

Abstract
In the last twenty years, protein kinases have been identified as important targets for cancer therapy. In order to prevent unexpected toxicity, medicinal chemists have always focused on discovering selective protein kinase inhibitors. However, cancer is a multifactorial process and its formation and progression depend on different stimuli. Therefore, it is imperative to develop anticancer therapy that targets multiple kinases associated cancer progression. In this research a series of hybrid compounds was designed and synthesized successfully with the aim of producing anticancer activity through the induction of multiple protein kinase inhibition. The designed derivatives comprise isatin and pyrrolo[2,3-d]pyrimidine scaffolds in their structures with a hydrazine linking the two pharmacophores. Antiproliferative and kinase inhibition assays revealed promising anticancer and multi-kinase inhibitory effects of compound 7 with comparable results with the reference standards. Moreover, compound 7 suppressed cell cycle progression and induced apoptosis in HepG2 cells. Finally, molecular docking simulation was performed to investigate the potential types of interactions between the protein kinase enzymes and the designed hybrid compounds. The results of this research indicated the promising anticancer effect of compound 7 through the inhibition of a number of protein kinase receptors and the suppression of cell cycle and the induction of apoptosis.
Keywords: Antitumor; Isatin; Hybrids design; Pyrrolo[2,3-d]pyrimidine; Protein kinase inhibitor
1. Introduction
Cancer is considered a huge public health issue worldwide with more individuals being diagnosed every year (Alanazi et al., 2021b, Zhou et al., 2022). Globally, there were 18.1 million new cases of invasive cancers with 9.9 deaths happened in 2020 (Baak et al., 2022, Jayaseelan, 2020). Unfortunately, drug resistance, significant side effects, and poor selectivity limit chemotherapy's efficacy as a primary cancer treatment (He and Shi, 2013). Additionally, Cancer cell survival, invasiveness, and treatment resistance have all been associated with protein kinase overactivation in several cell signaling pathways (Kim et al., 2023). Therefore, inhibiting protein kinases are becoming highly demanded (Alanazi et al., 2023, Krug and Hilgeroth, 2008).
Protein tyrosine kinases (PTKs) are critical components in cell division (Alkahtani et al., 2020). PTKs affect protein function by phosphorylating their amino acid residues which lead to a conformational change in the proteins’ 3D structure (El-Husseiny et al., 2020). PTKs’ dysfunctions have a crucial role in cancer development and progression. Therefore, targeting PTKs by small molecule inhibitors is critical in most types of cancers (El-Azab et al., 2020). Among those PTKs, the tyrosine kinase receptors such as epidermal growth factor receptor (EGFR), human epidermal receptor (Her2) and vascular endothelial growth factor receptor 2 (VEGFR2) which are key proteins in cell overexpression and metastasis. Therefore, EGFR, Her2 and VEGFR2 are vital receptors for the development of new anticancer drugs (Alanazi et al., 2021a).
Indole‐2,3‐dione (isatin) derivatives have proved a broad therapeutic effects including anti-inflammatory, antibacterial, antimalarial, antitubercular, and antitumor activities (Eldehna et al., 2015, Han et al., 2014). Moreover, Isatin containing compounds demonstrated the capability to modify multiple cellular targets including tyrosine kinases, modulating the level of certain apoptosis‐specific proteins which ultimately lead to apoptosis (Al-Sanea et al., 2021, Alkahtani et al., 2019). Sunitinib is a USFDA approved drug with an isatin based pharmacophore for the treatment of renal cell carcinoma and gastrointestinal stromal cancer (Alkahtani et al., 2019). Another isatin containing compound, semaxanib, is under clinical trial for the treatment of different types malignancies (Fig. 1) (Ding et al., 2020). On the other hand, pyrrolo[2,3-d]pyrimidine heterocycles drew much attention during the last few year as they have been revealed to exert a wide range of pharmacological activities such as anti-chemotherapeutic and anti-diabetic effects. Moreover, pyrrolo[2,3-d]pyrimidine scaffolds have been well studied as protein kinase inhibitors and resulted in several USFDA approved anticancer drugs such as Ribociclib, Ruxolitinib, Tofacitinib and Baricitinib (Fig. 1) (Sroor et al., 2019).
Fig. 1.

Chemical structures of protein kinase inhibitors containing isatin or pyrrolo[2,3-d]pyrimidine.
Each cell of human body comprises various genetics because of the variations in the inactive tumor suppressor genes and the active oncogenes. A single anticancer drug may effectively kill cancer cells holding a single alteration, nevertheless, other cells motivated by additional signaling will persist (Bhat et al., 2015). The complex cellular signaling pathways negatively affect the successfulness of the traditional single treatment strategy. Alternatively, modulating multiple targets is essential to beat tumor cells (Lineham et al., 2017). Thus, the purpose of this research is to design and synthesize novel protein kinase inhibitors with multiple fighting mechanisms against uncontrolled growth of cancer cells. Hybrid compounds combining different moieties from two different anticancer agents; the isatin (indole‐2,3‐dione) containing compounds and pyrrolo[2,3-d]pyrimidine bearing compounds were designed (Fig. 2) as both pharmacophores are parts of some USFDA approved anticancer drugs and; therefore, they may have the potential to suppress antitumor activity with a multi-kinase inhibition effect.
Fig. 2.

Rational design of isatin-pyrrolo[2,3-d]pyrimidine hybrid compounds.
2. Materials and methods
2.1. Chemistry and analysis
2.1.1. General procedure for synthesis of compound (2)
Hydrazine hydrate was reacted and refluxed with 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (1.5 gm, 10 mmol) for 2 h. The reaction was then left to cool and mixed with cold water. Afterwards, The reaction mixture was filtered, rinsed with water and left to dry to obtain the 4-hydrazineyl-7H-pyrrolo[2,3-d]pyrimidine 2 (Alkahtani et al., 2019).
4-hydrazineyl-7H-pyrrolo[2,3-d]pyrimidine (2)

Yield 84.94%; m.p. 259–260 °C; 1H NMR (700 MHz, DMSO‑d6): δ 11.51 (s, 1H), 8.86 – 8.33 (m, 1H), 8.13 (s, 1H), 7.06 (d, J = 3.2 Hz, 1H), 6.79 – 6.38 (m, 1H), 4.45 (s, 2H). 13C NMR (176 MHz, DMSO‑d6) δ 158.37 (C), 151.06 (CH), 150.44 (C), 121.11 (CH), 101.44 (C), 99.91 (CH). LC-MS m/z 148.00 (M-) 150.10 (M+).
2.1.2. General procedure for synthesis of compounds (3–8)
In 20 ml absolute ethanol, A mixture of 4-hydrazinyl-7H-pyrrolo[2,3-d]pyrimidine (0.3 gm, 2 mmol), a substituted isatin (0.7 mmol) and glacial acetic acid (1 ml) was refluxed for about 3–4 h. The reaction mixture was cooled and added to ice water. The formed precipitate was then filtered off, washed with water and recrystallized from a proper solvent to obtain the desired Schiff bases (Alkahtani et al., 2019, Mohameda et al., 2010, Musumeci et al., 2017).
3-(2-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)hydrazineylidene)indolin-2-one (3)

Yield 98.59 %; m.p. > 300 °C; 1H NMR (700 MHz, DMSO‑d6): δ 13.02 (s, 1H), 12.19 (m, 1H), 11.22 (s, 1H), 10.51 (d, 1H), 8.42 (m, 1H), 8.13 (m, 1H), 7.69 – 7.13 (m, 4H). 13C NMR (176 MHz, DMSO‑d6) δ 166.61 (C), 164.43 (C), 153.40 (CH), 151.14 (C), 142.65 (C), 133.07 (C), 130.49 (CH), 125.79 (CH), 122.86 (CH), 121.79 (CH), 121.00 (CH), 120.36 (C), 111.41 (C) 103.13 (CH). LC-MS m/z 276.9 (M-).
3-(2-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)hydrazineylidene)-5-chloroindolin-2-one (4)

Yield 86.03%; m.p. > 300 °C; 1H NMR (700 MHz, DMSO‑d6): δ 12.29 (m, 2H), 10.59 (s, 1H), 8.48 (s, 1H), 8.15 (s, 1H), 7.45 – 7.09 (m, 3H), 7.11 – 6.71 (m, 1H). 13C NMR (176 MHz, DMSO‑d6) δ 166.44 (C), 163.65 (C), 153.34 (CH), 151.16 (C), 140.32 (C), 131.92 (C), 130.05 (CH), 125.99 (C), 122.48 (CH), 119.90 (CH), 112.91 (CH), 111.31 (C), 103.21 (C), 102.29 (CH). LC-MS m/z 310.8 (M-).
3-(2-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)hydrazineylidene)-5-fluoroindolin-2-one (5)

Yield 87.66%; m.p. > 300 °C; 1H NMR (700 MHz, DMSO‑d6): δ 12.27(m, 2H), 11.23 (s, 1H), 10.60 (s, 1H), 8.25 (m, 2H), 7.76 – 6.50 (m, 3H). 13C NMR (176 MHz, DMSO‑d6) δ 166.74 (C), 163.71 (C), 158.67 (C), 153.00 (CH), 150.93 (C), 132.54 (C), 125.92 (CH), 123.82 (C), 122.04 (CH), 117.02 (CH), 112.36 (CH), 110.54 (C), 103.14 (C), 102.21 (CH). LC-MS m/z 294.9 (M-) 297.1 (M+).
3-(2-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)hydrazineylidene)-5-methylindolin-2-one (6)
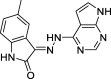
Yield 75.26%; m.p. > 300 °C; 1H NMR (700 MHz, DMSO‑d6): δ 13.03 (m, 1H), 12.17 (m, 2H), 10.40 (m, 1H), 8.27 (m, 2H), 7.68 – 6.48 (m, 3H), 2.77 – 2.10 (m, 3H). 13C NMR (176 MHz, DMSO‑d6) δ 166.67 (C), 163.61 (C), 153.33(C), 151.02(CH), 147.56 (C), 131.23 (C), 125.63 (CH), 123.29 (C), 120.83 (CH), 119.14 (CH), 111.07 (CH), 109.82 (C), 105.93 (C), 103.64 (CH), 21.20 (CH3). LC-MS m/z 293.1 (M+).
3-(2-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)hydrazineylidene)-5-methoxyindolin-2-one (7)
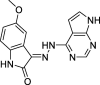
Yield 94.84%; m.p. > 300 °C; 1H NMR (700 MHz, DMSO‑d6): δ 13.05 (s, 1H), 12.15 (s, 1H), 10.96 (m, 1H), 10.31 (s, 1H), 8.32 (m, 2H), 7.76 – 6.43 (m, 3H), 4.24–3.48 (m, 3H). 13C NMR (176 MHz, DMSO‑d6) δ 166.65 (C), 163.68 (C), 155.82(C), 153.27 (CH), 150.81 (C), 136.41 (C), 125.81 (CH), 121.57 (C), 116.77 (CH), 112.18 (CH), 110.44 (CH), 105.77 (C), 103.00 (C), 102.13 (CH), 56.17 (CH3). LC-MS m/z 306.9 (M-) 309.1 (M+).
3-(2-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)hydrazineylidene)-5-nitroindolin-2-one (8)
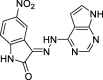
Yield 97.70 %; m.p. > 300 °C; 1H NMR (700 MHz, DMSO‑d6): δ 13.60 (s, 1H), 12.93 (s, 1H), 11.86 (s, 1H), 11.24 (s, 1H), 9.28 (m, 1H), 8.85 – 7.75 (m, 3H), 7.11 (m, 1H). 13C NMR (176 MHz, DMSO‑d6) δ 166.96 (C), 163.91 (C), 153.74 (C), 152.79 (CH), 150.92 (C), 147.48 (C), 143.22 (CH), 131.42 (C), 126.49 (CH), 124.61 (CH), 111.95 (CH), 109.96 (C), 103.44 (C), 102.11 (CH). LC-MS m/z 321.9 (M-) 324.1 (M+).
2.2. Biological evaluation
2.2.1. MTT assay
The antiproliferative activity of all synthesized hybrids 3–8 were evaluated against HepG2, MCF-7, MDA-MB-231 and HeLa human cancer cell lines by 3-(4,5- dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay according to a previously reported method (Alanazi et al., 2023; M. M. Alanazi et al., 2023).
2.2.2. In vitro protein kinase assays
The protein kinase inhibitory activity of compound 7 against CDK2, EGFR, Her2, and VEGFR-2 enzymes was evaluated according to a previously reported method (Alanazi et al., 2023, Alanazi et al., 2023, Riccardi and Nicoletti, 2006, Wang and Lenardo, 2000).
2.2.3. Analysis of cell cycle distribution
Effect of compounds 7 on liver HepG2 cell cycle distribution was evaluated according to a previously reported method (Alanazi et al., 2023, Alanazi et al., 2023).
2.2.4. Apoptosis assay by flow cytometry
The effect of compound 7 on the early and late apoptosis on HepG2 cells was evaluated according to a previously reported method (Alanazi et al., 2023, Alanazi et al., 2023).
2.2.5. Anti-apoptotic and pro-apoptotic protein assays
The protein levels of BAX, Bcl-2, Caspase 3 and Caspase 9 were measured according to a previously reported method (Alhakamy et al., 2022, Zhu et al., 2015).
2.2.6. Molecular docking study
Molecular docking study was conducted according to the method described previously using the X-ray crystal structures of erlotinib, sorafenib, and lapatinib in a complex with EGFR (PDB ID: 4HJO), VEGFR2 (PDB ID: 4ASD), and Her2 (PDB ID: 3RCD), respectively (Alanazi et al., 2023, Alanazi et al., 2023).
3. Results
3.1. Chemistry
The isatin-pyrrolo[2,3-d]pyrimidine derivatives 3–8 have been synthesized as shown in scheme 1 using chemistry previously reported by Alanazi et al (A. S. Alanazi et al., 2023).
Scheme 1.

Synthesis of isatin-pyrrolo[2,3-d]pyrimidine hybrid compounds 3–8. Reagents and conditions: i. excess of hydrazine hydrate, reflux, 2 h. ii. appropriate isatin, AcOH, EtOH, reflux, 3–7 h.
First, hydrazine hydrate was reacted under reflux with 4-chloro-7H-pyrrolo[2,3-d]pyrimidine for 2 h. The reaction was then left to cool and mixed with cold water. Afterwards, The reaction mixture was filtered, rinsed with water and left to dry to achieve the 4-hydrazineyl-7H-pyrrolo[2,3-d]pyrimidine 2. The later intermediate was obtained with a yield of 84.94 % and elucidated by 1H NMR, 13C NMR and LCMS in order to be involved in the following reaction. Then, a mixture of intermediate 2, different substituted Indole‐2,3‐dione and glacial acetic acid was refluxed in absolute ethanol for 3–7 h. The resultant mixture was left to cool at room temperature and poured into chilly water. The resultant turbid solution was passed through a proper filter, rinsed with cold water and recrystallized from the appropriate organic solvent to achieve the target product. All target hybrids were achieved with yields ranging from 75.26 to 98.59% and elucidated by 1H NMR, 13C NMR and LCMS (supplementary data).
3.2. Biological evaluation
3.2.1. In vitro antiproliferative assay
The cytotoxic effect of all derivatives 3–8 was evaluated on four different human cancer cells namely, Hepatocellular Carcinoma (HepG2), Mammary Gland (MCF-7), Breast Cancer (MDA-MB-231) and Epithelioid Cervix Carcinoma (HeLa) using MTT assay and doxorubicin as a reference compound. The results were expressed as IC50 values (µM) (Table 1).
Table 1.
Isatin- pyrrolo[2,3-d]pyrimidine hybrid compounds 3–8 MTT assay results on HepG2, MCF-7, MDA-MB-231 and HeLa cells.
 | |||||
|---|---|---|---|---|---|
| Compound | R | In vitro Cytotoxicity IC50 (µM) |
|||
| HepG2 | MCF-7 | MDA-MB-231 | HeLa | ||
| 3 | H | 19.06 ± 1.4 | 14.20 ± 1.2 | 7.86 ± 0.6 | 9.86 ± 0.7 |
| 4 | Cl | 28.45 ± 2.1 | 33.92 ± 2.2 | 21.07 ± 1.8 | 24.22 ± 1.8 |
| 5 | F | 37.28 ± 2.5 | 41.61 ± 2.6 | 32.53 ± 2.1 | 29.10 ± 2.0 |
| 6 | CH3 | 11.70 ± 0.9 | 27.56 ± 2.0 | 15.19 ± 1.1 | 19.75 ± 1.3 |
| 7 | OCH3 | 8.39 ± 0.7 | 9.08 ± 0.7 | 5.28 ± 0.3 | 6.37 ± 0.3 |
| 8 | NO2 | 52.11 ± 2.9 | 48.47 ± 2.8 | 36.94 ± 2.4 | 37.98 ± 2.2 |
| Doxorubicin | – | 4.50 ± 0.2 | 4.17 ± 0.2 | 3.18 ± 0.1 | 5.57 ± 0.4 |
Results are expressed as the mean ± SD of three independent experiments.
3.2.2. In vitro kinase inhibitory activity
Kinase inhibitory effects of compound 7 were evaluated against EGFR, Her2, VEGFR2 and CDK2 protein kinases. The results were described as IC50 (μM) in comparison to reference drugs (Table 2).
Table 2.
Protein kinase inhibition assays of compound 7 against multiple kinases.
| Kinase Protein | Compound | IC50 (μM) |
|---|---|---|
| CDK2 | 7 | 0.302 ± 0.016 |
| Ribociclib | 0.063 ± 0.003 | |
| EGFR | 7 | 0.166 ± 0.010 |
| Erlotinib | 0.041 ± 0.003 | |
| Her2 | 7 | 0.157 ± 0.022 |
| Lapatinib | 0.060 ± 0.018 | |
| VEGFR2 | 7 | 0.224 ± 0.012 |
| Sorafenib | 0.045 ± 0.002 |
Results are expressed as the mean ± SD of three independent experiments.
3.2.3. Molecular docking
Molecular simulation studies were performed by docking compound 7 into the active pockets of EGFR (Fig. 3), VEGFR2 (Fig. 4), and Her2 (Fig. 5).
Fig. 3.

Two- and three-dimensional representations of the docking simulation of erlotinib (A) and compound 7 (B) within the active binding site of EGFR (PDB ID: 4HJO).
Fig. 4.

Two- and three-dimensional representations of the docking simulation of sorafenib (A) and compound 7 (B) within the active binding site of VEGFR2 (PDB ID: 4ASD).
Fig. 5.

Two- and three-dimensional representations of the docking simulation of lapatinib (A) and compound 7 (B) within the active binding site of Her2 (PDB ID: 3RCD).
3.2.4. Analysis of cell cycle distribution
The flow cytometry assay was conducted to measure the cellular DNA content and the analysis of the cell cycle following treatment of HepG2 cells with compound 7 (Table 3, Fig. 6). The followed protocol for DNA measurement has been described by Wang et al. (Riccardi and Nicoletti, 2006, Wang and Lenardo, 2000).
Table 3.
Cell cycle progression after exposure to compound 7.
| Compound/ cell line | DNA content (%) |
|||
|---|---|---|---|---|
| %Sub–G1 | %G1 | %S | %G2/M | |
| Compound 7/HepG2 | 32.34 | 54.22 | 34.76 | 11.02 |
| Cont. HepG2 | 1.86 | 43.97 | 41.12 | 14.91 |
Results are presented as means of three different experiments.
Fig. 6.

Cell cycle phases of HepG2 cells treated with compound 7.
3.2.5. Rate of apoptosis
Annexin-V/propidium iodide (PI) double staining assay revealed the effect of compound 7 on the apoptosis rate after incubation with HepG2 cells for 24 h (Table 4).
Table 4.
Apoptotic effect compound 7 on HepG2 cells.
| Sample | Apoptosis |
|||
|---|---|---|---|---|
| Total | Early | Late | Necrosis | |
| Compound 7/HepG2 | 32.34 | 9.91 | 15.92 | 6.51 |
| Cont. HepG2 | 1.86 | 0.62 | 0.13 | 1.11 |
Results are presented as means of three independent experiments.
3.2.6. Anti-apoptotic and pro-apoptotic protein assays
The effect of compound 7 on the expression of BAX, Bcl-2, Caspase 3 and Caspase 9 was studied by incubating 8.39 μM of compound 7 with HepG2 cells for 24 h (Table 5). The results demonstrated that the treated cells had elevated levels of pro-apoptotic proteins and a lower level of Bcl-2 (anti-apoptotic protein).
Table 5.
Effect of compound 7 on gene expression of Bax, Bcl-2, Caspase 3 and Caspase 9.
| Compound/cell line | Apoptotic protein | Folds of gene expression (Normalized to β-actin) |
|---|---|---|
| 7/HepG2 | Bax | 0.293 |
| Control HepG2 | 1 | |
| 7/HepG2 | Bcl-2 | 4.913 |
| Control HepG2 | 1 | |
| 7/HepG2 | Caspase 3 | 5.421 |
| Control HepG2 | 1 | |
| 7/HepG2 | Caspase 9 | 3.295 |
| Control HepG2 | 1 |
All percentages are presented as means of three different experiments.
3.2.7. Physiochemical properties and ADMET profiles
As shown in Table 6, the in-silico analysis of the physicochemical properties and the ADMET profiles of the target compounds were predicted using pkCSM website.
Table 6.
In silico ADMET profiles of the target compounds.
| Parameters Compound | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|
| Molecular properties* | ||||||
| Molecular weight | 278.27 | 312.71 | 296.26 | 292.30 | 308.30 | 323.27 |
| LogP | 1.7262 | 2.3796 | 1.8653 | 2.0346 | 1.7348 | 1.6344 |
| H-acceptor | 5 | 5 | 5 | 5 | 6 | 7 |
| H-donors | 3 | 3 | 3 | 3 | 3 | 3 |
| Surface area | 118.669 | 128.973 | 122.835 | 125.034 | 130.148 | 133.322 |
| Absorption* | ||||||
| Water solubility | −2.897 | −3.002 | −2.956 | −2.95 | −3.25 | −2.998 |
| Intestinal absorption (human) | 83.399 | 84.65 | 83.754 | 83.942 | 85.147 | 80.341 |
| Skin permeability | −2.769 | −2.753 | −2.753 | −2.755 | −2.74 | −2.74 |
| Distribution* | ||||||
| BBB permeability | −0.874 | −1.054 | −1.092 | −0.9 | −1.06 | −1.108 |
| CNS permeability | −3.083 | −3.097 | −3.128 | −3.075 | −3.183 | −2.861 |
| Metabolism* | ||||||
| CYP1A2 inhibitor CYP2C19 inhibitor CYP2C9 inhibitor CYP2D6 inhibitor CYP3A4 inhibitor |
YES NO NO NO NO |
YES NO NO NO NO |
YES NO NO NO NO |
YES NO NO NO NO |
YES NO NO NO NO |
YES NO NO NO NO |
| Excretion * | ||||||
| Total clearance | 0.256 | 0.066 | 0.272 | 0.251 | 0.405 | 0.362 |
| Renal OCT2 substrate | NO | NO | NO | NO | NO | NO |
| Toxicity * | ||||||
| Max. tolerated dose (human) | 0.24 | 0.32 | 0.339 | 0.277 | 0.369 | 0.393 |
| Oral rate acute toxicity (LD50) | 2.48 | 2.489 | 2.442 | 2.52 | 2.498 | 3.344 |
| Oral rate chronic toxicity (LOAEL) | 1.203 | 1.118 | 1.199 | 1.071 | 1.132 | 1.847 |
| Hepatotoxicity | YES | YES | YES | YES | YES | YES |
| Skin sensitization | NO | NO | NO | NO | NO | NO |
4. Discussion
In the cytotoxicity assay, compounds 4, 5 and 8 were markedly less toxic on all tested human cancer cell lines compared with the cytotoxicity observed for other compounds (3, 6 and 7). Compound 7 substituted with methoxy showed the most potent antiproliferative activity against the examined cell lines; HepG2, MCF-7, MDA-MB-231 and HeLa with IC50 values 8.39 ± 0.7, 9.08 ± 0.7, 5.28 ± 0.3 and 6.37 ± 0.3 µM, respectively. In addition, both compounds 3 and 6 showed moderate to strong inhibitory activities against all tested cell lines with IC50 values ranging from 7.86 ± 0.6 to 27.56 ± 2.0 µM. Indeed, compounds 3 and 7 showed very promising results compared with the reference compound doxorubicin, a well-known anticancer agent.
Structure–activity relationship of the investigated hybrids revealed that; compounds 4, 5 and 8 substituted with electron withdrawing groups were markedly less cytotoxic towards HepG2, MCF-7, MDA-MB-231 and HeLa cell lines compared with the cytotoxic effects observed for compounds 3, 6 and 7. However, compound 4 with a Cl group at R position showed better cytotoxicity compared to compounds 5 and 8 with F and NO2 groups, respectively. This finding may be explained by that compound 4 is more lipophilic (cLogP = 1.44) than other compounds (cLogP = 0.874 for compound 5 and cLogP = 0.474 for compound 8).
In fact, compounds 3, 6 and 7 (R = H, R = CH3 and OCH3) showed good cytotoxic activity against the tested cell lines compared to the reference drug with compound 7 being the most potent compound against all the tested cancer cells. It seems that introducing a methoxy group at R position in compound 7 enables a hydrogen bonding interaction with its target sites in the kinase enzymes which in turn increases the cytotoxic activity. Therefore, compound 7 was selected for additional biological evaluation such as kinase inhibitory assays and molecular docking to investigate the possible mechanism of action.
As compound 7 demonstrated a promising antiproliferative active, kinase inhibition effects against four protein kinase were assessed. The results revealed that compound 7 exhibited potent kinase inhibitory effects against EGFR, Her2, VEGFR2 and CDK2. In fact, compound 7 exhibited comparable inhibitory results to the reference drugs with IC50 values in the nanomolar range. These results supported the promising activity of isatin-pyrrolo[2,3-d]pyrimidine hybrid compounds as protein kinase inhibitors and cytotoxic agents.
To explain the antiproliferative activities and estimate the potential binding interactions between the target hybrids and the investigated protein kinases, molecular simulation studies were performed by docking compound 7 into the active pockets of EGFR, VEGFR2, and Her2. Erlotinib, sorafenib, and lapatinib, the co-crystalized ligands, were used as reference standards for EGFR, VEGFR2, and Her2, respectively. First, docking erlotinib into the active site of EGFR resulted in one hydrogen bond with Met769 and several non-hydrogen bonds with the amino acid residues of the active site of EGFR. Contrarywise, compound 7 formed four hydrogen bonds with Asp831, Thr716, and Met769 (two). Moreover, compound 7 demonstrated a similar pose and alignment to erlotinib in the active site of EGFR and made almost similar types of hydrophobic interactions (Fig. 3 and Figure S1 in the Supplementary Materials). The calculated binding affinities of erlotinib and compound 7 with the EGFR were found − 9.5 and − 7.9 Kcal/mol, respectively. Second, sorafenib made four hydrogen bonds with Cys919 (two hydrogen bonds), Asp1046, and Glu885 and multiple non-hydrogen bonding with the amino acids of the ATP-binding site of VEGFR2. Similarly, compound 7 made four hydrogen bonds with Cys919, Asp1046, Val914, and Lys868. Even though, compound 7 was superimposed on sorafenib in the active site of VEGFR2, compound 7 was stabilized by less hydrophobic interactions and scored a lower binding energy than sorafenib (-8.9 Kcal/mol vs −12.1 Kcal/mol, respectively) (Fig. 4 and Figure S2 in the Supplementary Materials). Finally, the potential binding interactions between compound 7 and Her2 receptor was investigated by docking the co-crystalized ligand lapatinib and compound 7 into the active binding site of Her2 kinase enzyme. lapatinib performed hydrogen bonds with Met801, Thr798, and Thr862, two halogen bonds with Ser783 and Arg784 and five hydrophobic interactions with Arg784, Leu785, Leu796, Val734, and Lys753. Likewise, compound 7 interacted with the amino acid residues of Ala751, Leu796, Asp863, and Thr862 by hydrogen bonds, with Thr798 by van der waals interactions and with Ala751, Leu796, Lys753, Val734, and Leu785 by hydrophobic interactions. Compound 7 was also superimposed on the pose of lapatinib in the active site of Her2 and scored docking energy of −9.1 Kcal/mol versus −8.8 kcal/mol for lapatinib (Fig. 5 and Figure S3 in the Supplementary Materials).
Furthermore, to investigate the effect of compound 7 on the cell cycle progression, the DNA content was evaluated by exposing HepG2 cells to 8.39 μM of compound 7 for 24 h and compared with the untreated HepG2 cells. According to the results, compound 7 revealed a significant increase of the DNA in the sub-G1 (32.34%, 17-fold) compared to untreated cells (1.86%). On the other hand, cell death promoted by compound 7 was assessed after 24 h incubation with HepG2 cells by applying annexin-V/propidium iodide (PI) double staining assay. The results showed an increase in the apoptotic cells in the early (9.91%) and late (15.92%) stages compared to the control cells (Table 4).
Additionally, as compound 7 induced both early and late apoptosis, the effect of compound 7 on the expression of BAX, Bcl-2 Caspase 3 and Caspase 9 was studied. The protein expression of BAX, Caspase 3 and Caspase 9 increased by 4.91, 5.42 and 3.3 folds, respectively. Furthermore, the level of Bcl-2 protein expression reduced by 0.3-fold compared to the untreated cells.
Finally, in order to evaluate the physicochemical properties and the ADMET profiles of the target compounds, all compounds were subjected to in silico analysis using pkCSM and applied to Lipinski’s rule of five. All compounds obey Lipinski’s rule of five and therefore, they may have good oral bioavailability. As demonstrated in Table 6, all compounds have good intestinal absorption which may indicate the capability of these compounds to penetrate various biological membranes. Furthermore, the target compounds are expected to have no or mild CNC adverse effects since the BBB permeability is very low for the target compounds. All compounds were noticed to inhibit CYP1A2 while they had no effects on CYP2C19, CYP2C9, CYP2D6 and CYP3A4. On the other hand, all compounds seem to have high total clearance which may increase the daily dosing intervals. Moreover, the toxicity profile revealed that all compounds have good maximum tolerated doses which indicate that these compounds may have a wide therapeutic index. Finally, all tested compounds may cause hepatotoxicity and may not cause skin sensitization. In summary, these results indicate that compound 7 exhibited promising results as anticancer agent.
5. Conclusions
In conclusion, a series of isatin-pyrrolo[2,3-d]pyrimidine hybrid compounds was designed, synthesized and evaluated for the antiproliferative and kinase inhibition effects. A number of those hybrids exerted potent antitumor effects against four tested cancer cell lines. Moreover, compound 7 as compared to the reference drugs demonstrated good multi-kinase inhibitory activities in nanomolar range against EGFR, Her2, VEGFR2 and CDK2 protein kinases. Further evaluation of compound 7 revealed the ability to suppress cycle progression and induce programmed cell death. Finally, molecular docking was studied to predict the possible interactions made between compound 7 and the investigated protein kinase enzymes. This work revealed interesting new anticancer hybrids with multitargeting mechanisms which need further investigation toward deep mechanistic studies.
Funding
This research was funded by the Deanship of Scientific Research at Princess Nourah bint Abdulrahman University, through the Research Funding Program, Grant No. (FRP-1443–28).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
This research was funded by the Deanship of Scientific Research at Princess Nourah bint Abdulrahman University, through the Research Funding Program, Grant No. (FRP-1443-28).
Footnotes
Peer review under responsibility of King Saud University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jsps.2023.05.003.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- Al-Sanea, M.M., Obaidullah, A.J., Shaker, M.E., Chilingaryan, G., Alanazi, M.M., Alsaif, N.A., Alkahtani, H.M., Alsubaie, S.A., Abdelgawad, M.A., 2021. A New CDK2 Inhibitor with 3-Hydrazonoindolin-2-One Scaffold Endowed with Anti-Breast Cancer Activity: Design, Synthesis, Biological Evaluation, and In Silico Insights. Mol. 2021, Vol. 26, Page 412 26, 412. https://doi.org/10.3390/MOLECULES26020412. [DOI] [PMC free article] [PubMed]
- Alanazi, A.S., Mirgany, T.O., Alsfouk, A.A., Alsaif, N.A., Alanazi, M.M., 2023. Antiproliferative Activity, Multikinase Inhibition, Apoptosis- Inducing Effects and Molecular Docking of Novel Isatin–Purine Hybrids. Med. 2023, Vol. 59, Page 610 59, 610. https://doi.org/10.3390/MEDICINA59030610. [DOI] [PMC free article] [PubMed]
- Alanazi M.M., Alaa E., Alsaif N.A., Obaidullah A.J., Alkahtani H.M., Al-Mehizia A.A., Alsubaie S.M., Taghour M.S., Eissa I.H. Discovery of new 3-methylquinoxalines as potential anti-cancer agents and apoptosis inducers targeting VEGFR-2: design, synthesis, and in silico studies. J. Enzyme Inhib. Med. Chem. 2021;36:1732–1750. doi: 10.1080/14756366.2021.1945591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alanazi, M.M., Aldawas, S., Alsaif, N.A., 2023. Design, Synthesis, and Biological Evaluation of 2-Mercaptobenzoxazole Derivatives as Potential Multi-Kinase Inhibitors. Pharm. 2023, Vol. 16, Page 97 16, 97. https://doi.org/10.3390/PH16010097. [DOI] [PMC free article] [PubMed]
- Alanazi M.M., Mahdy H.A., Alsaif N.A., Obaidullah A.J., Alkahtani H.M., Al-Mehizia A.A., Alsubaie S.M., Dahab M.A., Eissa I.H. New bis([1,2,4]triazolo)[4,3-a:3′,4′-c]quinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, in silico studies, and anticancer evaluation. Bioorg. Chem. 2021;112 doi: 10.1016/J.BIOORG.2021.104949. [DOI] [PubMed] [Google Scholar]
- Alhakamy, N.A., Badr-Eldin, S.M., Ahmed, O.A.A., Aldawsari, H.M., Okbazghi, S.Z., Alfaleh, M.A., Abdulaal, W.H., Neamatallah, T., Al-Hejaili, O.D., Fahmy, U.A., 2022. Green Nanoemulsion Stabilized by In Situ Self-Assembled Natural Oil/Native Cyclodextrin Complexes: An Eco-Friendly Approach for Enhancing Anticancer Activity of Costunolide against Lung Cancer Cells. Pharm. 2022, Vol. 14, Page 227 14, 227. https://doi.org/10.3390/PHARMACEUTICS14020227. [DOI] [PMC free article] [PubMed]
- Alkahtani H.M., Abdalla A.N., Obaidullah A.J., Alanazi M.M., Almehizia A.A., Alanazi M.G., Ahmed A.Y., Alwassil O.I., Darwish H.W., Abdel-Aziz A.A.M., El-Azab A.S. Synthesis, cytotoxic evaluation, and molecular docking studies of novel quinazoline derivatives with benzenesulfonamide and anilide tails: Dual inhibitors of EGFR/HER2. Bioorg. Chem. 2020;95 doi: 10.1016/J.BIOORG.2019.103461. [DOI] [PubMed] [Google Scholar]
- Alkahtani H.M., Alanazi M.M., Aleanizy F.S., Alqahtani F.Y., Alhoshani A., Alanazi F.E., Almehizia A.A., Abdalla A.N., Alanazi M.G., El-Azab A.S., Abdel-Aziz A.-A.-M. Synthesis, anticancer, apoptosis-inducing activities and EGFR and VEGFR2 assay mechanistic studies of 5,5-diphenylimidazolidine-2,4-dione derivatives: Molecular docking studies. Saudi Pharm. J. 2019;27:682–693. doi: 10.1016/j.jsps.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baak J.P.A., Li H., Guo H. Clinical and biological interpretation of survival curves of cancer patients, exemplified with stage IV non-small cell lung cancers with long follow-up. Front. Oncol. 2022;12:36. doi: 10.3389/FONC.2022.837419/TEXT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat, M., Robichaud, N., Hulea, L., Sonenberg, N., Pelletier, J., Topisirovic, I., 2015. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015 144 14, 261–278. https://doi.org/10.1038/nrd4505. [DOI] [PubMed]
- Ding Z., Zhou M., Zeng C. Recent advances in isatin hybrids as potential anticancer agents. Arch. Pharm. (Weinheim) 2020;353:1900367. doi: 10.1002/ARDP.201900367. [DOI] [PubMed] [Google Scholar]
- El-Azab A.S., Abdel-Aziz A.A.M., AlSaif N.A., Alkahtani H.M., Alanazi M.M., Obaidullah A.J., Eskandrani R.O., Alharbi A. Antitumor activity, multitarget mechanisms, and molecular docking studies of quinazoline derivatives based on a benzenesulfonamide scaffold: cell cycle analysis. Bioorg. Chem. 2020;104 doi: 10.1016/J.BIOORG.2020.104345. [DOI] [PubMed] [Google Scholar]
- El-Husseiny W.M., El-Sayed M.A.A., El-Azab A.S., AlSaif N.A., Alanazi M.M., Abdel-Aziz A.A.M. Synthesis, antitumor activity, and molecular docking study of 2-cyclopentyloxyanisole derivatives: mechanistic study of enzyme inhibition. J. Enzyme Inhib. Med. Chem. 2020;35:744–758. doi: 10.1080/14756366.2020.1740695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldehna W.M., Altoukhy A., Mahrous H., Abdel-Aziz H.A. Design, synthesis and QSAR study of certain isatin-pyridine hybrids as potential anti-proliferative agents. Eur. J. Med. Chem. 2015;90:684–694. doi: 10.1016/J.EJMECH.2014.12.010. [DOI] [PubMed] [Google Scholar]
- Han K., Zhou Y., Liu F., Guo Q., Wang P., Yang Y., Song B., Liu W., Yao Q., Teng Y., Yu P. Design, synthesis and in vitro cytotoxicity evaluation of 5-(2-carboxyethenyl)isatin derivatives as anticancer agents. Bioorg. Med. Chem. Lett. 2014;24:591–594. doi: 10.1016/J.BMCL.2013.12.001. [DOI] [PubMed] [Google Scholar]
- He Q., Shi J. MSN anti-cancer nanomedicines: chemotherapy enhancement, overcoming of drug resistance, and metastasis inhibition. Adv. Mater. 2013;26:391–411. doi: 10.1002/ADMA.201303123. [DOI] [PubMed] [Google Scholar]
- Jayaseelan V.P. Emerging role of exosomes as promising diagnostic tool for cancer. Cancer Gene Ther. 2020;27:395–398. doi: 10.1038/s41417-019-0136-4. [DOI] [PubMed] [Google Scholar]
- Kim, H.J.;, Park, J.W.;, Seo, S ;, Cho, K.-H.;, Alanazi, M.M.;, Bang, E.-K.;, Keum, G ;, El-Damasy, A K, Perrotti, N., Yost, B.B., Kim, H.J., Woo Park, J., Seo, Sangjae, Cho, K.-H., Alanazi, M.M., Bang, E.-K., Keum, Gyochang, El-Damasy, Ashraf K, 2023. Discovery of New Quinolone-Based Diarylamides as Potent B-RAFV600E/C-RAF Kinase Inhibitors Endowed with Promising In Vitro Anticancer Activity. Int. J. Mol. Sci. 2023, Vol. 24, Page 3216 24, 3216. https://doi.org/10.3390/IJMS24043216. [DOI] [PMC free article] [PubMed]
- Krug M., Hilgeroth A. Recent advances in the development of multi-kinase inhibitors. Mini-Rev. Med. Chem. 2008;8:1312–1327. doi: 10.2174/138955708786369591. [DOI] [PubMed] [Google Scholar]
- Lineham E., Spencer J., Morley S.J. Dual abrogation of MNK and mTOR: a novel therapeutic approach for the treatment of aggressive cancers. Future Med. Chem. 2017;9:1539–1555. doi: 10.4155/FMC-2017-0062/ASSET/IMAGES/LARGE/FIGURE5.JPEG. [DOI] [PubMed] [Google Scholar]
- Mohameda M.S., Kamel M.M., Kassem E.M., Abotaleb N., AbdEl-Moez S.I., Ahmed M.F. Novel 6,8-dibromo-4(3H)quinazolinone derivatives of anti-bacterial and anti-fungal activities. Eur. J. Med. Chem. 2010;45:3311–3319. doi: 10.1016/J.EJMECH.2010.04.014. [DOI] [PubMed] [Google Scholar]
- Musumeci F., Fallacara A.L., Brullo C., Grossi G., Botta L., Calandro P., Chiariello M., Kissova M., Crespan E., Maga G., Schenone S. Identification of new pyrrolo[2,3-d]pyrimidines as Src tyrosine kinase inhibitors in vitro active against Glioblastoma. Eur. J. Med. Chem. 2017;127:369–378. doi: 10.1016/j.ejmech.2016.12.036. [DOI] [PubMed] [Google Scholar]
- Riccardi, C., Nicoletti, I., 2006. Analysis of apoptosis by propidium iodide staining and flow cytometry. https://doi.org/10.1038/nprot.2006.238 [DOI] [PubMed]
- Sroor F.M., Basyouni W.M., Tohamy W.M., Abdelhafez T.H., El-awady M.K. Novel pyrrolo[2,3-d]pyrimidine derivatives: design, synthesis, structure elucidation and in vitro anti-BVDV activity. Tetrahedron. 2019;75 doi: 10.1016/J.TET.2019.130749. [DOI] [Google Scholar]
- Wang J., Lenardo M.J. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J. Cell Sci. 2000;113:753–757. doi: 10.1242/JCS.113.5.753. [DOI] [PubMed] [Google Scholar]
- Zhou B., Zang R., Zhang M., Song P., Liu L., Bie F., Peng Y., Bai G., Gao S. Worldwide burden and epidemiological trends of tracheal, bronchus, and lung cancer: a population-based study. eBioMedicine. 2022;78 doi: 10.1016/J.EBIOM.2022.103951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z., Song L., He J., Sun Y., Liu X., Zou X. Int. J. Clin. Exp; Pathol: 2015. Ectopic expressed long non-coding RNA H19 contributes to malignant cell behavior of ovarian cancer. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.