from a variety of cellular sources.
3. Neuronal Senescence and Neurodegeneration
Aging is the single greatest risk factor for the most frequent neurodegenerative diseases (NDs) including Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) (Sah et al., 2021). NDs are particularly devastating – and their therapies severely limited – due to the restricted regenerative capacity of the aging brain (Sah et al., 2021). Senescent cells are considered to be an ND contributor by favoring or exacerbating protein aggregation (Walker et al., 2015), oxidative stress (Jurk et al., 2012), and inflammatory phenotypes associated with various NDs (Lye et al., 2019). For example, SASP is suspected to promote neurodegeneration progression through a paracrine effect, activating further senescence in adjacent glia – and vice versa – and inducing apoptosis or senescence in neuronal cells (Ungerleider et al., 2021). In response to DNA damage, neurons can re-enter the cell cycle and undergo apoptosis, which contributes to neuronal loss in neurodegenerative disease (Guerrero et al., 2021). However, some neurons develop a senescence-like phenotype rather than activating apoptosis (Jurk et al., 2012), thereby increasing the number of senescence-like neurons as aging progresses (Herdy et al., 2022). Undergoing senescence instead of apoptosis promotes inflammation but preserves cell numbers with limited functionality and regenerative capacity (Sapieha et al., 2018). Thus, senescence may also play a role in prolonging brain function at a limited capacity. While many observations have linked NDs with senescent phenotypes, it remains unclear whether senescence is a cause or a consequence of aging and neurodegeneration (Saez-Atienzar et al., 2020). We will focus here on the strongest links between ND and neuronal senescence, which have been studied in the contexts of AD and ALS.
3.1. Senescence and Alzheimer’s Disease
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder, which is accompanied by the death of neurons and loss of synapses (Asadi et al., 2021). Cellular and molecular hallmarks of AD include intracellular microtubule tau protein aggregation in neurofibrillary tangles (NFT), extracellular plaques of abnormally folded amyloid-β (Aβ), local inflammation, activation of microglia and astrocytes, and lysosome dysfunction (Guerrero et al., 2021; Martínez-Cué et al., 2020; Saez-Atienzar et al., 2020; Zhang et al., 2019). The relationship between neuronal senescence and AD phenotypes has been known for several years, and many thorough reviews have been written specifically on this topic (Flanary et al., 2007; Han et al., 2020; Liu, 2022; Saez-Atienzar et al., 2020). Herein we will briefly discuss the potential role of senescence in neurons and glia in the development of AD pathology.
Early Aβ pathology triggers senescence in neurons and/or astrocytes secreting SASP which induces SIPS in adjacent cells, and senescence releases more SASP to induce neuroinflammation to further aggravate AD (DiBattista et al., 2020; Martínez-Cué et al., 2020). High levels of expression of hyperphosphorylated tau and elevated stress levels lead to senescent glial cell accumulation and subsequent neurodegeneration (DiBattista et al., 2020). A direct relationship between oxidative stress and cellular senescence has been hypothesized in AD. Oxidative stress (Ott et al., 2018), neuroinflammation (Wilcock et al., 2013), and cellular senescence (Wei et al., 2016) are critical in the development of amyloid plaques and neurofibrillary tangles (NFTs) which expedite aging and cognitive impairment in AD. Increased senescence has been detected in several cell types in the AD brain including astrocytes, microglia, and neurons, characterized by increased SA-β-gal activity (He et al., 2013), p53 expression (Yates et al., 2015), an increase in the release of SASP (Erusalimsky, 2009), DNA damage (Myung et al., 2008), telomere attrition or damage (Flanary et al., 2004), and senescence-like morphological changes (Streit et al., 2004). Also, in AD, mitochondrial function and structure are impaired, which in turn are important in the development of SIPS (Cadonic et al., 2016). The increased ROS production in AD results in reduced ATP synthesis and leaves DNA damage caused by oxidative stress, with insufficient energy for repair (Monti et al., 1992).
Contributing to the complex neurotoxic landscape, protein aggregation such as tau NFTs and amyloid plaque deposition may themselves act as pro-senescence stimuli (McHugh et al., 2017; Saez-Atienzar et al., 2020). Senescent cell accumulation during aging has been proposed to promote or aggravate the early events of neuroinflammation (Heneka et al., 2015). Local inflammation and toxic protein aggregation result in a greater accumulation of stressors (Zhang et al., 2019). The consequent cellular damage and the initiation of local senescence may exacerbate the aging phenotype and further contribute to chronic senescence (Saez-Atienzar et al., 2020). Accumulation of tau in neurons has been shown to induce senescence in astrocytes as well as microglia, and elimination of senescent glia ameliorates neurodegeneration (Bussian et al., 2018; Musi et al., 2018). Long-term exposure to SASPs causes tissue and organ declines that are associated with aging and neurodegeneration (Baker et al., 2013). SASP in neurons as well as mitotic cells are regulated transcriptionally by nuclear factor κB (NFκB) and CCAAT/enhancer-binding protein β (CEBPβ) (Acosta et al., 2008). The SASPs activate innate immune responses, which in turn target and eliminate senescent cells (Xue et al., 2007), at least in mitotic cell senescence (Acosta et al., 2008; Wajapeyee et al., 2008). In this way, protein aggregates like tau might exacerbate senescence by triggering SASP and inflammation during aging, resulting in clearance of senescent neurons and neuronal loss, though those connections are difficult to disentangle experimentally and remain to be proven.
The toxicity caused by tau aggregation might contribute to cellular senescence and inflammation as well (Saez-Atienzar et al., 2020). In a mouse model, senescent astrocytes and microglia stimulate the hyperphosphorylation of tau, which is a hallmark of AD that is critical for tau aggregation and NFT formation (Alonso et al., 1997). Tau aggregation and subsequent NFT production activate a cellular homeostatic mechanism to relieve the stress induced by the proteinopathy itself (Ramsden et al., 2005). The acute stress-induced NFTs protect neurons from apoptosis in the early pathogenic phases, but later in life promote neurodegeneration via senescence-like mechanisms by modifying brain bioenergetic state and SASP upregulation (Musi et al., 2018). NFT-containing neurons may upregulate Cdkn1a and Cdkn2a expression to enter senescence and prevent apoptosis, and the Cdkn1a and Cdkn2a expression activates the SASP (Saez-Atienzar et al., 2020). So, as NFTs increase, so too do stimuli that push neurons further toward senescence.
Glial cells also play a role in senescence and neurodegenerative phenotypes in AD. Astrocytes produce Aβ and release cytokines that enable bidirectional communication with glial cells, neurons, and the blood-brain barrier (BBB) (Linnerbauer et al., 2020). Senescent glial cells have been observed in association with AD pathologies. For example, a subset of reactive and disease-associate astrocytes have been shown to display senescence phenotypes, including increased expression of MMP-1, p16, p21, and SA-β-gal (Bhat et al., 2012; Ogrodnik et al., 2021), and production of SASP including IL-6 (Turnquist et al., 2016). Aging microglia that are dystrophic and lipofuscin-positive – indicative of aged cells with lysosomal turnover defects (Safaiyan et al., 2016) – also have been observed to have senescent features including elevated expression of p16, p21, SASP, and lipofuscin (Ogrodnik et al., 2021). A possible explanation is that microglial cells become senescent first and then transmit senescence to astrocytes (Guerrero et al., 2021). Microglia are activated by Aβ or SASPs and contribute to the clearance of both soluble and fibrillar Aβ forms (Hunter et al., 2013; Lee et al., 2010). Activation of microglia may increase the rate of Aβ phagocytosis (Kakimura et al., 2002); however, microglia can also contribute to Aβ deposition in transgenic mice (Wegiel et al., 2004). Inflammation can also accelerate microglial activation and alter microglial responses to Aβ (Von Bernhardi, 2007). Oligodendrocyte progenitor cells (OPCs) are the major proliferating cells in the brain mobilized in response to neuronal injury and demyelination (Zhang et al., 2019). OPCs have been observed to accumulate near amyloid plaques in the inferior parietal cortex of AD patients, and OPC accumulation triggers OPC senescence with features of CDKN1A and CDKN2A proteins expression and SA-β-gal activities and contributes to local inflammation and associated damage to a neuron which exacerbates Aβ pathology (Zhang et al., 2019). These various connections between senescent glia and AD phenotypes support the idea that glia may propagate senescence among themselves and to neurons in the AD brain, though further research is needed to clarify these cause-and-effect relationships.
3.2. Senescence and ALS
Amyotrophic lateral sclerosis (ALS) is a progressive motor neurodegenerative disease with no existing cure (Maximova et al., 2021). ALS-induced degeneration in upper and lower motor neurons causes muscular atrophy and gradual paralysis (Dudman et al., 2020). ALS results in severe abnormalities in the function of astrocytes, microglia, motor neurons, and other central nervous system (CNS) cells, resulting in excessive neuroinflammation and neurodegeneration (Maximova et al., 2021). Less is known about the role of senescence in ALS than in AD, however, some reviews have previously covered this topic (Maximova et al., 2021; Sah et al., 2021; Si et al., 2021).
The two most prevalent ALS-related mutations are in chromosome 9 open reading frame 72 (C9orf72) and superoxide dismutase (SOD1) (Maximova et al., 2021), and both have been associated with senescence. The C9orf72 gene accounts for half of the familial inheritance (fALS) and 7% of sporadic ALS (sALS) (Masrori et al., 2020). Spinal cord astrocytes generated from induced pluripotent stem cells (iPSCs) of ALS patients with a C9orf72 mutation showed an elevated SA-β-gal expression and restricted cell proliferation, implying a role for senescence in ALS (Birger et al., 2019). In the human spinal cord and motor cortex, the production of toxic soluble molecules and astrocytic proinflammatory marker C3 overexpression by ALS patient astrocytes have been linked to the development of ALS-related symptoms in the SOD1G93A mouse model of ALS, and further increases ROS load (Guttenplan et al., 2020). ALS also increases proinflammatory cytokine production, which contributes to SASP and thereby to senescence. Thus, an intricate relationship exists between prevalent ALS mutations and senescence phenotypes in the disease state, though the details of the mechanism(s) of pathogenesis remain to be elucidated.
Glial cell senescence has also been linked to the development of ALS. In a rat model, the paralysis progression of ALS is defined by the emergence of abundant microglia, astrocytes, and motor neurons exhibiting phenotypic characteristics of senescence (Trias et al., 2019). The tumor suppressor p53 also plays a role in the onset of cellular senescence in glial cells in ALS (Turnquist et al., 2016). Senescent microglia with possible genotoxicity may emerge as a consequence of microglial activation (Spittau, 2017). Senescent microglia display an increase in size and arrested cell cycle (Trias et al., 2019). ALS-affected glial cells share similar phenotypes with senescent glial cells, and senescent glial cells are a consequence of exacerbated cell damage or genotoxic stress, rather than aging (Thonhoff et al., 2012). Here again, while circumstantial evidence abounds to implicate glial cell activation and senescence in the development of ALS, mechanistic insights remain to be elucidated.
4. Stress Granules: Links to Senescence and Neurodegeneration
4.1. Stress Granule Phenotypes in Senescent Cells
Stress granules (SGs) are cytoplasmic aggregates made up of RNAs, RNA binding proteins (RPBs), translation factors, signaling components, and proteins of the small ribosomal subunit (Kedersha et al., 1999, 2000, 2002, 2103; Anderson and Kedersha, 2009; Riggs et al., 2020). SGs are formed by RNA-mediated condensation of the RBPs G3BP1 and G3BP2 through liquid-liquid phase separation under external stress including heat shock, oxidative stress, hyperosmolarity, viral infection, and UV irradiation (Riggs et al., 2020; Anderson et al., 2009; Cabral et al., 2022; Dudman et al., 2020; Farny et al., 2009; Guillén-Boixet et al., 2020). Polysome disassembly triggers the formation of SGs through protein-protein and protein-RNA interactions, and the granules grow in size throughout the stress event through additional interactions that promote the fusion of these granules (Cao et al., 2020). Recently, several researchers have discovered specific connections between senescence and the formation of SGs (Table 2).
Table 2.
Existing knowledge on the relationship between SGs assembly and senescence.
| Citation | Cell type | Senescence Inducing Condition | Type of Senescence | Results Relative to SG Formation |
|---|---|---|---|---|
| (Lian et al., 2009) | IDH4 | Removing dexamethasone from growth media, and fetal bovine serum (FBS) was replaced by charcoal-stripped FBS | Telomere-initiated senescence (replicative senescence) | The number of SGs was 5-fold and 2-fold higher in late senescent cells and early senescent cells compared with proliferative cells, respectively. |
| WI-38 | Serial passages until reaching the population doubling number of 39 or more | Telomere-initiated senescence (replicative senescence) | ||
| (Li et al., 2016) | Human corneal epithelium cells | treated with TGF-β1 or indicated chemicals for 3 days | SASP | Excess TGF-β1 causes senescence, triggers the RNA stress response, and increases SGs in HCECs. |
| (Moujaber et al., 2017) | LLC-PK1 kidney cells | 10mM sodium butyrate for 5 days | Chromatin perturbation-induced senescence |
|
| 40uM lopinavir for 3 days | SIPS | |||
| (Omer et al., 2018) | IDH4 | Removing dexamethasone from growth media and replacing DMEM containing FBS with charcoal-stripped FBS | Telomere-initiated senescence (replicative senescence) | More SGs formed in PRO cells than PRE or SEN. The number of SGs in PRE and SEN cells decreased by ~2- and 4-fold, respectively. |
| WI-38 primary human fetal lung fibroblasts | short exposure to ionizing radiation (10 Gy) | DNA-damage-inducedsenescence | ||
| (Omer et al., 2020) | IDH4 | removing dexamethasone from the media and replacing DMEM containing FBS with charcoal-stripped FBS | Telomere-initiated senescence (replicative senescence) |
|
| (Al-Rushadi, 2022) | Primary fibroblast and U2OS | Ionizing radiation (10Gy 320kV, 9mA, 4min for primary fibroblast cells; 4Gy 320kV, 8mA, 110s for U2OS cells). Cells were harvested after 9 days | DNA-damage-induced senescence |
|
| Doxorubicin (200nM for 48h), the harvest after 7 days |
Sodium arsenite (SA) is a well-studied oxidative stress-inducing agent that promotes canonical SG assembly (Kedersha et al., 1999, 2000, 2002; McEwen et al., 2004; Aulas and Vande Velde 2015). SGs induced acutely by SA are typically treated with a range of SA from 100 – 500 μM for 30 – 60 minutes (Riggs et al., 2020). However, longer treatments at lower doses have been associated with the formation of chronic SGs (Reineke et al., 2019). To understand the relationship between senescence and SGs triggered by oxidative stress such as SA, several groups measured SG formation in senescent cells. Replicative senescence can be induced in IDH4 cells by growing cells in dexamethasone-free (dex) media, which inhibits SV40 T-antigen expression (Lian et al., 2009; Omer et al., 2018; Wang et al., 2001). After dex withdrawal, senescence cells can be fixed into 3 stages: proliferating stage (PRO), presenescent stage (PRE), and senescent stage (SEN) (Omer et al., 2018). The stages correspond roughly to the proliferative capacity of the cell population. In the PRO stage (days 0–3), the majority of the cells are able to divide. In the PRE stage (days 4–6), 30%−70% of cells are able to divide. In the SEN stage (days 7–10), less than 30% of cells are able to divide. SG formation was examined in the PRO, PRE, and SEN states using marker proteins FMRP and G3BP1. In an early study on the relationship of SGs to senescence, it was observed that the number of SGs formed in response to SA was 2- and 5-fold higher in the PRE stage and SEN stage compared to the PRO stage (Lian et al., 2009); however, these SGs were smaller than SA-induced SGs in non-senescent cells. A later study using the same cell types and dex-withdrawal protocol showed SGs formed in response to brief acute SA (500 μM for 30 minutes) in a substantial number of PRO cells, but in PRE and SEN cells the number of SGs fell by ~2- and 4-fold, respectively (Omer et al., 2018). These contradictory conclusions may result from a lack of consensus about what constitutes a SG and how to count SG-positive cells, or may be the result of methodological variations.
Further adding to the complexity of the findings, the mechanism behind the impairment of SGs varies with the mechanism of senescence induction (Moujaber et al., 2017). Cells induced by sodium butyrate have been shown to reduce the level of TIA-1/TIAR, eIF4G, and SP1; however, cells induced by lopinavir have been shown to have no significant change in the level of TIA-1/TIAR, eIF4G, and SP1 (Moujaber et al., 2017). Also, induction time for different senescence-inducing reagents varies. For example, 10mM sodium butyrate takes 5 days (Moujaber et al., 2017); 40uM lopinavir takes 3 days; X-ray ionizing radiation takes a few minutes (Al-Rushadi, 2022). With huge variations in induction time among the reagents, the previously defined senescence stages (PRO, PRE, SEN) are hard to distinguish in all models of senescence. Further, these methodological differences will mean that the resulting senescent cells will have different subsets of senescent phenotypes, or may express phenotypes to different degrees (Table 1).
Considering all known studies on the subject (Table 2), the consensus seems to be that SGs are smaller but more numerous in senescent cells, but that the effects of senescence on SGs may depend upon the mechanism and duration of senescence. The specific senescence phenotypes may also play a role. For example, it has been shown that the increased cell size that occurs during senescence causes cytoplasmic dilution, as both RNA and protein synthesis do not scale linearly with cell size past a certain threshold (Neurohr et al., 2019). As both protein and RNA concentrations are critical for the formation of SGs (Yang et al., 2020), it may be the case that dilution of SG factors in the cytoplasm of senescent cells causes many small SGs that fail to fuse or enlarge, or a decrease in overall SG formation, depending upon the amount of dilution as well as the presence of other contributing stress factors. These variables will inevitably be dependent upon the mechanism of senescence, particularly whether or not the enlarged cell phenotype is present. This complex landscape will no doubt be the subject of future studies.
4.2. Stress Granules and Neurodegeneration
The formation of SGs has direct relationships to a broad range of diseases including cancer (Anderson et al., 2015), viral infection (Malinowska et al., 2016), cardiovascular disease (Wang et al., 2022), and neurodegenerative disease (Wolozin and Ivanov, 2019). Age-related progressive protein homeostasis failure, stress (such as acute oxidative stress), and mitochondrial dysfunction lead to functional defects in cells and tissues and impact SG assembly (Cao et al., 2020). The aging-dependent decline in chaperone expression levels and activity contributes to the susceptibility of aged neuronal cells to misfolded proteins, which likely contribute to neurodegenerative diseases (Yang et al., 2014). Misfolded proteins caused by severe stress and aging could accumulate and aggregate within SGs, which may lead to a change in SG composition, SG dynamics impairment, and consequent abnormal liquid-like to solid-like state conversion (Ganassi et al., 2016). Many reviews have discussed the relationship of SGs and related RBPs to NDs, mainly focused on AD and ALS (Asadi et al., 2021; Li et al., 2013; Vanderweyde et al., 2013; Wang et al., 2022; Wolozin, 2012). Therefore, we will only summarize this information here, and refer the reader to these excellent and thorough reviews for more information. Aging-associated mitochondrial dysfunction and inactive metabolism might lead to limited regulation of this process and aberrant SGs (Cao et al., 2020). Under normal conditions, autophagy removes SGs after brief acute stresses and eliminates aberrant SG accumulation; however, during aging, disturbed protein quality control and defects in autophagy can have a deleterious influence on the elimination of aberrant SGs (Buchan et al., 2013). Chronic cellular stress, such as aging, can lead to persistent SGs which function as a seeding mechanism, resulting in RBPs accumulation (Wolozin and Ivanov, 2019). Acute SGs are dynamic with a pro-survival role (Arimoto et al., 2008), and dissemble upon alleviation of stress. Chronic SGs, on the other hand, are static with a pro-death role (Reineke et al., 2019). Chronic SGs disrupt assembly and interact with pathological aggregates, which results in neuron degeneration (Asadi et al., 2021). SGs are therefore hypothesized to function as a seeding mechanism for pathological protein aggregates, and may thereby represent a unifying contribution to neurodegenerative diseases (Wolozin and Ivanov, 2019).
4.2.1. Stress Granules and Alzheimer’s Disease
The two major pathological features of Alzheimer’s disease (AD) are extracellular plaques primarily consisting of amyloid-β (Aβ), and intracellular neurofibrillary tangles (NFT) formed by the accumulation of hyperphosphorylated tau protein (Rojo et al., 2006; Yu et al., 2022). Other characteristics of AD include local inflammation, activated microglia and astrocytes, lysosome dysfunction, and neuronal and synaptic loss (Guerrero et al., 2021; Martínez-Cué et al., 2020; Saez-Atienzar et al., 2020; Vanderweyde et al., 2013; Zhang et al., 2019). Normally, tau promotes the assembly and stability of microtubules. Non-fibrillized, abnormally hyperphosphorylated tau sequesters normal tau and disrupts microtubules (Iqbal et al., 2009). The increase in tau phosphorylation causes tau dissociation from the plasma membrane (Maas et al., 2000), which restricts the binding of tau to microtubules (Rojo et al., 2006). Disrupting microtubule function is known to cause a failure in fusion of SGs during their early biogenesis (Wheeler et al., 2016), which causes a significant decrease in the occurrence of large SGs (Ivanov et al., 2003), a decrease in the average size of SGs (Sablina et al., 2012), and a concurrent increase in SG numbers in cells. Hyperphosphorylated tau also accumulates in the soma and dendrites of neurons (Hoover et al., 2010), where it interacts with SG-associated RBPs (Cruz et al., 2019; Stamer et al., 2002). Key SG proteins TIA-1 and TTP directly bind to tau protein and interact with tau, playing a role in disease progression (Vanderweyde et al., 2012). TIA-1 stimulates tau aggregation and toxicity in both cultured cells and AD model mice by promoting tau-positive SG formation (Apicco et al., 2018; Vanderweyde et al., 2016). Stress promotes tau phosphorylation and mislocalization, which accelerates SG assembly (Hoover et al., 2010). Since RNA stimulates tau aggregation in vitro (Kampers et al., 1996), RNA implicated in SG assembly might accelerate tau aggregation. In other words, the evidence suggests that tau protein aggregation promotes SG formation, and SG formation in turn promotes tau aggregation, causing a feed-forward loop of pathogenic protein aggregation and exacerbation of the disease state in AD.
Tau protein binds to ribosomes and decreases protein synthesis to inhibit protein translation (Meier et al., 2016). SG formation is characterized by translation arrest and RBPs binding to mRNA (Asadi et al., 2021). Although there is evidence that larger tau aggregates recruit TIA-1 colocalization, the relationship between RBP aggregation and SG formation with tau aggregates remains unclear (Vanderweyde et al., 2012). One possible hypothesis is that tau aggregations might drive the dynamic equilibrium of SGs toward assembly or to SGs disequilibrium and consequent tau protein accumulation (Asadi et al., 2021). Since tau plays a crucial role in stress-mediated translational regulation, chronic stress-induced tau hyperphosphorylation might lead to permanent SGs (Advani et al., 2020). Conversely, genetic reduction of the SG nucleating protein TIA-1 improves lifespan and tau pathology in AD model mice (Apicco et al., 2018), lending further credence to a causative role for SGs in AD. Tau induces astrocyte senescence and secretes SASPs. These dysregulations manifest in the brain as an increase in neuroinflammation, increased BBB permeability, loss of neuronal synapses, demyelination, and dysregulated metabolism which is associated with impaired cognition, and clearance of senescent cells as a therapeutic strategy has shown to reduce pathology, inflammation, and neuronal dysfunction (Kritsilis et al., 2018; Martínez-Cué et al., 2020; Sah et al., 2021).
4.2.2. Stress Granules and ALS
Many strong experimental links have been made between SG formation and the accumulation of pathogenic protein inclusions in ALS, and many good reviews have been written specifically on the relationship of SGs to ALS (Li et al., 2013; Monahan et al., 2016; Fernandes et al., 2018; Desai and Bandopadhyay 2020; Zhang et al., 2021; Li et al., 2022; Fan et al., 2016; Dudman and Qi 2020). Here we will only briefly highlight this vast and growing literature.
ALS-associated mutant forms of SOD1, TDP-43, and FUS have been linked to dysregulation of SG dynamics, resulting in delayed SG clearance and chronic SG persistence. SGs are cleared in part by the autophagy pathway (Buchan et al., 2013), SOD1, TDP-43, and FUS mutations that impair autophagy in ALS (Morimoto et al., 2007; Soo et al., 2015; Xia et al., 2016) also prevent SG clearance and exacerbate protein aggregation (Mateju et al., 2017; Baradaran-Heravi et al., 2020; Fernandes et al., 2020). Notably, many proteins found in ALS pathological protein inclusions are also components of SGs even in unaffected cells (Monahan et al., 2016), including wild-type TDP-43 and FUS. SGs are enriched in RNA-binding proteins and other proteins with intrinsically disordered protein domains, which overlap significantly with the ALS inclusion proteome (Markmiller et al., 2018; Markmiller et al., 2021). Thus, the process of normal responses to stress via SGs, and the pathogenic effects of proteins seen in ALS, are linked through their dynamic interactions and overlapping proteomes.
The pathological seeding hypothesis (Wolozin and Ivanov 2019) of SGs and ND would predict that inducing SGs may exacerbate ALS pathology, whereas suppressing SG formation may be therapeutic for ALS. For example, repeated induction of SGs in a controlled optogenetic system led to the persistence of aggregates containing pathogenic ALS markers, such as phosphorylated TDP-43 and SQSTM (Zhang et al., 2019), supporting the seeding hypothesis as a critical mechanism in ALS pathology development. Inversely, mutations and interventions that suppress SGs and/or disfavor biomolecular condensation tend to decrease ALS pathology (Alexander et al., 2018; Daigle et al., 2016; Hofweber et al., 2018). The efficacy of SG suppression as a therapeutic strategy for ALS has yet to be fully examined. However, the close relationship between SGs and ALS proteins suggests further investigation in this area is warranted.
5. Connecting the Dots Between Senescence, Stress Granules, and Neurodegeneration
While the information gathered here draws out the many connections between aging, SGs, senescence, and ND, the mechanisms and cause-and-effect relationships between these various processes remain unclear. We propose that senescence may be a source of SG formation in neurons, which would increase the opportunity for pathogenic seeding and progression into pathogenic SGs and disease-associated protein aggregation. In this way, the age-related onset of senescence in neurons may be linked to SGs and ultimately to NDs. In accordance with the SG seeding hypothesis of ND (Wolozin and Ivanov 2019), we hypothesize that the increased number of small SGs associated with many senescence mechanisms may provide an additional opportunity for seeding pathological lesions in NDs, thereby linking the processes of SG formation, protein aggregation, and senescence in a “perfect storm” of neuronal dysfunction and decay (Figure 3). Senescence may be an additional early source of stress that favors pathogenic seeding, a process that remains entirely unclear in the pathogenesis of NDs broadly.
Figure 3. Hypothetical model of senescence, SGs, and NDs.
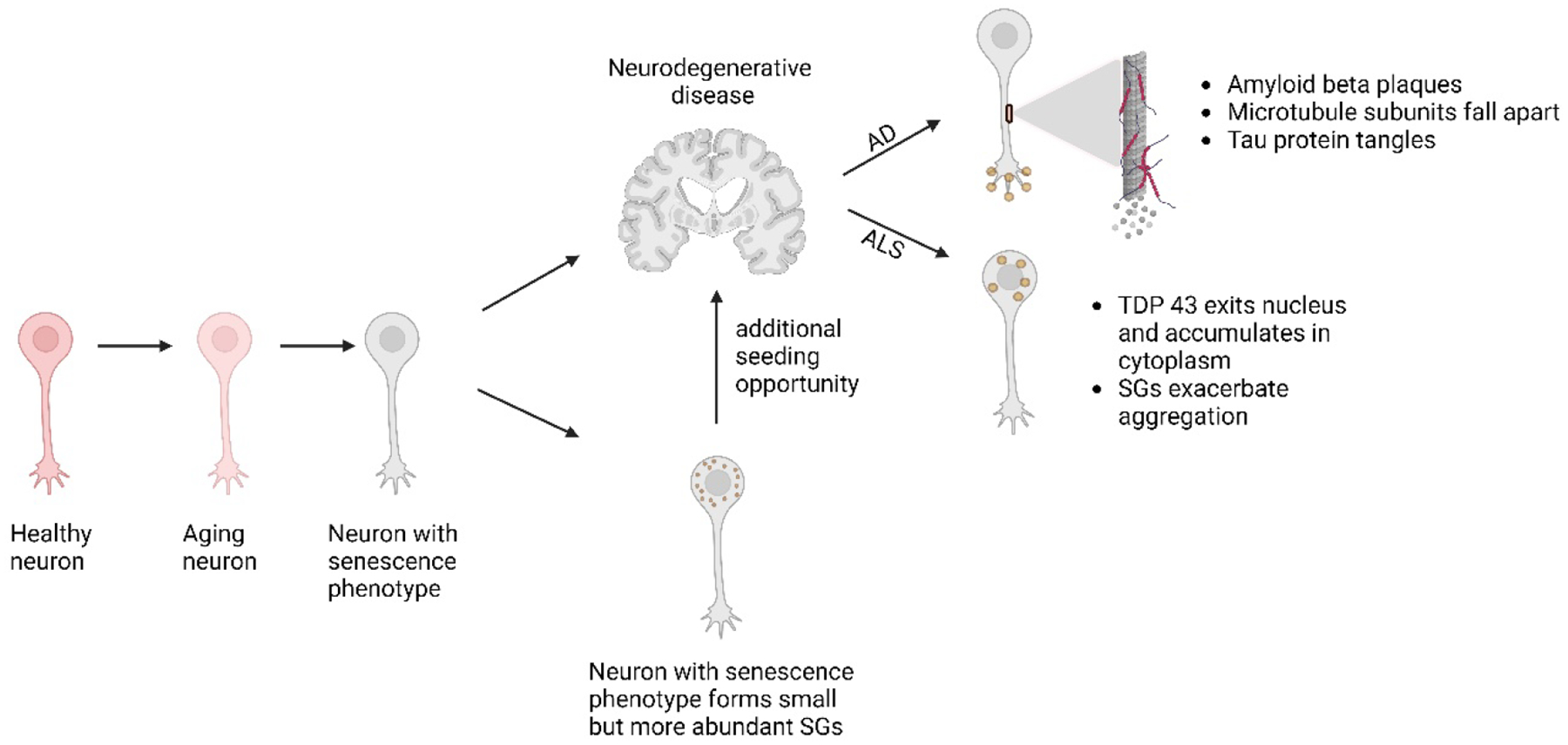
Post-mitotic neurons accumulate senescent phenotypes during aging. Among these phenotypes, senescent neurons may produce smaller but more abundant SGs, which may provide additional opportunities for aggregate seeding and thereby further exacerbate NDs. See text for additional details.
How specifically might senescence contribute to the formation of chronic SGs in neurons? In neurons, senescence does not cause an enlarged cell phenotype (Table 1). So, cytoplasmic dilution of aggregation-prone protein and mRNA, which would disfavor SG development, may not be a factor. There is evidence that the protein synthesis rate declines in aging and senescent neurons (Schimanski and Barnes, 2010). The combined result of low-level chronic stress due to aging and decreased translational efficiency during senescence may result in more untranslated mRNPs in the cytoplasm, which would favor SG assembly. As discussed above, we predict these SGs to be small but numerous, and to act as seeds that promote pathogenic aggregation. Several key pieces of evidence are missing from this model and would need to be determined experimentally.
As a first step, SG phenotypes have not yet been characterized in neurons, and it is unknown whether evidence of SG seeding can be detected in senescent neurons in vitro or in vivo. It will then be important to examine – in mouse models of ALS and AD but also in human samples when available – whether pathogenic SG formation is more prevalent in senescent cells (indicated by SA-β-gal staining and SASP). Further, there have been advances in the detection of sub-microscopic aggregate seeds (Marmor-Kollet et al., 2020; Cirillo et al., 2020; Streit et al., 2022) which could be applied to examine the relative levels of pre-SG seeding in senescent and non-senescent neurons. A careful study of inducible genetic mouse models of AD and ALS in which SGs, senescence, and disease pathology are all studied in parallel may also shed light on these relationships, but have not yet been performed.
There are indications that AD and ALS have a propagation component, that is, that TDP-43 and Aβ secreted by affected neurons may propagate their pathogenic phenotypes to neighboring cells (Liu et al., 2012; Sanders et al., 2014; Smethurst et al., 2016), thereby spreading the disease. With Aβ oligomers, this propagation triggers the formation of pathogenic SGs and is directly associated with neurodegeneration (Jiang et al., 2019). SASP has been linked to tau pathology propagation (Ungerleider et al., 2021), but more work remains to be done to determine if SASP is a common feature of ND propagation. Further, it will be illuminating to see whether suppression of senescence or elimination of senescent neurons and glia with senotherapeutics – therapies targeted to senescent cells (Park and Shin, 2022) – would slow ND progression. Several animal studies have shown promise in the application of senotherapeutics to NDs (Bussian et al., 2018; Zhang et al., 2019; Ogrodnik et al., 2021). The application of senotherapeutic approaches to the treatment of NDs (AD in particular) is already in clinical trials (Boccardi and Mecocci, 2021) but has not yet been studied mechanistically in relation to the formation of pathogenic SGs and disease aggregate seeding.
6. Conclusions and Future Directions
Aging plays a key role in many diseases. Aging promotes stress granule formation through mitochondrial dysfunction, the escalation of chronic stress, and a decline in protein homeostasis. Stress granule formation is linked to multiple NDs through pathological protein aggregation and the seeding hypothesis. During aging, DNA damage accumulation, shortened telomeres, oncogene activation, and oxidative stress all contribute to senescence. Senescent cells secrete SASP, which induces senescence in nearby cells and causes neuroinflammation, a major risk factor for neurodegenerative diseases. However, the exact relationship between SG formation and senescence in the context of post-mitotic cells during brain aging is poorly understood. In this Review, we propose that senescence might be a precursor of NDs, and that enhanced SG seeding in senescent cells is a potential mechanism linking senescence to NDs. Post-mitotic neurons acquire senescence phenotypes by a paracrine effect from neighboring senescent cells during aging. Then, neurons with senescent-like phenotypes secrete SASP, causing neuroinflammation and contributing to neurodegenerative diseases, which directly relates to SGs formation by seeding pathological protein aggregates such as tau. Since the SG formation pathway is impaired during senescence, more SGs will be formed, which provides more opportunity for the seeding mechanism and further aggravates NDs. If indeed senescence is a critical early step in ND pathogenesis, then senescent markers could be a promising tool for detecting NDs at the early stage.
It has been hypothesized that cumulative, low-level stresses in the aging brain contribute to the formation of chronic pathogenic SGs which, in turn, further seed disease-associated aggregation. Senescence, too, is triggered by the accumulation of stress and cellular damage over time. The most significant limitation in our current understanding of the relationship of senescence to ND is that it is not yet possible to determine whether senescence is a cause of pathogenic protein aggregation or a consequence (Saez-Atienzar and Masliah, 2020). We propose that the onset of senescence exacerbates the formation of SG seeds, and may act as a molecular “last straw” in the initiation of pathogenic SG formation. As early SG seeding is proposed to be an upstream initiating event to the later formation of pathogenic SGs and disease-related protein aggregation, determining whether early SGs are present in senescent neurons or glia may permit us to address the question of the causal or consequential role of senescence in the events of ND onset and progression. Examining SG formation in the context of senescence may therefore provide the answer to the mechanistic relationship between senescence and ND.
There remain many challenges in addressing the connections between senescence and SGs in the progression of NDs. First, there is no entirely unique and universally acknowledged set of markers or phenotypes to identify senescent cells and differentiate them from quiescent cells. Currently, senescent cell detection requires a combination of multiple markers, and the relevance of all markers to neurons and glia is an active area of investigation. Second, we proposed that senescent cells secrete an excess amount of SASPs, which leads to neuroinflammation and consequent NDs. However, whether eradicating senescent cells eliminates or delays ND is not well studied, particularly in human models. Third, to date, there are only five cell types studied that induce SG formation under senescence, and most of the cell lines are fibroblasts. Whether neurons displaying senescence phenotypes could form SGs is currently unknown. Even in fibroblasts, the mechanism of impaired SGs remains unclear. What is clear is that, given the broad prevalence of NDs, the potential relationships between senescence, SGs, and ND will be an important area of investigation for future therapeutic development and should be further explored.
Highlights:
Senescence in neurons and glia contributes to neurodegenerative diseases (NDs).
Stress granules (SGs) are stress-induced cytoplasmic biomolecular condensates.
NDs like Alzheimer’s Disease and ALS are linked to both SGs and senescence.
Senescence alters SG dynamics, causing smaller but more abundant SGs.
SG seeding may be a potential mechanism linking senescence to NDs.
Acknowledgments
The authors thank members of the Farny lab for feedback and support. Figures were assembled using BioRender.
Funding Support
This work was funded in part by NIH R03AG077140 to NGF, and new faculty start-up funds from WPI to NGF.
Abbreviations List
- AD
Alzheimer’s Disease
- ALS
Amyotrophic Lateral Sclerosis
- DDR
DNA damage response
- LLPMC
long lived post-mitotic cells
- ND
neurodegenerative disease
- NFT
neurofibrillary tangles
- ROS
reactive oxygen species
- SA-β-gal
senescence associated beta galactosidase
- SAHF
senescence-associated heterochromatin foci
- SASP
senescence associated secretory phenotype
- SIPS
stress-induced premature senescence
- SG
stress granules
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The authors declare no competing interests.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CREDIT Author Statement
YM: Conceptualization (supporting); Visualization (lead); Writing – Original Draft Preparation (lead); Writing – Review & Editing (supporting).
NGF: Conceptualization (lead); Funding Acquisition (lead); Project Administration (lead); Supervision (lead); Visualization (supporting); Writing – Original Draft Preparation (supporting); Writing – Review & Editing (lead).
References
- Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, … Gil J (2008). Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell, 133(6), 1006–1018. doi: 10.1016/j.cell.2008.03.038 [DOI] [PubMed] [Google Scholar]
- Advani VM, & Ivanov P (2020). Stress granule subtypes: an emerging link to neurodegeneration. Cellular and Molecular Life Sciences, 77(23), 4827–4845. doi: 10.1007/s00018-020-03565-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander EJ, Ghanbari Niaki A, Zhang T, Sarkar J, Liu Y, Nirujogi RS, Pandey A, Myong S, Wang J. Ubiquilin 2 modulates ALS/FTD-linked FUS-RNA complex dynamics and stress granule formation. Proc Natl Acad Sci U S A. 2018. Dec 4;115(49):E11485–E11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Mashhadi S, Simpson JE, Heath PR, Dickman M, Forster G, Matthews FE, … Wharton SB (2015). Oxidative Glial Cell Damage Associated with White Matter Lesions in the Aging Human Brain. Brain Pathol, 25(5), 565–574. doi: 10.1111/bpa.12216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Rushadi M, Musabah Rashid. (2022). Investigation of the role of G3BP in stress granule formation in senescence. Doctoral thesis, Durham University. Available from: http://etheses.dur.ac.uk/14347/ [Google Scholar]
- Allan JM, & Travis LB (2005). Mechanisms of therapy-related carcinogenesis. Nat Rev Cancer, 5(12), 943–955. doi: 10.1038/nrc1749 [DOI] [PubMed] [Google Scholar]
- Allen NJ, & Barres BA (2009). Glia — more than just brain glue. Nature, 457(7230), 675–677. doi: 10.1038/457675a [DOI] [PubMed] [Google Scholar]
- Alonso AD, Grundke-Iqbal I, Barra HS, & Iqbal K (1997). Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A, 94(1), 298–303. doi: 10.1073/pnas.94.1.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, & Kedersha N (2009). Stress granules. Curr Biol, 19(10), R397–398. doi: 10.1016/j.cub.2009.03.013 [DOI] [PubMed] [Google Scholar]
- Anderson P, Kedersha N, & Ivanov P (2015). Stress granules, P-bodies and cancer. Biochimica et biophysica acta, 1849(7), 861–870. doi: 10.1016/j.bbagrm.2014.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonangeli F, Zingoni A, Soriani A, & Santoni A (2019). Senescent cells: Living or dying is a matter of NK cells. J Leukoc Biol, 105(6), 1275–1283. [DOI] [PubMed] [Google Scholar]
- Apicco DJ, Ash PEA, Maziuk B, LeBlang C, Medalla M, Al Abdullatif A, … Wolozin B (2018). Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nature Neuroscience, 21(1), 72–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimoto K, Fukuda H, Imajoh-Ohmi S, Saito H, Takekawa M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat Cell Biol. 2008. Nov;. doi: 10.1038/s41593-017-0022-z [DOI] [PubMed] [Google Scholar]
- Asadi MR, Sadat Moslehian M, Sabaie H, Jalaiei A, Ghafouri-Fard S, Taheri M, & Rezazadeh M (2021). Stress Granules and Neurodegenerative Disorders: A Scoping Review. Front Aging Neurosci, 13, 650740. doi: 10.3389/fnagi.2021.650740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash PE, Vanderweyde TE, Youmans KL, Apicco DJ, & Wolozin B (2014). Pathological stress granules in Alzheimer’s disease. Brain Res, 1584, 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulas A, Vande Velde C. Alterations in stress granule dynamics driven by TDP-43 and FUS: a link to pathological inclusions in ALS?. Frontiers in cellular neuroscience. 2015. Oct 23;9:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, & Petersen RC (2018). Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J Clin Invest, 128(4), 1208–1216. doi: 10.1172/jci95145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Weaver RL, & van Deursen JM (2013). p21 both attenuates and drives senescence and aging in BubR1 progeroid mice. Cell Rep, 3(4), 1164–1174. doi: 10.1016/j.celrep.2013.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist CJ, & Kastan MB (2015). Chromatin perturbations during the DNA damage response in higher eukaryotes. DNA Repair, 36, 8–12. doi: 10.1016/j.dnarep.2015.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baradaran-Heravi Y, Van Broeckhoven C, van der Zee J. 2020. Stress granule mediated protein aggregation and underlying gene defects in the FTD-ALS spectrum. Neurobiol Dis 134:104639. [DOI] [PubMed] [Google Scholar]
- Beck J, Turnquist C, Horikawa I, & Harris C (2020). Targeting cellular senescence in cancer and aging: roles of p53 and its isoforms. Carcinogenesis, 41(8), 1017–1029. doi: 10.1093/carcin/bgaa071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU, … Torres C (2012). Astrocyte senescence as a component of Alzheimer’s disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birger A, Ben-Dor I, Ottolenghi M, Turetsky T, Gil Y, Sweetat S, … Reubinoff B (2019). Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine, 50, 274–289. doi: 10.1016/j.ebiom.2019.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccardi V, Mecocci P. Senotherapeutics: Targeting senescent cells for the main age-related diseases. Mechanisms of Ageing and Development. 2021. Jul 1;197:111526. [DOI] [PubMed] [Google Scholar]
- Buchan JR, Kolaitis R-M, Taylor JP, & Parker R (2013). Eukaryotic Stress Granules Are Cleared by Autophagy and Cdc48/VCP Function. Cell, 153(7), 1461–1474. doi: 10.1016/j.cell.2013.05.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, & Baker DJ (2018). Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature, 562(7728), 578–582. doi: 10.1038/s41586-018-0543-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral AJ, Costello DC, & Farny NG (2022). The Enigma of UV Stress Granules: Research Challenges and New Perspectives. 2022.2010.2011.511743. doi: 10.1101/2022.10.11.511743 %J bioRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadonic C, Sabbir MG, & Albensi BC (2016). Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease. Mol Neurobiol, 53(9), 6078–6090. doi: 10.1007/s12035-015-9515-5 [DOI] [PubMed] [Google Scholar]
- Campisi J (2001). Cellular senescence as a tumor-suppressor mechanism. Trends in Cell Biology, 11(11), S27–S31. [DOI] [PubMed] [Google Scholar]
- Campisi J, & d’Adda di Fagagna F (2007). Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol, 8(9), 729–740. doi: 10.1038/nrm2233 [DOI] [PubMed] [Google Scholar]
- Cao X, Jin X, & Liu B (2020). The involvement of stress granules in aging and aging-associated diseases. Aging Cell, 19(4), e13136. doi: 10.1111/acel.13136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee D, & Chakrabarti O (2022). Role of stress granules in modulating senescence and promoting cancer progression: Special emphasis on glioma. Int J Cancer, 150(4), 551–561. doi: 10.1002/ijc.33787 [DOI] [PubMed] [Google Scholar]
- Chernov KG, Barbet A, Hamon L, Ovchinnikov LP, Curmi PA, & Pastré D (2009). Role of Microtubules in Stress Granule Assembly. Journal of Biological Chemistry, 284(52), 36569–36580. doi: 10.1074/jbc.M109.042879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Lieu CA, DeMaria M, Laberge R-M, Campisi J, & Andersen JK (2013). Environmental stress, ageing and glial cell senescence: a novel mechanistic link to Parkinson’s disease? Journal of Internal Medicine, 273(5), 429–436. doi: 10.1111/joim.12029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo L, Cieren A, Barbieri S, Khong A, Schwager F, Parker R, & Gotta M (2020). UBAP2L forms distinct cores that act in nucleating stress granules upstream of G3BP1. Current Biology, 30(4), 698–707. [DOI] [PubMed] [Google Scholar]
- Collado M, Blasco MA, & Serrano M (2007). Cellular Senescence in Cancer and Aging. Cell, 130(2), 223–233. doi: 10.1016/j.cell.2007.07.003 [DOI] [PubMed] [Google Scholar]
- Cruz A, Verma M, & Wolozin B (2019). The Pathophysiology of Tau and Stress Granules in Disease. In Takashima A, Wolozin B, & Buee L (Eds.), Tau Biology (pp. 359–372). Singapore: Springer Singapore. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Adda di Fagagna F (2008). Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer, 8(7), 512–522. doi: 10.1038/nrc2440 [DOI] [PubMed] [Google Scholar]
- Daigle JG, Krishnamurthy K, Ramesh N, Casci I, Monaghan J, McAvoy K, Godfrey EW, Daniel DC, Johnson EM, Monahan Z, Shewmaker F, Pasinelli P, Pandey UB. Pur-alpha regulates cytoplasmic stress granule dynamics and ameliorates FUS toxicity. Acta Neuropathol. 2016. Apr;131(4):605–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai P, Bandopadhyay R. Pathophysiological implications of RNP granules in frontotemporal dementia and ALS. Neurochem Int. 2020. Nov;140:104819. [DOI] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, … d’Adda di Fagagna F (2006). Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature, 444(7119), 638–642. doi: 10.1038/nature05327 [DOI] [PubMed] [Google Scholar]
- DiBattista AM, Sierra F, & Masliah E (2020). NIA workshop on senescence in brain aging and Alzheimer’s disease and its related dementias. GeroScience, 42(2), 389–396. doi: 10.1007/s11357-020-00153-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudman J, & Qi X (2020). Stress Granule Dysregulation in Amyotrophic Lateral Sclerosis. Front Cell Neurosci, 14, 598517. doi: 10.3389/fncel.2020.598517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erusalimsky JD (2009). Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol (1985), 106(1), 326–332. doi: 10.1152/japplphysiol.91353.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CS, & Holzbaur ELF (2019). Autophagy and mitophagy in ALS. Neurobiol Dis, 122, 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan AC, & Leung AK (2016). RNA Granules and Diseases: A Case Study of Stress Granules in ALS and FTLD. Adv Exp Med Biol, 907, 263–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farny NG, Kedersha NL, & Silver PA (2009). Metazoan stress granule assembly is mediated by P-eIF2alpha-dependent and -independent mechanisms. Rna, 15(10), 1814–1821. doi: 10.1261/rna.1684009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay MM, Columbo D, Cotter C, Friend C, Henry S, Hoppe M, … Farny NG (2021). Bisphenol A promotes stress granule assembly and modulates the integrated stress response. Biol Open, 10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes N, Eshleman N, Buchan JR. Stress Granules and ALS: A Case of Causation or Correlation? Adv Neurobiol. 2018;20:173–212. [DOI] [PubMed] [Google Scholar]
- Fernandes N, Nero L, Lyons SM, Ivanov P, Mittelmeier TM, Bolger TA, Buchan JR. 2020. Stress granule assembly can facilitate but is not required for TDP-43 cytoplasmic aggregation. Biomolecules 10:1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielder E, von Zglinicki T, & Jurk D (2017). The DNA Damage Response in Neurons: Die by Apoptosis or Survive in a Senescence-Like State? J Alzheimers Dis, 60(s1), S107–s131. doi: 10.3233/jad-161221 [DOI] [PubMed] [Google Scholar]
- Firsanov DV, Solovjeva LV, & Svetlova MP (2011). H2AX phosphorylation at the sites of DNA double-strand breaks in cultivated mammalian cells and tissues. Clin Epigenetics, 2(2), 283–297. doi: 10.1007/s13148-011-0044-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer HG, Morawski M, Brückner MK, Mittag A, Tarnok A, & Arendt T (2012). Changes in neuronal DNA content variation in the human brain during aging. Aging Cell, 11(4), 628–633. doi: 10.1111/j.1474-9726.2012.00826.x [DOI] [PubMed] [Google Scholar]
- Flanary BE, Sammons NW, Nguyen C, Walker D, & Streit WJ (2007). Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res, 10(1), 61–74. doi: 10.1089/rej.2006.9096 [DOI] [PubMed] [Google Scholar]
- Flanary BE, & Streit WJ (2004). Progressive telomere shortening occurs in cultured rat microglia, but not astrocytes. Glia, 45(1), 75–88. doi: 10.1002/glia.10301 [DOI] [PubMed] [Google Scholar]
- Freyter BM, Abd Al-Razaq MA, Isermann A, Dietz A, Azimzadeh O, Hekking L, … Rübe CE (2022). Nuclear Fragility in Radiation-Induced Senescence: Blebs and Tubes Visualized by 3D Electron Microscopy. Cells, 11(2). doi: 10.3390/cells11020273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funayama R, & Ishikawa F (2007). Cellular senescence and chromatin structure. Chromosoma, 116(5), 431–440. doi: 10.1007/s00412-007-0115-7 [DOI] [PubMed] [Google Scholar]
- Ganassi M, Mateju D, Bigi I, Mediani L, Poser I, Lee HO, … Carra S (2016). A Surveillance Function of the HSPB8-BAG3-HSP70 Chaperone Complex Ensures Stress Granule Integrity and Dynamism. Molecular Cell, 63(5), 796–810. doi: 10.1016/j.molcel.2016.07.021 [DOI] [PubMed] [Google Scholar]
- Ghosh S, & Geahlen RL (2015). Stress Granules Modulate SYK to Cause Microglial Cell Dysfunction in Alzheimer’s Disease. EBioMedicine, 2(11), 1785–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero A, De Strooper B, & Arancibia-Cárcamo IL (2021). Cellular senescence at the crossroads of inflammation and Alzheimer’s disease. Trends in Neurosciences, 44(9), 714–727. doi: 10.1016/j.tins.2021.06.007 [DOI] [PubMed] [Google Scholar]
- Guillén-Boixet J, Kopach A, Holehouse AS, Wittmann S, Jahnel M, Schlüßler R, … Franzmann TM (2020). RNA-Induced Conformational Switching and Clustering of G3BP Drive Stress Granule Assembly by Condensation. Cell, 181(2), 346–361.e317. doi: 10.1016/j.cell.2020.03.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttenplan KA, Weigel MK, Adler DI, Couthouis J, Liddelow SA, Gitler AD, & Barres BA (2020). Knockout of reactive astrocyte activating factors slows disease progression in an ALS mouse model. Nature Communications, 11(1), 3753. doi: 10.1038/s41467-020-17514-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Zhang T, Liu H, Mi Y, & Gou X (2020). Astrocyte Senescence and Alzheimer’s Disease: A Review. Front Aging Neurosci, 12, 148. doi: 10.3389/fnagi.2020.00148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley CB, Futcher AB, & Greider CW (1990). Telomeres shorten during ageing of human fibroblasts. Nature, 345(6274), 458–460. doi: 10.1038/345458a0 [DOI] [PubMed] [Google Scholar]
- He N, Jin WL, Lok KH, Wang Y, Yin M, & Wang ZJ (2013). Amyloid-β(1–42) oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis, 4(11), e924. doi: 10.1038/cddis.2013.437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, Khoury JE, Landreth GE, Brosseron F, Feinstein DL, … Kummer MP (2015). Neuroinflammation in Alzheimer’s disease. The Lancet Neurology, 14(4), 388–405. doi: 10.1016/S1474-4422(15)70016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbig U, Jobling WA, Chen BP, Chen DJ, & Sedivy JM (2004). Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell, 14(4), 501–513. doi: 10.1016/s1097-2765(04)00256-4 [DOI] [PubMed] [Google Scholar]
- Herdy JR, Traxler L, Agarwal RK, Karbacher L, Schlachetzki JCM, Boehnke L, … Gage FH (2022). Increased post-mitotic senescence in aged human neurons is a pathological feature of Alzheimer’s disease. Cell Stem Cell, 29(12), 1637–1652.e1636. doi: 10.1016/j.stem.2022.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann S, Kedersha N, Anderson P, & Ivanov P (2021). Molecular mechanisms of stress granule assembly and disassembly. Biochim Biophys Acta Mol Cell Res, 1868(1), 118876. doi: 10.1016/j.bbamcr.2020.118876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofweber M, Hutten S, Bourgeois B, Spreitzer E, Niedner-Boblenz A, Schifferer M, Ruepp MD, Simons M, Niessing D, Madl T, Dormann D. Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell. 2018. Apr 19;173(3):706–719.e13 [DOI] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, … Liao D (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron, 68(6), 1067–1081. doi: 10.1016/j.neuron.2010.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Hickson LJ, Eirin A et al. Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrol 18, 611–627 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter S, Arendt T, & Brayne C (2013). The Senescence Hypothesis of Disease Progression in Alzheimer Disease: an Integrated Matrix of Disease Pathways for FAD and SAD. Molecular Neurobiology, 48(3), 556–570. doi: 10.1007/s12035-013-8445-3 [DOI] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong C-X, A. d. Alonso C, & Grundke-Iqbal I (2009). Mechanisms of tau-induced neurodegeneration. Acta Neuropathologica, 118(1), 53–69. doi: 10.1007/s00401-009-0486-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ, Cairns NJ, Grossman M, McMillan CT, Lee EB, Van Deerlin VM, … Trojanowski JQ (2015). Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol, 129(4), 469–491. doi: 10.1007/s00401-014-1380-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov PA, Chudinova EM, & Nadezhdina ES (2003). Disruption of microtubules inhibits cytoplasmic ribonucleoprotein stress granule formation. Exp Cell Res, 290(2), 227–233. doi: 10.1016/S0014-4827(03)00290-8 [DOI] [PubMed] [Google Scholar]
- Jacome Burbano MS, & Gilson E (2020). Long-lived post-mitotic cell aging: is a telomere clock at play? Mech Ageing Dev, 189, 111256. doi: 10.1016/j.mad.2020.111256 [DOI] [PubMed] [Google Scholar]
- Jiang L et al. TIA1 regulates the generation and response to toxic tau oligomers. Acta Neuropathol. 137, 259–277 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, … Cameron K (2012). Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell, 11(6), 996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakimura J-I, Kitamura Y, Takata K, Umeki M, Suzuki S, Shibagaki K, … Smith MA (2002). Microglial activation and amyloid-β clearance induced by exogenous heat-shock proteins. The FASEB Journal, 16(6), 601–603. [DOI] [PubMed] [Google Scholar]
- Kampers T, Friedhoff P, Biernat J, Mandelkow EM, & Mandelkow E (1996). RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett, 399(3), 344–349. doi: 10.1016/s0014-5793(96)01386-5 [DOI] [PubMed] [Google Scholar]
- Kang C (2019). Senolytics and Senostatics: A Two-Pronged Approach to Target Cellular Senescence for Delaying Aging and Age-Related Diseases. Mol Cells, 42(12), 821–827. doi: 10.14348/molcells.2019.0298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha NL, Gupta M, Li W, Miller I, Anderson P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2α to the assembly of mammalian stress granules. The Journal of cell biology. 1999. Dec 27;147(7):1431–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. The Journal of cell biology. 2000. Dec 11;151(6):1257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Chen S, Gilks N, Li W, Miller IJ, Stahl J, Anderson P. Evidence that ternary complex (eIF2-GTP-tRNAi Met)–deficient preinitiation complexes are core constituents of mammalian stress granules. Molecular biology of the cell. 2002. Jan 1;13(1):195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Ivanov P, & Anderson P (2013). Stress granules and cell signaling: more than just a passing phase? Trends Biochem Sci, 38(10), 494–506. doi: 10.1016/j.tibs.2013.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Panas MD, Achorn CA, Lyons S, Tisdale S, Hickman T, … Anderson P (2016). G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. The Journal of cell biology, 212(7), 845–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood TB, & Austad SN (2000). Why do we age? Nature, 408(6809), 233–238. doi: 10.1038/35041682 [DOI] [PubMed] [Google Scholar]
- Kotsantis P, Petermann E, & Boulton SJ (2018). Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov, 8(5), 537–555. doi: 10.1158/2159-8290.Cd-17-1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kritsilis M, V. R. S, Koutsoudaki PN, Evangelou K, Gorgoulis VG, & Papadopoulos D (2018). Ageing, Cellular Senescence and Neurodegenerative Disease. Int J Mol Sci, 19(10). doi: 10.3390/ijms19102937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Mooi WJ, & Peeper DS (2010). The essence of senescence. Genes Dev, 24(22), 2463–2479. doi: 10.1101/gad.1971610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBrasseur NK, Tchkonia T, & Kirkland JL (2015). Cellular Senescence and the Biology of Aging, Disease, and Frailty. Nestle Nutr Inst Workshop Ser, 83, 11–18. doi: 10.1159/000382054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CY, & Landreth GE (2010). The role of microglia in amyloid clearance from the AD brain. J Neural Transm (Vienna), 117(8), 949–960. doi: 10.1007/s00702-010-0433-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YR, King OD, Shorter J, & Gitler AD (2013). Stress granules as crucibles of ALS pathogenesis. Journal of Cell Biology, 201(3), 361–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Liu X, Liu M. Stress Granule Homeostasis, Aberrant Phase Transition, and Amyotrophic Lateral Sclerosis. ACS Chem Neurosci. 2022. Aug 17;13(16):2356–2370.372. doi: 10.1083/jcb.201302044 [DOI] [PubMed] [Google Scholar]
- Li ZY, Chen ZL, Zhang T, Wei C, & Shi WY (2016). TGF-β and NF-κB signaling pathway crosstalk potentiates corneal epithelial senescence through an RNA stress response. Aging (Albany NY), 8(10), 2337–2354. doi: 10.18632/aging.101050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian XJ, & Gallouzi IE (2009). Oxidative Stress Increases the Number of Stress Granules in Senescent Cells and Triggers a Rapid Decrease in p21waf1/cip1 Translation. J Biol Chem, 284(13), 8877–8887. doi: 10.1074/jbc.M806372200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnerbauer M, Wheeler MA, & Quintana FJ (2020). Astrocyte Crosstalk in CNS Inflammation. Neuron, 108(4), 608–622. doi: 10.1016/j.neuron.2020.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L et al. Trans-synaptic spread of tau pathology in vivo. PLOS ONE 7, e31302 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RM (2022). Aging, Cellular Senescence, and Alzheimer’s Disease. Int J Mol Sci, 23(4). doi: 10.3390/ijms23041989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C, Blasco MA, Partridge L, Serrano M, & Kroemer G (2013). The hallmarks of aging. Cell, 153(6), 1194–1217. doi: 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lye JJ, Latorre E, Lee BP, Bandinelli S, Holley JE, Gutowski NJ, … Harries LW (2019). Astrocyte senescence may drive alterations in GFAPα, CDKN2A p14(ARF), and TAU3 transcript expression and contribute to cognitive decline. GeroScience, 41(5), 561–573. doi: 10.1007/s11357-019-00100-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas T, Eidenmüller J, & Brandt R (2000). Interaction of Tau with the Neural Membrane Cortex Is Regulated by Phosphorylation at Sites That Are Modified in Paired Helical Filaments *. Journal of Biological Chemistry, 275(21), 15733–15740. doi: 10.1074/jbc.M000389200 [DOI] [PubMed] [Google Scholar]
- Mahboubi H, & Stochaj U (2014). Nucleoli and stress granules: connecting distant relatives. Traffic, 15(10), 1179–1193. doi: 10.1111/tra.12191 [DOI] [PubMed] [Google Scholar]
- Malinowska M, Niedźwiedzka-Rystwej P, Tokarz-Deptuła B, & Deptuła W (2016). Stress granules (SG) and processing bodies (PB) in viral infections. Acta Biochim Pol, 63(2), 183–188. doi: 10.18388/abp.2015_1060 [DOI] [PubMed] [Google Scholar]
- Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo EC, Krach F, Yang D, Sen A, Fulzele A, Wozniak JM, Gonzalez DJ, Kankel MW, Gao FB, Bennett EJ, Lécuyer E, Yeo GW. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell. 2018. Jan 25;172(3):590–604.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmiller S, Sathe S, Server KL, Nguyen TB, Fulzele A, Cody N, Javaherian A, Broski S, Finkbeiner S, Bennett EJ, Lécuyer E, Yeo GW. Persistent mRNA localization defects and cell death in ALS neurons caused by transient cellular stress. Cell Rep. 2021. Sep 7;36(10):109685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmor-Kollet H, Siany A, Kedersha N, Knafo N, Rivkin N, Danino YM, … & Hornstein E (2020). Spatiotemporal proteomic analysis of stress granule disassembly using APEX reveals regulation by SUMOylation and links to ALS pathogenesis. Molecular cell, 80(5), 876–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Cué C, & Rueda N (2020). Cellular Senescence in Neurodegenerative Diseases. Front Cell Neurosci, 14, 16. doi: 10.3389/fncel.2020.00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mas-Bargues C, Borrás C, & Viña J (2021). Bcl-xL as a Modulator of Senescence and Aging. Int J Mol Sci, 22(4). doi: 10.3390/ijms22041527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masrori P, & Van Damme P (2020). Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol, 27(10), 1918–1929. doi: 10.1111/ene.14393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateju D, Franzmann TM, Patel A, Kopach A, Boczek EE, Maharana S, Lee HO, Carra S, Hyman AA, Alberti S. 2017. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J 36:1669–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximova A, Werry EL, & Kassiou M (2021). Senolytics: A Novel Strategy for Neuroprotection in ALS? Int J Mol Sci, 22(21). doi: 10.3390/ijms222112078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D, & Gil J (2017). Senescence and aging: Causes, consequences, and therapeutic avenues. Journal of Cell Biology, 217(1), 65–77. doi: 10.1083/jcb.201708092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier S, Bell M, Lyons DN, Rodriguez-Rivera J, Ingram A, Fontaine SN, … Abisambra JF (2016). Pathological Tau Promotes Neuronal Damage by Impairing Ribosomal Function and Decreasing Protein Synthesis. J Neurosci, 36(3), 1001–1007. doi: 10.1523/jneurosci.3029-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan Z, Shewmaker F, & Pandey UB (2016). Stress granules at the intersection of autophagy and ALS. Brain Res, 1649(Pt B), 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti D, Grassilli E, Troiano L, Cossarizza A, Salvioli S, Barbieri D, … et al. (1992). Senescence, immortalization, and apoptosis. An intriguing relationship. Ann N Y Acad Sci, 673, 70–82. doi: 10.1111/j.1749-6632.1992.tb27438.x [DOI] [PubMed] [Google Scholar]
- Moreno-Blas D, Gorostieta-Salas E, Pommer-Alba A, Muciño-Hernández G, Gerónimo-Olvera C, Maciel-Barón LA, … Castro-Obregón S (2019). Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging (Albany NY), 11(16), 6175–6198. doi: 10.18632/aging.102181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgunova GV, Kolesnikov AV, Klebanov AA, & Khokhlov AN (2015). Senescence-associated β-galactosidase—A biomarker of aging, DNA damage, or cell proliferation restriction? Moscow University Biological Sciences Bulletin, 70(4), 165–167. doi: 10.3103/S0096392515040082 [DOI] [Google Scholar]
- Morimoto N, Nagai M, Ohta Y, Miyazaki K, Kurata T, Morimoto M, Murakami T, Takehisa Y, Ikeda Y, Kamiya T, Abe K. Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res., 1167 (2007), pp. 112–117 [DOI] [PubMed] [Google Scholar]
- Moujaber O, Mahboubi H, Kodiha M, Bouttier M, Bednarz K, Bakshi R, … & Stochaj U (2017). Dissecting the molecular mechanisms that impair stress granule formation in aging cells. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, 1864(3), 475–486. [DOI] [PubMed] [Google Scholar]
- Muñoz-Espín D, & Serrano M (2014). Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol, 15(7), 482–496. [DOI] [PubMed] [Google Scholar]
- Munro J, Barr NI, Ireland H, Morrison V, & Parkinson EK (2004). Histone deacetylase inhibitors induce a senescence-like state in human cells by a p16-dependent mechanism that is independent of a mitotic clock. Exp Cell Res, 295(2), 525–538. doi: 10.1016/j.yexcr.2004.01.017 [DOI] [PubMed] [Google Scholar]
- Musi N, Valentine JM, Sickora KR, Baeuerle E, Thompson CS, Shen Q, & Orr ME (2018). Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell, 17(6), e12840. doi: 10.1111/acel.12840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung NH, Zhu X, Kruman II, Castellani RJ, Petersen RB, Siedlak SL, … Lee HG (2008). Evidence of DNA damage in Alzheimer disease: phosphorylation of histone H2AX in astrocytes. Age (Dordr), 30(4), 209–215. doi: 10.1007/s11357-008-9050-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurohr GE, Terry RL, Lengefeld J, Bonney M, Brittingham GP, Moretto F, … Amon A (2019). Excessive Cell Growth Causes Cytoplasm Dilution And Contributes to Senescence. Cell, 176(5), 1083–1097 e1018. doi: 10.1016/j.cell.2019.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogrodnik M, Evans SA, Fielder E, Victorelli S, Kruger P, Salmonowicz H, … Jurk D (2021). Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell, 20(2), e13296. doi: 10.1111/acel.13296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omer A, Patel D, Lian XJ, Sadek J, Di Marco S, Pause A, … Gallouzi IE (2018). Stress granules counteract senescence by sequestration of PAI-1. EMBO Rep, 19(5). doi: 10.15252/embr.201744722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omer A, Patel D, Moran JL, Lian XJ, Di Marco S, & Gallouzi IE (2020). Autophagy and heat-shock response impair stress granule assembly during cellular senescence. Mech Ageing Dev, 192, 111382. doi: 10.1016/j.mad.2020.111382 [DOI] [PubMed] [Google Scholar]
- Ott BR, Jones RN, Daiello LA, de la Monte SM, Stopa EG, Johanson CE, … Grammas P (2018). Blood-Cerebrospinal Fluid Barrier Gradients in Mild Cognitive Impairment and Alzheimer’s Disease: Relationship to Inflammatory Cytokines and Chemokines. Front Aging Neurosci, 10, 245. doi: 10.3389/fnagi.2018.00245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Shin DW. Senotherapeutics and Their Molecular Mechanism for Improving Aging. Biomolecules & Therapeutics 2022; 30(6): 490–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatnitskaia S, Takahashi M, Kitaura H, Katsuragi Y, Kakihana T, Zhang L, … Fujii M (2019). USP10 is a critical factor for Tau-positive stress granule formation in neuronal cells. Sci Rep, 9(1), 10591. doi: 10.1038/s41598-019-47033-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plygawko A (2016). A Comparative Analysis of Stress Granule Assembly in Replicative and Stress-Induced Premature Senescence. Durham University, [Google Scholar]
- Prasad A, Bharathi V, Sivalingam V, Girdhar A, & Patel BK (2019). Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Frontiers in Molecular Neuroscience, 12. doi: 10.3389/fnmol.2019.00025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, … Ashe KH (2005). Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci, 25(46), 10637–10647. doi: 10.1523/jneurosci.3279-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM (2016). How neuroinflammation contributes to neurodegeneration. Science, 353(6301), 777–783. [DOI] [PubMed] [Google Scholar]
- Rao KS (2009). Free radical induced oxidative damage to DNA: relation to brain aging and neurological disorders. Indian J Biochem Biophys, 46(1), 9–15. [PubMed] [Google Scholar]
- Rayess H, Wang MB, & Srivatsan ES (2012). Cellular senescence and tumor suppressor gene p16. International journal of cancer, 130(8), 1715–1725. doi: 10.1002/ijc.27316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reineke LC, & Neilson JR (2019). Differences between acute and chronic stress granules, and how these differences may impact function in human disease. Biochem Pharmacol, 162, 123–131. doi: 10.1016/j.bcp.2018.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs CL, Kedersha N, Ivanov P, Anderson P. Mammalian stress granules and P bodies at a glance. J Cell Sci. 2020. Sep 1;133(16):jcs242487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzolio F, Lucchetti C, Caligiuri I, Marchesi I, Caputo M, Klein-Szanto AJ, … Giordano A (2012). Retinoblastoma tumor-suppressor protein phosphorylation and inactivation depend on direct interaction with Pin1. Cell Death Differ, 19(7), 1152–1161. doi: 10.1038/cdd.2011.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, & Campisi J (2011). Four faces of cellular senescence. The Journal of cell biology, 192(4), 547–556. doi: 10.1083/jcb.201009094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo L, Sjöberg MK, Hernández P, Zambrano C, & Maccioni RB (2006). Roles of cholesterol and lipids in the etiopathogenesis of Alzheimer’s disease. Journal of Biomedicine and Biotechnology, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roninson IB (2003). Tumor cell senescence in cancer treatment. Cancer Res, 63(11), 2705–2715. [PubMed] [Google Scholar]
- Sablina AA, Chudinova EM, Nadezhdina ES, & Ivanov PA (2012). [Stress granules in the cells with intact and discrupted microtubules: analysis with new algorithm of image processing]. Tsitologiia, 54(7), 560–565. [PubMed] [Google Scholar]
- Saez-Atienzar S, & Masliah E (2020). Cellular senescence and Alzheimer disease: the egg and the chicken scenario. Nature Reviews Neuroscience, 21(8), 433–444. doi: 10.1038/s41583-020-0325-z [DOI] [PubMed] [Google Scholar]
- Safaiyan S, Kannaiyan N, Snaidero N, Brioschi S, Biber K, Yona S, … Simons M (2016). Age-related myelin degradation burdens the clearance function of microglia during aging. Nature Neuroscience, 19(8), 995–998. doi: 10.1038/nn.4325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah E, Krishnamurthy S, Ahmidouch MY, Gillispie GJ, Milligan C, & Orr ME (2021). The Cellular Senescence Stress Response in Post-Mitotic Brain Cells: Cell Survival at the Expense of Tissue Degeneration. Life, 11(3), 229. Retrieved from https://www.mdpi.com/2075-1729/11/3/229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama R, Sadaie M, Hoare M, Narita MJG, & development. (2014). Cellular senescence and its effector programs. 28(2), 99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DW et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82, 1271–1288 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapieha P, & Mallette FA (2018). Cellular Senescence in Postmitotic Cells: Beyond Growth Arrest. Trends in Cell Biology, 28(8), 595–607. doi: 10.1016/j.tcb.2018.03.003 [DOI] [PubMed] [Google Scholar]
- Schimanski LA, Barnes CA. Neural Protein Synthesis during Aging: Effects on Plasticity and Memory. Front Aging Neurosci. 2010. Aug 6;2:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher B, Pothof J, Vijg J, & Hoeijmakers JHJ (2021). The central role of DNA damage in the ageing process. Nature, 592(7856), 695–703. doi: 10.1038/s41586-021-03307-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, & Blasco MA (2001). Putting the stress on senescence. Curr Opin Cell Biol, 13(6), 748–753. doi: 10.1016/s0955-0674(00)00278-7 [DOI] [PubMed] [Google Scholar]
- Sharpless NE, & Sherr CJ (2015). Forging a signature of in vivo senescence. Nature Reviews Cancer, 15(7), 397–408. doi: 10.1038/nrc3960 [DOI] [PubMed] [Google Scholar]
- Si Z, Sun L, & Wang X (2021). Evidence and perspectives of cell senescence in neurodegenerative diseases. Biomedicine & Pharmacotherapy, 137, 111327. doi: 10.1016/j.biopha.2021.111327 [DOI] [PubMed] [Google Scholar]
- Sierra A, Gottfried-Blackmore AC, McEwen BS, & Bulloch K (2007). Microglia derived from aging mice exhibit an altered inflammatory profile. Glia, 55(4), 412–424. doi: 10.1002/glia.20468 [DOI] [PubMed] [Google Scholar]
- Sikora E, Bielak-Zmijewska A, Dudkowska M, Krzystyniak A, Mosieniak G, Wesierska M, & Wlodarczyk J (2021). Cellular Senescence in Brain Aging. Front Aging Neurosci, 13, 646924. doi: 10.3389/fnagi.2021.646924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smethurst P et al. In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol Dis. 96, 236–247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Song Y, Kincaid B, Bossy B, & Bossy-Wetzel E (2013). Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: neuroprotection by SIRT3 and PGC-1α. Neurobiol Dis, 51, 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soo KY, Halloran M, Sundaramoorthy V, Parakh S, Toth RP, Southam KA, McLean CA, Lock P, King A, Farg MA, Atkin JD. Rab1-dependent ER-Golgi transport dysfunction is a common pathogenic mechanism in SOD1, TDP-43 and FUS-associated ALS. Acta Neuropathol., 130 (2015), pp. 679–697 [DOI] [PubMed] [Google Scholar]
- Spittau B (2017). Aging Microglia-Phenotypes, Functions and Implications for Age-Related Neurodegenerative Diseases. Front Aging Neurosci, 9, 194. doi: 10.3389/fnagi.2017.00194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit L, Kuhn T, Vomhof T et al. Stress induced TDP-43 mobility loss independent of stress granules. Nat Commun 13, 5480 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamer K, Vogel R, Thies E, Mandelkow E, & Mandelkow EM (2002). Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. The Journal of cell biology, 156(6), 1051–1063. doi: 10.1083/jcb.200108057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Sammons NW, Kuhns AJ, & Sparks DL (2004). Dystrophic microglia in the aging human brain. Glia, 45(2), 208–212. doi: 10.1002/glia.10319 [DOI] [PubMed] [Google Scholar]
- Takahashi M, Higuchi M, Matsuki H, Yoshita M, Ohsawa T, Oie M, & Fujii M (2013). Stress Granules Inhibit Apoptosis by Reducing Reactive Oxygen Species Production. Mol Cell Biol, 33(4), 815–829. doi: 10.1128/MCB.00763-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Kitaura H, Kakita A, Kakihana T, Katsuragi Y, Nameta M, … Fujii M (2018). USP10 Is a Driver of Ubiquitinated Protein Aggregation and Aggresome Formation to Inhibit Apoptosis. iScience, 9, 433–450. doi: 10.1016/j.isci.2018.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thonhoff JR, Gao J, Dunn TJ, Ojeda L, & Wu P (2012). Mutant SOD1 microglia-generated nitroxidative stress promotes toxicity to human fetal neural stem cell-derived motor neurons through direct damage and noxious interactions with astrocytes. Am J Stem Cells, 1(1), 2–21. [PMC free article] [PubMed] [Google Scholar]
- Tran JR, Chen H, Zheng X, & Zheng Y (2016). Lamin in inflammation and aging. Curr Opin Cell Biol, 40, 124–130. doi: 10.1016/j.ceb.2016.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trias E, Beilby PR, Kovacs M, Ibarburu S, Varela V, Barreto-Núñez R, … Barbeito L (2019). Emergence of Microglia Bearing Senescence Markers During Paralysis Progression in a Rat Model of Inherited ALS. Front Aging Neurosci, 11, 42. doi: 10.3389/fnagi.2019.00042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnquist C, Horikawa I, Foran E, Major EO, Vojtesek B, Lane DP, … Harris CC (2016). p53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell Death & Differentiation, 23(9), 1515–1528. doi: 10.1038/cdd.2016.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerleider K, Beck J, Lissa D, Turnquist C, Horikawa I, Harris BT, & Harris CC (2021). Astrocyte senescence and SASP in neurodegeneration: tau joins the loop. Cell Cycle, 20(8), 752–764. doi: 10.1080/15384101.2021.1909260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen JM (2014). The role of senescent cells in ageing. Nature, 509(7501), 439–446. doi: 10.1038/nature13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PEA, Cook C, Lummertz da Rocha E, … Wolozin B (2016). Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Reports, 15(7), 1455–1466. doi: 10.1016/j.celrep.2016.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T, Youmans K, Liu-Yesucevitz L, & Wolozin B (2013). Role of Stress Granules and RNA-Binding Proteins in Neurodegeneration: A Mini-Review. Gerontology, 59(6), 524–533. doi: 10.1159/000354170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T, Yu H, Varnum M, Liu-Yesucevitz L, Citro A, Ikezu T, … Wolozin B (2012). Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J Neurosci, 32(24), 8270–8283. doi: 10.1523/jneurosci.1592-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis Vassilis, A. PD, Alimonti Andrea, Bennett Dorothy C., Bischof Oliver, Bishop Cleo, Campisi Judith, Collado Manuel, Evangelou Konstantinos, Ferbeyre Gerardo, Gil Jesús, Hara Eiji, Krizhanovsky Valery, Jurk Diana, Maier Andrea B., Narita Masashi, Niedernhofer Laura, Passos João F., Robbins Paul D., Schmitt Clemens A., Sedivy John, Vougas Konstantinos, von Zglinicki Thomas, Zhou Daohong, Serrano Manuel, Demaria Marco,. (2019). Cellular Senescence: Defining a Path Forward,. Cell, 179(4), 813–827. Retrieved from 10.1016/j.cell.2019.10.005 [DOI] [PubMed] [Google Scholar]
- Vicencio JM, Galluzzi L, Tajeddine N, Ortiz C, Criollo A, Tasdemir E, … Kroemer G (2008). Senescence, apoptosis or autophagy? When a damaged cell must decide its path--a mini-review. Gerontology, 54(2), 92–99. doi: 10.1159/000129697 [DOI] [PubMed] [Google Scholar]
- Von Bernhardi R (2007). Glial cell dysregulation: a new perspective on Alzheimer disease. Neurotoxicity research, 12, 215–232. [DOI] [PubMed] [Google Scholar]
- von Kobbe C (2018). Cellular senescence: a view throughout organismal life. Cell Mol Life Sci, 75(19), 3553–3567. doi: 10.1007/s00018-018-2879-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajapeyee N, Serra RW, Zhu X, Mahalingam M, & Green MR (2008). Oncogenic BRAF Induces Senescence and Apoptosis through Pathways Mediated by the Secreted Protein IGFBP7. Cell, 132(3), 363–374. doi: 10.1016/j.cell.2007.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L, McAleese KE, Thomas AJ, Johnson M, Martin-Ruiz C, Parker C, … Attems J (2015). Neuropathologically mixed Alzheimer’s and Lewy body disease: burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol, 129(5), 729–748. doi: 10.1007/s00401-015-1406-3 [DOI] [PubMed] [Google Scholar]
- Wang J, Gan Y, Cao J, Dong X, & Ouyang W (2022). Pathophysiology of stress granules: An emerging link to diseases (Review). Int J Mol Med, 49(4). doi: 10.3892/ijmm.2022.5099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Yang X, Cristofalo VJ, Holbrook NJ, & Gorospe M (2001). Loss of HuR is linked to reduced expression of proliferative genes during replicative senescence. Mol Cell Biol, 21(17), 5889–5898. doi: 10.1128/mcb.21.17.5889-5898.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel J, Imaki H, Wang K-C, Wegiel J, & Rubenstein R (2004). Cells of monocyte/microglial lineage are involved in both microvessel amyloidosis and fibrillar plaque formation in APPsw tg mice. Brain Research, 1022(1–2), 19–29. [DOI] [PubMed] [Google Scholar]
- Wei Z, Chen XC, Song Y, Pan XD, Dai XM, Zhang J, … Zhu YG (2016). Amyloid β Protein Aggravates Neuronal Senescence and Cognitive Deficits in 5XFAD Mouse Model of Alzheimer’s Disease. Chin Med J (Engl), 129(15), 1835–1844. doi: 10.4103/0366-6999.186646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler JR, Matheny T, Jain S, Abrisch R, & Parker R (2016). Distinct stages in stress granule assembly and disassembly. eLife, 5, e18413. doi: 10.7554/eLife.18413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, & Griffin WS (2013). Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J Neuroinflammation, 10, 84. doi: 10.1186/1742-2094-10-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, … Campisi J (2016). Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab, 23(2), 303–314. doi: 10.1016/j.cmet.2015.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, Ravikumar B, & Rubinsztein DC (2006). Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol, 76, 89–101. [DOI] [PubMed] [Google Scholar]
- Wolozin B (2012). Regulated protein aggregation: stress granules and neurodegeneration. Molecular Neurodegeneration, 7(1), 56. doi: 10.1186/1750-1326-7-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B, & Ivanov P (2019). Stress granules and neurodegeneration. Nat Rev Neurosci, 20(11), 649–666. doi: 10.1038/s41583-019-0222-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, & Felsher DW (2007). Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A, 104(32), 13028–13033. doi: 10.1073/pnas.0701953104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Q, Wang H, Hao Z, Fu C, Hu Q, Gao F, Ren H, Chen D, Han J, Ying Z, Wang G. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J, 35 (2016), pp. 121–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, … Lowe SW (2007). Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature, 445(7128), 656–660. doi: 10.1038/nature05529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, Mathieu C, Kolaitis RM, Zhang P, Messing J, Yurtsever U, Yang Z, Wu J, Li Y, Pan Q, Yu J. G3BP1 is a tunable switch that triggers phase separation to assemble stress granules. Cell. 2020. Apr 16;181(2):325–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Huang S, Gaertig Marta A., Li X-J, & Li S (2014). Age-Dependent Decrease in Chaperone Activity Impairs MANF Expression, Leading to Purkinje Cell Degeneration in Inducible SCA17 Mice. Neuron, 81(2), 349–365. doi: 10.1016/j.neuron.2013.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates SC, Zafar A, Rabai EM, Foxall JB, Nagy S, Morrison KE, … Nagy Z (2015). The effects of two polymorphisms on p21cip1 function and their association with Alzheimer’s disease in a population of European descent. PLoS One, 10(1), e0114050. doi: 10.1371/journal.pone.0114050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HM, Zhao YM, Luo XG, Feng Y, Ren Y, Shang H, … Wang XY (2012). Repeated lipopolysaccharide stimulation induces cellular senescence in BV2 cells. Neuroimmunomodulation, 19(2), 131–136. doi: 10.1159/000330254 [DOI] [PubMed] [Google Scholar]
- Yu Q-Y, Ye L-Q, & Li H-L (2022). Molecular interaction of stress granules with Tau and autophagy in Alzheimer’s disease. Neurochemistry International, 157, 105342. doi: 10.1016/j.neuint.2022.105342 [DOI] [PubMed] [Google Scholar]
- Yuan J, Luo K, Zhang L, Cheville JC, & Lou Z (2010). USP10 regulates p53 localization and stability by deubiquitinating p53. Cell, 140(3), 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S, … Mattson MP (2019). Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nature Neuroscience, 22(5), 719–728. doi: 10.1038/s41593-019-0372-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Fan B, Yang P, Temirov J, Messing J, Kim HJ, Taylor JP. Chronic optogenetic induction of stress granules is cytotoxic and reveals the evolution of ALS-FTD pathology. Elife. 2019. Mar 20;8:e39578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gu J, Sun Q. Aberrant Stress Granule Dynamics and Aggrephagy in ALS Pathogenesis. Cells. 2021. Aug 30;10(9):2247. [DOI] [PMC free article] [PubMed] [Google Scholar]