

Summary
ATG5 is a part of the E3 ligase directing lipidation of ATG8 proteins, a process central to membrane atg8ylation and canonical autophagy. Loss of Atg5 in myeloid cells causes early mortality in murine models of tuberculosis. This in vivo phenotype is specific to ATG5. Here we show using human cell lines that absence of ATG5 but not of other ATGs directing canonical autophagy promotes lysosomal exocytosis and secretion of extracellular vesicles, and, in murine Atg5fl/fl LysM-Cre neutrophils, their excessive degranulation. This is due to lysosomal disrepair in ATG5 knockout cells and the sequestration by an alternative conjugation complex, ATG12-ATG3, of ESCRT protein ALIX which acts in membrane repair and exosome secretion. These findings reveal a previously undescribed function of ATG5 in its host-protective role in murine experimental models of tuberculosis and emphasize the significance of the branching aspects of the atg8ylation conjugation cascade beyond the canonical autophagy.
eTOC blurb
Wang et al., describe properties of a core autophagy protein ATG5 that fall outside of its canonical autophagy functions. Loss of ATG5 dysregulates lysosomal repair, increases exocytosis and exosome release, and in neutrophils causes hyperactivation and degranulation, partly explaining the function of ATG5 in mouse models of tuberculosis
Graphical Abstract
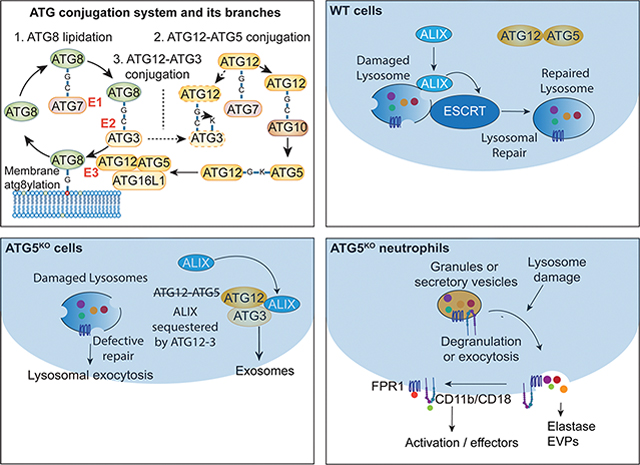
Introduction
Autophagy in mammalian cells is a complex set of homeostatic processes 1, including canonical degradative 2,3 and noncanonical processes 4,5. During ATG-dependent canonical autophagy, membranes from the constitutive secretory pathway 6–16 contribute to the formation of phagophores through fusion with endosomal organelles 17,18. Nascent phagophores enlarge via lipid transfer and close into double membrane autophagosomes 2. At the end-stage, canonical autophagosomes fuse with lysosomes to form autolysosomes 19.
A hallmark of canonical 6,20–23 and noncanonical autophagy 4,5 is lipidation of mammalian ATG8 proteins (mATG8s), mirroring lipidation of Atg8 in yeast 24,25. Whereas one of the mATG8s, LC3B, is commonly used to identify autophagosomes 26, it is also present on single membranes other than autophagosomes 4,27–36. Among the functions of mATG8s are the enhancement of cargo sequestration into autophagosomes 37, membrane remodeling 38 perturbation 39 and expansion 40,41 during autophagosome biogenesis, autophagosome-lysosome fusion 19, and more general membrane stress responses 5.
The modification of autophagic and other membranes by mATG8s, a process referred to as membrane atg8ylation 5, depends on conjugation of ubiquitin-like protein ATG12 to ATG5 by way of successive reactions involving E1-like enzyme ATG7 and E2-like enzyme ATG10 20. The resulting ATG12-ATG5 conjugate in a complex with Atg16L1, forms the Atg12-Atg5-Atg16L1 E3 ligase which guides lipidation of mATG8s 20. In a parallel cascade, mATG8s are activated at their exposed C-terminal Gly residue by the E1-like ATG7 and the E2-like ATG3 enzymes 20. A cross-over between the ATG12 conjugation and mATG8 conjugation branches has been reported, whereby a noncanonical ATG12-ATG3 conjugate is generated 42 with unconventional outputs via the ESCRT protein ALIX action in exosome secretion 43. ALIX also participates in the repair of lysosomal membranes thus maintaining acidification and functionality of lysosomes 44–47, whereas robustness and quality of the lysosomes is a determinant of effective autophagy 19.
The maturation of autophagosomes into autolysosomes where the captured cargo is degraded has many physiological outputs 48. Among such functions are the control of inflammation and direct elimination of intracellular pathogens 49. In macrophage infection models, induction of autophagy has been reported as a mechanism of controlling intracellular Mycobacterium tuberculosis (Mtb) 50. In murine models of Mtb infection, genetic knockout of Atg5 in myeloid cells caused neutrophilic inflammation and increased mortality 51–53, whereas a loss of other Atg genes had no apparent consequences in short term studies 53. Collectively, a conundrum has emerged of whether canonical autophagy acts here as a pathway and if all ATGs contribute to control of Mtb. In this study, we show that ATG5 has unique properties and functionalities outside of the canonical autophagy pathway. In the absence of ATG5, but not in the absence of other canonical autophagy factors, lysosomes are uncapable of recruiting ALIX and ESCRT repair machinery and are more sensitive to damage. The increased vulnerability of lysosomal membranes in ATG5 knockout cells contributes to a range of exocytic and secretory phenotypes. The lysosomal handicap impacts degranulation and other secretory functions of neutrophils, the inflammatory cells that have been previously linked to Mtb pathogenesis 51–54.
Results
Loss of ATG5 but not of other ATGs renders lysosomes excessively susceptible to damage
The initial reports testing in vivo role of autophagy genes in Mtb infection have shown that myeloid-specific Atg5 knockout, using Atg5fl/fl LysM-Cre transgenic mice, renders mice more susceptible to respiratory Mtb infection 51,52. Follow-up short-term infection studies (80 days) have reported that this Atg5 phenotype was unique among Atg genes and that a similar myeloid knockout in Atg7 (Atg7fl/fl LysM-Cre) transgenic mice was not sensitized to Mtb 53. We assessed these observations and extended them by employing a standard long-term low dose chronic Mtb infection model (aerosol exposure) 55–57 by monitoring mice for up to 200 days (Figure 1A). The results affirmed prior short-term observations with statistically significant differences in mortality between Atg5fl/fl LysM-Cre+ vs. Atg5fl/fl LysM-Cre− mice but not between Atg7fl/fl LysM-Cre+ vs. Atg7fl/fl LysM-Cre− mice (Figure 1A). We ascribe an early high mortality in WT (Atg5fl/fl LysM-Cre−) mice to the metagenomic effects since we used only littermates in infection experiments 58.
Figure 1. Loss of ATG5 but not of other ATGs renders lysosomes excessively susceptible to damage.
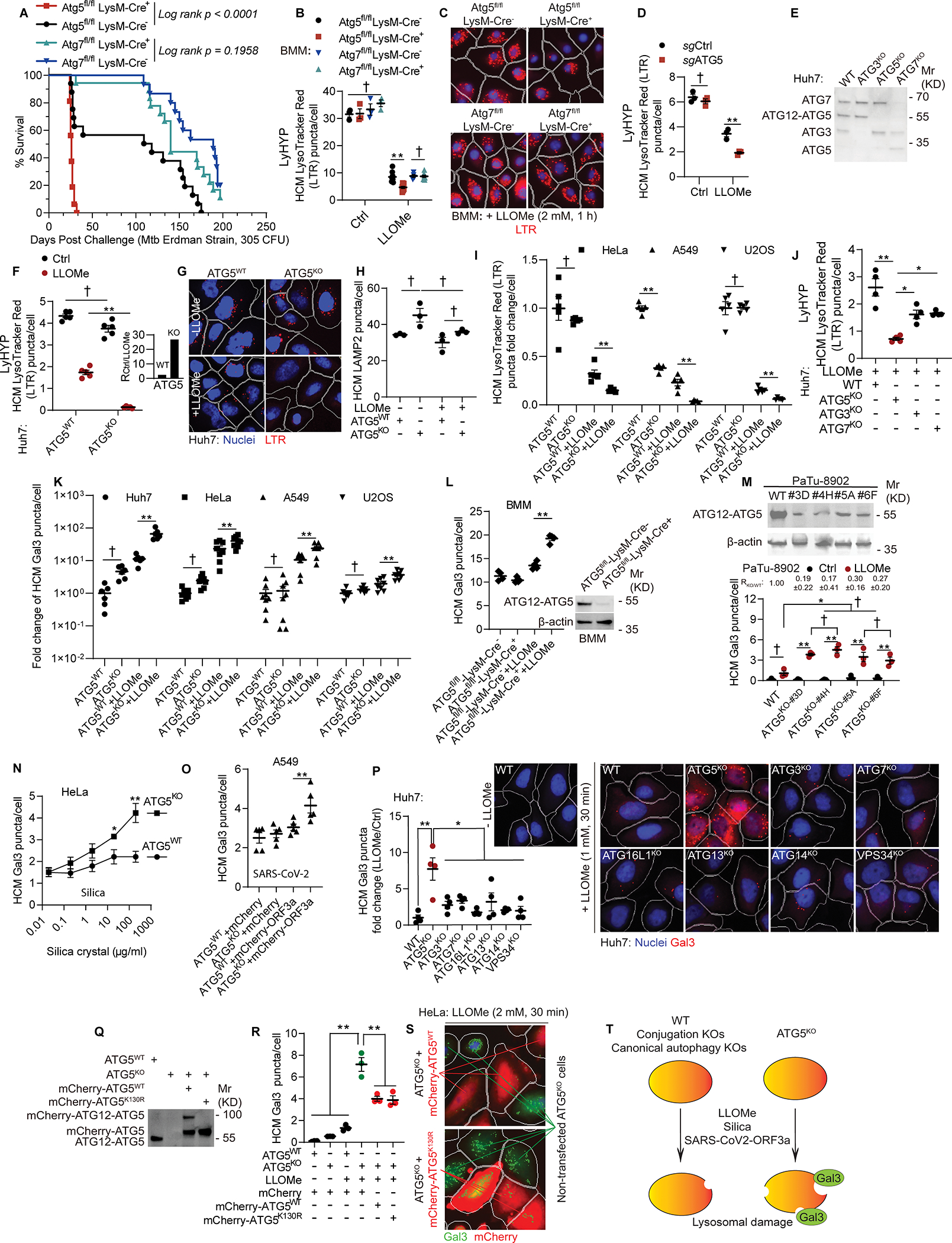
A, Survival curves (mice) post aerosol challenge with Mtb (Erdman; initial lung deposition of 305 CFU). Log-rank (Montel-Cox) test. B, Quantification and C, representative images of LysoTracker Red (LTR) puncta in bone marrow derived macrophage (BMMs) isolated from Atg5fl/fl LysM-Cre−, Atg5fl/fl LysM-Cre+, Atg7fl/fl LysM-Cre−, Atg7fl/fl LysM-Cre+ mice treated with 2 mM of LLOMe for 1 h using High Content Microscopy (HCM). Data, means ± SE (n≥4); unpaired t-test. Each data point represents independent biological replicates with BMMs from different animals. D, HCM quantification of LTR in BMMs subjected to CRISPR inactivation of Atg5 gene (Atg5BMM-CRISPR-KO) compared to Atg5BMM-WT control guide RNA treated cells. Data, means ± SE (n=3); unpaired t-test. E, Immunoblots, Huh7 and mutant cells. F, HCM quantification and G, representative image of LTR puncta in Huh7ATG5-WT and Huh7ATG5-KO cells treated with 1 mM LLOMe for 30 min. Data, means ± SE (n=5); two-way ANOVA with Tukey’s multiple comparisons. Inset, ratio of LTR puncta in Huh7ATG5-WT and Huh7ATG5-KO cells treated:untreated with LLOMe. H, HCM quantification of LAMP2 puncta in Huh7ATG5-WT and Huh7ATG5-KO cells treated with 1 mM LLOMe for 30 min. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. I, HCM quantification of LTR puncta in HeLa (2 mM LLOMe, 30 min), A549 and U2OS (1 mM LLOMe, 30 min) and mutant cells. Data, means ± SE (n=5); unpaired t-test. J, HCM quantification of LTR puncta in Huh7 and mutant (1 mM LLOMe, 30 min). Data, means ± SE (n=4); one-way ANOVA with Tukey’s multiple comparisons. K, HCM quantification of Gal3 puncta in cell lines and under conditions as in panel I. Data, means ± SE (n=6); two-way ANOVA with Tukey’s multiple comparisons. L, HCM quantification of Gal3 puncta in BMMs from Atg5fl/fl LysM-Cre− and Atg5fl/fl LysM-Cre+ mice treated with 2 mM LLOMe for 1 h. Data, means ± SE (n=4); two-way ANOVA with Tukey’s multiple comparisons. Each point represents independent biological replicate from BMMs derived from bone marrows of different animals. M, Immunoblot (top) and HCM quantification (bottom) of Gal3 puncta in PaTu-8902WT and PaTu-8902ATG5-KO cells treated or not with 1.5 mM LLOMe for 30 min. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. N, HCM quantification of Gal3 puncta in HeLaATG5-WT and HeLaATG5-KO cells treated with silica. Data, means ± SE (n=6); unpaired t-test. O, HCM quantification of Gal3 puncta in A549ATG5-WTand A549ATG5-KO cells overexpressing mCherry or mCherry-ORF3aSARS-CoV-2. Data, means ± SE (n=5); two-way ANOVA with Tukey’s multiple comparisons. P, HCM quantification and representative image of Gal3 puncta in Huh7 and mutant cells (1 mM LLOMe, 30 min). Data, means ± SE (n=4); One-way ANOVA with Tukey’s multiple comparisons. Q, Immunoblot of Huh7WT and Huh7ATG5-KO cells overexpressing or not overexpressing mCherry-ATG5WT or mCherry-ATG5K130R. R, HCM quantification and S, representative image of Gal3 puncta in HeLaATG5-WT and HeLaATG5-KO cells control or transfected with mCherry-ATG5WT or mCherry-ATG5K130R (2 mM LLOMe, 30 min). Data, means ± SE (n=3); unpaired t-test. T, Summary of the findings in Figure 1. In all HCM graphs, each individual data point represents independent biological replicate, with values based on 49–64 fields/well, >500 primary objects (cells) per well, 5 wells per sample (used only for sampling error) with bars denoting the mean for the biological replicates and standard errors of the mean. Statistical significance symbols in all panels, †p≥0.05, *p<0.05, **p<0.01.
Prior work has also indicated alterations in inflammatory mediators linked to properties of macrophages and neutrophils 51–53 that remain to be fully characterized. Our recent studies and studies by others have indicated that mammalian ATG factors participate in the processes of atg8ylation 5, ESCRT-dependent repair 59, and canonical autophagy 2,60, which among other effects contribute to lysosomal quality control. Lysosomes are important degradative organelles 61, of particular significance in professional phagocytic cells. Hence, we tested whether there are differences in lysosomal quality between primary bone marrow-derived macrophages (BMMs) from Atg5fl/fl LysM-Cre+ and Atg7fl/fl LysM-Cre+ mice. In these experiments we used LysoTracker Red (LTR) as a measure of the lysosomal status, and quantified LTR+ profiles by high content microscopy (HCM). HCM depends on epifluorescence mode of image capture to encompass all cellular profiles in a volume and allows unbiased machine-driven image acquisition and data analysis independently of the operator (each experiment consists of machine-identified 500 valid primary objects/cells per well, ≥5 wells/sample, in biological replicates of n≥3) 18,47,59,62,63. We treated ex vivo BMMs from Atg5fl/fl LysM-Cre+ and Atg7fl/fl LysM-Cre+ mice with a standard Cathepsin C-dependent lysosome-damaging agent Leu-Leu-OMe (LLOMe), which enters cells and endomembranous organelles by diffusion and specifically in lysosomes is converted into membrane-damaging polymers via a reverse dipeptidase action of cathepsins 47,64–68 (Figure S1A). The Atg5fl/fl LysM-Cre+ BMMs showed lysosomal hypersensitivity (LyHYP) phenotype when treated with LLOMe in comparison to BMMs from Atg7fl/fl LysM-Cre+ mice, evidenced by the reduced number of LTR+ profiles of LTR+ compartments per cell (Figures 1B and 1C). Additionally, we confirmed dependence on ATG5 of the LyHYP phenotype in BMMs, by ex vivo downregulating ATG5 in BMMs from Atg5fl/fl LysM-Cre− by employing an optimized strategy for CRISPR-dependent gene disruption in primary myeloid cells 69. BMMs from Atg5fl/fl LysM-Cre− mice subjected to CRISPR with sgATG5 and Cas9 protein displayed LyHYP phenotype relative to BMMs treated with control guide RNA and Cas9 (Figures 1D and S1B).
To further dissect the lysosomal quality variances between ATG5- and ATG7-deficient cells, we tested whether the LyHYP phenotype was applicable to other cells. We generated ATG5 (Huh7ATG5-KO) and ATG7 (Huh7ATG7-KO) CRISPR knockouts in human hepatocyte-derived carcinoma cell line Huh7 (Figure 1E). We observed that lysosomes in Huh7ATG5-KO cells are more sensitive to damage, i.e. express LyHYP phenotype, relative to their Huh7WT parental cells (Figures 1F, 1G and S1C; note ratios in Figure 1F inset). To ensure that the cellular content of lysosomes did not change in cells displaying lower number of LTR+ profiles, we quantified lysosomal content (LAMP2+ organelles) did not significantly differ between Huh7ATG5-KO and Huh7WT parental cells treated with LLOMe (Figures 1H and S1D). We next generated ATG5KO in other human cell lines, HeLa, U2OS, and A549 (Figure S1E), and confirmed a LyHYP phenotype in all of the tested ATG5KO lines (Figures 1I and S1O). No differences in lysosomal marker (LAMP1) profiles were observed in HeLaATG5-KO compared to HeLaWT treated with LLOMe (Figures S1F and S1G). LLOMe at concentrations used did not induce cell death or plasma membrane permeabilization as quantified by PI staining, with only 1–3 % of LLOMe-treated adherent cells being PI+ whether ATG5 WT or KO (Figures S1H–S1J). We ruled out that LyHYP could be explained by elevated Cathepsin C cellular levels, which were not increased in the absence of ATG5 in all cell types used herein (Figures S1K and S1L). LyHYP could not be explained by changes in endocytosis, as Dextran-TxR-3000 endocytic uptake was not altered in ATG5 KO relative to ATG5 WT cells (Figures S1M and S1N).
When we compared Huh7ATG7-KO with Huh7ATG5-KO isogenic Huh7 cell lines we observed that, like Atg7fl/fl LysM-Cre+ vs Atg5fl/fl LysM-Cre+ BMMs, Huh7ATG7-KO and Huh7ATG5-KO displayed different sensitivity to lysosomal damage. Huh7ATG5-KO had a more pronounced loss of LTR+ profiles relative to Huh7ATG7-KO (Figure 1J). Moreover, another KO mutant affecting atg8ylation conjugation cascade, ATG3 (Figure 1E), tracked with the milder effects of ATG7 loss relative to the highly pronounced effect of ATG5 loss on increased LyHYP (Figure 1J).
We employed another marker of lysosomal damage, galectin 3 (Gal3), a β-galactoside-binding cytosolic lectin that is recruited to lysosomes during their membrane damage by recognizing exposed lumenal glycans 64, and can be used for quantitative assessment of lysosomal damage by HCM as previously described 47. Upon treatment with LLOMe, all cell lines tested (Huh7, HeLa, A549, and U2OS) showed a quantifiable Gal3 response (Figures 1K and S1P). Further, Gal3 puncta response to LLOMe treatment was higher in ATG5KO derivatives relative to parental ATG5 WT cells (Figures 1K and S1P). Primary macrophages mirrored these properties, as Atg5fl/fl LysM-Cre+ BMMs showed stronger Gal3 response to LLOMe treatment than Atg5fl/fl LysM-Cre− BMMs (Figures 1L and S1Q). We generated partial ATG5KO clones of pancreatic PaTu-8902 carcinoma cell line 70,71, and observed that Gal3 response was elevated in CRISPR clones with 20–30% residual ATG12-ATG5 conjugate (Figures 1M and S1R).
The above relationships were observed using lysosomal damaging agents other than LLOMe, including silica crystals 47,72–74 and ORF3aSARS-CoV-2 75,76, which elicited elevated Gal3 responses in ATG5KO cells (Figures 1N, 1O, S1S, and S1T). We additionally verified the effects of ATG5 loss on ORF3aSARS-CoV-2-induced lysosomal damage by LysoTracker Green (LTG) staining, and observed LyHYP in A549ATG5-KO relative to A549WT (Figure S1U and S1V).
Further, we generated a series of isogenic cell lines derived from Huh7 cells, adding ATG16L1, ATG13, ATG14 and VPS34 (Figure S1W) to the ATG7 and ATG3 KOs described above (Figure 1E) and assessed their sensitivity to lysosomal damage using Gal3 for quantification of their responses (Figure 1P). Huh7ATG7-KO and Huh7ATG5-KO isogenic cell lines had different Gal3 responses to LLOMe, with Huh7ATG5-KO cells showing more Gal3 puncta than Huh7ATG7-KO (Figure 1P) in keeping with ATG5 having stronger impact on lysosomal status and quality control than ATG7. The KOs of all other ATGs tested showed less pronounced Gal3 response compared to ATG5KO cells (Figure 1P). Thus, ATG5 displays unique properties among all ATGs tested in rendering lysosomes more susceptible to damage.
We next tested whether LyHYP can be complemented by ATG5. We used two alleles of ATG5, ATG5WT and its conjugation-deficient mutant, ATG5K130R (Figure 1Q). We transfected HeLaATG5-KO cells with mCherry-ATG5WT or mCherry-ATG5K130R and assessed LyHYP upon LLOMe treatment by quantifying Gal3 and LTG responses by gating on transfected cells (Figures 1R, 1S, S1X, and S1Y). Both ATG5WT and ATG5K130R complemented HeLaATG5-KO cells and rescued lysosomal hypersensitivity phenotype (Figures 1R, 1S, S1X, and S1Y). Whereas it may appear surprising that both WT and conjugation-deficient ATG5 complemented the ATG5KO, this was in keeping with the observations that conjugation machinery (represented by ATGs such as ATG3, ATG7, and ATG16L1) did not strongly affect LyHYP (Figure 1P). Thus, ATG5 affects lysosomal sensitivity to damage independent of conjugation and canonical autophagy machinery (Figure 1T).
ATG5 is required for recruitment of ESCRTs to repair damaged lysosomes
Lysosomal homeostasis and membrane maintenance involve ESCRT-dependent mechanisms with protein ALIX playing a major role in lysosomal membrane repair 44,47,77,78. Thus, we tested whether ATG5 has an effect on ALIX recruitment to damaged lysosomes. In Huh7ATG5-WT cells, ALIX puncta were induced and ALIX colocalized with lysosomes upon 30 min of LLOMe-exposure, whereas in Huh7ATG5-KO this response was diminished (Figures 2A–2C and S2A–S2C). This was not due to a decrease in ALIX protein levels (Figure S2D). In contrast to ALIX, another ESCRT-I component, TSG101, known to participate in lysosomal damage repair 44,77, was detectable at very low levels as sparse puncta, and it too was affected (a measurable decrease) by the absence of ATG5 (Figures S2E and S2F).
Figure 2. ATG5 is required for recruitment of ESCRTs to repair damaged lysosomes.
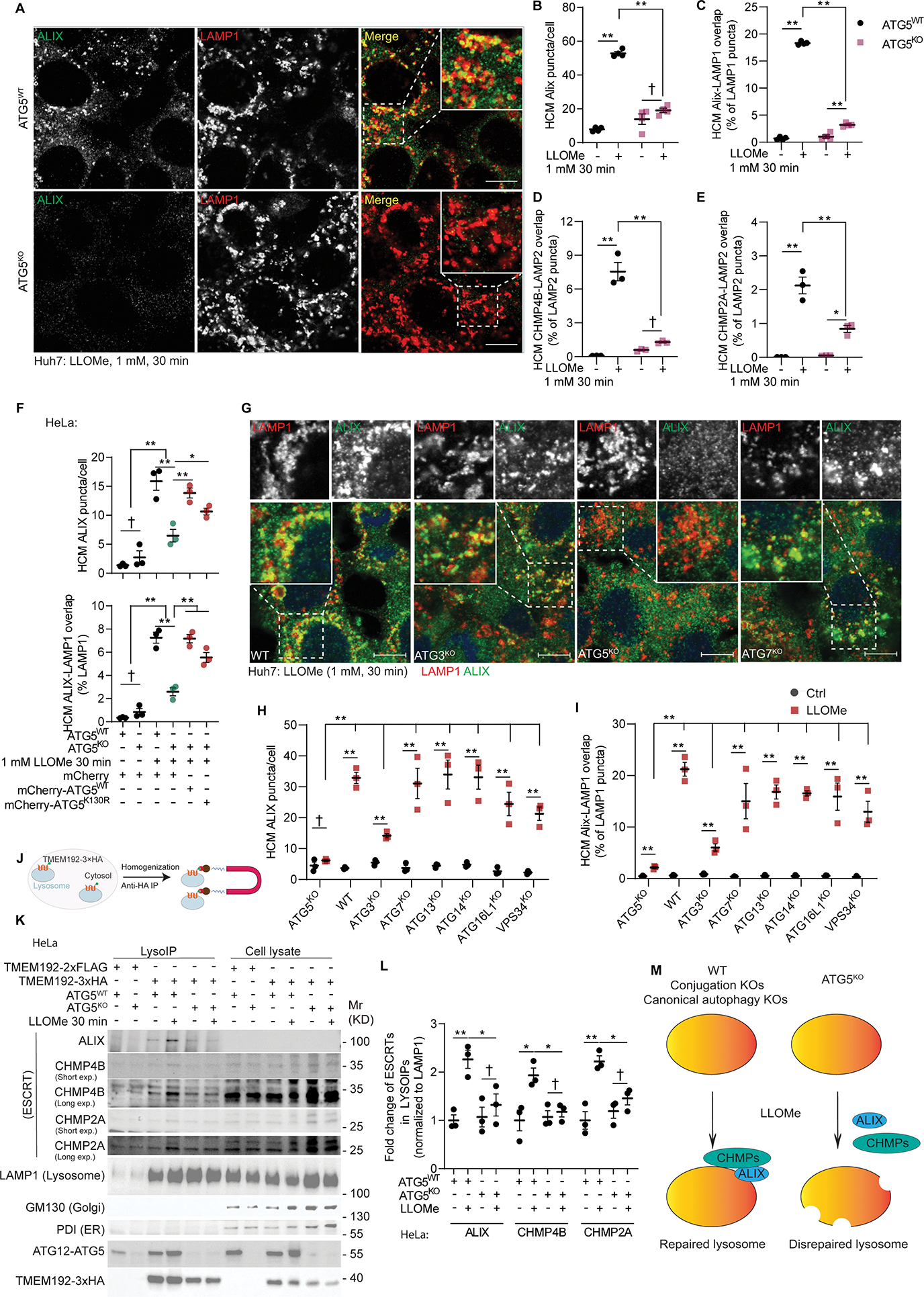
A, Representative confocal images of ALIX and LAMP1 staining in Huh7ATG5-WT and Huh7ATG5-KO cells treated with 1 mM LLOMe for 30 min. Scale bars, 10 μm. B&C, HCM quantification of ALIX puncta and ALIX-LAMP1 overlap in Huh7ATG5-WT and Huh7ATG5-KO cells treated with or without 1 mM LLOMe for 30 min. Data, means ± SE (n=4); two-way ANOVA with Tukey’s multiple comparisons. D&E, HCM quantification of CHMP4B-LAMP2 overlap and CHMP4B-LAMP2 overlap in Huh7ATG5-WT and Huh7ATG5-KO cells treated as panel B. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. F, HCM quantification of ALIX puncta and ALIX-LAMP1 overlap in HeLaATG5-WT and HeLaATG5-KO cells transfected with mCherry, mCherry-ATG5WT, or mCherry-ATG5K130R followed by treatment with or without 2 mM LLOMe for 30 min. Data, means ± SE (n=3); unpaired t-test. G, Representative confocal images and H&I, quantification (HCM) of ALIX puncta and ALIX-LAMP1 overlap in Huh7 and mutant cells treated as in panel B. Data, means ± SE (n=3); unpaired t-test. Scale bars, 10 μm. J, Schematic description, K, Immunoblot, and L, quantification of lysosome ESCRT components by anti-HA immunoprecipitation (LysoIP; TMEM192–3xHA) from HeLaATG5-WT and HeLaATG5-KO cells treated as in panel F. Long exp (long exposure) and Short exp (short exposure) correspond to the same blot. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. M, Summary of the findings in Figure 2. All HCM graphs represent independent biological replicates as in detailed in Figure 1. Statistical significance symbols in all panels, †p≥0.05, *p<0.05, **p<0.01.
ESCRT-III components, CHMP4B and CHIMP2A, known to act downstream of ALIX in lysosomal repair 44,47,77, were also diminished in response and recruitment to lysosomes in Huh7ATG5-KO cells relative to the parental Huh7ATG5-WT cells (Figures 2D, 2E and S2G–S2L). The reduced ALIX response phenotype (overall puncta and their overlap with lysosomal marker LAMP2) was observed in ATG5KO vs ATG5WT U2OS and A549 cells (Figures S2M–S2T). We also confirmed the loss of ALIX-LAMP1 overlap phenotype associated with ATG5KO in HL-60 cell line which can be differentiated into polymorphonuclear neutrophil (PMN)-like cells. We had to generate fresh ATG5KO cells (which could not be maintained in passages) for each experiment (n=3). Differentiated HL-60s ATG5WT and ATG5KO cells were analyzed as nonadherent cells in an AMNIS imaging flow cytometer. ATG5KO differentiated HL-60 cells displayed reduced Similarity Bright Detail parameter (an indicator of marker colocalization; IDEAS 6.2 software) for ALIX-LAMP1 overlap (Figure S2U).
We next complemented the loss of ALIX response phenotype in ATG5KO cells, by transfecting them with pDEST-mCherry-ATG5 alleles (Figures 2F and S2V). As in the case of LyHYP complementation with both conjugation-competent (ATG5WT) and conjugation-disabled ATG5K130R alleles, the lack of ALIX response phenotype was complemented by both pDEST-mCherry-ATG5WT and pDEST-mCherry-ATG5K130R (Figures 2F and S2V), suggesting that the effects of ATG5 on ALIX recruitment to damaged lysosomes was independent of the conjugation cascade. In keeping with this, the effects of KOs of ATG proteins important for the conjugation-lipidation cascade (ATG3, ATG7, and ATG16L1), which normally work with ATG5 in the canonical autophagy pathway 20, lacked pronounced effects on ALIX puncta response and overlaps with damaged lysosomes (Figures 2G–2I and S2W). Further, knockouts of other factors involved in canonical autophagy 2, ATG13, ATG14, and VPS34, did not display an appreciable ALIX-deficiency phenotype compared to ATG5KO (Figures 2H, 2I, S2W and S2X).
Finally, we confirmed by immunoblotting of purified lysosomes (LysoIP; Figure 2J) 79 the defect in ALIX recruitment to lysosomes caused by the absence of ATG5. Purified lysosomes from HeLaATG5-KO had diminished ALIX and downstream ESCRT-III components (CHMP4B and CHMP2A) relative to HeLaATG5-WT cells exposed to lysosomal damage by LLOMe (Figures 2K and 2L). We conclude from the collective data presented above that in the absence of ATG5, ALIX and other ESCRTs cannot be efficiently recruited to lysosomes (Figure 2M), despite the presence of molecules that normally recruit ALIX to damaged lysosomes such as Ca2+ efflux and Gal3 44,47,77,78.
Proximity biotinylation proteomics of ATG5 interactors
The observations that ATG5, independently of the conventional conjugation/atg8ylation cascade, affects the ability of the cell to maintain functional lysosomes, prompted us to carry out unbiased proteomic analyses of ATG5 interactors. We employed proximity biotinylation 80 and LC/MS/MS quantitative DIA approach 47,63,81. After generating stable Flip-In HeLa cell lines expressing (Tet-On) APEX2 fusions, with the ATG5 alleles ATG5WT and ATG5K130R, we validate functionality of APEX2 fusions by detecting APEX2-ATG5WT in conjugates with ATG12 that were absent in APEX2-ATG5K130R expressing cells (Figure 3A(i)).
Figure 3. Proximity biotinylation proteomics of ATG5 interactors.
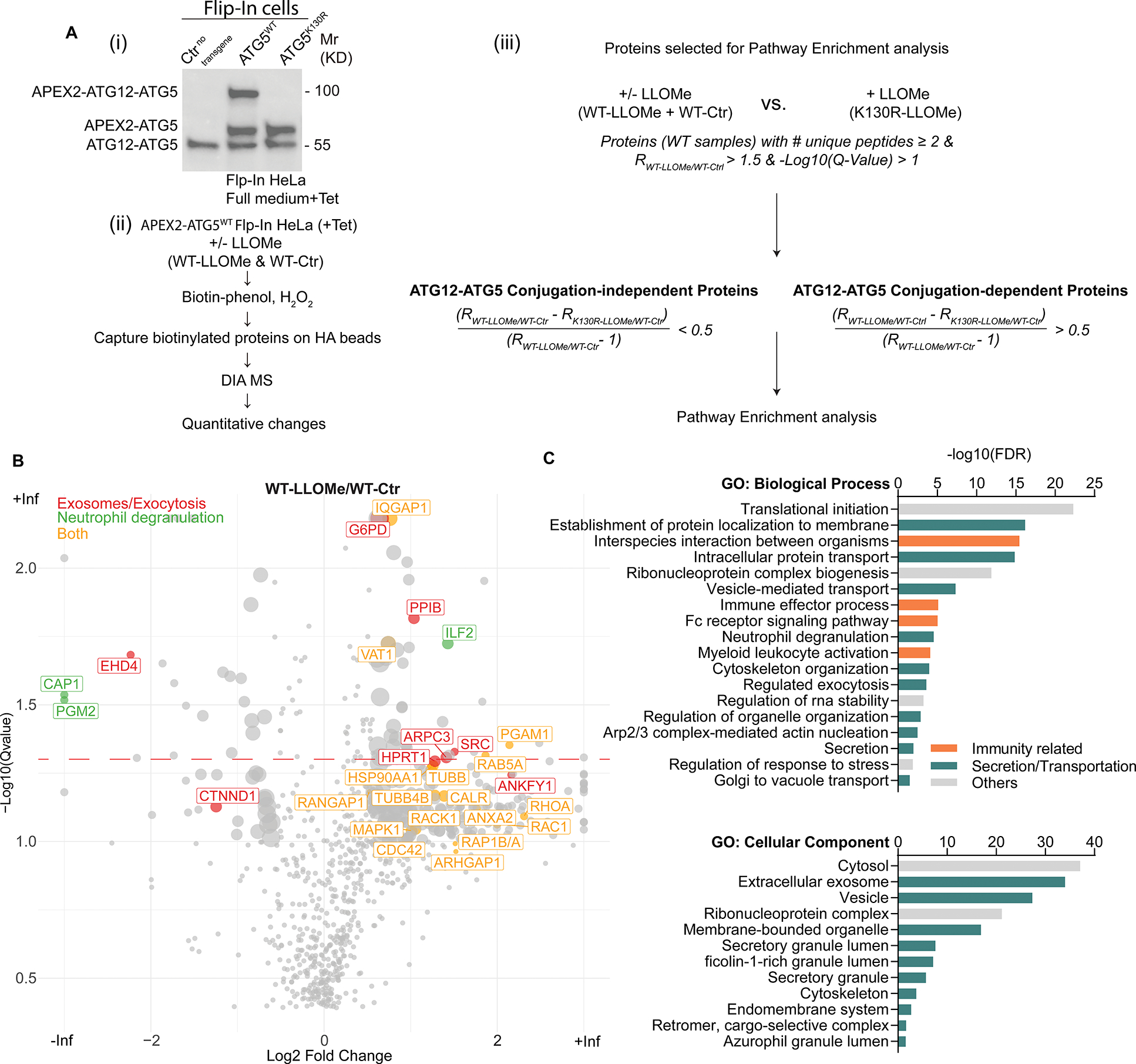
. A, (i) Stable FlpIn-HeLaAPEX2-ATG5-WT or FlpIn-HeLaAPEX2-ATG5-K130R cells were (ii) incubated with or without 2 mM LLOMe in full medium supplemented with 2 μg/mL tetracycline for 30 min. Biotinylated proteins were captured by HA beads and subjected to DIA MS. (iii), conjugation-independent or conjugation-dependent interactors of ATG5 were determined by relative fold change of FlpIn-HeLaAPEX2-ATG5-K130R and FlpIn-HeLaAPEX2-ATG5-WT cells treated with LLOMe compared with FlpIn-HeLaAPEX2-ATG5-WT cells without LLOMe incubation. B, Volcano plot of mass spec result of FlpIn-HeLaAPEX2-ATG5-WT incubated with or without LLOMe. C, GO terms, pathway enrichment analysis of conjugation-independent proteins.
The above cells were subjected to lysosomal damage or not, and proximity biotinylation performed followed by LC/MS/MS and quantitative DIA analysis (Table S1, Tabs 1–10). Among the potential ATG5WT proximity interactors with changes upon lysosomal damage (Table S1, Tabs 2,3) an enrichment of proteins associated with exosomes and exocytosis/degranulation (Figure 3B) was observed. In addition to all ATG5 proximity interactors (Figure 3B. Table S1, Tabs 1–3), we next applied an algorithm (Figure 3A(iii)) that discriminates potential ATG5 partners comparing conjugation-competent (ATG5WT) and conjugation-disabled (ATG5K130R) ATG5-interactor subsets. Gene Ontology (GO) biological processes and cellular component global analyses of potential interactors that are in the vicinity of ATG5 independently of ATG5’s conjugation capacity (Figure 3C, Table S1) corroborated the general trend observed in the volcano plot (Figure 3B) of the cumulative proteomic data. GO analyses of the data within conjugation-dependent ATG5 interactor group (Figure S3A, Table S1) indicated that they are globally different from conjugation-independent subset and showed no enrichment in secretory components (Figure S3A). Thus, the unique properties of ATG5, i.e. those independent of its participation in conventional conjugation with ATG12, appear to be associated, directly or indirectly, with sets of proteins participating in intracellular trafficking and vectorial transport, exocytosis, degranulation, and exosome secretion.
Lysosomal damage induces release of extracellular vesicles and particles and neutrophil degranulation
We next wanted to address whether ATG5 affects release of extracellular vesicles and particles (EVPs) and other exocytic events based on the following rationale: (i) ATG5 deficiency results in more vulnerable lysosomes; (ii) this effect is independent of canonical conjugation; and (iii) functional enrichment analyses of proteomic data that implicated exocytic processes were independent of ATG5’s conjugation capacity. We first established whether lysosomal damage elicits exocytic processes. Using nanoparticle tracking analysis (NTA; NanoSight), we observed extracellular particles in EVP-enrichment preparations 82,83 from supernatants of HeLa cells, which increased upon treatment with LLOMe (Figures 4A and 4B), with some shift in their size (Figure S4A). NTA analysis was also applied to cells subjected to lysosomal damage caused by overexpressing ORF3aSARS-CoV-2, known to cause lysosomal damage 76 and recently reported to play a role in secretory autophagy (the type termed SALI) via effects on autophagosomal maturation 84, and it showed increased particle release (Figures 4C and 4D). These observations were followed up in imaging-flow cytometry (AMNIS; ImageStream), whereby we detected CD63+ extracellular vesicles (EVs) (Figure S4B) released from cells subjected to lysosomal damage either by LLOMe (Figure 4E) or ORF3aSARS-CoV-2 (Figure 4F).
Figure 4. Lysosomal damage induces extracellular vesicle release and neutrophil degranulation.
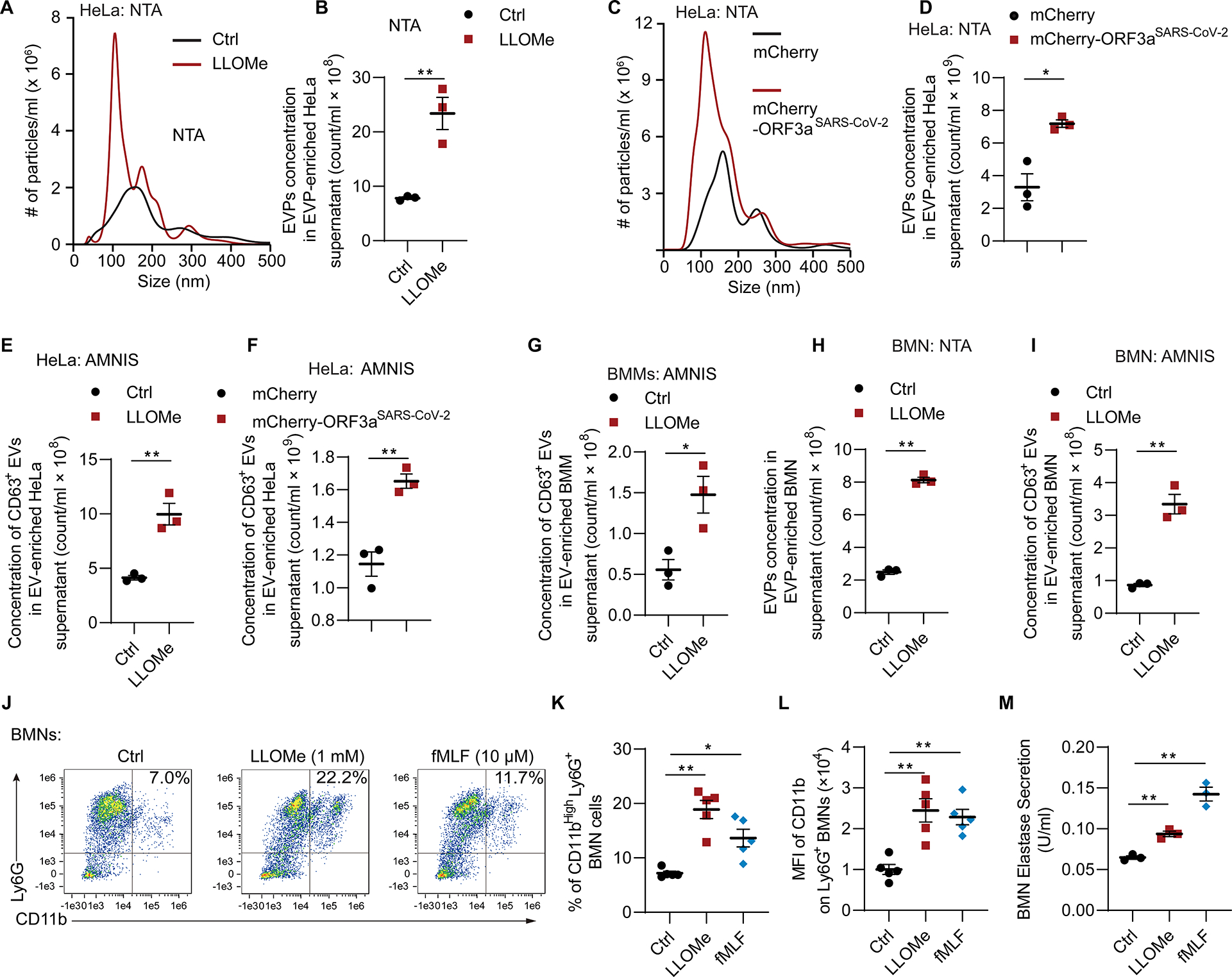
A, Representative graph of concentration (y-axis), size distribution (x-axis) and B, quantification of EVPs from supernatant of HeLa cells treated with or without 2 mM LLOMe for 1 h using nanoparticle tracking analysis (NTA). Data, means ± SE (n=3); unpaired t-test. C, Representative tracing of concentration, size distribution and D, quantification of EVPs from supernatants of HeLa cells expressing mCherry or mCherry-ORF3a (transient transfection for 24 h) using NTA. Data, means ± SE (n=3); unpaired t-test. E, Quantification of CD63+ extracellular vesicles from supernatant of HeLa cells treated as in panel A using AMNIS. Data, means ± SE (n=3); unpaired t-test. F, Quantification of CD63+ extracellular vesicles from supernatant of HeLa cells transfected with mCherry or mCherry-ORF3a for 42 h using AMNIS. Data, means ± SE (n=3); unpaired t-test. G, Quantification of CD63+ extracellular vesicles from supernatants of BMMs treated as in panel A using AMNIS. Data, means ± SE (n=3); unpaired t-test. H, Quantification of EVPs from supernatant of BMNs (murine bone marrow derived neutrophils) treated with or without 1 mM LLOMe for 1 h using NTA. Data, means ± SE (n=3); unpaired t-test. I, Quantification of CD63+ extracellular vesicles extracted from supernatant of BMNs treated as in panel H using AMNIS. Data, means ± SE (n=3); unpaired t-test. J, Representative flow cytometry and K, quantification of % CD11bHigh Ly6G+ BMNs treated with 1 mM LLOMe for 30 min, or 10μg/ml dihydrocytochalasin B for 5 min followed by 10 μM fMLF for 15 min. Data, means ± SE (n=5); one-way ANOVA with Tukey’s multiple comparisons. L, Mean fluorescence intensity (MFI) of CD11b on BMNs treated as in panel J. Data, means ± SE (n=5); one-way ANOVA with Tukey’s multiple comparisons. M, Quantification of elastase activity in supernatant of BMNs treated as in panel J. Data, means ± SE (n=3); one-way ANOVA with Tukey’s multiple comparisons. In all panels, each data point represents independent biological replicate. For BMM and BMN experiments, each point represents independent biological replicates from different animals. Statistical significance symbols in all panels, †p≥0.05, *p<0.05, **p<0.01.
Release of CD63+ EV was elicited by lysosomal damage in primary cells as well. This included macrophages, i.e. BMMs (Figure 4G), and bone marrow derived neutrophils (BMNs) (Figures 4H, 4I and S4C). Furthermore, we tested levels of CD11b (ITGAM) on Ly6G+ BMNs as a measure of neutrophil activation and degranulation 85–88. These membrane proteins become exposed on the cell surface upon granule exocytosis. We found that the percent of CD11bHi Ly6G+ BMNs increased upon LLOMe treatment (Figures 4J–4L). This increase was comparable (Figures 4J–4L) to the effects of the conventional inducer of BMN degranulation, fMLF 89,90. Thus, lysosomal damage elicits exocytosis of EVPs and degranulation in PMNs. The latter also evident from increase in extracellular levels of elastase, a component of neutrophil primary granules 91 (Figure 4M).
ATG5 knockout enhances EVP release in response to lysosomal damage
Having established that lysosomal damage elicits exocytic processes including EV release, we tested whether ATG5 status affects this phenomenon. ATG5-deficient HeLa cells displayed appreciable increase in release of CD63+ EVs and total EVPs relative to HeLaWT cells (Figures 5A and 5B). This ATG5-defficient phenotype was partially complemented in transfections with either mCherry-ATG5WT or its conjugation-incompetent mutant mCherry-ATG5K130R (Figures S5A and S5B). Quantitative immunoblotting analysis of enriched exosomes from the supernatants of cells treated with LLOMe indicated elevated markers/components of exosomes (ALIX, TSG101, and syntenin-1; 92,93) in ATG5-deficient HeLa cells (Figure 5C). A similar increase in exosomal markers was observed in ATG5-deficient cells expressing ORF3aSARS-CoV-2 (Figures 5D–5F).
Figure 5. ATG5 knockout enhances EV release in response to lysosomal damage.
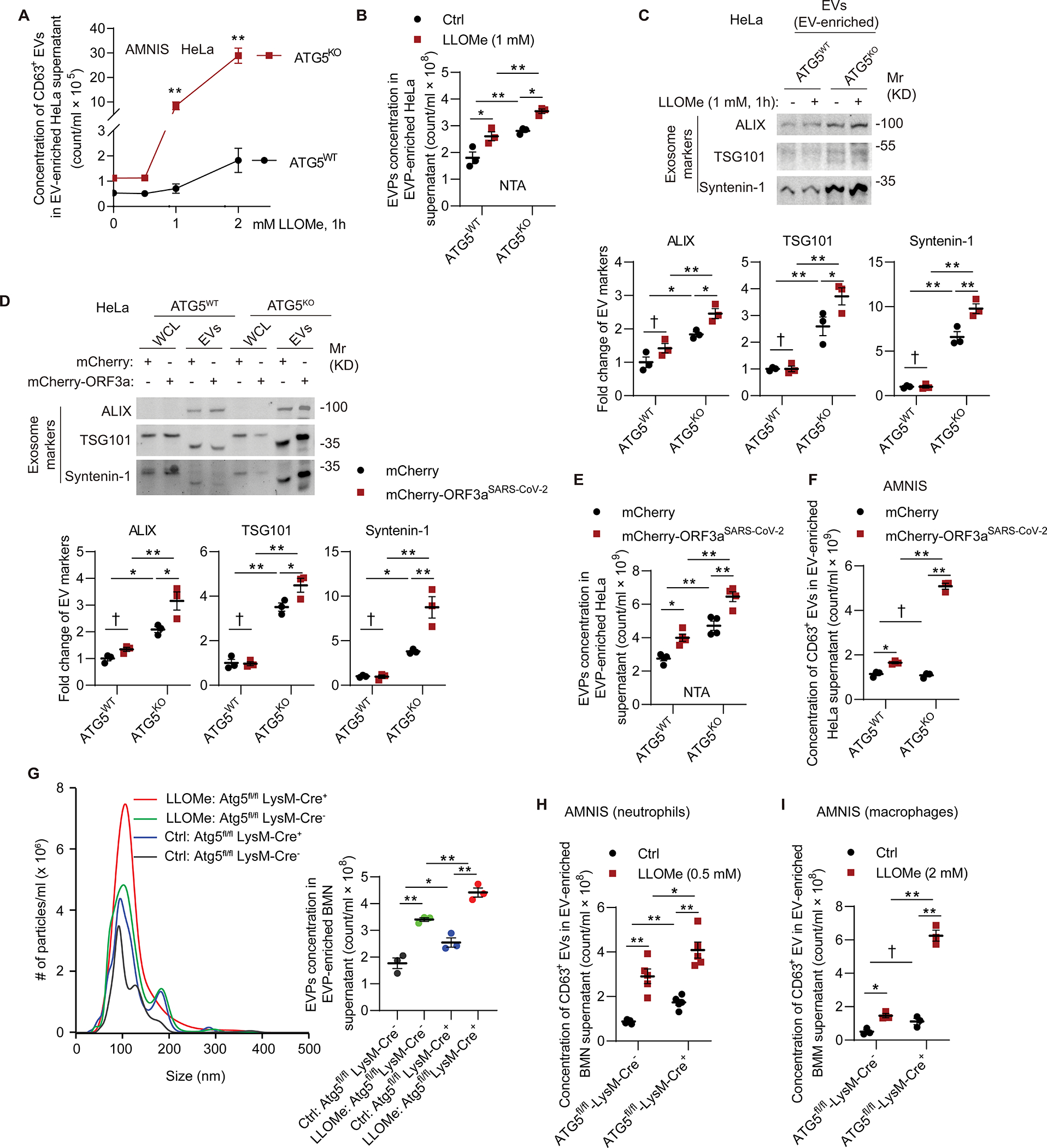
A, Flow cytometry quantification of CD63+ EVs extracted from supernatant of HeLaATG5-WT and HeLaATG5-KO cells incubated with indicated concentration of LLOMe for 1 h. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. B, Quantification of EVPs extracted from supernatant of HeLaATG5-WT and HeLaATG5-KO cells treated with or without 2 mM LLOMe for 1 h using NTA. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. C, Immunoblot and quantification of exosomal markers in EVs isolated from supernatant of HeLaATG5-WT and HeLaATG5-KO cells incubated with 1 mM LLOMe for 1 h. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. D, Immunoblot and quantification of exosomal markers in EVs isolated from supernatant of HeLaATG5-WT and HeLaATG5-KO cells transfected with mCherry or mCherry-ORF3a for 42 h. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. E, Quantification (NTA) of EVPs from supernatant as in panel D. Data, means ± SE (n=4); two-way ANOVA with Tukey’s multiple comparisons. F, Flow cytometry quantification of CD63+ EVs extracted from supernatant as in panel D. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. G, Representative graph of concentration, size distribution and quantification (NTA) of EVPs from supernatant of BMNs from Atg5fl/fl LysM-Cre− and Atg5fl/fl LysM-Cre+ mice incubated with or without 0.5 mM LLOMe for 1 h. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. H, Flow cytometry quantification of CD63+ EVs from supernatant as in panel G. Data, means ± SE (n=5); two-way ANOVA with Tukey’s multiple comparisons. I, Flow cytometry quantification of CD63+ EVs from supernatant of Atg5fl/fl LysM-Cre− and Atg5fl/fl LysM-Cre+ BMMs incubated with or without 2 mM LLOMe for 1 h. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. For BMM and BMN experiments, each point represents independent biological replicates from different. Statistical significance symbols in all panels, †p≥0.05, *p<0.05, **p<0.01.
We next tested primary neutrophils and compared EVPs released by murine BMNs derived from bone marrow of Atg5fl/fl LysM-Cre+ and Atg5fl/fl LysM-Cre− transgenic mice. By NTA (NanoSight) analysis of particles released from these BMNs, we observed increased release of EVPs (Figures 5G). This was followed up by imaging-flow cytometry (AMNIS ImageStream) of enriched CD63+ exosomes, showing that both neutrophils/BMNs (Figure 5H) and macrophages/BMMs (Figure 5I) lacking ATG5 displayed augmented release of EVPs (including exosomes) in response to lysosomal damage.
ATG5 knockout enhances PMN degranulation and lysosomal exocytosis in response to lysosomal damage
The BMNs derived from Atg5fl/fl LysM-Cre+ mice displayed increased parameters of exocytosis of secretory vesicles and granules (degranulation) in response to lysosomal damage. This was manifested as increase in CD11b, a cell surface marker of exocytosis, which was elevated on live Atg5fl/fl LysM-Cre+ but not on Atg7fl/fl LysM-Cre+ BMNs treated with LLOMe (Figures 6A–6C and S5C). The % of CD11bHi Ly6G+ cells were compatible with previously reported % levels with bone marrow derived neutrophils 94,95 vis-à-vis the customarily higher % levels found in peripheral blood PMNs. The markers of degranulation/exocytosis were also increased in Atg5fl/fl LysM-Cre+ BMNs upon treatment with the conventional inducer fMLF (Figures 6A and S5C), indicating that the receptor system for detection of fMLF was likely elevated on the surface on Atg5fl/fl LysM-Cre+ BMNs, compatible with increased basal release of secretory vesicles where fMLF receptors are principally localized 96. This was corroborated by the augmented cell surface FPR1 levels in Atg5fl/fl LysM-Cre+ BMNs compared with Atg5fl/fl LysM-Cre− BMNs, reaching significance under LLOMe-treatment conditions (Figures 6D and S5D). Neutrophil elastase (a component of primary/azurophilic granules) was released in response to lysosomal damage in higher amounts from Atg5fl/fl LysM-Cre+ BMNs than from Atg5fl/fl LysM-Cre− BMN (Figure 6E). Thus, we conclude that neutrophils lacking ATG5 undergo increased exocytosis and degranulation in response to stimulation by physiological agonists (fMLF) or lysosomal damage.
Figure 6. ATG5 knockout enhances PMN degranulation in response to lysosomal damage.
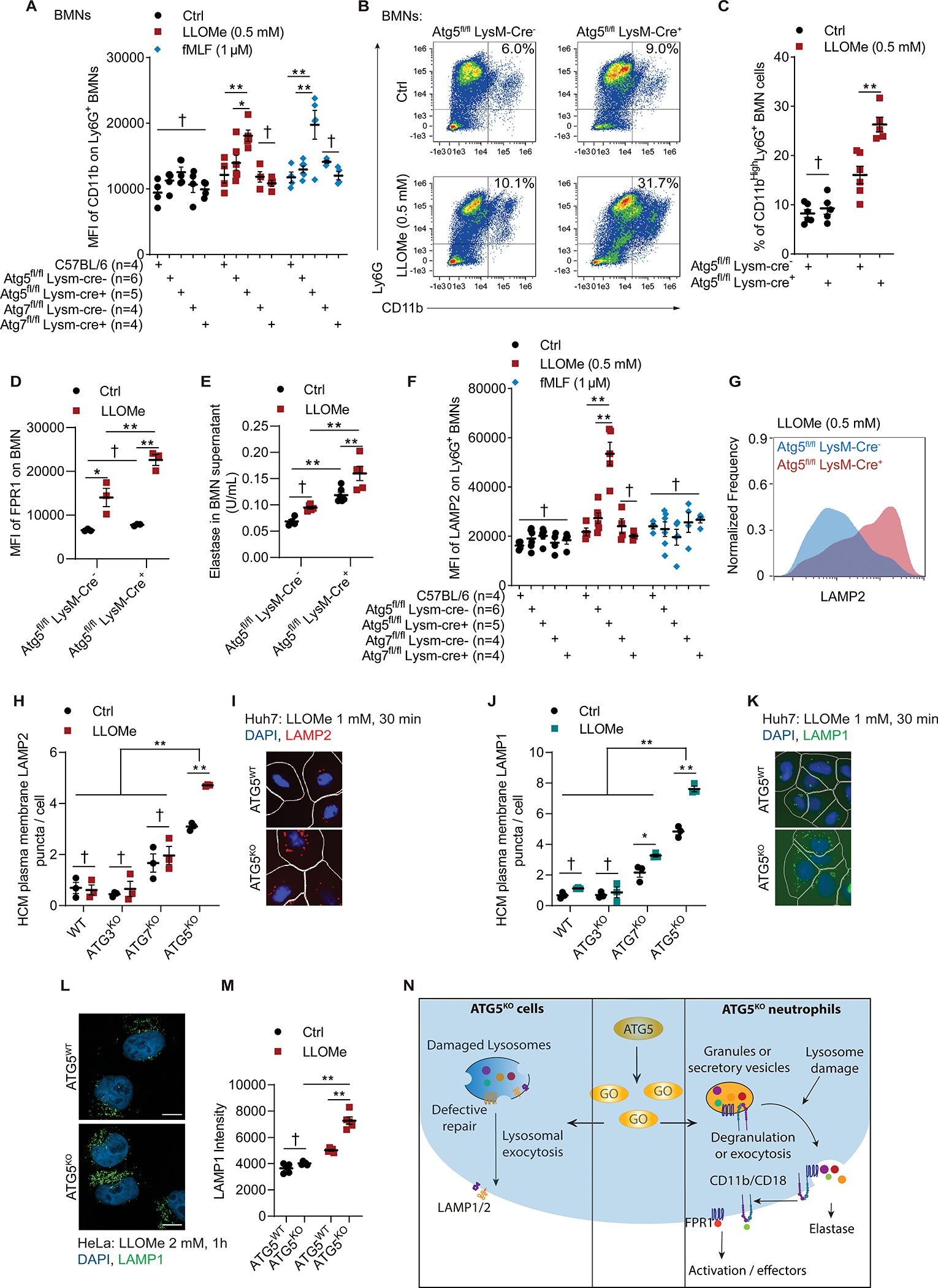
A, MFI of CD11b on BMNs incubated with 0.5 mM LLOMe for 30 min, or 10μg/ml dihydrocytochalasin B for 5 min followed by 1 μM fMLF for 15 min. Data, means ± SE (n=4–6); two-way ANOVA with Tukey’s multiple comparisons. B, Representative flow cytometry of BMNs and C, quantification of % CD11bHigh Ly6G+ BMNs from Atg5fl/fl LysM-Cre− and Atg5fl/fl LysM-Cre+ mice incubated with or without 0.5 mM LLOMe for 30 min. Data, means ± SE (n=5–6); two-way ANOVA with Tukey’s multiple comparisons. D, MFI of FPR1 on BMNs treated as in panel C. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. E, Quantification of elastase activity in supernatant of BMNs from Atg5fl/fl LysM-Cre− and Atg5fl/fl LysM-Cre+ mice treated as in panel C. Data, means ± SE (n=4–6); two-way ANOVA with Tukey’s multiple comparisons. F, MFI of LAMP2 on Atg5fl/fl LysM-Cre− and Atg5fl/fl LysM-Cre+ BMNs treated as in panel A. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. G, Representative flow cytometry of LAMP2 intensity from Atg5fl/fl LysM-Cre− and Atg5fl/fl LysM-Cre+ treated as in panel A. H, HCM quantification and I, representative images of plasma membrane LAMP2 puncta on Huh7WT, Huh7ATG3-KO, Huh7ATG7-KO, and Huh7ATG5KO cells treated with or without 1 mM LLOMe for 30 min. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. J, HCM quantification and K, representative images of plasma membrane LAMP1 puncta on Huh7 and mutant cells treated with or without 1 mM LLOMe for 30 min. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. L, Representative confocal images and M, quantification of plasma membrane LAMP1 puncta on HeLaATG5-WT, and HeLaATG5-KO cells treated with or without 2 mM LLOMe for 30 min. Scale bars, 10 μm. Data, means ± SE (n=4); two-way ANOVA with Tukey’s multiple comparisons. Fluorescence intensity was calculated based on ≥20 fields (with a total ≥100 cells) per biological replicate. N. Summary of data in Figure 6. GO (middle panel), ATG5 interactors identified in proteomic analyses. All HCM graphs represent independent biological replicates as in detailed in Figure 1. For BMMs and BMNs experiments, each point represents independent biological replicates with BMNs from different animals. Statistical significance symbols in all panels, †p≥0.05, *p<0.05, **p<0.01.
Lysosomal exocytosis was increased in Atg5fl/fl LysM-Cre+ BMNs relative to Atg5fl/fl LysM-Cre− BMNs (Figure 6F). For this we used LAMP2 normally present in lysosomes but translocating to plasma membrane where it is exofacially exposed upon fusion of lysosomes with the cell surface. By comparison to activation of lysosomal exocytosis (proxied by cell surface LAMP2) upon lysosomal damage, fMLF did not elicit this response (Figure 6F). For flow cytometry carried out with live neutrophils, sodium azide was used to block reuptake and endocytosis (Figures 6F and 6G) 97–99. To complement this by HCM with adherent cells, we allowed endocytosis (if any) to take place during staining of live and unpermeabilized Huh7 cells with anti-LAMP2 lumenal domain mAb and quantified LAMP2 puncta. By this method, Huh7ATG5-KO cells exposed to LLOMe showed increased number of LAMP2 puncta relative to untreated Huh7ATG5-KO cells, whereas Huh7ATG7-KO cells did not have that phenotype (Figures 6H and 6I). This was confirmed by staining for LAMP1 using the same procedure with live cells (Figures 6J and 6K) and confocal microscopy (Figures 6L and 6M).
In conclusion, ATG5 defect promotes lysosomal exocytosis and elicits degranulation in PMNs in response to damage and stimulation by physiological ligands (Figure 6N).
Alternative conjugation contributes to lysosomal vulnerability and exocytic processes
Recent studies have indicated the existence of an alternative conjugation pathway whereby ATG12 can form a sidestep conjugate ATG12-ATG3 that contributes to increased exosome secretion 42,43. We detected increased levels of ATG12-ATG3 conjugates in ATG5KO cells (Figures S6A and S6B). Furthermore, we observed that ATG3 puncta are increased in Huh7ATG5-KO cells vs. Huh7WT irrespective of LLOMe treatment, whereas distinct, large ALIX puncta are elicited by lysosomal damage in Huh7WT cells and do not appear in Huh7ATG5-KO cells (Figures 7A–7E). Moreover, colocalization between ATG3 and ALIX significantly increases in response to LLOMe in the absence of ATG5, i.e., in Huh7ATG5-KO cells, whereas this phenomenon is absent in Huh7WT cells (Figures 7E and S6D). ALIX was increased in protein complexes with ATG3, which included elevated ATG12-ATG3 conjugate (Figure S6C). A knockout of ATG3 alone did not alter the EV release in response to LLOMe (Figures 7F, 7G, and S6E; HeLaATG3-KO vs. HeLaWT). However, a knockout of ATG3 in HeLaATG5-KO cells, thus generating double KO cells (HeLaATG3/5-DKO), abrogated the elevated EVPs release in response to LLOMe treatment seen in single, ATG5KO cell line (Figures 7F, 7G and S6E; HeLaATG5-KO vs. HeLaATG3/5-DKO). This was confirmed by measuring CD63+ EVs using AMNIS in Huh7 cells (Figures S6F and S6G). Thus, ablation of ATG3 in the absence of ATG5 results in normalization of exocytic processes.
Figure 7. Alternative conjugation contributes to lysosomal vulnerability and exocytic processes.
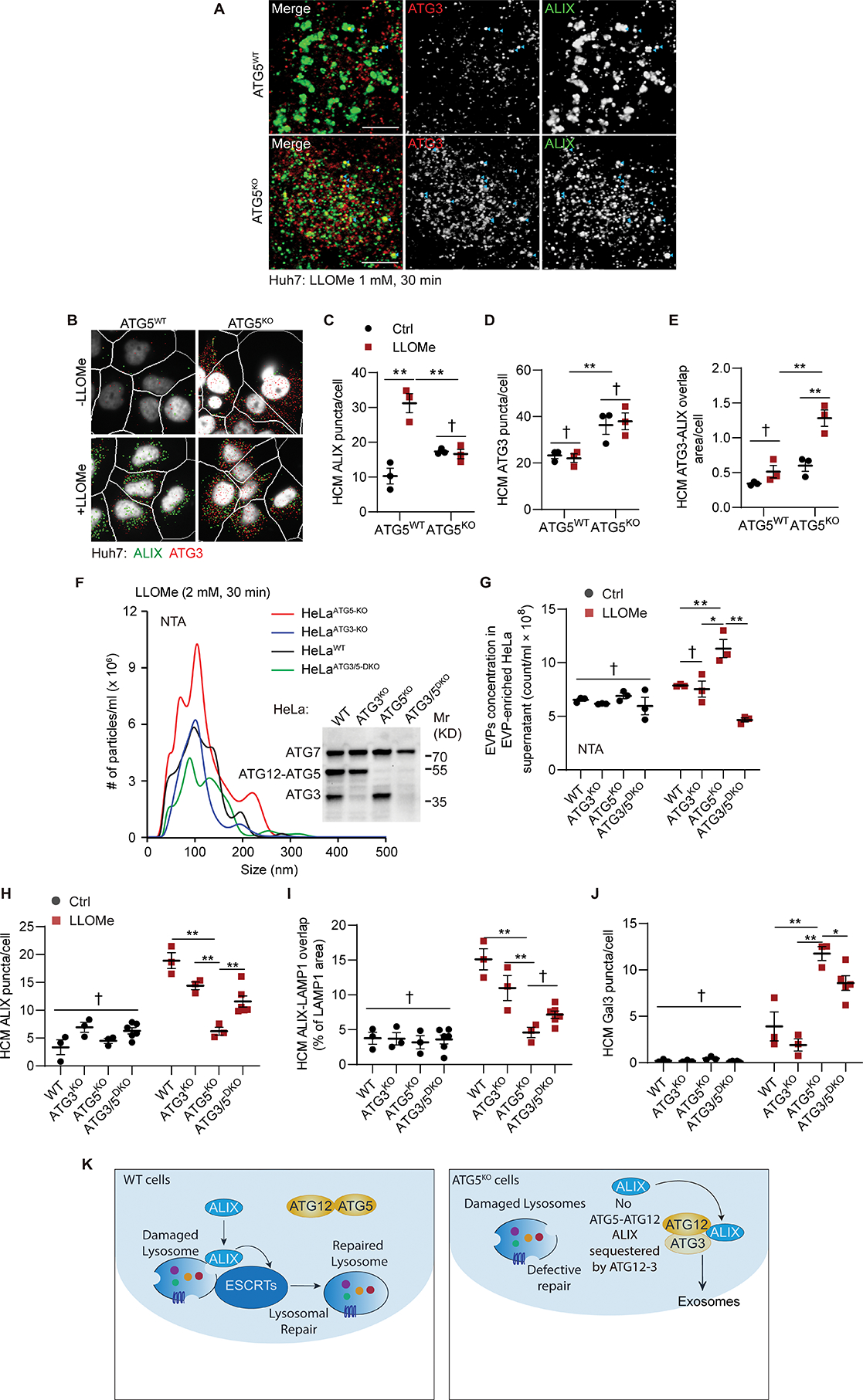
A, Representative confocal images of Huh7ATG5-WT and Huh7ATG5-KO cells incubated with 1 mM LLOMe for 30 min. Scale bars, 5 μm. B-E, Representative images (B) and quantifications of ALIX puncta (C), ATG3 puncta (D), and ATG3-ALIX overlap area (E) in Huh7ATG5-WT and Huh7ATG5-KO cells treated as panel A. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. F, Representative graphs and G, quantification of EVPs from supernatant of HeLa and mutant cells treated with 2 mM LLOMe for 30 min. Data, means ± SE (n=3); two-way ANOVA with Tukey’s multiple comparisons. Inset: Immunoblot of HeLa and mutant cells. H-J, HCM quantification of ALIX puncta (H), ALIX-LAMP1 overlap area (I), and Gal3 puncta (J) in Huh7 and mutant cells treated as in panel A. Data, means ± SE (n≥3); two-way ANOVA with Tukey’s multiple comparisons. K, Summary of the mechanistic findings in Figure 7. All HCM graphs represent independent biological replicates as in detailed in Figure 1. Statistical significance symbols in all panels, †p≥0.05, *p<0.05, **p<0.01.
Furthermore, the number of ALIX puncta and ALIX-LAMP1 colocalization following LLOMe treatment, which decreased in Huh7ATG5-KO cells, were partially recovered by ATG3 KO in Huh7ATG5-KO cells (Huh7ATG3/5-DKO cells vs Huh7ATG5-KO; Figures 7H, 7I and S6H, top row). Mirroring the partial recovery of ALIX recruitment in Huh7ATG3/5-DKO relative to Huh7ATG5-KO cells in response to lysosomal damage, Gal3 as a marker of the extent of the damage was partially suppressed in Huh7ATG3/5-DKO cells (Figures 7J and S6H, bottom row). This is indicative of improvement in the ability of cells lacking both ATG5 and ATG3 to repair lysosomes. This paradoxical observation is consistent with a model (Figure 7K) whereby ATG12-ATG3 increases in the absence of ATG5, whereas ATG12 is normally tied up in ATG12-ATG5 conjugates when ATG5 is available at normal levels. In the absence of ATG5, the alternative ATG12-ATG3 conjugate contributes to ALIX’s mislocalization and diminishes its function in the lysosome repair.
Discussion
In this study, we uncovered unanticipated aspects of the protein conjugation and lipidation cascade commonly associated with autophagy. Whereas ATG5 has been primarily studied as a part of the E3 ligase leading to mATG8s lipidation, here we show that ATG5 is positioned at a critical intersection with other conjugation branches (Figure S6I) affecting lysosomal quality, exocytosis, and neutrophil degranulation independently of the conventional mATG8 lipidation and canonical autophagy. In the absence of ATG5, ESCRT proteins are not efficiently recruited to the membrane repair sites on damaged lysosomes. Under these conditions, lysosomes, storage granules, secretory vesicles, and EVPs are prone to exocytosis. The dysregulated exocytic processes in neutrophils may contribute to infection outcomes in Mtb murine models exceeding ATG5’s role in canonical autophagy.
We find that a loss of ATG5, and the resulting ATG12-ATG3 conjugate formation favored in the absence of ATG5, diminish available ALIX thus compromising lysosomal membrane repair 44,47,77. This results in a plethora of downstream effects such as lysosomal hypersensitivity to damage and includes roles in exosome formation 43 and other processes. These phenomena are different from the recently described eructophagy 100 since the latter depends on ATG genes other than ATG5. Other studies propose additional ATG5-specific effects, including measurable metabolic consequences 101.
Reduced levels of ATG5 may occur in physiological and disease states. Lower ATG5 levels are a risk factor in Parkinson’s diseases (PD) 102–105. Changes in ATG5 levels have been reported in diabetes 106 and cancer 107–111. Lysosomal status has been linked specifically to pancreatic cancer cells 112 and lysosomal degradation of plasma membrane MHC-I molecules 113. ATG5 is implicated in autoimmunity and inflammation 49. Loss of ATG5 affects cardiac capacity 114. Thus, changes in ATG5 may affect a variety of diseases.
Our present study uncovers effects of ATG5 knockout on neutrophils, first responders to bacterial infection 115,116. When neutrophils are hyperactivated, they can cause tissue damage 117,118. Neutrophils are activated via receptors 119,120 such as FPR1 and CD11b/CD18 associated with neutrophil degranulation, and were increased in ATG5 knockouts. Granule components are found in neutrophil NETs released during NETosis 121. Although ATG5 does not alter NETosis 122, NETs contain granule components 123 and may be affected by an ATG5 loss. In human populations, active tuberculosis disease is associated with persistent neutrophilic signatures of inflammation 54; we postulate that this is linked to the findings in our study concerning neutrophil degranulation, exocytosis, and EVP release.
Limitations of the study
Limitations of our study include a focus on neutrophils and a diversity of exocytic processes altered in ATG5-deficient cells that could not be easily parsed for effects in vivo. It remains to be determined which subsets are responsible for increased susceptibility to Mtb in mice with Atg5-deficient myeloid cells. This is compounded by the duality of phenotypes downstream of the ATG5 deficiency, i.e., lysosomal damage vs. exocytosis and exosomal secretion, and a partial link of both aspects to the sequestration of ALIX. There are additional effects of ATG5 on the lysosomal status beyond the sequestration of ALIX by ATG12-ATG3, and these processes remain to be delineated.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Vojo Deretic (vderetic@salud.unm.edu).
Materials availability
Plasmids and cell lines generated in this study are available from the lead contact.
Data and code availability
Raw MS DIA/DDA data have been deposited at the MassIVE proteomics repository MassIVE (MSV000090348) and Proteome Exchange (PXD036850).
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Atg5fl/fl LysM-Cre−, Atg5fl/fl LysM-Cre+, Atg7fl/fl LysM-Cre−, and Atg7fl/fl LysM-Cre+ mice were previously described 51,125.
Housing and husbandry conditions of experimental animals
All mice were housed in AAALAC-accredited Animal Research Facility (ARF) of the University of New Mexico Health Sciences Center (UNM HSC) and institutionally approved husbandry conditions and approved breeding protocols were followed. M. tuberculosis-infected animals were housed in a separate ABSL3 suite within the UNM HSC ARF facility and all staff followed strict ABSL3, BSL3, and animal protocols approved by the UNM HSC Biosafety committee and the Institutional Animal Care and Use Committee. The study was compliant with all relevant ethical guidelines for animal research.
Cells and cell line models
HEK293T, HeLa, A549 and U2OS cells were from ATCC. Huh7 cells were from Rocky Mountain Laboratories. Patu-8902 cells were from Alec Kimmelman. Murine primary cells (Bone marrow derived macrophages and neutrophils; BMMs and BMNs) were used for degranulation, exosome secretion, and lysosomal damage studies. BMMs were isolated from femurs Atg5/7fl/fl LysM-Cre− and Atg5/7fl/fl LysM-Cre+ mice and cultured in DMEM supplemented with mouse macrophage colony stimulating factor (mM-CSF, #5228, CST) for 5 to 10 days before use. BMNs were isolated from femurs Atg5/7fl/fl LysM-Cre− and Atg5/7fl/fl LysM-Cre+ mice according to 126 and gating strategy according to 127. Briefly, mouse bone marrow cells were collected, filtered, and centrifuged, followed by 0.2% NaCl lysis red blood cells and gradient centrifugation with Histopaque 1119 and 1077. Cells were then washed and recovered for 1 hour before use. Flp-In cell lines were generated using constructs from Terje Johansen, cell lines for LysoIP were generated using constructs obtained from Addgene, and knockout cell lines were generated by CRISPR/Cas9-mediated knockout system as detailed below.
METHOD DETAILS
Antibodies
Antibodies from Abcam were ATG5 (1:2000 for Western blot (WB), ab108327), ATG7 (1:2000 for WB, ab52472), mCherry (1:1000 for WB, ab183628), Syntenin-1 (1:1000 for WB, ab19903), TSG101 (1:1000 for WB; 1:200 for immunofluorescence (IF), ab83), VPS34 (1:1000 for WB, ab124905).
Antibodies from Biolegend were ALIX (1:400 for IF, #634502), Galectin-3 (1:200 for IF, #125402).
Antibodies from Cell Signaling Technology were ATG13 (1:1000 for WB, #13468), GM130 (1:1000 for WB, #12480), HA-tag (1:1000 for WB, #3724), LAMP1 (1:1000 for WB; 1:1000 for IF, #9091), PDI (1:1000 for WB, #3501).
Antibodies from Proteintech were ALIX (1:500 for WB, 12422-1-AP), CHMP2A (1:500 for WB; 1:500 for IF, 10477-1-AP), and CHMP4B (1:500 for WB; 1:500 for IF, 13683-1-AP). Antibodies from Sigma Aldrich were ATG3 (1:1000 for WB; 1:300 for IF, #A3231) and CD63 (1:400 for IF, SAB4301607).
Other antibodies used in this study were from the following sources: beta-Actin (1:500 for WB, sc-47778), Cathepsin C (1:500 for WB, sc-74590) and GAPDH (1:500 for WB, sc-47724) from Santa Cruz Biotechnology; LAMP2 (1:1000 for IF, H4B4) from DSHB of University of Iowa; FITC-FPR1 (1:400 for IF, FPR1-FITC) from Fabgennix; ATG14 (1:500 for WB, PD026) and ATG16L1 (1:500 for IF, PM040) from MBL International; CD11b (1:500 for IF, NB110-89474) from Novus Biological; Ly-6G (1:500 for IF, #60031) from STEMCELL Technologies; and ATG12 (1:1000 for WB, 366400) from Thermofisher. Alexa Fluor 488, 568, 647 (1:500 for IF) and IgG-HRP (1:10000 for WB) secondary antibodies were from ThermoFisher Scientific. IgG Polyclonal Antibody (IRDye® 800CW and IRDye® 680RD) (1:10000 for WB) secondary antibodies were from LI-COR Biosciences.
Plasmids, siRNAs, and transfection
Plasmids used in this study, such as ATG5 cloned into pDONR221 using BP cloning, and expression vectors were made utilizing LR cloning (Gateway, ThermoFisher) in appropriate pDEST vectors for immunoprecipitation assay.
ATG5 mutants were generated utilizing the QuikChange site-directed mutagenesis kit (Agilent) and confirmed by sequencing (Genewiz). Plasmid transfections were performed using the ProFection Mammalian Transfection System, FuGENE® HD Transfection Reagent (Promega), or Lipofectamine 2000 Transfection Reagent (ThermoFisher Scientific).
Generation of CRISPR mutant cells
Knockout cells (HeLaATG5-KO, HeLaATG3-KO, HeLaATG3/5-DKO, Huh7ATG5-KO, Huh7ATG3-KO, Huh7ATG7-KO, Huh7ATG13-KO, Huh7ATG14-KO, Huh7ATG16L1-KO, Huh7VPS34-KO, Huh7ATG3/5-DKO, U2OSATG5-KO, and A549ATG5-KO) were generated by CRISPR/Cas9-mediated knockout system. The lentiviral vector lentiCRISPRv2 carrying both Cas9 enzyme and a gRNA targeting ATG5 (gRNA-puro: AAGAGTAAGTTATTTGACGT), ATG3 (gRNA-Hygro: GTGAAGGCATACCTACCAAC), ATG7 (gRNA-puro: CTTCCGTGACCGTACCATGC), ATG13 (gRNA-puro: CTGTCCCAACACGAACTGTC), ATG14 (gRNA-puro: CTACTTCGACGGCCGCGACC), ATG16L1 (gRNA-puro: ACCAAATGCAGCGGAAGGAC), VPS34 (gRNA-puro: ACAGACATTTGTAGATCGGT) (VectorBuilder) were transfected into HEK293T cells together with the packaging plasmids psPAX2 and pCMV-VSV-G at the ratio of 5:3:2. Two days after transfection, the supernatant containing lentiviruses was collected. Cells were infected by the lentiviruses with 8–10 μg/mL polybrene. 36 h after infection, the cells were selected with puromycin (1–10 μg/mL) for one week in order to select knockout cells. All knockouts were confirmed by western blot. Selection of single clones was performed by dilution in 96-well.
Generation of CRISPR mutant in primary cells
CRISPR knockout of ATG5 in primary murine BMMs was performed as described 69. Briefly, monocyte-derived macrophages were cultured in macrophage medium (DMEM high glucose supplemented with 10% FBS [Gibco], 2 mMGlutaMAX, 100U/ml penicillin, 100 μg/ml streptomycin, and M-CSF 100 ng/ml [Peprotech]). Medium was changed every 2–3 d by adding half the volume of medium. gRNAs (IDT) were mixed with Cas9 (IDT Alt-R S.p. Cas9 Nuclease V3) at a molar ratio of 2:1 and incubated at room temperature for≥20 min. Macrophages were detached (Detachin, Genlantis), washed, counted and resuspended in 20 μl of P3 primary nucleofection solution (Lonza) per 106 cells. The cells were then added to the Cas9-gRNAs complex and mix by pipetting. The mix was the loaded into the nucleofector cassette strip (Lonza) and nuclefected using the Buffer P3, CM-137 condition in the Lonza 4D-Nucleofector (4D-Nucleofector Core Unit: Lonza, AAF-1002B; 4D-Nucleofector X Unit: AAF-1002X). The cells were then cultured and KO efficiency assayed by Western blot.
Generating Flp-In-ATG5-WT/K130RTet-ON cell line
Transfect Hela Flp-In host cells with ATG5/K130R reconstructed plasmid and the pOG44 expression plasmid at ration of 9:1. 24 h after transfection, wash the cells and add fresh medium to the cells. 48 h after transfection, split the cells into fresh medium around 25 % confluent. Incubate the cells at 37 °C for 2–3 h until they have attached to the culture dish. Then them medium was removed and added with fresh medium containing hygromycin. Feed the cells with selective medium every 3–4 days until single cell clone can be identified. Pick hygromycin-resistant clones and expand each clone to test. The tested clones incubated in the medium containing 1–2 μg/mL tetracycline for 24 h were determined by western blot for the expressing of ATG5.
LysoIP assay
Lentiviruses constructs for generating stable LysoIP cells were purchased from Addgene. HEK293T cells were transfected with pLJC5-TMEM192–3xHA or pLJC5-TMEM192–2XFLAG constructs in combination with pCMV-VSV-G and psPAX2 packaging plasmids, 60 h after transfection, the supernatant containing lentiviruses was collected and centrifuged to remove cells and then frozen at −80 °C. To establish LysoIP stably expressing cell lines, cells were plated in 10cm dish in DMEM with 10 % FBS and infected with 500μL of virus-containing media overnight, then add puromycin for selection.
Selected cells in 15 cm plates with 90 % confluency were used for each LysoIP. Cells with or without treatment were quickly rinsed twice with PBS and then scraped in1mL of KPBS (136 mM KCl, 10 mM KH2PO4, pH7.25 was adjusted with KOH) and centrifuged at 3000 rpm for 2 min at 4 °C. Pelleted cells were resuspended in 950 μL KPBS and reserved 25 μL for further processing of the whole-cell lysate. The remaining cells were gently homogenized with 20 strokes of a 2 mL homogenizer. The homogenate was then centrifuged at 3000 rpm for 2 min at 4 °C and the supernatant was incubated with 100 μL of KPBS prewashed anti-HA magnetic beads (ThermoFisher) on a gentle rotator shaker for 15 min. Immunoprecipitants were then gently washed three times with KPBS and eluted with 2 × Laemmli sample buffer (Bio-Rad) and subjected to immunoblot analysis.
High content microscopy (HCM)
Cells in 96 well plates were fixed in 4 % paraformaldehyde for 5 min. Cells were then permeabilized with 0.1 % saponin in 3 % Bovine serum albumin (BSA) for 30 min followed by incubation with primary antibodies for 2 h and secondary antibodies for 1h. Hoechst 33342 staining was performed for 3 min. High content microscopy with automated image acquisition and quantification was carried out using a Cellomics HCS scanner and iDEV software (ThermoFisher Scientific). Automated epifluorescence image collection was performed for a minimum of 500 cells per well. Epifluorescence images were machine analyzed using preset scanning parameters and object mask definitions. Hoechst 33342 staining was used for autofocus and to automatically define cellular outlines based on background staining of the cytoplasm. Primary objects were cells, and regions of interest (ROI) or targets were algorithm-defined by shape/segmentation, maximum/minimum average intensity, total area and total intnsity, etc., to automatically identify puncta or other profiles within valid primary objects. Each experiment (independent biological repeats; n≥3) consists of machine-identified 500 valid primary objects/cells per well, ≥5 wells/sample. All data collection, processing (object, ROI, and target mask assignments) and analyses were computer driven independently of human operators.
Co-immunoprecipitation assay
Cells transfected with 8–10 μg of plasmids were lysed in NP-40 buffer (ThermoFisher Scientific) supplemented with protease inhibitor cocktail (Roche, 11697498001) and 1 mM PMSF (Sigma, 93482) for 30 min on ice. Supernatants were incubated with (2–3 μg) antibodies overnight at 4 °C. The immune complexes were captured with Dynabeads (ThermoFisher Scientific), followed by three times washing with 1X PBS. Proteins bound to Dynabeads were eluted with 2×Laemmli sample buffer (Bio-Rad) and subjected to immunoblot analysis.
Immunofluorescence confocal microscopy and analysis
Cells were plated onto coverslips in 6-well plates. After treatment, cells were fixed in 4% paraformaldehyde for 5 min followed by permeabilization with 0.1% saponin in 3% BSA for 30 min. Cells were then incubated with primary antibodies for 2 h and appropriate secondary antibodies Alexa Fluor 488 or 568 (ThermoFisher Scientific) for 1 h at room temperature. Coverslips were mounted using Prolong Gold Antifade Mountant (ThermoFisher Scientific). Images were acquired using a confocal microscope (META; Carl Zeiss) equipped with a 63 3/1.4 NA oil objective, camera (LSM META; Carl Zeiss), and AIM software (Carl Zeiss).
Flow cytometry
BMNs after treatment were washed with 3% BSA in PBS supplemented with 0.1% of NaN3 before staining for surface markers. After staining, cells were then resuspended with 3% BSA in PBS supplemented with 0.1% of NaN3 until acquisition on Amins ImageStreamx MKII (ISx, EMD Millipore, Seattle, WA, USA). For neutrophil degranulation studies, Ly6G+ neutrophils were gated for analysis of surface CD11b.
Exosome isolation
Exosomes are isolated using SmartSEC™ (SSEC200A-1) and ExoQuick-TC (EXOTC10A-1) according to manufacturer’s instructions from supernatant of cells. For mCherry-ATG5WT and mCherry-ATG5K130R transfection experiments, cells were incubated in LLOMe in a capped Eppendorf tube for 1 h in 37°C after transfection for 48 h. For mCherry-ORF3aSARS-CoV-2, supernatant was collected from 6 h after transfection when lipofectamine 2000 was washed away and new medium was added until 48 h after transfection.
ImageStreamx small particle acquisition and analysis
Multispectral imaging flow-cytometric acquisition of EVs and small particle calibrators was performed using Amins ImageStreamx MKII (ISx, EMD Millipore, Seattle, WA, USA) with fluidics set at low speed, sensitivity set to high, magnification at 60×, core size 7 μm, and the “Hide Beads” option unchecked prior to every acquisition in order to visualize speed beads in analyses. All parameters are stored in acquisition template except the latter, which requires unchecking prior to each acquisition. The ISx was equipped with the following lasers run at maximal power to ensure maximal sensitivity: 405 nm (120 mW), 488 nm (200 mW), 561 nm (200 mW), and 642 nm (150 mW). Upon each startup, the instrument calibration tool ASSIST® was performed to optimize performance and consistency. Each of the two charged couple device (CCD) cameras with which the ISx is equipped have six channels of detection. Two channels (Ch01 and Ch09) were set to brightfield (BF), permitting spatial coordination between cameras. Channel 12 was set to side-scatter (SSC), and further fluorescence channels were used for antibody detection as required. The advanced fluidic control of ISx, coupled with the presence of continuously running speed beads enable cell/particle enumeration using the “objects per mL” feature within the IDEAS® data analysis software. To avoid the risk of coincident particle detection, EV samples were not run at concentrations greater than 1010 objects/mL. All samples were acquired using INSPIRE® software, with a minimum of 5,000 G1 events collected, or as dictated by the type of analysis to be undertaken. Data analyses were performed, and spectral compensation matrices produced, using ISx Data Exploration and Analysis Software (IDEAS®).
Nanoparticle tracking analysis
Isolated EVPs (as described above) from cell culture supernatants were diluted in PBS and used for nanoparticle tracking analysis using a Nanosight NS300 instrument (NanoSight), followed by evaluation using the Nanoparticle Tracking Analysis (NTA) software. Conditions were as follows: camera type, sCMOS; detection threshold, 5; recording for 800 frames at 25 frames per s.
APEX2 labelling and streptavidin enrichment for LC–MS/MS analysis
HeLaAPEX2-ATG5-WT and HeLaAPEX2-ATG5-K130R cells were incubated in complete medium supplemented with 500 μM biotin–phenol (AdipoGen) with or without 2 mM LLOMe for 30 min. A 1-min pulse with 1 mM H2O2 at room temperature was stopped with quenching buffer (10 mM sodium ascorbate, 10 mM sodium azide and 5 mM Trolox in PBS). All samples were washed twice with quenching buffer, and twice with PBS for 1 min. For LC–MS/MS analysis, cell pellets were lysed in 500 μl ice-cold lysis buffer (6 M urea, 0.3 M NaCl, 1 mM EDTA, 1 mM EGTA, 10 mM sodium ascorbate, 10 mM sodium azide, 5 mM Trolox, 1% glycerol and 25 mm Tris-HCl, pH 7.5) for 30 min by gentle pipetting. Lysates were clarified by centrifugation and protein concentrations were determined using Pierce 660 nm protein assay reagent. Streptavidin-coated magnetic beads (Pierce) were washed with lysis buffer. A total of 1 mg of each sample was mixed with 100 μl of streptavidin beads. The suspensions were gently rotated at 4 °C overnight to bind biotinylated proteins. The flow-through after enrichment was removed and the beads were washed in sequence with 1 ml IP buffer (150 mM NaCl, 10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100) twice; 1 ml 1 M KCl; 1 ml of 50 mM Na2CO3; 1 ml 2 M urea in 20 mM Tris HCl (pH 8.0); and 1 ml IP buffer. Biotinylated proteins were eluted and processed for mass spectrometry.
Sample preparation for LC-MS/MS
Protein samples on magnetic beads were washed four times with 200 μl of 50 mM Triethyl ammonium bicarbonate (TEAB) with a twenty-minute shake time at 4 °C in between each wash. Roughly 2.5 μg of trypsin was added to the bead and TEAB mixture and the samples were digested over night at 800 rpm shake speed. After overnight digestion the supernatant was removed and the beads were washed once with enough 50 mM ammonium bicarbonate to cover. After 20 min at a gentle shake the wash is removed and combined with the initial supernatant. The peptide extracts are reduced in volume by vacuum centrifugation and a small portion of the extract is used for fluorometric peptide quantification (Thermo scientific Pierce). One microgram of sample based on the fluorometric peptide assay was loaded for each LC-MS analysis.
Liquid chromatography-tandem mass spectrometry
Peptides were desalted and trapped on a Thermo PepMap trap and separated on an Easy-spray 100 μm × 25 cm C18 column using a Dionex Ultimate 3,000 nUPLC at 200 nL/min. Solvent A = 0.1% formic acid, Solvent B = 100% Acetonitrile 0.1% formic acid. Gradient conditions = 2% B to 50% B over 60 min, followed by a 50%–99% B in 6 min and then held for 3 min than 99% B to 2% B in 2 min and total run time of 90 min using Thermo Scientific Fusion Lumos mass spectrometer. The samples were run in DIA mode; mass spectra were acquired using a collision energy of 35, resolution of 30 K, maximum inject time of 54 ms and an AGC target of 50K, using staggered isolation windows of 12 Da in the m/z range 400–1,000 m/z.
DIA Quantification and Statistical Analysis
DIA data was analyzed using Spectronaut 14.10 (Biognosys Schlieren, Switzerland) using the directDIA workflow with the default settings. Briefly, protein sequences were downloaded from Uniprot (Human Proteome UP000005640), ATG5 from Uniprot and common laboratory contaminant sequences from https://thegpm.org/crap/. Trypsin/P specific was set for the enzyme allowing two missed cleavages. Fixed Modifications were set for Carbamidomethyl, and variable modification were set to Acetyl (Protein N-term) and Oxidation. For DIA search identification, PSM and Protein Group FDR was set at 1%. A minimum of 2 peptides per protein group were required for quantification. Proteins known to be endogenously biotinylated were excluded from consideration. Pathway enrichment analysis and gene ontology of Biological Process and Cellular Component of the ATG12-ATG5 conjugation-independent or conjugation-dependent protein subsets were performed using STRING (https://string-db.org/).
Quantification and statistical analysis
Data in this study are presented as means ± SEM (n ≥ 3). Data were analyzed with either analysis of variance (ANOVA) with Tukey’s HSD post-hoc test, or a two-tailed Student’s t-test. For HCM, n ≥ 3 includes in each independent experiment: 500 valid primary objects/cells per well, from ≥ 5 wells per plate per sample. Statistical significance was defined as: † (not significant) p ≥ 0.05 and *p < 0.05, **p < 0.01.
Supplementary Material
Supplementary Table S1. Proximity biotinylation proteomics of ATG5 interactors continued (related to Figure 3). Tabs 1–10 in the excel sheet show raw data and subset lists and analyses as indicated in tab titles.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Anti-APG5L/ATG5 (EPR1755(2)) | Abcam | ab108327 |
| Rabbit Anti-ATG7 (EP1759Y) | Abcam | ab52472 |
| Rabbit Anti-mCherry | Abcam | ab183628 |
| Rabbit Anti-Syntenin-1 | Abcam | ab19903 |
| Rabbit Anti-TSG101 (4A10) | Abcam | ab83 |
| Rabbit Anti-VPS34 - N-terminal | Abcam | ab124905 |
| Mouse Anti-ALIX | BioLegend | #634502 |
| Mouse Anti-Galectin-3 | BioLegend | #125402 |
| Rabbit ATG13(E1Y9V) | Cell Signaling Technology | #13468 |
| Rabbit Gm130 (D6B1) | Cell Signaling Technology | #12480S |
| Rabbit HA-Tag (C29F4) | Cell Signaling Technology | #3724 |
| Rabbit Lamp1 (D2D11) | Cell Signaling Technology | #9091S |
| Rabbit Pdi (C81H6) | Cell Signaling Technology | #3501S |
| Mouse LAMP2 | DSHB of University of Iowa | H4B4 |
| Rabbit FPR1 Antibody FITC | Fabgennix | FPR1-FITC |
| Rabbit Anti-Atg14 | MBL International | PD026 |
| Rabbit Anti-ATG16L1 | MBL International | PM040 |
| Rabbit CD11b | Novus Biological | NB110-89474 |
| Rabbit Alix | Proteintech | 12422-1-AP |
| Rabbit CHMP2A | Proteintech | 10477-1-AP |
| Rabbit CHMP4B | Proteintech | 13683-1-AP |
| Rabbit beta-Actin (C4) | Santa Cruz Biotechnology | sc-47778 |
| Mouse Anti-cathepsin C (D-6) | Santa Cruz Biotechnology | sc-74590 |
| Mouse Anti-GAPDH (0411) | Santa Cruz Biotechnology | sc-47724 |
| Rabbit Anti-ATG3 | Sigma Aldrich | #A3231 |
| Rabbit Anti-CD63 | Sigma Aldrich | SAB4301607 |
| Rat Ly-6G (Clone 1A8) | STEMCELL Technologies | #60031 |
| Mouse ATG12 | ThermoFisher | 366400 |
| Alexa Fluor 488 Goat anti-Rabbit secondary antibody | ThermoFisher | #A-11034 |
| Alexa Fluor 488 Goat anti-Mouse secondary antibody | ThermoFisher | #A-11029 |
| Alexa Fluor 568 Goat anti-Rabbit secondary antibody | ThermoFisher | #A-11011 |
| Alexa Fluor 568 Goat anti-Mouse secondary antibody | ThermoFisher | #A-11004 |
| Alexa Fluor 647 Goat anti-Rabbit secondary antibody | ThermoFisher | #A27040 |
| Alexa Fluor 647 Goat anti-Rat secondary antibody | ThermoFisher | #A-21247 |
| Goat anti-rabbit IgG-HRP secondary antibody | ThermoFisher | #31460 |
| Goat anti-mouse IgG-HRP secondary antibody | ThermoFisher | #31430 |
| Goat Anti-Rabbit IgG Polyclonal Antibody (IRDye® 800CW) | LI-COR Biosciences | 926-32211 |
| Goat Anti-Mouse IgG Polyclonal Antibody (IRDye® 680RD), | LI-COR Biosciences | 926-68070 |
| Bacterial and virus strains | ||
| Mycobacterium tuberculosis Erdman | Manzanillo et al. 124 | PMID: 22607800 |
| NEB 5-alpha Competent E.coli (High Efficiency) | New England Biolabs | C2987 |
| One Shot Mach1 Phage-Resistant Competent E.coli | ThermoFisher | C862003 |
| Chemicals, peptides, and recombinant proteins | ||
| Leu-Leu-methyl ester hydrobromide (LLOMe) | Sigma Aldrich | L7393 |
| Pierce™ Streptavidin Magnetic Beads | ThermoFisher | 88816 |
| Tetracycline hydrochloride | Sigma Aldrich | T3383 |
| Puromycin dihydrochloride | Sigma Aldrich | P9620 |
| Hygromycin B | Sigma Aldrich | H0654 |
| Silica crystal | US Silica | MIN-U-SIL-15 |
| mouse macrophage colony stimulating factor (mM-CSF) |
Cell Signaling Technology | 5228 |
| Hoechst 33342 | ThermoFisher | H3570 |
| DAPI | Sigma Aldrich | D9542 |
| Prolong Gold Antifade Mountant with DAPI | ThermoFisher | P36931 |
| Polybrene | Sigma Aldrich | TR-1003-G |
| LysoTracker™ Red DND-99 | ThermoFisher | L7528 |
| Biotinyl tyramide | AdiopoGen Life Sciences | CDX-B0270-M500 |
| X-tremeGENE™ HP DNA Transfection Reagent | Sigma Aldrich | 6366236001 |
| Histopaque®-1119 | Sigma Aldrich | 11191 |
| Histopaque®-1077 | Sigma Aldrich | 10771 |
| N-Formyl-Met-Leu-Phe | Sigma Aldrich | 47729-10MG-F |
| LR Clonase Plus Enzyme Mix | ThermoFisher | 11791100 |
| BP Clonase Plus Enzyme Mix | ThermoFisher | 11789100 |
| Nuclease-Free Duplex Buffer | Integrated DNA Technologies | 11-05-01-03 |
| Alt-R® S.p. Cas9 Nuclease V3 | Integrated DNA Technologies | 1081059 |
| Detachin | Genlantis | T00100 |
| Sodium ascorbate | Sigma Aldrich | A7631 |
| Sodium azide | Sigma Aldrich | S2002 |
| Trolox | Sigma Aldrich | 238813 |
| Dextran, Texas Red™, 3000 MW, Lysine Fixable | ThermoFisher | D3328 |
| Critical commercial assays | ||
| QuikChange II Site-Directed Mutagenesis Kit | Agilent | 200523 |
| TidyBlot | BioRad | STAR209 |
| P3 Primary Cell 4D-Nucleofector® X Kit L | Lonza | V4XP-3024 |
| SmartSEC™ Single for EV Isolation | SBI System Biosciences | SSEC200A-1 |
| ExoQuick-TC | SBI System Biosciences | EXOTC10A-1 |
| Gateway Vector Conversion System | ThermoFisher | 11828-029 |
| Lipofectamine 2000 Transfection Reagent | ThermoFisher | 12566014 |
| Molecular Probes EnzChek Elastase Assay Kit | ThermoFisher | E12056 |
| Deposited data | ||
| Raw MS DIA data | https://massive.ucsd.edu | MSV000090348 |
| Raw MS DIA data | http://www.proteomexchange.org | Proteome Exchange = PXD036850 |
| Experimental models: Cell lines | ||
| HeLa WT | ATCC | CCL-2 |
| HeLa ATG5KO | This paper | N/A |
| HeLa ATG3KO | This paper | N/A |
| HeLa ATG3/5DKO | This paper | N/A |
| Huh7 WT | Rocky Mountain laboratories | N/A |
| Huh7 ATG5KO | This paper | N/A |
| Huh7 ATG3KO | This paper | N/A |
| Huh7 ATG3/5DKO | This paper | N/A |
| Huh7 ATG7KO | This paper | N/A |
| Huh7 ATG13KO | This paper | N/A |
| Huh7 ATG14KO | This paper | N/A |
| Huh7 ATG16L1KO | This paper | N/A |
| Huh7 VPS34KO | This paper | N/A |
| A549 WT | ATCC | CCL-185 |
| A549 ATG5KO | This paper | N/A |
| U2OS WT | ATCC | HTB-96 |
| U2OS ATG5KO | This paper | N/A |
| HEK293T | ATCC | CRL-3216 |
| HeLa WT-TMEM192-3XHA | This paper | N/A |
| HeLa ATG5KO -TMEM192-3XHA | This paper | N/A |
| HeLa WT-TMEM192-2XFLAG | This paper | N/A |
| HeLa ATG5KO -TMEM192-2XFLAG | This paper | N/A |
| Flp-In-HeLa APEX2-ATG5WT | This paper | N/A |
| Flp-In-HeLa APEX2-ATG5K130R | This paper | N/A |
| HL-60 WT | This paper | N/A |
| HL-60 ATG5KO | This paper | N/A |
| Patu-8902WT | Alec Kimmelman | N/A |
| Patu-8902ATG5-KO | This paper | N/A |
| Experimental models: Organisms/strains | ||
| C57BL/6 | The Jackson Laboratory | Strain #000664 |
| ATG5fl/fl LysM-Cre-negative mice | Castillo et al. 51 | PMID: 23040807 |
| ATG5fl/fl LysM-Cre-positive mice | Castillo et al. 51 | PMID: 23040807 |
| ATG7fl/fl LysM-Cre-negative mice | Komatsu et al.125 | PMID: 15866887 |
| ATG7fl/fl LysM-Cre-positive mice | Komatsu et al.125 | PMID: 15866887 |
| Oligonucleotides | ||
| ATG5 CRISPR gRNA: AAGAGTAAGTTATTTGACGT | VectorBuilder | gRNA#1544 |
| ATG3 CRISPR gRNA: GTGAAGGCATACCTACCAAC | VectorBuilder | gRNA#1119 |
| ATG7 CRISPR gRNA: CTTCCGTGACCGTACCATGC | VectorBuilder | gRNA#6556 |
| ATG13 CRISPR gRNA: CTGTCCCAACACGAACTGTC | VectorBuilder | gRNA#4570 |
| ATG14 CRISPR gRNA: CTACTTCGACGGCCGCGACC | VectorBuilder | gRNA#58 |
| ATG16L1 CRISPR gRNA: ACCAAATGCAGCGGAAGGAC | VectorBuilder | gRNA#1540 |
| VPS34 CRISPR gRNA: ACAGACATTTGTAGATCGGT | VectorBuilder | gRNA#5287 |
| ATG5 primers for gateway cloning: Forward: GGGGACAAGTTTGTACAAAAAAGCAGGCTTCACAGATGACAAAGATGTGCTTCGAG Reverse: GGGG-AC-CAC-TTT-GTA-CAA-GAA-AGC-TGG-GTC-TCAATCTGTTGGCTGTGGGATG |
Integrated DNA Technologies | N/A |
| ATG5 primers for K130R mutation: Sense: 5'-GCAATTGAAGCTCATTTTATGTCATGTATGAGAGAAGCTGATGCTTTA-3' Antisense: 5'-TAAAGCATCAGCTTCTCTCATACATGACATAAAATGAGCTTCAATTGC-3' |
Integrated DNA Technologies | N/A |
| Alt-R® CRISPR-Cas9 guide RNA for ATG5 sgRNA1: TCCATCCAAGGATGCGGTTG sgRNA2: AAGAGTCAGCTATTTGACGT |
Integrated DNA Technologies | Mm.Cas9.ATG5.1.AA Mm.Cas9.ATG5.1.AB |
| Recombinant DNA | ||
| pDEST-Flp-In-APEX2-ATG5WT | This paper | N/A |
| pDEST-Flp-In-APEX2-ATG5K130R | This paper | N/A |
| pDEST-mCherry-ATG5WT | This paper | N/A |
| pDEST-mCherry-ATG5K130R | This paper | N/A |
| pDEST-mCherry-SARS-CoV-2ORF3a | This paper | N/A |
| pCMV-VSV-G | Addgene | #8454 |
| psPAX2 | Addgene | #12260 |
| pOG44 | This paper | N/A |
| pLJC5-TMEM192-3xHA | Addgene | #102930 |
| pLJC5-TMEM192-2xFLAG | Addgene | #102929 |
| Software and algorithms | ||
| iDEV software | ThermoFisher | N/A |
| Zen 3.4 | Carl Zeiss | N/A |
| Spectronaut software | Biognosys Inc | N/A |
| Prism 8 | GraphPad | https://www.graphpad.com/ |
| Image Lab | BIO-RAD | N/A |
| IDEAS 6.2 | Luminex | N/A |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| NTA 3.3 | Malvern Panalytical | N/A |
| R (v4.1.2) | The R Foundation | https://www.r-project.org |
| Other | ||
| NP40 Cell Lysis Buffer | ThermoFisher | FNN0021 |
| Protease Inhibitor Cocktail Tablets | Roche | 11697498001 |
| PMSF | Sigma Aldrich | 93482 |
| Anti-HA Magnetic Beads | ThermoFisher | 88836 |
| Dynabeads Protein G | ThermoFisher | 10003D |
Highlights.
ATG5 is at a cardinal junction between canonical and noncanonical ATG conjugates
Loss of ATG5 favors a sidestep conjugate ATG12-ATG3 which sequesters ESCRT ALIX
Loss of ATG5 causes lysosomal hypersensitivity to damage and increased exocytosis
The noncanonical role of ATG5 affects neutrophils and tuberculosis pathogenesis
Acknowledgments
We thank M. Campen and G. Herbert for nanoparticle tracking analysis, and A. Kimmelman for pancreatic carcinoma cell line PaTu-8902. This work was supported by NIH grants R37AI042999 and R01AI111935, and center grant P20GM121176 to V.D. Mass spectrometry analysis, B.P., and M.S., were supported by NIH shared instrumentation grant S10OD021801.
Footnotes
Competing interests
The authors declare no competing interests.
Additional Information: Supplementary information is available for this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mizushima N, Levine B, Cuervo AM, and Klionsky DJ (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morishita H, and Mizushima N (2019). Diverse Cellular Roles of Autophagy. Annu Rev Cell Dev Biol 35, 453–475. 10.1146/annurev-cellbio-100818-125300. [DOI] [PubMed] [Google Scholar]
- 3.Levine B, and Kroemer G (2019). Biological Functions of Autophagy Genes: A Disease Perspective. Cell 176, 11–42. 10.1016/j.cell.2018.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galluzzi L, and Green DR (2019). Autophagy-Independent Functions of the Autophagy Machinery. Cell 177, 1682–1699. 10.1016/j.cell.2019.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deretic V, and Lazarou M (2022). A guide to membrane atg8ylation and autophagy with reflections on immunity. J Cell Biol 221. 10.1083/jcb.202203083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mizushima N, Yoshimori T, and Ohsumi Y (2011). The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27, 107–132. 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 7.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, and Yamamoto A (2009). A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 11, 1433–1437. 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 8.Tooze SA, and Yoshimori T (2010). The origin of the autophagosomal membrane. Nat Cell Biol 12, 831–835. ncb0910–831 [pii] 10.1038/ncb0910-831. [DOI] [PubMed] [Google Scholar]
- 9.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, and Ktistakis NT (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182, 685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, and Mizushima N (2008). FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 181, 497–510. 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Itakura E, and Mizushima N (2010). Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6. 12709 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Itakura E, and Mizushima N (2011). p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. The Journal of cell biology 192, 17–27. 10.1083/jcb.201009067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishimura T, Tamura N, Kono N, Shimanaka Y, Arai H, Yamamoto H, and Mizushima N (2017). Autophagosome formation is initiated at phosphatidylinositol synthase-enriched ER subdomains. EMBO J 36, 1719–1735. 10.15252/embj.201695189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ge L, Melville D, Zhang M, and Schekman R (2013). The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife 2, e00947. 10.7554/eLife.00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ge L, Zhang M, and Schekman R (2014). Phosphatidylinositol 3-kinase and COPII generate LC3 lipidation vesicles from the ER-Golgi intermediate compartment. Elife 3, e04135. 10.7554/eLife.04135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ge L, Zhang M, Kenny SJ, Liu D, Maeda M, Saito K, Mathur A, Xu K, and Schekman R (2017). Remodeling of ER-exit sites initiates a membrane supply pathway for autophagosome biogenesis. EMBO Rep 18, 1586–1603. 10.15252/embr.201744559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar S, Gu Y, Abudu YP, Bruun JA, Jain A, Farzam F, Mudd M, Anonsen JH, Rusten TE, Kasof G, et al. (2019). Phosphorylation of Syntaxin 17 by TBK1 Controls Autophagy Initiation. Dev Cell. 10.1016/j.devcel.2019.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar S, Javed R, Mudd M, Pallikkuth S, Lidke KA, Jain A, Tangavelou K, Gudmundsson SR, Ye C, Rusten TE, et al. (2021). Mammalian hybrid pre-autophagosomal structure HyPAS generates autophagosomes. Cell 184, 5950–5969 e5922. 10.1016/j.cell.2021.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao YG, and Zhang H (2019). Autophagosome maturation: An epic journey from the ER to lysosomes. J Cell Biol 218, 757–770. 10.1083/jcb.201810099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mizushima N (2020). The ATG conjugation systems in autophagy. Curr Opin Cell Biol 63, 1–10. 10.1016/j.ceb.2019.12.001. [DOI] [PubMed] [Google Scholar]
- 21.Xin Y, Yu L, Chen Z, Zheng L, Fu Q, Jiang J, Zhang P, Gong R, and Zhao S (2001). Cloning, expression patterns, and chromosome localization of three human and two mouse homologues of GABA(A) receptor-associated protein. Genomics 74, 408–413. 10.1006/geno.2001.6555. [DOI] [PubMed] [Google Scholar]
- 22.He H, Dang Y, Dai F, Guo Z, Wu J, She X, Pei Y, Chen Y, Ling W, Wu C, et al. (2003). Post-translational modifications of three members of the human MAP1LC3 family and detection of a novel type of modification for MAP1LC3B. J Biol Chem 278, 29278–29287. 10.1074/jbc.M303800200. [DOI] [PubMed] [Google Scholar]
- 23.Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, and Elazar Z (2010). LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. The EMBO journal 29, 1792–1802. 10.1038/emboj.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, et al. (2000). A ubiquitin-like system mediates protein lipidation. Nature 408, 488–492. [DOI] [PubMed] [Google Scholar]
- 25.Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, and Ohsumi Y (1998). A protein conjugation system essential for autophagy. Nature 395, 395–398. [DOI] [PubMed] [Google Scholar]
- 26.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, and Yoshimori T (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J 19, 5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, and Green DR (2007). Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257. [DOI] [PubMed] [Google Scholar]
- 28.Ulferts R, Marcassa E, Timimi L, Lee LC, Daley A, Montaner B, Turner SD, Florey O, Baillie JK, and Beale R (2021). Subtractive CRISPR screen identifies the ATG16L1/vacuolar ATPase axis as required for non-canonical LC3 lipidation. Cell Rep 37, 109899. 10.1016/j.celrep.2021.109899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee C, Lamech L, Johns E, and Overholtzer M (2020). Selective Lysosome Membrane Turnover Is Induced by Nutrient Starvation. Dev Cell 55, 289–297 e284. 10.1016/j.devcel.2020.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loi M, Raimondi A, Morone D, and Molinari M (2019). ESCRT-III-driven piecemeal micro-ER-phagy remodels the ER during recovery from ER stress. Nat Commun 10, 5058. 10.1038/s41467-019-12991-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo H, Chitiprolu M, Roncevic L, Javalet C, Hemming FJ, Trung MT, Meng L, Latreille E, Tanese de Souza C, McCulloch D, et al. (2017). Atg5 Disassociates the V1V0-ATPase to Promote Exosome Production and Tumor Metastasis Independent of Canonical Macroautophagy. Dev Cell 43, 716–730 e717. 10.1016/j.devcel.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 32.Leidal AM, Huang HH, Marsh T, Solvik T, Zhang D, Ye J, Kai F, Goldsmith J, Liu JY, Huang YH, et al. (2020). The LC3-conjugation machinery specifies the loading of RNA-binding proteins into extracellular vesicles. Nat Cell Biol 22, 187–199. 10.1038/s41556-019-0450-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakamura S, Shigeyama S, Minami S, Shima T, Akayama S, Matsuda T, Esposito A, Napolitano G, Kuma A, Namba-Hamano T, et al. (2020). LC3 lipidation is essential for TFEB activation during the lysosomal damage response to kidney injury. Nat Cell Biol 22, 1252–1263. 10.1038/s41556-020-00583-9. [DOI] [PubMed] [Google Scholar]
- 34.Kumar S, Jain A, Choi SW, da Silva GPD, Allers L, Mudd MH, Peters RS, Anonsen JH, Rusten TE, Lazarou M, and Deretic V (2020). Mammalian Atg8 proteins and the autophagy factor IRGM control mTOR and TFEB at a regulatory node critical for responses to pathogens. Nat Cell Biol 22, 973–985. 10.1038/s41556-020-0549-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goodwin JM, Walkup W.G.t., Hooper K, Li T, Kishi-Itakura C, Ng A, Lehmberg T, Jha A, Kommineni S, Fletcher K, et al. (2021). GABARAP sequesters the FLCN-FNIP tumor suppressor complex to couple autophagy with lysosomal biogenesis. Sci Adv 7, eabj2485. 10.1126/sciadv.abj2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar S, Jia J, and Deretic V (2021). Atg8ylation as a general membrane stress and remodeling response. Cell Stress 5, 128–142. 10.15698/cst2021.09.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamark T, and Johansen T (2021). Mechanisms of Selective Autophagy. Annu Rev Cell Dev Biol 37, 143–169. 10.1146/annurev-cellbio-120219-035530. [DOI] [PubMed] [Google Scholar]
- 38.Weidberg H, Shpilka T, Shvets E, Abada A, Shimron F, and Elazar Z (2011). LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Developmental cell 20, 444–454. 10.1016/j.devcel.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 39.Maruyama T, Alam JM, Fukuda T, Kageyama S, Kirisako H, Ishii Y, Shimada I, Ohsumi Y, Komatsu M, Kanki T, et al. (2021). Membrane perturbation by lipidated Atg8 underlies autophagosome biogenesis. Nat Struct Mol Biol 28, 583–593. 10.1038/s41594-021-00614-5. [DOI] [PubMed] [Google Scholar]
- 40.Xie Z, Nair U, and Klionsky DJ (2008). Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell 19, 3290–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen TN, Padman BS, Usher J, Oorschot V, Ramm G, and Lazarou M (2016). Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol 215, 857–874. 10.1083/jcb.201607039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Radoshevich L, Murrow L, Chen N, Fernandez E, Roy S, Fung C, and Debnath J (2010). ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 142, 590–600. S0092–8674(10)00787–7 [pii] 10.1016/j.cell.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murrow L, Malhotra R, and Debnath J (2015). ATG12-ATG3 interacts with Alix to promote basal autophagic flux and late endosome function. Nat Cell Biol 17, 300–310. 10.1038/ncb3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skowyra ML, Schlesinger PH, Naismith TV, and Hanson PI (2018). Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 360. 10.1126/science.aar5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhen Y, Radulovic M, Vietri M, and Stenmark H (2021). Sealing holes in cellular membranes. EMBO J 40, e106922. 10.15252/embj.2020106922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bohannon KP, and Hanson PI (2020). ESCRT puts its thumb on the nanoscale: Fixing tiny holes in endolysosomes. Curr Opin Cell Biol 65, 122–130. 10.1016/j.ceb.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jia J, Claude-Taupin A, Gu Y, Choi SW, Peters R, Bissa B, Mudd MH, Allers L, Pallikkuth S, Lidke KA, et al. (2020). Galectin-3 Coordinates a Cellular System for Lysosomal Repair and Removal. Dev Cell 52, 69–87 e68. 10.1016/j.devcel.2019.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mizushima N, and Levine B (2020). Autophagy in Human Diseases. N Engl J Med 383, 1564–1576. 10.1056/NEJMra2022774. [DOI] [PubMed] [Google Scholar]
- 49.Deretic V, and Levine B (2018). Autophagy balances inflammation in innate immunity. Autophagy, 1–9. 10.1080/15548627.2017.1402992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, and Deretic V (2004). Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766. 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 51.Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, Delgado-Vargas M, Timmins GS, Bhattacharya D, Yang H, et al. (2012). Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proceedings of the National Academy of Sciences of the United States of America 109, E3168–3176. 10.1073/pnas.1210500109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Watson RO, Manzanillo PS, and Cox JS (2012). Extracellular M. tuberculosis DNA Targets Bacteria for Autophagy by Activating the Host DNA-Sensing Pathway. Cell 150, 803–815. 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, Virgin HW, and Stallings CL (2015). Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 528, 565–569. 10.1038/nature16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, et al. (2010). An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466, 973–977. 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MacMicking JD, Taylor GA, and McKinney JD (2003). Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science 302, 654–659. [DOI] [PubMed] [Google Scholar]
- 56.Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T, Choi SW, Peters R, Mandell M, Bruun JA, et al. (2016). TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-direct Autophagy in Endomembrane Damage Homeostasis. Dev Cell 39, 13–27. 10.1016/j.devcel.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Orme IM, and Collins FM (1983). Protection against Mycobacterium tuberculosis infection by adoptive immunotherapy. Requirement for T cell-deficient recipients. J Exp Med 158, 74–83. 10.1084/jem.158.1.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stappenbeck TS, and Virgin HW (2016). Accounting for reciprocal host-microbiome interactions in experimental science. Nature 534, 191–199. 10.1038/nature18285. [DOI] [PubMed] [Google Scholar]
- 59.Claude-Taupin A, Jia J, Bhujabal Z, Garfa-Traore M, Kumar S, da Silva GPD, Javed R, Gu Y, Allers L, Peters R, et al. (2021). ATG9A protects the plasma membrane from programmed and incidental permeabilization. Nat Cell Biol 23, 846–858. 10.1038/s41556-021-00706-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, and Yoshimori T (2013). Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 32, 2336–2347. 10.1038/emboj.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ballabio A, and Bonifacino JS (2020). Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol 21, 101–118. 10.1038/s41580-019-0185-4. [DOI] [PubMed] [Google Scholar]
- 62.Jia J, Abudu YP, Claude-Taupin A, Gu Y, Kumar S, Choi SW, Peters R, Mudd MH, Allers L, Salemi M, et al. (2018). Galectins Control mTOR in Response to Endomembrane Damage. Mol Cell 70, 120–135 e128. 10.1016/j.molcel.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jia J, Bissa B, Brecht L, Allers L, Choi SW, Gu Y, Zbinden M, Burge MR, Timmins G, Hallows K, et al. (2020). AMPK, a Regulator of Metabolism and Autophagy, Is Activated by Lysosomal Damage via a Novel Galectin-Directed Ubiquitin Signal Transduction System. Mol Cell. 10.1016/j.molcel.2019.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aits S, Kricker J, Liu B, Ellegaard AM, Hamalisto S, Tvingsholm S, Corcelle-Termeau E, Hogh S, Farkas T, Holm Jonassen A, et al. (2015). Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 11, 1408–1424. 10.1080/15548627.2015.1063871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thiele DL, and Lipsky PE (1990). Mechanism of L-leucyl-L-leucine methyl ester-mediated killing of cytotoxic lymphocytes: dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc Natl Acad Sci U S A 87, 83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eriksson I, Waster P, and Ollinger K (2020). Restoration of lysosomal function after damage is accompanied by recycling of lysosomal membrane proteins. Cell Death Dis 11, 370. 10.1038/s41419-020-2527-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bonet-Ponce L, Beilina A, Williamson CD, Lindberg E, Kluss JH, Saez-Atienzar S, Landeck N, Kumaran R, Mamais A, Bleck CKE, et al. (2020). LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci Adv 6. 10.1126/sciadv.abb2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tan JX, and Finkel T (2022). A phosphoinositide signalling pathway mediates rapid lysosomal repair. Nature. 10.1038/s41586-022-05164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Freund EC, Lock JY, Oh J, Maculins T, Delamarre L, Bohlen CJ, Haley B, and Murthy A (2020). Efficient gene knockout in primary human and murine myeloid cells by non-viral delivery of CRISPR-Cas9. J Exp Med 217. 10.1084/jem.20191692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Elsasser HP, Lehr U, Agricola B, and Kern HF (1993). Structural analysis of a new highly metastatic cell line PaTu 8902 from a primary human pancreatic adenocarcinoma. Virchows Arch B Cell Pathol Incl Mol Pathol 64, 201–207. 10.1007/BF02915113. [DOI] [PubMed] [Google Scholar]
- 71.Daemen A, Peterson D, Sahu N, McCord R, Du X, Liu B, Kowanetz K, Hong R, Moffat J, Gao M, et al. (2015). Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc Natl Acad Sci U S A 112, E4410–4417. 10.1073/pnas.1501605112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mossman BT, and Churg A (1998). Mechanisms in the pathogenesis of asbestosis and silicosis. Am J Respir Crit Care Med 157, 1666–1680. 10.1164/ajrccm.157.5.9707141. [DOI] [PubMed] [Google Scholar]
- 73.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, and Latz E (2008). Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9, 847–856. ni.1631 [pii] 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jia J, Abudu YP, Claude-Taupin A, Gu Y, Kumar S, Choi SW, Peters R, Mudd MH, Allers L, Salemi M, et al. (2019). Galectins control MTOR and AMPK in response to lysosomal damage to induce autophagy. Autophagy 15, 169–171. 10.1080/15548627.2018.1505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yue Y, Nabar NR, Shi CS, Kamenyeva O, Xiao X, Hwang IY, Wang M, and Kehrl JH (2018). SARS-Coronavirus Open Reading Frame-3a drives multimodal necrotic cell death. Cell Death Dis 9, 904. 10.1038/s41419-018-0917-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jia J, Wang F, Bhujabal Z, Peters R, Mudd M, Duque T, Allers L, Javed R, Salemi M, Behrends C, et al. (2022). Stress granules and mTOR are regulated by membrane atg8ylation during lysosomal damage. J Cell Biol 221. 10.1083/jcb.202207091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Radulovic M, Schink KO, Wenzel EM, Nahse V, Bongiovanni A, Lafont F, and Stenmark H (2018). ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J 37. 10.15252/embj.201899753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shukla S, Larsen KP, Ou C, Rose K, and Hurley JH (2022). In vitro reconstitution of calcium-dependent recruitment of the human ESCRT machinery in lysosomal membrane repair. bioRxiv, 2022.2003.2030.486420. 10.1101/2022.03.30.486420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, Freinkman E, and Sabatini DM (2017). Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813. 10.1126/science.aan6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lam SS, Martell JD, Kamer KJ, Deerinck TJ, Ellisman MH, Mootha VK, and Ting AY (2015). Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods 12, 51–54. 10.1038/nmeth.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Searle BC, Pino LK, Egertson JD, Ting YS, Lawrence RT, MacLean BX, Villén J, and MacCoss MJ (2018). Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nature Communications 9, 5128. 10.1038/s41467-018-07454-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shao H, Im H, Castro CM, Breakefield X, Weissleder R, and Lee H (2018). New Technologies for Analysis of Extracellular Vesicles. Chem Rev 118, 1917–1950. 10.1021/acs.chemrev.7b00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hirschberg Y, Boonen K, Schildermans K, van Dam A, Pintelon I, Vandendriessche C, Velimirovic M, Jacobs A, Vandenbroucke RE, Nelissen I, et al. (2022). Characterising extracellular vesicles from individual low volume cerebrospinal fluid samples, isolated by SmartSEC. Journal of Extracellular Biology 1, e55. 10.1002/jex2.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Solvik TA, Nguyen TA, Tony Lin YH, Marsh T, Huang EJ, Wiita AP, Debnath J, and Leidal AM (2022). Secretory autophagy maintains proteostasis upon lysosome inhibition. J Cell Biol 221. 10.1083/jcb.202110151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Soehnlein O, Oehmcke S, Ma X, Rothfuchs AG, Frithiof R, van Rooijen N, Morgelin M, Herwald H, and Lindbom L (2008). Neutrophil degranulation mediates severe lung damage triggered by streptococcal M1 protein. Eur Respir J 32, 405–412. 10.1183/09031936.00173207. [DOI] [PubMed] [Google Scholar]
- 86.Sengelov H, Kjeldsen L, Diamond MS, Springer TA, and Borregaard N (1993). Subcellular localization and dynamics of Mac-1 (alpha m beta 2) in human neutrophils. J Clin Invest 92, 1467–1476. 10.1172/JCI116724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lau D, Mollnau H, Eiserich JP, Freeman BA, Daiber A, Gehling UM, Brummer J, Rudolph V, Munzel T, Heitzer T, et al. (2005). Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc Natl Acad Sci U S A 102, 431–436. 10.1073/pnas.0405193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Martin-Martin B, Nabokina SM, Blasi J, Lazo PA, and Mollinedo F (2000). Involvement of SNAP-23 and syntaxin 6 in human neutrophil exocytosis. Blood 96, 2574–2583. [PubMed] [Google Scholar]
- 89.Bentwood BJ, and Henson PM (1980). The sequential release of granule constitutents from human neutrophils. J Immunol 124, 855–862. [PubMed] [Google Scholar]
- 90.Fumagalli L, Zhang H, Baruzzi A, Lowell CA, and Berton G (2007). The Src family kinases Hck and Fgr regulate neutrophil responses to N-formyl-methionyl-leucyl-phenylalanine. J Immunol 178, 3874–3885. 10.4049/jimmunol.178.6.3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sheshachalam A, Srivastava N, Mitchell T, Lacy P, and Eitzen G (2014). Granule protein processing and regulated secretion in neutrophils. Front Immunol 5, 448. 10.3389/fimmu.2014.00448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kugeratski FG, Hodge K, Lilla S, McAndrews KM, Zhou X, Hwang RF, Zanivan S, and Kalluri R (2021). Quantitative proteomics identifies the core proteome of exosomes with syntenin-1 as the highest abundant protein and a putative universal biomarker. Nature Cell Biology 23, 631–641. 10.1038/s41556-021-00693-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ghossoub R, Lembo F, Rubio A, Gaillard CB, Bouchet J, Vitale N, Slavík J, Machala M, and Zimmermann P (2014). Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat Commun 5, 3477. 10.1038/ncomms4477. [DOI] [PubMed] [Google Scholar]
- 94.Bae GH, Kim YS, Park JY, Lee M, Lee SK, Kim JC, Kim JG, Shin YJ, Lee H, Kim SY, et al. (2022). Unique characteristics of lung-resident neutrophils are maintained by PGE2/PKA/Tgm2-mediated signaling. Blood 140, 889–899. 10.1182/blood.2021014283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lauterbach M, O’Donnell P, Asano K, and Mayadas TN (2008). Role of TNF priming and adhesion molecules in neutrophil recruitment to intravascular immune complexes. J Leukoc Biol 83, 1423–1430. 10.1189/jlb.0607421. [DOI] [PubMed] [Google Scholar]
- 96.Sengelov H, Boulay F, Kjeldsen L, and Borregaard N (1994). Subcellular localization and translocation of the receptor for N-formylmethionyl-leucyl-phenylalanine in human neutrophils. Biochem J 299 ( Pt 2), 473–479. 10.1042/bj2990473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sato K, Nagai J, Mitsui N, Ryoko Y, and Takano M (2009). Effects of endocytosis inhibitors on internalization of human IgG by Caco-2 human intestinal epithelial cells. Life Sci 85, 800–807. 10.1016/j.lfs.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 98.Subramanya S, Hardin CF, Steverding D, and Mensa-Wilmot K (2009). Glycosylphosphatidylinositol-specific phospholipase C regulates transferrin endocytosis in the African trypanosome. Biochem J 417, 685–694. 10.1042/BJ20080167. [DOI] [PubMed] [Google Scholar]
- 99.Shi JM, Zhu L, Lan X, Zhao DW, He YJ, Sun ZQ, Wu D, and Li HY (2020). Endocytosis Is a Key Mode of Interaction between Extracellular β-Amyloid and the Cell Membrane. Biophys J 119, 1078–1090. 10.1016/j.bpj.2020.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Greene CJ, Nguyen JA, Cheung SM, Arnold CR, Balce DR, Wang YT, Soderholm A, McKenna N, Aggarwal D, Campden RI, et al. (2022). Macrophages disseminate pathogen associated molecular patterns through the direct extracellular release of the soluble content of their phagolysosomes. Nat Commun 13, 3072. 10.1038/s41467-022-30654-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang Y, Gomez M, Marsh T, Poillet-Perez L, Sawant A, Chen L, Park NR, Jackson SR, Hu Z, Alon N, et al. (2022). Autophagy in PDGFRalpha+ mesenchymal cells is essential for intestinal stem cell survival. Proc Natl Acad Sci U S A 119, e2202016119. 10.1073/pnas.2202016119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Han J, Feng G, Wu J, Zhang Y, Long Z, and Yao X (2022). Association of ATG5 gene polymorphism with Parkinson’s disease in a Han Chinese population. Acta Neurol Belg 122, 1049–1056. 10.1007/s13760-021-01814-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hu ZY, Chen B, Zhang JP, and Ma YY (2017). Up-regulation of autophagy-related gene 5 (ATG5) protects dopaminergic neurons in a zebrafish model of Parkinson’s disease. J Biol Chem 292, 18062–18074. 10.1074/jbc.M116.764795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang L, Chen X, Chang M, and Jiao B (2021). MiR-30c-5p/ATG5 Axis Regulates the Progression of Parkinson’s Disease. Front Cell Neurosci 15, 644507. 10.3389/fncel.2021.644507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, and Mizushima N (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. nature04724 [pii] 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 106.Yassin R, Tadmor H, Farber E, Igbariye A, Armaly-Nakhoul A, Dahan I, Nakhoul F, and Nakhoul N (2021). Alteration of autophagy-related protein 5 (ATG5) levels and Atg5 gene expression in diabetes mellitus with and without complications. Diab Vasc Dis Res 18, 14791641211062050. 10.1177/14791641211062050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu H, He Z, von Rutte T, Yousefi S, Hunger RE, and Simon HU (2013). Downregulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci Transl Med 5, 202ra123. 10.1126/scitranslmed.3005864. [DOI] [PubMed] [Google Scholar]
- 108.Frangez Z, Gerard D, He Z, Gavriil M, Fernandez-Marrero Y, Seyed Jafari SM, Hunger RE, Lucarelli P, Yousefi S, Sauter T, et al. (2021). ATG5 and ATG7 Expression Levels Are Reduced in Cutaneous Melanoma and Regulated by NRF1. Front Oncol 11, 721624. 10.3389/fonc.2021.721624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cho DH, Jo YK, Kim SC, Park IJ, and Kim JC (2012). Down-regulated expression of ATG5 in colorectal cancer. Anticancer Res 32, 4091–4096. [PubMed] [Google Scholar]
- 110.An CH, Kim MS, Yoo NJ, Park SW, and Lee SH (2011). Mutational and expressional analyses of ATG5, an autophagy-related gene, in gastrointestinal cancers. Pathol Res Pract 207, 433–437. 10.1016/j.prp.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 111.Gorgulu K, Diakopoulos KN, Ai J, Schoeps B, Kabacaoglu D, Karpathaki AF, Ciecielski KJ, Kaya-Aksoy E, Ruess DA, Berninger A, et al. (2019). Levels of the Autophagy-Related 5 Protein Affect Progression and Metastasis of Pancreatic Tumors in Mice. Gastroenterology 156, 203–217 e220. 10.1053/j.gastro.2018.09.053. [DOI] [PubMed] [Google Scholar]
- 112.Gupta S, Yano J, Mercier V, Htwe HH, Shin HR, Rademaker G, Cakir Z, Ituarte T, Wen KW, Kim GE, et al. (2021). Lysosomal retargeting of Myoferlin mitigates membrane stress to enable pancreatic cancer growth. Nat Cell Biol 23, 232–242. 10.1038/s41556-021-00644-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, Sohn ASW, Mukhopadhyay S, Lin EY, Parker SJ, et al. (2020). Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581, 100–105. 10.1038/s41586-020-2229-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ljubojevic-Holzer S, Kraler S, Djalinac N, Abdellatif M, Voglhuber J, Schipke J, Schmidt M, Kling KM, Franke GT, Herbst V, et al. (2022). Loss of autophagy protein ATG5 impairs cardiac capacity in mice and humans through diminishing mitochondrial abundance and disrupting Ca2+ cycling. Cardiovasc Res 118, 1492–1505. 10.1093/cvr/cvab112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nauseef WM, and Borregaard N (2014). Neutrophils at work. Nat Immunol 15, 602–611. 10.1038/ni.2921. [DOI] [PubMed] [Google Scholar]
- 116.Németh T, Sperandio M, and Mócsai A (2020). Neutrophils as emerging therapeutic targets. Nature Reviews Drug Discovery 19, 253–275. 10.1038/s41573-019-0054-z. [DOI] [PubMed] [Google Scholar]
- 117.Mollinedo F (2019). Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends in Immunology 40, 228–242. 10.1016/j.it.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 118.Rosales C (2018). Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Frontiers in Physiology 9. 10.3389/fphys.2018.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Leslie J, Millar BJ, Del Carpio Pons A, Burgoyne RA, Frost JD, Barksby BS, Luli S, Scott J, Simpson AJ, Gauldie J, et al. (2020). FPR-1 is an important regulator of neutrophil recruitment and a tissue-specific driver of pulmonary fibrosis. JCI Insight 5. 10.1172/jci.insight.125937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lakschevitz FS, Hassanpour S, Rubin A, Fine N, Sun C, and Glogauer M (2016). Identification of neutrophil surface marker changes in health and inflammation using high-throughput screening flow cytometry. Experimental Cell Research 342, 200–209. 10.1016/j.yexcr.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 121.Eichelberger KR, and Goldman WE (2020). Manipulating neutrophil degranulation as a bacterial virulence strategy. PLoS Pathog 16, e1009054. 10.1371/journal.ppat.1009054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Germic N, Stojkov D, Oberson K, Yousefi S, and Simon HU (2017). Neither eosinophils nor neutrophils require ATG5-dependent autophagy for extracellular DNA trap formation. Immunology 152, 517–525. 10.1111/imm.12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gupta S, and Kaplan MJ (2016). The role of neutrophils and NETosis in autoimmune and renal diseases. Nat Rev Nephrol 12, 402–413. 10.1038/nrneph.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Manzanillo PS, Shiloh MU, Portnoy DA, and Cox JS (2012). Mycobacterium Tuberculosis Activates the DNA-Dependent Cytosolic Surveillance Pathway within Macrophages. Cell host & microbe 11, 469–480. 10.1016/j.chom.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. (2005). Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 169, 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Swamydas M, Luo Y, Dorf ME, and Lionakis MS (2015). Isolation of Mouse Neutrophils. Curr Protoc Immunol 110, 3 20 21–23 20 15. 10.1002/0471142735.im0320s110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.McGill CJ, Lu RJ, and Benayoun BA (2021). Protocol for analysis of mouse neutrophil NETosis by flow cytometry. STAR Protoc 2, 100948. 10.1016/j.xpro.2021.100948. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Proximity biotinylation proteomics of ATG5 interactors continued (related to Figure 3). Tabs 1–10 in the excel sheet show raw data and subset lists and analyses as indicated in tab titles.
Data Availability Statement
Raw MS DIA/DDA data have been deposited at the MassIVE proteomics repository MassIVE (MSV000090348) and Proteome Exchange (PXD036850).
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.