

Abstract
The sirtuin deacetylase SIRT5 plays important roles in regulating multiple metabolic pathways, and potentially represents an attractive target for the treatment of several human diseases, especially cancer. In this study, we report the identification of the hit compound 11 bearing a 2-hydroxybenzoic acid functional group as a novel SIRT5-selective inhibitor via our medium-throughput thermal shift screening assay. Hit 11 stabilizes SIRT5 in a dose-dependent manner and shows moderate inhibitory activity against SIRT5 and high subtype selectivity over SIRT1, 2, and 3 in a trypsin coupled enzyme-based assay. The carboxylic acid and the adjacent hydroxyl group of 11 are essential for maintaining activity. To further improve the potency of compound 11, a lead optimization was carried out, resulting in compound 43 with a 10-fold improved potency. Overall, compound 11 represents a promising new chemical scaffold for further investigation to develop SIRT5-selective inhibitors.
Keywords: SIRT5 inhibitors, Structure-activity relationship, 2-Hydroxybenzoic acid, Molecular docking, Protein-ligand interactions fingerprinting
1. Introduction
Sirtuins are nicotinamide adenine dinucleotide (NAD+) - dependent protein deacylases which are involved in regulating important biological functions, including life span, transcription, DNA repair, protein secretion, and metabolism [1–3]. There are seven sirtuin members encoded in mammalian genomes, SIRT1 to SIRT7, which are distinguished by subcellular localization, biochemical activities, and biological functions [4, 5]. SIRT5, which primarily resides in the mitochondrial matrix, has robust desuccinylation, demalonylation, and deglutarylation activities in vitro and in vivo, while having very weak deacetylase activity [6–8]. SIRT5 has attracted increasing attention in recent years due to its role in regulating ammonia detoxification, fatty acid oxidation, cellular respiration, ketone body formation, tricarboxylic acid cycle, glycolysis, and reactive oxygen species metabolism [9–16]. Dysregulation of SIRT5 is found in multiple cancers [17–22], such as non-small cell lung cancer, colorectal cancer, breast cancer, hepatocellular carcinoma and melanoma. The development of SIRT5 modulators is likely to be an attractive strategy for the treatment of cancer and metabolic diseases.
To date, only a handful of SIRT5 inhibitors (Fig. 1) have been reported. Among them, some are peptide-based, which generally suffer from limited stability and poor membrane permeability. Several small molecules inhibitors are reported, and many with weak activity and poor selectivity [23–34]. Therefore, the development of novel, potent, and selective SIRT5 inhibitors is highly desirable.
Fig. 1.
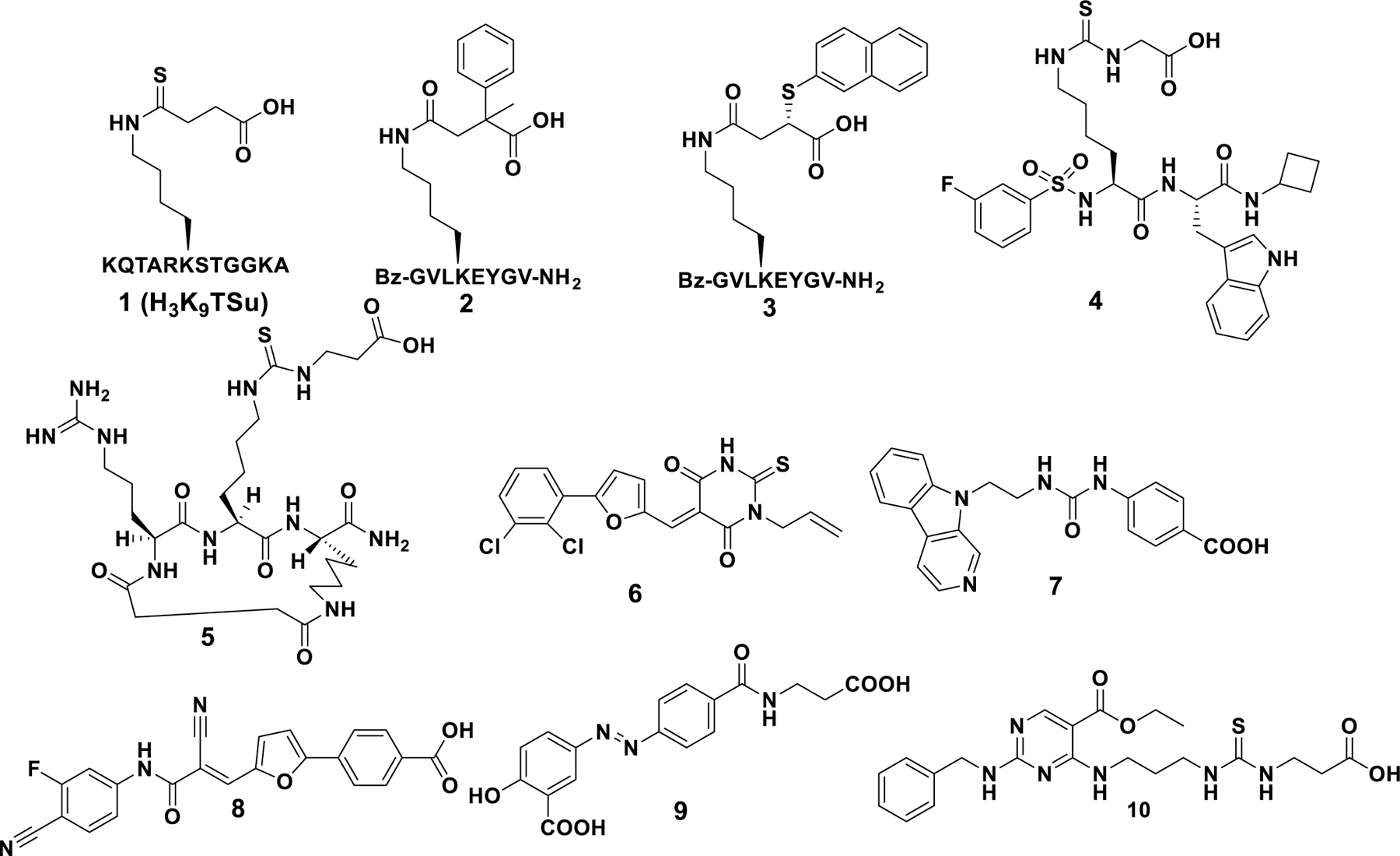
Reported SIRT5 inhibitors.
Herein, we report the identification of hit compound 11 (Fig. 2) containing 2-hydroxybenzoic acid functional group as a selective SIRT5 inhibitor using a medium-throughput thermal shift screening. Hit 11 dose-dependently stabilized SIRT5 and showed moderate inhibitory activity against SIRT5 and substantial subtype selectivity over SIRT1, 2, and 3 in a trypsin coupled fluorescence assay. Starting with 11, a lead optimization guided by molecular docking was conducted that resulted in the identification of compound 43. Compound 43 has drug-like properties, inhibits SIRT5 in low micromolar range, and is 10 times more potent than the hit 11.
Fig. 2.
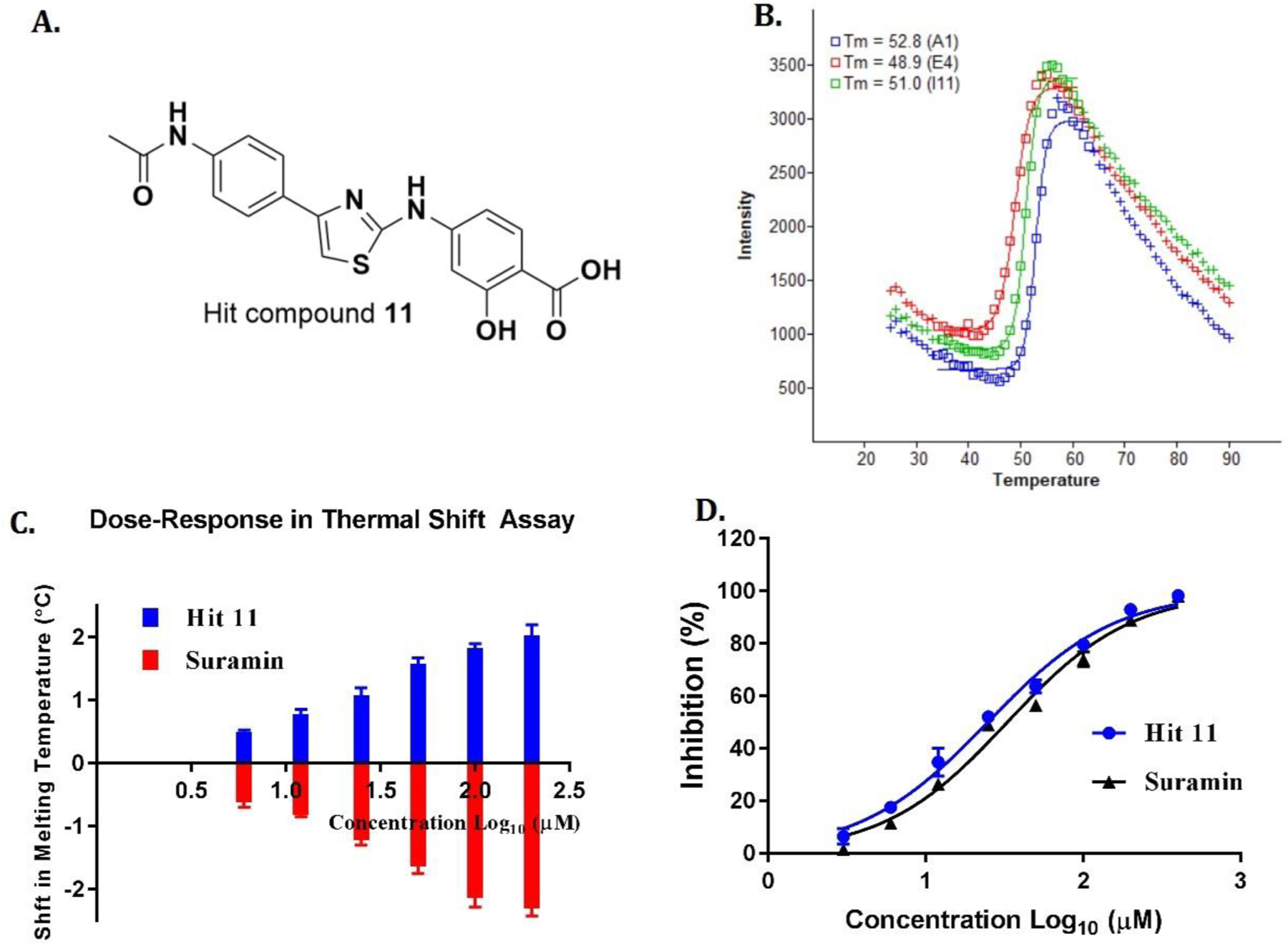
(A) Structure of hit compound 11. (B) Thermal melting curve of 11 at 100 μM versus reference (DMSO) control and a positive control (suramin). A1 (blue curve): sample well contains 11, wild type SIRT5, dye, and buffer; I11 (green curve): reference (DMSO) control well contains DMSO, wild type SIRT5, dye, and buffer; E4 (red curve): positive control well contains suramin, wild type SIRT5, dye, and buffer. The melting curve is presented as fluorescence intensity versus temperature. (C) Concentration-dependent stabilization of SIRT5 by 11. The ΔTm was measured as above using SIRT5 and compound 11 (6–200 μM). The curve represents an exponential fit to the ΔTm values plotted against the compound concentration. (D) Concentration-dependent inhibition of SIRT5 by hit 11. All measurements were done in quadruplicates. Reported SIRT5 inhibitor suramin was used as a positive control.
2. Results and discussion
2.1. Identification of hit compound 11 as a SIRT5 inhibitor
We developed and optimized a medium throughput thermal shift assay specific for SIRT5 and screened a drug-like library of 5000 inhouse compounds. In this screening campaign, hit compound 11 (Fig. 2A) was identified as a novel SIRT5 inhibitor. Compound 11 stabilized SIRT5 protein with a temperature shift of 1.8 °C (Fig. 2B) in a concentration-dependent manner (Fig. 2C). To further validate above findings, we tested compound 11 in a trypsin coupled fluorescence assay as reported previously [35,36]. As shown in Fig. 2D, compound 11 displayed moderate inhibitory activity against SIRT5 with an IC50 of 26.4 ± 0.8 μM. However, 11 showed high selectivity for SIRT5 over SIRT1, 2, and 3 (see Table S1). Even at the highest concentration of 400 μM, no inhibitions against SIRT1–3 were observed.
2.2. Structure-activity relationships of analogues of compound 11
To optimize 11 for further studies, we performed molecular docking studies using crystal structure of SIRT5 (PDB ID: 2NYR) [37]. As shown in Fig. 3A, the carboxylate of 11 forms bidentate salt bridge with Arg105 as well as H-bond to Tyr102, in the deep end of the substrate binding pocket. The hydroxyl group forms a H-bond with Val221 and acetyl carbonyl group forms another H-bond with Tyr255. The compound also forms pi-pi interactions with Phe223 and Tyr255.
Fig. 3.
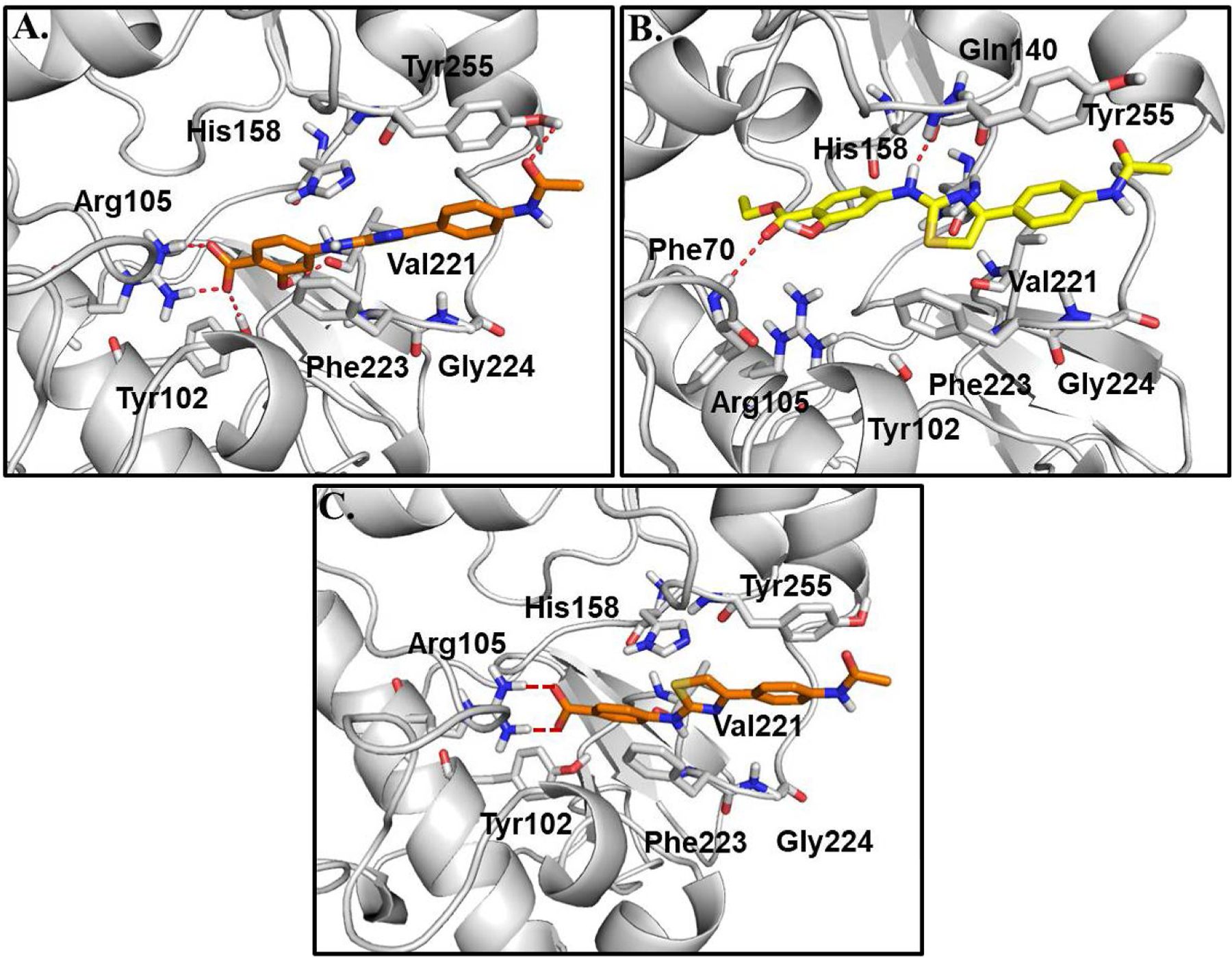
(A) The predicted binding modes of compound 11 with SIRT5. (B & C) The predicted binding modes of inactive compounds 12 and 13, respectively with SIRT5.
To assess the effect of the carboxylic acid on the enzymatic inhibitory activities of 11, compound 12 was tested for its inhibitory activity against SIRT5, in which the carboxylic acid was masked. As shown in Table 1, compound 12 lost nearly all inhibitory activity against SIRT5, confirming the significance of the carboxylic acid group. This result can easily be understood by the docking model (Fig. 3B). Compound 12 does not form a salt bridge and H-bond interactions with key residues, such as Arg105 and Tyr102. To explore the impact of the hydroxyl on efficacy, we synthesized compound 13 in which the hydroxyl group was replaced by a hydrogen atom. Compound 13 is about 8-fold less potent than 11, highlighting the importance of the hydroxyl group. Although compound 13 forms a salt bridge with Arg105 and pi-pi interactions with Phe223 and Tyr255, it is missing any interactions with Val221, Gly224, and Tyr102. These results validated our model and that the predicted binding mode of 11 with SIRT5 is reasonable and can be used for further optimization.
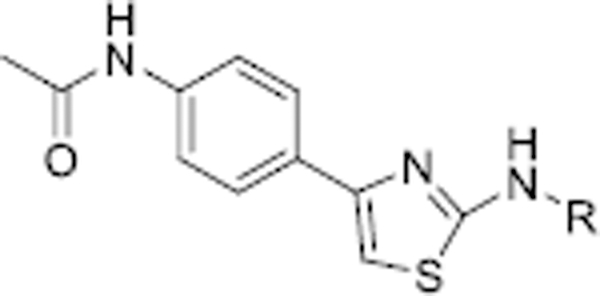
Table 1.
The SIRT5 inhibitory activities of analogues 11–13a.
| |||||
|---|---|---|---|---|---|
| Cpd. | R | SIRT5 IC50 (μM) | bΔTm (◦C) | ccLogP | dLE |
| 11 |
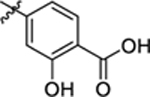
|
26.4 ± 0.8 | 1.8 ± 0.1 | 4.04 | 0.25 |
| 12 |
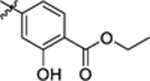
|
>400 | 0.12 ± 0.08 | 4.64 | – |
| 13 |
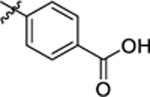
|
200.6 ± 8.9 | 0.53 ± 0.17 | 3.97 | 0.21 |
Suramin was used as a positive control with an IC50 of 28.4 ± 2.5 μM.
The ΔTm values were calculated from the thermal shift assay at a compound concentration of 100 μM.
cLogP values were calculated using ChemDraw Ultra 12.0.
Calculated LE = 1.4 pIC50 (M)/N, where N is the number of non-hydrogen atom.
Having found that the carboxylic acid and the adjacent hydroxyl of 11 are essential for the inhibitory activity against SIRT5, we retained these features and modified the remainder of the molecule. Compounds containing a thiourea group could form stalled intermediates with ADP-ribose in the SIRT5 active sites and effectively inhibit its enzymatic activity. Previously, Olsen’s group disclosed the crystal structures of the zebrafish Sirt5 in complex with stalled bicyclic intermediate of an inhibitor containing a thiourea group (PDB ID 6EO0) [30]. Zheng’s group reported selective thiourea-based SIRT5 inhibitory warheads [38,39]. Inspired by these studies, we embedded a thiourea functionality into 11 to improve water solubility and SIRT5 inhibitory activity. This strategy led to analogue 14, with a lower cLogP of 2.74 by comparison with hit 11 (cLogP = 4.04). Analogue 14 is 2 times more potent than 11 against SIRT5 (Table 2). In our molecular docking study, compound 14 binds in a similar fashion to compound 11 (Fig. 4A), forming salt bridge and H-bond interactions with Arg105 and Tyr102. Compound 14 also forms H-bond interactions with Val221 and Tyr255 as well as pi-pi interactions with Phe223 and Tyr255. Accordingly, we selected analogue 14 for further optimization.
Table 2.
The SIRT5 inhibitory activities of analogues 14–43a.
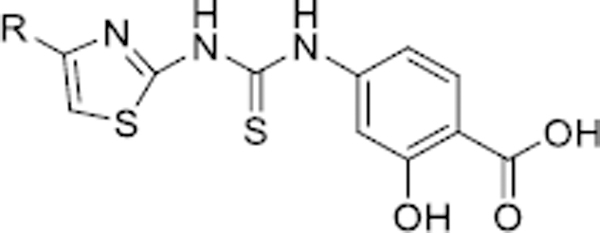
| |||||
|---|---|---|---|---|---|
| Cpd. | R | SIRT5 IC50 (μM) | bΔTm (°C) | ccLogP | dLE |
| 14 |
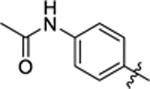
|
12.4 ± 0.6 | 2.5 ± 0.2 | 2.74 | 0.24 |
| 15 |
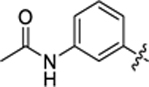
|
35.6 ± 1.3 | 1.5 ± 0.2 | 2.74 | 0.21 |
| 16 |
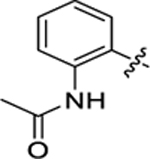
|
55.6 ± 5.5 | 0.9 ± 0.1 | 1.71 | 0.20 |
| 17 |
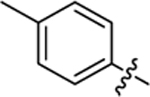
|
23.4 ± 0.9 | 1.4 ± 0.1 | 4.04 | 0.25 |
| 18 |
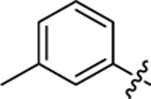
|
27.7 ± 1.0 | 1.2 ± 0.2 | 4.04 | 0.25 |
| 19 |
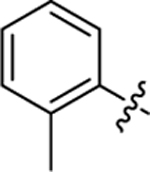
|
34.0 ± 2.2 | 1.1 ± 0.2 | 3.74 | 0.24 |
| 20 |
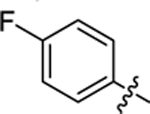
|
17.3 ± 0.3 | 1.3 ± 0.2 | 3.69 | 0.26 |
| 21 |
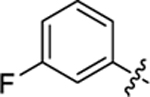
|
18.3 ± 1.0 | 1.22 ± 0.1 | 3.69 | 0.26 |
| 22 |
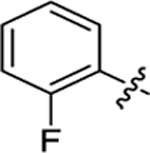
|
26.2 ± 2.5 | 1.0 ± 0.2 | 3.69 | 0.25 |
| 23 |
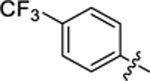
|
24.3 ± 2.5 | 1.2 ± 0.1 | 4.43 | 0.22 |
| 24 |
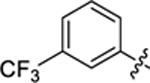
|
26.8 ± 1.4 | 1.1 ± 0.1 | 4.43 | 0.22 |
| 25 |
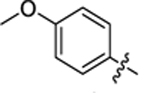
|
29.2 ± 2.9 | 0.9 ± 0.2 | 3.56 | 0.24 |
| 26 |
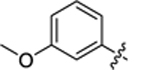
|
41.6 ± 3.8 | 0.9 ± 0.1 | 3.56 | 0.23 |
| 27 |
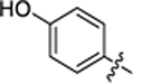
|
42.5 ± 3.8 | 0.8 ± 0.1 | 3.08 | 0.24 |
| 28 |
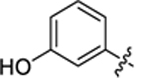
|
32.8 ± 3.9 | 0.72 ± 0.1 | 3.08 | 0.24 |
| 29 |
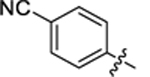
|
11.4 ± 0.5 | 2.1 ± 0.3 | 2.98 | 0.26 |
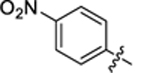
| 30 |
|
4.3 ± 0.3 | 1.8 ± 0.2 | 3.29 | 0.27 |
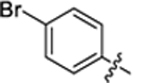
| 31 |
|
18.6 ± 0.2 | 1.2 ± 0.1 | 4.40 | 0.25 |
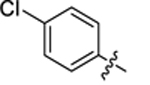
| 32 |
|
18.4 ± 0.3 | 1.1 ± 0.1 | 4.26 | 0.25 |
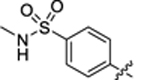
| 33 |
|
38.8 ± 3.1 | 0.9 ± 0.1 | 2.53 | 0.21 |
| 34 |
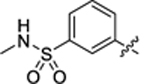
|
26.1 ± 1.9 | 0.8 ± 0.2 | 2.53 | 0.21 |
| 35 |
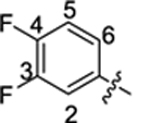
|
16.1 ± 0.7 | 1.8 ± 0.1 | 3.76 | 0.25 |
| 36 |
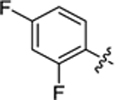
|
19.3 ± 0.9 | 1.9 ± 0.2 | 3.83 | 0.24 |
| 37 |
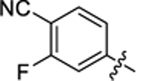
|
12.8 ± 0.5 | 1.8 ± 0.1 | 3.13 | 0.24 |
| 38 |
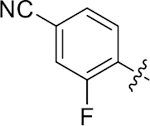
|
14.8 ± 0.8 | 1.9 ± 0.1 | 3.13 | 0.24 |
| 39 |
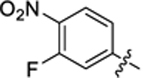
|
14.9 ± 1.3 | 1.8 ± 0.1 | 3.14 | 0.23 |
| 40 |
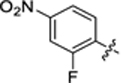
|
23.8 ± 2.7 | 1.6 ± 0.1 | 3.44 | 0.22 |
| 41 |
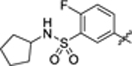
|
24.9 ± 0.5 | 1.3 ± 0.2 | 4.32 | 0.18 |
| 42 |
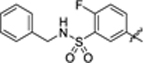
|
8.2 ± 1.3 | 1.9 ± 0.1 | 4.61 | 0.19 |
| 43 |
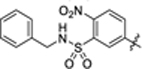
|
2.5 ± 0.2 | 1.8 ± 0.1 | 4.38 | 0.20 |
Suramin was used as a positive control with an IC50 of 28.4 ± 2.5 μM.
The ΔTm values were calculated from the thermal shift assay at a compound concentration of 100 μM.
cLogP values were calculated using ChemDraw Ultra 12.0.
Calculated LE = 1.4 pIC50 (M)/N, where N is the number of nonhydrogen atom.
Fig. 4.
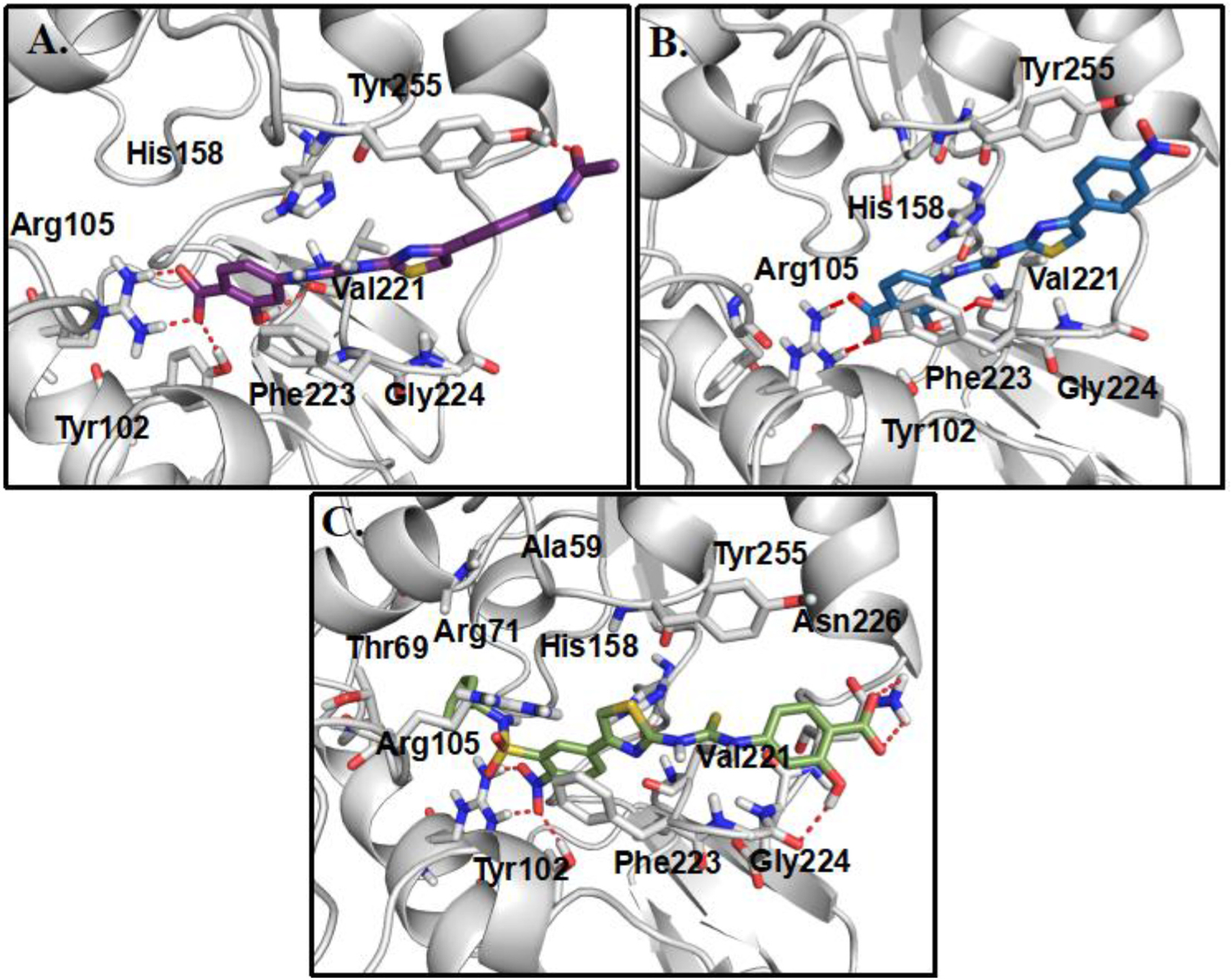
(A) The predicted binding modes of compounds 14 (A), 30 (B) and 43 (C) with SIRT5, respectively.
To investigate whether modifications on the phenyl group of 14 can improve potency, a series of analogues 15–34 with a variety of functional groups at different positions of the benzene ring were designed and synthesized. The biochemical activity results showed that the acetamide group at the meta- (15) or ortho-position (16) of the benzene ring decreased the inhibitory activities against SIRT5 compared with analogue 14, indicating that substitution at para-position of the benzene ring is preferred over other positions. A similar trend was observed in other methyl, fluorine, trifluoromethyl or methoxy substituted derivatives (Table 2). Given the notion that the benzene ring makes pi-pi interactions with Tyr255 in the molecular modeling of compound 14 (Fig. 4A), we assumed that the electron density of the aromatic ring would have a great influence on the interaction intensity between inhibitor and SIRT5. To test the validity of this hypothesis, compounds 17, 25, and 20 were synthesized, in which the acetamide was replaced by electron-donating group methyl, methoxy, and electron-withdrawing group fluorine, respectively. Among these three compounds, compound 20 bearing fluoro-substitution is the most potent, indicating that incorporating an electron-withdrawing group on the benzene ring is beneficial for potency enhancement. Replacement of the fluorine with bromine (31) or chlorine (32) maintained potency in comparison to 20, suggesting that substituent size may not be a contributing factor. Based on these results, we designed and synthesized compounds 23, 29, and 30, in which the acetamide was replaced by stronger electron-withdrawing group trifluoromethyl, cyano, and nitryl, respectively. As anticipated, compound 30 bearing a nitro substitution at para-position of the benzene ring is the most potent, displaying a 6-fold increased potency in comparison with 11. The molecular docking revealed that carboxylate of compound 30 forms salt bridge with Arg105, and hydroxylate forms H-bond with Val221 (Fig. 4B). Compound 30 also forms a pi-pi interaction with Tyr255. There is also a strong possibility of H-bond interaction between Tyr255 OH group and NO2 group of compound 30 in other docking poses. According to the predicted binding mode of 30 with SIRT5 (Fig. 4B), we speculated that incorporation of additional binding moieties at the meta- or ortho-position of the benzene ring may improve potency by increasing hydrophobic interactions with surrounding residues. Therefore, a series of di-substituted analogues 35–43 were designed and synthesized for further SAR studies. To explore the optimal di-substituted positions, 35 bearing 3,4-difluoropheny substituent and 36 bearing 2,4-difluorophenyl substituent were designed and synthesized. Compound 35 is more potent than 36, indicating that di-substitution at the 3 and 4 positions of the benzene ring is preferred, which may be due to the steric effects. Therefore, more 3,4 di-substituted compounds (41–43) were designed and synthesized. As anticipated, 43 was the most potent, showing a 10-fold increased potency in comparison with 11, in which a nitro substitution and a N-benzylsulfamoyl were incorporated at para-position and meta-position of the benzene ring, respectively. The molecular docking studies (Fig. 4C) indicated that compound 43 docked in a flipped position in such a way that NO2 group forms salt bridge and H-bond with Arg105 and Tyr102. The benzyl amino sulphonyl group is placed in an inner hydrophobic cavity formed by Thr69, Arg71, and Ala59. While the hydroxyl group of compound 43 forms H-bond interaction with Gly224, and the carboxylate forms weak H-bond with Asn226. We plotted the docking scores (GOLD score) vs pIC50 values for all compounds (Fig. 5) and observed a highly significant correlation (R2 = 0.71). A protein-ligand interactions fingerprinting was plotted to visualize interactions between key active site residues and all ligands (Fig. 6). All ligands are organized into 5 major clusters. Majority of active compounds (pIC50 > 4.8) are in clusters 1 and 2 (C1 and C2), moderately active compounds (4.8 ≤ pIC50 ≥ 4.5) are in cluster 3 (C3), while inactive compounds (pIC50< 4.5) are in clusters 4 and 5 (C4 and C5). The protein-ligand interaction fingerprints plot revealed the importance of residues Arg105, Tyr102, Val221, Phe223, Gly224, and Tyr255 in SIRT5-ligand binding. Salt bridge with Arg105 is critical for this class of compounds to be active. Hydrogen bonds with either Val221, Tyr102, or Gly224 are advantageous for the activity of compounds.
Fig. 5.
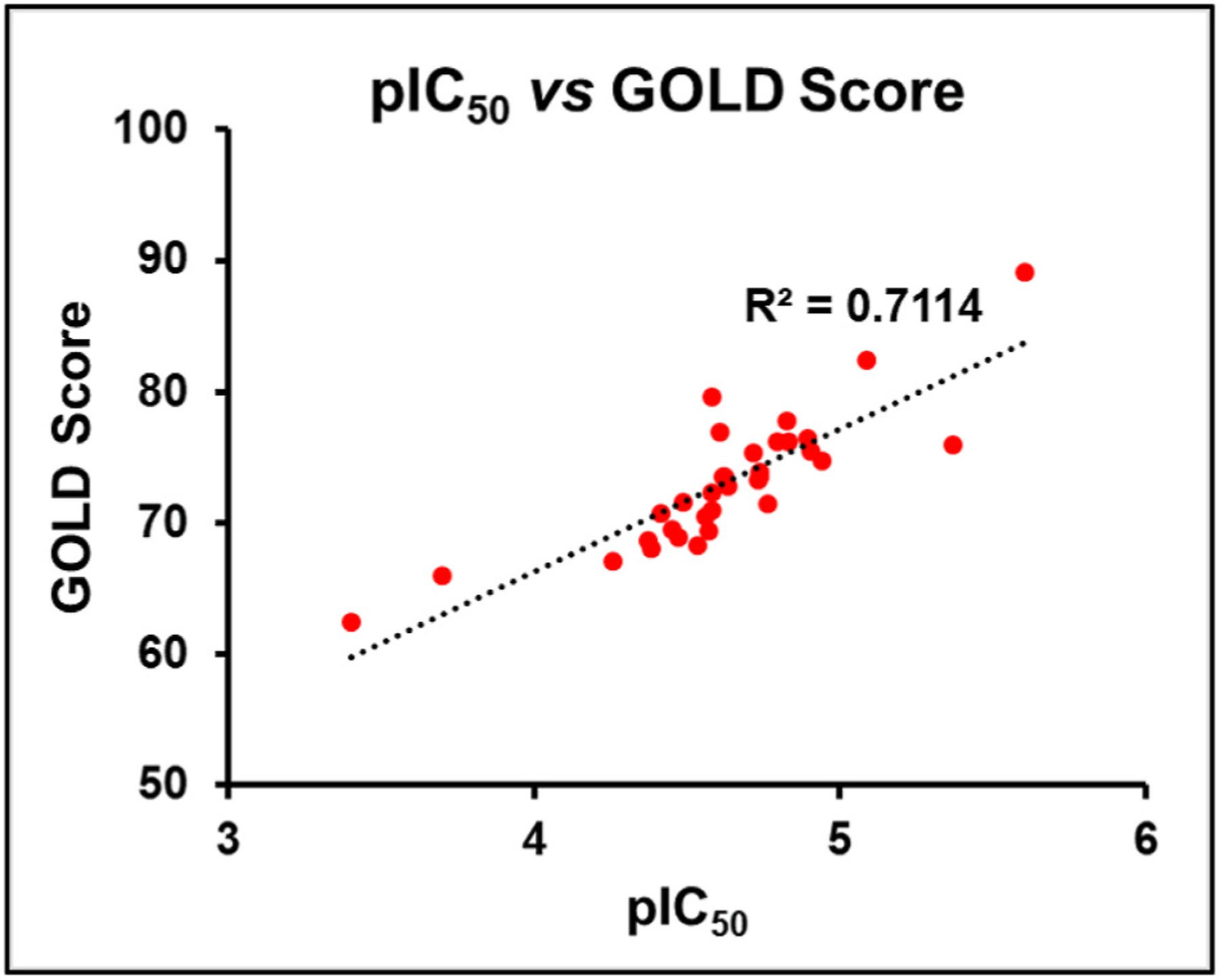
Docking score (GOLD score) vs pIC50 values.
Fig. 6.
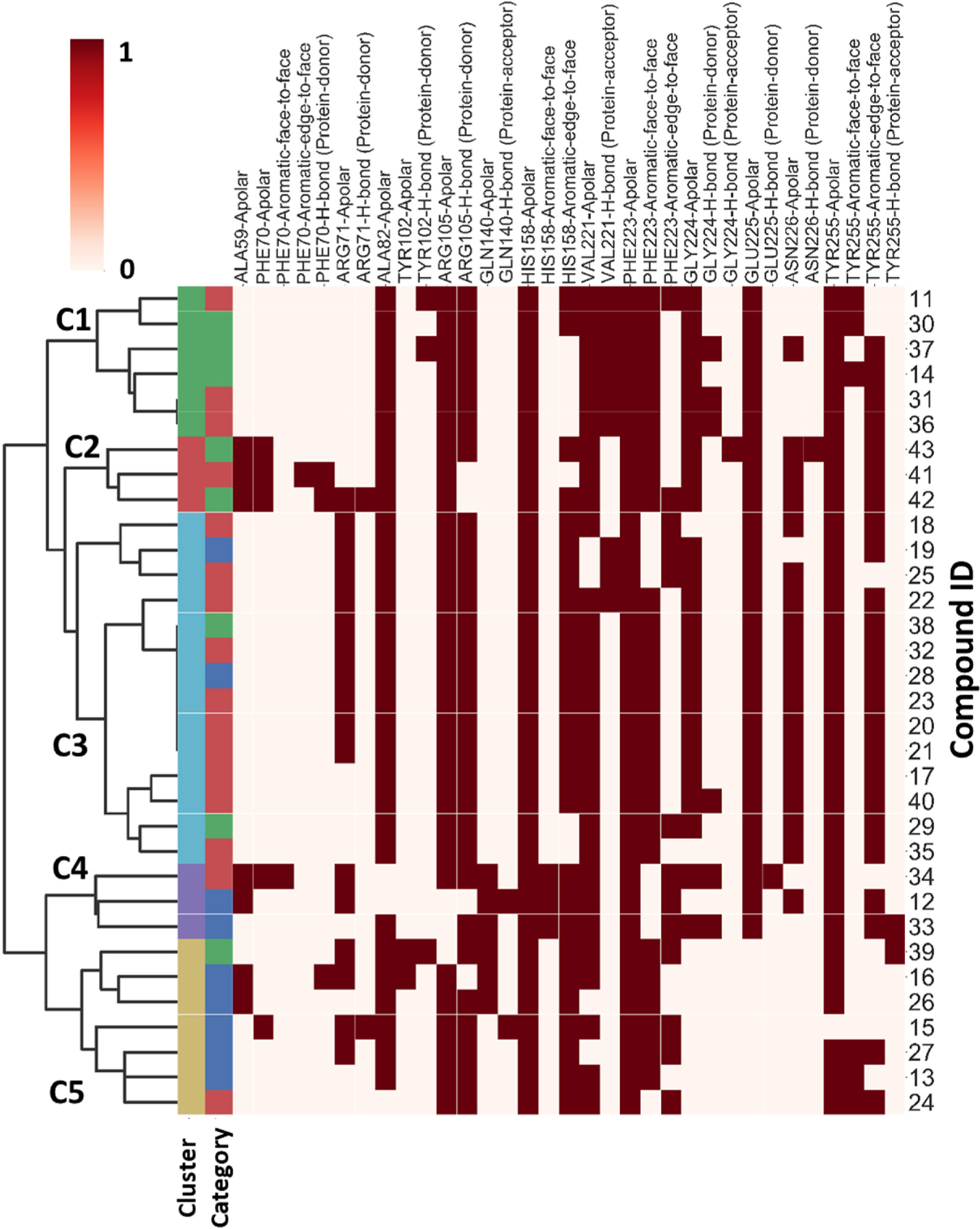
Protein-ligand interactions fingerprinting of compounds 11–43 with the key residues of SIRT5 protein. At the top, the x-axis represents key residues with seven different types of interactions. If the interactions are present, then the cells are in red, otherwise no color. On the left, a dendrogram is drawn to show the relationship between different clusters (C1–C5). The first column at the left represents color-coded clusters of fingerprints. Second column from the left represents category of compounds based on enzymatic activity; actives (pIC50 > 4.8), moderately actives (4.8 ≤ pIC50 ≥ 4.5) and inactives (pIC50 < 4.5).
To investigate the selectivity profile, representative compounds 14 and 43 were tested against SIRT1, SIRT2, and SIRT3. We observed that these two compounds displayed no inhibition against these family members even at the highest concentration of 400 μM, showing substantial selectivity for SIRT5 (see Table S1).
2.3. Chemistry
Hit compound 11 and its ethylester 12 were prepared according to the procedure shown in Scheme 1. Reaction of 4-amino-2-hydroxybenzoic acid with thiophosgene in hydrochloric acid gave isothiocyanate 46 in 86% yield. Treatment of 46 with ammonium hydroxide afforded thiourea 47, which was then reacted with 48 to generate compound 11 in 54% yield. Compound 11 was subjected to sulfuric acid in ethanol to give intermediate 49. Condensation of 49 with acetyl chloride afforded compound 12.
Scheme 1.
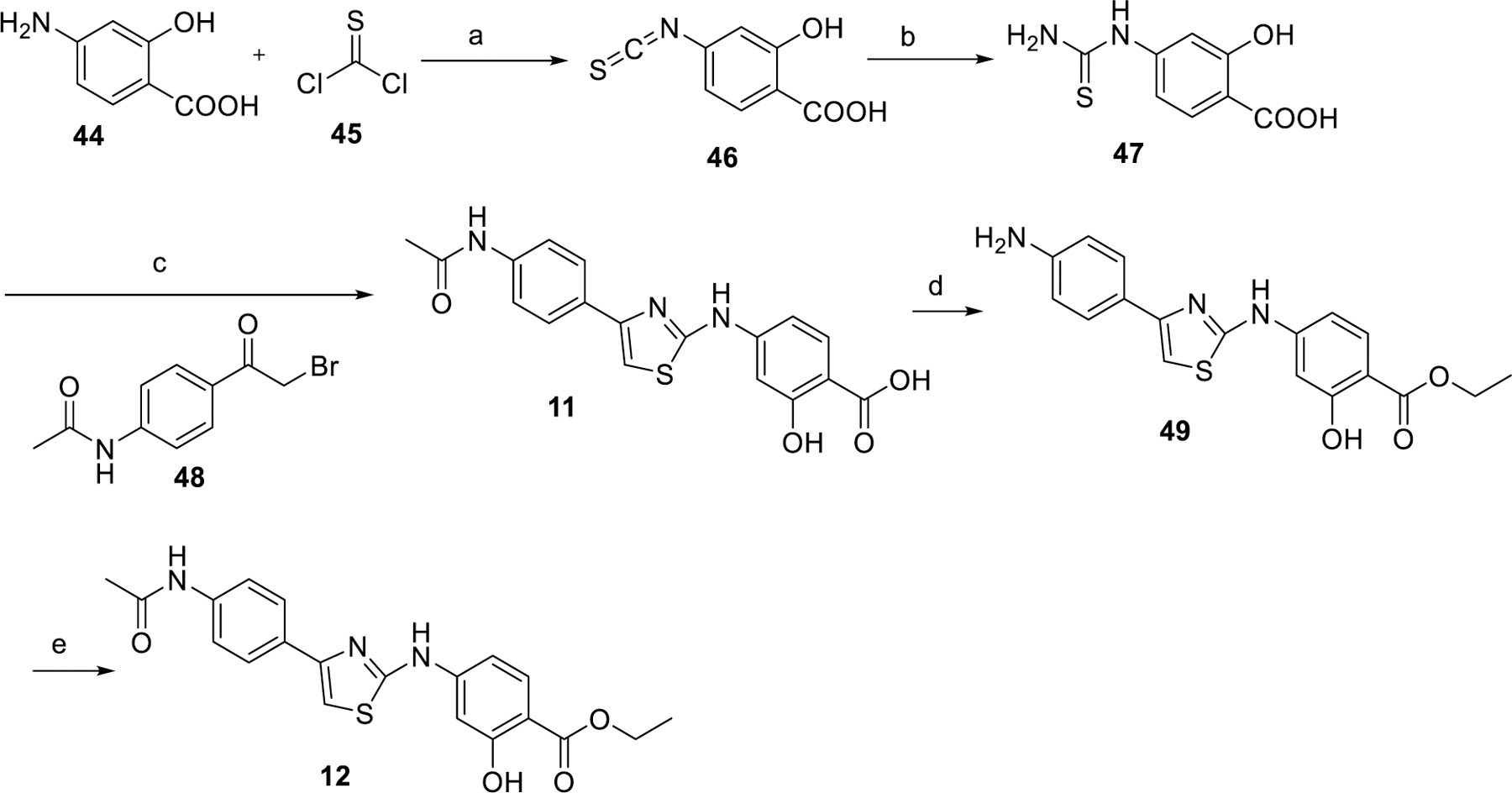
Synthesis of hit compound 11 and 12a
aReaction conditions: (a) HCl, H2O, rt; (b) aqueous ammonia, rt; (c) N-(4-(2-bromoacetyl)phenyl)acetamide, EtOH, reflux; (d) H2SO4, EtOH, reflux; (e) acetylchloride, DMF, 0 °C to rt.
Synthesis of compound 13 is outlined in Scheme 2. Buchwald coupling of 54 with tert-butyl 4-iodobenzoate 55 afforded intermediate 56 Tert-butyl deprotection of 56 gave compound 13.
Scheme 2.
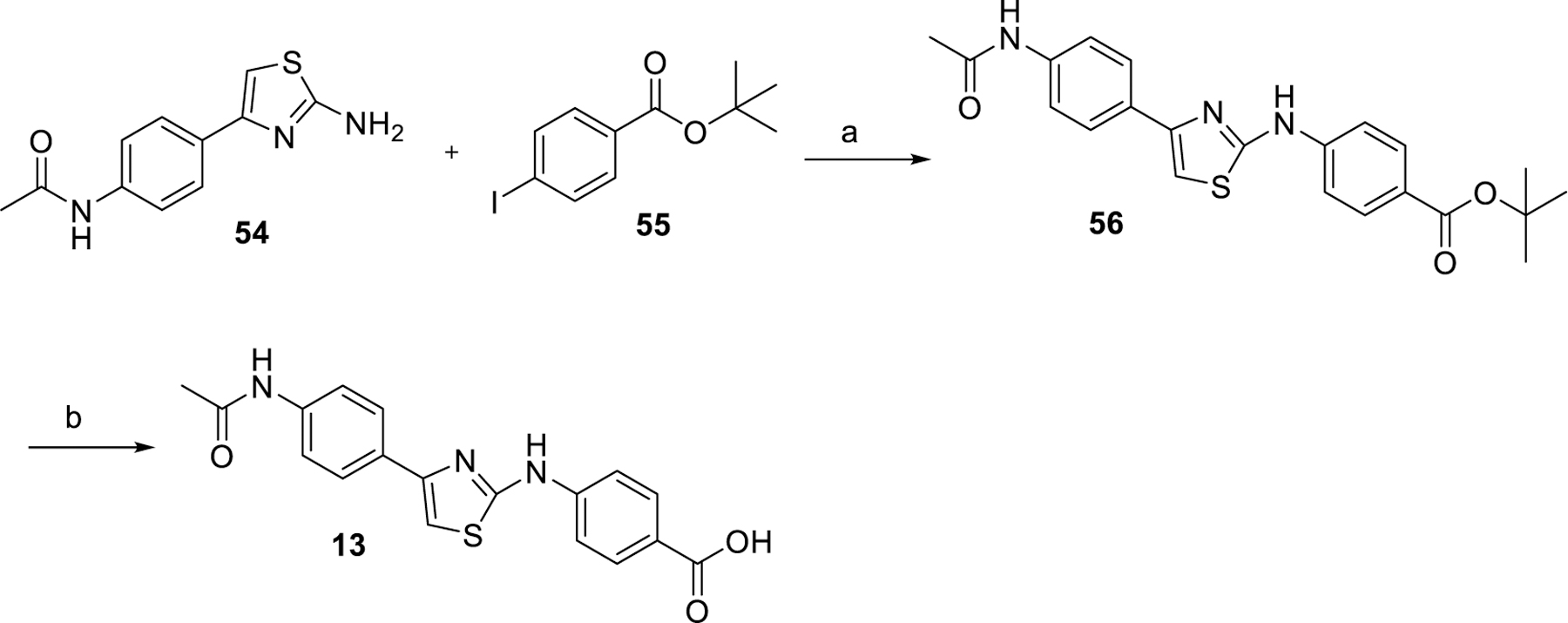
Synthesis of compound 13a
aReagents and conditions: (a) Pd(OAc)2, BINAP, Cs2CO3, DMF, 100 °C; (b) TFA, DCM, rt.
The synthetic route to compounds 14–40 is shown in Scheme 3. The isothiocyanate 46 was reacted with amine 54 to produce compound 14. Compounds 15–40 were prepared by reacting the common intermediate 46 with different amines using the same method as was used for 14.
Scheme 3.
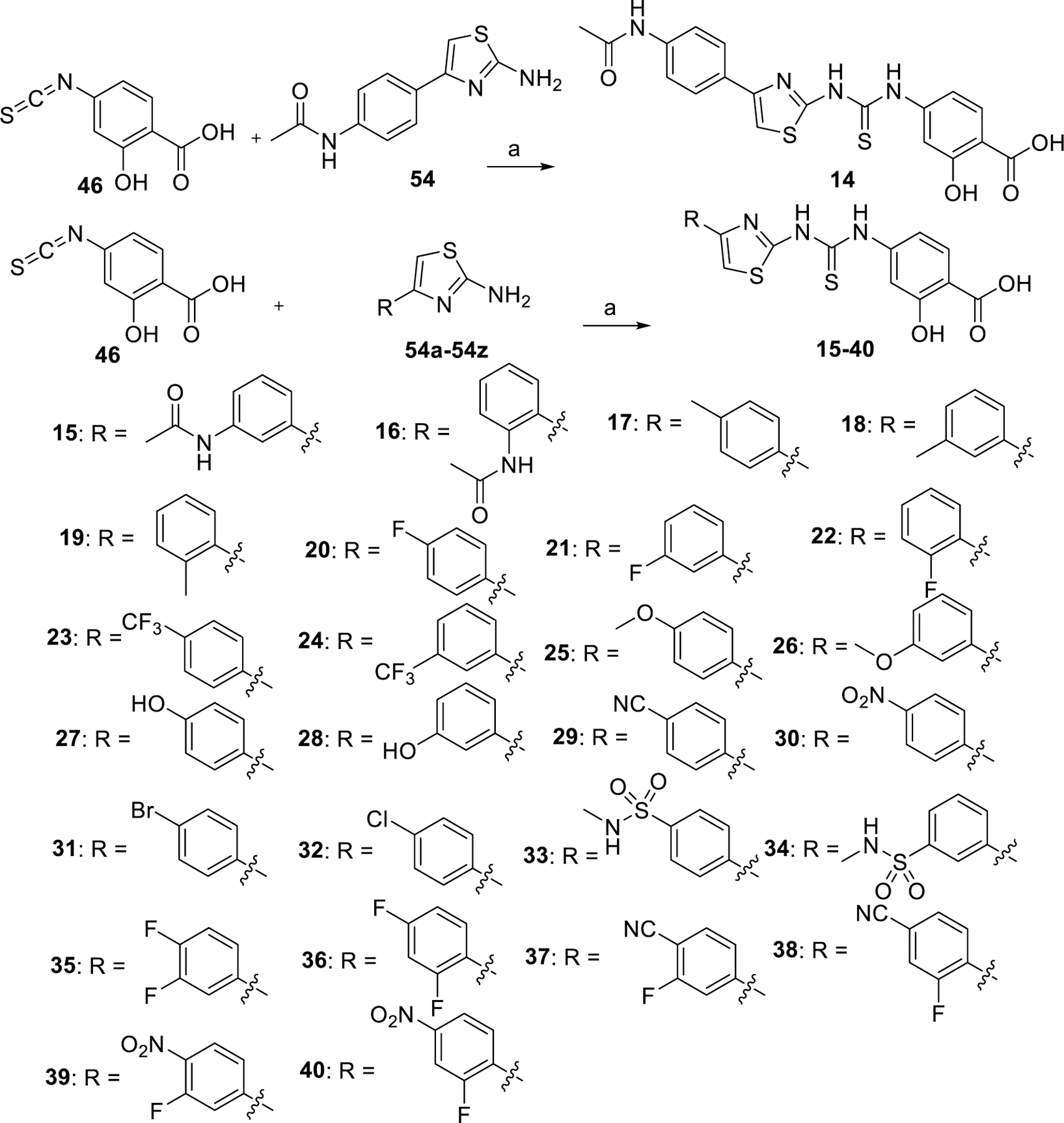
Synthesis of compound 14–40a
aReaction conditions: (a) pyridine, 60 °C.
Synthesis of compounds 41–43 is shown in Scheme 4. Coupling reaction of compound 57a with benzylthiol afforded the intermediate 58a. Treatment of 58a with 1,3-dichloro-5,5-dimethylimidazolidine-2,4-dione 59 gave sulfonyl chloride 60a. Condensation of 60a with cyclopentanamine generated the intermediate 62c. Bromination of 62c followed by thiazole cyclization reaction afforded the intermediate 65c. Reacting the intermediate 65c with 46 afforded compound 41. Compounds 42 and 43 were obtained using the same procedure as was used for 41.
Scheme 4.
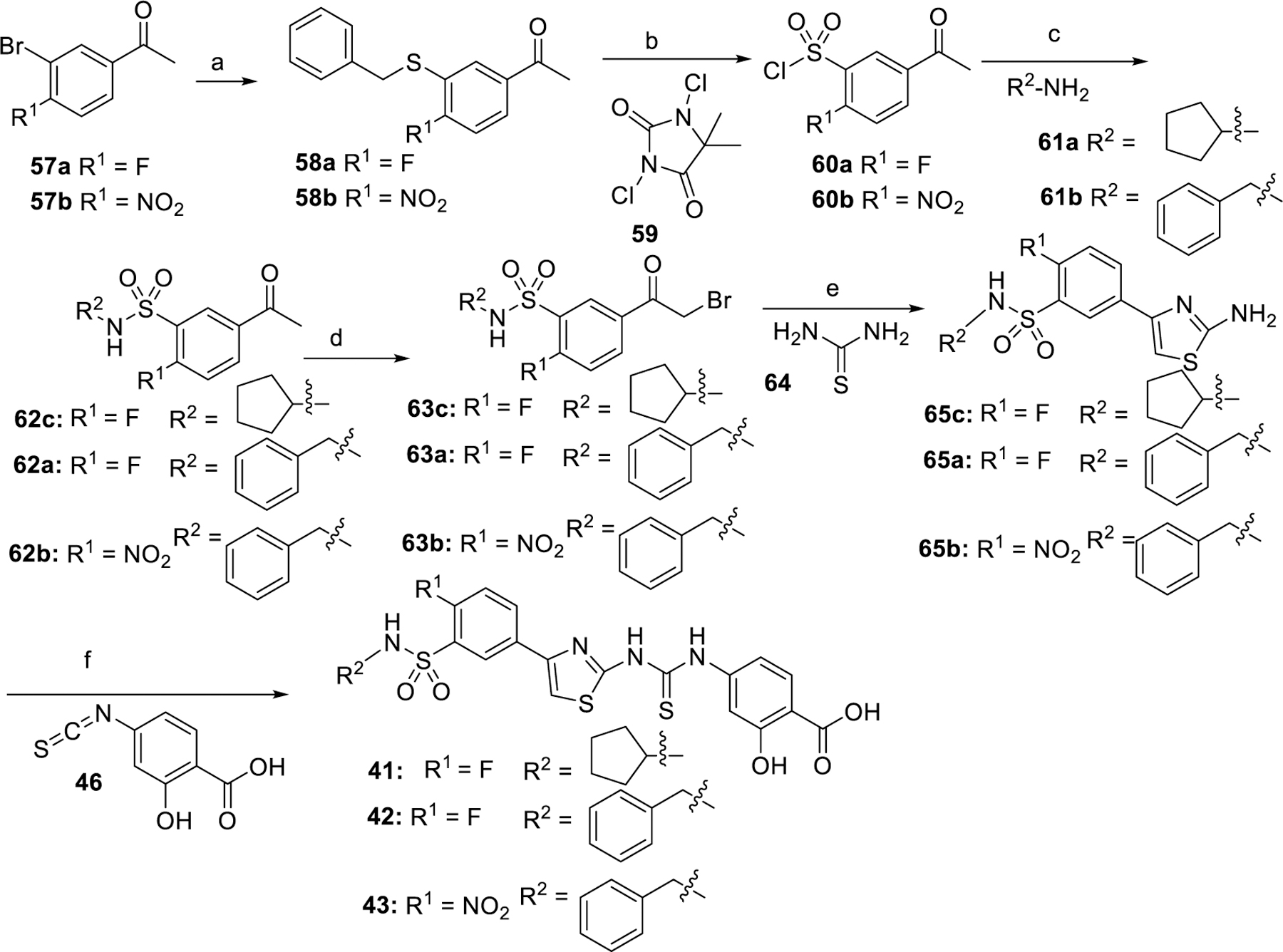
Synthesis of compound 41–43a
aReaction conditions: (a) DIPEA, 1,4 dioxane, Pd2(dba)3, Xantphos, benzylthiol, reflux; (b) CH3CN, H2O, AcOH, 0 °C; (c) TEA, DCM, rt; (d) Bu4NBr3, CH3CN, 60 °C; (e) EtOH, reflux; (f) pyridine, 60 °C.
3. Conclusions
In this study, we designed and optimized a medium throughput thermal shift assay specific for SIRT5 and identified the hit compound 11 as a novel SIRT5 inhibitor. Compound 11 stabilizes SIRT5 in a dose-dependent manner and inhibits SIRT5 enzymatic activity with an IC50 value of 26.4 ± 0.8 μM. In a trypsin coupled fluorescence assay, compound 11 was more potent against SIRT5 as compared to SIRT1, SIRT2, and SIRT3. Molecular docking studies indicate that the carboxylate group forms electrostatic interactions and H-bonds with unique residues Arg105 and Tyr102 of SIRT5 in the deep end of substrate binding pocket. The adjacent hydroxyl group of 11 forms a H-bond with Val221. These observations were validated by in vitro enzymatic assay, highlighting the necessity of 2-hydroxybenzoic acid moiety for maintaining SIRT5 inhibition. These results indicate that 2-hydroxybenzoic acid could serve as a warhead for the design of selective SIRT5 inhibitors. To improve potency of 11, a total of 34 new derivatives were designed and synthesized, among which compound 43 is the most potent and displayed a 10-fold increased potency in comparison with hit 11. In summary, we identified 11 as a promising novel chemical scaffold to target SIRT5 and indicate that the thermal shift assay is a useful method to discover novel small-molecule SIRT5 inhibitors.
4. Experimental section
4.1. Chemistry
All reagents and anhydrous solvents were purchased from commercial sources and used without purification, unless specified. Reaction progress was monitored by UV absorbance using thin-layer chromatography (TLC). Flash chromatography was performed using a Biotage Isolera chromatography system equipped with 10 and 25 g Ultra-SNAP Cartridge columns (25 μM spherical silica). 1H NMR spectra were obtained using a Bruker (300 or 400 MHz) instrument. A Shimadzu LCMS 2020 system was utilized for generating HPLC traces, obtaining mass spectrometry data, and evaluating purity. Purity of final compounds was assessed at 254 nm. Reverse-phase preparative purifications were performed on a Shimadzu LC-20 modular HPLC system. This system utilized a PDA detector and a Kinetex 5 μm XB-C18 100 Å, 150 mm × 21.2 mm column. The purity of all final compounds was >95%.
2-hydroxy-4-isothiocyanatobenzoic acid (46).
4-Aminosalicylic acid (3.06 g, 0.02 mol) was suspended in 35 mL of water and 6.7 mL of concentrated hydrochloric acid added and the mixture was stirred at room temperature. To this suspension another 35 mL portion of water was added followed by thiophosgene (2.7 g, 0.024 mol). After 3 h of stirring, the orange color of the thiophosgene was dissipated indicating completion of the reaction. The solid product was filtered; the filter cake was washed multiple times with water and dried over phosphorus pentoxide. This white solid product (3.35 g, 86% yield) was recrystallized from toluene. The pure product was slightly unstable to light. 1H NMR (400 MHz, DMSO‑d6) δ 7.83 (d, J = 8.4 Hz, 1H), 7.00 (d, J = 2.0 Hz, 1H), 6.96 (dd, J = 8.4, 2.1 Hz, 1H).
2-hydroxy-4-thioureidobenzoic acid (47).
Compound 46 (3.756 g, 0.019 mol) was dissolved in 70 mL of aqueous ammonia (28%) and the solution stirred overnight. The white solid product (2.98 g, 73% yield) was recrystallized from aqueous ethanol. 1H NMR (400 MHz, DMSO‑d6) δ 11.38 (s, 1H), 9.97 (s, 1H), 8.07 (s, 1H), 7.70 (d, J = 8.7 Hz, 1H), 7.38 (d, J = 2.1 Hz, 1H), 6.96 (dd, J = 8.7, 2.2 Hz, 1H).
4-((4-(4-acetamidophenyl)thiazol-2-yl)amino)-2-hydroxybenzoic acid (11).
To a solution of compound 47 (424 mg, 2.0 mmol) in ethanol was added N-(4-(2-bromoacetyl)phenyl)acetamide 48 (512 mg, 2.0 mmol). The reaction mixture was refluxed for 3 h. Preparative HPLC purification afforded the pure product 11 as a white solid (400 mg, 54% yield). 1H NMR (300 MHz, DMSO‑d6) δ 10.72 (s, 1H), 10.10 (s, 1H), 7.84 (d, J = 8.5 Hz, 2H), 7.74 (d, J = 8.7 Hz, 1H), 7.67 (d, J = 8.5 Hz, 2H), 7.56 (d, J = 2.1 Hz, 1H), 7.34 (s, 1H), 7.10 (dd, J = 8.9, 2.1 Hz, 1H), 2.07 (s, 3H). 13C NMR (150 MHz, DMSO‑d6) δ 172.3, 168.8, 163.2, 162.5, 150.6, 147.6, 139.4, 131.8, 129.7, 126.6, 119.5, 108.9, 105.9, 103.6, 103.5, 24.5. HRMS (ESI): m/z [M + H]+ calculated for C18H16N3O4S, 370.0856; found, 370.0860. HPLC purity at 254 nm, 99.5%.
Ethyl 4-((4-(4-aminophenyl)thiazol-2-yl)amino)-2-hydroxybenzoate (49).
To a solution of compound 11 (370 mg, 1 mmol) in ethanol (3 mL) was added conc. H2SO4 (0.2 mL). The reaction mixture was refluxed for 8 h. Preparative HPLC purification afforded the pure product 49 as a white solid (200 mg, 56% yield). 1H NMR (400 MHz, DMSO‑d6) δ 10.88 (s, 1H), 10.70 (s, 1H), 7.84–7.80 (m, 2H), 7.77 (d, J = 8.8 Hz, 1H), 7.59 (d, J = 2.1 Hz, 1H), 7.30 (s, 1H), 7.12 (dd, J = 8.8, 2.2 Hz, 1H), 7.02 (d, J = 8.5 Hz, 2H), 4.35 (q, J = 7.1 Hz, 2H), 1.35 (t, J = 7.1 Hz, 3H). MS (ESI) 356 [M + H]+.
Ethyl 4-((4-(4-acetamidophenyl)thiazol-2-yl)amino)-2-hydroxybenzoate (12).
Compound 49 (30 mg, 0.085 mmol) was dissolved in anhydrous DMF (2 mL). Acetylchloride (10 μL, 0.141 mmol) and DIPEA (40 μL, 0.242 mmol) were added sequentially at 0 °C. The reaction mixture was then allowed to reach ambient temperature and stirred for 2 h. Preparative HPLC purification afforded the pure product 12 as a white solid (29 mg, 86% yield). 1H NMR (400 MHz, DMSO‑d6) δ 10.87 (s, 1H), 10.71 (s, 1H), 10.06 (s, 1H), 7.86 (d, J = 8.7 Hz, 2H), 7.78 (d, J = 8.8 Hz, 1H), 7.67 (d, J = 8.8 Hz, 2H), 7.57 (d, J = 2.1 Hz, 1H), 7.36 (s, 1H), 7.15 (dd, J = 8.8, 2.2 Hz, 1H), 4.35 (q, J = 7.1 Hz, 2H), 2.07 (s, 3H), 1.35 (t, J = 7.1 Hz, 3H). LCMS (ESI) 398 [M + H]+, 396 [M − H]−. HPLC purity at 254 nm, 99.6%.
4-((4-(4-acetamidophenyl)thiazol-2-yl)amino)benzoic acid (13).
To a solution of compound N-(4-(2-aminothiazol-4-yl)phenyl) acetamide 54 (233 mg, 1 mmol) in anhydrous DMF (4 mL) was added Pd (OAc)2 (22 mg, 0.1 mmol), BINAP (124 mg, 0.2 mmol), Cs2CO3 (390 mg, 1.2 mmol) and tert-butyl 4-iodobenzoate 55 (234 μL, 1.2 mmol). The resulting mixture was stirred at 100 °C for 16 h. The reaction mixture was then allowed to reach ambient temperature, filtered and concentrated. The crude product was purified by flash column chromatography to provide product 56 (188 mg, 46% yield) as a yellowish solid. The intermediate 56 (41 mg, 0.1 mmol) was dissolved in DCM (2 mL) and TFA (1 mL). After stirring for 2 h, the solvent was evaporated and the residue was purified by HPLC to give the product 13 as a white solid (34 mg, 95% yield). 1H NMR (400 MHz, DMSO‑d6) δ 10.67 (s, 1H), 10.05 (s, 1H), 7.97–7.92 (m, 2H), 7.90–7.80 (m, 4H), 7.70–7.63 (m, 2H), 7.32 (s, 1H), 2.07 (s, 3H). LCMS (ESI) 354 [M + H]+, 352 [M − H]−. HPLC purity at 254 nm, 99.0%.
4-(3-(4-(4-acetamidophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (14).
Intermediate 46 (20 mg, 0.1 mmol) and amine 54 (23 mg, 0.1 mmol) were dissolved in anhydrous pyridine (2 mL). The reaction mixture was stirred at 60 °C overnight and then purified by HPLC to isolate the product as a slightly yellow solid (26 mg, 60% yield). 1H NMR (400 MHz, DMSO‑d6) δ 11.40 (s, 1H), 10.08 (s, 1H), 7.99–7.03 (m, 8H), 2.08 (d, J = 2.4 Hz, 3H). LCMS (ESI) 429 [M + H]+, 427 [M − H]−. HPLC purity at 254 nm, 99.5%.
4-(3-(4-(3-acetamidophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (15).
Compound 15 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 10.05 (s, 1H), 8.26–7.10 (m, 8H), 2.08 (s, 3H). LCMS (ESI) 429 [M + H]+, 427 [M − H]−. HPLC purity at 254 nm, 99.9%.
4-(3-(4-(2-acetamidophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (16).
Compound 16 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 13.57 (s, 1H), 12.07 (s, 1H), 11.35 (s, 1H), 8.29–6.87 (m, 8H), 2.16–2.02 (m, 3H). LCMS (ESI) 429 [M + H]+, 427 [M − H]−. HPLC purity at 254 nm, 98.2%.
2-hydroxy-4-(3-(4-(p-tolyl)thiazol-2-yl)thioureido)benzoic acid (17).
Compound 17 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 7.81–7.60 (m, 4H), 7.46 (d, J = 2.1 Hz, 1H), 7.29 (t, J = 7.5 Hz, 3H), 2.34 (s, 3H). LCMS (ESI) 386 [M + H]+, 384 [M − H]−. HPLC purity at 254 nm, 99.5%.
2-hydroxy-4-(3-(4-(m-tolyl)thiazol-2-yl)thioureido)benzoic acid (18).
Compound 18 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 7.83–7.02 (m, 8H), 2.37 (s, 3H). LCMS (ESI) 386 [M + H]+, 384 [M − H]−. HPLC purity at 254 nm, 99.5%.
2-hydroxy-4-(3-(4-(o-tolyl)thiazol-2-yl)thioureido)benzoic acid (19).
Compound 19 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 11.37 (s, 1H), 7.73–7.60 (m, 2H), 7.53–7.45 (m, 1H), 7.40–7.24 (m, 4H), 7.07 (s, 1H), 2.38 (s, 3H). LCMS (ESI) 386 [M + H]+, 384 [M − H]−. HPLC purity at 254 nm, 99.9%.
4-(3-(4-(4-fluorophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (20).
Compound 20 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 11.41 (s, 1H), 10.80 (s, 1H), 7.93 (ddd, J = 9.1, 5.2, 2.2 Hz, 2H), 7.74 (dd, J = 18.1, 8.6 Hz, 1H), 7.55 (m,2H), 7.42–7.15 (m, 3H). LCMS (ESI) 390 [M + H]+, 388 [M − H]−. HPLC purity at 254 nm, 99.9%.
4-(3-(4-(3-fluorophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (21).
Compound 21 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 12.02 (s, 1H),δ 11.43 (s, 1H), 10.73 (s, 1H),δ 7.75 (q, J = 11.0, 9.9 Hz, 4H), 7.63–7.42 (m, 2H), 7.21 (d, J = 9.8 Hz, 2H). LCMS (ESI) 390 [M + H]+, 388 [M − H]−. HPLC purity at 254 nm, 99.4%.
4-(3-(4-(2-fluorophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (22).
Compound 22 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 12.05 (s, 1H),δ 11.41 (s, 1H), 10.89 (s, 1H), 7.99 (s, 1H), 7.75 (dd, J = 17.9, 8.8 Hz, 1H), 7.63 (d, J = 2.1 Hz, 1H), 7.54–7.30 (m, 4H), 7.20 (s, 1H). LCMS (ESI) 390 [M + H]+, 388 [M − H]−. HPLC purity at 254 nm, 99.9%.
2-hydroxy-4-(3-(4-(4-(trifluoromethyl)phenyl)thiazol-2-yl)thioureido)benzoic acid (23).
Compound 23 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 12.12 (s, 1H),δ 11.42 (s, 1H), 10.77 (s, 1H),δ 8.12 (d, J = 8.0 Hz, 2H), 7.81 (dd, J = 19.1, 8.4 Hz, 4H), 7.63 (d, J = 2.1 Hz, 1H), 7.19 (s, 1H). LCMS (ESI) 440 [M + H]+, 438 [M − H]−. HPLC purity at 254 nm, 96.4%.
2-hydroxy-4-(3-(4-(3-(trifluoromethyl)phenyl)thiazol-2-yl)thioureido)benzoic acid (24).
Compound 24 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 12.12 (s, 1H),δ 11.41 (s, 1H), 10.80 (s, 1H),δ 8.28–8.19 (m, 2H), 7.87–7.69 (m, 4H), 7.63 (d, J = 2.1 Hz, 1H), 7.18 (s, 1H). LCMS (ESI) 440 [M + H]+, 438 [M − H]−. HPLC purity at 254 nm, 98.9%.
2-hydroxy-4-(3-(4-(4-methoxyphenyl)thiazol-2-yl)thioureido) benzoic acid (25).
Compound 25 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 11.39 (s, 1H), 7.86–7.71 (m, 3H), 7.63 (s, 1H), 7.33 (s, 2H), 7.04 (dd, J = 8.6, 6.6 Hz, 2H), 3.82 (s, 3H). LCMS (ESI) 402 [M + H]+, 400 [M − H]−. HPLC purity at 254 nm, 99.8%.
2-hydroxy-4-(3-(4-(3-methoxyphenyl)thiazol-2-yl)thioureido) benzoic acid (26).
Compound 26 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 11.40 (s, 1H), 10.81 (s, 1H), 7.76 (d, J = 8.7 Hz, 1H), 7.64–7.52 (m, 2H), 7.49–7.35 (m, 3H), 7.28 (s, 1H), 6.99–6.91 (m, 1H), 3.84 (s, 3H). LCMS (ESI) 402 [M + H]+, 400 [M − H]−. HPLC purity at 254 nm, 99.8%.
2-hydroxy-4-(3-(4-(4-hydroxyphenyl)thiazol-2-yl)thioureido) benzoic acid (27).
Compound 27 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 11.38 (s, 1H), 9.73 (s, 1H), 7.80–7.56 (m, 4H), 7.50–7.12 (m, 2H), 6.85 (dd, J = 8.6, 6.5 Hz, 2H). LCMS (ESI) 388 [M + H]+, 386 [M − H]−. HPLC purity at 254 nm, 99.9%.
2-hydroxy-4-(3-(4-(3-hydroxyphenyl)thiazol-2-yl)thioureido) benzoic acid (28).
Compound 28 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 11.39 (s, 1H), 9.61 (s, 1H), 7.76 (d, J = 8.7 Hz, 1H), 7.63 (d, J = 2.1 Hz, 1H), 7.50–7.37 (m, 1H), 7.35–7.18 (m, 4H), 6.80 (d, J = 7.7 Hz, 1H). LCMS (ESI) 388 [M + H]+, 386 [M − H]−. HPLC purity at 254 nm, 99.5%.
4-(3-(4-(4-cyanophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (29).
Compound 29 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 12.07 (s, 1H), 11.36 (d, J = 34.0 Hz, 1H), 10.76 (s, 1H), 8.13–8.05 (m, 2H), 7.93 (dd, J = 8.2, 5.5 Hz, 2H), 7.85 (d, J = 6.5 Hz, 1H), 7.79 (d, J = 8.6 Hz, 1H), 7.62 (d, J = 2.1 Hz, 1H), 7.18 (s, 1H). LCMS (ESI) 397 [M + H]+, 395 [M − H]−. HPLC purity at 254 nm, 98.5%.
2-hydroxy-4-(3-(4-(4-nitrophenyl)thiazol-2-yl)thioureido)benzoic acid (30).
Compound 30 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 12.12 (s, 1H), 11.41 (s, 1H), 10.75 (s, 1H), 8.39–8.30 (m, 2H), 8.21–8.15 (m, 2H), 7.95 (s, 1H), 7.80 (d, J = 8.6 Hz, 1H), 7.62 (d, J = 2.1 Hz, 1H), 7.17 (s, 1H). LCMS (ESI) 417 [M + H]+, 415 [M − H]−. HPLC purity at 254 nm, 99.2%.
4-(3-(4-(4-bromophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (31).
Compound 31 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 12.05 (s, 1H), 11.40 (s, 1H), 10.81 (s, 1H), 7.89–7.83 (m, 2H), 7.77 (d, J = 8.7 Hz, 1H), 7.72–7.54 (m, 4H), 7.21 (s, 1H). LCMS (ESI) 450 [M + H]+. HPLC purity at 254 nm, 99.9%.
4-(3-(4-(4-chlorophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (32).
Compound 32 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 12.03 (s, 1H),δ 11.40 (s, 1H), 10.81 (s, 1H), 7.98–7.88 (m, 2H), 7.77 (d, J = 8.7 Hz, 1H), .69–7.49 (m, 4H), 7.21 (s, 1H). LCMS (ESI) 406 [M + H]+, 404 [M − H]−. HPLC purity at 254 nm, 99.0%.
2-hydroxy-4-(3-(4-(4-(N-methylsulfamoyl)phenyl)thiazol-2-yl) thioureido)benzoic acid (33).
Compound 33 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 12.06 (s, 1H), 11.41 (s, 1H), 10.79 (s, 1H), 8.11 (d, J = 8.2 Hz, 2H), 7.92–7.72 (m, 4H), 7.63 (d, J = 2.1 Hz, 1H), 7.54–7.05 (m, 2H), 2.45 (dd, J = 5.0, 2.6 Hz, 3H). LCMS (ESI) 465 [M + H]+, 463 [M − H]−. HPLC purity at 254 nm, 98.9%.
2-hydroxy-4-(3-(4-(3-(N-methylsulfamoyl)phenyl)thiazol-2-yl) thioureido)benzoic acid (34).
Compound 34 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 12.06 (s, 1H), 11.41 (s, 1H), 10.72 (s, 1H), 8.34 (s, 1H), 8.16 (d, J = 7.6 Hz, 1H), 7.87–7.66 (m, 5H), 7.49 (dd, J = 7.4, 3.4 Hz, 1H), 7.16 (d, J = 20.1 Hz, 1H), 2.46 (dd, J = 5.0, 1.7 Hz, 3H). LCMS (ESI) 465 [M + H]+, 463 [M − H]−. HPLC purity at 254 nm, 98.8%.
4-(3-(4-(3,4-difluorophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (35).
Compound 35 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 12.03 (s, 1H), 11.40 (s, 1H), 10.79 (s, 1H), 7.94 (t, J = 9.9 Hz, 1H), 7.84–7.40 (m, 5H), 7.18 (s, 1H). LCMS (ESI) 408 [M + H]+, 406 [M − H]−. HPLC purity at 254 nm, 99.5%.
4-(3-(4-(2,4-difluorophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (36).
Compound 36 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 11.39 (s, 1H), 8.02 (s, 1H), 7.75 (dd, J = 19.7, 8.5 Hz, 1H), 7.62 (d, J = 2.1 Hz, 1H), 7.41 (d, J = 13.1 Hz, 2H), 7.20 (d, J = 31.9 Hz, 2H). LCMS (ESI) 408 [M + H]+, 406 [M − H]−. HPLC purity at 254 nm, 99.0%.
4-(3-(4-(4-cyano-3-fluorophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (37).
Compound 37 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 12.14 (s, 1H), 11.39 (s, 1H), 10.73 (s, 1H), 8.01–7.93 (m, 4H), 7.76 (dd, J = 18.3, 8.6 Hz, 1H), 7.59 (d, J = 2.1 Hz, 1H), 7.20–7.13 (m, 1H). LCMS (ESI) 415 [M + H]+, 413 [M − H]−. HPLC purity at 254 nm, 98.8%.
4-(3-(4-(4-cyano-2-fluorophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (38).
Compound 38 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 12.08 (s, 1H), 11.40 (s, 1H), 10.77 (s, 1H), 8.19 (q, J = 7.8 Hz, 1H), 8.05–7.94 (m, 1H), 7.87–7.68 (m, 3H), 7.60 (d, J = 2.1 Hz, 1H), 7.15 (d, J = 9.2 Hz, 1H). LCMS (ESI) 415 [M + H]+, 413 [M − H]−. HPLC purity at 254 nm, 99.9%.
4-(3-(4-(3-fluoro-4-nitrophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (39).
Compound 39 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 12.09 (s, 1H), 11.40 (s, 1H), 10.73 (s, 1H), 8.33–8.22 (m, 1H), 8.14–7.91 (m, 3H), 7.77 (dd, J = 18.4, 8.5 Hz, 1H), 7.60 (d, J = 2.1 Hz, 1H), 7.25–7.03 (m, 1H). LCMS (ESI) 435 [M + H]+, 433 [M − H]−. HPLC purity at 254 nm, 99.9%.
4-(3-(4-(2-fluoro-4-nitrophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (40).
Compound 40 was synthesized following the procedure used to prepare compound 14. 1H NMR (400 MHz, DMSO‑d6) δ 12.15 (s, 1H), 11.41 (s, 0H), 10.74 (s, 1H), 8.40–8.00 (m, 4H), 7.85–7.75 (m, 1H), 7.60 (d, J = 2.1 Hz, 1H), 7.15 (d, J = 9.0 Hz, 1H). LCMS (ESI) 435 [M + H]+, 433 [M − H]−. HPLC purity at 254 nm, 99.8%.
1-(3-(benzylthio)-4-fluorophenyl)ethan-1-one (58a).
To a flask were added 1-(3-bromo-4-fluorophenyl)ethan-1-one 57a (217 mg, 1 mmol), i-Pr2NEt (0.4 mL, 2 mmol) and dry 1,4-dioxane. The mixture was evacuated and backfilled with nitrogen (3 cycles). Catalyst Pd2(dba)3 (23 mg, 0.025 mmol), Xantphos (29 mg, 0.05 mmol) and the benzylthiol (0.12 mL, 1 mmol) were added and then the mixture was degassed twice more. The mixture was heated under reflux overnight. The reaction mixture was then allowed to reach ambient temperature, filtered and concentrated. The crude product was purified by flash column chromatography to provide pure product 58a (250 mg, 96% yield) as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 7.90–7.79 (m, 2H), 7.34–7.23 (m, 5H), 7.13 (t, J = 8.7 Hz, 1H), 4.16 (s, 2H), 2.51 (s, 3H). MS (ESI) 261 [M + H]+.
1-(3-(benzylthio)-4-nitrophenyl)ethan-1-one (58b).
Compound 58b was synthesized following the procedure used to prepare compound 58a. 1H NMR (400 MHz, DMSO‑d6) δ 8.29 (d, J = 8.5 Hz, 1H), 8.12 (d, J = 1.7 Hz, 1H), 7.86 (dd, J = 8.6, 1.7 Hz, 1H), 7.51–7.44 (m, 2H), 7.40–7.25 (m, 3H), 4.50 (s, 2H), 2.65 (s, 3H). MS (ESI) 288 [M + H]+.
5-acetyl-N-cyclopentyl-2-fluorobenzenesulfonamide (62c).
To an ice-cold solution of compound 58a (223 mg, 0.858 mmol) in CH3CN-HOAc-H2O (10 mL-0.6 mL–0.4 mL) was added portion-wise 2,4-dichloro-5,5-dimethylhydantoin (339 mg, 1.72 mmol). The reaction mixture was stirred at 0 °C for 2 h and concentrated to near dryness under vacuum. The crude product was diluted with CH2Cl2 (16 mL), and the solution cooled down to 0 °C. 5% NaHCO3 aqueous solution (18 mL) was added slowly at 0 °C. The mixture was stirred at 0 °C for 15 min, and the lower organic layer was washed once more with 10% brine solution at < 10 °C. The lower organic layer was dried over MgSO4 and filtered and concentrated to afford the product 60a as a yellow oil, which was used in the next reaction without further purification.
A solution of 5-acetyl-2-fluorobenzenesulfonyl chloride 60a (78 mg, 0.33 mmol) and cyclopentanamine (39 μL, 0.396 mmol) in dichloromethane was treated with triethylamine (92 μL, 0.66 mmol) and stirred at 0 °C for 2 h. The mixture was purified by flash column chromatography to provide pure product 62c (75 mg, 80% yield) as a yellow solid. 1H NMR (400 MHz, DMSO‑d6) δ 8.32–8.26 (m, 2H), 8.16 (d, J = 7.6 Hz, 1H), 7.65–7.56 (m, 1H), 3.50–3.53 (m, 1H), 2.63 (s, 3H), 1.70–1.50 (m, 4H), 1.45–1.28 (m, 4H). MS (ESI) 286 [M + H]+.
5-acetyl-N-benzyl-2-fluorobenzenesulfonamide (62a).
Compound 62a was synthesized following the procedure used to prepare compound 62c. 1H NMR (300 MHz, DMSO‑d6) δ 8.70 (t, J = 6.4 Hz, 1H), 8.17 (t, J = 7.2 Hz, 2H), 7.50 (t, J = 9.2 Hz, 1H), 7.20 (d, J = 4.0 Hz, 5H), 4.15 (d, J = 6.2 Hz, 2H), 2.59 (d, J = 1.3 Hz, 3H). MS (ESI) 308 [M + H]+.
5-acetyl-N-benzyl-2-nitrobenzenesulfonamide (62b).
Compound 62b was synthesized following the procedure used to prepare compound 62c. 1H NMR (400 MHz, DMSO‑d6) δ 8.84 (s, 1H), 8.27 (dd, J = 8.3, 1.8 Hz, 1H), 8.22 (d, J = 1.8 Hz, 1H), 8.07 (d, J = 8.2 Hz, 1H), 7.24–7.12 (m, 5H), 4.19 (s, 2H), 2.61 (s, 3H). MS (ESI) 335 [M + H]+.
5-(2-aminothiazol-4-yl)-N-cyclopentyl-2-fluorobenzenesulfonamide (65c).
The intermediate 62c (14.3 mg, 0.05 mmol) was dissolved in acetonitrile (2 mL) followed by addition of tetrabutylammonium-tribromide (25 mg, 0.05 mmol). The reaction mixture was stirred at 60 °C overnight. The solvent was removed; the residue was extracted with dichloromethane and washed with water. The organic layers were combined and concentrated under vacuum to provide the crude product 63c, which was used in the next step without further purification.
The intermediate 63c was dissolved in ethanol (2 mL) followed by addition of thiourea (4 mg, 0.05 mmol). The reaction mixture was refluxed for 3 h and then purified by HPLC to isolate the product 65c as a white solid (11 mg, 62% yield). 1H NMR (400 MHz, DMSO‑d6) δ 8.24 (dd, J = 7.2, 2.2 Hz, 1H), 8.07 (ddd, J = 8.7, 4.6, 2.3 Hz, 1H), 7.97 (d, J = 7.6 Hz, 1H), 7.45 (dd, J = 10.1, 8.6 Hz, 1H), 7.17 (d, J = 1.7 Hz, 1H), 3.20–3.13 (m, 1H), 1.61–1.52 (m, 4H), 1.36 (tt, J = 12.4, 6.7 Hz, 4H). MS (ESI) 342 [M + H]+.
5-(2-aminothiazol-4-yl)-N-benzyl-2-fluorobenzenesulfonamide (65a).
Compound 65a was synthesized following the procedure used to prepare compound 65c. 1H NMR (400 MHz, DMSO‑d6) δ 8.51 (t, J = 5.6 Hz, 1H), 8.19 (dt, J = 7.2, 2.2 Hz, 1H), 8.02 (ddd, J = 8.6, 4.7, 2.3 Hz, 1H), 7.37 (t, J = 9.3 Hz, 1H), 7.30–7.16 (m, 5H), 7.13 (d, J = 2.4 Hz, 1H), 4.12 (d, J = 6.3 Hz, 2H). MS (ESI) 364 [M + H]+.
5-(2-aminothiazol-4-yl)-N-benzyl-2-nitrobenzenesulfonamide (65b).
Compound 65b was synthesized following the procedure used to prepare compound 65c. 1H NMR (400 MHz, DMSO‑d6) δ 8.55 (t, J = 6.2 Hz, 1H), 8.31 (d, J = 1.8 Hz, 1H), 8.13 (dd, J = 8.4, 1.8 Hz, 1H), 7.95 (d, J = 8.4 Hz, 1H), 7.35 (s, 1H), 7.32–7.10 (m, 5H), 4.21 (d, J = 6.2 Hz, 2H). MS (ESI) 391 [M + H]+.
4-(3-(4-(3-(N-cyclopentylsulfamoyl)-4-fluorophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (41).
Intermediate 2-hydroxy-4-isothiocyanatobenzoic acid 46 (20 mg, 0.1 mmol) and amine 65c (34 mg, 0.1 mmol) were dissolved in anhydrous pyridine (2 mL). The reaction mixture was stirred at 60 °C overnight and then purified by HPLC to isolate the product as a white solid (43 mg, 80% yield). 1H NMR (400 MHz, DMSO‑d6) δ 12.13 (s, 1H), 11.43 (s, 1H), 10.78 (s, 1H), 8.36 (d, J = 6.8 Hz, 1H), 8.19 (ddd, J = 8.8, 4.6, 2.4 Hz, 1H), 8.03 (t, J = 7.6 Hz, 1H), 7.82–7.65 (m, 3H), 7.61–7.46 (m, 1H), 7.14 (s, 1H), 3.55 (q, J = 7.3 Hz, 1H), 1.69–1.49 (m, 4H), 1.45–1.30 (m, 4H). LCMS (ESI) 537 [M + H]+, 535 [M − H]−. HPLC purity at 254 nm, 99.9%.
4-(3-(4-(3-(N-benzylsulfamoyl)-4-fluorophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (42).
Compound 42 was synthesized following the procedure used to prepare compound 41. 1H NMR (400 MHz, DMSO‑d6) δ 12.12 (s, 1H), 11.42 (s, 1H), 10.80 (s, 1H), 8.57 (q, J = 6.2 Hz, 1H), 8.28 (s, 1H), 8.13 (s, 1H), 7.82–7.64 (m, 3H), 7.51–7.41 (m, 1H), 7.27–7.03 (m, 6H), 4.15 (d, J = 6.2 Hz, 2H). LCMS (ESI) 559 [M + H]+, 557 [M − H]−. HPLC purity at 254 nm, 99.9%.
4-(3-(4-(3-(N-benzylsulfamoyl)-4-nitrophenyl)thiazol-2-yl)thioureido)-2-hydroxybenzoic acid (43).
Compound 43 was synthesized following the procedure used to prepare compound 41. 1H NMR (400 MHz, DMSO‑d6) δ 12.17 (s, 1H), 11.41 (s, 1H), 10.77 (s, 1H), 8.57 (t, J = 6.2 Hz, 1H), 8.36 (d, J = 1.9 Hz, 1H), 8.24 (dd, J = 8.3, 2.0 Hz, 1H), 8.03 (dd, J = 8.3, 4.3 Hz, 1H), 7.88 (s, 1H), 7.81–7.63 (m, 2H), 7.28–7.06 (m, 6H), 4.24 (d, J = 6.2 Hz, 2H). 13C NMR (125 MHz, DMSO‑d6) δ 171.9, 162.1, 146.4, 145.3, 142.9, 140.8, 137.6, 134.7, 131.3, 130.3, 129.8, 128.6, 128.0, 127.6, 127.1, 125.8, 119.2, 118.6, 115.7, 115.6, 113.4, 113.4, 108.9, 46.8. HRMS (ESI): m/z [M + H]+ calculated for C24H20N5O7S3, 586.0519; found, 586.0517. HPLC purity at 254 nm, 97.7%.
Thermal Shift Assay.
Thermal shift assay was performed using the ThermoFluor instrument from Johnson and Johnson. Briefly, all reactions were carried out in a buffered solution of 20 mM Bis-Tris propane pH = 7.5, 150 mM NaCl at a final concentration of 0.2 mg/ml SIRT5, 0.1 mM 1,8-ANS, and 100 μM compound or DMSO as a vehicle control. The reaction mixtures were prepared in a 384-well microplate with a total volume of 5 μL and then layered with 1.5 μL silicon oil. The plate was sealed with aluminum sealing film and spun at 1000 rpm for 1 min and then heated from 25 °C to 90 °C under continuous ramp heating mode with the ThermoFluor instrument. Each reaction was repeated in quadruplicate. Melt curves were analyzed with the ThermoFluor ++ software.
Fluorescence-Based Inhibition Assay.
The inhibition activities of compounds against human SIRT5, SIRT1, SIRT2, and SIRT3 were measured using a fluorescence-based assay as reported [35,36]. For details, see Supporting Information.
Molecular Docking.
Compounds were docked onto the active site of SIRT5 (PDB ID: 2NYR) using GOLD (Genetic Optimization for Ligand Docking) software package, version 4.0 (Cambridge Crystallographic Data Centre, Cambridge, U.K.) [40]. Prior to docking, compounds were ionized, energy-minimized, and ten possible conformers were generated with Omega from OpenEye software using the MMFF94S force field [41]. GOLD uses a genetic algorithm to explore the conformational space of a compound inside the binding site of a protein [42,43]. Docking studies were performed using the standard default settings with 100 genetic algorithm (GA) runs on each molecule. For each of the 100 independent GA runs, a maximum of 100,000 operations was performed on a set of five groups with a population of 100 individuals. With respect to ligand flexibility, special care was taken by including options such as flipping of ring corners, amides, pyramidal nitrogens, secondary and tertiary amines, and rotation of carboxylate groups, as well as torsion angle distribution and post-process rotatable bonds as default. The annealing parameters were used as default cutoff values of 3.0 Å for hydrogen bonds and 4.0 Å for van der Waals interactions. Hydrophobic fitting points were calculated to facilitate the correct starting orientation of the compound for docking by placing the hydrophobic atoms appropriately in the corresponding areas of the active site. When the top three solutions attained root-mean-square deviation (rmsd) values within 1.5 Å, docking was terminated. GOLD-Score, a scoring function within the software, is a dimensionless fitness value that takes into account the intra- and intermolecular hydrogen bonding interaction energy, van der Waals energy, and ligand torsion energy [42,43]. Protein-ligand interactions fingerprinting was generated using PyPLIF, a python program, which converts three-dimensional (3D) protein-ligand interactions into one-dimensional (1D) bitstrings [44]. There are seven types of possible interactions between ligand and every residue: (i) Apolar (van der Waals), (ii) aromatic face to face, (iii) aromatic edge to face, (iv) hydrogen bond (protein as hydrogen bond donor), (v) hydrogen bond (protein as hydrogen bond acceptor), (vi) electrostatic interaction (protein positively charged), and (vii) electrostatic interaction (protein negatively charged) [44]. Fingerprinting then clustered and plotted using matplotlib and seaborn (python packages for heat map, clustering, and plotting).
Supplementary Material
Acknowledgments
Supported by NIH awards R01CA253986 (to DL/NN) and R01GM101171 (to DL), DOD awards NF170044 (to DL/NN) and CA190267 (to DL), and Melanoma Research Alliance Team Science Award #396513.
Abbreviations
- SIRT5
Sirtuin 5
- SAR
structure activity relationship
- LCMS
liquid chromatography–mass spectrometry
- DMF
N,N-dimethylformamide
- DCM
dichloromethane
- EtOH
ethanol
- DIPEA
diisopropylethylamine
- TFA
trifluoroacetic acid
- TEA
triethylamine
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2022.114623.
Data availability
Data will be made available on request.
References
- [1].Tanny JC, Dowd GJ, Huang J, Hilz H, Moazed D, An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing, Cell 99 (1999) 735–745, 10.1016/S0092-8674(00)81671-2. [DOI] [PubMed] [Google Scholar]
- [2].Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, Wolberger C, Prolla TA, Weindruch R, Alt FW, Guarente L, SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic β cells, Cell 126 (2006) 941–954, 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- [3].Meter MV, Mao Z, Gorbunova V, Seluanov A, Repairing split ends: SIRT6, mono-ADP ribosylation and DNA repair, Aging 3 (2011) 829–835, 10.18632/aging.100389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Frye RA, Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins, Biochem. Biophys. Res. Commun 273 (2000) 793–798. 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- [5].Giblin W, Skinner ME, Lombard DB, Sirtuins: guardians of mammalian healthspan, Trends Genet 30 (2014) 271–286, 10.1016/j.tig.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kumar S, Lombard DB, Mitochondrial sirtuins and their relationships with metabolic disease and cancer, Antioxidants Redox Signal 22 (2015) 1060–1077, 10.1089/ars.2014.6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, Cerione RA, Auwerx J, Hao Q, Lin H, SIRT5 is a NAD-dependent protein lysine demalonylase and desuccinylase, Science 334 (2011) 806–809, 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Parihar P, Solanki I, Mansuri ML, Parihar MS, Mitochondrial sirtuins: emerging roles in metabolic regulations, energy homeostasis and diseases, Exp. Gerontol 61 (2015) 130–141, 10.1016/j.exger.2014.12.004. [DOI] [PubMed] [Google Scholar]
- [9].Nakagawa T, Lomb DJ, Haigis MC, Guarente L, SIRT5 deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle, Cell 137 (2009) 560–570, 10.1016/j.cell.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BMM, Skinner ME, Lombard DB, Zhao Y, SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways, Mol. Cell 50 (2013) 919–930, 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nishida Y, Rardin MJ, Carrico C, He W, Sahu AK, Gut P, Najjar R, Fitch M, Hellerstein M, Gibson BW, Verdin E, SIRT5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target, Mol. Cell 59 (2015) 321–332, 10.1016/j.molcel.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Polletta L, Vernucci E, Carnevale I, Arcangeli T, Rotili D, Palmerio S, Steegborn C, Nowak T, Schutkowski M, Pellegrini L, Sansone L, Villanova L, Runci A, Pucci B, Morgante E, Fini M, Mai A, Russo MA, Tafani M, SIRT5 regulation of ammonia-induced autophagy and mitophagy, Autophagy 11 (2015) 253–270, 10.1080/15548627.2015.1009778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gertz M, Steegborn C, Function and regulation of the mitochondrial sirtuin isoform SIRT5 in mammalia, Biochim. Biophys. Acta 1804 (2010) 1658–1665, 10.1016/j.bbapap.2009.09.011. [DOI] [PubMed] [Google Scholar]
- [14].Lin Z-F, Xu H-B, Wang J-Y, Lin Q, Ruan Z, Liu F-B, Jin W, Huang H-H, Chen X, SIRT5 desuccinylates and activates SOD1 to eliminate ROS, Biochem. Biophys. Res. Commun 441 (2013) 191–195, 10.1016/j.bbrc.2013.10.033. [DOI] [PubMed] [Google Scholar]
- [15].Jing H, Lin H, Sirtuins in epigenetic regulation, Chem. Rev 115 (2015) 2350–2375, 10.1021/cr500457h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bringman-Rodenbarger LR, Guo AH, Lyssiotis CA, Lombard DB, Emerging roles for SIRT5 in metabolism and cancer, Antioxidants Redox Signal 28 (2018) 677–690, 10.1089/ars.2017.7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lu W, Zuo Y, Feng Y, Zhang M, SIRT5 facilitates cancer cell growth and drug resistance in non-small cell lung cancer, Tumor Biol 35 (2014) 10699–10705, 10.1007/s13277-014-2372-4. [DOI] [PubMed] [Google Scholar]
- [18].Giblin W, Bringman-Rodenbarger L, Guo AH, Kumar S, Monovich AC, Mostafa AM, Skinner ME, Azar M, Mady AS, Chung CH, Kadambi N, Melong K-A, Lee H-J, Zhang L, Sajjakulnukit P, Trefely S, Varner EL, Iyer S, Wang M, Wilmott JS, Soyer HP, Sturm RA, Pritchard AL, Andea AA, Scolyer RA, Stark MS, Scott DA, Fullen DR, Bosenberg MW, Chandrasekaran S, Nikolovska-Coleska Z, Verhaegen ME, Snyder NW, Rivera MN, Osterman AL, Lyssiotis CA, Lombard DB, The deacylase SIRT5 supports melanoma viability by influencing chromatin dynamics, J. Clin. Invest 131 (2021), 138926, 10.1172/jci138926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yang X, Wang Z, Li X, Liu B, Liu M, Liu L, Chen S, Ren M, Wang Y, Yu M, Wang B, Zou J, Zhu W-G, Yin Y, Gu W, Luo J, SHMT2 desuccinylation by SIRT5 drives cancer cell proliferation, Cancer Res 78 (2018) 372–386, 10.1158/0008-5472.CAN-17-1912. [DOI] [PubMed] [Google Scholar]
- [20].Wang Y-Q, Wang H-L, Xu J, Tan J, Fu L-N, Wang J-L, Zou T-H, Sun D-F, Gao Q-Y, Chen Y-X, Fang J-Y, Sirtuin5 contributes to colorectal carcinogenesis by enhancing glutaminolysis in a deglutarylation-dependent manner, Nat. Commun 9 (2018) 545, 10.1038/s41467-018-02951-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shi L, Yan H, An S, Shen M, Jia W, Zhang R, Zhao L, Huang G, Liu J, SIRT5-mediated deacetylation of LDHB promotes autophagy and tumorigenesis in colorectal cancer, Mol. Oncol 13 (2019) 358–375, 10.1002/1878-0261.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bai L, Yang Z-X, Ma P-F, Liu J-S, Wang D-S, Yu H-C, Overexpression of SLC25A51 promotes hepatocellular carcinoma progression by driving aerobic glycolysis through activation of SIRT5, Free Radic. Biol. Med 182 (2022) 11–22, 10.1016/j.freeradbiomed.2022.02.014. [DOI] [PubMed] [Google Scholar]
- [23].He B, Du J, Lin H, Thiosuccinyl peptides as SIRT5-specific inhibitors, J. Am. Chem. Soc 134 (2012) 1922–1925, 10.1021/ja2090417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Roessler C, Nowak T, Pannek M, Gertz M, Nguyen GTT, Scharfe M, Born I, Sippl W, Steegborn C, Schutkowski M, Chemical probing of the human sirtuin 5 active site reveals its substrate acyl specificity and peptide-based inhibitors, Angew. Chem., Int. Ed 53 (2014) 10728–10732, 10.1002/anie.201402679. [DOI] [PubMed] [Google Scholar]
- [25].Maurer B, Rumpf T, Scharfe M, Stolfa DA, Schmitt ML, He W, Verdin E, Sippl W, Jung M, Inhibitors of the NAD+-dependent protein desuccinylase and demalonylase SIRT5, ACS Med. Chem. Lett 3 (2012) 1050–1053, 10.1021/ml3002709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Suenkel B, Fischer F, Steegborn C, Inhibition of the human deacylase Sirtuin 5 by the indole GW5074, Bioorg. Med. Chem. Lett 23 (2013) 143–146, 10.1016/j.bmcl.2012.10.136. [DOI] [PubMed] [Google Scholar]
- [27].Yang L-L, He Y-Y, Chen Q-L, Qian S, Wang Z-Y, Design and synthesis of new 9-substituted norharmane derivatives as potential SIRT5 inhibitors, J. Heterocycl. Chem 54 (2017) 1457–1466, 10.1002/jhet.2732. [DOI] [Google Scholar]
- [28].Liu S, Ji S, Yu Z-J, Wang H-L, Cheng X, Li W-J, Jing L, Yu Y, Chen Q, Yang L-L, Li G-B, Wu Y, Structure-based discovery of new selective small-molecule sirtuin 5 inhibitors, Chem. Biol. Drug Des 91 (2018) 257–268, 10.1111/cbdd.13077. [DOI] [PubMed] [Google Scholar]
- [29].Yang L, Ma X, He Y, Yuan C, Chen Q, Li G, Chen X, Sirtuin 5: a review of structure, known inhibitors and clues for developing new inhibitors, Sci. China, Life Sci 60 (2017) 249–256, 10.1007/s11427-016-0060-7. [DOI] [PubMed] [Google Scholar]
- [30].Rajabi N, Auth M, Troelsen KR, Pannek M, Bhatt DP, Fontenas M, Hirschey MD, Steegborn C, Madsen AS, Olsen CA, Mechanism-based inhibitors of the human sirtuin 5 deacylase: structure-activity relationship, biostructural, and kinetic insight, Angew. Chem., Int. Ed 56 (2017) 14836–14841, 10.1002/anie.201709050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kalbas D, Liebscher S, Nowak T, Meleshin M, Pannek M, Popp C, Alhalabi Z, Bordusa F, Sippl W, Steegborn C, Schutkowski M, Potent and selective inhibitors of human sirtuin 5, J. Med. Chem 61 (2018) 2460–2471, 10.1021/acs.jmedchem.7b01648. [DOI] [PubMed] [Google Scholar]
- [32].Glas C, Dietschreit JCB, Wossner N, Urban L, Ghazy E, Sippl W, Jung M, Ochsenfeld C, Bracher F, Identification of the subtype-selective Sirt5 inhibitor balsalazide through systematic SAR analysis and rationalization via theoretical investigations, Eur. J. Med. Chem 206 (2020), 112676, 10.1016/j.ejmech.2020.112676. [DOI] [PubMed] [Google Scholar]
- [33].Yang F, Su H, Deng J, Mou L, Wang H, Li R, Dai Q-Q, Yan Y-H, Qian S, Wang Z, Li G-B, Yang L, Discovery of new human Sirtuin 5 inhibitors by mimicking glutaryl-lysine substrates, Eur. J. Med. Chem 225 (2021), 113803, 10.1016/j.ejmech.2021.113803. [DOI] [PubMed] [Google Scholar]
- [34].Jiang Y, Zheng W, Cyclic tripeptide-based potent and selective human SIRT5 inhibitors, Med. Chem 16 (2020) 358–367, 10.2174/1573406415666190603101937. [DOI] [PubMed] [Google Scholar]
- [35].Madsen AS, Olsen CA, Substrates for efficient fluorometric screening employing the NAD-dependent sirtuin 5 lysine deacylase (KDAC) enzyme, J. Med. Chem 55 (2012) 5582–5590, 10.1021/jm300526r. [DOI] [PubMed] [Google Scholar]
- [36].Wang H-L, Liu S, Yu Z-J, Wu C, Cheng L, Wang Y, Chen K, Zhou S, Chen Q, Yu Y, Li G-B, Interactions between sirtuins and fluorogenic small-molecule substrates offer insights into inhibitor design, RSC Adv 7 (2017) 36214–36222, 10.1039/C7RA05824A. [DOI] [Google Scholar]
- [37].Schuetz A, Min J, Antoshenko T, Wang C-L, Allali-Hassani A, Dong A, Loppnau P, Vedadi M, Bochkarev A, Sternglanz R, Plotnikov AN, Structural basis of inhibition of the human NAD+-dependent deacetylase SIRT5 by suramin, Structure 15 (2007) 377–389, 10.1016/j.str.2007.02.002. [DOI] [PubMed] [Google Scholar]
- [38].Zang W, Hao Y, Wang Z, Zheng W, Novel thiourea-based sirtuin inhibitory warheads, Bioorg. Med. Chem. Lett 25 (2015) 3319–3324, 10.1016/j.bmcl.2015.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hirsch BM, Hao Y, Li X, Wesdemiotis C, Wang Z, Zheng W, A mechanism-based potent sirtuin inhibitor containing Nε-thiocarbamoyl-lysine (TuAcK), Bioorg. Med. Chem. Lett 21 (2011), 10.1016/j.bmcl.2011.06.069, 4753–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].GOLD Software Package, Version 5.3, Cambridge Crystallographic Data Centre, Cambridge, U.K., https://www.ccdc.cam.ac.uk. [Google Scholar]
- [41].OMEGA 3.1.2.2: OpenEye Scientific Software, Santa Fe, NM. http://www.eyesopen.com.
- [42].Jones G, Willett P, Docking small-molecule ligands into active sites, Curr. Opin. Biotechnol 6 (1995) 652–656, 10.1016/0958-1669(95)80107-3. [DOI] [PubMed] [Google Scholar]
- [43].Jones G, Willett P, Glen RC, Leach AR, Taylor R, Development and validation of a genetic algorithm for flexible docking, J. Mol. Biol 267 (1997) 727–748, 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- [44].Radifar M, Yuniarti N, Istyastono EP, PyPLIF: python-based protein-ligand interaction fingerprinting, Bioformation 9 (2013) 325–328, 10.6026/97320630009325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.