

Abstract
Purpose:
To describe a case of primary coenzyme Q10 deficiency in a child manifesting as early-onset renal failure, retinal dystrophy, and optic atrophy leading to progressive vision loss.
Methods:
Clinical presentation and workup including visual fields, electroretinogram, and optical coherence tomography are presented. Genetic testing was performed.
Results:
An eight-year-old female with nephropathy requiring renal transplantation subsequently developed progressive cone-rod dystrophy and optic atrophy. The patient had negative results on a targeted next-generation sequencing retinal dystrophy panel but whole-exome sequencing revealed two variants in COQ2 (likely biallelic), consistent with a diagnosis of primary coenzyme Q10 deficiency.
Conclusions:
Primary coenzyme Q10 deficiency is a rare disorder with variable systemic and ocular findings; there is also genetic heterogeneity. Genetic testing aids in the diagnosis of this condition, and variants in the COQ2 and PDSS1 genes appear to have the strongest association with ocular manifestations. Oral supplementation of coenzyme Q10 may slow progression of disease. This case highlights the utility of whole-exome sequencing in the diagnosis of a rare syndromic form of ocular disease and reports a novel phenotypic association for this condition.
Keywords: Primary coenzyme Q10 deficiency, Cone-rod retinal dystrophy, Optic atrophy, Whole-exome genetic sequencing
Introduction
Primary coenzyme Q10 (CoQ10) deficiency is a genetic disorder of the mitochondrial respiratory chain inherited in an autosomal recessive fashion.1 The systemic manifestations are widespread and involve neurologic, renal, and cardiac findings, reflecting the ubiquity of oxidative phosphorylation. The condition is also genetically heterogeneous with pathogenic variants in nine genes identified to date.1 There are no formal diagnostic criteria and, as such, diagnosis generally relies on a high index of suspicion and subsequent genetic testing. Patients with primary CoQ10 deficiency may also present with retinal dystrophy and, occasionally, optic atrophy, although reports of these are limited in number.2–7 We report a case of a child with progressive vision loss due to cone-rod retinal dystrophy and optic atrophy of both eyes confirmed to have primary CoQ10 deficiency on whole-exome sequencing.
Case Report
An eight-year-old female with best-corrected visual acuity (BCVA) of 20/40 in both eyes (OU) one year prior to presentation developed decreased vision in both eyes over six months. The patient was born healthy after an uncomplicated full-term pregnancy but developed focal segmental glomerulonephritis at age two thought to be due to minimal change disease and C1q nephropathy. She subsequently developed renal failure, requiring kidney transplantation at age three. Past medical history was additionally significant for attention deficit hyperactivity disorder, dyslexia, and short stature. Family history was noncontributory.
BCVA was 20/250 in the right eye (OD) and 20/150 in the left eye (OS) with normal intraocular pressures and no relative afferent pupillary defect. The patient identified 3 of 14 Ishihara color plates OD and 2 of 14 OS. Confrontation visual fields were full, but Goldmann visual fields showed an incomplete midperipheral scotoma OS; there was asymmetric constriction to the I4e target with enlargement of the physiological blind spot in each eye (Figure 1). Slit lamp examination was unremarkable, but dilated fundus examination revealed moderate optic nerve pallor and vascular attenuation OU with a subtle bull’s eye pattern of retinal pigmentary changes (Figure 2A–B).
Figure 1.
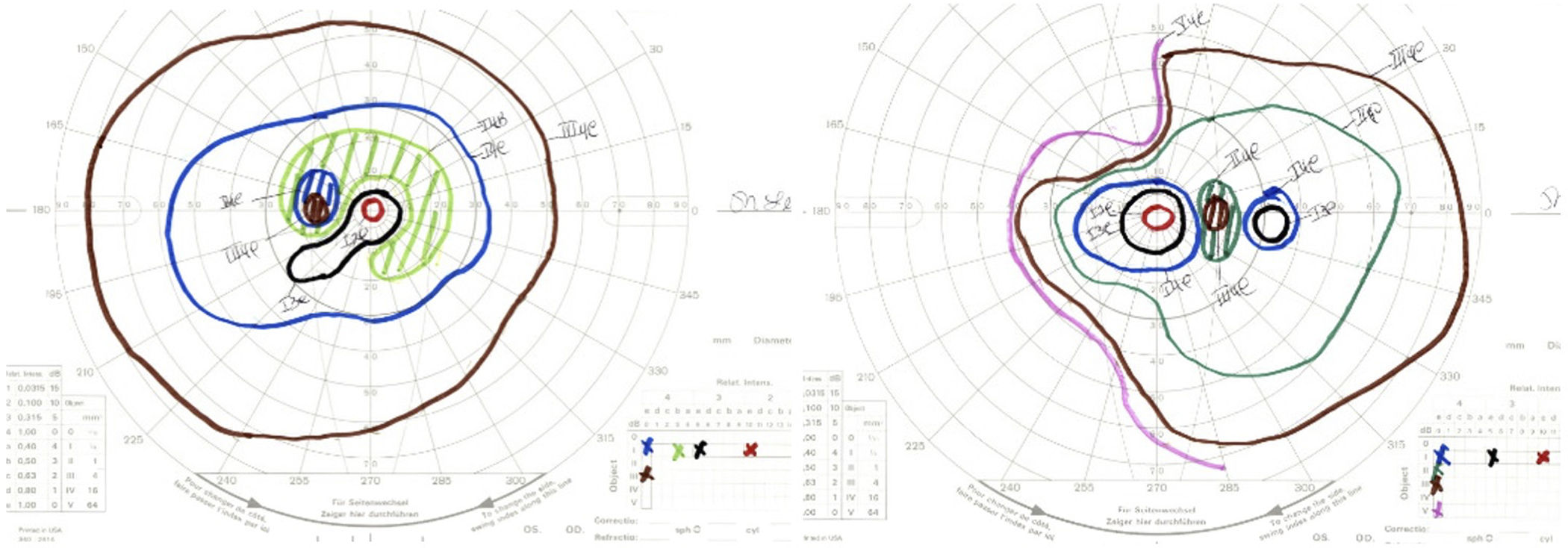
Goldmann visual fields showed moderate constriction to the I4e isopter in the right eye and a paracentral scotoma to the I4b isopter in the left eye with enlargement of the physiological blind spot in each eye.
Figure 2A-B.
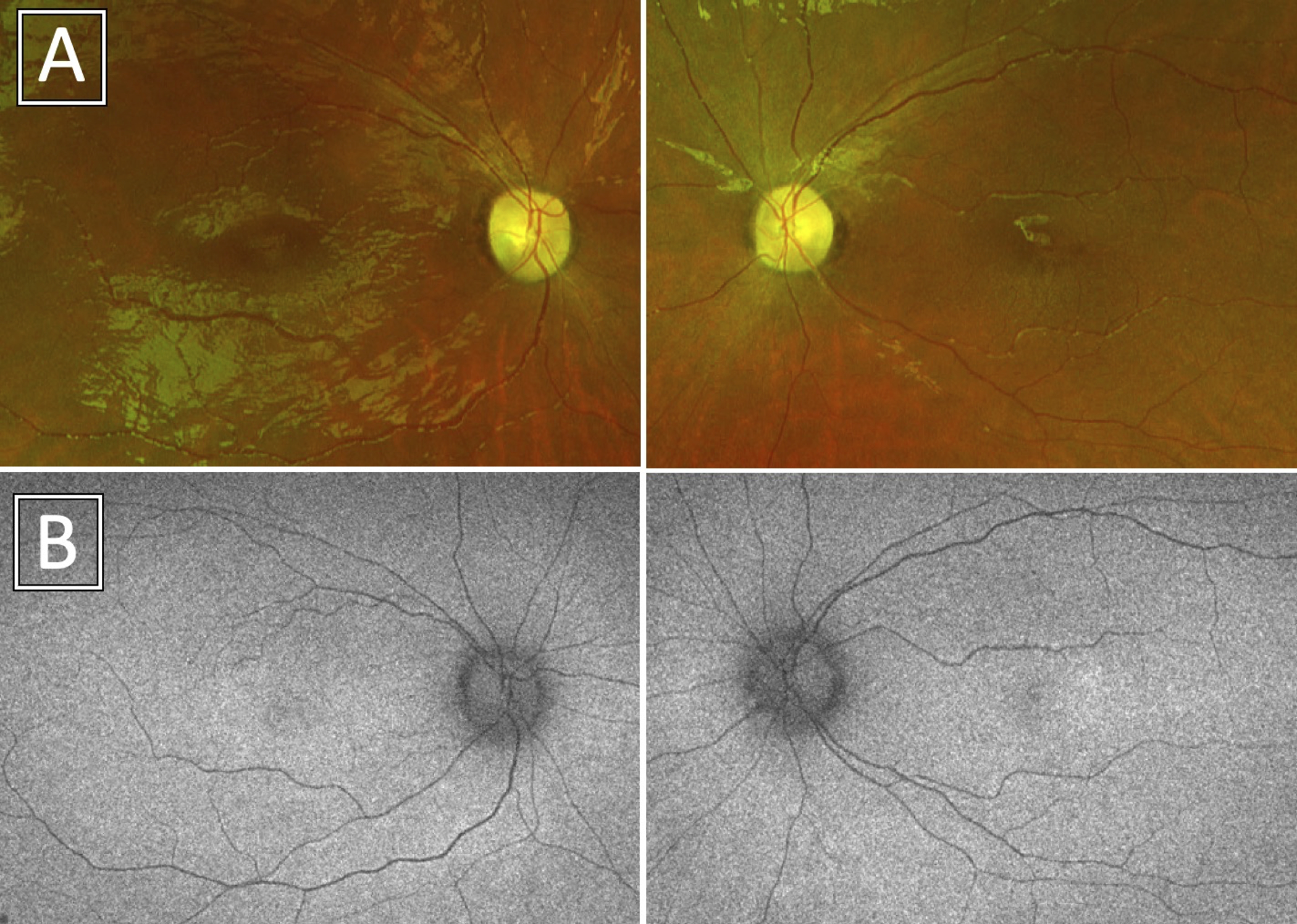
Ultrawidefield color (A) and fundus autofluorescence (B) photos demonstrated optic nerve pallor and vascular attenuation with a bull’s eye maculopathy pattern in each eye.
Macular optical coherence tomography (OCT) revealed parafoveal intraretinal cysts overlying ellipsoid zone loss in a bull’s eye pattern OU (Figure 3A), while optic nerve OCT scans showed diffuse retinal nerve fiber layer thinning, worse OD than OS (Figure 3B). An awake electroretinogram was performed (according to International Society for Clinical Electrophysiology of Vision standard using Burian-Allen bipolar contact lens electrodes), which showed moderate cone greater than rod dysfunction bilaterally (Figure 4A–B). Magnetic resonance imaging of the brain and orbits, performed at an outside facility, was normal by report. Next-generation sequencing was performed with a targeted 285-gene retinal dystrophy panel (Blueprint Genetics, Espoo, Finland, through the My Retina Tracker Institutional Review Board-approved protocol) and was non-diagnostic.
Figure 3.
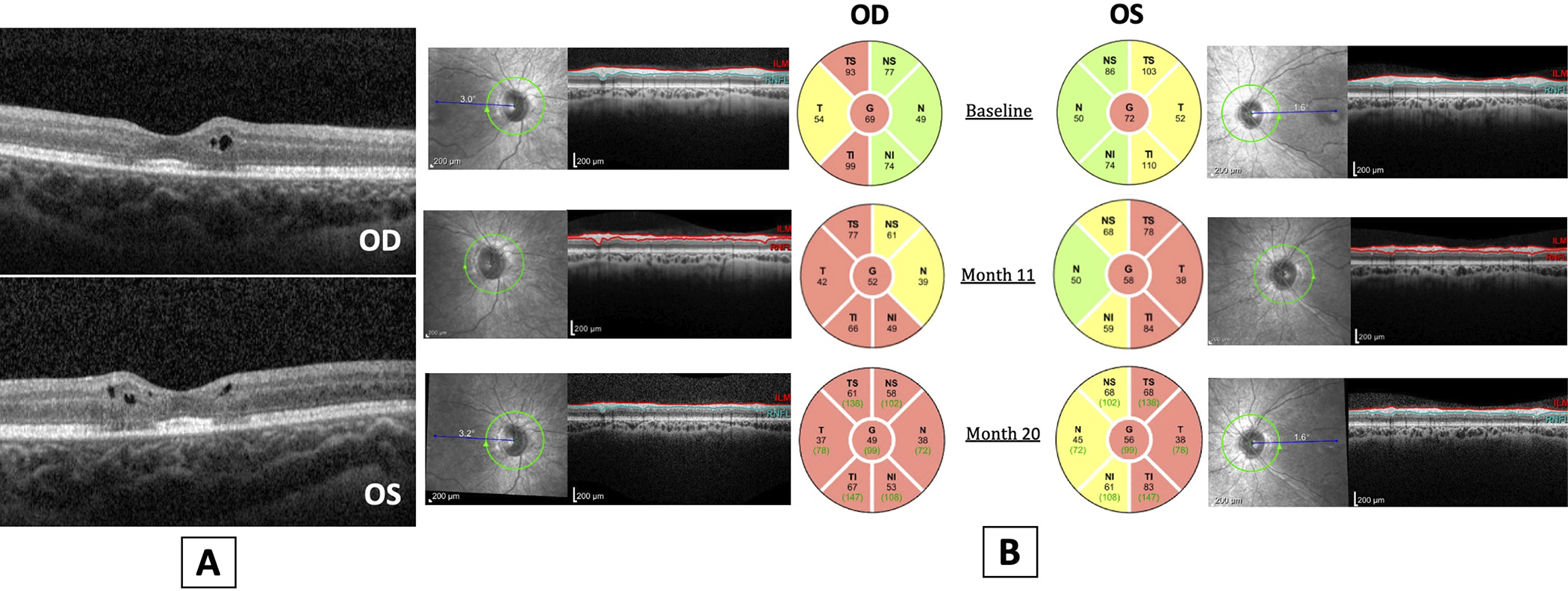
A. Optical coherence tomography of the macula showed parafoveal intraretinal cysts overlying reduced reflectivity of the ellipsoid zone in a bull’s eye pattern. B. OCT of the retinal nerve fiber layer over 20 months demonstrated progressive nerve fiber layer thinning, worse in the right eye than the left eye.
Figure 4A-D.
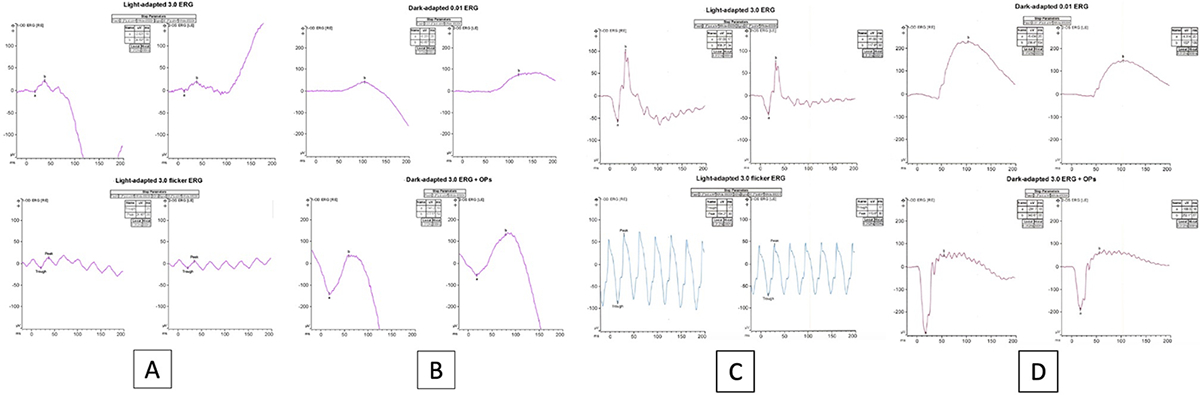
Full-field electroretinogram traces showed photopic (4A) greater than scotopic (4B) dysfunction, although blink artifact contaminated the scotopic responses making it impossible to assess scotopic function reliably in each eye. Photopic, cone-mediated responses were reduced by about 60% below the lower limits of normal in amplitude and delayed in timing in both eyes. A normal full-field electroretinogram is displayed in panels 4C and 4D for comparison.
Over the course of 20 months, the patient’s BCVA progressively declined to 20/400 OD. BCVA OS remained stable. She additionally exhibited progressive optic nerve atrophy on exam and OCT. Whole-exome sequencing was performed as a clinical test approximately 1.5 years after initial presentation and uncovered two variants in COQ2 (NCBI Ref Seq ID: NM_015697.7, OMIM: 609825). The first variant, c.683A>G, was a maternally-inherited, rare (gnomAD allele frequency: 0.01%)8 missense substitution (p.Asn228Ser) that has multiple lines of evidence supporting pathogenicity.9–12 As a result, it was classified as likely pathogenic by the reference laboratory (see ClinVar Entry: VCV000001439.5 for additional details).13 The second variant detected, c.518 G>A, was a similarly rare (gnomAD allele frequency: 0.01%)8 missense substitution (p.Arg173His) with some evidence supporting pathogenicity.14, 15 However, it was classified as a variant of unknown significance by the testing laboratory given that it has never been definitively detected in trans with a pathogenic variant. The c.518 G>A variant was not detected in the patient’s mother, but the father was unavailable for testing. Given that the variant segregates in the general population,8 it was most likely inherited from the patient’s father rather than occurring de novo. This indicates that the patient is likely compound heterozygous for two rare, missense variants in COQ2.
Given these genetic findings coupled with strong phenotypic overlap, the patient was diagnosed with primary CoQ10 deficiency. Additional diagnostic testing was considered, specifically muscle biopsy and measurement of CoQ10 concentration plus mitochondrial respiratory chain activity levels.16, 17 However, the patient’s biochemical screening labs (serum lactate, acylcarnitine profile, plasma amino acids, and urine organic acids) were normal, and she lacked systemic findings typical for a mitochondrial myopathy. As a result, the sensitivity of this invasive testing was uncertain, and a negative/inconclusive result was unlikely to change clinical management. Therefore, muscle biopsy was deferred for now. Of note, serum CoQ10 levels are not reflective of intracellular concentrations and thus have very limited utility in diagnosing primary CoQ10 deficiency (our patient’s serum CoQ10 was normal).16 Given the presumed diagnosis, oral supplementation with CoQ10 was initiated, with stability of ocular findings over the following six months. The patient was evaluated by neurology and cardiac specialists and found to have no additional systemic pathology.
Discussion
We report a case of primary CoQ10 deficiency characterized by nephropathy, progressive cone-rod dystrophy, and optic atrophy associated with biallelic variants in the COQ2 gene (c.683A>G, c.518G>A).
CoQ10 plays a key role in the oxidative phosphorylation pathway of the mitochondrial respiratory chain, primarily acting to transfer reducing equivalents but also serving as a transmembrane hydrogen carrier and antioxidant.1 The CoQ2 gene encodes the enzyme involved in the second step of CoQ10 biosynthesis.3, 18 To date, pathogenic variants in the genes COQ2–9 and PDSS1–2, all of which encode proteins involved in the direct biosynthesis of CoQ10, have been identified as a cause for primary CoQ10 deficiency.1
Primary CoQ10 deficiency is associated with a wide range of clinical phenotypes. The systemic hallmark is steroid-resistant nephrotic syndrome presenting with proteinuria in early childhood; left untreated, this condition usually progresses to end-stage renal disease requiring kidney transplantation.7 Neurologic findings are common but vary widely and range from severe encephalomyopathy (similar to multiple-systemic atrophy) to mild intellectual disability.6 Other systemic manifestations, reported to occur on a more variable basis, include hypertrophic cardiomyopathy, muscle weakness, and sensorineural hearing loss.1
Few reports of eye findings in primary CoQ10 deficiency describe retinopathy and optic atrophy as the primary ocular manifestations (Supplementary Table 1). Retinopathy is more commonly reported, and often appears similar to retinitis pigmentosa with abnormal rod greater than cone function.4, 5, 19 In a case report of primary CoQ10 deficiency caused by variants in the COQ2 gene, all three affected siblings demonstrated extensive intraretinal pigment migration in the periphery and encircling the posterior poles with rod-cone dysfunction and undetectable scotopic responses.7 All cases of retinopathy in primary CoQ10 deficiency have been reported in the setting of mutations in the COQ2 gene, although none of these reported cases were associated with the pathogenic variants identified in the current case.5–7 In general, reports of COQ2-related CoQ10 deficiency are rare; of these, only a handful of patients have been reported to have associated retinopathy since COQ2 was identified as the first gene associated with primary CoQ10 deficiency in 2006.5–7, 18, 20
Optic atrophy may occur in conjunction with the retinopathy associated with biallelic variants in COQ2,6, 7 as described in our present case, or it may occur in the absence of retinal pathology, such as in cases of primary CoQ10 deficiency due to variants in PDSS1.2, 3 However, because precise molecular diagnosis is a relatively recent development, older case reports do not describe the exact genetic cause. Note, onset of vision loss is variable and reported to range from infancy, presenting as nystagmus, to young adulthood.5, 6
While the c.683A>G variant has strong evidence supporting pathogenicity, its implication in cases with ophthalmic manifestations is more rare.9–12 Diomedi-Camassei et al. report a case of a child with the c.683A>G variant but do not report the presence of retinal disease or optic atrophy.9 The second variant implicated in the current case, c.518G>A, was recently reported in a case of isolated adult-onset retinitis pigmentosa with Leber hereditary optic neuropathy; notably, the patient lacked any systemic findings consistent with primary CoQ10 deficiency.15 A patient with childhood onset nephropathy was reported to carry both the c.683A>G and c.518G>A variants identified in the current case; despite the same genotype, however, the patient was not reported to have any ocular pathology (although the patient was only two years old at the time of the report).14 As such, this is the first report to implicate these two variants associated with retinal and optic nerve disease in the setting of primary CoQ10 deficiency with systemic findings.
The diagnosis of primary CoQ10 deficiency was historically established by enzymatic assessment of tissue: a reduction in CoQ10 concentration and mitochondrial respiratory chain activity levels have been documented.3, 6 That said, the sensitivity and specificity of such testing is uncertain [PMID: 29781757], particularly in patients that lack systemic findings consistent with severe mitochondrial dysfunction.17 Therefore, the advent of genetic testing has greatly improved ease and accessibility of diagnosis. Whole-exome sequencing, in particular, facilitates the diagnosis of this rare condition that is not routinely assessed on targeted gene sequencing panels. Early diagnosis is important, as supplementation with oral CoQ10 has been reported to improve encephalomyopathy and rarely nephropathy, with the suggestion that earlier treatment may be of greater benefit.4, 6, 21 In the previously mentioned case report by Abdelhakim and colleagues, treatment with CoQ10 over six months did not improve ERG findings in all three siblings; however, BCVA and areas of retinal atrophy remained stable, suggesting treatment may aid in slowing progression of ocular disease.7
The differential diagnosis for child presenting with steroid-resistant nephropathy, retinopathy, and optic atrophy includes AVIL-related nephrotic syndrome although optic atrophy has not been reported in this condition.22 Additionally, Pierson syndrome may be considered although in addition to retinal changes, the ocular phenotype encompasses significant anterior segmental abnormalities of the cornea, lens, and ciliary body.23 Given the limited understanding of the phenotypic diversity of these conditions, whole exome or genome sequencing ultimately provides timely and definitive diagnosis and improved characterization of disease.
Conclusions
We present a case of primary CoQ10 deficiency likely caused by biallelic variants in the COQ2 gene. The patient’s phenotype is characterized by steroid-resistant nephropathy, cone-rod retinal dystrophy, and optic atrophy with normal neurologic function. Primary CoQ10 deficiency should be considered in cases of retinal degeneration or optic nerve atrophy associated with classic systemic findings. The increased affordability and availability of whole-exome sequencing will facilitate this diagnosis and lead to prompt treatment of this rare condition.
Supplementary Material
Acknowledgements and Disclosures
Funding:
This research was supported, in part, by the UCSF Vision Core shared resource of the NIH/NEI P30 EY002162, and by an unrestricted grant from Research to Prevent Blindness, New York, NY. The sources support general research efforts in the Ophthalmology department at UCSF and had no role in influencing the study design, data analysis or result interpretation.
Footnotes
Conflicts of interest:
JLD: (C): AGTC, Astellas, California Institute for Regenerative Medicine, DTx Pharma, Editas Medicine, Eloxx, Eyevensys, Foundation Fighting Blindness, Gyroscope, Helios, ProQR Therapeutics, PYC Therapeutics, Relay Therapeutics, SparingVision, Spark Therapeutics, Vedere Bio. (S): Allergan, California Institute for Regenerative Medicine, Biogen/NightstaRx, Foundation Fighting Blindness, Neurotech USA, Inc.
The following authors have no financial disclosures: JYS, DRB, AS, ATM, AGD
Authorship: All authors attest that they meet the current ICMJE criteria for Authorship.
Patient Consent
Written consent to publish this case has not been obtained. This report does not contain any personal identifying information.
References
- 1.Salviati L, Trevisson E, Doimo M, Navas P. Primary coenzyme Q10 deficiency. In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews. Seattle, WA: University of Washington, 2017. [PubMed] [Google Scholar]
- 2.Nardecchia F, De Giorgi A, Palombo F, et al. Missense PDSS1 mutations in coenzyme Q10 synthesis cause optic atrophy and sensorineural deafness. Ann Clin Transl Neurol 2021;8(1):247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mollet J, Giurgea I, Schlemmer D, et al. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J Clin Invest 2007;117(3):765–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rötig A, Appelkvist EL, Geromel V, et al. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet 2000;356(9227):391–5. [DOI] [PubMed] [Google Scholar]
- 5.Hara K, Momose Y, Tokiguchi S, et al. Multiplex families with multiple system atrophy. Arch Neurol 2007;64(4):545–51. [DOI] [PubMed] [Google Scholar]
- 6.Salviati L, Sacconi S, Murer L, et al. Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: a CoQ10-responsive condition. Neurology 2005;65(4):606–8. [DOI] [PubMed] [Google Scholar]
- 7.Abdelhakim AH, Dharmadhikari AV, Ragi SD, et al. Compound heterozygous inheritance of two novel COQ2 variants results in familial coenzyme Q deficiency. Orphanet J Rare Dis 2020;15(1):320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581(7809):434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol 2007;18(10):2773–80. [DOI] [PubMed] [Google Scholar]
- 10.McCarthy HJ, Bierzynska A, Wherlock M, et al. Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 2013;8(4):637–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bezdíčka M, Štolbová Š, Seeman T, et al. Genetic diagnosis of steroid-resistant nephrotic syndrome in a longitudinal collection of Czech and Slovak patients: a high proportion of causative variants in NUP93. Pediatr Nephrol 2018;33(8):1347–63. [DOI] [PubMed] [Google Scholar]
- 12.Desbats MA, Morbidoni V, Silic-Benussi M, et al. The COQ2 genotype predicts the severity of coenzyme Q10 deficiency. Hum Mol Genet 2016;25(19):4256–65. [DOI] [PubMed] [Google Scholar]
- 13.Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 2018;46(D1):D1062–d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sadowski CE, Lovric S, Ashraf S, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol 2015;26(6):1279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurata K, Hosono K, Takayama M, et al. Retinitis pigmentosa with optic neuropathy and COQ2 mutations: A case report. Am J Ophthalmol Case Rep 2022;25:101298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trevisson E, DiMauro S, Navas P, Salviati L. Coenzyme Q deficiency in muscle. Curr Opin Neurol 2011;24(5):449–56. [DOI] [PubMed] [Google Scholar]
- 17.Yubero D, Montero R, Santos-Ocaña C, et al. Molecular diagnosis of coenzyme Q(10) deficiency: an update. Expert Rev Mol Diagn 2018;18(6):491–8. [DOI] [PubMed] [Google Scholar]
- 18.Quinzii C, Naini A, Salviati L, et al. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet 2006;78(2):345–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boitier E, Degoul F, Desguerre I, et al. A case of mitochondrial encephalomyopathy associated with a muscle coenzyme Q10 deficiency. J Neurol Sci 1998;156(1):41–6. [DOI] [PubMed] [Google Scholar]
- 20.Mutations in COQ2 in familial and sporadic multiple-system atrophy. N Engl J Med 2013;369(3):233–44. [DOI] [PubMed] [Google Scholar]
- 21.Montini G, Malaventura C, Salviati L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N Engl J Med 2008;358(26):2849–50. [DOI] [PubMed] [Google Scholar]
- 22.Rao J, Ashraf S, Tan W, et al. Advillin acts upstream of phospholipase C ϵ1 in steroid-resistant nephrotic syndrome. J Clin Invest 2017;127(12):4257–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pierson M, Cordier J, Hervouuet F, Rauber G. An unusual congenital and familial congenital malformation combination involving the eye and kidney. J Genet Hum 1963;12:184–213. [PubMed] [Google Scholar]
- 24.Mitsui J, Koguchi K, Momose T, et al. Three-year follow-up of high-dose ubiquinol supplementation in a case of familial multiple system atrophy with compound heterozygous COQ2 mutations. Cerebellum 2017;16(3):664–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.