

Abstract
Structurally diverse indole-3-pyrazole-5-carboxamide analogues (10–29) were designed, synthesized, and evaluated for their antiproliferative activity against three cancer cell lines (Huh7, MCF-7, and HCT116) using the sulforhodamine B assay. Some of the derivatives showed anticancer activities equal to or better than sorafenib against cancer cell lines. Compounds 18 showed potent activity against the hepatocellular cancer (HCC) cell lines, with IC50 values in the range 0.6–2.9 μM. Compound 18 also exhibited moderate inhibitory activity against tubulin polymerization (IC50 = 19 μM). Flow cytometric analysis of cultured cells treated with 18 also demonstrated that the compound caused cell cycle arrest at the G2/M phase in both Huh7 and Mahlavu cells and induced apoptotic cell death in HCC cells. Docking simulations were performed to determine possible modes of interaction between 18 and the colchicine site of tubulin and quantum mechanical calculations were performed to observe the electronic nature of 18 and to support docking results.
Keywords: indole-pyrazole, cancer, hepatocellular carcinoma, tubulin polymerization inhibitor
Graphical Abstract
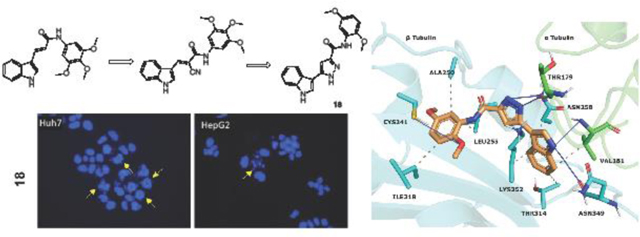
1. Introduction
Cancer is considered one of the main life-threatening diseases worldwide. The American Cancer Society estimates that the number of new deaths due to liver and intrahepatic bile duct cancer in the United States in 2022 will be approximately 30,000. The 5-year relative survival rate for liver cancer is about 20%, very low in comparison with prostate cancer (98%) and breast cancer in women (90%) [1]. Around 80% of liver cancer cases are hepatocellular cancer (HCC), and the remaining cases are intrahepatic cholangiocarcinoma [2].
Chemotherapy, despite the low survival rate, is the best available treatment in advanced HCC. In the last decade new anticancer agents like sorafenib and lenvatinib were approved by the FDA as first-line treatments for advanced HCC, while nivolumab, regorafenib and cabozantinib were approved as second-line agents for advanced HCC [3]. These anticancer agents are critical for treatment of the disease, but side effects and drug resistance create an urgent need to explore novel drugs with reduced side effects and better efficacy [4].
Microtubules play an important role in different processes in cells, including cell division, cell shape, endothelial cell biology and intracellular transport, and several conventional and newer anticancer agents target microtubules [5, 6]. There is great continuing interest in the discovery and design of new agents that inhibit microtubule polymerization [7]. It has been reported that Combrestatin A-4 (CA-4), a natural cis-stilbene, exhibits antitumoral activity by binding strongly to the colchicine binding site of tubulins and inhibiting the polymerization of tubulins [4, 8]. Although CA-4 has been proven to have strong antitumoral activity against many cancer cell lines, including cancer cell lines that are resistant to combined drug therapy, it has been found to be inactive in in vivo studies due to its low water solubility. In vivo studies with disodium phosphate prodrug form (CA4P) have been shown to reduce tumor vascularity. Researchers synthesized cis-combretastatin analogs to improve the water solubility of CA-4 with a prodrug approach [9]. Many potent CA-4 derivatives have been developed in recent years, but only a few of these, such as fosbretabulin, Oxi4503, and ombrabulin, have progressed to clinical studies (Figure 1) [10].
Figure 1.

CA-4 and its derivatives in clinical trials; pyrazole and indole containing structures with anticancer activity; lead tubulin inhibitors and title compounds.
Structure–activity relationship studies of CA-4 and its derivatives have shown that the methoxyphenyl ring “Ring A” is crucial for antitubulin activity, but, based on the precise fit into the active site, this moiety affects tubulin polymerization depending on the number and locations of the methoxy moieties. Additionally, the olefinic bond and “Ring B” can be modified to design multiple derivatives [11]. The olefinic bond can be replaced by heterocyclic structures, such as imidazole, indole, pyrazole, triazole, pyrrole, and lactams, and non-heterocyclic cores, such as ethers, olefins, ketones, sulfonamides, sulfonates, amine, amide derivatives and cyclopentanes, to tweak the properties of the parent molecule [12–16]. The B ring system on CA-4 can also be modified by adding a 5-membered heterocyclic ring to the main and/or modified core of CA-4, and pyrazole and indole rings were broadly used in such modification strategies [17]. Pyrazole derivatives possess various pharmacological activities, such as inflammatory, antipyretic, analgesic [18, 19], antiviral [20], antibacterial [21], and anticancer [22, 23] activities. Additionally, the indole is a biologically important heterocyclic structure, and various compounds carrying this skeleton have been reported to have antitumor effects [24–27]. Indole-pyrazole hybrids are of interest because they have the potential to show synergistic pharmacological effects compared to compounds containing only one of the individual pharmacophores (indole or pyrazole). This makes these hybrids a promising area for further research in medicinal chemistry. Many different types of indole-pyrazole hybrids have been reported to show a range of biological activities including, antimicrobial, antitumor, anti-inflammatory, anti-oxidant, among others. These results suggest that indole-pyrazole hybrids have potential as therapeutic agents in various disease areas [28]. Various studies were focused on the hybridization strategy to create compounds that have pyrazole with indole and/or methoxyphenyl moieties to develop and evaluate their antitumor activity as CA-4 analogs. Compound 1 (Figure 1) is a pyrazole-trimethoxyphenyl hybrid with potent anticancer activity against the Huh7, Mahlavu, HepG2 and SNU-475 cancer cell lines [23]. Compound 2, is a combination of pyrazoline, indole and methoxyphenyl with potent anticancer activity against the A549, MCF-7 and HepG2 cancer cell lines and showed potent activity as an inhibitor of tubulin polymerization [29]. Kamal et al. [30] designed compound 3, which possesses pyrazole as a replacement of bridge groups, a cis double bond and an indole as ring B. Studies of their cytotoxicity against selected human cancer cell lines and of their effects on tubulin inhibition have shown that these types of compounds have potent activity in both assays. Our research team previously synthesized a series of indole-acrylamide derivatives that contain as substitutes indole for ring B and acrylamide for the olefinic group. Compound 4 has potent anticancer activity against both Raji and HL-60 cancer cells and showed tubulin inhibition activity with an IC50 value of 17 μM. Moreover, its cyano-substituted counterpart, compound 5 showed similar activity as a tubulin inhibitor [6, 31].
The literature and our ongoing anticancer program [4–6, 22, 23, 31–36] created the idea for the design, synthesis and biological evaluation of new indole-pyrazole hybrid derivatives that possesses pyrazole as a replacement of bridge groups. In this study, we synthesized twenty new indole-pyrazole hybrid (10–29) derivatives and evaluated their biological effects at the molecular level. The novelty of this study is based on different factors, including synthesis of novel series of compounds, evaluation of their antiproliferative activities by using various biological methods, and in silico techniques to investigate possible binding interactions between the most potent compounds and the tubulin target accordingly.
2. Material and methods
2.1. Chemistry
All chemicals were purchased from local suppliers. Reactions were followed by analytical thin-layer chromatography (TLC) on precoated silica plated aluminum sheets (Silica gel 60 F254, Merck). Purification by flash chromatography was performed with a Teledyne-Isco Combiflash®Rf automated flash chromatography system using prepacked RediSep disposable columns (Lincoln, NE, USA) using DCM-MeOH, hexane-EtOAc or DCM-EtOAc solvent gradients. Purity was determined as >97% for all final compounds by the UPLC/MS method using a water/AcCN solvent gradient containing 0.1% formic acid (1% → 90%) on an Aquity BEH C18 column (2.1 × 100 mm, 1.7 μm) with flow rate = 0.3 ml/min. The 1H- and 13C-NMR were recorded with a Varian Mercury (Agilent) 400 MHz FT-NMR spectrometer (Agilent Technologies, Santa Clara, CA, USA) in DMSO-d6 using tetramethylsilane as internal standard at the NMR facility of the Faculty of Pharmacy, Ankara University. All chemical shifts were recorded as δ (ppm). Chemical shifts were given in δ (ppm), coupling constants as Hertz. IR spectra was obtained using a Perkin Elmer Spectrum 400 FTIR/FTNIR spectrometer equipped with a Universal ATR Sampling Accessory. High resolution mass spectra (HRMS) were recorded using a Waters LCT Premier XE Mass Spectrometer using the ESI (+) or ESI (−) method, which is coupled to an AQUITY Ultra Performance Liquid Chromatography system (Waters Corporation, Milford, MA, USA). Melting points were determined with SMP-II Digital Melting Point Automatic Apparatus (Schorpp Geaetetechnik, Überlingen, Germany) and are uncorrected. Elemental analyses were performed with a LECO-932 (C, H, N, S-Elemental Analyzer) at the Faculty of Pharmacy, Ankara University. Ethyl 4-(1H-indol-3-yl)-2,4-dioxobutanoate (2) and ethyl 3-(1H-indol-3-yl)-pyrazole-5-carboxylate (3) were synthesized as reported previously [37].
2.1.1. General procedure for 3-(1H-indol-3-yl)-1H-pyrazole-5-carboxylic acid synthesis (9)
The intermediate (8) (5.50 mmol; 1.25g) was dissolved in a methanol – THF (1:1) mixture in a round bottom flask, and LiOH (55.01 mmol; 2.30 g) in water was added dropwise at room temperature. After the addition of LiOH, reaction was then refluxed for 4 h. The reaction was monitored using TLC and after the complete consumption of starting material, reaction mixture was cooled to room temperature. Solvent was then removed by evaporation under reduced pressure, and The residue was taken in water and then acidified (pH 4–5) by addition of 2 N HCl. The solid formed was removed by filtration, then crude productwas purified by flash column chromatography using a DCM:MeOH (90:10) solvent system. M.p. 295–296 °C, yield: 83%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.10 (1H, bs), 11.36 (1H, s), 7.94 (1H, d, J = 7.2 Hz), 7.81 (1H, d, J = 2.4 Hz), 7.44 (1H, d, J = 7.6 Hz), 7.16–7.07 (2H, m), 6.99 (1H, s). 13C-NMR (DMSO-d6, 100 MHz) δC: 162.32, 136.32, 124.50, 123.63, 121.61, 119.65, 111.73, 106.25, 103.99. IR (FT-IR/ATR) cm−1: 3372 (N-H), 3149–2617 (O-H), 1686 (C=O). HRMS (m/z): [M+H]+ calcd. for C12H9N2O3 228.0773, found m/z 228.0779 (Cas No: 914351–46-5).
2.1.2. General procedure for synthesis of 3-(1H-indol-3-yl)-1H-pyrazole-3-carboxamide derivatives (10–29)
The intermediate (9) (1.5 mmol) was dissolved in DCM (15 mL). After adding DMAP (0.3 mmol) and EDC (1.8 mmol), the mixture was stirred under argone at room temperature for 1 h. Appropriate aniline derivative (1.8 mmol) was added to the mixture and stirred for 24–78 h. At the end of the reaction, DCM was evaporated, and the residue was dissolved in EtOAc, and then extracted with 1% NaHCO3 and brine. The organic layer was dried with Na2SO4 and evaporated. The crude was purified by flash chromatography and/or by crystallization utilizing an appropriate solvent system [38, 39].
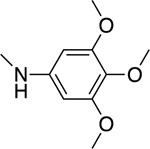
2.1.3. 3-(1H-Indol-3-yl)-N-(3,4,5-trimethoxyphenyl)-1H-pyrazole-5-carboxamide 10
Automated flash chromatography was performed using a DCM: MeOH (96: 4) solvent system. M.p. 239.5–240.5 °C, yield: 31%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.48 (0.2H, bs), 13.42 (0.8H, s), 11.51 (0.8H, s), 11.25 (0.2H, bs), 10.11 (0.2H, bs), 9.87 (0.8H, s), 7.85–7.83 (2H, m), 7.47 (1H, d, J = 7.6 Hz), 7.32 (2H, s), 7.20–7.09 (2H, m), 7,03 (1H, s), 3.76 (6H, s), 3.62 (3H, s). 13C-NMR (DMSO-d6 + 1 drop TFA, 100 MHz) δC: 160.10, 158.62 (q, J = 38.7 Hz, TFA-CO), 153.08, 145.91, 141.23, 136.67, 136.65, 135.28, 134.05, 124.89, 124.07, 124.03, 122.26, 120.30, 115.30 (q, J = 285.8 Hz, TFA-CF3), 112.34, 105.50, 102.20, 98.32, 60.36, 56.04. IR (FT-IR/ATR) cm−1: 3394–3171 (N-H), 2992 (C-H), 1657 (C=O). HRMS (m/z): [M+H]+ calcd. for C21H21N4O4 393.1563, found 393.1561. Elemental analyses for C21H20N4O4.0.2MeOH calculated: C, 63.85; H, 5.26; N, 14.05; found: C, 63.62; H, 5.26; N, 14.15.
2.1.4. N-(4-(tert-Butyl)phenyl)-3-(1H-indol-3-yl)-1H-pyrazole-5-carboxamide 11
Automated flash chromatography was performed using a DCM: MeOH (94:6) solvent system, followed by crystallization from a mixture of acetone: H2O. M.p. 305.8–306.4 °C, yield: 61%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.41 (1H, s), 11.49 (0.8H, s), 11.26 (0.2H, bs), 10.11 (0.2H, bs), 9.91 (0.8H, s), 7.84 (2H, m), 7.72 (2H, d, J = 8 Hz), 7.46 (1H, d, J = 7.6 Hz), 7.34 (2H, d, J = 7.6 Hz), 7.19–7.11 (2H, m), 7.04 (1H, s), 1.27 (9H, s). 13C-NMR (DMSO-d6, 100 MHz) δC: 160.39, 152.04, 147.54, 145.64, 139.22, 136.31, 125.15, 124.34, 123.75, 121.85, 119.95, 119.06, 111.96, 109.50, 104.38, 101.69, 34.00, 31.10. IR (FT-IR/ATR) cm−1: 3371–3188 (N-H), 2964 (C-H), 1636 (C=O). HRMS (m/z): [M+H]+ calcd. for C22H23N4O 359.1872, found 359.1871. Elemental analyses for C22H23N4O calculated: C, 73.72; H, 6.19; N, 15.63; found: C, 73.61; H, 6.19; N, 15.69.
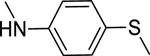
2.1.5. 3-(1H-Indol-3-yl)-N-(4-(methylthio)phenyl)-1H-pyrazole-5-carboxamide 12
Automated flash chromatography was performed using solvent system DCM: MeOH (94:6). M.p. 260–261 °C, yield: 37%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.43 (1H, s), 11.50 (0.8H, s), 11.26 (0.2H, bs), 10.18 (0.2H, bs), 10.03 (0.8H, s), 8.10 (0.2H, bs), 7.85–7.79 (4H, m), 7.46 (1H, d, J = 7.6 Hz), 7.26 (2H, d, J = 8.4 Hz), 7.19–7.11 (2H, m), 7.04 (1H, s), 2.45 (3H, s). 13C-NMR (DMSO-d6, 100 MHz) δC: 160.95, 147.92, 139.74, 136.79, 132.36, 127.40, 126.86, 124.81, 124.29, 122.37, 121.28, 120.45, 119.55, 112.45, 102.19, 15.99. IR (FT-IR/ATR) cm−1: 3370–3310 (N-H), 2974 (C-H), 1623 (C=O). HRMS (m/z): [M+H]+ calcd. for C19H17N4OS 349.1123, found 349.1127. Elemental analyses for C19H16N4OS calculated: C, 65.50; H, 4.63; N, 16.08; S, 9.20; found: C, 65.68; H, 4.63; N, 16.19; S, 9.27.
2.1.6. 3-(1H-Indol-3-yl)-N-(3,4,5-trimethoxybenzyl)-1H-pyrazole-5-carboxamide 13
Automated flash chromatography was performed using solvent system DCM: MeOH (96:4), followed by crystallization with a acetone: H2O mixture. M.p. 249.8–250.3 °C, yield: 39%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.26 (1H, s), 11.43 (1H, s), 8.55 (1H, s), 7.83–7.79 (2H, m), 7.44 (1H, d, J = 7.2 Hz), 7.16–7.11 (2H, m), 6.93 (1H, s), 6.66 (2H, s), 4.38 (2H, d, J = 6 Hz), 3.74 (6H, s), 3.62 (3H, s). 13C-NMR (DMSO-d6 + 1 drop TFA, 100 MHz) δC: 161.23, 158.58 (q, J = 38.9 Hz, TFA-CO), 153.27, 145.13, 141.39, 137.06, 136.67, 135.60, 124.89, 124.03, 122.21, 120.25, 119.63, 115.26 (q, J = 285.5 Hz, TFA-CF3), 112.30, 110.98, 105.52, 105.44, 101.98, 60.18, 56.10, 42.66. IR (FT-IR/ATR) cm−1: 3413–3137 (N-H), 2935 (C-H), 1627 (C=O). HRMS (m/z): [M-H]− calcd. for C22H21N4O4 405.1563, found 405.1562. Elemental analyses for C22H22N4O4 calculated: C, 65.01; H, 5.46; N, 13.78; found: C, 65.56; H, 5.40; N, 13.89.
2.1.7. 3-(1H-Indol-3-yl)-N-phenyl-1H-pyrazole-5-carboxamide 14
Automated flash chromatography was performed using a DCM: MeOH (94:6) solvent system. M.p. 272.5–273 °C, yield: 67%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.39 (1H, bs), 11.45 (1H, bs), 10.02 (1H, bs), 7.91 (1H, d, J = 6.8 Hz), 7.83–7.81 (3H, m), 7.46 (1H, d, J = 7.6 Hz), 7.33 (2H, t, J = 7.8 Hz), 7.19–7.05 (4H, m). 13C-NMR (DMSO-d6, 100 MHz) δC: 160.47, 157.04, 139.32, 136.79, 129.03, 124.94, 124.10, 123.84, 122.28, 120.32, 119.87, 119.82, 112.40, 102.30. IR (FT-IR/ATR) cm−1: 3370–3165 (N-H), 1623 (C=O). HRMS (m/z): [M+H]+ calc. for C18H15N4O 303.1246, found 303.1246. Elemental analyses for C18H14N4O.0.5MeOH calculated: C, 69.80; H, 5.07; N, 17.60; found: C, 69.60; H, 4.75; N, 17.87.
2.1.8. 3-(1H-Indol-3-yl)-N-(4-methoxyphenyl)-1H-pyrazole-5-carboxamide 15
Automated flash chromatography was performed using hexane:ethyl acetate (50:50) solvent system followed by crystallization with a hexane: EtOAc mixture. M.p. 265–266 °C, yield: 36%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.48 (0.2H, bs),13.40 (0.8H, s), 11.50 (0.8H, s), 11.25 (0.2H, bs), 10.09 (0.2H, bs), 9.89 (0.8H, s), 7.85 (2H, m), 7.72 (2H, d, J = 8 Hz), 7.47 (1H, d, J = 7.2 Hz), 7.18–7.14 (2H, m), 7.03 (1H, s), 6.90 (2H, d, J = 8.8 Hz), 3.73 (3H, s). 13C-NMR (DMSO-d6 + 1 drop TFA, 100 MHz) δC: 159.74, 158.61 (q, J = 38.7 Hz, TFA-CO), 155.94, 145.67, 136.66, 132.14, 124.90, 124.03, 122.24, 122.17, 120.28, 119.72, 115.29 (q, J = 288.7 Hz, TFA-CF3), 114.10, 112.32, 111.00, 105.58, 102.21, 55.38. IR (FT-IR/ATR) cm-1: 3317–3120 (N-H), 2929 (C-H), 1600 (C=O). HRMS (m/z): [M+H]+ calculated for C19H17N4O2 333.1352, found 333.1351. Elemental analyses for C19H16N4O2.0.2hexane calculated: C, 69.40; H, 5.42; N, 16.03; found: C, 69.25; H, 5.45; N, 16.04.
2.1.9. N-(3,5-Dimethoxyphenyl)-3-(1H-indol-3-yl)-1H-pyrazole-5-carboxamide 16
Automated flash chromatography was performed using a DCM: MeOH (96:4) solvent system, followed by crystallization with a acetone: H2O mixture. M.p. 126–127 °C, yield: 94%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.50 (0.2H, bs),13.43 (0.8H, s), 11.50 (0.8H, s), 11.25 (0.2H, bs), 10.11 (0.2H, bs), 9.90 (0.8H, s), 8.10 (0.15H, bs) 7.85 (1.7H, s), 7.71 (0.15H, bs), 7.46 (1H, d, J = 7.6 Hz), 7.18–7.14 (4H, m), 7.04 (1H, s), 6.23 (1H, s), 3.72 (6H, s). 13C-NMR (DMSO-d6, 100 MHz) δC: 160.82, 147.91, 141.02, 139.77, 136.77, 124.80, 124.31, 122.40, 120.47, 119.52, 112.48, 104.80, 102.18, 98.86, 95.84, 55.55. IR (FT-IR/ATR) cm−1: 3540–3265 (N-H), 2945 (C-H), 1668 (C=O). HRMS (m/z): [M+H]+ calculated for C20H19N4O3 363.1457, found 363.1459. Elemental analyses for C20H18N4O3.1H2O calculated: C, 63.15; H, 5.30; N, 14.73; found: C, 63.19; H, 5.32; N, 15.00.
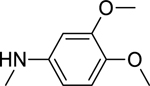
2.1.10. N-(3,4-Dimethoxyphenyl)-3-(1H-indol-3-yl)-1H-pyrazole-5-carboxamide 17
Automated flash chromatography was performed by using a DCM: MeOH solvent system (96:4), followed by crystallization from a mixture of acetone: H2O. M.p. 215–216 °C, yield: 28%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.44 (0.2H, s), 13.39 (0.8H, s), 11.50 (0.8H, s), 11.25 (0.2H, bs), 10.06 (0.2H, bs), 9.84 (0.8H, s), 8.11 (0.2H, s) 7.85 (1.6H, s), 7.70 (0.2H, bs), 7.54 (1H, s), 7.46 (1H, d, J = 8 Hz), 7.38 (1H, d, J = 8 Hz), 7.18–7.12 (2H, m), 7.03 (1H, s), 6.90 (1H, d, J = 8.8 Hz), 3.75 (3H, s), 3.72 (3H, s). 13C-NMR (DMSO-d6, 100 MHz) δC: 161.54, 149.81, 148.90, 146.29, 140.50, 137.63, 133.81, 125.67, 125.06, 123.20, 121.27, 120.33, 113.38, 106.84, 105.72, 102.90, 57.08, 56.74. IR (FT-IR/ATR) cm−1: 3338–3165 (N-H), 2931 (C-H), 1650 (C=O). HRMS (m/z): [M-H]+ calculated for C20H19N4O3 363.1457, found 363.1459. Elemental analyses for C20H18N4O3.0.25H2O calculated: C, 65.47; H, 5.08; N, 15.27; found: C, 65.50; H, 4.99; N, 15.38.
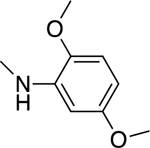
2.1.11. N-(2,5-Dimethoxyphenyl)-3-(1H-indol-3-yl)-1H-pyrazole-5-carboxamide 18
Automated flash chromatography was performed using a DCM: MeOH (96:4) solvent system, followed by crystallization with a mixture of acetone: H2O. M.p. 197.5–198.5 °C, yield: 76%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.51 (1H, s), 11.53 (1H, s), 9.43 (1H, s), 8.06 (1H, d, J = 2.8 Hz), 7.86–7.84 (2H, m), 7.47 (1H, d, J = 7.2 Hz), 7.20–7.11 (2H, m), 7.02–6.99 (2H, m), 6.63 (1H, dd, J = 8.6, 2.4 Hz), 3.86 (3H, s), 3.71 (3H, s). 13C-NMR (DMSO-d6, 100 MHz) δC: 159.52, 153.11, 146.93, 142.09, 139.92, 136.25, 127.88, 124.17, 123.98, 121.90, 120.04, 118.97, 111.97, 111.39, 107.04, 105.75, 104.14, 101.28, 56.33, 55.26. IR (FT-IR/ATR) cm−1: 3388–3199 (N-H), 2942 (C-H), 1656 (C=O). HRMS (m/z): [M+H]- calculated for C20H17N4O3 361.1301, found 361.1299. Elemental analyses for C20H18N4O3 calculated: C, 66.29; H, 5.01; N, 15.46; found: C, 66.12; H, 4.97; N, 15.66.
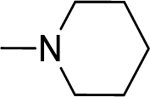
2.1.12. (3-(1H-Indol-3-yl)-1H-pyrazol-5-yl)(piperidin-1-yl)methanone 19
Automated flash chromatography was performed using a DCM: MeOH (96:4) solvent system, followed by crystallization with a mixture of hexane: EtOAc. M.p. 205–206 °C, yield: 87%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.18 (1H, s), 11.45 (0.8H, s), 11.21 (0.2H, bs), 7.81 (1H, d), 7.77 (1H, s), 7.43 (1H, d, J = 7.6 Hz), 7.10–7.15 (2H, m), 6.77 (1H, s), 3.85–3.58 (4H, m), 1.63–1.52 (6H, m). 13C-NMR (DMSO-d6 + 1 drop TFA, 100 MHz) δC: 162.16, 158.61 (q, J = 38.2 Hz, TFA-CO), 145.43, 140.23, 136.82, 124.85, 124.02, 122.19, 120.25, 119.80, 115.43 (q, J = 228.4 Hz, TFA-CF3), 112.31, 105.58, 103.23, 47.85, 43.14, 26.73, 25.83, 24.52. IR (FT-IR/ATR) cm−1: 3406–3115 (N-H), 2998 (C-H), 1577 (C=O). HRMS (m/z): [M+H]+ calculated for C17H19N4O 295.1559, found 295.1556. Elemental analyses for C17H18N4O calculated: C, 69.37; H, 6.16; N, 19.03; found: C, 69.74; H, 6.71; N, 18.89.
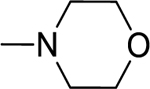
2.1.13. (3-(1H-Indol-3-yl)-1H-pyrazol-5-yl)(morpholino)methanone 20
Automated flash chromatography was performed using a DCM: MeOH (90:10) solvent system, followed by crystallization with EtOAc. M.p. 235–236.5 °C, yield: 72%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.25 (1H, s), 11.46 (0.8H, s), 11.21 (0.2H, bs) 8.11 (0.2H, bs), 7.79 (1.8H, s), 7.44 (1H, d, J = 6.8 Hz), 7.16–7.10 (2H, m), 6.83 (1H, s), 4.02, 3.63 (8H, m). 13C-NMR (DMSO-d6, 100 MHz) δC: 162.80, 147.66, 138.45, 136.75, 124.74, 124.13, 122.33, 120.41, 119.51, 112.43, 104.84, 103.94, 67.01, 66.72, 47.60, 42.80. IR (FT-IR/ATR) cm−1: 3255–3132 (N-H), 2924–2859 (C-H), 1592 (C=O). HRMS (m/z): [M+H]+ calculated for C16H17N4O2 297.1352, found 297.1364. Elemental analyses for C16H16N4O2 calculated: C, 64.85; H, 5.44; N,18.91; found: C, 64.86; H, 5.41; N, 18.71.
2.1.14. N-Butyl-3-(1H-indol-3-yl)-1H-pyrazole-5-carboxamide 21
Automated flash chromatography was performed using a DCM: MeOH (90:10) solvent system, followed by crystallization with a hexane: EtOAc mixture. M.p. 165–166 °C, yield: 70%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.21 (1H, s), 11.42 (0.8H, s), 11.21 (0.2H, bs), 8.03 (1H, s), 7.82–7.77 (2H, m), 7.43 (1H, d, J = 7.6 Hz), 7.15–7.10 (2H, m), 6.89 (1H, s), 3.24–3.21 (2H, m), 1.49 (2H, q, J = 7.4 Hz), 1.32 (2H, q, J = 7.4 Hz), 0.89 (3H, t, J = 7.2 Hz). 13C-NMR (DMSO-d6 + 1 drop TFA, 100 MHz) δC: 160.49, 158.58 (q, J = 39.0 Hz, TFA-CO), 144.89, 141.59, 136.67, 124.88, 124.18, 122.24, 120.29, 115.25 (q, J = 228.7 Hz, TFA-CF3) 112.33, 110.97, 105.36, 101.83, 38.53, 31.66, 19.92, 13.72. IR (FT-IR/ATR) cm−1: 3378–3111 (N-H), 2962–2931 (C-H), 1619 (C=O). HRMS (m/z): [M+H]+ calculated for C16H19N4O 283.1559, found 283.1557. Elemental analyses for C16H18N4O calculated: C, 68.06; H, 6.43; N, 19.84; found: C, 68.09; H, 6.60; N, 19.61.
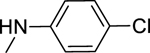
2.1.15. N-(4-Chlorophenyl)-3-(1H-indol-3-yl)-1H-pyrazole-5-carboxamide 22
Automated flash chromatography was performed using a DCM: MeOH (92:8) solvent system, followed by crystallization from a mixture of DCM: MeOH. M.p. 220–220.5 °C, yield: 56%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.44 (1H, s), 11.49 (0.8H, s), 11.24 (0.2H, bs), 10.27 (0.2H, bs), 10.17 (0.8H, s), 8.09 (0.3H, bs), 7.89–7.85 (3.7H, m), 7.46 (1H, d, J = 8 Hz), 7.38 (2H, d, J = 8.8 Hz), 7.20–7.11 (2H, m), 7.05 (1H, s). 13C-NMR (DMSO-d6, 100 MHz) δC: 161.16, 147.77, 139.29, 138.38, 136.81, 128.88, 127.39, 124.84, 124.33, 122.10, 120.47, 119.54, 112.47, 104.81, 102.28. IR (FT-IR/ATR) cm−1: 3370–3109 (N-H), 1624 (C=O). HRMS (m/z): [M+H]+ calculated for C18H14N4OCl 337.0856, found 337.0859. Elemental analyses for C18H13N4OCl calculated: C, 64.19; H, 3.89; N, 16.64; found: C, 63.75; H, 3.90; N, 16.74.
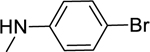
2.1.16. N-(4-Bromophenyl)-3-(1H-indol-3-yl)-1H-pyrazole-5-carboxamide 23
Automated flash chromatography was performed using a DCM: MeOH (92:8) solvent system, followed by crystallization from a mixture of DCM: MeOH. M.p. 326–326.5 °C, yield: 60%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.55 (0.2H, bs), 13.45 (0.8H, s), 11.50 (0.8H, s), 11.25 (0.2H, bs), 10.30 (0.2H, bs), 10.18 (0.8H, s), 8.11 (0.3H, bs), 7.85–7.82 (3.7H, m), 7.50 (2H, d, J = 8.4 Hz), 7.46 (1H, d, J = 7.6 Hz), 7.20–7.12 (2H, m), 7.04 (1H, s). 13C-NMR (DMSO-d6, 100 MHz) δC: 161.16, 147.75, 139.79, 138.82, 136.76, 131.78, 124.79, 124.33, 122.54, 122.40, 120.47, 119.53, 115.42, 112.48, 104.78, 102.27. IR (FT-IR/ATR) cm−1: 3368–3107 (N-H), 1623 (C=O). HRMS (m/z): [M+H]- calculated for C18H12N4OBr 379.0194, found 379.0194. Elemental analyses for C18H13N4OBr calculated: C, 56.71; H, 3.44; N, 14.70; found: C, 56.21; H, 3.51; N, 14.94.
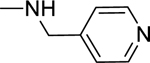
2.1.17. 3-(1H-Indol-3-yl)-N-(pyridin-4-ylmethyl)-1H-pyrazole-5-carboxamide 24
Automated flash chromatography was performed using a DCM: MeOH (90:10) solvent system. M.p. 169–171 °C, yield: 92%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.37 (0.2H, bs), 13.34 (0.8H, s), 11.47 (0.8H, s), 11.22 (0.2H, bs), 9.09 (0.2H, bs), 8.83 (0.8H, t, J = 5.2 Hz), 8.48 (2H, d, J = 5.2 Hz), 8.07 (0.2H, bs), 7.83–7.81 (1.6H, m), 7.67 (0.2H, bs), 7.45 (1H, d, J = 8.4 Hz), 7.29 (2H, d, J = 5.2 Hz), 7.19–7.10 (2H, m), 6.94 (1H, s), 4.46 (2H, d, J = 6 Hz). 13C-NMR (DMSO-d6, 100 MHz) δC: 162.70, 149.90, 149.36, 147.59, 139.41, 136.76, 124.78, 124.13, 122.59, 122.35, 120.42, 119.50, 112.44, 104.94, 101.83, 41.57. IR (FT-IR/ATR) cm−1: 3367–3318 (N-H), 2848 (C-H), 1626 (C=O). HRMS (m/z): [M+H]+ calculated for C18H16N5O 318.1355, found 318.1350. Elemental analyses for C18H15N5O calculated: C, 68.13; H, 4.76; N, 22.07; found: C, 68.17; H, 4.84; N, 21.62.
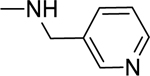
2.1.18. 3-(1H-Indol-3-yl)-N-(pyridin-3-ylmethyl)-1H-pyrazole-5-carboxamide 25
Automated flash chromatography was performed using a DCM: MeOH (90:10) solvent system, followed by crystallization with a DCM: MeOH mixture. M.p. 278–281 °C, yield: 82%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.28 (1H, s), 11.42 (0.8H, s), 11.17 (0.2H, bs), 9.00 (0.2H, bs), 8.75 (0.8H, s), 8.56 (1H, s), 8.44 (1H, d, J = 4 Hz), 8.05 (0.3H, bs), 7.80 (1.7H, s), 7.72 (1H, d, J = 8 Hz), 7.45 (1H, d, J = 7.6 Hz), 7.34–7.31 (1H, m), 7.16–7.11 (2H, m), 6.93 (1H, s), 4.47 (2H, d, J = 5.6 Hz). 13C-NMR (DMSO-d6, 100 MHz) δC: 162.12, 150.86, 148.80, 147.87, 147.18, 138.82, 136.26, 135.30, 135.02, 124.29, 123.53, 123.27, 121.76, 119.80, 118.94, 111.84, 104.43, 101.26. IR (FT-IR/ATR) cm−1: 3414–3312 (N-H), 2884 (C-H), 1653 (C=O). HRMS (m/z): [M+H]+ calculated for C18H16N5O 318.1355, found 318.1350. Elemental analyses for C18H15N5O calculated: C, 68.13; H, 4.76; N, 22.07; found: C, 68.37; H, 4.83; N, 21.88.
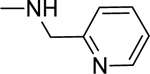
2.1.19. 3-(1H-Indol-3-yl)-N-(pyridin-2-ylmethyl)-1H-pyrazole-5-carboxamide 26
Automated flash chromatography was performed using DCM: MeOH (90:10). M.p. 129–131 °C, yield: 84%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.29 (1H, s), 11.43 (0.8H, s), 11.17 (0.2H, bs), 9.00 (0.2H, bs), 8.65 (0.8H, s), 8.51 (1H, d, J = 4.8 Hz), 8.07 (0.3H, bs), 7.84–7.83 (1.7H, m), 7.74 (1H, td, J = 8, 1.6 Hz), 7.45 (1H, d, J = 8 Hz), 7.33 (1H, d, J= 8 Hz), 7.26–7.10 (3H, m), 7.10 (1H, s), 4.57 (2H, d, J = 5.6 Hz). 13C-NMR (DMSO-d6, 100 MHz) δC: 162.58, 159.05, 149.20, 147.78, 139.45, 137.09, 136.83, 124.86, 124.13, 122.44, 121.42, 120.40, 119.51, 112.43, 109.98, 105.03, 101.80, 44.40. IR (FT-IR/ATR) cm−1: 3377–3167 (N-H), 2918 (C-H), 1626 (C=O). HRMS (m/z): [M+H]- calculated for C18H14N5O 318.1355, found 318.1354. Elemental analyses for C18H15N5O.0.2ethyl acetate calculated: C, 67.41; H, 4.99; N, 20.91; found: C, 67.13; H, 5.28; N, 21.30.
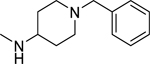
2.1.20. N-(1-Benzylpiperidin-4-yl)-3-(1H-indol-3-yl)-1H-pyrazole-5-carboxamide 27
Automated flash chromatography was performed using a DCM: MeOH (90:10) solvent system, followed by crystallization with a hexane - EtOAc mixture. M.p. 235–236 °C, yield: 71%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.19 (1H, s), 11.41 (0.8H, s), 11.15 (0.2H, bs), 8.16–7.65 (3H, m), 7.45 (1H, d, J = 8 Hz), 7.32–7.10 (7H, m), 6.90 (1H, s), 3.78–3.76 (1H, m), 3.45 (2H, s), 2.79, 2.03, 1.75, 1.62 (8H, m). 13C-NMR (DMSO-d6, 100 MHz) δC: 161.68, 148.06, 139.28, 139.12, 136.80, 129.16, 128.55, 127.24, 124.86, 124.08, 122.32, 120.36, 119.51, 112.40, 109.98, 105.04, 101.72, 62.59, 52.66, 45.54, 32.02. IR (FT-IR/ATR) cm−1: 3404–3156 (N-H), 2939–2816 (C-H), 1643 (C=O). HRMS (m/z): [M+H]+ calculated for C24H26N5O 400.2137, found 400.2136. Elemental analyses for C24H25N5O calculated: C, 72.16; H, 6.31; N, 17.53; found: C, 72.11; H, 6.54; N, 17.34.
2.1.21. (3-(1H-Indol-3-yl)-1H-pyrazol-5-yl)(4-(4-(trifluoromethyl)phenyl)piperazin-1yl)methanone 28
Automated flash chromatography was performed using a DCM: MeOH (92:8) solvent system, followed by crystallization with a mixture of acetone-H2O. M.p. 298–298.5 °C, yield: 78%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.30 (1H, s), 11.48 (0.8H, s), 11.23 (0.2H, bs), 8.13 (0.2H, bs), 7.83–7.80 (1.8H, m), 7.51 (2H, d, J = 8.8 Hz), 7.45 (1H, d, J = 7.6 Hz), 7.17–7.06 (4H, m), 6.87 (1H, s), 4.18 (2H, s), 3.79 (2H, s), 3.37 (4H, m). 13C-NMR (DMSO-d6, 100 MHz) δC: 162.28, 152.95, 147.24, 137.97, 136.25, 126.12 (q, 3JC-F = 3.6 Hz), 124.23 (q, 1JC-F = 245.6 Hz), 123.61, 123.53, 120.40, 119.90, 117.99, 114.25, 111.91, 109.45, 104.32, 103.45, 47.55, 46.81, 45.77, 41.46. IR (FT-IR/ATR) cm−1: 3406–3120 (N-H), 2922–2856 (C-H), 1595 (C=O). HRMS (m/z): [M+H]+ calculated for C23H21F3N5O 440.1698, found 440.1695. Elemental analyses for C23H20F3N5O calculated: C, 62.86; H, 4.59; N, 15.94; found: C, 63.09; H, 4.87; N, 15.47.
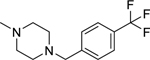
2.1.22. (3-(1H-Indol-3-yl)-1H-pyrazol-5-yl)(4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)methanone 29
Automated flash chromatography was performed using a DCM: MeOH (90:10) solvent system, followed by crystallization with an acetone-H2O mixture. M.p. 229.4–230 °C, yield: 95%. 1H-NMR (DMSO-d6, 400 MHz) δH: 13.26 (1H, s), 11.49 (1H, s), 7.84–7.80 (2H, m), 7.70 (2H, d, J = 8.4 Hz), 7.57 (2H, d, J = 8.4 Hz), 7.46 (1H, d, J = 7.6 Hz), 7.18–7.12 (2H, m), 6.83 (1H, s), 4.02 (2H, s), 3.66–3.62 (4H, m), 2.46–2.44 (4H, m). 13C-NMR (DMSO-d6 + 1 drop TFA, 100 MHz) δC: 162.38, 158.68 (q, J = 30.8 Hz, TFA-CO), 145.93, 139.51, 136.82, 136.66, 134.29, 132.66, 130.53 (q, 2JC-F = 32.1 Hz), 126.13 (q, 3JC-F = 3.7 Hz), 124.77 (q, 1JC-F = 245.6 Hz), 123.27, 122.31, 120.37, 118.84, 115.25 (q, J = 228.7 Hz, TFA-CF3), 114.26, 112.40, 111.98, 105.02, 104.29, 58.60, 51.59, 43.71. IR (FT-IR/ATR) cm−1: 3311–3107 (N-H), 2951–2830 (C-H), 1591 (C=O). HRMS (m/z): [M+H]+ calculated for C24H23F3N5O 454.1855, found 454.1853. Elemental analyses for C24H22F3N5O calculated: C, 63.57; H, 4.89; N, 15.44; found: C, 63.88; H, 5.06; N, 15.48.
2.2. Biological evaluation
2.2.1. Cell culture
MCF-7, Huh7, Mahlavu, HepG2, and HCT116 were grown in DMEM supplemented with 10% FBS (fetal bovine serum), 0.1 mM NEAA (non-essential amino acids) (GIBCO, Invitrogen), 100 units/mL penicillin and streptomycin (Invitrogen GIBCO) whereas SNU475 was grown in RPMI medium containing 10% FBS, 2 mM L-glutamine, and 100 units/mL penicillin and streptomycin in a 37 °C, 5% CO2 incubator.
2.2.2. NCI-60 SRB assay
The SRB assay was performed as described previously [35]. Cells were seeded into 96-well cell culture plates, and treated with each compound in increasing concentrations (40, 20, 10, 5, 2.5 μM) in triplicates. After 72 h, cells were fixed with 10% TCA and stained with SRB (0.04 g/10 mL, Sigma-Aldrich). Absorbance measurements were done at 515 nm after addition of 10 mM Tris-base solution into each well.
2.2.3. Real-time cell monitoring of HCC cells
xCELLigence System (Agilent Technologies) was used to determine real-time cell growth inhibition in HCC cell lines, as described previously [35]. HCC cells were seeded into 96-well E-plates and treated with the selected compounds at the indicated concentrations in triplicates after 24 h. The cell index (CI) values were recorded every 30 min. Time-dependent cell growth curves were generated for each compound relative to DMSO.
2.2.4. Tubulin polymerization
Electrophoretically homogenous tubulin obtained from bovine brains [40] was used to evaluate effects on tubulin assembly and on the binding of [3H]colchicine to tubulin. In the assembly studies, tubulin and various compound concentrations were initially incubated at 30 °C for 15 min and then chilled on ice, followed by addition of 10 μL of 0.01 M GTP was added. Baselines were obtained at 350 nm, and the temperature was raised to 30 °C over 30 s. Reactions were followed by development of turbidity, measured by an apparent change in absorbance at 350 nm, and the IC50 was the compound concentration that inhibited the increase in turbidity by 50% at 20 min. Detailed methods have been presented [41]. The method is generally most reliable at compound concentrations no greater than 20 μM. At higher concentrations, interference with the turbidity development caused by tubulin polymerization might occurred.
2.2.5. Inhibition of colchicine binding
Evaluation of inhibition of the binding of [3H]colchicine to tubulin was determined as previously reported [42, 43] with modifications in the reaction mixtures which contained 1 μM tubulin, 5 μM [3H]colchicine and 5 μM and/or 50 μM test compound.
2.2.6. Detection of apoptosis
Nuclear structures indicating apoptotic cell death were visualized fluorescently via Hoechst 33258 staining, as described previously [35]. Briefly, cells that were seeded onto cover slips inside 6 well plates were treated with the IC100 concentrations of compounds the next day. After 48 h of incubation, cells were fixed with 100% cold methanol. 1 μg/mL Hoechst dye solution was added onto each well to stain cell nuclei which were visualized using fluorescence microscopy with a blue filter (340–380 nm).
2.2.7. Flow cytometry for cell cycle analysis
Cell cycle analysis upon treatment with selected compounds was performed as described previously [35]. Cells were seeded into 6-well culture plates. The next day, cells were treated with the IC50 or a 10 μM concentrations of compounds for 72 h. Then cells were fixed with 70% cold EtOH and stained with a propidium iodide staining solution (MUSE Cell cycle kit, Millipore) to be analyzed by flow cytometry.
2.3. Computational studies
2.3.1. Molecular docking studies
The reported structures were sketched by Maestro 12.9 [44] and prepared at pH 7.0 ± 2.0 with LigPrep [45]. Protein Preparation Wizard [46] was utilized for parameterizing tubulin (PDB code 5LYJ [47]). Atom types were set with OPLS4 force field for ligands and protein. Molecular docking simulations were conducted with Glide 9.2[48–50]. Compounds were docked into the colchicine site that is located between the α1 and β1 tubulins. The active site coordinates were assigned by using CA-4 that is co-crystallized with the protein. The Van der Waals radius scaling factor was used as 1.0, and partial charge cutoff value was used as 0.25. The simulations were performed in extra precision mode (GlideScore XP). The docking scores and calculated descriptors for 10, and 18 (Table S1) were selected by the top-ranking docking results. The docking pose of 18 was visualized (Figure 6) at the colchicine binding region. Protein-ligand contacts were classified with PLIP 2.2.2 [51], and figures were rendered with PyMOL 2.4.1 [52].
Figure 6.

18 visualized within the colchicine site. α tubulin is shown as green sticks, β tubulin as cyan sticks and 18 as orange sticks. Interacting residues and interaction types were identified with PLIP 2.2.2 [51], and the image was prepared with PyMOL 2.4.1 [52].
2.3.2. Quantum chemistry
We utilized density functional theory (DFT) calculations to map electrostatic potential surface map (Figure 7 A) and frontier orbitals based on the docking generated binding orientation compound 18. The calculations were run with Jaguar 11.3 [53]. The binding pose of the ligand was extracted from the binding cavity and the bond orders were optimized by the software with B3LYP-D3 theory and 6–31G** basis set in accurate mode and electrostatic potential surfaces were mapped with by using default settings of the software. Later the figures were generated with PyMOL 2.4.1 [52].
Figure 7.

Binding mode of 18 extracted from the colchicine binding site. A) Visualization of electrostatic potential surface occurred by the partial charge distributions. Representations of B) HOMO, C) HOMO −1, D) LUMO and E) LUMO +1 for compound 18, generated with its bound conformation. Red indicates negative electron density and blue indicates positive.
3. Results and Discussion
3.1. Chemistry
The synthesis of indole-pyrazole hybrids 10–29 was achieved as presented in Scheme 1. Firstly, ethyl 4-(1H-indol-3-yl)-2,4-dioxobutanoate 7 [37, 54] and ethyl 3-(1H-indol-3-yl)pyrazole-5-carboxylate 8 [30] were synthesized according to previously published methods. Then, by using lithium hydroxide, the intermediate 8 was hydrolyzed into 3-(1H-indol-3-yl)pyrazole-5-carboxylic acid 9 [37]. Finally, to provide the carboxamides 10–29, DMAP as covalent nucleophilic catalyst [55] and EDC as activating agent and were used to activate the carboxylic acid, and then it was treated with different amines [38, 39].
Scheme 1.

Synthesis of title compounds. Reagents and conditions: a) THF/NaOEt and diethyl oxalate; b) EtOH and NH2NH2.H2O; c) MeOH/THF/H2O, LiOH; d) aniline or amine derivative, EDC/DMAP in DCM (R groups are the aniline or amine derivatives that is listed in Table 1).
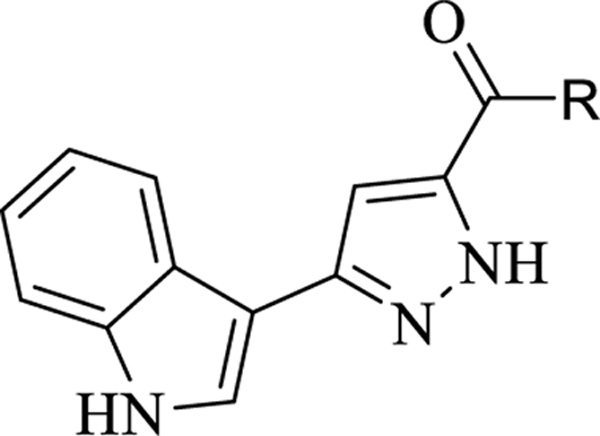
For final compounds 10-29, the IR spectra showed strong bands caused by carbonyl groups in the region between 1691–1577 cm−1. In the 1H-NMR spectrum of all synthesized indole-pyrazole-carboxamide derivatives, all protons gave appropriate cleavage and chemical shift values consistent with their chemical structures. In addition, some protons displayed two different chemical shift values in most indole-pyrazole carboxamide derivatives due to the tautomerism in the pyrazole ring. According to the literature, the chemical shift values for the pyrazole ring NH proton in the 1H-NMR spectra of the compounds were recorded as two separate singlets, such as 30 (Figure 2) [56]. It has been reported that the proton on the nitrogen atom at the 1st or 2nd position (N1-H form or N2-H form; Figure 2) is affected by the group at the 5th position of the pyrazole. Electron withdrawing groups such as BH2, COOH and CHO on the 5th position of the pyrazole ring stabilize the N1-H form, while electron donating groups such as OH, NH2, Cl, CONH2, CN and CH3 stabilize the N2-H form (Figure 2) [57].
Figure 2.

Compound VI and the tautomer forms of the pyrazole ring relative to the group in position 5 of the pyrazole (N1-H form R = BH2, COOH, COOEt and CHO; N2-H form R2 = OH, NH2, Cl, CONH2, CN and CH3).
According to the literature [56, 57], it can be speculated that compound 9 (3-(1H-indol-3-yl)-1H-pyrazole-5-carboxylic acid), which has a COOH group on the 5th position of the pyrazole ring, stabilizes the N1-H form, while final products that contain an amide group stabilize the N2-H form. Two separate signals were observed for all compounds except 9, 13, 14, 18, and 29, which may mean that compound 4 strongly stabilizes the N1-H form, while compounds 13, 14, 18, and 29 strongly stabilize the N2-H form. The remainder of the final compounds stabilize both forms because of different substituted groups on the phenyl ring. For example, two separate signals in 16 (for each NH) and just one signal for the pyrazole NH of 18 were observed (Figure S1) in their 1H-NMR spectra.
Meanwhile, the NH protons of the pyrazole ring were observed as one or two separate singlets, in the range of 13.48–13.18 ppm. The proton at the 4th position of the pyrazole was observed as a singlet between 7.66–7.03 ppm. The indole N-H proton was observed in the range of 11.51–11.10 ppm, and then the NH proton belonging to the amide group was observed at 10.11–8.03 ppm. For methoxy groups carrying derivatives (10, 13–17), methoxy protons were observed as singlets in the range of 3.86–3.62 ppm. The signals of carbonyl carbons in the 13C-NMR spectrums of final compounds in DMSO-d6 were observed at 160–159 ppm. 13C-NMR spectra of some derivatives were not clear due to tautomerism. Thus, we used trifluoroacetic acid (TFA) for running 13C-NMR spectra for compounds 10, 13, 15, 19, 21, and 29.
3.2. Biological evaluations
3.2.1. Cytotoxicity evaluation of the title compounds
Title compounds 10–29 were initially evaluated against breast (MCF-7), colon (HCT116) and liver (Huh7) cancer cell lines by the sulforhodamine B (SRB) assay. The IC50 values after 72 h treatments with each compound were calculated in comparison with the positive control sorafenib as shown in Table 1.
Table 1.
Cytotoxicity of the target compounds 10–29 indicated with their IC50 values in three human cancer cell lines
| ||||
|---|---|---|---|---|
| IC50 (μM) ± S.D. | ||||
| Compound | R | Huh7 | MCF-7 | HCT116 |
| 10 |
|
1.0±0.2 | 2.1±0.1 | 2.5±0.3 |
| 11 |
|
0.6±0.4 | 1.0±0.1 | 1.2±0.5 |
| 12 |
|
6.3±1.6 | 2.0±0.2 | 9.0±1.9 |
| 13 |
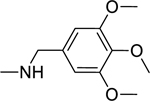
|
17.2±2.1 | 17.9±1.0 | 31.0±2.4 |
| 14 |
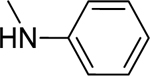
|
16.8±3.8 | 22.2±1.8 | NI |
| 15 |
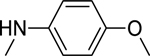
|
8.6±2.1 | 5.5±0.5 | 11.9±1.0 |
| 16 |
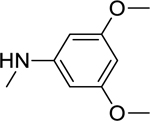
|
4.5±0.1 | 2.6±0.5 | 6.2±1.0 |
| 17 |
|
2.8±1.2 | 1.7±0.4 | 10.1±0.7 |
| 18 |
|
0.6±0.1 | 3.6±0.4 | 1.4±0.5 |
| 19 |
|
19.1±1.8 | 21.7±2.0 | 23.0±3.5 |
| 20 |
|
NI | NI | NI |
| 21 |
|
NI | 26.7±0.5 | 23.1±5.1 |
| 22 |
|
NI | 29.8±3.1 | NI |
| 23 |
|
NI | 39.3±1.0 | NI |
| 24 |
|
30.4±1.4 | 31.2±2.6 | 33.2±1.0 |
| 25 |
|
29.1±1.0 | 36.5±4.0 | 26.8±2.1 |
| 26 |
|
35.4±1.8 | NI | 34.0±1.1 |
| 27 |
|
6.8±0.7 | 8.6±0.2 | 8.9±0.7 |
| 28 |
|
16.2±3.2 | 16.1±1.7 | 27.6±2.4 |
| 29 |
|
1.7±1.8 | 3.0±0.8 | 4.1 ±2.0 |
| Sorafenib | 6.5±0.7 | 13.8±1.2 | 12.0±0.6 | |
NI: no inhibition (maximum concentration examined, 50 μM)
Compound 11 was the most potent compound against all three lines, with an IC50 range of 0.6–1.2 μM, while compound 10, which has a 3,4,5-trimethoxyphenyl moiety, also had potent activity, with an IC50 range of 1.0–2.5 μM. The IC50 values were between 0.6–11.9 μM for 10–12, 15–18, 27 and 29. Compounds 13, 14, 19-26 and 28 were less active than sorafenib, while compounds 10–12, 15–18, 27, and 29 were more effective than sorafenib against the three cell lines.
Generally, the most potent compounds (such as 10 and 11) had a phenyl moiety linked directly to the amide with electron donating substituents on the phenyl ring, including tert-butyl, methoxy, and methylthio groups. In contrast, activity was decreased when the compounds had one carbon atom between the phenyl and amide moieties (cf. 13 with 10). Both an unsubstituted phenyl ring (14) or one bearing electron withdrawing groups (22, Cl or 23, Br) in the amide moiety caused a decrease in antiproliferative activity. When the amide nitrogen was substituted with aliphatic groups instead of aromatic groups (19, 20), a significant decrease in activity was again observed. In conclusion, for better activity, the phenyl ring should be directly attached to the amide group and substituted with alkyl or methoxy groups (electron donating groups).
Three compounds (10, 11 and 18) were chosen for further anticancer evaluation against the Huh7, HepG2, Mahlavu and SNU475 cell lines in comparison with compound IV. They showed potent anticancer activity against these HCC lines, with IC50 values lower than 5 μM, which were all lower than IC50 values of sorafenib (Table 2).
Table 2.
IC50 values of compounds 10, 11 and 18, and lead compound 4 for HCC cell lines
| IC50 (μM) ± S.D. | ||||
|---|---|---|---|---|
|
| ||||
| Compound | Huh7 | HepG2 | Mahlavu | SNU475 |
| 10 | 1.0±0.2 | 2.9±0.1 | 2.7±0.4 | 4.1±2.1 |
| 11 | 0.6±0.4 | 2.7±0.9 | 3.4±0.8 | 4.3±0.9 |
| 18 | 0.6±0.2 | 1.3±0.09 | 2.9±0.8 | 2.9±1.13 |
| 4 | 0.5±0.2 | 3.6±0.6 | 1.0±0.1 | 0.3±0.1 |
| Sorafenib | 6.5±0.7 | 4.4 ± 0.8 | 8.3 ± 1.0 | 9.6 ± 0.5 |
These four compounds showed potent antiproliferative activity against the Huh7 cancer cell line, with IC50 values ≤1.0 μM. These compounds also showed potent antiproliferative activity against the HepG2 line, with IC50 values below 3 μM, which was better than our lead compound 4 (Figure 1). We also observed significant antiproliferative activity against the Mahlavu and SNU-475 cancer cell lines.
3.2.2. Inhibition of tubulin polymerization and colchicine binding
Based on their structural resemblance to CA-4, we considered tubulin as a potential target for our active compounds. To investigate whether the antiproliferative activity of these compounds is due to an interaction with tubulin, we evaluated the effects of 10, 11, and 18 on the polymerization of purified tubulin, using the highly potent CA-4 as a positive control [41]. Only 18 inhibited tubulin polymerization, with an IC50 value below 20 μM. Compound 18, with a 2,5-dimethoxyphenyl substituent on the amide moiety, had an IC50 value of 19 μM. This agreed with 18 having antiproliferative activity against the six cancer cell lines examined, with IC50 values in the range of 0.6–3.6μM. Compounds 10, 11, and 18 were also examined for their inhibitory effects at 5 and/or 50 μM on the binding of 5 μM [3H]colchicine to 1 μM tubulin (Table 3) [43]. Inhibition was observed with 10 and 18 at 50 μM. Thus, the agents with the greatest antiproliferative effects on cancer cell growth had only modest effects in interacting with purified tubulin.
Table 3.
Inhibition of tubulin polymerization and colchicine binding by 10, 11 and 18 as compared with CA-4
| Tubulin assemblya IC50 (μM) ± SD | Colchicine bindingb % inhibition ± SD |
||
|---|---|---|---|
| Compound | 50μM | 5 μM | |
| 10 | > 20 | 26 ± 7 | - |
| 11 | > 20 | 9.4 ± 2 | - |
| 18 | 19 | 23 ± 3 | - |
| CA-4 | 0.54 ± 0.06 | - | 98 ± 0.1 |
Tubulin was at 10 μM;
Tubulin and colchicine were at 1 and 5 μM concentrations, respectively.
3.2.3. Real-time cellular response of HCC cells treated with 10, 11, and 18
Time-dependent cytotoxic activities of indole-pyrazole hybrids were performed with real time cell electronic sensing (RT-CES) [59] on Huh7 and Mahlavu cells. The RT-CES experiments were performed to determine the effects of the selected compounds as a function of time and concentration. In Huh7 cells, all compounds caused decrease in the cell growth (indicated as CI: cell index) in a dose dependent manner compared to the control group (Figure 3). In the mesenchymal-like Mahlavu cells, only compound 11 caused dose-dependent growth inhibition with respect to the control group. Compound 10 could display a noticeable inhibition in cell growth in 6 μM concentration. Finally, 3 and 6 μM of compound 18 resulted in similar levels of growth inhibition, for which dose-dependent response could not be observed as in Huh7 cells. This difference in effective doses of compounds against two liver cancer cell lines could be related to the aggressive phenotype of Mahlavu cells.
Figure 3.

RT-CES analysis of HCC cell lines treated with compounds 10, 11 and 18 at increasing concentrations and with a DMSO control (0.1%) for 72 h. Graphs indicate normalized cell index (CI) values with respect to time.
3.2.4. Compounds 10, 11 and 18 induced cell cycle arrest
The effects of selected compounds 10, 11, and 18 on cell cycle phase arrest in Huh7 and Mahlavu cells were evaluated by flow cytometry. Huh7 and Mahlavu cells were treated with the compounds for 72 h at the indicated concentrations, then fixed and stained with propidium iodide. Compound 10 caused arrest in the G0/G1 phase in Huh7 cells, whereas compounds 11 and 18 did not cause a significant change in these cells (Figure 4A). In contrast, 11 caused arrest in the S phase, while 18 caused arrest in the G2/M phase in Mahlavu cells, compatible with its tubulin inhibitory effect (Figure 4B). To understand the effects of these compounds at higher concentrations, we also treated cells with 10 μM 11 or 18. In these higher concentration studies, 11 caused arrest in the S phase and 18 caused arrest in the G2/M phase in both Huh7 and Mahlavu cells.
Figure 4.

Effects of selected compounds on cell cycle of HCC cells. a) Cell cycle analysis of Huh7 and b) Mahlavu cells after treatment with compounds 10, 11, and 18 and DMSO controls following 72 h of treatment with either the corresponding IC50 values (Huh7, 10: 1.0 μM, 11: 0.6 μM, 18: 0.6 μM, Mahlavu, 10: 2.7 μM, 11: 3.4 μM, 18: 2.9 μM) or with 10 μM concentrations (indicated with *). Results are represented as stacked column charts indicating different phases of the cell cycle.
3.2.5. Compounds 10, 11 and 18 induced apoptosis
Hoechst staining experiments [60] were performed on Huh7, HepG2, Mahlavu and SNU475 cells treated with compounds 10, 11, or 18 for 48 h to determine the cell death mechanism induced by the potent compounds, compared to DMSO controls. Condensed nuclei and blebbing were identified in these cells, which indicates apoptotic cell death. Compounds 10, 11 and 18 caused visible nuclear blebs or deformed nuclei in all four lines of the HCC cells (Figure 5).
Figure 5.

Detection of apoptosis using fluorescence microscopy; Hoechst33258 staining of Huh7, HepG2, Mahlavu and SNU-475 cells treated with compounds 10, 11, or 18 for 48 h with IC100 concentrations. Arrows indicate apoptotic nuclei and nuclear blebs caused by the compound treatments.
3.3. Computational Studies
We applied in silico approaches to evaluate possible binding properties of the reported compounds to the colchicine site of tubulin. As reported before [61, 62], we know that forming polar interactions with Val181 and Cys241 is required for achieving potent tubulin polymerization inhibitors that bind into the colchicine binding site. However, the most potent tubulin polymerization inhibitor 18 was, surprisingly, obtained by replacing the 3,4,5-trimethoxyphenyl of 10 with a 2,5 dimethoxyphenyl group. We, therefore, anticipated that there would be a better steric adaptation of 18 into the binding site. In agreement with that prospect, docking results showed that para substituents in the phenyl ring resulted in steric clashes with the binding site. This led to a score of −8.24 with 18 as compared with a score of −1.12 with 10. Thus, the docking score of 10 was heavily penalized because of the steric restrictions in the binding pocket. A complete list of the calculated Glide XP descriptors is presented in Table S1. The docking-derived binding mode of 18 (Figure 6) shows that the pyrazole ring forms hydrogen bonds with αThr179 and βAsn258; the indole ring forms hydrogen bonds with αVal181 and βAsn349 and hydrophobic contacts with αVal181, βAsn258, βThr314, βLys352; and the methoxy group of ring A forms a hydrogen bond with βCys241 and Van der Waals contacts with βAla250, βLeu255 and βIle318. Finally, we invite the readers to see the work of Bissantz et al [63] for detailed information about the nature of the protein-ligand interactions.
Later on, docking derived binding mode of 18 was issued with quantum chemical calculations to visualize the electrostatic potential surface (EPS, Figure 7. A) and important orbitals of the atoms (Figure 7. B–E). EPS visualizes the partial charges of the atoms on their surface. The highest occupied molecular orbital (HOMO) represents electron donors and the lowest unoccupied molecular orbital (LUMO) visualizes electron acceptors. In agreement with the interactions observed by docking, as stated in the previous paragraph, the polar hydrogen atom on the indol group acts as a hydrogen bond donor and interacts with the backbone atom of αVal181. Another significant interaction is formed by the oxygen atom at the fifth position of the 2,5 dimethoxyphenyl fragment of 18, which acts as a hydrogen bond acceptor against βCys241. Both interactions were found strong due the electrostatic potentials and HOMO/LUMO analysis (Figure 7) and also the XP scores based on each interacting residue.
4. Conclusion
In this study, we synthesized a series of indole-3-pyrazole-5-carboxamide derivatives and evaluated their anticancer activities against different human cancer cell lines in comparison with the positive control sorafenib. All newly synthesized compounds were structurally characterized by modern techniques, and their biological activities and mechanisms of action were studied by experiments performed mostly on HCC cell lines. The tests aimed firstly to evaluate the cytotoxic effects of the newly synthesized compounds, and active compounds were evaluated by RT-CES, apoptosis studies and cell cycle analysis. Some of the synthesized compounds were similar to or more effective than sorafenib in their antitumor activity against Huh7, MCF-7 and HCT116 cancer cells. Furthermore, compounds 11 and 18 showed a very strong antiproliferative effect against the Huh7 HCC line (IC50 value, 0.6 μM). Mechanism studies revealed that compound 18 effectively inhibited tubulin polymerization in vitro (IC50 = 19 μM) and should therefore disrupt microtubule dynamics, as manifested by its induction of cell cycle arrest in the G2/M phase in both Huh7 and Mahlavu cells. A docking-derived binding mode of 18 showed interactions between 18 and the colchicine site of tubulin. In future studies, we plan to evaluate the detailed cellular networks that are affected using highthroughput genomic screening methods, including transcriptome analysis with next-generation sequencing in the presence of selected compounds. This may allow identification of additional molecular targets involved in the cell cycle for targeted drug design and development for cancer treatment.
Supplementary Material
Highlights.
Synthesis, characterization and biological evaluation of indole-pyrazole derivatives as anticancer agents.
Compounds 10, 11 and 18 showed potent activity against the hepatocellular cancer cell lines, with IC50 values in the range 0.6–4.3 μM.
Compound 18 showed significant inhibitory activity against tubulin polymerization with IC50 value of 19 μM.
The Molecular docking studies showed that pyrazole, indole, and methoxy sections of compound 18 forms hydrogen bonds with various amino acids in the colchicine binding site.
Acknowledgment
We greatly acknowledge The Scientific and Technological Research Council of Turkey (TUBITAK) for financial support to S.N.B. (Grant No. 113S973).
Disclaimer
This research was supported in part by the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute, which includes federal funds under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
CRediT Authorship Contribution Statement
S.N.B. designed the compounds and their synthesis. M.H. performed the chemical synthesis and characterized the compounds. R.C.A. and D.C.K. designed the biological experiments. D.C.K. and S.G.E. performed the experiments for biological evaluation of the compounds on cell lines. E.H. performed the tubulin assays. A.O. performed the molecular docking studies. S.N.B., M.H., D.C.K. E.H. and A.O. contributed to the writing of the manuscript. All authors have given approval to the final version of the manuscript.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.The American Cancer Society. https://cancerstatisticscenter.cancer.org/?_ga=2.161823868.1998619257.16294483331315443985.1627639273#!/cancer-site/Liver%20and%20intrahepatic%20bile%20duct, (Accessed on 25 January 2022), 2022.
- 2.Bray F, et al. , Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J. Clin, 2018. 68(6): p. 394–424. [DOI] [PubMed] [Google Scholar]
- 3.Couri T. and Pillai A, Goals and targets for personalized therapy for HCC. Hepatol. Int, 2019. 13(2): p. 125–137. [DOI] [PubMed] [Google Scholar]
- 4.Hawash M. and Baytas SN, Antiproliferative activities of some biologically important scaffold. FABAD J. Pharm. Sci, 2017. 43(1): p. 59–77. [Google Scholar]
- 5.Karahalil B, Yardım-Akaydin S, and Baytas SN, An overview of microtubule targeting agents for cancer therapy. Arh. Hig. Rada. Toksikol, 2019. 70(3): p. 160–172. [DOI] [PubMed] [Google Scholar]
- 6.Baytas SN, et al. , Synthesis, biological evaluation and molecular docking studies of trans-indole-3-acrylamide derivatives, a new class of tubulin polymerization inhibitors. Bioorg. Med. Chem , 2014. 22(12): p. 3096–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamal A, et al. , Synthesis and biological evaluation of 3,5-diaryl isoxazoline/isoxazole linked 2,3-dihydroquinazolinone hybrids as anticancer agents. Eur. J. Med. Chem, 2011. 46(2): p. 691–703. [DOI] [PubMed] [Google Scholar]
- 8.Nathan P, et al. , Phase I trial of combretastatin A4 phosphate (CA4P) in combination with bevacizumab in patients with advanced cancer. Clin. Cancer Res, 2012. 18(12): p. 1–12. [DOI] [PubMed] [Google Scholar]
- 9.Hura N, et al. , Combretastatin-inspired heterocycles as antitubulin anticancer agents. ACS Omega, 2018. 3: p. 9754–9769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi BR, et al. , Effect of 4-chloro-7-trifluoromethyl-10H-benzo[4,5]furo[3,2-b]indole-1-carboxylic acid on the intraurethral pressure in a rat model of benign prostatic hyperplasia. Int J Urol, 2016. 23(3): p. 259–65. [DOI] [PubMed] [Google Scholar]
- 11.Li L, et al. , Design, synthesis, and biological evaluation of 1-substituted-2-aryl imidazoles targeting tubulin polymerization as potential anticancer agents. Eur. J. Med. Chem, 2019. 184: p. 111732. [DOI] [PubMed] [Google Scholar]
- 12.Wang L, et al. , Potent, orally active heterocycle-based combretastatin A-4 analogues: synthesis, structure-activity relationship, pharmacokinetics, and in vivo antitumor activity evaluation. J. Med. Chem, 2002. 45: p. 1697–1711. [DOI] [PubMed] [Google Scholar]
- 13.Patel VK and Rajak H, Synthesis, biological evaluation and molecular docking studies of 2-amino-3, 4, 5-trimethoxyaroylindole derivatives as novel anticancer agents. Bioorg. Med. Chem. Lett, 2016. 26(9): p. 2115–2118. [DOI] [PubMed] [Google Scholar]
- 14.Semenova MN, et al. , Sea urchin embryo model as a reliable in vivo phenotypic screen to characterize selective antimitotic molecules. Comparative evaluation of combretapyrazoles, -isoxazoles, −1,2,3-triazoles, and -pyrroles as tubulin-binding agents. ACS Comb. Sci, 2018. 20 p. 700–721. [DOI] [PubMed] [Google Scholar]
- 15.Al-Mansury S, et al. , Synthesis and anti-colon cancer activity of 1,2,4-triazole derivatives with aliphatic S-substituents. Orient. J. Chem, 2019. 35(1): p. 77. [Google Scholar]
- 16.Elmeligie S, et al. , Synthesis and cytotoxic activity of certain trisubstituted azetidin-2-one derivatives as a cis-restricted combretastatin A-4 analogues. Arch. Pharm. Res, 2017. 40: p. 13–24. [DOI] [PubMed] [Google Scholar]
- 17.Medarde M, et al. , Synthesis and pharmacological activity of diarylindole derivatives. Cytotoxic agents based on combretastatins. Bioorg. Med. Chem. Lett, 1999. 9(16): p. 2303–2308. [DOI] [PubMed] [Google Scholar]
- 18.Assali M, et al. , Synthesis, biological activity, and molecular modeling studies of pyrazole and triazole derivatives as selective COX-2 inhibitors. J. Chem, 2020. 2020.
- 19.Baytas S, et al. , Synthesis, anti-inflammatory, antiplatelet and in silico evaluations of (E)-3-(3-(2,3-dihydro-3-methyl-2-oxo-3H-benzoxazole-6-yl)-1-phenyl-1H-pyrazole-4-yl)acrylamides. Turk. J. Chem, 2012. 36: p. 367–382. [Google Scholar]
- 20.Shih S-R, et al. , Pyrazole compound BPR1P0034 with potent and selective anti-influenza virus activity. J. Biomed. Sci, 2010. 17(1): p. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanitame A, et al. , Synthesis and antibacterial activity of a novel series of DNA gyrase inhibitors: 5-[(E)-2-arylvinyl] pyrazoles. Bioorg. Med. Chem. Lett, 2005. 15(19): p. 4299–4303. [DOI] [PubMed] [Google Scholar]
- 22.Demiroglu‐Zergeroglu A, et al. , Investigation of potent anticarcinogenic activity of 1, 3-diarylpyrazole acrylamide derivatives in vitro. J. Pharm. Pharmacol, 2018. 70(12): p. 1619–1629. [DOI] [PubMed] [Google Scholar]
- 23.Hawash MM, et al. , Synthesis and biological evaluation of novel pyrazolic chalcone derivatives as novel hepatocellular carcinoma therapeutics. Eur. J. Med. Chem, 2017. 129: p. 12–26. [DOI] [PubMed] [Google Scholar]
- 24.De Martino G, et al. , Arylthioindoles, potent inhibitors of tubulin polymerization. J. Med. Chem, 2004. 47: p. 6120–6123. [DOI] [PubMed] [Google Scholar]
- 25.Tantak MP, et al. , 2-(3’-Indolyl)-N-arylthiazole-4-carboxamides: synthesis and evaluation of antibacterial and anticancer activities. Bioorg. Med. Chem. Lett, 2015. 25(19): p. 4225–4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamath PR, et al. , Some new indole-coumarin hybrids; synthesis, anticancer and Bcl-2 docking studies. Bioorg. Chem, 2015. 63: p. 101–109. [DOI] [PubMed] [Google Scholar]
- 27.Das Mukherjee D, et al. , Development of Novel Bis(indolyl)-hydrazide-Hydrazone Derivatives as Potent Microtubule-Targeting Cytotoxic Agents against A549 Lung Cancer Cells. Biochemistry, 2016. 55: p. 3020–3035. [DOI] [PubMed] [Google Scholar]
- 28.Fabitha K, et al. , Recent Developments in the Synthesis of Indole‐Pyrazole Hybrids. ChemistrySelect, 2022. 7(21): p. e202201064. [Google Scholar]
- 29.Yang M-R, et al. , Synthesis, biological evaluation and molecular docking studies of novel 1-(4,5-dihydro-1H-pyrazol-1-yl)ethanone-containing 1-methylindol derivatives as potential tubulin assembling inhibitors. R. Soc. Chem, 2016. 6(36): p. 30412–30424. [Google Scholar]
- 30.Kamal A, et al. , Synthesis of (Z)-(arylamino)-pyrazolyl/isoxazolyl-2-propenones as tubulin targeting anticancer agents and apoptotic inducers. Org. Biomol. Chem, 2015. 13(11): p. 3416–3431. [DOI] [PubMed] [Google Scholar]
- 31.Hawash M, et al. , Design and synthesis of novel substituted indole-acrylamide derivatives and evaluation of their anti-cancer activity as potential tubulin-targeting agents. J. Mol. Struct, 2022. 1254: p. 132345. [Google Scholar]
- 32.Baytas SN, Inceler N, and Yılmaz A, Synthesis, cytotoxicity, and molecular properties prediction of novel 1,3-diarylpyrazole derivatives. Med. Chem. Res, 2013. 22: p. 4893–4908. [Google Scholar]
- 33.Inceler N, Yılmaz A, and Baytas SN, Synthesis of ester and amide derivatives of 1-phenyl-3-(thiophen-3-yl)-1H-pyrazole-4-carboxylic acid and study of their anticancer activity. Med. Chem. Res, 2013. 22: p. 3109–3118. [Google Scholar]
- 34.Inceler N, et al. , Design, synthesis and biological evaluation of novel 1,3-diarylpyrazoles as cyclooxygenase inhibitors, antiplatelet and anticancer agents. Med. Chem. Commun, 2018. 9: p. 795–811. [DOI] [PMC free article] [PubMed]
- 35.Hawash M, et al. , Induction of apoptosis in hepatocellular carcinoma cell lines by novel indolylacrylamide derivatives: synthesis and biological evaluation. Chem. Biodivers, 2021: p. e2001037. [DOI] [PubMed]
- 36.Hawash M, et al. , Synthesis of novel indole-isoxazole hybrids and evaluation of their cytotoxic activities on hepatocellular carcinoma cell lines. BMC Chem., 2021. 15: p. 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang D, et al. , Synthesis and biological evaluation of 3-(1H-indol-3-yl)pyrazole-5-carboxylic acid derivatives. Arch. Pharm. Res, 2011. 34(3): p. 343–355. [DOI] [PubMed] [Google Scholar]
- 38.Iwaszkiewicz-Grzes D, et al. , Synthesis and biological activity of mycophenolic acid-amino acid derivatives. Eur. J. Med. Chem, 2013. 69: p. 863–871. [DOI] [PubMed] [Google Scholar]
- 39.Kim IH, et al. , Structure–activity relationships of substituted oxyoxalamides as inhibitors of the human soluble epoxide hydrolase. Bioorg. Med. Chem, 2014. 22: p. 1163–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hamel E. and Lin CM, Separation of active tubulin and microtubule-associated proteins by ultracentrifugation and isolation of a component causing the formation of microtubule bundles. Biochemistry, 1984. 23: p. 4173–4184. [DOI] [PubMed] [Google Scholar]
- 41.Hamel E, Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem. Biophys, 2003. 38: p. 1–22. [DOI] [PubMed] [Google Scholar]
- 42.Hamel E. and Lin CM, Stabilization of the colchicine binding activity of tubulin by organic acids. Biochim. Biophys. Acta, 1981. 675: p. 226–231. [DOI] [PubMed] [Google Scholar]
- 43.Verdier-Pinard P, et al. , Structure-activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol. Pharmacol, 1998. 53: p. 62–76. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Q, et al. , Highly Potent Triazole-Based Tubulin Polymerization Inhibitors. Journal of Medicinal Chemistry, 2007. 50: p. 749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang D, et al. , Synthesis and cytotoxic activity of novel 3-(1H-indol-3-yl)-1H-pyrazole-5-carbohydrazide derivatives. European Journal of Medicinal Chemistry, 2011. 46(12): p. 5868–5877. [DOI] [PubMed] [Google Scholar]
- 46.Abdellatif KR, Lamie PF, and Omar HA, 3-methyl-2-phenyl-1-substituted-indole derivatives as indomethacin analogs: design, synthesis and biological evaluation as potential anti-inflammatory and analgesic agents. Journal of Enzyme Inhibition and Medicinal Chemistry, 2016. 31(2): p. 318–324. [DOI] [PubMed] [Google Scholar]
- 47.Gaspari R, et al. , Structural basis of cis - and trans -combretastatin binding to tubulin. Chem., 2017. 2(1): p. 102–113. [Google Scholar]
- 48.Friesner RA, et al. , Glide: A new approach for rapid, accurate docking and scoring. 1. method and assessment of docking accuracy. J. Med. Chem, 2004. 47(7): p. 1739–1749. [DOI] [PubMed] [Google Scholar]
- 49.Halgren TA, et al. , Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem, 2004. 47(7): p. 1750–9. [DOI] [PubMed] [Google Scholar]
- 50.Bian J, et al. , Discovery of N-hydroxy-4-(1H-indol-3-yl)butanamide as a histone deacetylase inhibitor. Drug Discov Ther, 2016. [DOI] [PubMed]
- 51.Salentin S, et al. , PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Res., 2015. 43(W1): p. W443–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Modzelewska A, et al. , Anticancer activities of novel chalcone and bis-chalcone derivatives. Bioorganic & Medicinal Chemistry, 2006. 14(10): p. 3491–3495. [DOI] [PubMed] [Google Scholar]
- 53.Bochevarov AD, et al. , Jaguar: A high‐performance quantum chemistry software program with strengths in life and materials sciences. International Journal of Quantum Chemistry, 2013. 113(18): p. 2110–2142. [Google Scholar]
- 54.Burke TR, et al. , Compounds to treat HIV infection and AIDS, in Related US. Application Data. 2006: United States. p. 1–36.
- 55.Udayasri A, et al. , Ecofriendly Synthesis of Ribociclib Intermediate Using Regioselective Hydrodechlorination and DMAP Catalyzed Ester Hydrolysis. Topics in Catalysis, 2022: p. 1–6.
- 56.Wen J, et al. , Identification of N-(6-mercaptohexyl)-3-(4-pyridyl)-1H-pyrazole-5-carboxamide and its disulfide prodrug as potent histone deacetylase inhibitors with in vitro and in vivo anti-tumor efficacy. Eur. J. Med. Chem, 2016. 109: p. 350–359. [DOI] [PubMed] [Google Scholar]
- 57.Jarończyk M, Dobrowolski JC, and Mazurek AP, Theoretical studies on tautomerism and IR spectra of pyrazole derivatives. J. Mol. Struct, 2004. 673(1–3): p. 17–28. [Google Scholar]
- 58.Jarończyk M, Dobrowolski JC, and Mazurek AP, Theoretical studies on tautomerism and IR spectra of pyrazole derivatives. Journal of Molecular Structure: Theochem, 2004. 673(1–3): p. 17–28. [Google Scholar]
- 59.La Regina G, et al. , Design and Synthesis of 2-Heterocyclyl-3-arylthio-1H-indoles as Potent Tubulin Polymerization and Cell Growth Inhibitors with Improved Metabolic Stability. Journal of Medicinal Chemistry, 2011. 54(24): p. 8394–8406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Durmaz I, et al. , Liver cancer cells are sensitive to Lanatoside C induced cell death independent of their PTEN status. Phytomedicine, 2016. 23(1): p. 42–51. [DOI] [PubMed] [Google Scholar]
- 61.Baytas SN, et al. , Synthesis, biological evaluation and molecular docking studies of trans-indole-3-acrylamide derivatives, a new class of tubulin polymerization inhibitors. Bioorganic & Medicinal Chemistry, 2014. [DOI] [PMC free article] [PubMed]
- 62.Hawash M, et al. , Design and synthesis of novel substituted indole-acrylamide derivatives and evaluation of their anti-cancer activity as potential tubulin-targeting agents. Journal of Molecular Structure, 2022. 1254. [Google Scholar]
- 63.Bissantz C, Kuhn B, and Stahl M, A medicinal chemist’s guide to molecular interactions. J Med Chem, 2010. 53(14): p. 5061–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.