

Abstract
Hintergrund
Entzündliche Erkrankungen der Muskulatur sind wichtige, zum Teil schwer verlaufende Erkrankungen, die oft zu einer erheblichen Beeinträchtigung der Lebensqualität führen. Neben der Muskelschwäche gibt es oft eine Mitbeteiligung anderer Organe wie Herz, Lunge oder Speiseröhre mit Symptomen wie Atemnot oder Schluckstörung.
Fragestellung
Nur durch eine frühe und zuverlässige Diagnosestellung, die den aktuellen nationalen und internationalen Standards entspricht, ist eine schnelle und effektive Therapie möglich.
Methoden
Zum diagnostischen Repertoire gehören u. a. Autoantikörperbestimmung, Bildgebung, Muskelbiopsie, Erfassung extramuskulärer Manifestationen z. B. durch eine hochauflösende Lungencomputertomographie und eine individualisierte Tumorsuche. Nur durch eine gute Kooperation von Fächern wie Neurologie bzw. Pädiatrie, Rheumatologie, Dermatologie, Neuropathologie sowie Pulmonologie oder Kardiologie ist eine optimale Therapie und die Vermeidung irreversibler Schäden wie etwa ein Verlust der Gehfähigkeit möglich.
Ergebnisse
Neben der Standardimmunsuppression mit Glukokortikosteroiden, Azathioprin oder Methotrexat ist eine Eskalationstherapie mit Rituximab mittlerweile sehr gut etabliert. Zur interdisziplinären Therapie gehört die Vorstellung an einem entsprechend qualifizierten Zentrum und die Orientierung an nationalen und internationalen Standards wie Leitlinien zur Myositis.
Diskussion
Als hilfreiche Ressource steht das MYOSITIS NETZ e. V. (www.myositis-netz.de) und die International Myositis Society (iMyoS; www.imyos.org) zur Verfügung.
Schlüsselwörter: Entzündliche Myopathie, Muskelschwäche, Interstitielle Lungenerkrankung, Dysphagie, Myositisautoantikörper
Abstract
Background
Inflammatory diseases of the skeletal muscle are important, often severe diseases with a considerable impact on the quality of life. In addition to muscle weakness there is often involvement of other organs, such as the heart, lungs and esophagus with symptoms such as dyspnea or dysphagia.
Purpose
A fast and effective treatment is only possible by an early and reliable diagnosis according to current national and international standards.
Methods
The diagnostic repertoire includes autoantibody testing, imaging, muscle biopsy, detection of extramuscular manifestations, e.g., by high-resolution lung computed tomography (CT) and an individualized tumor search. An optimal treatment and the avoidance of irreversible damage, such as a loss of walking ability, are only possible through a good interdisciplinary cooperation including neurology or pediatrics, rheumatology, dermatology, neuropathology, pulmonology and cardiology.
Results
In addition to standard immunosuppression with glucocorticosteroids, azathioprine or methotrexate, escalation treatment with rituximab is now well established. Interdisciplinary treatment according to national and international standards, such as guidelines on myositis, should be coordinated at qualified centers of excellence.
Discussion
Helpful resources are the MYOSITIS NETZ (www.myositis-netz.de) and the International Myositis Society (iMyoS; www.imyos.org).
Keywords: Inflammatory myopathy, Muscle weakness, Interstitial lung disease, Dysphagia, Myositis autoantibodies
Muskelentzündungen können schwer und rasch verlaufen. Schnelles und interdisziplinäres Handeln ist erforderlich. Bei manchen Unterformen können extramuskuläre Organmanifestationen im Vordergrund stehen und eine Muskelschwäche oder eine Kreatinkinase(CK)-Erhöhung sogar vollständig fehlen, z. B. bei der MDA-5-positiven amyopathischen Dermatomyositis, die mit einer hohen Mortalität durch eine Atemstörung infolge einer interstitiellen Lungenerkrankung (ILD) einhergeht. Oft können nur eine sehr zügige Myositisdiagnose und ein sofortiger Therapiebeginn helfen. Aus diesen Gründen ist es wichtig, das ganze Spektrum der Myositis und die Grundzüge der Behandlung zu kennen.
Klassifikation der Myositis
Myositis – Synonym: entzündliche Muskelerkrankung, idiopathische entzündliche Myopathie – ist eine Gruppe autoimmun bedingter Entzündungen der Muskulatur [1, 2], die in allen Altersgruppen auftritt und Schmerzen und Schwäche in proximalen Muskeln verursachen kann [1, 3]. Die häufigste Unterform der Myositis ist die Dermatomyositis (DM), die durch eine Kombination von Haut- und Muskelentzündung charakterisiert ist (Tab. 1). Die DM kommt als Erkrankung des Erwachsenen (adulte DM) und juvenile Dermatomyositis (JDM; jährliche Inzidenz ca. 3/Mio.) vor.
| Klassische, autoimmune Unterformen der Myositis | Nichtklassische Unterformen der Myositis |
|---|---|
|
Dermatomyositis (DM) Juvenile Dermatomyositis (JDM) Polymyositis (PM) Antisynthetasesyndrom (ASS) Immunvermittelte nekrotisierende Myopathie (IMNM) Einschlusskörpermyositis (IBM) Myositis bei Overlap-Syndrom (OM) bei SLE, Sjögren etc. |
Tumorassoziierte Myositis Infektiöse Myositis (Borreliose, COVID-19 o. a.) Parainfektiöse oder postinfektiöse Myositis Sarkoidose Myositis Immuncheckpoininhibitorassoziierte Myositis Brachiozervikale entzündliche Myopathie (BCIM) Eosinophile Myositis oder Fasziitis; andere Fasziitis Systemische Vaskulitis mit Muskelbeteiligung wie Polyarteritis nodosa oder ANCA-assoziierte Vaskulitis |
COVID-19 „coronavirus disease 2019“, SLE systemischer Lupus erythematodes
Erheblich seltener ist die Polymyositis (PM), bei der Hautsymptome fehlen. Häufiger sind die Antisynthetasesyndrome (ASS), die durch Antikörper gegen tRNA-Synthetasen charakterisiert sind und die neben der Myositis charakteristische Symptome der Haut und Gelenke zeigen. Bei dem Myositis-Overlap-Syndrom –Synonym: Overlap-Myositis (OM) – kommt neben der Muskelentzündung auch eine Kollagenose wie z. B. ein Lupus erythematodes (SLE) oder Sjögren-Syndrom gleichzeitig vor. Eine oft sehr rasch verlaufende Form der Myositis ist die immunvermittelte nekrotisierende Myopathie (IMNM) – Synonym: nekrotisierende autoimmune Myositis (NAM) oder nekrotisierende Myositis (NM).
Die Einschlusskörpermyositis („inclusion body myositis“, IBM) betrifft Menschen ab 45 Jahren und verläuft deutlich langsamer. Typisch ist hier eine Asymmetrie und die Beteiligung distaler ebenso wie proximaler Muskeln, insbesondere der tiefen Fingerbeuger und der Oberschenkelstrecker. Seltenere Formen der Myositis, wie z. B. durch Sarkoidose oder eine Infektionserkrankung bedingt, sind exemplarisch in Tab. 1 dargestellt.
Übersicht der Symptome und Organbeteiligung
Skelettmuskulatur
Bei fast allen Unterformen außer der IBM zeigen sich symmetrische, proximal betonte, oft schmerzhafte Paresen der Extremitäten. Der Verlauf ist typischerweise subakut bis akut progredient und kann in wenigen Wochen bis zur Tetraparese führen (Tab. 2).
| Dermatomyositis | Juvenile Dermatomyositis | Polymyositis | Antisynthetasesyndrome | Overlap-Syndrom | Nekrotisierende Myositis | Einschlusskörpermyositis | |
|---|---|---|---|---|---|---|---|
| Alter | Mittleres Erwachsenenalter | Kindheit bis Jugend | Mittleres Erwachsenenalter | Mittleres Erwachsenenalter | Mittleres Erwachsenenalter | Mittleres Erwachsenenalter | Über 45 Jahre |
| Verlauf | Subakut/akut | Subakut/akut | Subakut/akut | Subakut/akut | Subakut/akut | Subakut/akut | Chronisch |
| Paresen | Proximal betont | Proximal betont | Proximal betont | Proximal betont | Proximal betont | Proximal betont | Proximal und distal, v. a. tiefe Fingerbeuger und Quadrizeps |
| Typische Symptome | Hautbeteiligung | Hautbeteiligung | Paresen im Vordergrund | Raynaud Syndrom, Arthritis, Mechaniker-Hände | Je nach zusätzlicher Erkrankung wie Sklerodermie, SLE oder Sjögren-Syndrom | Paresen im Vordergrund | Paresen im Vordergrund |
| Mögliche Organbeteiligung, andere Besonderheiten | Hautbeteiligung kann fehlen (DM sine dermatitis), Paresen können fehlen (amyopathische DM), ILD, Dysphagie, Myokarditis |
Kalzinose der Haut Gastrointestinaltrakt |
Dysphagie, Myokarditis, ILD | ILD, Arthritis, Myokarditis | Je nach zusätzlicher Erkrankung sind Myokarditis, ILD, Arthritis, Nephritis möglich | ILD, v. a. bei SRP-AK, Myokarditis | Dysphagie (kann Erstsymptom sein) |
| Tumorassoziation | Häufig, vor allem bei Nachweis von NXP-2- und TIF-1γ-AK | Nein | Möglich | Sehr selten | Möglich | Häufig, vor allem bei seronegativen und HMGCR-AK | Sehr selten |
| Kreatinkinase | Meistens erhöht; typisch: 5‑ bis 50fach über der Norm | Meistens erhöht; typisch: 5‑ bis 50fach über der Norm | Meistens erhöht; typisch 5‑ bis 50fach über der Norm | Meistens erhöht; typisch: 5‑ bis 50fach über der Norm | Meistens erhöht; typisch: 5‑ bis 50fach über der Norm | Meistens erhöht; typisch: 5‑ bis 50fach über der Norm | Meistens erhöht; typisch: 2‑ bis 5fach über der Norm |
| Auto-AK | Mi‑2, SAE, MDA‑5, NXP‑2, TIF-1γ | Mi‑2, MDA‑5, NXP‑2, TIF-1γ | – | Jo‑1, PL‑7, PL-12, Ha, OJ, KS, Zo, EJ | SSA/Ro, SSB/La, RNP, Pm-Scl, Ku | SRP, HMGCR | cN1A |
| Biopsie | Perifaszikuläre Atrophie, Kapillarverlust, Komplementbindung, Perifaszikuläre Entzündung mit MHC-I- und MxA-Nachweis | Ähnlich der DM | Endomysiale Entzündung mit zytotoxischen T‑Zellen, Makrophagen und Einzelfasernekrosen | Perifaszikuläre Nekrosen | Variabel, auch abhängig von der Koerkrankung | Einzelfasernekrosen mit sekundärer Makrophageninfiltration | Endomysiale Entzündung mit zytotoxischen T‑Zellen, ubiquitäre MHC-I-Expression, geränderte Vakuolen, Proteinakkumulation mit p62 und TDP-43 Nachweis, COX-Defizienz |
AK Antikörper, COX Cytochrom-C-Oxidase, DM Dermatomyositis, ILD interstitielle Lungenerkrankung, MHC „major histocompatibility complex“
Allgemeinbefinden
Insbesondere bei Kindern fällt eine deutliche Beeinträchtigung des Allgemeinbefindens auf. Die Kinder sind oft unleidig und gereizt.
Haut
Die Hautsymptome ähneln sich bei DM und JDM. Sie umfassen:
ein Schmetterlingserythem im Gesicht (vor allem im Bereich der Augen, der Nase und der Wangen),
Erytheme am Dekolleté, Nacken sowie an den Streckseiten der Extremitäten bis zum Handrücken,
Atrophien der Haut (Poikilodermie) bei längerer Dauer,
flache Papeln über proximalen Finger- und Handgelenke (Gottron-Papeln),
oft ausgedehnte subkutane Kalzinosen bei JDM,
rissige, verdickte Haut der Hände („Mechaniker-Hände“) vor allem bei ASS, deutlich seltener bei DM,
Raynaud-Phänomen bei ASS.
Schlucken
Alle Myositiden können zu einer Schluckstörung – Dysphagie – führen, die von leichten Symptomen bis zur vollständigen Dysfunktion reichen. Vor allem bei einem langsam-chronischen Verlauf wie bei der IBM werden die Symptome oft auch auf einfache Fragen nicht berichtet und müssen sehr gezielt eruiert werden, z. B. ob beim Essen oft geräuspert oder sogar gehustet werden muss (Tab. 3). Eine Schluckstörung muss konsequent behandelt werden, um eine Aspiration bzw. eine Mangelernährung zu vermeiden.
| Symptome | Gezielte Fragen | Diagnostik | Fragebogen/Skala (Details siehe Text; Skalen siehe auch bei www.myositis-netz.de) |
|---|---|---|---|
|
Räuspern beim Essen Verschlucken bei festem Essen/Flüssigkeiten Husten beim Essen Vermeidung bestimmter Speisen (z. B. Steak) Langsames Essen, Veränderung der Essgewohnheiten |
Müssen Sie sich oft räuspern beim Essen? Husten Sie beim Essen? Verschlucken Sie sich beim Essen/Trinken? Bleibt Essen „im Halse stecken“? Müssen Sie mehrmals nachschlucken? Vermeiden Sie bestimmtes Essen? Essen Sie sehr viel langsamer als üblich oder haben Sie ihre Essgewohnheiten verändert? |
Fragebogen/gezielte Fragen FEES Videofluoroskopie Ggf. orale Manometrie Ggf. Ultraschall der Mundboden- und Halsmuskulatur Ggf. Echtzeit-MRT des Schluckens |
Swallowing Quality of Life (Swal-Quol) Functional Oral Intake Scale (FOIS) MD Anderson Dysphagia Inventory Sidney Swallowing Questionnaire (SSQ) sIFA |
FEES endoskopische Evaluation des Schluckens, MRT Magnetresonanztomographie, sIFA sporadic Inclusion Body Myositis Physical Functioning Assessment
Atmung
Die Atmung kann durch eine interstitielle Lungenerkrankung (ILD) bei den meisten Formen einer Myositis beeinträchtig werden (Tab. 2), insbesondere sind ASS und bestimmte Varianten der DM wie z. B. die amyopathische DM damit assoziiert. Die ILD ist eine schwere Erkrankung, die über die Erkrankungsdauer mit einer Mortalität von 13 % einhergeht und eine intensive (Eskalations‑)Therapie erfordert (s. unten; [4]). Eine schwere restriktive Ventilationsstörung ist deutlich seltener.
Herz
Eine Herzbeteiligung im Sinne einer Myokarditis kann bei mehreren Unterformen der Myositis vorkommen (Tab. 2). Bei entsprechenden klinischen Anzeichen sollte eine umgehende kardiologische Diagnostik und Therapie erfolgen (s. unten).
Tumoren
Eine Myositis kann beim Erwachsenen sowohl autoimmun als auch paraneoplastisch bedingt sein (Tab. 2). Im Kindes‑/Jugendalter spielen tumorassoziierte Formen hingegen keine Rolle. Am häufigsten sind Tumoren der Brustdrüse und der Lunge mit Myositis assoziiert. Wegen der therapeutischen Konsequenzen sollte die Tumorerkrankung frühestmöglich identifiziert werden. Wichtig: eine Tumorerkrankung kann vor oder nach einer Myositisdiagnose auftreten, am häufigsten innerhalb der ersten 3 Monate bis zu einem Jahr, aber auch noch später, wenngleich seltener [5].
Weitere Organmanifestation
Eine Arthritis kommt beim ASS häufig vor. Bei der OM hängt die Beteiligung anderer Organe wie Gelenke, Nieren oder auch Gastrointestinaltrakt sehr von der rheumatologischen Grunderkrankung wie dem Sjögren-Syndrom, der Sklerodermie oder einem SLE ab.
Diagnostik und aktuelle Kriterien
Allgemeines und Klassifikationskriterien
Da die Myositis eine komplexe, oft über den Muskel hinausgehende Erkrankung ist, sollten entsprechend den Standards der neuromuskulären Zentren der Deutschen Gesellschaft für Muskelkranke e.V. (DGM e. V; www.dgm.org) und den Leitlinien für die Myositis [2] Diagnostik und Behandlung in einem multidisziplinären Team erfolgen [6]. (1) Klinischer Befund, (2) Laborbefunde und Myopathologie, (3) Bildgebung der Muskulatur und apparative Diagnostik der betroffenen Organe (Abb. 1, siehe unten für Details) sind unerlässlich und untrennbar synergistisch erforderlich für eine zuverlässige Diagnose. Für die aktuellen Klassifikationskriterien, die von den rheumatologischen Fachgesellschaften European League Against Rheumatism (EULAR) und American College of Rheumatology (ACR) akzeptiert sind [7], ist ein Onlinekalkulator verfügbar: http://www.imm.ki.se/biostatistics/calculators/iim/. Obwohl es einige erhebliche Unzulänglichkeiten dieser Kriterien gibt, weisen sie eine gute Spezifität und Sensitivität auf und werden aktuell auch in klinischen Studien verwendet. Die Kriterien von Hoogendijk et al. können hingegen auch die NM abgrenzen [8]. Für die IBM haben sich die Kriterien nach Rose gut bewährt [9]. Details finden sich auch in den Myositis-Leitlinien und aktuellen Übersichten [1, 2].
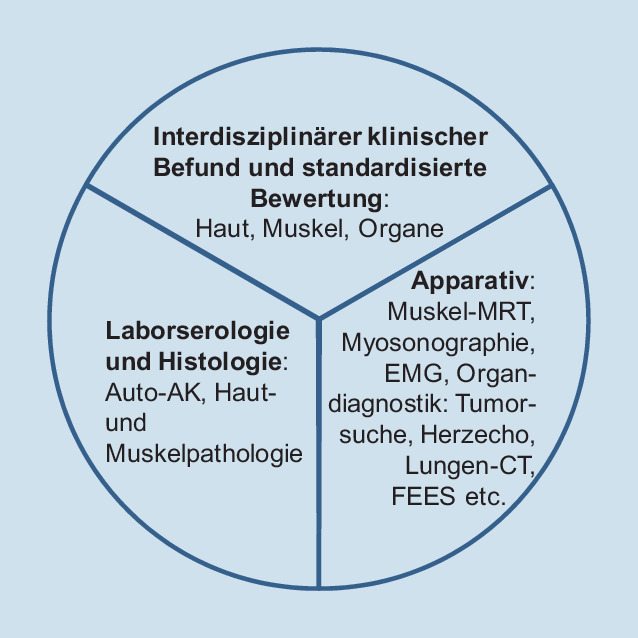
Klinischer Befund
Neben der Anamnese sollte die klinische Untersuchung sowohl die Muskulatur als auch die extramuskuläre Mitbeteiligung erfassen (Tab. 2 und 3). Die Verwendung entsprechender Skalen hilft, den Verlauf besser nachvollziehen zu können ([10]; z. B. MRC[Medical Research Council]-Summenwert, Schluckskalen, organspezifische Skalen, siehe auch www.myositis-netz.de für Details). Muskelsymptome fehlen bei einer amyopathischen DM.
Laborserologie und Myositisautoantikörper
Die Labordiagnostik sollte alle klassischen Entzündungsparameter wie Blutsenkungsgeschwindigkeit (BSG), C‑reaktives Protein (CRP), Vaskulitisparameter sowie die Zellzerfallsenzyme CK, Aspartat-Aminotransferase (AST)/Alanin-Aminotransferase (ALT), Laktatdehydrogenase (LDH) und ein breites Myositis-Auto-Antikörper(AK)-Suchpanel, das alle derzeit bekannten Myositis-AK einschließt (Tab. 4), beinhalten. Das Ausmaß des Auto-AK-Nachweises (Titer bzw. semiquantitative Beurteilung) spiegeln nicht unbedingt die Aktivität der Erkrankung wider.
| Unterform der Myositis | Autoantikörper | Häufigkeit, Assoziation, Besonderheit |
|---|---|---|
| Antisynthetasesyndrom (ASS) | Jo‑1, PL‑7, PL-12, EJ, OJ, Ha (YRS, Tyr), Zo, KS |
Antikörpernachweis obligat Jo‑1 bis 20 %, alle anderen < 5 % |
| Myositis-Overlap-Syndrom (OM) | SSA/Ro, SSB/La, RNP, Pm-Scl, Ku |
Praktisch alle Patienten Auto-AK positiv SSA/Ro: SLE oder Sjögren-Syndrom; SSB/La: Sjögren-Syndrom; RNP, PmScl, Ku: Sklerodermie Alle AK: Assoziation mit interstitieller Lungenerkrankung (ILD) |
| Dermatomyositis (DM) | Mi‑2, SAE, MDA5, NXP‑2, TIF-1γ |
Seronegativ: ~ 50 % DM Mi-2: ~ 20–30 % klassische DM SAE: < 5 % klassische DM MDA-5: < 5 % DM mit > 80 % amyopathisch und ILD NXP-2: < 5 % DM mit ~ 70 % Tumor; TIF-1γ ~ 20 % DM mit ~ 80 % Tumor |
| Juvenile Dermatomyositis (JDM) | NXP‑2, TIF-1γ, MDA‑5, Mi‑2 |
Alle AK: keine Assoziation mit Tumoren TIF-1γ: ~ 20–35 % NXP-2: ~ 20 %, hohes Risiko für Kalzinose der Haut MDA-5: ~ 5 %, oft mit ILD assoziiert Mi-2: < 5 % mit klassischer JDM |
| Nekrotisierende Myopathie (NM) | SRP, HMGCR |
Seronegativ: ~ 20 % NM mit ~ 60 % Tumor SRP: ~ 20–30 % NM mit ~ 30 % ILD HMGCR: ~ 10–20 % NM mit ~ 40 % Tumor und > 60 % Statinexposition |
| Einschlusskörpermyositis (IBM) | cN1A |
Seronegativ: > 60 % IBM cN1A: ~ 30 % IBM |
Auto-AK Autoantikörper
Eine CK-Erhöhung unterschiedlicher Höhe (2- bis 50fach über der Norm) findet sich meistens bei allen Unterformen (Tab. 2). Jedoch schließt eine normale CK auch eine floride Myositis nicht aus. Eine funktionelle Beeinträchtigungen auf molekularer Ebene kann ohne Zelluntergang zur Muskelschwäche führen [11]. Die Myositis-Auto-AK geben Hinweise auf (1) den Subtyp der Myositis, (2) eine mögliche Tumorassoziation und (3) die Häufigkeit anderer Organbeteiligungen (Tab. 4). Autoantikörper reichen alleine nicht aus, um eine Diagnose zu etablieren, da sie mitunter auch bei muskelgesunden Personen vorkommen.
Haut- und Muskelbiopsie
Bei einer Hautbeteiligung sollte eine Hautbiopsie entnommen werden, um die Art und Genese der Entzündung zu differenzieren. Einen elementaren Bestandteil der Diagnostik stellt zumindest im Erwachsenenalter die Muskelbiopsie dar, die neben dem Nachweis der Muskelentzündung, auch zur Klassifikation beiträgt (Tab. 2; Abb. 1 und 2). Der isolierte histologische Nachweis einer Myositis reicht allerdings NICHT aus, um eine Diagnose zu stellen. Eine Vielzahl nicht entzündlicher Erkrankungen kann ein „myositisähnliches“ Bild in der Histologie aufweisen [1, 12, 13].
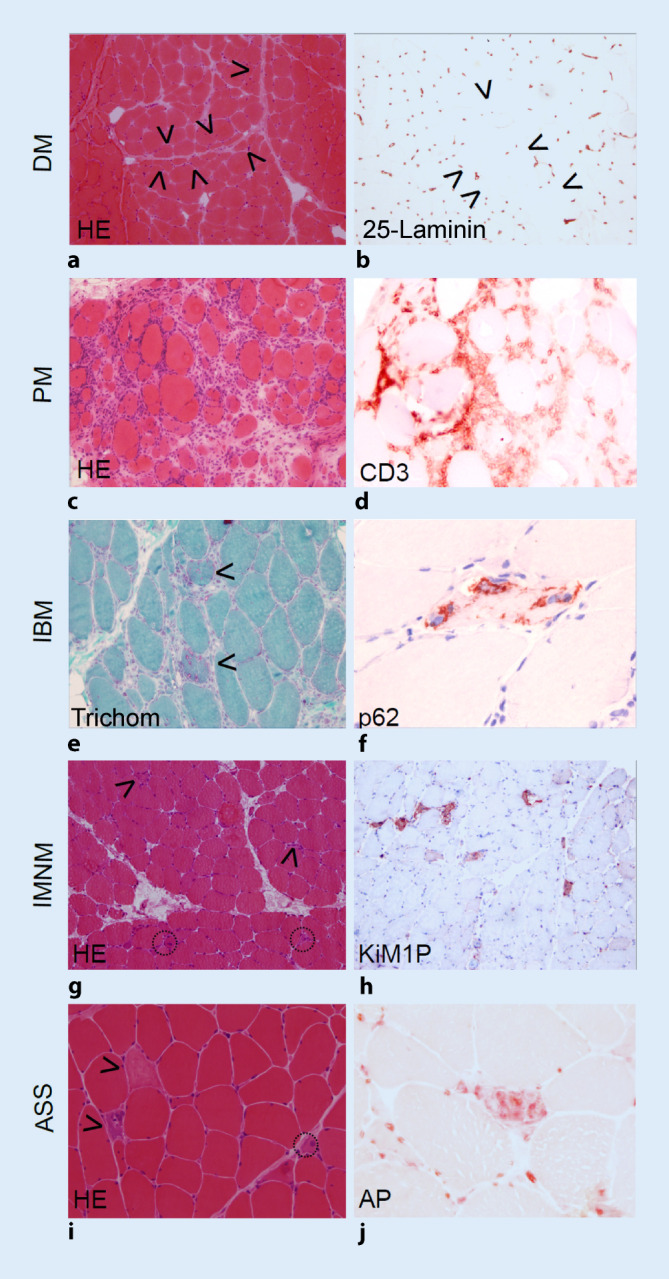
Im Kindesalter kann die Muskelbiopsie in klinisch nicht eindeutigen Fällen zum Nachweis anderer neuromuskulärer Erkrankungen sinnvoll sein, wobei hier molekulargenetische Methoden inzwischen deutlich im Vordergrund stehen. Der Stellenwert der Muskelbiopsie wird von einzelnen Zentren unterschiedlich eingeschätzt.
Magnetresonanztomographie und Muskelsonographie
Eine Magnetresonanztomographie (MRT) der Muskulatur ist hilfreich für:
den Nachweis akuter Veränderungen wie ein Muskelödem,
die Festlegung einer Biopsiestelle (Abb. 3),
die Identifikation des Ausmaßes der (sub‑)klinisch betroffenen Muskeln,
die Dokumentation des lipomatös-myosklerotischen Umbaus,
ein Therapiemonitoring,

Im Kindesalter muss die Indikation insbesondere in Fällen, in denen eine Narkose notwendig wäre, kritisch erwogen werden.
Auch muskelsonographisch kann ein Umbau der Muskulatur nachgewiesen werden (Abb. 3). Dieses ist vor allem für den Nachweis der (subklinischen) Mitbeteiligung der tiefen Fingerbeuger eine wertvolle diagnostische Hilfe für die Erkennung einer IBM [15]. Bei der JDM dient die Sonographie auch der Dokumentation und Verlaufskontrolle einer Kalzinose.
Elektromyographie und Elektroneurographie
Oft lässt sich in der Elektromyographie (EMG) eine myopathische Schädigung nachweisen, jedoch kann nicht hinreichend zwischen einer Myositis und einer Muskeldystrophie unterschieden werden. Bei einer floriden Myositis bzw. im Schub lassen sich mitunter eine pathologische Spontanaktivität im EMG ableiten. Dadurch können chronische Läsionen oder eher metabolisch bedingte Schädigung wie z. B. bei einer Steroidmyopathie abgegrenzt werden. Die Elektroneurographie dient zur Abgrenzung einer Polyneuropathie.
Organdiagnostik
Tumorassoziation
Bei den meisten Formen der Myositis muss wegen der möglichen Tumorassoziation zeitnah nach der Diagnosestellung eine Tumorsuche durchgeführt werden (Abb. 1; Tab. 2). Da bei Erwachsenen die Tumorassoziation auch nach einem Jahr noch hoch ist, sollte auch im Verlauf eine entsprechende Diagnostik erfolgen. Neben der Bildgebung müssen ggf. eine Koloskopie, Gastroskopie sowie urologische/gynäkologische Diagnostik erfolgen [2]. Ob eine Computertomographie (CT) ausreicht oder eine Positronenemissionstomographie(PET)-CT indiziert ist, muss individuell entschieden werden und ist derzeit nicht unumstritten [16].
Im Kindes- und Jugendalter ist eine Tumorsuche bei Dermatomyositis in der Regel nicht notwendig
Die Tumorassoziation gilt nicht für das Kindesalter (selbst beim Vorliegen der oben genannten, häufig mit Tumoren verbundenen Autoantikörper). Im Kindes- und Jugendalter gibt es deshalb keine generelle Empfehlung zur Tumorsuche.
ILD
Beim Verdacht auf eine Lungenbeteiligung (Tab. 2) muss eine entsprechende Stufendiagnostik durchgeführt werden. Eine Lungenfunktionsdiagnostik sollte unverzüglich erfolgen (einschließlich Untersuchung der Diffusionskapazität) und beim Hinweis auf eine Beeinträchtigte Atmung bzw. Oxygenierung sollte ein hochauflösendes Lungen-CT durchgeführt werden.
Myokarditis
Bei dem Verdacht auf eine Myokarditis müssen neben dem Labor (für CK-MB, Troponin‑T und -I) eine Elektrokardiographie (EKG) und Herzecho durchgeführt werden, gegebenenfalls zusätzlich eine kardiale MRT-Diagnostik.
Dysphagie
Nach einer detaillierten Anamnese sollte sich eine flexible endoskopische Evaluation des Schluckens (FEES) anschließen und ggf. eine Videofluoroskopie. Hierbei kann z. B. bei der IBM eine funktionelle Stenose im Bereich des M. cricopharyngeus demonstriert werden (siehe oben und unten; [17]).
Therapiestandards und Studienausblick
Allgemeines zur Therapie
Die Behandlung der Myositis sollte entsprechend nationaler und internationaler Standards in jedem Fall interdisziplinär erfolgen unter Miteinbeziehung von Rheumatologie, Neurologie, pädiatrischer Neurologie, pädiatrischer Rheumatologie, Dermatologie sowie je nach Organbeteiligung auch Kardiologie, Pulmonologie oder Hals-Nasen-Ohren Heilkunde [2, 6].
Nichtpharmakologische Therapie aller Unterformen der Myositis
Parallel zu der pharmakologischen Therapie muss sofort eine nichtpharmakologische Therapie etabliert werden (Infobox 1). Dazu gehören Physiotherapie, Ergotherapie, (Neuro‑)Psychologie sowie eine Sozialberatung, Rehabilitation und Hilfsmittelversorgung. Die Behandlung muss in der Aktuphase ebenso wie in der Stabilisierung erfolgen. Bei einer Schluckstörung kann die vorübergehende Anlage einer nasogastralen Sonde erforderlich sein.
Symptomatische Therapie aller Unterformen der Myositis
Insbesondere bei Schmerzen sollte eine Schmerztherapie entsprechend dem WHO(World Health Organization)-Schema erfolgen mit einem Beginn mit klassischen nichtsteroidalen Antirheumatika (NSAR).
Pharmakologische Behandlung der DM, JDM, PM, ASS, OM, NM
Die kausale Behandlung basiert auf einer Immunsuppression mit Glukokortikosteroiden [2, 18]. Es werden sowohl eine orale Dauertherapie, Pulsschemata oder eine Kombination aus beiden verwendet. Im Erwachsenenalter wird häufig mit einer intravenösen Pulstherapie mit 250–1000 mg Prednisolonäquivalent pro Tag für 3 bis 5 Tage begonnen (Infobox 1). Anschließend erfolgt üblicherweise eine orale Fortsetzung der Behandlung mit 1 mg/kg Prednisolonäquivalent pro Tag für etwa 6 bis 8 Wochen bzw. bis zur Besserung der Symptome. Im Kindesalter werden die Kortikosteroide initial entweder als Pulstherapie (20–30 mg/kgKG Prednisolonäquivalent in wöchentlichen Abständen, kombiniert mit einer Dauertherapie von 0,2–0,5 mg/kgKG Prednisolonäquivalent) oder als eine kontinuierliche Behandlung (initial 1–2 mg/kg KG) verabreicht [19].
Bei schweren Verläufen sollte frühzeitig ein geeignetes Immunsuppressivum hinzugenommen werden
Vor allem bei einem schweren Verlauf und immer, wenn mit einer längerfristigen Immunsuppression zu rechnen ist, sollte frühzeitig ein geeignetes Immunsuppressivum wie Azathioprin (1–2 mg/kg täglich), Methotrexat (15 mg/m2/Woche) oder Mycophenolatmofetil (10–20 mg/kg täglich) hinzugenommen werden. Bei Kindern und Jugendlichen mit schwerem Verlauf der JDM können intravenöse Immunglobuline (IVIG; 1–2 g/kg monatlich) zusätzlich zum Einsatz kommen. Insbesondere im Kindesalter fehlen kontrollierte Studien, die Therapie basiert auf Expertenempfehlungen.
Bei einem unzureichenden Therapieeffekt stehen Ciclosporin oder Tacrolimus zur Verfügung oder alternativ eine Kombination des Immunsuppressivums der 1. Wahl mit IVIG. Für die DM konnte kürzlich eine internationale Doppelblind-Therapiestudie (ProDerm-Studie) erfolgreich abgeschlossen werden [20], worauf die Zulassung für Octagam® für die DM erfolgt ist, für Fälle, in denen Immunsuppressiva unzureichend wirken bzw. Nebenwirkungen verursachen oder eine Kontraindikation für eine Immunsuppression besteht.
Vor allem bei einer Organbeteiligung wie einer ILD oder Myokarditis sollte neben der vorgenannten Eskalation vor allem auch eine alternative Behandlung mit Rituximab oder Cyclophosphamid oder eine Kombinationstherapie erwogen werden (Infobox 1).
Pharmakologische Behandlung der IBM
Bei der IBM ist entgegen den anderen Myositisformen eine Immunsuppression nicht indiziert [2, 18]. Eine Behandlung mit IVIG kann zumindest bei einem Teil der Betroffenen zur Stabilisierung beitragen und insbesondere die Symptome einer Schluckstörung verbessern (Übersicht in [13]). Bei einer alltagsrelevanten Dysphagie sollte auch eine Lokaltherapie erwogen werden: Hierfür kommen z. B. eine Myotomie als chirurgisches, nichtreversibles Verfahren oder rezidivierende endoskopische Botulinumtoxininjektionen in den M. cricopharyngeus als individuelle Heilversuche in Betracht [2, 13, 21].
Studienlage
Eine Reihe an klinischen Studien wird derzeit auf internationaler Ebene und auch mit deutscher Beteiligung durchgeführt zu praktisch allen Formen der Myositis. Es handelt sich um alle Stufen der Studien, von explorativ bis zur Zulassungsstudie [18, 22, 23].
Kinderwunsch und Schwangerschaft
Eine Myositis stellt keine grundsätzliche Kontraindikation zur Schwangerschaft dar. Die Lebensplanung kann jedoch durch die Erkrankung zumindest zeitweise beeinträchtigt werden. Bei einer Immunsuppression muss je nach Substanz eine entsprechende Aufklärung der Betroffenen erfolgen und ggf. neben einer Empfängnisverhütung auch eine Kryokonservierung von Samen‑/Eizellen in Erwägung gezogen werden.
Zusammenfassung
Es ist wichtig, die Diagnostik und Therapie der Myositis kontinuierlich interdisziplinär durchzuführen. Nur dadurch kann eine bestmögliche Diagnose und Therapie etabliert werden, die den nationalen und internationalen Standards entspricht. Für die Diagnose einer Myositis ist es zwingend erforderlich, sämtliche klinischen, serologischen, histologischen und organbezogenen Befunde als eine Einheit und in interdisziplinärer Weise zu bewerten. Diese Konstellation ist vor allem an spezialisierten Zentren möglich [6].
Infobox 1 Therapieschema (s. Details im Text)
Basistherapie:
Nichtpharmakologisch: (1) Physiotherapie; ggf. (2) Logopädie, Ergotherapie, Rehabilitation, Hilfsmittelversorgung, Sozialberatung, Psychologische Betreuung, Selbsthilfegruppen, Patientenregister.
Pharmakologisch kausal: (1) Glukokortikosteroide (z. B. Prednisolon): initial oft als i.v. Pulstherapie mit 250–1000 mg/Tag für 3 bis 5 Tage, danach 1 mg/kg p.o. bis zur Besserung, jedoch möglichst nicht in unveränderter Dosis für länger als 6 bis 8 Wochen ohne Langzeitimmunsuppressiva; (2) Langzeitimmunsuppressiva: üblicherweise früh zusätzlich zur Steroidbehandlung etablieren; erste Wahl: Azathioprin, Methotrexat, Mycophenolatmofetil; zweite Wahl: Cyclosporin, Tacrolimus oder Kombination mit IVIG; (3) bei Kindern oder Unverträglichkeit bzw. Kontraindikation für Immunsuppression auch primär IVIG erwägen.
Pharmakologisch supportiv: Schmerztherapie.
Eskalationstherapie (bei unzureichendem Effekt der Basistherapie oder früh bei schwerem Verlauf bzw. Organbeteiligung wie ILD):
Kombination mit IVIG
Kombination 1. Wahl Immunsuppressivum mit Ciclosporin oder Tacrolimus
Rituximab
Cyclophosphamid
Experimentell: z. B. Leflunomid, Tofazitinib, Barizitinib, Rapamycin
Infobox 2 Mehr Informationen zum Thema
Leitlinien für Myositis: https://dgn.org/leitlinie/143
MYOSITIS NETZ e. V.: www.myositis-netz.de
Diagnosegruppe Myositis (für Betroffene, Angehörige, aber auch für Fachpersonal z. B. um Informationsflyer kostenfrei zu bestellen): https://www.dgm.org/diagnosegruppe/myositis
Onlinekalkulator zur Myositisklassifikation: http://www.imm.ki.se/biostatistics/calculators/iim/
International Society for Myositis: www.imyos.org
Fazit für die Praxis
Eine Myositis verläuft oft schwer und kann die Lebensqualität erheblich beeinträchtigen. Neben der Schwäche an Armen und Beinen durch die Entzündung der Muskulatur gibt es oft eine Mitbeteiligung anderer Organe wie Herz, Lunge oder Speiseröhre.
Nur durch eine frühe und zuverlässige Diagnosestellung entsprechend aktueller internationaler Standards kann eine schnelle und effektive Therapie etabliert werden. Hierzu gehören die Synthese aus klinischen Parametern und Befunden der Muskelbiopsie und Autoantikörper sowie die entsprechende Organdiagnostik und eine Tumorsuche.
Essenziell ist die interdisziplinäre Behandlung durch Neurologen bzw. Pädiater, Rheumatologen, Dermatologen, Neuropathologen sowie Pulmonologen oder Kardiologen. Nur so lassen sich eine bestmögliche klinische Besserung durch Immunsuppressiva bzw. eine Eskalationstherapie erzielen und irreversible Schädigungen abwenden. Hierdurch soll eine dauerhafte Beeinträchtigung von Alltagsaktivitäten verhindert werden wie z. B. einem Verlust der Gehfähigkeit.
Acknowledgments
Danksagung
Wir danken Dr. Rachel Zeng (Klinik für Neurologie, UMG), Dr. Sabrina Zechel (Institut für Neuropathologie, UMG), Dr. Omar Al-Bourini und Prof. Ali Seif (Institut für diagnostische und interventionelle Radiologie, UMG) für die freundliche Bereitstellung von Bildmaterial.
Einhaltung ethischer Richtlinien
Interessenkonflikt
J. Schmidt hat Beraterhonorare, Reisekostenerstattungen oder Projektförderungen erhalten von Abcuro, Alnylam, Argenx, Biotest, CSL Behring, Euroimmun, Janssen, Kezar, LFB, Novartis, Octapharma, UCB. W. Müller-Felber hat Beraterhonorare, Reisekostenerstattungen und Vortragshonorare erhalten von Biogen, Novartis, Roche, Sarepta, PTC, Sanofi-Aventis.
Für diesen Beitrag wurden von den Autor/-innen keine Studien an Menschen oder Tieren durchgeführt. Für die aufgeführten Studien gelten die jeweils dort angegebenen ethischen Richtlinien.
Footnotes
J. Schmidt und W. Müller-Felber sind Mitglieder des Europäischen Referenznetzwerkes für seltene neuromuskuläre Erkrankungen ERN EURO-NMD.

QR-Code scannen & Beitrag online lesen
Literatur
- 1.Schmidt J. Current classification and management of inflammatory myopathies. J Neuromuscul Dis. 2018;5:109–129. doi: 10.3233/JND-180308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wiendl H, Schmidt J et al (2022) Myositissyndrome, S2k-Leitlinie; in: Deutsche Gesellschaft für Neurologie (Hrsg.), Leitlinien für Diagnostik und Therapie in der Neurologie. https://www.dgn.org/leitlinien. Zugegriffen: 23.05.2023
- 3.Senecal JL, Raynauld JP, Troyanov Y. Editorial: a new classification of adult autoimmune myositis. Arthritis Rheumatol. 2017;69:878–884. doi: 10.1002/art.40063. [DOI] [PubMed] [Google Scholar]
- 4.Johnson C, Pinal-Fernandez I, Parikh R, et al. Assessment of mortality in autoimmune myositis with and without associated interstitial lung diseas. Lung. 2016;194(5):733–737. doi: 10.1007/s00408-016-9896-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hida A, Yamashita T, Hosono Y, et al. Anti-TIF1-gamma antibody and cancer-associated myositis: a clinicohistopathologic study. Neurology. 2016;19(87):299–308. doi: 10.1212/WNL.0000000000002863. [DOI] [PubMed] [Google Scholar]
- 6.Korsten P, Rademacher JG, Seitz CS, et al. Interdisziplinäre Fallkonferenz als Chance für Myositis-Patienten? – Retrospektive Analyse des „Göttinger Modells“ zur interdisziplinären Diagnostik und Therapie. Nervenheilkunde. 2019;38:377–380. [Google Scholar]
- 7.Lundberg IE, Tjarnlund A, Bottai M, et al. 2017 European league against rheumatism/American college of rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Arthritis Rheumatol. 2017;69:2271–2282. doi: 10.1002/art.40320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoogendijk JE, Amato AA, Lecky BR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10–12 October 2003, Naarden, The Netherlands. Neuromuscul Disord. 2004;14:337–345. doi: 10.1016/j.nmd.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 9.Rose MR. 188th ENMC international workshop: inclusion body myositis, 2–4 december 2011, Naarden, The Netherlands. Neuromuscul Disord. 2013;23:1044–1055. doi: 10.1016/j.nmd.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 10.Aggarwal R, Rider LG, Ruperto N, et al. 2016 American College of Rheumatology/European League against Rheumatism criteria for minimal, moderate, and major clinical response in adult Dermatomyositis and Polymyositis: an international myositis assessment and clinical studies group/paediatric rheumatology international trials organisation collaborative initiative. Arthritis Rheumatol. 2017;69:898–910. doi: 10.1002/art.40064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller FW, Lamb JA, Schmidt J, et al. Risk factors and disease mechanisms in myositis. Nat Rev Rheumatol. 2018;14:255–268. doi: 10.1038/nrrheum.2018.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhai SF, Dimachkie MM, de Visser M. Is it really myositis? Mimics and pitfalls. Best Pract Res Clin Rheumatol. 2022;36:101764. doi: 10.1016/j.berh.2022.101764. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt K, Schmidt J. Inclusion body myositis: advancements in diagnosis, pathomechanisms, and treatment. Curr Opin Rheumatol. 2017;29:632–638. doi: 10.1097/BOR.0000000000000436. [DOI] [PubMed] [Google Scholar]
- 14.Ferrer I, Martin B, Castano JG, et al. Proteasomal expression, induction of immunoproteasome subunits, and local MHC class I presentation in myofibrillar myopathy and inclusion body myositis. J Neuropathol Exp Neurol. 2004;63:484–498. doi: 10.1093/jnen/63.5.484. [DOI] [PubMed] [Google Scholar]
- 15.Abdelnaby R, Mohamed KA, Elgenidy A, et al. Muscle sonography in inclusion body myositis: a systematic review and meta-analysis of 944 measurements. Cells. 2022;11:600. doi: 10.3390/cells11040600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korsten P, Seitz CS, Sahlmann CO, et al. Comment on: The temporal relationship between cancer and adult onset anti-transcriptional intermediary factor 1 antibody-positive dermatomyositis. Rheumatol (Oxford) 2019;58:2071–2073. doi: 10.1093/rheumatology/kez313. [DOI] [PubMed] [Google Scholar]
- 17.Olthoff A, Carstens PO, Zhang S, et al. Evaluation of dysphagia by novel real-time MRI. Neurology. 2016;87:2132–2138. doi: 10.1212/WNL.0000000000003337. [DOI] [PubMed] [Google Scholar]
- 18.Glaubitz S, Zeng R, Rakocevic G, et al. Update on myositis therapy: from today’s standards to tomorrow’s possibilities. Curr Pharm Des. 2022;28:863–880. doi: 10.2174/1381612827666211115165353. [DOI] [PubMed] [Google Scholar]
- 19.Pachman LM, Nolan BE, DeRanieri D, et al. Juvenile dermatomyositis: new clues to diagnosis and therapy. Curr Treatm Opt Rheumatol. 2021;7:39–62. doi: 10.1007/s40674-020-00168-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aggarwal R, Charles-Schoeman C, Schessl J, et al. Trial of intravenous immune globulin in dermatomyositis. N Engl J Med. 2022;387:1264–1278. doi: 10.1056/NEJMoa2117912. [DOI] [PubMed] [Google Scholar]
- 21.Zeng R, Schmidt J. Impact and management of dysphagia in inflammatory myopathies. Curr Rheumatol Rep. 2020;22:74. doi: 10.1007/s11926-020-00950-3. [DOI] [PubMed] [Google Scholar]
- 22.Zeng R, Glaubitz S, Schmidt J. Antibody therapies in autoimmune inflammatory myopathies: promising treatment options. Neurotherapeutics. 2022;19:911–921. doi: 10.1007/s13311-022-01220-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeng R, Glaubitz S, Schmidt J. Inflammatory myopathies: shedding light on promising agents and combination therapies in clinical trials. Expert Opin Investig Drugs. 2021;30:1125–1140. doi: 10.1080/13543784.2021.2003776. [DOI] [PubMed] [Google Scholar]