

Abstract

Nicotinamide adenine nucleotide (NADH) is involved in many biologically relevant redox reactions, and the photochemical regeneration of its oxidized form (NAD+) under physiological conditions is of interest for combined photo- and biocatalysis. Here, we demonstrate that tri-anionic, water-soluble variants of typically very lipophilic iridium(III) complexes can photo-catalyze the reduction of an NAD+ mimic in a comparatively efficient manner. In combination with a well-known rhodium co-catalyst to facilitate regioselective reactions, these iridium(III) photo-reductants outcompete the commonly used [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) photosensitizer in water by up to 1 order of magnitude in turnover frequency. This improved reactivity is attributable to the strong excited-state electron donor properties and the good chemical robustness of the tri-anionic iridium(III) sensitizers, combined with their favorable Coulombic interaction with the di-cationic rhodium co-catalyst. Our findings seem relevant in the greater context of photobiocatalysis, for which access to strong, efficient, and robust photoreductants with good water solubility can be essential.
Short abstract
Sulfonated cyclometalated iridium(III) compounds perform well in biologically relevant photoreduction reactions, largely because they offer a good balance between excited-state donor and ground-state acceptor properties.
Introduction
The emerging field of photobiocatalysis combines enzymatic reactions with photocatalysis and represents an attractive approach to selective and mild chemical transformations using visible light as the energy source.1−7 Natural photosynthesis as well as many other biologically relevant processes rely on nicotinamide adenine nucleotide (NADH) as a mediator of redox equivalents, and its oxidized NAD+ form is typically recycled by an enzymatic reaction,8 thereby allowing the catalytic use of this precious co-factor.1,9,10 Given its widespread importance in many different biotransformations, the chemical,11,12 enzymatic,13−17 electrochemical,18,19 and photochemical reduction of NAD+ to NADH using readily available and cheap reductants has received considerable attention. In biological systems, the regioselective regeneration of the 1,4-NADH isomer from NAD+ is orchestrated enzymatically, whereas in artificial settings this is typically achieved with an organometallic rhodium co-catalyst derived from [Cp*Rh(bpy)Cl]+, where Cp* = C5Me5– and bpy = 2,2′-bipyridine.1,20 The catalytically active [Cp*Rh(bpy)H]+ species can be photochemically generated by the consecutive transfer of two electrons from a sensitizer (Figure 1) along with a proton transfer from a suitable source. Such catalytic cycles necessitate robust photosensitizers that can act as very strong electron donors under physiological conditions.1,2 To date, several different types of heterogeneous photocatalysts,21−24 including graphene-based materials,21,22 semiconductors, and Cd-based nanocrystals, have been used for this purpose.23,24 Among homogeneous photocatalysts, derivatives of [Ru(bpy)3]2+,25−29 Zn porphyrins,30−32 or xanthene dyes33 have been popular. Aside from eosin Y, which can coordinate to the abovementioned rhodium co-catalyst,33 thus enabling efficient intramolecular electron transfer within the resulting photosensitizer-rhodium dyad, the turnover frequencies (TOF) for both heterogeneous and homogeneous conditions have remained somewhat modest in many cases, with TOF values typically below 20 h–1 at room temperature.
Figure 1.
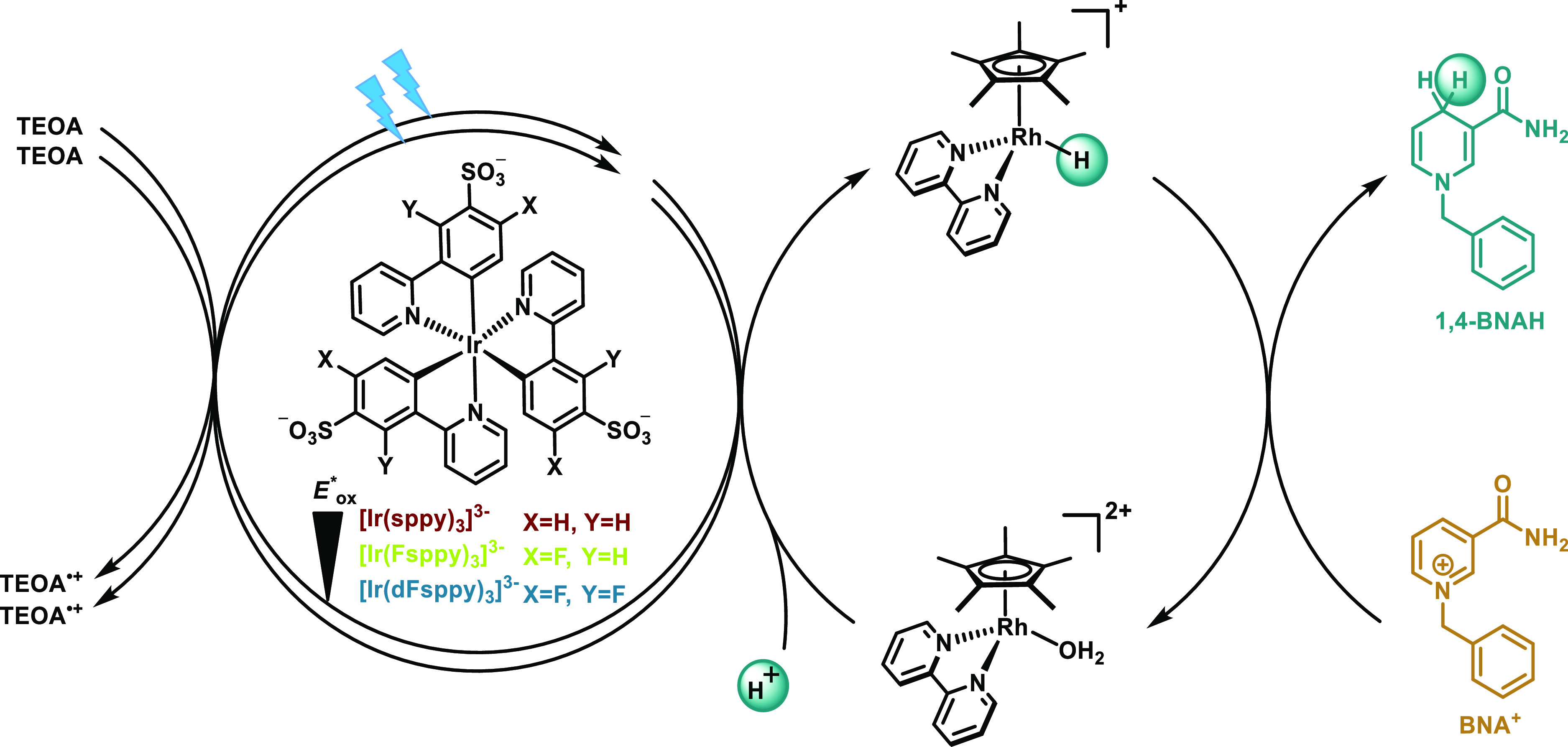
Photochemical regeneration of 1,4-BNAH using TEOA as a combined electron and proton source (oxidized form of TEOA),64 water-soluble iridium(III) photosensitizers, and a rhodium co-catalyst to accomplish the regioselective 1,4-reduction of BNA+.
Homoleptic tris(cyclometalated) iridium(III) complexes such as fac-[Ir(ppy)3] (ppy = 2-phenylpyridine) are promising photosensitizers for the regeneration of 1,4-NADH because they are strong and robust photoreductants with long-lived excited states.38−43 However, these charge-neutral homoleptic tris(cyclometalated) iridium(III) complexes are very lipophilic and therefore usually insoluble in water.44 Several different groups previously obtained water-soluble cyclometalated iridium(III) complexes, though commonly based on heteroleptic ligand frameworks,37,45−53 which are often less photo-reducing and less photorobust than the homoleptic versions.37,54,55 Our group recently developed a tri-sulfonated variant of fac-[Ir(ppy)3] with an excited-state oxidation potential (Eox*) of −1.89 V versus SCE in an aqueous phosphate buffer at pH 7 (Table 1, [Ir(sppy)3]3–).37,56,57 The excited-state electron donor properties turned out to be further tunable by ligand fluorination, resulting in [Ir(Fsppy)3]3– (Eox* = −1.85 V vs SCE, Table 1) and [Ir(dFsppy)3]3– (Eox* = −1.76 V vs SCE, Table 1). All three compounds (structures included in Figure 1) are very well soluble in water, and they seem to have suitably strong excited-state electron donor properties for the photochemical regeneration of 1,4-NADH from NAD+.36 1-Benzyl-1,4-dihydronicotinamide (1,4-BNAH, Figure 1) has emerged as an easy-to-handle and readily available small-molecule surrogate for NADH58−62 and hence we focused on the photochemical reduction of the oxidized form of this compound (BNA+) using our water-soluble iridium(III) compounds. Triethanolamine (TEOA) served as a sacrificial electron donor, and the abovementioned rhodium co-catalyst was used to facilitate regioselective 1,4-reduction.20 The co-catalyst enters the catalytic cycle in the form of [Cp*Rh(bpy)(H2O)]2+, and its two-electron reduction coupled to protonation leads to the catalytically active rhodium(III) hydride complex that reduces BNA+ to 1,4-BNAH.63
Table 1. Summary of Selected Electrochemical, Photochemical, and Photophysical Properties of the Photosensitizers Used in This Study.
Experimental Section
Unless otherwise indicated, the used chemicals were obtained commercially in high purity and were used as received. The investigated iridium(III) photosensitizers were available from some of our previous projects.36,57 The rhodium co-catalysts ([Cp*Rh(bpy)Cl]Cl)65 and BNACl66 were synthesized and characterized according to previously published procedures. 1,4-BNAH was synthesized by BNA+ reduction using Na2S2O4 and characterized according to a previous literature report.67 Ultrapure Millipore MilliQ water (specific resistance, 18.2 MΩ cm) was used for all spectroscopic measurements as well as for all photoinduced reductions. All solutions for optical spectroscopy and reductions were purged with argon (5.0, Pan Gas) for 5 min. To ensure stable pH conditions, a Tris-buffer (0.1 M, pH 8.8) or a phosphate buffer (0.1 M, pH 7) was used. Steady-state UV–vis absorption spectra were recorded with a Cary 5000 spectrometer (Varian). The luminescence lifetime and quenching measurements were performed on a LifeSpec II spectrometer (Edinburgh Instruments) using the time-correlated single-photon counting (TCSPC) technique. The excitation source was a 405 nm picosecond pulsed diode laser (ca. 60 ps pulse width). Laser flash photolysis was performed on an LP920-KS apparatus from Edinburgh Instruments with 420 nm laser pulsed excitation (14 mJ per pulse) using a frequency-tripled Nd/YAG laser (Quantel Brilliant, ca. 10 ns pulse width) equipped with an OPO from Opotek. The kinetics at a single wavelength were recorded using a photomultiplier tube.
Results and Discussion
To monitor the light-driven regeneration of the NADH mimic, a combination of UV–vis absorption and 1H NMR spectroscopy was employed. BNA+ and 1,4-BNAH both have distinct UV–vis absorption features that permit to follow the photoreaction progress over time. While both BNA+ and 1,4-BNAH show an absorption band below 300 nm (though with maxima at different wavelengths, inset of Figure 2), the reduced form (1,4-BNAH) displays a characteristic absorption ranging from 320 to 420 nm.26,30−32,68 Consequently, the photochemical formation of BNAH from BNA+ was monitored at 358 nm, complemented by 1H NMR experiments (see below) to confirm the 1,4-regioselectivity of the reaction. In an initial experiment performed with 1 mM BNA+, the [Ir(sppy)3]3– photosensitizer (10 μM) was used along with 0.1 mM [Cp*Rh(bpy)Cl]+ as a precursor complex to [Cp*Rh(bpy)(H2O)]2+, and TEOA (0.5 M) served as a sacrificial electron donor. Upon irradiation at 455 nm with the collimated output of an LED (1.1 W), the formation of BNAH was readily observed within 30 min (dashed upward arrow in Figure 2A). Since [Ir(sppy)3]3– and [Cp*Rh(bpy)(H2O)]2+ both have non-negligible extinction at 358 nm, the BNAH yields were estimated based on the observable optical density changes (before and after irradiation). Based on the molar extinction coefficient at 358 nm (ε358) of 5070 M–1 cm–1 (Figure S1), a BNAH yield of 48% after an irradiation time of 150 min is estimated (red trace in Figure 2B).
Figure 2.

Monitoring visible-light-driven BNAH regeneration by UV–vis absorption spectroscopy. (A) Photoinduced absorption changes of a reaction mixture containing BNA+ (1.0 mM), [Cp*Rh(bpy)Cl]+ (0.1 mM), TEOA (0.5 M), and [Ir(sppy)3]3– (10 μM) in deaerated phosphate buffer (0.1 M, pH 7) at 22 °C. The dashed vertical arrow marks the increase of the diagnostic BNAH absorption band maximum at 358 nm as a function of irradiation time with a 455 nm collimated LED. The inset contains calibrated UV–vis absorption spectra of BNA+ (orange trace) and 1,4-BNAH (green trace) in deaerated phosphate buffer (0.1 M, pH 7) at 22 °C. (B) Visible-light-driven BNAH regeneration from BNA+ using identical conditions as in (A) with four different photosensitizers and in the absence of any sensitizer (control experiment). The red circles connected by the red lines represent the data from the main plot of (A); the yields were calculated based on optical density changes (before and after irradiation) at 358 nm with ε358 = 5070 M–1 cm–1 for 1,4-BNAH.
Aside from the 1,4-BNAH reduction product, the single-electron transfer reaction promoted by the photosensitizer can in principle lead to regioisomers or the well-known BNA dimer,69 which absorbs around 356 nm.70,71 To confirm the regioselective regeneration of 1,4-BNAH, its light-driven formation was monitored by 1H NMR spectroscopy using a solution containing BNA+ (10 mM), [Cp*Rh(bpy)Cl]+ (1.0 mM), [Ir(sppy)3]3– (0.1 mM), and TEOA (1.0 M). To limit deuterium incorporation into the reduction product,63 a 1:1 mixture of CD3CN and non-deuterated phosphate buffer (0.1 M, pH 7) was used as a solvent. The characteristic 1H NMR signals of BNA+ decrease as a function of irradiation time, concomitant with the sole formation of 1,4-BNAH (Figures S2–S4). After 6 h of illumination, the yield of 1,4-BNAH was 82% based on the integration of NMR resonances characteristic for BNA+ and BNAH. The cleanliness of the conversion (with the starting material reacting directly to one clearly dominant product) validates the approach of using UV–vis absorption spectroscopy for reaction monitoring.
In the following, we monitored the photochemical reduction of BNA+ to BNAH with different photosensitizers by UV–vis spectroscopy at 358 nm. The formation of BNAH was observed for all employed sensitizers when irradiating the reaction mixture with a 455 nm collimated LED (Figure S5). The yield of BNAH using different sensitizers was traced over 150 min (Figure 2B) and increased in the order of [Ir(sppy)3]3– (46%) < [Ir(Fsppy)3]3– (56%) < [Ir(dFsppy)3]3– (64%), whereas [Ru(bpy)3]2+ only gave 13% yield of BNAH (Table 2). When comparing the initial TOF within the first 30 min of irradiation (Table 2 and Supporting Information Page S11–S12), the iridium(III) photosensitizers show comparatively high initial TOFs. Specifically, they range from 80 h–1 ([Ir(sppy)3]3–), 111 h–1 ([Ir(Fsppy)3]3–) to 146 h–1 ([Ir(dFsppy)3]3–),25,30−33 outperforming [Ru(bpy)3]2+ with a TOF of only 16 h–1 (Table 2). Evidently, the [Ir(dFsppy)3]3– sensitizer provides a roughly 10-fold TOF enhancement compared to [Ru(bpy)3]2+, whereas the BNAH yield after 150 min only shows a 5-fold improvement with [Ir(dFsppy)3]3– relative to [Ru(bpy)3]2+ (64% vs 13%). This discrepancy between TOF and BNAH yield improvements is likely due to the non-negligible re-oxidation of BNAH at longer reaction times and the limited stability of BNAH.73 These greatly enhanced TOFs and BNAH yields when using iridium(III) sensitizers instead of [Ru(bpy)3]2+ are all the more remarkable given the fact that the latter absorbs the excitation light at 455 nm more than 15 times better. (The pertinent molar extinction coefficients are ε455 < 860 M–1 cm–1 (Table 1)36 for the iridium(III) sensitizers compared to ε455 ∼ 14,600 M–1 cm–1 (Table 1) for [Ru(bpy)3]2+).35 The high TOFs and BNAH yields achievable with the iridium(III) sensitizers likely reflect their enhanced excited state electron donor properties, attractive Coulombic interactions with the cationic [Cp*Rh(bpy)(H2O)]2+ co-catalyst, and longer excited-state lifetimes compared to [Ru(bpy)3]2+ (see discussion below).
Table 2. Yields and TOFs for the Photochemical Regeneration of BNAH, Gibbs Free Energy (ΔG) for Photoinduced Electron Transfer from the Sensitizers to [Cp*Rh(bpy)(H2O)]2+, Excited-State Quenching Rate Constants (kq) for Bimolecular Reactions with [Cp*Rh(bpy)(H2O)]2+, and Quenching Efficiencies (η) at a Given [Cp*Rh(bpy)(H2O)]2+ Concentration.
| yield/% | TOF/h–1 | ΔGET,1a/eV | kqb/108 M–1s–1 | η([Cp*Rh(bpy)(H2O)]2+)c/% | ΔGET,2d/eV | η(TEOA)e/% | initial TONf (PS) | |
|---|---|---|---|---|---|---|---|---|
| [Ru(bpy)3]2+ | 13 | 16 | –0.07 | 1.2 | 0.77 | –0.53 | n/d | 26 |
| [Ir(sppy)3]3– | 46 | 80 | –1.15 | 41.7 | 40.4 | 0.00 | n/d | 92 |
| [Ir(Fsppy)3]3– | 56 | 111 | –1.11 | 41.2 | 47.1 | –0.15 | n/d | 113 |
| [Ir(dFsppy)3]3– | 64 | 146 | –1.02 | 41.3 | 46.6 | –0.29 | 6.3 | 128 |
Gibbs free energies (ΔG) were calculated based on ΔG = −e × (Ered0 – Eox*), where e is the elementary charge, Eox* is the excited state oxidation potential of the used sensitizers (Table 1, column 4), and Ered0 is the ground-state reduction potential of the Rh catalyst (−0.74 V vs SCE).72
Determined by time-resolved luminescence spectroscopy using the TCSPC technique with solutions containing individual photosensitizers (50 μM) and [Cp*Rh(bpy)Cl]Cl (0 to 500 μM) in deaerated Tris-buffer (0.1 M, pH 8.8) at 20 °C.
The quenching efficiency η was estimated based on η = (τ0 – τ)/τ0 = (kq × [Q])/(τ0–1 + kq × [Q]), where kq is the reaction rate constant, τ0 is the natural excited-state lifetime of the sensitizers (Table 1, column 5), and [Q] is the concentration of the [Cp*Rh(bpy)(H2O)]2+ quencher under reaction conditions (0.1 mM).
Gibbs free energies (ΔG) were calculated based on ΔG = −e × (Ered0 – Eox0), where e is the elementary charge, Eox0 is the oxidation potential of TEOA, and Ered0 is the acceptor reduction potential (Table 1, column 3). For the calculation of ΔGET,2, an oxidation potential of 0.76 V versus SCE was used for TEOA.65
The quenching efficiency η was determined based on η = (τ0 – τ)/τ0, where τ0 is the natural excited-state lifetime (Table 1), and τ is the lifetime measured in the presence of 1 M TEOA (Figure S6).
For details, see Supporting Information Section 3.
In principle, the photochemical regeneration of BNAH can proceed via several different reaction mechanisms. In the reductive excited-state quenching mechanism (Figure 3, right), the excited state of the photosensitizer accepts an electron from the sacrificial donor TEOA, followed by onward electron transfer from the reduced photosensitizer ([Ir(sppy)3]4–) to [Cp*Rh(bpy)(H2O)]2+. In the oxidative excited-state quenching mechanism (Figure 3, left), the excited photosensitizer donates an electron to [Cp*Rh(bpy)(H2O)]2+ and forms its oxidized species ([Ir(sppy)3]2–), which subsequently accepts an electron from TEOA to regenerate the initial state of the photosensitizer ([Ir(sppy)3]3–).
Figure 3.

Latimer diagram for [Ir(sppy)3]3– presenting the possible oxidative and reductive quenching cycles for the photochemical regeneration of BNA+.
To assess the dominant pathway in Figure 3, time-resolved luminescence quenching experiments were performed with the different photosensitizers (Figure S6). TEOA does not quench the luminescence of [Ir(sppy)3]3–, [Ir(Fsppy)3]3–, and [Ru(bpy)3]2+ to an appreciable extent (Figure S6A,B,D), whereas for [Ir(dFsppy)3]3–, a quenching efficiency η of 6.3% is obtained with 1 M of TEOA in 0.1 M deaerated phosphate buffer (pH 7) at 20 °C (Figure S6C). This observation is in line with the relatively high oxidation potential of TEOA (Eox = +0.76 V vs SCE)65 and the estimated relatively low excited-state reduction potentials (Ered*) of the [Ir(sppy)3]3–, [Ir(Fsppy)3]3– and [Ru(bpy)3]2+ sensitizers. For [Ru(bpy)3]2+, the excited-state reduction potential (Ered*) is +0.84 V versus SCE in aqueous solution, whereas for the iridium(III) complexes, we have been unable to determine the respective reduction potentials in water. Using the Ered* value of the neutral analog fac-[Ir(ppy)3] in acetonitrile as a proxy (+0.31 V vs SCE),38 we estimate a reaction free energy for reductive excited-state quenching ΔGET,3 of +0.45 eV (= e × (Eox – Ered*), with e the elementary charge). Thus, it seems that the respective photoreactions with [Ir(sppy)3]3– and [Ir(Fsppy)3]3– are too exergonic, whereas the two-fold fluoro-substitution in [Ir(dFsppy)3]3– seems to increase the electron-accepting capacity to the extent that some quenching (<6.3%) at very high TEOA concentrations (1.0 M) can occur. Thus, even for TEOA concentrations up to 1.0 M, the reductive excited-state quenching pathway remains negligible for all four photosensitizers considered here.
In contrast, the luminescent excited-state of all three iridium(III) photosensitizers is strongly quenched by [Cp*Rh(bpy)(H2O)]2+, yielding diffusion-controlled bimolecular reaction rate constants (kq) around 4.1 × 109 M–1 s–1 (Table 2 and Figure S7). For [Ru(bpy)3]2+, a kq value of 1.2 × 108 M–1 s–1 is obtained (Figure S7D). Based on these rate constants (kq, Table 2), the natural excited-state lifetimes in absence of any quencher (τ0, Table 1), and the [Cp*Rh(bpy)(H2O)]2+ concentration ([Q] = 0.1 mM) used in the UV–vis experiments, the quenching efficiency η can be estimated from η = (τ0 – τ)/τ0 = (kq × [Q])/(τ0–1 + kq × [Q]). Under reaction conditions in which 0.1 mM [Cp*Rh(bpy)(H2O)]2+ is present, quenching efficiencies above 40% are estimated for all three iridium(III) photosensitizers (Table 2), whereas for [Ru(bpy)3]2+ a quenching efficiency of less than 1% is estimated. These marked differences in kq and η values between the iridium(III) complexes on the one hand and [Ru(bpy)3]2+ on the other hand are in line with the much lower excited state oxidation potentials of the iridium photosensitizers (−1.76 to −1.89 V vs SCE, Table 1) compared to [Ru(bpy)3]2+ (−0.86 V vs SCE, Table 1).34 Based on a reduction potential (Ered) of −0.74 V versus SCE72 for [Cp*Rh(bpy)(H2O)]2+ and the known excited-state oxidation potentials (Eox*)36,57 for the three iridium(III) photosensitizers, reaction free energies (ΔGET,1) of −1.0 eV ([Ir(dFsppy)3]3–), −1.11 eV ([Ir(dFsppy)3]3–), and −1.15 eV ([Ir(sppy)3]3–) are obtained (Table 2). For [Ru(bpy)3]2+, ΔGET,1 only amounts to −0.07 eV; hence, there is far less driving force for photoinduced electron transfer to [Cp*Rh(bpy)(H2O)]2+ in this case. The much higher driving force for photoinduced electron transfer from the iridium(III) complexes might be further aided by attractive Coulombic interactions between these tri-anionic photosensitizers and the di-cationic [Cp*Rh(bpy)(H2O)]2+ co-catalyst, in comparison with the repulsive Coulombic interactions in the case of the di-cationic [Ru(bpy)3]2+.72 Furthermore, the substantially longer excited-state lifetimes of the iridium(III) complexes (1625 to 2150 ns, Table 1) compared to [Ru(bpy)3]2+ (600 ns)35 are helpful in promoting photoinduced electron transfer. Given the findings summarized in Table 2, it seems plausible that oxidative excited-state quenching by [Cp*Rh(bpy)(H2O)]2+ represents the dominant reaction pathway.35
At first glance, it might seem intriguing that among the three iridium(III) photosensitizers, the TOF does not correlate with the excited-state oxidation potential. The TOF of the complex with the strongest excited-state electron donor properties, [Ir(sppy)3]3– (Eox* = −1.89 V vs SCE, Table 1), is roughly a factor of two lower than the TOF of [Ir(dFsppy)3]3– (80 h–1 vs 146 h–1), even though the twofold fluorinated complex is a weaker excited-state donor (Eox* = −1.76 V vs SCE, Table 1). Taking into account that [Ir(sppy)3]3– absorbs more strongly at 455 nm (ε455 = 887 M–1 cm–1, Table 1) than [Ir(dFsppy)3]3– (ε455 = 232 M–1 cm–1, Table 1), the observed reactivity order might seem even more puzzling.36
Therefore, the possibility of a triplet–triplet energy transfer (TTET) reaction mechanism was considered (Figure S8), whereby the excitation energy would be transferred from the triplet-excited iridium(III) sensitizers to [Cp*Rh(bpy)(H2O)]2+. The resulting triplet excited state of the latter could then in principle react with TEOA to afford [Cp*Rh(bpy)(H2O)]+, which could ultimately lead to the reduction of BNA+.63,74 In principle, the supposed TTET seems plausible because the triplet energies of the used iridium(III) sensitizers are high (up to 2.81 eV) and because the BNAH regeneration yields and TOFs correlate with the triplet energies of the used iridium(III) sensitizers. The triplet energy of [Cp*Rh(bpy)(H2O)]2+ is not known, but it seems reasonable to use the value of 2.5 eV75 determined previously for [Rh(NH3)6]3+ as a proxy, yielding estimated TTET driving forces (ETIr(III) – ETRh(III)) between −0.15 eV ([Ir(sppy)3]3–) and −0.31 eV ([Ir(dFsppy)3]3–). Significantly differing excited state quenching rate constants (kq) can typically be observed for TTET in this driving-force range,76 but time-resolved luminescence quenching experiments (Figure S7) showed almost no differences between the kq values obtained for the three different iridium(III) sensitizers when using [Cp*Rh(bpy)(H2O)]2+ as a quencher. Given this observation and the facts that (i) electron transfer from the excited iridium(III) sensitizers to [Cp*Rh(bpy)Cl]+ has far higher driving forces (ΔGET,1 < −1.0 eV), and (ii) the observable excited state quenching rate constants approach the diffusion limit (kdiff ∼ 6.5 × 109 M–1 s–1),77 TTET does not seem to be a major reaction pathway. The oxidative pathway illustrated by the left part of Figure 3 instead seems to be of key importance.
The experimentally accessible TOF and BNAH yields naturally represent measures of the overall reaction performance resulting from several different elementary steps,27,28 among which the initial excited-state electron transfer process is not necessarily the decisive factor. The recovery of the initial state of the photosensitizer after one-electron oxidation likely becomes a key step for the iridium(III) complexes considered here. Based on the (ground state) iridium(IV/III) redox potentials of the photosensitizers (Table 1)36 and the oxidation potential of TEOA (Eox = +0.76 V vs SCE),65 the reaction free energies (Table 2, ΔGET,2) for reduction of the individual iridium(IV) species by the sacrificial electron donor can be estimated. This simple analysis reveals that ΔGET,2 ≈ 0 eV for [Ir(sppy)3]2– and then becomes increasingly negative for [Ir(Fsppy)3]2– (ΔGET,2 ≈ −0.15 eV) and [Ir(dFsppy)3]2– (ΔGET,2 ≈ −0.29 eV). In other words, the regeneration of the initial photosensitizer from its oxidized iridium(IV) form back to the iridium(III) state is associated with a considerably higher driving force in [Ir(dFsppy)3]3– than in [Ir(sppy)3]3–. It seems plausible that this is a main factor determining the order of the TOFs among the iridium(III) complexes considered here.
Conclusions
Our comparative study of three water-soluble iridium(III) photosensitizers and [Ru(bpy)3]2+ with [Cp*Rh(bpy)Cl]Cl as a co-catalyst for the light-driven regeneration of the nucleotide co-factor mimic BNAH provides the following key insights: (1) the newly used iridium(III) photosensitizers feature initial TOFs (88–146 h–1, Table 2) that are up to 10-fold higher than those obtained for [Ru(bpy)3]2+ (16 h–1), in line with the much stronger electron donor properties of the photoexcited iridium(III) complexes compared to [Ru(bpy)3]2+. (2) Among the three iridium(III) photosensitizers, the TOF correlates with the ease of iridium(IV) to iridium(III) reduction in the electronic ground state, indicating that the regeneration of the initial photosensitizer redox state by the sacrificial electron donor TEOA can become performance limiting. Collectively, these two key insights illustrate the importance of balancing the excited-state donor and ground-state acceptor properties of the photosensitizer.
The anionic nature of our tri-sulfonated iridium(III) complexes stands in contrast to [Ru(bpy)3]2+ and many other cationic transition metal-based photosensitizers. For the rhodium co-catalyzed reduction considered here, this could play an important role in the sense that electrostatic interactions between the negatively charged iridium(III) photosensitizers and the positively charged rhodium co-catalyst could further facilitate the desired photoinduced electron transfer reaction. Such pre-aggregation effects that promote photochemical reactions beyond mere diffusion control could indeed play an underappreciated role in photocatalysis.78−80 Furthermore, it seems that the development of water-soluble strong photoreductants lags considerably behind recent advances made in that regard for organic solvents.81−83 We hope that the insights gained herein and our potent water-soluble iridium(III) photoreductants will be useful for future developments in photobiocatalysis.84
Acknowledgments
This work was supported by the Swiss National Science Foundation through the NCCR Molecular Systems Engineering and through grant number 200020_207329. B.P. acknowledges a fellowship from the National Research Fund, Luxembourg (Ph.D. grant 14583224).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.2c03100.
Additional UV–vis data, NMR data, time-resolved luminescence data, and details of mechanistic studies (PDF)
Author Contributions
§ M.R.S. and B.P. contributed equally to this work.
The authors declare no competing financial interest.
Dedication
This work is dedicated to Professor Wolfgang Weigand on the occasion of his 65th birthday.
Supplementary Material
References
- Lee S. H.; Kim J. H.; Park C. B. Coupling Photocatalysis and Redox Biocatalysis Toward Biocatalyzed Artificial Photosynthesis. Chem.—Eur. J. 2013, 19, 4392–4406. 10.1002/chem.201204385. [DOI] [PubMed] [Google Scholar]
- Maciá-Agulló J. A.; Corma A.; Garcia H. Photobiocatalysis: The Power of Combining Photocatalysis and Enzymes. Chem.—Eur. J. 2015, 21, 10940–10959. 10.1002/chem.201406437. [DOI] [PubMed] [Google Scholar]
- Seel C. J.; Gulder T. Biocatalysis Fueled by Light: On the Versatile Combination of Photocatalysis and Enzymes. ChemBioChem 2019, 20, 1871–1897. 10.1002/cbic.201800806. [DOI] [PubMed] [Google Scholar]
- Lee S. H.; Choi D. S.; Kuk S. K.; Park C. B. Photobiocatalysis: Activating Redox Enzymes by Direct or Indirect Transfer of Photoinduced Electrons. Angew. Chem., Int. Ed. 2018, 57, 7958–7985. 10.1002/anie.201710070. [DOI] [PubMed] [Google Scholar]
- Schmermund L.; Jurkaš V.; Özgen F. F.; Barone G. D.; Büchsenschütz H. C.; Winkler C. K.; Schmidt S.; Kourist R.; Kroutil W. Photo-Biocatalysis: Biotransformations in the Presence of Light. ACS Catal. 2019, 9, 4115–4144. 10.1021/acscatal.9b00656. [DOI] [Google Scholar]
- Sandoval B. A.; Hyster T. K. Emerging strategies for expanding the toolbox of enzymes in biocatalysis. Curr. Opin. Chem. Biol. 2020, 55, 45–51. 10.1016/j.cbpa.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesana P. T.; Li B. X.; Shepard S. G.; Ting S. I.; Hart S. M.; Olson C. M.; Martinez Alvarado J. I.; Son M.; Steiman T. J.; Castellano F. N.; Doyle A. G.; MacMillan D. W. C.; Schlau-Cohen G. S. A biohybrid strategy for enabling photoredox catalysis with low-energy light. Chem 2022, 8, 174–185. 10.1016/j.chempr.2021.10.010. [DOI] [Google Scholar]
- Barber J. Photosynthetic energy conversion: natural and artificial. Chem. Soc. Rev. 2009, 38, 185–196. 10.1039/b802262n. [DOI] [PubMed] [Google Scholar]
- Quinto T.; Köhler V.; Ward T. R. Recent Trends in Biomimetic NADH Regeneration. Top. Catal. 2014, 57, 321–331. 10.1007/s11244-013-0187-y. [DOI] [Google Scholar]
- Wu H.; Tian C.; Song X.; Liu C.; Yang D.; Jiang Z. Methods for the regeneration of nicotinamide coenzymes. Green Chem. 2013, 15, 1773–1789. 10.1039/c3gc37129h. [DOI] [Google Scholar]
- Soldevila-Barreda J. J.; Habtemariam A.; Romero-Canelón I.; Sadler P. J. Half-sandwich rhodium(III) transfer hydrogenation catalysts: Reduction of NAD+ and pyruvate, and antiproliferative activity. J. Inorg. Biochem. 2015, 153, 322–333. 10.1016/j.jinorgbio.2015.10.008. [DOI] [PubMed] [Google Scholar]
- Soldevila-Barreda J. J.; Bruijnincx P. C. A.; Habtemariam A.; Clarkson G. J.; Deeth R. J.; Sadler P. J. Improved Catalytic Activity of Ruthenium–Arene Complexes in the Reduction of NAD+. Organometallics 2012, 31, 5958–5967. 10.1021/om3006307. [DOI] [Google Scholar]
- Seelbach K.; Riebel B.; Hummel W.; Kula M.-R.; Tishkov V. I.; Egorov A. M.; Wandrey C.; Kragl U. A Novel, Efficient Regenerating Method of NADPH Using a New Formate Dehydrogenase. Tetrahedron Lett. 1996, 37, 1377–1380. 10.1016/0040-4039(96)00010-X. [DOI] [Google Scholar]
- Ding H.-T.; Liu D.-F.; Li Z.-L.; Du Y.-Q.; Xu X.-H.; Zhao Y.-H. Characterization of a thermally stable and organic solvent-adaptative NAD+ -dependent formate dehydrogenase from Bacillus sp. F1. J. Appl. Microbiol. 2011, 111, 1075–1085. 10.1111/j.1365-2672.2011.05124.x. [DOI] [PubMed] [Google Scholar]
- Köhler V.; Wilson Y. M.; Dürrenberger M.; Ghislieri D.; Churakova E.; Quinto T.; Knörr L.; Häussinger D.; Hollmann F.; Turner N. J.; Ward T. R. Synthetic cascades are enabled by combining biocatalysts with artificial metalloenzymes. Nat. Chem. 2013, 5, 93–99. 10.1038/nchem.1498. [DOI] [PubMed] [Google Scholar]
- Okamoto Y.; Köhler V.; Paul C. E.; Hollmann F.; Ward T. R. Efficient In Situ Regeneration of NADH Mimics by an Artificial Metalloenzyme. ACS Catal. 2016, 6, 3553–3557. 10.1021/acscatal.6b00258. [DOI] [Google Scholar]
- Paul C. E.; Gargiulo S.; Opperman D. J.; Lavandera I.; Gotor-Fernández V.; Gotor V.; Taglieber A.; Arends I. W. C. E.; Hollmann F. Mimicking Nature: Synthetic Nicotinamide Cofactors for C=C Bioreduction Using Enoate Reductases. Org. Lett. 2013, 15, 180–183. 10.1021/ol303240a. [DOI] [PubMed] [Google Scholar]
- Canivet J.; Süss-Fink G.; Štěpnička P. Water-Soluble Phenanthroline Complexes of Rhodium, Iridium and Ruthenium for the Regeneration of NADH in the Enzymatic Reduction of Ketones. Eur. J. Inorg. Chem. 2007, 2007, 4736–4742. 10.1002/ejic.200700505. [DOI] [Google Scholar]
- Song H.-K.; Lee S. H.; Won K.; Park J. H.; Kim J. K.; Lee H.; Moon S.-J.; Kim D. K.; Park C. B. Electrochemical Regeneration of NADH Enhanced by Platinum Nanoparticles. Angew. Chem., Int. Ed. 2008, 47, 1749–1752. 10.1002/anie.200703632. [DOI] [PubMed] [Google Scholar]
- Lo H. C.; Buriez O.; Kerr J. B.; Fish R. H. Regioselective Reduction of NAD+ with [Cp*Rh(bpy)H]+: Structure - Activity Relationships and Mechanistic Aspects in the Formation of the 1,4-NADH Derivatives. Angew. Chem., Int. Ed. 1999, 38, 1429–1432. . [DOI] [PubMed] [Google Scholar]
- Yadav R. K.; Baeg J.-O.; Oh G. H.; Park N. J.; Kong K.-J.; Kim J.; Hwang D. W.; Biswas S. K. A Photocatalyst-Enzyme Coupled Artificial Photosynthesis System for Solar Energy in Production of Formic Acid from CO2. J. Am. Chem. Soc. 2012, 134, 11455–11461. 10.1021/ja3009902. [DOI] [PubMed] [Google Scholar]
- Choudhury S.; Baeg J.-O.; Park N.-J.; Yadav R. K. A Photocatalyst/Enzyme Couple That Uses Solar Energy in the Asymmetric Reduction of Acetophenones. Angew. Chem., Int. Ed. 2012, 51, 11624–11628. 10.1002/anie.201206019. [DOI] [PubMed] [Google Scholar]
- Park C. B.; Lee S. H.; Subramanian E.; Kale B. B.; Lee S. M.; Baeg J.-O. Solar energy in production of L-glutamate through visible light active photocatalyst-redox enzyme coupled bioreactor. Chem. Commun. 2008, 5423–5425. 10.1039/b808256a. [DOI] [PubMed] [Google Scholar]
- Nam D. H.; Lee S. H.; Park C. B. CdTe, CdSe, and CdS Nanocrystals for Highly Efficient Regeneration of Nicotinamide Cofactor Under Visible Light. Small 2010, 6, 922–926. 10.1002/smll.201000077. [DOI] [PubMed] [Google Scholar]
- Wienkamp R.; Steckhan E. Selective Generation of NADH by Visible Light. Angew. Chem., Int. Ed. 1983, 22, 497. 10.1002/anie.198304971. [DOI] [Google Scholar]
- Mengele A.; Weixler D.; Amthor S.; Eikmanns B. J.; Seibold G. M.; Rau S. Transforming Escherichia coli Proteomembranes into Artificial Chloroplasts Using Molecular Photocatalysis. Angew. Chem., Int. Ed. 2022, 61, e202114842 10.1002/anie.202114842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zedler L.; Müller C.; Wintergerst P.; Mengele A. K.; Rau S.; Dietzek-Ivanšić B. Influence of the Linker Chemistry on the Photoinduced Charge-Transfer Dynamics of Hetero-dinuclear Photocatalysts. Chem.—Eur. J. 2022, 28, e20220049 10.1002/chem.202200490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zedler L.; Wintergerst P.; Mengele A. K.; Müller C.; Li C.; Dietzek-Ivanšić B.; Rau S. Outpacing conventional nicotinamide hydrogenation catalysis by a strongly communicating heterodinuclear photocatalyst. Nat. Commun. 2022, 13, 2538. 10.1038/s41467-022-30147-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco A.; Zaffagnini M.; Bergamini G. Mediator-free NADH photochemical regeneration with the aid of the amino acid L-cysteine. Sustainable Energy Fuels 2022, 6, 4393–4397. 10.1039/d2se01045c. [DOI] [Google Scholar]
- Oppelt K. T.; Wöß E.; Stiftinger M.; Schöfberger W.; Buchberger W.; Knör G. Photocatalytic Reduction of Artificial and Natural Nucleotide Co-Factors with a Chlorophyll-Like Tin-Dihydroporphyrin Sensitizer. Inorg. Chem. 2013, 52, 11910–11922. 10.1021/ic401611v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H.; Nam D. H.; Park C. B. Screening Xanthene Dyes for Visible Light-Driven Nicotinamide Adenine Dinucleotide Regeneration and Photoenzymatic Synthesis. Adv. Synth. Catal. 2009, 351, 2589–2594. 10.1002/adsc.200900547. [DOI] [Google Scholar]
- Nam D. H.; Park C. B. Visible Light-Driven NADH Regeneration Sensitized by Proflavine for Biocatalysis. ChemBioChem 2012, 13, 1278–1282. 10.1002/cbic.201200115. [DOI] [PubMed] [Google Scholar]
- Lee S. H.; Nam D. H.; Kim J. H.; Baeg J.-O.; Park C. B. Eosin Y-Sensitized Artificial Photosynthesis by Highly Efficient Visible-Light-Driven Regeneration of Nicotinamide Cofactor. ChemBioChem 2009, 10, 1621–1624. 10.1002/cbic.200900156. [DOI] [PubMed] [Google Scholar]
- Kohtani E.; Miyabe S.; Yoshioka E.; Tanaka H. Photocatalytic Cascade Carbon-Carbon Bond-Forming Radical Reaction in Aqueous Media. Synlett 2013, 24, 1578–1582. 10.1055/s-0033-1339182. [DOI] [Google Scholar]
- Kerzig C.; Wenger O. S. Sensitized triplet-triplet annihilation upconversion in water and its application to photochemical transformations. Chem. Sci. 2018, 9, 6670–6678. 10.1039/c8sc01829d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfund B.; Steffen D. M.; Schreier M. R.; Bertrams M.-S.; Ye C.; Börjesson K.; Wenger O. S.; Kerzig C. UV Light Generation and Challenging Photoreactions Enabled by Upconversion in Water. J. Am. Chem. Soc. 2020, 142, 10468–10476. 10.1021/jacs.0c02835. [DOI] [PubMed] [Google Scholar]
- Schreier M. R.; Guo X.; Pfund B.; Okamoto Y.; Ward T. R.; Kerzig C.; Wenger O. S. Water-Soluble Tris(cyclometalated) Iridium(III) Complexes for Aqueous Electron and Energy Transfer Photochemistry. Acc. Chem. Res. 2022, 55, 1290–1300. 10.1021/acs.accounts.2c00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias-Rotondo D. M.; McCusker J. K. The photophysics of photoredox catalysis: a roadmap for catalyst design. Chem. Soc. Rev. 2016, 45, 5803–5820. 10.1039/c6cs00526h. [DOI] [PubMed] [Google Scholar]
- DiLuzio S.; Connell T. U.; Mdluli V.; Kowalewski J. F.; Bernhard S. Understanding Ir(III) Photocatalyst Structure-Activity Relationships: A Highly Parallelized Study of Light-Driven Metal Reduction Processes. J. Am. Chem. Soc. 2022, 144, 1431–1444. 10.1021/jacs.1c12059. [DOI] [PubMed] [Google Scholar]
- Mills I. N.; Porras J. A.; Bernhard S. Judicious Design of Cationic, Cyclometalated Ir(III) Complexes for Photochemical Energy Conversion and Optoelectronics. Acc. Chem. Res. 2018, 51, 352–364. 10.1021/acs.accounts.7b00375. [DOI] [PubMed] [Google Scholar]
- Lo K. K.-W. Luminescent Rhenium(I) and Iridium(III) Polypyridine Complexes as Biological Probes, Imaging Reagents, and Photocytotoxic Agents. Acc. Chem. Res. 2015, 48, 2985–2995. 10.1021/acs.accounts.5b00211. [DOI] [PubMed] [Google Scholar]
- Bevernaegie R.; Wehlin S. A. M.; Elias B.; Troian-Gautier L. A Roadmap Towards Visible Light Mediated Electron Transfer Chemistry with Iridium(III) Complexes. ChemPhotoChem 2021, 5, 217–234. 10.1002/cptc.202000255. [DOI] [Google Scholar]
- Shon J.-H.; Kim D.; Rathnayake M. D.; Sittel S.; Weaver J.; Teets T. S. Photoredox catalysis on unactivated substrates with strongly reducing iridium photosensitizers. Chem. Sci. 2021, 12, 4069–4078. 10.1039/d0sc06306a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jespersen D.; Keen B.; Day J. I.; Singh A.; Briles J.; Mullins D.; Weaver J. D. III Solubility of Iridium and Ruthenium Organometallic Photoredox Catalysts. Org. Process Res. Dev. 2019, 23, 1087–1095. 10.1021/acs.oprd.9b00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGoorty M. M.; Khnayzer R. S.; Castellano F. N. Enhanced photophysics from self-assembled cyclometalated Ir(III) complexes in water. Chem. Commun. 2016, 52, 7846–7849. 10.1039/c6cc03932d. [DOI] [PubMed] [Google Scholar]
- McGoorty M. M.; Singh A.; Deaton T. A.; Peterson B.; Taliaferro C. M.; Yingling Y. G.; Castellano F. N. Bathophenanthroline Disulfonate Ligand-Induced Self-Assembly of Ir(III) Complexes in Water: An Intriguing Class of Photoluminescent Soft Materials. ACS Omega 2018, 3, 14027–14038. 10.1021/acsomega.8b02034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. P.-Y.; Liu H.-W.; Zhang K. Y.; Lo K. K.-W. Modification of Luminescent Iridium(III) Polypyridine Complexes with Discrete Poly(ethylene glycol) (PEG) Pendants: Synthesis, Emissive Behavior, Intracellular Uptake, and PEGylation Properties. Chem.—Eur. J. 2010, 16, 8329–8339. 10.1002/chem.201000474. [DOI] [PubMed] [Google Scholar]
- Newman B.; Chen L.; Henderson L. C.; Doeven E. H.; Francis P. S.; Hayne D. J. Water-Soluble Iridium(III) Complexes Containing Tetraethylene-Glycol-Derivatized Bipyridine Ligands for Electrogenerated Chemiluminescence Detection. Front. Chem. 2020, 8, 583631. 10.3389/fchem.2020.583631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr E.; Doeven E. H.; Barbante G. J.; Connell T. U.; Donnelly P. S.; Wilson D. J.; Ashton T. D.; Pfeffer F. M.; Francis P. S. Blue Electrogenerated Chemiluminescence from Water-Soluble Iridium Complexes Containing Sulfonated Phenylpyridine or Tetraethylene Glycol Derivatized Triazolylpyridine Ligands. Chem.—Eur. J. 2015, 21, 14987–14995. 10.1002/chem.201502037. [DOI] [PubMed] [Google Scholar]
- Lier R. C. W.; Bruijn A. D.; Roelfes G. A Water-Soluble Iridium Photocatalyst for Chemical Modification of Dehydroalanines in Peptides and Proteins. Chem.—Eur. J. 2021, 27, 1430–1437. 10.1002/chem.202002599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi S.; Lu Z.; Zhang J.; Wang J.; Xie Z.; Hou L. Amphiphilic Gemini Iridium(III) Complex as a Mitochondria-Targeted Theranostic Agent for Tumor Imaging and Photodynamic Therapy. ACS Appl. Mater. Interfaces 2019, 11, 15276–15289. 10.1021/acsami.9b01205. [DOI] [PubMed] [Google Scholar]
- Fernandez-Hernandez J. M.; Longhi E.; Cysewski R.; Polo F.; Josel H.-P.; De Cola L. Photophysics and Electrochemiluminescence of Bright Cyclometalated Ir(III) Complexes in Aqueous Solutions. Anal. Chem. 2016, 88, 4174–4178. 10.1021/acs.analchem.6b00312. [DOI] [PubMed] [Google Scholar]
- Coe B. J.; Helliwell M.; Raftery J.; Sánchez S.; Peers M. K.; Scrutton N. S. Cyclometalated Ir(III) complexes of deprotonated N-methylbipyridinium ligands: effects of quaternised N centre position on luminescence. Dalton Trans. 2015, 44, 20392–20405. 10.1039/c5dt03753k. [DOI] [PubMed] [Google Scholar]
- Connell T. U.; Fraser C. L.; Czyz M. L.; Smith Z. M.; Hayne D. J.; Doeven E. H.; Agugiaro J.; Wilson D. J. D.; Adcock J. L.; Scully A. D.; Gómez D. E.; Barnett N. W.; Polyzos A.; Francis P. S. The Tandem Photoredox Catalysis Mechanism of [Ir(ppy)2(dtb-bpy)]+ Enabling Access to Energy Demanding Organic Substrates. J. Am. Chem. Soc. 2019, 141, 17646–17658. 10.1021/jacs.9b07370. [DOI] [PubMed] [Google Scholar]
- Teegardin K.; Day J. I.; Chan J.; Weaver J. Advances in Photocatalysis: A Microreview of Visible Light Mediated Ruthenium and Iridium Catalyzed Organic Transformations. Org. Process Res. Dev. 2016, 20, 1156–1163. 10.1021/acs.oprd.6b00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X.; Okamoto Y.; Schreier M. R.; Ward T. R.; Wenger O. S. Reductive Amination and Enantioselective Amine Synthesis by Photoredox Catalysis. Eur. J. Org. Chem. 2020, 2020, 1288–1293. 10.1002/ejoc.201900777. [DOI] [Google Scholar]
- Guo X.; Okamoto Y.; Schreier M. R.; Ward T. R.; Wenger O. S. Enantioselective synthesis of amines by combining photoredox and enzymatic catalysis in a cyclic reaction network. Chem. Sci. 2018, 9, 5052–5056. 10.1039/c8sc01561a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauzerall D.; Westheimer F. H. 1-Benzyldihydronicotinamide—A Model for Reduced DPN. J. Am. Chem. Soc. 1955, 77, 2261–2264. 10.1021/ja01613a070. [DOI] [Google Scholar]
- Fukuzumi S.; Koumitsu S.; Hironaka K.; Tanaka T. Energetic Comparison between Photoinduced Electron-Transfer Reactions from NADH Model Compounds to Organic and Inorganic Oxidants and Hydride-Transfer Reactions from NADH Model Compounds to p-Benzoquinone Derivatives. J. Am. Chem. Soc. 1987, 109, 305–316. 10.1021/ja00236a003. [DOI] [Google Scholar]
- Pellegrin Y.; Odobel F. Sacrificial electron donor reagents for solar fuel production. C. R. Chim. 2017, 20, 283–295. 10.1016/j.crci.2015.11.026. [DOI] [Google Scholar]
- Fukuzumi S.; Inada O.; Suenobu T. Mechanisms of Electron-Transfer Oxidation of NADH Analogues and Chemiluminescence. Detection of the Keto and Enol Radical Cations. J. Am. Chem. Soc. 2003, 125, 4808–4816. 10.1021/ja029623y. [DOI] [PubMed] [Google Scholar]
- Kobayashi A.; Konno H.; Sakamoto K.; Sekine A.; Ohashi Y.; Iida M.; Ishitani O. Transition Metal Complexes Coordinated by an NAD(P)H Model Compound and their Enhanced Hydride-Donating Abilities in the Presence of a Base. Chem.—Eur. J. 2005, 11, 4219–4226. 10.1002/chem.200401211. [DOI] [PubMed] [Google Scholar]
- Lo H. C.; Leiva C.; Buriez O.; Kerr J. B.; Olmstead M. M.; Fish R. H. Bioorganometallic Chemistry. 13. Regioselective Reduction of NAD+ Models, 1-Benzylnicotinamde Triflate and β-Nicotinamide Ribose-5′-methyl Phosphate, with in Situ Generated [Cp*Rh(Bpy)H]+: Structure-Activity Relationships, Kinetics, and Mechanistic Aspects in the Formation of the 1,4-NADH Derivatives. Inorg. Chem. 2001, 40, 6705–6716. 10.1021/ic010562z. [DOI] [PubMed] [Google Scholar]
- DeLaive P. J.; Foreman T. K.; Giannotti C.; Whitten D. G. Photoinduced Electron Transfer Reactions of Transition-Metal Complexes with Amines. Mechanistic Studies of Alternate Pathways to Back Electron Transfer. J. Am. Chem. Soc. 1980, 102, 5627–5631. 10.1021/ja00537a037. [DOI] [Google Scholar]
- Ghosh T.; Slanina T.; König B. Visible light photocatalytic reduction of aldehydes by Rh(III)-H: a detailed mechanistic study. Chem. Sci. 2015, 6, 2027–2034. 10.1039/c4sc03709j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H.; Wang J.; Zhang Y.; Xing Y.; Sun Q.; Li R. Tandem synthesis of substituted 2,7-naphthyridin-1(7H)-ones via Reissert reaction/intramolecular nucleophilic addition/oxidation dehydrogenation. Tetrahedron 2013, 69, 8299–8304. 10.1016/j.tet.2013.05.105. [DOI] [Google Scholar]
- van Schie M.; Paul C. E.; Arends I.; Hollmann F. Photoenzymatic epoxidation of styrenes. Chem. Commun. 2019, 55, 1790–1792. 10.1039/c8cc08149b. [DOI] [PubMed] [Google Scholar]
- Fukuzumi S.; Suenobu T.; Patz M.; Hirasaka T.; Itoh S.; Fujitsuka M.; Ito O. Selective One-Electron and Two-Electron Reduction of C60 with NADH and NAD Dimer Analogues via Photoinduced Electron Transfer. J. Am. Chem. Soc. 1998, 120, 8060–8068. 10.1021/ja9813459. [DOI] [Google Scholar]
- McSkimming A.; Colbran S. B. The coordination chemistry of organo-hydride donors: new prospects for efficient multi-electron reduction. Chem. Soc. Rev. 2013, 42, 5439–5488. 10.1039/c3cs35466k. [DOI] [PubMed] [Google Scholar]
- Pac C.; Miyauchi Y.; Ishitani O.; Ihama M.; Yasuda M.; Sakurai H. Redox-Photosensitized Reactions. 11. Ru(bpy)32+-Photosensitized Reactions of 1-Benzyl-1,4-dihydronicotinamide with Aryl-Substituted Enones, Derivatives of Methyl Cinnamate, and Substituted Cinnamonitriles: Electron-Transfer Mechanism and Structure-Reactivity Relationships. J. Org. Chem. 1984, 49, 26–34. 10.1021/jo00175a006. [DOI] [Google Scholar]
- Fukuzumi S.; Ohkubo K.; Fujitsuka M.; Ito O.; Teichmann M. C.; Maisonhaute E.; Amatore C. Photochemical Generation of Cyclopentadienyliron Dicarbonyl Anion by a Nicotinamide Adenine Dinucleotide Dimer Analogue. Inorg. Chem. 2001, 40, 1213–1219. 10.1021/ic0009627. [DOI] [PubMed] [Google Scholar]
- Ryu J.; Nam D. H.; Lee S. H.; Park C. B. Biocatalytic Photosynthesis with Water as an Electron Donor. Chem.—Eur. J. 2014, 20, 12020–12025. 10.1002/chem.201403301. [DOI] [PubMed] [Google Scholar]
- Hentall P. L.; Flowers N.; Bugg T. D. Enhanced acid stability of a reduced nicotinamide adenine dinucleotide (NADH) analogue. Chem. Commun. 2001, 20, 2098–2099. 10.1039/b107634p. [DOI] [PubMed] [Google Scholar]
- Kölle U.; Grützel M. Organometallic Rhodium(III) Complexes as Catalysts for the Photoreduction of Protons to Hydrogen on Colloidal TiO2. Angew. Chem., Int. Ed. 1987, 26, 567–570. 10.1002/anie.198705671. [DOI] [Google Scholar]
- Petersen J. D.; Ford P. C. Photochemistry of Rhodium(III) Complexes. Ligand Field Excitation of Hexaaminerhodium(III) and Characteristics of Nonradiative Deactivation Paths. J. Phys. Chem. 1974, 78, 1144–1149. 10.1021/j100605a003. [DOI] [Google Scholar]
- Murtaza Z.; Graff D. K.; Zipp A. P.; Worl L. A.; Jones W. E. J.; Bates W. D.; Meyer T. J. Energy Transfer in the Inverted Region: Calculation of Relative Rate Constants by Emission Spectral Fitting. J. Phys. Chem. 1994, 98, 10504–10513. 10.1021/j100092a020. [DOI] [Google Scholar]
- Montalti M.; Credi A.; Prodi L. P.; Gandolfi M. T.. Handbook of Photochemistry; Taylor & Francis, 2006. [Google Scholar]
- Giedyk M.; Narobe R.; Weiß S.; Touraud D.; Kunz W.; König B. Photocatalytic activation of alkyl chlorides by assembly-promoted single electron transfer in microheterogeneous solutions. Nat. Catal. 2019, 3, 40–47. 10.1038/s41929-019-0369-5. [DOI] [Google Scholar]
- Haimerl J.; Ghosh I.; König B.; Vogelsang J.; Lupton J. M. Single-molecule photoredox catalysis. Chem. Sci. 2019, 10, 681–687. 10.1039/c8sc03860k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser F.; Wenger O. S. Red Light-Based Dual Photoredox Strategy Resembling the Z-Scheme of Natural Photosynthesis. JACS Au 2022, 2, 1488–1503. 10.1021/jacsau.2c002658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Wenger O. S. Photophysics of Perylene Diimide Dianions and Their Application in Photoredox Catalysis. Angew. Chem., Int. Ed. 2022, 61, e202110491 10.1002/anie.202110491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z.; Niu F.; Li P.; Wang H.; Zhang Z.; Meyer G. J.; Hu K. Visible Light Generation of a Microsecond Long-Lived Potent Reducing Agent. J. Am. Chem. Soc. 2022, 144, 7043. 10.1021/jacs.2c00422. [DOI] [PubMed] [Google Scholar]
- Kim D.; Teets T. S. Strategies for accessing photosensitizers with extreme redox potentials. Chem. Phys. Rev. 2022, 3, 021302. 10.1063/5.0084554. [DOI] [Google Scholar]
- Nau R. E. P.; Bösking J.; Pannwitz A. ChemPhotoChem 2022, 6, e202200158. 10.1002/cptc.202200158. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.