

Abstract
Brain metastases are the most lethal progression event, in part because the biological processes underpinning brain metastases are poorly understood. There is a paucity of realistic models of metastasis, as current in vivo murine models are slow to manifest metastasis. We set out to delineate metabolic and secretory modulators of brain metastases by utilizing two models consisting of in vitro microfluidic devices: 1) a blood brain niche (BBN) chip that recapitulates the blood-brain-barrier and niche; and 2) a migration chip that assesses cell migration. We report secretory cues provided by the brain niche that attract metastatic cancer cells to colonize the brain niche region. Astrocytic Dkk-1 is increased in response to brain-seeking breast cancer cells and stimulates cancer cell migration. Brain-metastatic cancer cells under Dkk-1 stimulation increase gene expression of FGF-13 and PLCB1. Further, extracellular Dkk-1 modulates cancer cell migration upon entering the brain niche.
Keywords: cancer, blood brain barrier, metastasis, microenvironment, secretions, metabolites, dkk-1, microfluidic, secretome
Graphical Abstract
Culture in a synthetic blood brain barrier revealed the tumor micro-environment remodels differentially based on metastatic nature of the tumor cells. Moreover, analysis of the secretions, genes and pathways involved identified Dkk1 as promoting remodeling of the pro-metastatic brain niche. Treatment with Dkk1 antibodies and FGF13 knockdowns confirm its role.
1. Introduction
Brain metastasis is the most lethal secondary cancer progression for most tumor types, including breast, and occurs in 10–20% of cases with a median survival of 5–20 months, depending on the subtype [1]. Treatment of other metastatic sites has improved [2], but many treatments do not cross the blood brain barrier or are ineffective at treating brain metastases [3]. Although a subset of metastatic cancer can extravasate through the blood brain barrier (BBB), the molecular processes that regulate this are poorly understood. Even though many cancer cells succeed in crossing the BBB, the majority are unable to colonize the brain niche microenvironment and instead die or senesce. According to the classical seed-and-soil hypothesis, for metastatic cancer cells to colonize they must interact with brain niche components and secretions to promote a hospitable environment [4]. The unknown factors contributing to cancer cell death or survival in the brain niche could directly influence the development of therapeutics and are of interest to the cancer community. Two resident cells of the brain niche, astrocytes and microglia, appear to react to cancer cells entering the brain space and have been detected surrounding brain metastases [5]. However, limited data is available to understand the influence of secretory molecules emanating from the brain niche cells in regulating brain metastasis.
Astrocytes are vital regulators of homeostasis in the normal brain niche and support the roles of endothelial and pericyte cells in forming the BBB through multi-modal interactions [6]. Astrocytes adopt reactive phenotypes upon sensing homeostatic deviations, such as brain injury or infection [7]. Reactive astrocytes in the presence of cancer cells have recently emerged as cellular components complicit in the formation of brain metastases [6, 8]. A partial panel of astrocyte secretions produced in reaction to cancer cells in the brain have been identified [9]. Brain metastatic cancer cells increase reactive astrocytic secretion of plasminogen activators (PA), tissue-PA (tPA), and urokinase (uPA). These PAs induce high levels of plasmin in the brain niche, which promote cell death in many cancer cells reducing BBB colonization rates. To thrive in the brain niche with elevated PAs, brain-metastatic cancer cells secrete PA inhibitors, SerpinI1 and SerpinB2 [10]. Conversely, in a beneficial manner astrocytic secretion of IL-6 and MCP-1 (CCL-2) influence extravasated cancer cells to form tumors in proximity to the BBB, forming blood tumor barriers (BTB) through S1P3 signaling [11].
Microglia are the resident macrophage of the brain niche that actively survey the CNS for damage and infectious agents, including cancer cells [12]. Like astrocytes, microglia adopt different phenotypes depending on the environment. Classical M1 microglia destroy infiltrating tumors through production of cytotoxic factors and proinflammatory cytokines (IL-1β, TNF-α, ROS, IFN-γ, IL-12). Conversely, activated M2 microglia elicit a pro-tumor response through secretion of anti-inflammatory cytokines, immunomodulatory mediators, and growth factors (IL-10, TGB-β, VEGF, and MMPs) [13]. In the context of brain metastasis, heterogeneous populations of microglia surround and infiltrate tumors [14].
In this study, we utilized both a migration and a blood brain niche (BBN) microfluidic chip to characterize alterations to the brain niche and cancer cell metastatic progression. Our goal is to characterize phenotypic and secretory cues provided by individual cellular residents of the brain niche – astrocytes and microglia – that attract metastatic cancer cells. We characterized these behaviors in two breast cancer cell lines, MDA-MB-231 (TNBC) and JIMT1 (Her2+), as well as two brain-metastatic derivatives of these lines, MDA-MB-231-BR and JIMT1-BR [15]. Fluctuations in metastatic cancer cell secretions (cytokines and metabolites) after stimulation with individual brain niche cellular or secretory components are used to establish secretory profiles of tumor cells that promote brain niche remodeling. Conversely, variations in astrocyte, microglia, and brain microvascular endothelial cell secretions when stimulated with cancer cellular or secretory components are used to identify unique cytokine and metabolite profiles. We examine if individual secretions can be inhibited to modulate remodeling of the brain niche. Specifically, we observe if neutralization of Dkk-1 secretion alters the cancer cell induced remodeling within the brain niche. Finally, we examined the impact on two genes (FGF-13, PLCB1) that are important to Dkk-1 related pathways and study their impact on cell migration in Dkk-1 gradients.
2. Results
2.1. Astrocyte cells promote metastatic cancer cell extravasation through the BBB
To examine the influence of astrocytes and microglia cells on cancer cell extravasation through the blood brain barrier, we utilized an established microfluidic BBN chip to observe and characterize cancer cell extravasation and colonization of the BBN (Figure 1a) [16]. The details of the BBN chip and the method used for analysis (confocal tomography) are discussed in the methods and supplemental methods. All the data in Figure 1 is derived by measuring the positions (centroids) of cancer cells and brain resident cells (astrocytes and microglia) relative to the blood brain barrier (endothelial cells) in the device. These were measured from fluorescent 3D confocal images. To image the entire device a motorized stage was used to stitch images from 9 adjacent fields of view along the 14 mm length of the channel. At each location, 50 images were taken at 10 μm steps along the z axis. The depth of the device is 500 μm with 200 μm imaged above the BBB membrane and 300 μm below it. Taking ~50 images at 10 μm steps enabled accurate 3D volumetric reconstruction of the cells within the device. The cells were segmented and measured as described in the supplemental section and in prior work. This synthetic BBN system was utilized to test the ability of astrocytes or microglia to attract parental and brain-metastatic (BR) cancer cells to the brain region after 2- and 9-days of interaction (Figure 1).
Figure. 1. Brain-metastatic cancer cells extravasate towards astrocyte cells.
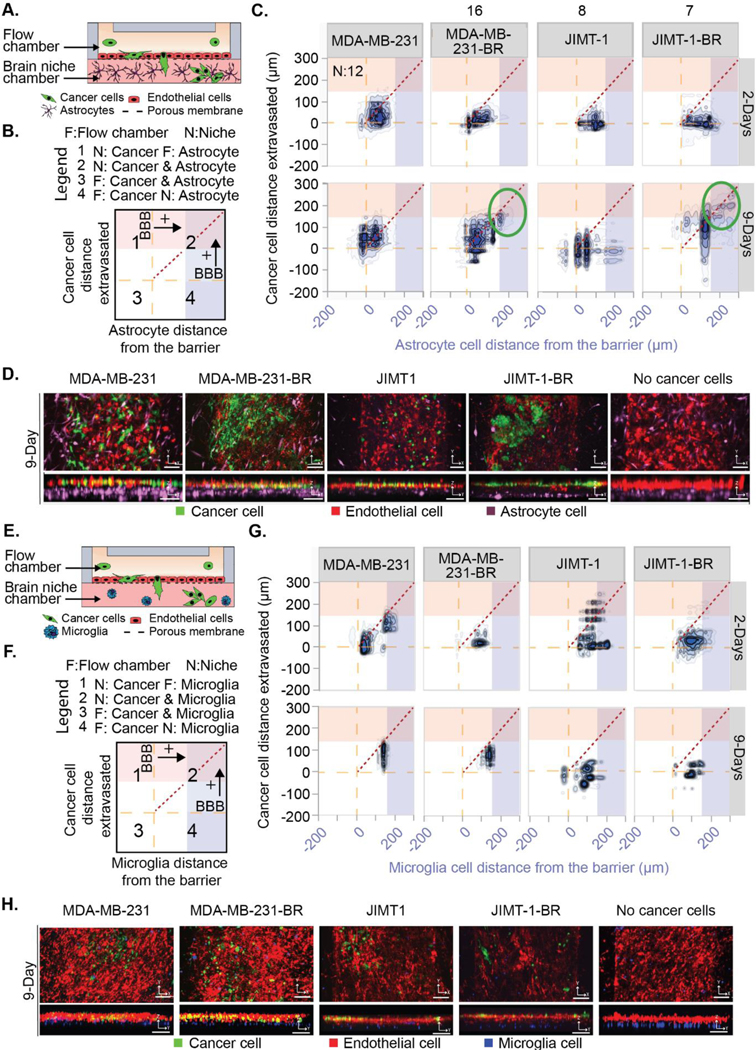
(A and E) Schematic of a BBNiche with (A) astrocytes or (E) microglia in the brain niche chamber. Cancer cells in the top flow chamber interact with the BBNiche for 2- and 9-Days. (B and F) Legend for (C) and (G) respectively. Each quadrant shows interactions between cancer cells and brain niche cells. The yellow dashed line represents the endothelial barrier. Red and blue overlay denote a distance from barrier > 150 μm. (C and G) 2-D density plot of cancer cell and (C) astrocyte or (G) microglia interaction for MDA-MB-231, MDA-MB-231-BR, JIMT-1, JIMT-1-BR for 2- and 9-Days. (D and H) Representative fluorescent confocal images of cancer cells in BBN chips with (D) astrocytes or (H) microglia after 9-Days. Images are in the XY and YZ planes, scale bar = 125 μm. A list of replicates per condition are given in Table S1.
Figure 1c displays the interaction between individual astrocytes and cancer cells represented as a density plot (parental:MDA-MB-231, JIMT1 and brain metastatic: MDA-MB-231-BR, JIMT1-BR) whereas Figure 1b describes how to interpret the plot shown in Figure 1c. The X axis shows the position of the cancer cells relative to the BBB while the Y axis shows the position of the astrocytes or microglia relative to the BBB. Each measurement is for a pair of cancer cells and astrocytes in the device. Cells were paired if the cancer cell was directly above or below a nearby astrocyte. In this way the relationship of the effect of astrocytes on cancer cells is observed locally and then transformed into a 2D plot with the BBB as a reference. These plots are divided into four quadrants that describe where in the BBN device the cells are located. Quadrant 1 shows cancer cells in the brain side of the device and astrocytes on the flow chamber side. Quadrant 2 shows cancer and astrocyte cells in the brain side of the device. Quadrant 3 shows cancer and astrocyte cells on the flow chamber side of the device. Finally, quadrant 4 shows cancer cells in the flow chamber and astrocyte cells in the brain side of the device. The proximity of the measured interaction can be visualized by the distance away from the short, dotted diagonal line. When the two cells are touching, they will be plotted on the diagonal line and the further apart they get from each other, the further from the diagonal line they will be. The red overlays in quadrants 1 & 2 show when cancer cells are in the lower 50% of the brain chamber. The blue overlays in quadrants 2 & 4 show when the astrocyte cells are in the lower 50% of the brain chamber. In the figure the endothelial barrier is represented by the long-dashed lines. Because this is a density plot of pairs of cancer cells and astrocytes from along the length of the device we can appreciate the overall change in how cancer cell type alters the positions of the astrocytes and vice-versa.
Astrocyte cells supplemented in the niche uniquely attracted some MDA-MB-231-BR and JIMT1-BR and formed distinct clustered micrometastases spanning the niche after 9 days of culture (Figure 1c circled region). The MDA-MB-231-BR average distance extravasated across the time points did not significantly vary, but the overall distribution of metastatic cancer cells differed significantly between brain-metastatic and parental cancer cells. Most parental MDA-MB-231 cells remained concentrated at the endothelial barrier with a small subset of cells that migrated deeply into the brain niche. JIMT1-BR and parental JIMT1 also maintained proximity to the endothelial barrier after 2-days (Figure 1). However, after 9-days, both MDA-MB-231-BR and JIMT1-BR exhibited a large proportion of fully extravasated cells into the brain niche compartment and contained subsets of migratory cells positioned farther within the brain niche. The distributions of distance extravasated measurements of MDA-MB-231 and JIMT1 cancer cells were significantly different when comparing between 1) brain-metastatic and parental lines, and 2) across timepoints (Figure 1c).
Simultaneous monitoring of the astrocytes revealed they reposition themselves within the BBNiche. The presence of brain-metastatic MDA-MB-231-BR cells influenced astrocytes towards the endothelial barrier, touching the extravasated MDA-MB-231-BR cancer cells at 2 days and sustaining contacts after 9 days (Figure 1c). Astrocytes appeared to minimize contact with JIMT1-BR; JIMT1 were positioned the farthest away from the barrier at both 2 days and 9 days (Figure 1c). Figure 1d shows representative images at 9 days, images with an enlarged z axis are provided in Figure S1a for 2 and 9 days.
2.2. Microglia cells influence metastatic cancer cells to remain in proximity to the BBB
Using the same methodology (Figure 1e–h), we sought to describe the effects of microglia on cancer cell metastasis. In contrast to astrocytes, there was limited remodeling of the microglia in the BBNiche chips containing microglia cells (Figure 1g). For the MDA-MB-231 cells the microglia cells transition from two groups (13 μm and 150 μm) to a single cluster at (150 μm) at 9 days. However, the MDA-MB-231 cells have reduced their dispersion at 2 days. The JIMT-1 parental line does show a larger change in the cancer cells moving from deep within the device towards the endothelial layer. Contrasting this to the MDA-MB-231-BR cells the microglia again move towards 150 μm away from the BBB with exceptions. The JIMT-1-BR tighten behavior but show no major changes between 2 and 9 days. It is also observed that the microglia reposition into thin layers in most cases. Figure 1h. shows representative images at 9 days, images with an enlarged z axis are provided in Figure S1b for 2 and 9 days.
2.3. BBN secretions influence cancer cell migration and extravasation
Given the differences in cancer cell extravasation when exposed to astrocyte and microglia cells, we hypothesized that basal secretions within the brain niche alone may influence cancer cell migration and extravasation. To examine the effect of brain niche cell secretions on metastatic breast cancer cell migration, we used a microfluidic chip that permits quantitative monitoring of linear cell migration [17], which is described in the supplemental data (Figure S2a) and methods. We found astrocyte secretions could alter migratory behavior or MDA-MB-231-BR cells.
We next used a microfluidic BBN chip composed of a cell-free collagen brain niche chamber infused with either basal astrocyte or microglia secretions (Figure 2a, b). This created a brain niche rich in cytokines protected by an endothelial cell barrier. We monitored cancer cell extravasation towards astrocyte or microglia secretions after both 2 and 9 days. Controls using nonconditioned media are shown in Figure S3.
Figure 2. Astrocyte and endothelial secretions influence brain-metastatic cancer cell migration.
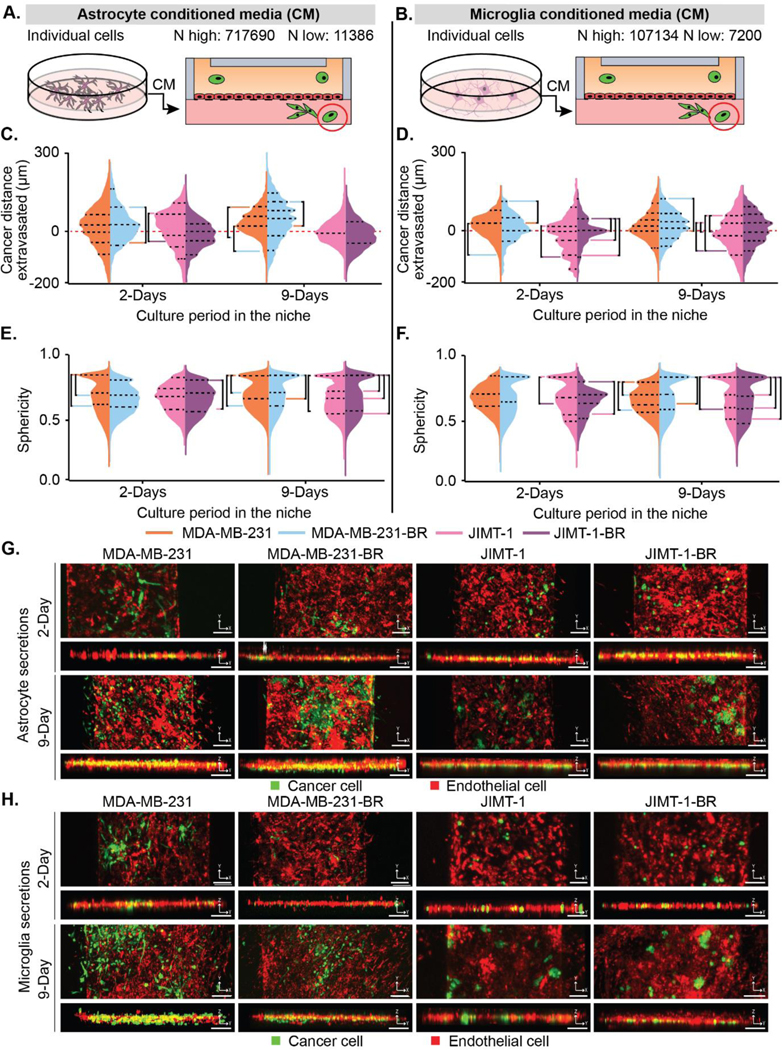
(A-B) Depiction of how (A) astrocyte and (B) microglia conditioned media was collected and placed in the device. The highest (N highest) and lowest (N lowest) count of cells from all replicates and conditions. (C-D) Cancer cell positions in BBN chips with (C) astrocyte and (D) microglia secretions plotted as distance (μm) to the endothelial barrier (0 μm). (E-F) Sphericity of cancer cells in BBN chips with (E) astrocyte and (F) microglia secretions. (G-H) Representative fluorescent confocal images of cancer cell extravasation in μBBN devices infused with (G) astrocyte or (H) microglia secretions after 2- and 9-Days, scale bar 125 μm. (C-F) p < 6.25*10–4 determined by Mann-Whitney significance tests with a Bonferroni correction. A list of replicates per condition are given in Table S1.
After 2 days, astrocyte secretions promoted cancer cell extravasation of both brain-seeking and parental cancer cell types, with most of the cancer cells surrounding the endothelial barrier (Figure 2c). At 9 days the parental MDA-MB-231 cells moved towards the barrier while the brain tropic cells migrated deeper into the device and developed into distinct sub-populations. Conversely, the JIMT-1 parental and JIMT-1-BR converged towards the endothelial barrier. However, the JIMT-1-BR maintained two population peaks. The trend towards a spherical shape (sphericity) increased for all four cell types between 2 and 9 days (Figure 2e).
Parental MDA-MB-231 and MDA-MB-231-BR cells cultured in BBN chips with microglia secretions at 2 days behaved similarly to astrocyte secretions clustering around the barrier with the brain tropic cells closer to the barrier, and showed qualitatively similar, but less aggressive, divergence at 9 days (Figure 2d). JIMT-1 cells however diverge in the presence of microglia secretions with brain tropic cells, by migrating deeper into the device in aggregate (Figure 2d). All cell types increased in sphericity, but differed at 2 days from the behavior observed in the presence of astrocyte secretions (Figure 2f). Representative images of each condition are shown in Figure 2g and 2h for astrocyte and microglia conditions, respectively. Enlarged images in the z direction are provided in Figure S4.
We extracted counts of BBN-relevant micro-metastasis or clusters of cancer cells in astrocyte and microglia secretions (Figure 3a–b). The range of extravasation for the clusters is smaller than the single cells (Figure 3c–d). We also observed a wide variation in the number of clusters for the astrocyte secretion condition, triplicate experiments between 128 to 1845 extravasated clusters. Parental MDA-MB-231 cells had a broader set of cluster positions while JIMT-1 cells were much more concentrated around the barrier. However, there were small groups at 2 days of deeply advancing clusters. Sphericity of clusters cultured in astrocyte secretions revealed clusters that are significantly less spherical than individual cells (Figure 2e vs Figure 3e). By 9 days, the brain seeking cell clusters for both brain tropic cell lines are more spherical than the parental cell clusters (Figure 3e).
Figure 3. Astrocyte and endothelial secretions influence brain-niche micro-metastasis migration.

(A-B) Depiction of how (A) astrocyte and (B) microglia conditioned media was collected and placed into the device. The highest (N highest) and lowest (N lowest) count of cells from all replicates and conditions. (C-D) Cancer cell BBN micro-metastasis positions in BBN chips with (C) astrocyte and (D) microglia secretions plotted as distance (μm) to the endothelial barrier (0 μm). (E-F) Sphericity of cancer cell BBN micro-metastasis in BBN chips with (E) astrocyte and microglia (F) secretions. (G-H) Rendering of the largest BBN micro-metastasis selected from random devices at 2-Days and 9-Days for (G) astrocyte secretions and (H) microglia secretions. Scale bars are 200 μm. (C-F) p < 6.25*10–4 determined by Mann-Whitney significance tests with a Bonferroni correction. A list of replicates per condition are given in Table S1.
Interestingly, in the presence of microglia secretions, the clusters for both cancer cell types cluster around the barrier (Figure 3d) yet maintain a small shift below the barrier for MDA-MB-231-BR and JIMT-1-BR cell types at 9 days compared to the parental cell types. The JIMT-1-BR cells at 9 days exhibit lower variance clusters when in microglia secretions than in the astrocyte secretions (Figure 3c–d). Compared to astrocyte secretions, dramatic shifts in cluster sphericity in the microglia condition occurred (Figure 3e–f). MDA-MB-231 parental clusters are initially more spherical at 2 days but shift at 9 days towards MDA-MB-231-BR clusters (Figure 3f). However, MDA-MB-231-BR clusters have a tail of non-spherical shapes. Inverse behavior was observed in the JIMT-1 cells. The JIMT-1-BR clusters, become more spherical by 9 days, which is also the reverse of their behavior in the astrocyte secretions (Figure 2f vs Figure 3f).
Representative images of the largest cluster found in a randomly selected channel for each cancer cell type and culture time condition are shown in Figure 3g and h for astrocyte and microglia conditioned media, respectively. The clusters of brain tropic cells are larger than those of the parental line in astrocyte secretions than in microglia secretions.
2.4. The cytokine response of the brain niche is influenced by interaction with MDA-MB-231-BR
Astrocyte, microglia, and endothelial cells are known to secrete cytokines within the normal brain niche to maintain homeostasis and to have the ability to rapidly fluctuate in response to invasive pathogens or damage [5, 18]. Given the unique astrocytic and microglial reorganization in the context of brain-metastatic cancer cells, we examined the secretory cytokine profiles of astrocytes and endothelial cells dependent on cancer cell interaction using dot blot arrays (Figure 4b). ELISA showed astrocytes increased secretion of IL-6, IL-8, Dkk-1, CXCL-5, Chitinase 3-like 1, VCAM-1, SerpinE1, and MCP-1 when stimulated with MDA-MB-231-BR (Figure 4c, Figure S5a). No significantly increased endothelial secretions were detected with MDA-MB-231-BR stimulation (Figure 4d, Figure S5b) using ELISA. A total of 9 cytokines were significantly elevated in microglial secretions when stimulated with MDA-MB-231-BR: Angiogenin, Dkk-1, GM-CSF, IL-8, Gro-a, Chitinase 3-like 1, IL-6, MCP-1, and SerpinE1(Figure S5c, S5d) using ELISA . However, the relative quantity of microglia secretion was lower compared to the output of cytokines by the astrocytic cytokines, thus, subsequently, the study focused on understanding the influence of astrocytic secretions on cancer cell metastasis.
Figure 4. Brain niche cytokine profiles are altered after interaction with cancer cells.

(A) Schematic showing collection of conditions used for cytokine profiling and distribution to a dot blot assay and ELISA. (B) Human XL cytokine dot blot array results for astrocyte and endothelial cell secretions; when stimulated with either MDA-MB-231-BR or parental MDA-MB-231 and normalized to a non-stimulated control. Heatmap colors indicate row comparisons of each cytokine level; blue (low) to red (high). Square size indicates overall cytokine protein levels. (C-D) ELISAs of (C) astrocyte or (D) endothelial secretions after stimulation with either cancer cells or paired secretions were normalized to unstimulated secretion controls. For all assays, orange outlines indicate statistically significant comparisons and t-tests with Holm-Šídák multiple comparisons correction were utilized. The assays were performed in triplicate.
We employed ELISAs as an orthogonal approach to validate and quantify the salient cytokines identified using dot blot arrays. Astrocytes or endothelial cells were stimulated with either cancer cells or their corresponding secretions. When astrocytes were stimulated with MDA-MB-231-BR cancer cell secretions alone, they significantly increased secretions of cytokines MCP-1, IL-8, and CXCL5, compared to stimulation with MDA-MB-231-BR cells, parental MDA-MB-231 cells, or parental MDA-MB-231 secretions (Figure 4c). Astrocytic stimulation with physical MDA-MB-231-BR cells, but not their secretions, only increased the astrocytic secretion of Dkk-1. Under stimulation with MDA-MB-231-BR cells but not with parental MDA-MB-231 cell stimulation, endothelial cells significantly increased the secretion of VEGF (Figure 4d). Endothelial stimulation with cancer secretions alone promoted an increase in cytokine production whose levels were not significantly different between stimulation with MDA-MB-231-BR and MDA-MB-231 secretions.
2.5. MDA-MB-231-BR and parental MDA-MB-231 cancer cells differentially modulate cytokine profiles in response to BBNiche stimulation
Cancer cell extravasation across the BBB and colonization of the brain niche requires cancer cells to remodel the brain niche to promote their survival. We hypothesized para-cellular communication through cytokines as a means cancer cells use to modify the brain niche into a suitable metastatic environment. We identified secretions from cancer cells with and without direct interaction from individual brain niche cells (astrocytes, microglia, or endothelial cells) using dot blot arrays (Figure S6a) to profile cytokines from the cancer cells which promote metastasis We observed significantly different levels of IL-6, SerpinE1, and Dkk-1 cytokines secreted by MDA-MB-231-BR when stimulated with astrocytes or microglia (Figure S6b).
2.6. Dkk-1 influences brain metastatic cancer cell migration and extravasation across the BBB
As astrocytic and endothelial secretions promoted a migratory phenotype (Figure S2), we postulated that a regulated and prevalent cytokine may influence cancer cell extravasation across the BBB. Dkk-1 was a cytokine that astrocytic and endothelial components upregulated in the presence of MDA-MB-231-BR stimulation. Dkk-1 is a known Wnt signaling inhibitor and influencer of cancer cell dormancy [19]. Dysregulation of Wnt signaling has commonly been implicated in cancer progression [20]. Thus, we interrogated the influence of Dkk-1 on metastatic cancer cell migration and extravasation into the brain niche and on linear 2D migration devices.
To uncover the role of astrocytic Dkk-1 secretion on cancer cell migration in 2D, MDA-MB-231-BR cells were subject to analysis in our microfluidic migration chip. Two Dkk-1 gradients were assessed alongside controls: 1) an increasing concentration of Dkk-1 in SFM to assess the effects of Dkk-1 as a chemoattractant; and 2) with Dkk-1 provided in SFM as a stimulus to the cancer cells across the migration chip, with 10% FBS as an external chemotactic gradient. Chemotactic gradients of SFM-Dkk-1 (20 ng/mL) do not promote MDA-MB-231-BR or MDA-MB-231 cancer cell migration (Figure 5a–c). Instead, Dkk-1 acted as a stimulatory factor and enhanced cancer cell migration towards external gradients of FBS, compared to FBS alone (Figure 5a–c). Thus, Dkk-1 does not act alone in a chemotactic manner; instead, cancer cells adopt a migratory phenotype due to direct stimulation of Dkk-1. No statistically significant cell growth was detected over 48 hrs in all gradient conditions (Mann-Whitney t-tests with Bonferroni correction, p < 6.25 * 10−3).
Figure 5. The Dkk-1 cytokine influences cancer cell extravasation and interaction with astrocytes.

(A-B) Purified Dkk-1 was tested as a chemoattractant/stimulant for cancer cell migration using microfluidic device. (A) Image of the microfluidic migration chip. Details in the supplemental methods. (B) MDA-MB-231 and (C) MDA-MB-231-BR cell migration in chemotactic gradients using combinations of: serum free media (SFM), FBS and 20 ng/mL Dkk-1. (D-H) Cancer cells in BBN chips with astrocytes treated with or without Dkk-1 neutralization antibody (10 μg/mL) for MDA-MB-231, MDA-MB-231-BR, JIMT-1, JIMT-1-BR. (D) Dkk-1 neutralizing antibody was administered in the brain niche chamber. (E) Distance (μm) between the cancer cells and the endothelial barrier in BBN chips compared to astrocyte cell position treated with or without Dkk-1 neutralization antibody (10 μg/mL) for MDA-MB-231, MDA-MB-231-BR, JIMT-1, JIMT-1-BR. (F-G) Difference between the samples treated with Dkk-1 neutralizing Ab and untreated samples for (F) MDA-MB-231-BR and MDA-MB-231 cells, and (G) JIMT-1-BR or JIMT-1. (H) Confocal images of cancer cell extravasation into BBN chips treated with 10 μg/mL Dkk-1 neutralization antibody, scale bar 125 μm. (B-C) **p < 6.25*10−3, ***p < 6.25*10−4, Mann-Whitney significance tests with a Bonferroni correction. A-B was performed with three biolgoical replicates each with three technical replicates,refer to Table S1 for list of replicates per condition in D-H.
To confirm Dkk-1s role in the BBN chip, after introducing cancer cells we used 10 μg/mL of Dkk-1 neutralization antibody in the brain niche space of an astrocyte laden microfluidic BBN chip twice daily. After 2-days cancer cell migration within the brain niche was markedly decreased in the μBBN chip with Dkk-1 neutralization, (Figure 5d–h). We overlayed the response of the cancer cell distance extravasated and astrocyte cell distance from the barrier shown in Figure 1 (blue) with the results from cells cultured in the Dkk-1 neutralization antibody (red) (Figure 5e). All MDA-MB-231 type cells moved away from the dashed diagonal line indicating differential remodeling away from each other (Figure 5e). The parental MDA-MB-231 cells do not migrate as deeply into the device and the astrocyte cells move further from the barrier. The MDA-MB-231-BR cells divide into groups where astrocytes move away from the barrier as cancer cells infiltrate. This results in a similar shift in distance but by a different mechanism. The JIMT-1 parental line shows the astrocytes congregating at 100 μm away from the barrier but a large proportion of cancer cells have moved back into the flow side of the chamber. Finally, the JIMT-1-BR cells exhibit mixed behavior where many astrocytes group 150 μm away from the barrier, but a second group moved towards the tumor cell location.
The average distance between cancer cells in astrocytic BBN chips with or without Dkk-1 neutralization (Figure 5f–g) confirm these interpretations. The kernel estimate histogram of an un-treated control was subtracted from the sample treated with the Dkk-1 neutralization antibody and plotted to show the change in cancer to astrocyte distance. In the presence of Dkk-1 neutralizing antibody (low Dkk-1 condition), the MDA-MB-231 parental line and astrocytes are ~150 μm apart on average. A similar trend is seen for the MDA-MB-231 brain tropic cells, but the cells are further apart (~175 μm). The JIMT-1 parental line also shifts away from the original bulk position of 50 μm to two subsets positioned at 150 μm and 175 μm. However, the brain tropic JIMT-1 cells have two small populations that move apart but have a broader set of cells across distances. Finally, we show representative images of the device with and without the Dkk-1 neutralizing antibody treatment in Figure 5h.
2.7. Brain-metastatic cancer cell metabolism is re-wired when stimulated with brain niche secretions
The influence of the brain niche on cancer cell migration and extravasation suggested that contact with brain niche secretions alter cancer cell metabolism. Changes in the extracellular metabolites of MDA-MB-231-BR and MDA-MB-231 under stimulation with brain niche secretions were quantified using mass spectrometry and NMR.
Alterations of the secreted metabolome were observed in amino acids (Figure 6a). The MDA-MB-231-BR secreted the highest levels of phenylalanine, succinate, and alanine when stimulated with brain niche secretions (Figure 6b). In contrast, the highest parental MDA-MB-231 secreted metabolites included histidine when stimulated with astrocyte or microglia secretions, and valine when stimulated with endothelial secretions (Figure 6b). The same measurements are provided for the metabolite changes in the brain niche cells (astrocytes, microglia, and endothelial cells) due to stimulation from the MDA-MB-231 and MDA-MB-231-BR secretions (Figure S8).
Figure 6. Cancer cell metabolism is influenced by brain niche secretions.

(A) Summary of changes in metabolic pathway enrichment between MDA-MB-231 and MDA-MB-231-BR cells in the presence of astrocytes. (B) Cancer secretion metabolites quantified using NMR. Each cancer type was stimulated with brain niche secretions. Heatmap colors indicate row comparisons of each metabolite level; blue (low) to red (high). The size of each square indicates overall metabolite levels. (C-D) Metabolic pathway impact in (C) MDA-MB-231 or (D) MDA-MB-231-BR under stimulation with astrocyte secretions, p < 0.05 Metaboanalyst pathway impact. NMR was performed once.
The MDA-MB-231-BR and MDA-MB-231 enriched several metabolic pathways under stimulation of brain niche cell secretions including (Figure 6c, 6d, Figure S9): urea cycle; aspartate metabolism; glucose-alanine; alanine metabolism; aspartate metabolism; glycine and serine metabolism; Warburg effect; valine, leucine, and isoleucine degradation; and glutathione metabolism. Astrocyte secretion stimulation of MDA-MB-231-BR and MDA-MB-231 uniquely increased arginine and proline metabolism compared to stimulation. MDA-MB-231-BRs and MDA-MB-231s stimulated with endothelial secretions promoted pathway enrichment of ammonia recycling, glutamate metabolism, amino sugar metabolism, and ethanol degradation. Microglia secretions did not promote any unique metabolomic pathways compared to astrocyte or endothelial secretions shared in both MDA-MB-231-BRs and MDA-MB-231s.
The MDA-MB-231-BR extracellular metabolomic profile under stimulation of brain niche cell secretions commonly expressed pathway enrichment of: glutamate metabolism; arginine and proline metabolism; and amino sugar metabolism. When MDA-MB-231-BR are stimulated with astrocyte secretions, the MDA-MB-231-BR uniquely increase: ammonia recycling; glutamate metabolism; arginine and proline metabolism; and amino sugar metabolism (Figure 6d). With microglia secretion stimulation, the pathways unique to MDA-MB-231-BR were: glucose-alanine cycle; Warburg effect; glycine and serine metabolism; amino sugar metabolism; alanine metabolism; glutamate metabolism; ethanol degradation; glutathione metabolism; arginine and proline metabolism; transfer of acetyl groups into mitochondria; and valine leucine and isoleucine degradation. Endothelial secretion stimulation of MDA-MB-231-BR resulted in signaling of: arginine and proline metabolism; transfer of acetyl groups into mitochondria; and methylhistidine metabolism.
No shared metabolic pathways were enriched in the parental MDA-MB-231 under brain niche cell secretions. The MDA-MB-231s under astrocyte secretion stimulation increased signaling of: ethanol degradation; transfer of acetyl groups into mitochondria; tryptophan metabolism; and folate metabolism. Endothelial secretions stimulated one unique metabolic pathway in MDA-MB-231s compared to MDA-MB-231-BR: arginine and proline metabolism. Microglia secretions did not yield enrichment of pathways that were uniquely expressed by the MDA-MB-231s and not MDA-MB-231-BR. Overall, interactions with the niche cells, especially astrocytes, yielded increases in critical amino acid metabolic pathways especially in brain trophic cells compared to parental controls, pointing to modulation of these pathways as reasonable potential avenues to help control metastatic growth in the brain niche.
2.8. Gene expression profiling of MDA-MB-231-BR and MDA-MB-231 under Dkk-1 stimulation
To characterize the cancer cell response to Dkk-1 at the molecular level, NanoString PanCancer Pathways gene expression profiles were compared between MDA-MB-231-BR and MDA-MB-231 with and without Dkk-1 stimulation for 24 hrs. A total of 30 significant differentially expressed genes were found to be expressed in the MDA-MB-231-BRs under Dkk-1 stimulation (Figure 7a, b). They showed increased expression of FGF-13, PLCB1, and MYC, and decreased expression of ITGB4, NGF, and PRKX. NanoString nSolver identified Wnt, Ras, PI3K, and MAPK signaling pathways as utilized by the MDA-MB-231-BRs under Dkk-1 stimulation (Figure 7c). Further detailed data are shown in Figure S10.
Figure 7. The role of astrocytic Dkk-1 can be regulated by modulating relevant MDA-MB-231-BR pathways.

(A-C) NanoString gene expression profiling of untreated and Dkk-1 treated MDA-MB-231-BR and MDA-MB-231 cells. (A) Volcano plots of fold change between cell types untreated and treated with Dkk-1 with adjusted p-values. (B) Venn diagram of significant differentially expressed genes, FDR threshold p < 0.05. (C) Fold change of significant differentially expressed genes under Dkk-1 stimulation categorized by signaling pathway. (D) ELISA showing Dkk1 found in the secretions of astrocyte stimulated MDA-MB-231 FGF13 KD and MDA-MB-231-BR FGF13 KD is lowered when FDF13 is KD. (E) Images of MDA-MB-231-BR and MDA-MB-231 cells migrating towards Dkk-1. Scale bars = 200 μm (F-G) Percent change in distance migrated between FGF-13 knockdown and pGIPZ scramble in Dkk-1 gradients for MDA-MB-231-BR and MDA-MB-231 at (E) 24 hr and (F) 48 hr. **p < 3.0*10−3, ****p < 3.0*10−4 t-tests with Holm-Šídák multiple comparisons test. A-D was performed with triplicate technical replicates. E-G was performed with three biological replicates each with three technical replicates.
FGF-13 was knocked down in both MDA-MB-231 cells to determine if it plays a pivotal role in regulating the effect of Dkk1 in the tumor micro-environment. The pGIPZ and FGF-13 knockdown variants were seeded into the migration device discussed in Figure S2 under the same conditions before being imaged and analyzed at 24 hrs and 48 hrs. First, to confirm the knockdown reduced Dkk1 expression undrer astrocytic stimulation ELISA (Figure 7d) was performed on the FGF-13 knockdown and controls. The expression levels were reduced an order of magnitude. The images in Figure 7e show the MDA-MB-231-BR and MDA-MB-231 cells exhibited different morphologies with the MDA-MB-231-BR cells elongated under an SFM-Dkk1 gradient. Finally, Figure 7f, 7g show the relative average distance FGF-13 shRNA knockdown cells migrated compared to pGIPZ scramble at 24 and 48 hrs, respectively. The error bars show the standard error. Significance was tested by t-tests with Holm-Šídák multiple comparisons test between the parental and brain seeking cells. At 24 hrs., the brain seeking cells differed from the parental cells but also had reduced migration when fully stimulated with Dkk1-Dkk1 relative to the control. In contrast, the parental knockdown was often more migratory than the control. At 48 hrs these differences often resolve with both cell types migrating in a similar manner to the control in the presences of Dkk1.
3. Discussion
3.1. Astrocyte cells and their secretions are integral for brain metastasis.
While previous work has identified the basic behavior of parental and brain tropic cells inside of the BBN, little is known about the interaction between the cancer cells and the individual cell components within the brain [21]. A great deal of work has shown that the tumor cells can evade the BBB although a mechanism is not fully understood [22]. Recent studies have begun to elucidate the role of astrocyte cells on brain tumor and brain metastasis development [23]. In murine models astrocytes are responsible for regulating tight junctions within the blood brain barrier. and may regulate the entire brain micro-environment. Zhang et al. has summarized these results showing multiple immunosuppressive effects such as paralyzing T cell migration, mediating proinflammatory molecules such as CD4 and CD8+ T cells activation [24]. In addition, Heiland et al. found that astrocytes may play a role in suppressing the immune system surrounding glioblastoma, in part by inducing T cell exhaustion through expression of PD-L1. Beyond protein secretions, astrocyte secretion of microRNAs has been shown to induce micro-environment PTEN loss, an important tumor suppressor [25].
Moreover, cancer cells may interact with astrocytes by either direct cell-cell interaction or via secretions. Several authors suggest that astrocytes may promote tumor formation by secretions. Liu et al. have shown that astrocytes promote medulloblastoma progression through hedgehog secretion [26]. Another possibility is that astrocytes secrete chemokines that promote extravasation. One example is the CCL2-CCR2 astrocyte-cancer cell receptor interaction found by Hajal et al. that demonstrated CCR2-deficient cancer cells exhibit significantly reduced extravasation in murine models. Neurotrophic factors secreted by astrocytes including TGF-α and CXCL12 may increase invasiveness of GBM cells. These examples indicate that the brain micro-environment is a complex set of interactions between all cells and system that result in promoting certain brain metastasis.
In this study, we utilized the BBN to unravel the physical and secretory interactions between cancer cells and the brain niche cells (endothelial, astrocyte and microglia). Our previous work has shown that there is differential behavior of the parental MDA-MB-231 and MDA-MB-231-BR cells in the niche that mimics known in vivo invasive behavior [16]. The MDA-MB-231-BR cells would degrade the endothelial barrier at a faster rate than the parental cells and grew more rapidly [16, 27]. In addition here, we found that the cooperative interaction between the cancer and brain niche cells induced remodeling of the BBN. Although this behavior was consistent, the way it was achieved differed according to which cell (cancer or astrocyte) moved the most relative to the endothelial barrier. This behavior suggests that the mechanisms driving these two remodeling schemes may differ. Because this behavior indicates that the cancer cells and astrocytes interact, an important question was if extravasation was driven by physical or chemical signals. Our data support the notion that both physical interaction with astrocytes and their cytokines are important to drive cancer progression in the brain. The data show that this occurs both as a migratory and invasive phenotype. Importantly, MDA-MB-231 brain tropic cells were primarily affected by astrocytes secretions and microglia secretions. However, the strongest effect was observed when the secretions were used to directly stimulate cells. This suggests that the cells require a certain level of cytokine stimulation, before being activated to migrate. Combined with our observation that brain tropic cells degrade the endothelial barrier at a greater rate may form a compelling argument for how these cells develop into more aggressive metastasis. It may be that brain tropic metastases degrade the endothelial layer and are then stimulated by released brain cytokines that draw the cancer cells and astrocytes towards one another.
In the BBN, cancer cells became more spherical when cultured with brain niche cell secretions. This may indicate, as previously reported, the migratory mechanism being utilized by cells that extravasate deeply into the device. Conversely, the clusters of cells (>10 cells by volume) become flatter, likely as they grow along the endothelial barrier for support. The fact that clusters stay closer to the endothelial barrier than individual cells also indicates that they may be driven by growth, supported by nearby cancer cells. This belies the idea that a single cell may rapidly extravasate and travel some distance before forming a deadly cluster. This observation may be of importance for therapeutics. First, it indicates that clusters of cells form near the blood brain barrier. Treatments targeting those cell clusters may leave single cells deeper in the brain niche space that then can proliferate. Second, our data indicates brain tropic cells degrade the BBB during the growth stage. Therefore, cancer cells may form a secondary barrier that reduces drug penetrations into the tumor. From our work, a model emerges in which major changes occur with only the modulation of secretions, so we pose that the barrier must be damaged prior to large scale invasion of cancer cells and clusters.
A compelling therapeutic target then is to disrupt crosstalk between cancer cells and brain niche cells that drives brain metastasis [28]. For example, the canonical Wnt-beta catenin pathway is known to control cell proliferation, migration, differentiation, and is critical for mediating astrocyte – neuronal crosstalk in the brain by regulating glutamate uptake for astrocytes [29]. Wnt signaling is more active in metastatic triple-negative breast cancer (TNBC) compared to non-basal subtypes of breast cancer and correlates with poor prognoses [30]. Yet it is unknown which pathway may be therapeutically targeted. This challenge motivated this study to identify secretion dependent cancer-astrocyte and cancer-microglia interactions. We observed using dual independent techniques, only Dkk-1is increased when brain niche cells were stimulated with both cancer cells and cancer cell secretions. Dkk-1 is a known modulator of the Wnt signaling pathway; where increased levels of Dkk-1 can block Wnt-signaling of β-catenin eventually reducing GS production and glutamine and glutamate metabolism [20]. Reducing Dkk-1 levels near invading tumor cells may increase metabolite production reducing extravasation.
Astrocytic Dkk-1 influences cancer cell migration and extravasation into the brain niche, as shown by both the migratory behavior of MDA-MB-231 cancer cells when stimulated with Dkk-1 and the significant change in the distance between cancer cells and astrocytes when the BBN is treated with Dkk-1 neutralizing antibody. The mean distance between cancer and astrocyte cells more than doubles because of neutralizing Dkk-1. This is a remarkable change that may highlight a strategy towards preventing the development of a pre-metastatic niche.
NMR analysis of extracellular MDA-MB-231-BR metabolites revealed an increase in glutamate metabolism, indicating the brain-tropic cell line’s inherent metabolic plasticity under brain niche secretion stimulation. Moreover, stimulation with endothelial secretions also increases glutamate metabolism of both MDA-MB-231-BR and MDA-MB-231 cancer cells. Knowing these pathways are enriched and critical to the role of Dkk-1 in the Wnt signaling pathway we finally observed if cancer gene expression signatures differed under Dkk-1 stimulation. By shRNA knock down of salient Wnt related genes (FGF-13) that increased in gene expression with Dkk-1 stimulation we found that cancer cell migration patterns were altered in the migration chip analysis in Dkk-1 gradients. Specifically, the MDA-MB-231-BR FGF-13 knockdown was less migratory both under direct stimulation and chemotaxis. The MDA-MB-231 FGF-13 knockdown initially showed mixed migratory behavior but after 48 hrs, it also exhibited dampened migration in the presence of Dkk-1. However, one concern is that the cells became more migratory under nominal conditions without serum. This could indicate that knocking down FGF-13 may make metabolically restricted cells more migratory.
Several limitations of our study require careful consideration. Exosomal crosstalk, including miRNA modulation was not evaluated. Moreover, metabolites were not separated from protein secretions, possibly confounding some of the results. Astrocytes and microglia were assessed separately but not cooperatively, and no pericytes were present in our brain niche model. Secretions from stimulated cells also contained the stimulating secretions. This limits the physiological relevance compared to a human model and would be an important next study.
4. Conclusion
In conclusion, using the BBN and migration device, we have highlighted one of the communication strategies that leads to remodeling of the brain niche in pre-metastatic states in the presence of brain tropic cancer cells. We have also identified key cytokines driving that remodeling process. Finally, we highlighted important changes to the metabolism and upregulated pathways utilized by cancer cells to induce extravasation and remodeling including key regulatory genes.
5. Experimental Section/Methods
Cell culture and reagents:
Triple negative breast cancer (TNBC) MDA-MB-231, the brain-metastatic derived line MDA-MB-231-BR-GFP, Her2+ breast cancer JIMT-1, and the brain-metastatic derived line JIMT-1-BR cells were obtained from Dr. Patricia Steeg. MDA-MB-231 and JIMT-1 lines were maintained in DMEM supplemented with 4.5 gL−1 glucose, 10% FBS, 2 mM L-glutamine, and 1X antibiotic-antimycotic. Normal human astrocytes (Lonza, CC-3186) were maintained in DMEM supplemented with 4.5 gL−1 glucose, 10% FBS, 2 mM glutamax, 2 mM sodium pyruvate, 1X N2 growth supplement, and 1X antibiotic-antimycotic. Human microglia line HMC3 (ATCC, CRL-3304) were maintained in EMEM supplemented with 10% FBS, 2 mM L-glutamine, 2mM sodium pyruvate, 1X non-essential amino acids, and 1X antibiotic-antimycotic. Brain microvascular endothelial cell line hCMEC/D3 (Millipore, SCC066) were maintained in endothelial cell growth medium 2 (PromoCell) with 1X antibiotic-antimycotic. All cells in routine cell culture and within microfluidic devices were grown at 37 °C in 5% CO2. MDA-MB-231, JIMT1, and JIMT1-BR with stable cytoplasmic GFP expression were generated by lentiviral transduction of pLL.EV-GFP empty vector, astrocytes were transduced with pLL3.7-smURFP (small ultra-red fluorescent protein), and microglia were transiently stained with efluor proliferation dye 450 according to manufacturer’s instruction. All non-fluorescent cells in each stable fluorescent cell lines were removed using fluorescence-activated cell sorting prior to experimentation (Moflo Astrios cell sorter). For shRNA knockdown studies, pGIPZ-FGF-13 (Dharmacon clone 367913, 409644), and pGIPZ-scrambled (Dharmacon catalog number RHS4346) were individually transduced into MDA-MB-231 and MDA-MB-231-BR cells.
Assessment of cancer cell extravasation using a microfluidic blood brain niche device:
A total of 4 blood brain niche channels per single chip for replicates. The bottom chamber of each blood brain niche channel is formed with a collagen solution (3 mg/mL, PureCol) in 1X MEM supplemented with 4.5 gL−1 glucose and sodium bicarbonate and allowed to gel beneath a polycarbonate membrane (5 μm pore size) separating the bottom and top chambers at 37 °C. This collagen mixture is infused with either 1) 125,000 astrocytes stably labeled to express cytoplasmic pLL3.7-small ultra-red fluorescent protein (astrocyte-smURFP), 2) or 18,750 microglia cells stained with eFluor 450 proliferation dye according to the manufacturers protocol (microglia-eF450), or 3) left as a collagen media mixture. Once the bottom chamber is solidified, Matrigel solution (2%, growth factor reduced with phenol red) diluted in endothelial cell complete media coats the top flow channel with the bottom lining membrane containing the brain niche. Brain microvascular endothelial cells stably labeled to express cytoplasmic dsRed (hCMEC/D3-dsRed) are then seeded and form a barrier on the matrigel coated membrane after 2 days of culture with media exchanged twice daily. Devices containing astrocytes in the brain niche are cultivated using a 50/50 mixture of complete astrocyte and endothelial media. Devices containing microglia in the brain niche are cultured using a 75/25 mixture of complete endothelial/microglia media. μBBN chips were cultured for 2 days to permit BBB formation prior to the addition of cancer cells. All μBBN chips containing no astrocyte or microglia cells were exchanged to contain astrocyte or microglia secretions at 1X concentration in the brain niche. μBBN chips containing astrocyte cells were treated with 10 μgmL−1 Dkk-1 neutralization antibody (R&D systems) in the bottom brain niche side of the chip twice daily. A total of 30,000 cancer cells stably expressing cytoplasmic GFP (pEV-GFP) in single cell suspension were seeded into each top flow chamber of a μBBN chip and cultured for up to 9- days post-seeding to visualize micrometastasis. All BBN chips containing astrocyte-smURFP cells were stimulated with 10 μM biliverdin diluted in the chip media as a co-factor to visualize smURFP fluorescence 24 hours prior to imaging each time point.
BBNiche chips are imaged 2- and 9- days after seeding cancer cells using a Nikon A1r fluorescent confocal microscope. Each channel is captured using a XY tiled z-stack. A previously reported confocal tomography technique was applied for analysis of cancer cell extravasation into the chip and modified to include monitoring of the BBNiche astrocyte and microglia cell positions at each timepoint [16, 27]. This approach extracts individual cellular volumes from the 3D confocal image and reports various cellular characteristics from them including: 1) cell position relative to the endothelial barrier, 2) sphericity as a metric for cell shape are reported, 3) distances between cancer cells and astrocytes or microglia, and 4) cell counts. These methods were expanded to include cluster analysis and structural analysis of the tumor micro-environment. Cluster analysis was done by filtering out 3D volumes less than ten times the median cell volume. The remaining larger volumes were labelled as clusters or BBNiche micrometastasis and the metrics were re-calculated. The volumes were also broken up using a BCC crystal lattice overlay sized on the median size of the cell population and the individual cells were annotated and added to the list of total cells. For both cells and clusters, resulting distributions for each metric were analyzed to identify sub-populations of cells within the device. This was done by unbiased identification of subpopulations. First the KDE bandwidth for each distribution was selected by scoring the troughs found between peaks along the KDE. The score was determined to identify troughs between peaks that were possible breaks between sub-populations. The score was established using the following equation:
| (1) |
is the summation of the prominence of each trough, G is the number of sub-populations expected, and is the average distance between troughs. This approach minimized the number of subpopulations while maximizing the prominence and spacing. Next a k-means fuzzy clustering was used on each cell to determine the probability it belonged to the list of sub-populations. Finally, boot strapping sampling was used to identify the mean, median and standard error of each sub-population. This enables comparison between sub-populations of cells within each device to sub-populations in other devices. Finally, interaction between the cells the niche was measured in two ways. First, the endothelial layer was found by fitting a planar or curved surface to the endothelial cell centroids in the device. Then the distance of cells or clusters to that barrier were measured. Second, the distance between both the centroid and bounding box edges of individual cells of different types were calculated. We performed a minimum of 6 biological replicates for each experimental condition, and each measurement included thousands of cells. Statistical significance was determined using a Smirnov-Kolmogorov test and Kruskal-Wallis Rank Sum Test (p < 0.05) for distributions and t-test with multiple comparison correction for peaks.
Interpreting 2D density remodeling plots:
The goal of these plots is to show how cancer cells and resident brain niche cells co-locate over time. The 2D density plots convey the interactions between pairs of cancer cells and brain resident cells at specific locations within the blood brain barrier niche. Figure 1b, 2c and 5c are the legend for interpreting the graph. Darker colors indicate more pairs of cells interacting at a specific location, while lighter colors indicate fewer pairs. These plots are divided into four quadrants that describe where in the BBN device the cells are located. Quadrant 1 shows that the cancer cell in a pair is on the brain side of the device and the astrocyte is on the flow chamber side. Quadrant 2 shows cancer and astrocyte cells are both on the brain side of the device. Quadrant 3 shows both cancer and astrocyte cells on the flow chamber side of the device. Finally, quadrant 4 shows cancer cells in the flow chamber and astrocyte cells in the brain side of the device. The proximity of the cells to each other can be visualized by the distance the pair is from the short, dotted diagonal line. When the two cells are touching, they will be plotted on the diagonal line and the further apart they get from each other, the further from the diagonal line they will be. The red overlays in quadrants 1 & 2 show when cancer cells are in the lower 50% of the brain chamber. The blue overlays in quadrants 2 & 4 show when the astrocyte cells are in the lower 50% of the brain chamber. The endothelial barrier is represented by the long-dashed lines. The “distance extravasated” axis indicates the distance (μm) between the cancer cells and the endothelial barrier. Distances ranging from −200 to 0 μm are cells that have remained in the microfluidic flow chamber space and have not extravasated through the endothelial barrier. A distance of −200 μm indicates cancer cells that reside in the top of the flow chamber, farthest away from the endothelial barrier positioned at 0 μm. Positive extravasated distances (greater than 0 μm) represent cells that have migrated into the brain niche compartment. Cells can maximally travel a distance of 300 μm in the brain niche compartment before reaching the bottom of the brain niche chip. While the “distance to the barrier” axis indicates the distance (μm) between the astrocyte cells and the blood brain barrier, astrocyte distance to the barrier ranged from 0 to 300 μm within the microfluidic chip brain niche component, with 300 μm representing the farthest distance from the endothelial barrier. In this way you can identify how many pairs of cancer cells and astrocytes cells are a certain distance from each other and the endothelial barrier providing a robust understanding of remodeling in the niche.
Assessment of Cell Migration using a Microfluidic Migration Device:
Briefly, the migration chip is constructed out of polydimethylsiloxane (PDMS) and positions single cells at one of the entrances of 40 migration channels that are 1000 μm in length, 20 μm wide and 10 μm tall (Figure S2a). Passive diffusion chemoattractant gradients were established, and cells were monitored microscopically at 0 and 24 hrs to visualize migration. Cancer cell migration was assessed towards two gradients: 1) an increasing concentration of astrocyte, microglia, or endothelial secretions in serum-free media (SFM) to address chemotactic qualities of the brain niche secretions, and 2) with each brain niche cell secretion in SFM provided as a stimulus to the cancer cells throughout the entirety of the migration chip, with 10% FBS as an external chemotactic gradient. Astrocyte and endothelial secretory chemotactic gradients did not affect MDA-MB-231-BR or MDA-MB-231 cells migration when provided as a chemoattractant compared to SFM alone. However, in comparison to SFM alone, a chemotactic gradient of microglia secretions significantly influenced parental MDA-MB-231 migration (Figure S2b). In fact, MDA-MB-231-BR significantly reduced their migratory capacity in the presence of microglia secretions compared to SFM only. We then provided the individual brain niche secretions as a potentially stimulatory factor to induce them to migrate towards an external gradient of FBS, to ascertain the ability of the secretions to convert non-migratory cancer cells towards a migratory phenotype. Astrocyte and endothelial secretions stimulated MDA-MB-231-BR migration towards an FBS gradient compared to baseline migration towards an FBS gradient alone (Figure S2c). Although microglia secretion gradients were sufficient to promote parental MDA-MB-231 migration, the microglia secretions did not prime parental MDA-MB-231s to increase their migration towards external FBS gradients. Importantly, no statistically significant cell growth was detected over the 24 hr. migration period in all gradient conditions (Figure S2d–e).
The migration chips design and utility has been described in previous reports and were built in the same manner out of polydimethylsiloxane (PDMS) with biopsy punches to form the inlets and outlets and bonded to glass slides using an oxygen plasma treatment [17, 31]. Degassed migration chips are coated with collagen I (1 μgmL−1) lining all channels for 18–24 hours in a tissue culture incubator then rinsed with HBSS under flow conditions to remove excess collagen. Single cell suspensions of cancer cells were seeded into the top left reservoir and flowed down towards the bottom left reservoir with the opportunity to attach to the entrances of the horizontal migration channels. Excess, non-attached cells were removed with excess complete media, then the cancer cells were incubated at 37 °C, 5% CO2 to permit cell adherence for 2 hours. The migration chips were washed with HBSS +Ca/Mg under flow then exchanged into appropriate experimental conditions to form 1) chemoattractant gradients of astrocyte, microglia, and endothelial secretions or 20 ngmL−1 purified human Dkk-1 (Peprotech) in serum free media and 2) secretion or 20 ngmL−1 Dkk-1 stimulation in serum free media with an FBS gradient. Epifluorescent phase contrast images were captured at 0 and 24 hrs. Using a Python script, cell migration was calculated as the distance (μm) in horizontal position of the cell to the beginning of the seeding channel to encompass non-migratory cell populations residing at the entrances to the migration channels. Plotted data represent 3–5 separate biological replicates, for an average of 215 cell migration distances per experimental condition (range: 147–419 cells). Statistical significance was determined by Mann-Whitney t-test and a Bonferroni correction (p < 0.00625).
Cytokine and metabolite profiling:
Secretion collection:
Astrocytes, microglia and endothelial cells were seeded into dishes at 60% confluence and cultured overnight. Each cell type was either left in basal media or stimulated with MDA-MB-231-BR cells, MDA-MB-231-BR secretions, MDA-MB-231 cells, or MDA-MB-231 secretions the following day. Empty dishes were also seeded with the same components used for stimulation: MDA-MB-231-BR cells, MDA-MB-231-BR secretions, MDA-MB-231 cells, MDA-MB-231 secretions or basal media and carried as controls. Conditioned media containing cellular secretions were collected after 24 hours of interaction with the cancer cells, centrifuged to remove any debris/particulates and frozen at −80 °C until use. Similarly, cancer cells seeded into dishes were left unstimulated or stimulated with astrocyte, microglia, and endothelial cells or paired secretions for collection after 24 hours. Again, empty dishes were also seeded with astrocyte, microglia, and endothelial cells or secretions and carried as controls.
Cytokine profile identification:
Assessment of cytokines secreted by all brain niche and cancer cells was performed using Proteome Profiler Human XL cytokine array (R&D Systems) according to the manufacturer’s protocol using 500 μL of each secretion. One biological replicate of each experimental sample was assayed in technical duplicates. All dot blots were exposed to film for 10 seconds before developing. The dot blot intensities were quantified using Quick Spots Tool (Western Vision Software). Fold change of cytokine expression in stimulated compared to unstimulated controls was calculated and plotted using the Morpheus software (Broad Institute). Statistical significance was determined using multiple t-tests with a Benjamini, Krieger, and Yekutieli false discovery rate correction in Prism.
Cytokine quantification:
Collected secretions were concentrated 10X using Amicon Ultra centrifugal filters with a 3 Kda molecular weight cut off and submitted to the University of Michigan Rogel Cancer Center Immunology core for ELISA quantification. Assays included: Human Dkk-1 (Quantikine kit, R&D Systems), MCP-1, IL-8, CXCL5, GM-CSF, IL-6, Gro-1, and VEGF. A total of 4 biological replicates were assayed in technical duplicates. Raw cytokine concentrations of control media used for stimulation were subtracted from each experimental sample accordingly, and corrected by total protein content (BCA, ThermoFisher). Resulting cytokine levels were normalized to their paired unstimulated control condition. Heatmaps were plotted using Morpheus software (Broad Institute) and statistical significance was determined using multiple t-tests with a Benjamini, Krieger, and Yekutieli false discovery rate correction in Prism.
Extracellular metabolite quantification:
A single biological replicate for each experimental sample was submitted to the University of Michigan College of Pharmacy NMR core for metabolite quantification. Nanosep centrifugal devices (Pall Corporation) with a 3 Kda molecular weight cut off were pre-rinsed x3 with ultrapure water, then x1 with deuterated water (Sigma Aldrich). Metabolite samples (500 μL per sample) were then filtered through the pre-rinsed devices to remove proteins according to manufacturer’s specifications (14,000xg at 4 °C for 20 minutes). After filtration, 50 μL deuterated water was applied to the top of the filter, vortexed, then centrifuged again to collect all metabolites in the filtrate. 50 μL of DSS internal standard (IS) was added to each sample, then samples were stored at −80 °C in cryovials until the NMR assay was performed. The 1D-1H-NMR spectrum of each filtered cell media sample was acquired on an Agilent, 500 MHz NMR spectrometer with a VNMRS console operated by host software VNMRJ 4.0 and equipped with a 5-mm One-Probe. The Chenomx internal standard (IS), DSS-d6 (3-(Trimethylsilyl)-1-propanesulfonic acid-d6 sodium salt) was used as a reference signal for the quantification of metabolites. On the day of the NMR experiments, samples were thawed, pH was measured and adjusted to be within 6.5–7.5 range and 32 scans were collected. The NMR experiment, which consists of the first increment of a 1H,1H-NOESY (commonly referred to as a 1D-NOESY or METNOESY) pulse sequence was as follows: a 1 s recovery delay, a 990 ms saturation pulse of 80 Hz (γB1) induced field strength empirically centered on the water resonance, 2 calibrated 90 ° pulses, a mixing time (tmix) of 100 ms, a final 90 ° pulse, and an acquisition period of 4 s. Optimal excitation pulse widths were obtained by utilizing an array of pulse lengths to determine the 360 ° pulse null for water, and dividing by 4 to obtain the 90° optimum. Spectra were acquired at a room temperature of 298 ± 0.3 K. The resulting NMR spectra were analyzed using Chenomx NMR Suite 8.3 (Chenomx, Inc.). The Processor module was used to phase shift and baseline correct each spectrum. Compounds were then identified and quantified in the Profiler module of the software, which accounts for the pH of the sample and the concentration of the IS and quantifies metabolite concentration relative to the IS. Metabolite identity was confirmed using the Chenomx Compound Library, which contains >300 compounds. Around 32 compounds resulted in the Chenomx library used on the cell media fluids to identify and quantify metabolites with various degrees of certainty. The difference in metabolite concentrations in conditioned cell culture media from concentrations in the control medium utilized for stimulation were corrected by total protein content (BCA, ThermoFisher). Resulting metabolite concentrations were normalized to their paired unstimulated control condition and analyzed using Metaboanalyst 4.0 software for metabolite pathway enrichment (Xia lab, McGill University). Heatmaps were plotted using the Morpheus software (Broad Institute).
Blood brain niche microfluidic device fabrication and assembly:
The blood brain niche design has been described in previous reports and were built in the same manner out of polydimethylsiloxane (PDMS) with biopsy punches to form the inlets and outlets [16, 27]. A polycarbonate 5 μm polymer porous membrane divides the upper and lower chambers on which the endothelial layer was cultured. The μBBN devices are bonded to 50 mm x 75 mm glass slides after oxygen plasma treatment (50 Watts, 30 seconds). P200 pipette tips are cut and used for media reservoirs in the blood brain niche chip.
NanoString gene expression profiling:
6-well plates were seeded with 0.15*106 MDA-MB-231-BR or MDA-MB-231 cells/well and allowed to attach overnight, then treated with 20 ngmL−1 purified human Dkk-1 for 24 hrs or left untreated. RNA isolations were performed using the Rneasy mini kit (Qiagen) and assessed for quantity and purity using a nanodrop (ThermoFisher). Gene expression was assessed using a NanoString Human PanCancer Pathways codeset according to manufacturer’s instruction and further analyzed using the nCounter Advanced Analysis Module which normalizes gene expression to a set of positive and negative controls and optimizes housekeeping genes using linearity across the dataset. nCounter software was utilized to convert raw gene counts to Log2 fold changes of MDA-MB-231-BR vs MDA-MB-231 with and without Dkk-1 treatment. Statistically significant p-values of differential expression were determined by the nCounter software using T-tests and a Benjamini-Hochberg correction, p < 0.05. Statistically significant differentially expressed genes that were exclusive to the Dkk-1 treatment were determined and plotted in R and were assorted by signaling pathway as dictated by the NanoString panel.
Statistical Analysis:
Statistical analysis was performed using R scripts (μBBN chip data) and Prism (2D migration, dot blot, ELISA analyses) and Python’s Scikit. Comparisons between the populations of cells within μBBN chips were made using Smirnov-Kolmogorov tests and Kruskal-Wallis Rank Sum Test (p < 0.05). Statistical significance of the average cell migration in 2D in the microfluidic migration chips was determined using multiple Mann-Whitney t-tests with a Bonferroni correction (p < 0.00625). For comparison between sub-populations within a distribution of a cells metrics (distance extravasated, sphericity) statistical significance was determined using a t-test with multiple comparison correction for peaks.
Comparisons between each chemotactic gradient of BBN secretions and the SFM-SFM control were performed. Stimulatory gradients of secretions were tested for statistical significance by comparing against the SFM-FBS and FBS-FBS controls. Relative changes between FGF-13 knockdowns and pGIPZ scramble controls used Holm-Šídák multiple comparisons test. The mean dot blot intensities and ELISA concentrations of MDA-MB-231-BR and parental MDA-MB-231 secretions under each stimulation condition were determined to be statistically significant using multiple t-tests with a Benjamini, Krieger, and Yekutieli false discovery rate correction. Similarly, brain niche cells stimulated with MDA-MB-231-BR or MDA-MB-231 were compared for significance for dot blot and ELISA assays. The values for n, p, and the specific statistical test performed for each experiment are included in the appropriate figure legend and main text.
Supplementary Material
Acknowledgements
General:
We would like to thank Dr. Patricia Steeg’s Lab, at the National Cancer Institute for the generous donation of MDA-MB-231-BR-GFP, JIMT1, and JIMT1-BR cells. pLenti-smURFP was a gift from Erik Rodriguez & Roger Tsien (Addgene plasmid #80349; http://n2t.net/addgene:80349; RRID:Addgene 80349). We thank several University of Michigan Biomedical Research Core Facilities for contributions to this work: The Vector core for subcloning and lentiviral preparation, Fluorescence-activated cell sorting was performed at the Flow Cytometry Core, and the Microscopy core for confocal microscopy of the BBN devices. Confocal microscopy was also performed at the University of Michigan Biointerface Institute (BI). We wish to thank Joel Whitfield of the University of Michigan Rogel Cancer Center Immunology Core for ELISA analysis, Dr. Larisa Yeomans at the University of Michigan College of Pharmacy NMR core for analysis and quantification of metabolites, Kelley Kidwell and Ryan Ross, University of Michigan School of Public Health, Department of Biostatistics for statistical consultation, and Dr. Abhinav Achreja for helpful suggestions.
Funding:
T.M.W. was partially supported by 1R21CA245597-01 and the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR002240. C.R.O. was partially supported by an NIH T-32 Training Fellowship (T32CA009676) and 1R21CA245597-01. Funding for materials and characterization was provided by National Cancer Institute of the National Institutes of Health under award number 1R21CA245597-01, P30CA046592, 5T32CA009676-23, CA196018, AI116482, METAvivor Foundation, and the Breast Cancer Research Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Trisha M. Westerhof, Michigan Medicine, Department of Internal Medicine, Division of Hematology/Oncology, University of Michigan Medical School, Ann Arbor, MI 48109, USA..
Benjamin A. Yang, School of Engineering, Department of Biomedical Engineering, University of Michigan, Ann Arbor, MI 48109, USA.
Nathan M. Merill, Michigan Medicine, Department of Internal Medicine, Division of Hematology/Oncology, University of Michigan Medical School, Ann Arbor, MI 48109, USA.
Dr. Joel A. Yates, Michigan Medicine, Department of Internal Medicine, Division of Hematology/Oncology, University of Michigan Medical School, Ann Arbor, MI 48109, USA.
Dr. Megan Altemus, Michigan Medicine, Department of Internal Medicine, Division of Hematology/Oncology, University of Michigan Medical School, Ann Arbor, MI 48109, USA..
Liam Russell, School of Engineering, Department of Biomedical Engineering, University of Michigan, Ann Arbor, MI 48109, USA..
Anna J. Miller, School of Engineering, Department of Biomedical Engineering, University of Michigan, Ann Arbor, MI 48109, USA.
Liwei Bao, Michigan Medicine, Department of Internal Medicine, Division of Hematology/Oncology, University of Michigan Medical School, Ann Arbor, MI 48109, USA..
Zhifen Wu, Michigan Medicine, Department of Internal Medicine, Division of Hematology/Oncology, University of Michigan Medical School, Ann Arbor, MI 48109, USA..
Dr. Peter J. Ulintz, Michigan Medicine, Department of Internal Medicine, Division of Hematology/Oncology, University of Michigan Medical School, Ann Arbor, MI 48109, USA.
Carlos A. Aguilar, School of Engineering, Department of Biomedical Engineering, University of Michigan, Ann Arbor, MI 48109, USA.
Aki Morikawa, Michigan Medicine, Department of Internal Medicine, Division of Hematology/Oncology, University of Michigan Medical School, Ann Arbor, MI 48109, USA.
Maria G. Castro, Michigan Medicine, Department of Neurosurgery, University of Michigan, Ann Arbor, MI 48109, USA. Michigan Medicine, Department of Cell and Developmental Biology, University of Michigan, Ann Arbor, MI 48109, USA.
Sofia D. Merajver, Michigan Medicine, Department of Internal Medicine, Division of Hematology/Oncology, University of Michigan Medical School, Ann Arbor, MI 48109, USA.
Christopher R. Oliver, Michigan Medicine, Department of Internal Medicine, Division of Hematology/Oncology, University of Michigan Medical School, Ann Arbor, MI 48109, USA.
Data availability
The data that support the findings of this study are openly available in Zenodo at doi:10.5281/zenodo.6376563[32].
References
- [1].Cagney DN, Martin AM, Catalano PJ, Redig AJ, Lin NU, Lee EQ, Wen PY, Dunn IF, Bi WL, Weiss SE, Haas-Kogan DA, Alexander BM, Aizer AA, Neuro Oncol 2017, 19 (11), 1511, 10.1093/neuonc/nox077; [DOI] [PMC free article] [PubMed] [Google Scholar]; Jin J, Gao Y, Zhang J, Wang L, Wang B, Cao J, Shao Z, Wang Z, BMC Cancer 2018, 18 (1), 446, 10.1186/s12885-018-4371-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ihle CL, Provera MD, Straign DM, Smith EE, Edgerton SM, Van Bokhoven A, Lucia MS, Owens P, J Immunother Cancer 2019, 7 (1), 293, 10.1186/s40425-019-0753-3; [DOI] [PMC free article] [PubMed] [Google Scholar]; Deng YI, Verron E, Rohanizadeh R, Anticancer Res 2016, 36 (11), 5639, 10.21873/anticanres.11147. [DOI] [PubMed] [Google Scholar]
- [3].Weidle UH, Niewohner J, Tiefenthaler G, Cancer Genomics Proteomics 2015, 12 (4), 167. [PubMed] [Google Scholar]
- [4].Achrol AS, Rennert RC, Anders C, Soffietti R, Ahluwalia MS, Nayak L, Peters S, Arvold ND, Harsh GR, Steeg PS, Chang SD, Nat Rev Dis Primers 2019, 5 (1), 5, 10.1038/s41572-018-0055-y. [DOI] [PubMed] [Google Scholar]
- [5].Lorger M, Felding-Habermann B, Am J Pathol 2010, 176 (6), 2958, 10.2353/ajpath.2010.090838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wasilewski D, Priego N, Fustero-Torre C, Valiente M, Front Oncol 2017, 7, 298, 10.3389/fonc.2017.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Liddelow SA, Barres BA, Immunity 2017, 46 (6), 957, 10.1016/j.immuni.2017.06.006. [DOI] [PubMed] [Google Scholar]
- [8].Priego N, Zhu L, Monteiro C, Mulders M, Wasilewski D, Bindeman W, Doglio L, Martinez L, Martinez-Saez E, Ramon YCS, Megias D, Hernandez-Encinas E, Blanco-Aparicio C, Martinez L, Zarzuela E, Munoz J, Fustero-Torre C, Pineiro-Yanez E, Hernandez-Lain A, Bertero L, Poli V, Sanchez-Martinez M, Menendez JA, Soffietti R, Bosch-Barrera J, Valiente M, Nat Med 2018, 24 (7), 1024, 10.1038/s41591-018-0044-4. [DOI] [PubMed] [Google Scholar]
- [9].Steeg PS, Nature Reviews Clinical Oncology 2021, 10.1038/s41571-021-00529-6. [DOI] [PubMed] [Google Scholar]
- [10].Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ, Chaft JE, Kris MG, Huse JT, Brogi E, Massague J, Cell 2014, 156 (5), 1002, 10.1016/j.cell.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gril B, Paranjape AN, Woditschka S, Hua E, Dolan EL, Hanson J, Wu X, Kloc W, Izycka-Swieszewska E, Duchnowska R, Pęksa R, Biernat W, Jassem J, Nayyar N, Brastianos PK, Hall OM, Peer CJ, Figg WD, Pauly GT, Robinson C, Difilippantonio S, Bialecki E, Metellus P, Schneider JP, Steeg PS, Nat Commun 2018, 9 (1), 2705, 10.1038/s41467-018-05030-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Soto MS, Sibson NR, Front Cell Neurosci 2018, 12, 414, 10.3389/fncel.2018.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wu SY, Watabe K, Front Biosci (Landmark Ed) 2017, 22, 1805, 10.2741/4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].You H, Baluszek S, Kaminska B, Front Immunol 2019, 10, 1941, 10.3389/fimmu.2019.01941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dun MD, Chalkley RJ, Faulkner S, Keene S, Avery-Kiejda KA, Scott RJ, Falkenby LG, Cairns MJ, Larsen MR, Bradshaw RA, Hondermarck H, Mol Cell Proteomics 2015, 14 (9), 2316, 10.1074/mcp.M114.046110; [DOI] [PMC free article] [PubMed] [Google Scholar]; Tanner M, Kapanen AI, Junttila T, Raheem O, Grenman S, Elo J, Elenius K, Isola J, Mol Cancer Ther 2004, 3 (12), 1585. [PubMed] [Google Scholar]
- [16].Oliver CR, Altemus MA, Westerhof TM, Cheriyan H, Cheng X, Dziubinski M, Wu Z, Yates J, Morikawa A, Heth J, Castro MG, Leung BM, Takayama S, Merajver SD, Lab Chip 2019, 19 (7), 1162, 10.1039/c8lc01387j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Allen SG, Chen YC, Madden JM, Fournier CL, Altemus MA, Hiziroglu AB, Cheng YH, Wu ZF, Bao L, Yates JA, Yoon E, Merajver SD, Sci Rep 2016, 6, 39190, 10.1038/srep39190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lanza M, Casili G, Campolo M, Paterniti I, Colarossi C, Mare M, Giuffrida R, Caffo M, Esposito E, Cuzzocrea S, Brain sciences 2021, 11 (4), 466, 10.3390/brainsci11040466; [DOI] [PMC free article] [PubMed] [Google Scholar]; Matias D, Balça-Silva J, da Graça GC, Wanjiru CM, Macharia LW, Nascimento CP, Roque NR, Coelho-Aguiar JM, Pereira CM, Dos Santos MF, Pessoa LS, Lima FRS, Schanaider A, Ferrer VP, Tania S Cristina Leite de Sampaio e, V. Moura-Neto, Frontiers in cellular neuroscience 2018, 12, 235, 10.3389/fncel.2018.00235; [DOI] [PMC free article] [PubMed] [Google Scholar]; Osipova ED, Semyachkina-Glushkovskaya OV, Morgun AV, Pisareva NV, Malinovskaya NA, Boitsova EB, Pozhilenkova EA, Belova OA, Salmin VV, Taranushenko TE, Noda M, Salmina AB, Reviews in the Neurosciences 2018, 29 (5), 567, 10.1515/revneuro-2017-0092. [DOI] [PubMed] [Google Scholar]
- [19].Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, de Stanchina E, Massague J, Cell 2016, 165 (1), 45, 10.1016/j.cell.2016.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shao YC, Wei Y, Liu JF, Xu XY, Am J Cancer Res 2017, 7 (9), 1754. [PMC free article] [PubMed] [Google Scholar]
- [21].O’Brien E, Howarth C, Sibson N, Frontiers in Cellular Neuroscience 2013, 7 (40), 10.3389/fncel.2013.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tiwary S, Morales JE, Kwiatkowski SC, Lang FF, Rao G, McCarty JH, Scientific Reports 2018, 8 (1), 8267, 10.1038/s41598-018-26636-6; [DOI] [PMC free article] [PubMed] [Google Scholar]; Wrobel JK, Toborek M, Molecular medicine (Cambridge, Mass.) 2016, 22, 32, 10.2119/molmed.2015.00207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang H, Zhou Y, Cui B, Liu Z, Shen H, Biomedicine & Pharmacotherapy 2020, 126, 110086, 10.1016/j.biopha.2020.110086. [DOI] [PubMed] [Google Scholar]
- [24].Priego N, Valiente M, Frontiers in Immunology 2019, 10 (1314), 10.3389/fimmu.2019.01314; [DOI] [PMC free article] [PubMed] [Google Scholar]; Henrik Heiland D, Ravi VM, Behringer SP, Frenking JH, Wurm J, Joseph K, Garrelfs NWC, Strähle J, Heynckes S, Grauvogel J, Franco P, Mader I, Schneider M, Potthoff A-L, Delev D, Hofmann UG, Fung C, Beck J, Sankowski R, Prinz M, Schnell O, Nature Communications 2019, 10 (1), 2541, 10.1038/s41467-019-10493-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhang L, Zhang S, Yao J, Lowery FJ, Zhang Q, Huang W-C, Li P, Li M, Wang X, Zhang C, Wang H, Ellis K, Cheerathodi M, McCarty JH, Palmieri D, Saunus J, Lakhani S, Huang S, Sahin AA, Aldape KD, Steeg PS, Yu D, Nature 2015, 527 (7576), 100, 10.1038/nature15376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu Y, Yuelling LW, Wang Y, Du F, Gordon RE, O’Brien JA, Ng JMY, Robins S, Lee EH, Liu H, Curran T, Yang Z-J, Cancer research 2017, 77 (23), 6692, 10.1158/0008-5472.CAN-17-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Oliver CR, Westerhof TM, Castro MG, Merajver SD, J Vis Exp 2020, (162), 10.3791/61654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dai W, Zhu H, Chen G, Gu H, Gu Y, Sun X, Zeng X, J Thorac Dis 2016, 8 (11), E1450, 10.21037/jtd.2016.11.11; [DOI] [PMC free article] [PubMed] [Google Scholar]; Martínez-Reyes I, Chandel NS, Nature Reviews Cancer 2021, 10.1038/s41568-021-00378-6. [DOI] [PubMed] [Google Scholar]
- [29].Lutgen V, Narasipura SD, Sharma A, Min S, Al-Harthi L, J Neuroinflammation 2016, 13 (1), 242, 10.1186/s12974-016-0691-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Klemm F, Bleckmann A, Siam L, Chuang HN, Rietkotter E, Behme D, Schulz M, Schaffrinski M, Schindler S, Trumper L, Kramer F, Beissbarth T, Stadelmann C, Binder C, Pukrop T, Carcinogenesis 2011, 32 (3), 434, 10.1093/carcin/bgq269. [DOI] [PubMed] [Google Scholar]
- [31].Chen YC, Allen SG, Ingram PN, Buckanovich R, Merajver SD, Yoon E, Sci Rep 2015, 5, 9980, 10.1038/srep09980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wsterhof TM, Benjamin AY, Merill NM, Yates JA, Altemus M, Russell L, Miller AJ, Bao L, Wu Z, Ulintz PJ, Aguilar CA, Morikawa A, Castro MG, Merajver SD, Oliver CR, (Preprint) v0.1, submitted. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are openly available in Zenodo at doi:10.5281/zenodo.6376563[32].