

Entry for the Table of Contents
Chiral γPNAs containing miniPEG side-chains can invade any sequence of double helical B-form DNA, with the recognition occurring through direct Watson-Crick base-pairing.
Keywords: DNA recognition, γPNA, strand invasion, Watson-Crick base-pairing
Graphical Abstract

Development of general principles for designing molecules to bind sequence-specifically to double-stranded DNA (dsDNA) has been a long-sought goal of bioorganic chemistry and molecular biology.[1–3] Pursuit of this goal, in the past, has generally been focused on the minor and major grooves—in large part, because of the ease of accessibility of the chemical groups that reside on these external parts of the double helix and the precedence for their recognition in nature.[2] It was long recognized that while Watson-Crick (W-C) base-pairing provides a more direct and specific means for establishing sequence-specific interactions with nucleic acid biopolymers such as DNA and RNA, it would be difficult to do so with intact double helical DNA because of the preexisting base-pairs.[4] This effort has so far led to the development of several major classes of antigene molecules, with the like of triplex-forming oligonucleotides,[5–7] minor-groove binding polyamides,[8–11] and major-groove binding zinc-finger peptides.[12–16] While they can be designed to bind sequence-specifically to dsDNA, there are still remaining issues with sequence selection, specificity and/or target length that have not yet been completely resolved[8, 13, 17–19]—although some progress has been made in recent years.
Over the past two decades, peptide nucleic acids (PNAs)[20]—a particular class of nucleic acid mimics comprised of a pseudopeptide backbone (Scheme 1A)—have been shown to be capable of invading dsDNA.[21] This finding is significant because, contrary to the traditional belief, it demonstrates that DNA double helix is relatively dynamic at physiological temperatures and that W-C base-pairing interactions can be established with intact dsDNA. Though promising as antigene reagents, because of the specificity of recognition and generality in sequence design, PNA binding is presently limited to mostly homopurine[22] and homopyrimidine targets.[23] Mixed-sequence PNAs have been shown to be capable of invading topologically constrained supercoiled plasmid DNA,[24–27] conformationally perturbed regions of genomics DNA[28] and duplex termini;[29] however, they are unable to invade the interior regions of double helical B-form DNA (B-DNA)—the most stable form of DNA double helix. “Tail-clamp”[30, 31] and “double-duplex invasion”[32] strategies have subsequently been developed, enabling mixed-sequence PNAs to invade B-DNA, but they are not without limitations.[33]
Scheme 1.

Chemical structures of (A) PNA, (B) L-alanine-derived γPNA (S-MeγPNA), and (C) (R)-MiniPEG-containing γPNA (R-MPγPNA). See ref. [36] for the synthesis of R-MPγPNA monomers; the methyl ether protecting group of miniPEG side-chain is removed in the final cleavage/deprotection step of oligomer synthesis.
Recently we showed that mixed-sequence PNAs, when preorganized into a right-handed helix by installing an (S)-Me stereogenic center at the γ-backbone (Scheme 1B), can invade B-DNA.[34] However, all of our studies so far are limited to a few selected sequences due to the poor water solubility and propensity of these first-generation S-MeγPNAs to aggregate and adhere to surfaces and other macromolecules, including DNA, in a nonspecific manner. This problem is exacerbated with increasing G/C content, making it difficult to characterize their invasion properties over a wide concentration range and broad sequence space. To rectify this problem, we replaced the methyl group with diethylene glycol (or ‘MiniPEG’, MP [Scheme 1C]). MP was chosen because of its relatively small size and hydrophilic nature, and because it has been shown to be effective in imparting water solubility and biocompatibility to non-biological systems.[35] Our initial study revealed that inclusion of MP indeed significantly improves the water solubility and biocompatibility of PNAs,[36] but whether these MP-modified γPNAs (R-MPγPNAs) can invade B-DNA has not yet been determined. Herein we show that R-MPγPNAs can invade any sequence of double helical B-DNA, ranging from 0 to 100% G/C content, with the recognition occurring in a highly sequence specificity through Watson-Crick base-pairing.
To assess binding, we performed gel-shift assays, comparing R-MPγPNA1 to that of S-MeγPNA1 and PNA1 (Table 1). A 291-bp DNA fragment containing an internal binding site was chosen as a target (Figure 1A, PM) because initial study showed that it is short enough to impart electrophoretic separation upon binding of γPNAs and long enough not to interfere with the invasion efficiency (Figure S1, Supporting Information). The target was incubated with different concentrations of oligos in 10 mM sodium phosphate (NaPi) buffer for 16 hrs, followed by electrophoretic separation and staining with SYBR-Gold for visualization. Our result showed that not only could R-MPγPNA1 bind dsDNA, but it did so with greater efficiency than S-MeγPNA, as evidenced by the relative intensity of the retarded bands (Figure 1B; compare lanes 5–7 to lanes 8–10, respectively). The dissociation constant (Kd) of R-MPγPNA1 to PM was determined to be 3.7 (±0.2) x 10−7 M. However, under identical conditions, no binding was observed for the achiral PNA (lanes 2–4). This result is consistent with the earlier finding.[37] Even at a high (100:1) oligo-to-DNA strand ratio, at which point all DNA incubated with S-MeγPNA (lane 7) and PNA1 (lane 4) had disappeared, presumably due to aggregation and nonspecific binding of S-MeγPNA and PNA to DNA, the presence of large excess R-MPγPNA1 had no effect on the mobility or intensity of the retarded band (lane 10). This result highlights the importance of MP at the γ-backbone in suppressing aggregation and nonspecific binding.
Table 1.
Oligonucleotides employed in this study.
| Oligonucleotide | Sequence | Length | G/C [%] |
|---|---|---|---|
|
| |||
| PNA1 | H-K-GACCACAGATCTAAG-K-NH2 | 15 | 47 |
| (S)-MeγPNA1 | H-K-GACCACAGATCTAAG-K-NH2 | 15 | 47 |
| (R)-MPγPNA1 | H-K-GACCACAGATCTAAG-K-NH2 | 15 | 47 |
| (R)-MPγPNA2a | H-K-CAGATCTAAG-K-NH2 | 10 | 40 |
| (R)-MPγPNA2b | H-K-CCACAGATCTAAG-K-NH2 | 13 | 46 |
| (R)-MPγPNA2c[a] | H-K-GACCACAGATCTAAG-K-NH2 | 15 | 47 |
| (R)-MPγPNA2d | H-K-GAGACCACAGATCTAAG-K-NH2 | 17 | 47 |
| (R)-MPγPNA2e | H-K-TATGAGACCACAGATCTAAG'K-NH2 | 20 | 40 |
| (R)-MPγPNA3 | H-K-ATTTAATAATATAAT-K-NH2 | 15 | 0 |
| (R)-MPγPNA4 | H-K-CTAAACTAATGTAAT-K-NH2 | 15 | 20 |
| (R)-MPγPNA5 | H-K-GATTACATAGATGTC-K-NH2 | 15 | 33 |
| (R)-MPγPNA6 | H-K-TGCGTGAGCATCAGTK-NH2 | 15 | 53 |
| (R)-MPγPNA7 | H-K-CAGTGTCACGCACGG-K-NH2 | 15 | 67 |
| (R)-MPγPNA8 | H-K-CGGACGCAG GCTCGC K-NH2 | 15 | 80 |
| (R)-MPγPNA9 | H-K-CGCCCGCCGCCCGCC-K-NH2 | 15 | 100 |
| (R)-MPγPNA8b | H-K-GCGAGCCTGCGTCCG-K-NH2 | 15 | 80 |
| (R)-MPγPNA8bm | H-K-CGGACGCACGCTCGC-K-NH2 | 15 | 80 |
(R)-MPγPNA2c and (R)-MPγPNA1 are the same oligonucleotides, and (R)-MPγPNAB and l-lysine. (R)-MPγPNABb are complementary to one another; K: L-lysine.
Figure 1.

Effects of backbone modification and target sequence on binding efficiency. (A) Schematic of the DNA targets. Results of the gel-shift assays following incubation of 0.1 μM DNA containing (B) PM binding site with different concentrations of PNA1, S-MeγPNA1, and R-MPγPNA1, and (C) PM, MM1, MM2, and MM3 binding sites with 2 μM R-MPγPNA1 in 10 mM NaPi buffer at 37 °C for 16 hrs. The samples were separated on 10%-nondenaturing PAGE gel and stained with SYBR-Gold.
Formation of the complex, in this case, occurred in a highly sequence-specific manner. No incubation of DNA, whether containing a single-base mismatch in the middle (MM1) or towards one end (MM2 or MM3), resulted in formation of the retarded band (Figure 1C). This finding was further corroborated by DNase-I footprinting, which revealed protection at the expected binding site (Figure 2A). The footprinting pattern was only observed with the perfect-match (lanes 2–4) and not with the single-base mismatches (lanes 5 and 6). The binding mode of R-MPγPNA1 was confirmed by diethyl pyrocarbonate (DEPC)-chemical probing assay (Figure 2B),[38, 39] which revealed selective cleavage at the adenine and, to a smaller extent, guanine sites on the homologous DNA strand, directly across from the binding site, following piperidine treatment. This result is consistent with binding occurring through a strand invasion mechanism.
Figure 2.
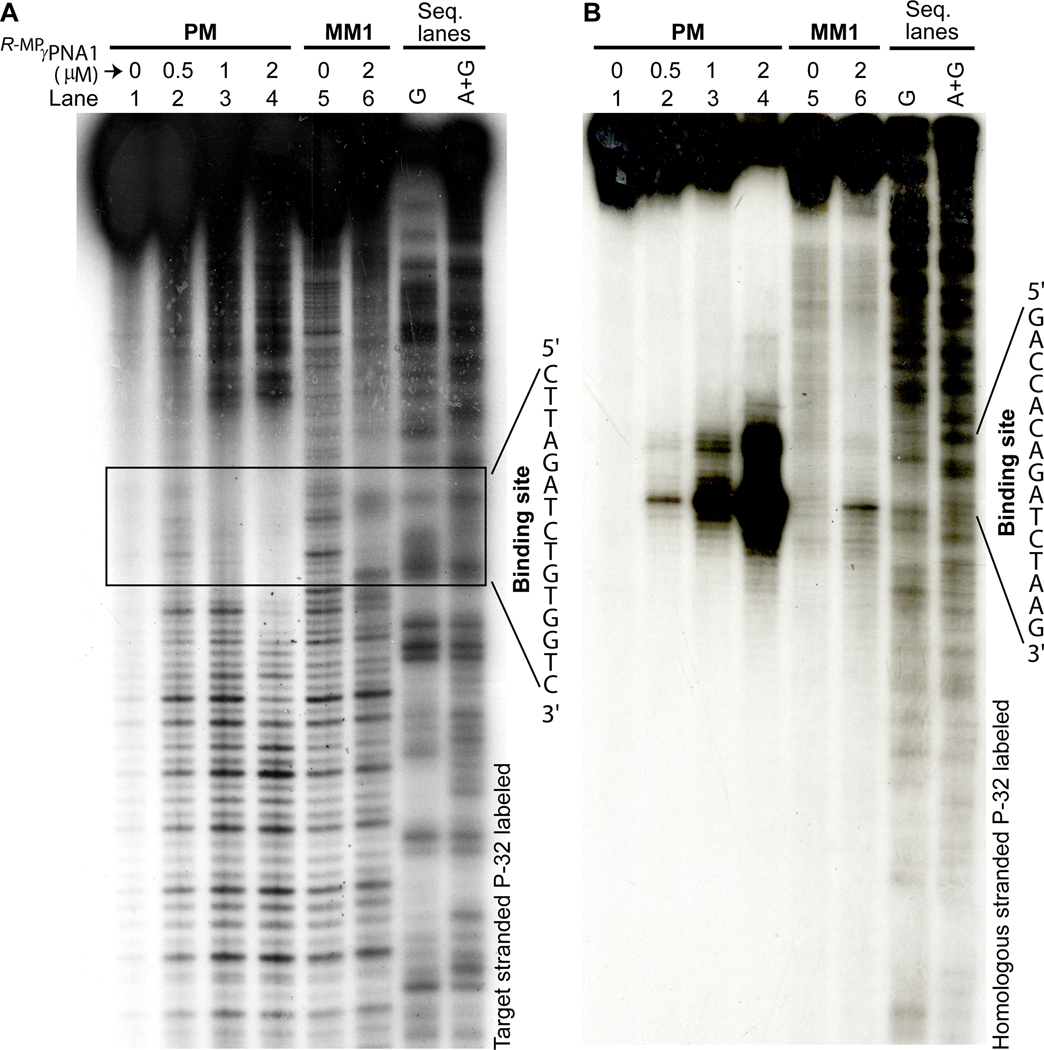
Results of (A) DNase-I footprinting and (B) DEPC-chemical probing assays following incubation of 171-bp DNA fragments containing PM and MM1 binding sites with different concentrations of R-MPγPNA1 in 10 mM NaPi buffer at 37 °C for 16 hrs. In (A) the target strand was labeled with P-32, whereas in (B) it was the homologous strand—both at the 3’-end.
Next, we determined the effects of temperature and ionic strength on the invasion efficiency. Our result showed that the invasion efficiency is strongly dependent on temperature (Figure 3A). This was an expected finding, because temperature has a direct effect on the rate of base-pair breathing (or opening). Strand invasion was extremely inefficient at 4 °C, in which case less than 10% of the invasion complex was formed after a 24 hr-incubation. The rate of strand invasion dramatically increased at 37 °C, reaching equilibrium within ~ 2 hrs. The rate obeys peudo-first order kinetics, with kps, 37 °C = 0.025 min−1 (Figure S2)— roughly 4 times higher than that of S-MeγPNA1 for the same target.[34] Further increase in temperature resulted in further increase in the rate of strand invasion, but the rate increase was less pronounced from 37 to 50 °C than from 22 to 37 °C. This result indicates that at physiological temperatures, DNA double helix is sufficiently dynamic to permit strand invasion provided that the required binding free energy can be met. Similarly, we found that the rate of strand invasion was strongly dependent on ionic strength (Figure 3B). No binding was observed at 100% physiologically simulated ionic strength (2 mM MgCl2, 150 mM KCl, 10 mM sodium phosphate; pH 7.4)[40] after a 16 hr-incubation at 37 °C. The lack of productive binding, in this case, is not due to the lack accessibility—the inability of R-MPγPNA1 to gain access to the nucleobase targets, but rather due to the lack of binding free energy—as demonstrated in a recent study.[41] Once bound, the complex dissociated rather slowly, with a half-life of ~ 2 hrs (Figure 3B, Inset). This result indicates that it should be possible to perform in vitro experiments under physiologically simulated conditions with the pre-bound complex, as long as the experiments can be carried out within this time frame.
Figure 3.

Effects of (A) temperature and (B) ionic strength on the invasion efficiency. The amounts of fraction bound was determined by gel-shift assays following incubation of 0.1 μM DNA containing PM binding site with 2 μM of R-MPγPNA1 in (A) 10 mM NaPi buffer for 16 hrs at the indicated temperatures, and (B) different percentages of physiological ionic strength (2 mM MgCl2, 150 mM KCl, 10 mM NaPi) at 37 °C for 16 hrs, followed by electrophoretic separation and SYBR-Gold staining. (B) Inset: Profile of the R-MPγPNA1•DNA complex dissociating as a function of time after reconstituting the sample with 100% physiological ionic strength. t1/2 is defined as the time it took to reach 50% binding.
To assess the generality of R-MPγPNA binding, we determined the effects of oligo length and sequence composition on the invasion efficiency. Our results showed that although a 10mer, R-MPγPNA2a, was unable to invade dsDNA, addition of just three nucleotides (R-MPγPNA2b) restored the binding (Figure 4A, lane 3), and the efficiency gradually increased with increasing oligo length (lanes 4 to 6). However, we do not expect this trend to continue indefinitely, because, at some point, intermolecular R-MPγPNA interactions would predominate, resulting in a gradual decrease in the invasion efficiency with further increase in oligo length. We do not consider the failure of short MPγPNAs to invade dsDNA to be a detriment to most biological applications, because targeting a unique site within a mammalian genome would statistically require a recognition site of ~ 17 nucleotides in length,[42] which is within the recognition repertoire of R-MPγPNAs, and, if necessary, these oligos could be further chemically modified to provide the necessary binding free energy.[37, 41, 43]
Figure 4.

Results of the gel-shift assays showing the effects of oligo (A) size and (B) sequence composition on the invasion efficiency. Gel-shift assays were performed under identical conditions as stated in the Figure 1 caption at a DNA:R-MPγPNA ratio of 20:1. The sequence of the R-MPγPNA oligos and corresponding DNA targets are shown in Table 1 and Table S1, respectively.
Interestingly, we observed that unlike PNAs, even with the latest design which still requires a minimum of 40% A/T content,[33] R-MPγPNAs can invade any sequence of B-DNA, ranging from 0 to 100% G/C content (Figure 4B). Formation of these complexes occurred in a highly-sequence specific manner. Neither cross-incubation of these oligos with the other DNA targets (Figure S3), nor incubation of the G/C-rich oligos (R-MPγPNA8, R-MPγPNA8b or R-MPγPNA8bm) with DNA containing single-base mismatches (Figure S4), resulted in formation of the invasion complex. However, it should be emphasized as in the case with any intermolecular recognition event, precaution must be exercised in designing the nucleobase sequence to minimize self-hybridization because of the strong R-MPγPNA-R-MPγPNA interaction, in order to achieve optimum invasion.
In summary, we have shown that (R)-MiniPEG-containing γPNAs, 13 to 20 nucleotides in length, can invade any sequence of double helical B-DNA. Recognition, in this case, occurs in a highly sequence-specific manner (vide infra) through Watson-Crick base-pairing. The crystal structure of a S-MeγPNA-DNA duplex has been determined,[44] showing the methyl groups projecting outward toward the solvent. We do not expect the MP side-chains to behave any differently. The main advantages of MP over Me are improvements in water solubility and biocompatibility, and suppression of aggregation and nonspecific binding. Though R-MPγPNA binding is presently limited to relatively low ionic strengths, like all other strand invading PNAs with the exception of triplex binding, this is not because they are unable to gain access to the nucleobase targets at physiological conditions (ionic strengths and temperatures), but rather because they are unable to compete with the native complementary DNA strand. This is predominantly a thermodynamic, rather than a kinetic, issue that could be resolved through molecular design. We have already demonstrated certain aspects of this design through covalent attachment of DNA intercalating agents[37] and replacement of natural nucleobases with synthetic analogues.[41] R-MPγPNAs are attractive, as compared to the other classes of antigene reagents that have been developed to date, because they are relatively easy to synthesize and they hybridize to their targets in a highly sequence specific manner in accordance with the simple rules of Watson-Crick base-pairing. Such antigene reagents could be employed in a number of biotechnology and genomic applications, including recombinant DNA, [27] genome mapping[45] and chromatin capture,[46] as well as in vivo, including gene regulation[28, 47] and gene correction.[48]
Supplementary Material
[**].
This work was funded by the National Institutes of Health (GM076251), the National Science Foundation (CHE-1012467), and the DSF Charitable Foundation.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- [1].Thuong NT, Helene C, Angew. Chem. Int. Ed 1993, 32, 666. [Google Scholar]
- [2].Nielsen PE, Chem. Eur. J 1997, 3, 505. [Google Scholar]
- [3].Denison C, Kodadek T, Chem. Biol 1998, 5, R129. [DOI] [PubMed] [Google Scholar]
- [4].Frank-Kamenetskii MD, Nature 1987, 328, 17. [DOI] [PubMed] [Google Scholar]
- [5].Moser HE, Dervan PB, Science 1987, 238, 645. [DOI] [PubMed] [Google Scholar]
- [6].Sun JS, Helene C, Curr. Opin. Struct. Biol 1993, 3, 345. [DOI] [PubMed] [Google Scholar]
- [7].Giovannangeli C, Helene C, Antisense Nucleic Acid Drug Dev. 1997, 7, 413. [DOI] [PubMed] [Google Scholar]
- [8].Kielkopf CL, White S, Szewczyk JW, Turner JM, Baird EE, Dervan PB, Rees DC, Science 1998, 282, 111. [DOI] [PubMed] [Google Scholar]
- [9].Gottesfeld JM, Neely L, Trauger JW, Baird EE, Dervan PB, Nature 1997, 387, 202. [DOI] [PubMed] [Google Scholar]
- [10].White S, Szewczyk JW, Turner JM, Baird EE, Dervan PB, Nature 1998, 391, 468. [DOI] [PubMed] [Google Scholar]
- [11].O’Hare CC, Mack D, Tandon M, Sharma SK, Lown JW, Kopka ML, Dickerson RE, Hartley JA, Proc. Nat. Acad. Sci. U.S.A 2002, 99, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Greisman HA, Pabo CO, Science 1997, 275, 657. [DOI] [PubMed] [Google Scholar]
- [13].Jantz D, Amann BT, Gatto GJ, Berg JM, Chem. Rev 2004, 104, 789. [DOI] [PubMed] [Google Scholar]
- [14].Beerli RR, Barbas CFI, Nat. Biotech 2002, 20, 135. [DOI] [PubMed] [Google Scholar]
- [15].Moore M, Klug A, Choo Y, Proc. Natl. Acad. Sci. USA 2001, 98, 1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Garvie CW, Wolberger C, Molecular Cell 2001, 8, 937. [DOI] [PubMed] [Google Scholar]
- [17].Gowers DM, Fox KR, Nucleic Acids Res. 1999, 27, 1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kelly JJ, Baird EE, Dervan PB, Proc. Natl. Acad. Sci. U.S.A 1996, 93, 6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kielkopf CL, Baird EE, Dervan PB, Rees DC, Nat. Struct. Biol 1998, 5, 104. [DOI] [PubMed] [Google Scholar]
- [20].Nielsen PE, Egholm M, Berg RH, Buchardt O, Science 1991, 254, 1497. [DOI] [PubMed] [Google Scholar]
- [21].Nielsen PE, Acc. Chem. Res 1999, 32, 624. [Google Scholar]
- [22].Egholm M, Nielsen PE, Buchardt O, Berg RH, J. Am. Chem. Soc 1992, 114, 9677. [Google Scholar]
- [23].Nielsen PE, Christensen L, J. Am. Chem. Soc 1996, 118, 2287. [Google Scholar]
- [24].Bentin T, Nielsen PE, Biochemistry 1996, 35, 8863. [DOI] [PubMed] [Google Scholar]
- [25].Smulevitch SV, Simmons CG, Norton JC, Wise TW, Corey DR, Nat. Biotechnol 1996, 14, 1700. [DOI] [PubMed] [Google Scholar]
- [26].Zhang X, Ishihara T, Corey DR, Nucleic Acids Res. 2000, 28, 3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Komiyama M, Aiba Y, Yamamoto Y, Sumaoka J, Nat. Protoc 2008, 3, 655. [DOI] [PubMed] [Google Scholar]
- [28].Janowski BA, Kaihatsu K, Huffman KE, Schwartz JC, Ram R, Hardy D, Mendelson CR, Corey DR, Nat. Chem. Biol 2005, 1, 210. [DOI] [PubMed] [Google Scholar]
- [29].Smolina IV, Demidov VV, Soldatenkov VA, Chasovskikh SG, Frank-Kameneteskii MD, Nucleic Acid Res. 2005, 33, e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bentin T, Larsen HJ, Nielsen PE, Biochemistry 2003, 42, 13987. [DOI] [PubMed] [Google Scholar]
- [31].Kaihatsu K, Shah RH, Zhao X, Corey DR, Biochemistry 2003, 42, 13996. [DOI] [PubMed] [Google Scholar]
- [32].Lohse J, Dahl O, Nielsen PE, Proc. Natl. Acad. Sci. U.S.A 1999, 96, 11804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Demidov VV, Protozanova E, Izvolsky KI, Price C, Nielsen PE, Frank-Kamenetskii MD, Proc. Natl. Acad. Sci. U.S.A 2002, 99, 5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].He G, Rapireddy S, Bahal R, Sahu B, Ly DH, J. Am. Chem. Soc 2009, 131, 12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Harris JM, Chess RB, Nat. Rev. Drug Discov 2003, 2, 214. [DOI] [PubMed] [Google Scholar]
- [36].Sahu B, Sacui I, Rapireddy S, Zanotti KJ, Bahal R, Armitage BA, Ly DH, J. Org. Chem 2011, 76, 5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rapireddy S, He G, Roy S, Armitage BA, Ly DH, J. Am. Chem. Soc 2007, 129, 15596. [DOI] [PubMed] [Google Scholar]
- [38].Scholten PM, Nordheim A, Nucleic Acids Res. 1986, 14, 3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Furlong JC, Lilley DMJ, Nucleic Acids Res. 1986, 14, 3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Santoro SW, Joyce GF, Proc. Nat. Acad. Sci. U.S.A 1997, 94, 4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rapireddy S, Bahal R, Ly DH, Biochemistry 2011, 50, 3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Liu Z, Venkatesh SS, Maley CC, BMC Genomics 2008, 9, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chenna V, Rapireddy S, Sahu B, Ausin C, Pedroso E, Ly DH, ChemBioChem 2008, 9, 2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yeh JI, Shivachev B, Rapireddy S, Crawford MJ, Gil RR, Du S, Madrid M, Ly DH, J. Am. Chem. Soc 2010, 132, 10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lin J, Lai Z, Aston C, Jing J, Anantharaman TS, Mishra B, White O, Daly MJ, Minton KW, Venter JC, Schwartz DC, Science 1999, 285, 1558. [DOI] [PubMed] [Google Scholar]
- [46].Dekker J, Rippe K, Dekker M, Kleckner N, Science 2002, 295, 1306. [DOI] [PubMed] [Google Scholar]
- [47].Mollegaard NE, Buchardt O, Egholm M, Nielsen PE, Proc. Natl. Acad. Sci. USA 1994, 91, 3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Vasquez KM, Narayanan L, Glazer PM, Science 2000, 290, 530. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.