


Background:
HCC is the most common primary liver cancer and a leading cause of cancer-related mortality. Gut microbiota is a large collection of microbes, predominately bacteria, that harbor the gastrointestinal tract. Changes in gut microbiota that deviate from the native composition, that is, “dysbiosis,” is proposed as a probable diagnostic biomarker and a risk factor for HCC. However, whether gut microbiota dysbiosis is a cause or a consequence of HCC is unknown.
Methods:
To better understand the role of gut microbiota in HCC, mice deficient of toll-like receptor 5 (TLR5, a receptor for bacterial flagellin) as a model of spontaneous gut microbiota dysbiosis were crossed with farnesoid X receptor knockout mice (FxrKO), a genetic model for spontaneous HCC. Male FxrKO/Tlr5KO double knockout (DKO), FxrKO, Tlr5KO, and wild-type (WT) mice were aged to the 16-month HCC time point.
Results:
Compared with FxrKO mice, DKO mice had more severe hepatooncogenesis at the gross, histological, and transcript levels and this was associated with pronounced cholestatic liver injury. The bile acid dysmetabolism in FxrKO mice became more aberrant in the absence of TLR5 due in part to suppression of bile acid secretion and enhanced cholestasis. Out of the 14 enriched taxon signatures seen in the DKO gut microbiota, 50% were dominated by the Proteobacteria phylum with expansion of the gut pathobiont γ-Proteobacteria that is implicated in HCC.
Conclusions:
Collectively, introducing gut microbiota dysbiosis by TLR5 deletion exacerbated hepatocarcinogenesis in the FxrKO mouse model.
INTRODUCTION
HCC accounts for 75%–85% of primary liver cancer cases, and its incidence and mortality rates are steadily continuing to rise.1 In accordance with the multiple parallel hits theory proposed for other hepatic diseases,2 several triggers are usually required for HCC progression, and the variety of etiological risk factors can result in genomic and phenotypical heterogeneity.3 For instance, obstructive jaundice due to a narrowed or blocked bile duct is a consequence in 5%–44% of HCC cases and is referred to as the “cholestatic type of HCC.”4 Moreover, inherited diseases like progressive familial intrahepatic cholestasis often correlate with increased HCC risk.5 The role of cholestasis in promoting HCC risk is not completely understood. It is likely due in part to bile acid-mediated inflammation resulting in fibrosis and cirrhosis that could lead to hepatocarcinogenesis.6
Bile acids are biomolecules generated in the liver that function to facilitate the absorption of dietary lipids and vitamins due to their detergent-like properties. Recent studies have revealed that they have many other functions, including the modulation of metabolic diseases and cancer through interaction with cellular receptors and immune cells. The farnesoid X receptor (FXR) is the major bile acid sensor expressed in the liver and small intestine that controls the enterohepatic circulation of bile acids; about 95% of bile acids are reabsorbed from the intestine.7 FXR deficiency results in cholemia (ie, high serum bile acids) and low-grade chronic inflammation at a young age and spontaneous HCC by 16 months of age.8–10 Decreased levels of hepatic FXR are correlated with malignant clinicopathologic characteristics seen in HCC patients,11 and FXR knockout (FxrKO) mice have spontaneous HCC that mimics human pathology.12 Using the FxrKO mouse model, we wanted to investigate possible risk factors that could contribute to bile acid-dependent HCC.
The gut microbiota is a collection of trillions of microorganisms found within the gastrointestinal tract. A change in the composition of resident commensal communities relative to the community found in healthy individuals can result in gut microbiota “dysbiosis.”13 Generally, gut microbiota dysbiosis is denoted by either a loss in beneficial microbes and/or an expansion in opportunistic pathogens. Gut microbiota comparisons made between disease and nondiseased states suggest a potential panel of microbial dysbiosis changes associated with HCC.14 This includes identifying a plausible HCC signature taxon for specific genus or species alterations in the gut microbiota. The diagnostic and prognostic potential of fecal microbiota analysis proposes it as a probable noninvasive biomarker for HCC.15 The recent discovery of tumor-associated microbiota in HCC tissues16–18 has expanded the concept of microbiota to also be a possible contributor to HCC.
The connection between gut microbiota and HCC is constantly growing but it is unknown whether dysbiosis is a causation or correlation factor in HCC progression. To better understand the role of gut microbiota in HCC, gut microbiota dysbiosis was induced in FxrKO mice by means of genetic intervention (through TLR5 deletion) and the spontaneous HCC pathogenesis characterized. Introducing gut microbiota dysbiosis in HCC-prone FxrKO mice potentiated cholestatic liver injury and exacerbated liver cancer pathogenesis. An expansion in gut pathobionts like γ-Proteobacteria and further dysregulation of bile acid metabolism potentially explains the increased HCC. Collectively, this study demonstrates that gut microbiota dysbiosis in tandem with bile acid dysmetabolism worsens HCC.
METHODS
Mice
Toll-like receptor 5-deficient (Tlr5KO) mice on a C57BL/6J background were generated by Dr. Shizuo Akira, Japan.19 Farnesoid X receptor–deficient [FxrKO, C57BL/6J background9] and Tlr5KO mice were bred with C57BL/6J wild-type (WT) mice to generate their WT littermates. Homozygous Tlr5KO and FxrKO mice were crossed to generate the heterozygous F1 generation, and continual breeding resulted in double knockout (DKO) mice, which was confirmed by genotyping. All mice were bred and maintained under specific pathogen-free conditions at the University of Toledo. Mice were housed in cages (n = 5 mice/cage) containing corncob bedding (Bed-O-Cob, The Andersons Co.) and nestlets (cat# CABFM00088, Ancare) at 23 °C under a 12-hour light/dark phase cycle. Mice were fed grain-based chow ad libitum (LabDiet 5020 for breeders and LabDiet 5001 for weaned mice) and given unrestricted access to regular, nonacidified drinking water. All animal studies described were conducted as per institutional animal care and use committee (IACUC) approval obtained from the University of Toledo (protocol number 108790) and followed ARRIVE guidelines (Animal Research: Reporting In Vivo experiments).20
Flagellin administration
Flagellin from Salmonella enterica subsp. enterica serovar Typhimurium (SL3201, fljB-) was purified through sequential cation and anion exchange HPLC, as described.21 Twelve-week-old male WT and FxrKO mice were intraperitoneally injected with flagellin (10 µg/mouse), and blood collected after 2 hours was analyzed for serum cytokine analysis.
Liver cancer study
FxrKO mice were reported to develop HCC at 15–16 months of age, and this happens spontaneously without the introduction of a carcinogen or dietary intervention.8,10 Accordingly, male WT (n = 5), Tlr5KO (n = 5), FxrKO (n = 16), and DKO (n = 18) mice were aged to 16 months (fed grain-based chow) and assessed for liver cancer, including tumor number and size (diameter, mm). The minimum number of mice was >10 in the FxrKO and DKO groups, which accounts for the known marked heterogeneity associated with experimental liver cancer.
Tissue and serum collection
At 16 months of age, WT, Tlr5KO, FxrKO, and DKO mice underwent 5-hour fasting and were euthanized by CO2 asphyxiation and blood collected by cardiac puncture using a BD microtainer (Becton, Dickinson). Hemolysis-free sera were obtained after centrifugation (10,000 rpm for 10 min at 4 °C) and stored at −80 °C until further analysis of cholestatic injury and liver cancer markers. Epididymal white adipose tissue and livers were collected for organ weight. Livers were further processed for quantitative real-time (RT)-PCR and histological analysis (Supplemental Materials and Methods, http://links.lww.com/HC9/A298). In a separate experiment, 3- and 16-month-old WT and FxrKO mice were euthanized, and livers were analyzed for TLR5 expression through RT-qPCR.
Serum total bile acid quantification
Total bile acids in hemolysis-free sera were measured using a total bile acid assay kit (enzyme cycling method; Diazyme Laboratories), according to the manufacturer’s protocol.
Serum lipids and transaminases measurement
Serum cholesterol, triglycerides, alanine aminotransferase (ALT), alkaline phosphatase (ALP), and total, conjugated, and unconjugated bilirubin were measured by using biochemical kits from Randox Laboratories (Kearneysville, WV).
ELISA
Serum IL-6, KC (alias chemokine ligand 1), and α-fetoprotein (AFP) were measured using the Duoset ELISA kit (R&D Systems; Minneapolis, MN), according to the manufacturer’s protocol.
Serum immunoreactivity to LPS and flagellin
Serum immunoreactivity to lipopolysaccharide (LPS) and flagellin was examined by ELISA. High-binding ELISA plates were coated overnight with purified flagellin (100 ng/well) or LPS (2 μg/well; from E. coli 0128: B12, Sigma) in a 9.6 pH bicarbonate buffer. Sera were diluted 1:500 and added to wells coated with flagellin or LPS. After incubation at 37 °C for 1 hour, the wells were washed and then incubated with HRP-conjugated anti-mouse IgG (1:1000). After washing, the peroxidase substrate tetramethylbenzidine was added to the wells, and after 5 minutes, optical density was read at 450 nm with an ELISA plate reader. Data are reported as optical density corrected by subtracting with the readings in blank samples.
Quantitative reverse-transcription PCR
Liver samples from 3-month-old and 16-month-old WT and FxrKO mice were analyzed for TLR5 expression. Liver samples from 16-month-old WT, Tlr5KO, FxrKO, and DKO mice were analyzed for inflammation, lipogenesis, fibrosis, and HCC markers. Liver and ileum tissues were also examined for the relative expression of genes encoding for bile acid metabolism, transport, and secretion. RNA was extracted from liver and ileum tissue using TRI reagent (Sigma) as per the manufacturer’s protocol. The cDNA was synthesized from 800 ng of purified RNA using the qScript cDNA Synthesis Kit (Quanta BioSciences). Quantitative RT-PCR was performed using the Step One Plus Real-Time PCR System (Applied Biosystems) in a 10 μL reaction mixture containing cDNA, SYBR Green Master Mix (Quanta BioSciences), and mouse-specific oligonucleotides (Supplemental Table S1, http://links.lww.com/HC9/A299). The 36b4 gene was used as an internal standard. The data were expressed as a relative fold-change in comparison with the WT control.
16S rRNA gene sequencing and analysis of microbiota composition
16S PCR Library Preparation, Clean-up, Normalization, and Pooling
QIAamp PowerFecal Pro DNA kit (QIAGEN) was used to extract gDNA from the fecal pellets (~40–50 mg) of WT, Tlr5KO, FxrKO, and DKO mice. The gDNA was eluted in low TE buffer (0.1 mM EDTA, Tris-HCl buffer, 10 mM, pH 8.5) instead of the AE buffer provided in the kit. DNA concentration was determined using NanoDrop, and samples were diluted to a final concentration of 5 ng/µL in low TE buffer. The Illumina User Guide has followed: “16S Metagenomic Sequencing Library Preparation-Preparing 16S Ribosomal RNA Gene Amplicons for the Illumina MiSeq System (Part # 15044223 Rev. B).” The 16S rRNA gene targeting the V3-V4 region was amplified by PCR using the Illumina sequencing primers: 5′ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG and 5′ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGGACTACHVGGGTWTCTAAT. For index PCR, the Nextera XT index kit (FC-131-1002) from Illumina was used to attach dual indices. Each 25 µL reaction mixture contained 2.5 µL of 10X reaction buffer (Invitrogen, Thermo Fisher Scientific, Waltham, MA), 0.5 µL of 10 mM dNTPs, 0.75 (for target PCR)/1 µL (for index PCR) of 50 mM MgCl2, 0.1 µL of 5 U/µL of HotTaq polymerase (Invitrogen), 1 µL of each primer (5 µM) and 2.5 µL of 5 ng/µL DNA. All samples were reconstituted in water for a final volume of 25 µL. Thermocycling was performed in a BioRad T100TM thermal cycler (Hercules, CA), and the cycling conditions were as follows: initial denaturation at 95 °C for 5 minutes, followed by 25 cycles of 95 °C for 30 seconds, 58 °C for 30 seconds, 72 °C for 30 seconds, and a final extension at 72 °C for 5 minutes for target PCR. Index PCR was carried out in 8 cycles, with an initial denaturation at 95 °C for 3 minutes, followed by 95 °C for 30 seconds, 55 °C for 30 seconds, 72 °C for 30 seconds, and a final extension at 72 °C for 5 minutes. Each PCR amplicon sample was purified in 2 rounds using AMPure XP beads (Beckman Coulter Inc. Brea, CA). Each concentration of purified index PCR products was measured using the Qubit dsDNA HS Assay kit with Qubit 3.0 fluorometer (Life Technologies, Carlsbad, CA). The 4 nM of each amplicon was pooled equally. Pooled library was checked for quality using a 2100 Bioanalyzer (Agilent, Santa Clara, CA) before sequencing. Library denaturing and MiSeq sample loading were done according to Illumina User Guide Illumina MiSeq System. The 10 pmol/L denatured and diluted library with 10% PhiX was loaded on an Illumina MiSeq V3 flow cell kit with 2 × 300 cycles.
Quality Filtering, amplicon sequence variant Picking, and Data Analysis
Chimeric sequences were identified and filtered using the Quantitative Insights in Microbial Ecology (QIIME II) software package (version 2021.11)22 with custom scripts for analyzing all samples and for filtering groups based on comparisons. All samples were rarified at 29746 sequencing reads. The amplicon sequence variants were subsequently picked using QIIME II, and taxonomy assignment was performed using Silva23 as the reference database. LEfSe (linear discriminant analysis effect size) (https://huttenhower.sph.harvard.edu/galaxy/) was used to visualize the differential enrichment of gut microbiota between groups. Raw data are deposited into the public database GenBank (BioProject ID: PRJNA941305, BioSample accessions: SAMN33601343, SAMN33601344, SAMN33601345, and SAMN33601346).
Analysis of bacterial composition
Fecal samples were aseptically collected from WT, Tlr5KO, FxrKO, and DKO mice. Total bacterial DNA was extracted from equal amounts of feces (~50 mg) using the QIAamp DNA Stool Mini Kit (Qiagen Inc., Valencia, CA). The total bacterial load was analyzed by quantitative PCR (Step One Plus Real-Time PCR System, Applied Biosystems) in a reaction mixture of fecal DNA, SYBR green master mix, and bacterial-specific 16S primers (Supplemental Table S1, http://links.lww.com/HC9/A299). The data were expressed as copy number in comparison to a generated E. coli standard. E. coli standards were prepared in serial dilution (10,000 pg-DNA to 0.01 pg-DNA), and the CT values were obtained by RT-qPCR. E. coli standard copy numbers were calculated from the CT values with standardized formulas.24
Statistical analysis
All data are presented as mean ± SEM. Statistical significance between 2 groups was calculated using unpaired, 2-tailed t test. Data from more than 2 groups were compared using a 1-way ANOVA followed by Tukey multiple comparison test (to compare the mean of each column with the mean of every other column). *p < 0.05 was considered statistically significant. All statistical analyses were performed with the GraphPad Prism 9.0 program (GraphPad).
Data availability
The data generated in this study are available on request from the corresponding author.
RESULTS
Tumor burden is increased in FxrKO mice after genetic disruption of TLR5
Toll-like receptor 5 (TLR5) is a pattern recognition receptor that recognizes bacterial flagellin from motile bacteria. Earlier studies revealed that TLR5-deficient (Tlr5KO) mice have spontaneous gut microbiota dysbiosis.25,26 Of note, FxrKO mice have increased hepatic Tlr5 expression at both precancer and cancer time points (Supplemental Figure S1A, B, http://links.lww.com/HC9/A300) and the innate immune response (ie, IL-6 and KC) to flagellin administration was greater in FxrKO mice (Supplemental Figure S1C, D, http://links.lww.com/HC9/A300). As such, we envisioned TLR5 as a relevant target to induce gut microbiota dysbiosis in FxrKO mice and investigate the potential changes in HCC severity. After crossing Tlr5KO and FxrKO mice, DKO mice and their single knockout counterparts with WT mice as healthy controls were aged to 16 months, which is the reported time point that tumors are found in FxrKO mice.8,10
Compared with Tlr5KO mice that were reported to develop indices of metabolic syndrome,26 DKO mice had lower body and fat pad weights comparable to WT mice (Supplemental Figure S2A–C, http://links.lww.com/HC9/A301). Yet, both DKO and FxrKO mice had hyperlipidemia as revealed by increased, but not significant, levels of serum triglycerides and cholesterol (Supplemental Figure S2E-F, http://links.lww.com/HC9/A301). Compared with FxrKO mice, DKO mice had more pronounced hepatomegaly (Supplemental Figure S2D, http://links.lww.com/HC9/A301) and greater tumor number (Figure 1A, B). Tumor size trended larger in DKO mice, but this trend did not reach statistical significance (Figure 1C). There were no indications of extrahepatic metastasis in the lungs, colon, and spleen at the gross and histological level, which accord with previous findings that the FxrKO mouse model exhibits only primary liver tumors. Histologic analysis and scoring confirmed that DKO mice have the most robust liver tumors and other prominent features, including multiple lymphocyte clusters, focal inflammation, anoxic vacuolation, bile duct hyperplasia, and more pigment-laden cells that exceeded the pathology seen in FxrKO mice (Figure 1D, E). The substantially elevated CK-19 immunohistochemical staining (Figure 1F), serum AFP levels (Figure 1G), and Gpc3 and Myc mRNAs (Figure 1H, I) further indicated that DKO mice had worsened tumor burden when compared with FxrKO mice.
FIGURE 1.

HCC is aggravated in FxrKO mice on Tlr5 deletion. (A–I) Male WT (n = 5), Tlr5KO (n = 5), FxrKO (n = 16), and DKO (n = 18) mice were aged for 16 months, and liver cancer parameters were analyzed. (A) Gross liver, (B) tumor number, (C) tumor size (diameter, mm), (D) H&E staining at scale bars of i. 2 mm and ii. 50 µm, (E) liver histology score, (F) cytokeratin 19 immunohistochemistry (CK19; scale bar, 50 μm), (G) serum α-fetoprotein (AFP), and quantitative RT-PCR of hepatic (H) Gpc3 and (I) Myc. 36b4 was used as internal control for RT-qPCR. Results are expressed as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Abbreviations: DKO, double knockout; RT-PCR, real-time PCR; WT, wild type.
Inflammation, steatosis, and fibrosis are comparable between DKO and FxrKO mice
Tlr5KO mice are known to have steatosis and inflammation,26 which we believed could be driving the increased HCC in DKO mice. As such, well-established markers for inflammation, steatosis, and fibrosis were analyzed. mRNA transcript levels of inflammatory markers (ie, Mcp-1 and Ylk-40) were higher in the DKO and FxrKO groups, but the levels did not differ between them (Supplemental Figure S3Ai, ii, http://links.lww.com/HC9/A302). In a similar manner, quantitative RT-PCR showed that the expression of lipogenic gene mRNAs (ie, Fas and Hmgcr) and fibrosis gene mRNAs (ie, Timp-1 and αSmc) remained unchanged between the DKO and FxrKO mice (Supplemental Figures S3Bi, ii, http://links.lww.com/HC9/A302 and S3Ci, ii, http://links.lww.com/HC9/A302). Histochemical staining further solidified the nonsignificant differences in inflammation, lipogenesis, and fibrosis when comparing DKO and FxrKO mice (Supplemental Figure S3Di, ii, http://links.lww.com/HC9/A302). Oil red O staining revealed that both DKO and FxrKO mice had macro and microsteatosis, but DKO mice were unique to exhibit fat deposits infiltrating the tumor region (Supplemental Figure S3Di, http://links.lww.com/HC9/A302). These data suggest that common HCC risk factors could not explain the exacerbated liver cancer in DKO mice.
DKO mice have more severe cholestatic liver injury
As FxrKO mice are known to exhibit bile acid-dependent cholestasis,8 we next measured the clinical parameters of cholestatic liver injury to determine whether this could explain the greater HCC severity in DKO mice. Notably, the liver injury marker ALT was significantly increased in DKO mice when compared with the modest elevation seen in FxrKO mice (Figure 2A). A similar observation between DKO and FxrKO mice was found with the cholestatic liver injury marker ALP (Figure 2B). Correspondingly, DKO mice had the most severe jaundice (ie, hyperbilirubinemia), whereas FxrKO mice had only a mild jaundice phenotype (Figure 2C, D). The abnormal systemic bilirubin accumulation in both DKO and FxrKO mice was marked by a predominance of conjugated bilirubin with minimal unconjugated bilirubin levels (Figure 2E, F). As expected, total serum bile acids were increased in FxrKO mice when compared to WT mice, but the levels in DKO mice were further increased by 27% (Figure 2G). Collectively, it appears that the exacerbated cholestatic liver injury in DKO mice may be associated with their greater HCC pathology.
FIGURE 2.

Cholestatic liver injury is aggravated in Fxr/Tlr5 DKO mice. (A–G) Male WT (n = 5), Tlr5KO (n = 5), FxrKO (n = 16), and DKO (n = 18) mice were aged for 16 months, and cholestatic liver injury parameters in the serum were analyzed. (A) Alanine aminotransferase (ALT), (B) alkaline phosphatase (ALP), (C) total bilirubin, (D) gross serum depicting hyperbilirubinemia (ie, jaundice, yellowing of the serum), (E) conjugated bilirubin, (F) unconjugated bilirubin, and (G) total bile acids. Results are expressed as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; DKO, double knockout; WT, wild type.
Bile acid metabolism is further dysregulated in FxrKO mice following TLR5 deletion
FXR deficiency results in altered bile acid metabolism marked with an overproduction of bile acids in the liver and their diminished transport into the gallbladder. Evidence of bile acids as a potential hepatic carcinogen6 supported the next step to measure a large panel of bile acid biosynthesis, feedback regulators, and transporters at both major sites of FXR signaling, the liver and ileum. We first analyzed the expression of genes associated with the classical and alternative pathways of bile acid biosynthesis in the liver. Hepatic Cyp7a1, mRNA encoding the rate-limiting enzyme in the classical pathway, was increased in DKO mice compared with FxrKO mice (Figure 3Ai). Cyp8b1 mRNA was significantly lower in the DKO mice (Figure 3Aii), suggesting that the classical pathway was directed toward producing more chenodeoxycholate rather than the main bile acid metabolite cholate. If this postulation was true, then it could explain why the 2 major alternative pathway enzymes (encoded by Cyp27a1 and Cyp7b1) for chenodeoxycholate synthesis were severely repressed in DKO mice (Figure 3Aiii, iv). Irrespective, the significant increase in hepatic Cyp7a1 expression emphasized that the DKO mice may have a greater aberrancy in the negative feedback loops from both liver and ileal FXR signaling. Indeed, the DKO mice had the most repression of hepatocyte nuclear factor 4α (Hnf4α) and small heterodimer partner (Shp) (Figure 3Bi, ii), which are the 2 main bile acid regulators that normally function to inhibit Cyp7a1 expression.7 Similarly, 3 downstream components of ileal FXR signaling, that is, ileal FGF 15 (Fgf15), hepatic FGF receptor 4 (Fgfr4) and hepatic β-Klotho (Klb) were either nearly undetectable or further suppressed in the DKO mice compared with FxrKO mice (Figure 3Biii, iv, and 3E). Collectively, DKO mice have a greater deficiency (ie, increased synthesis but decreased secretion) in bile acid regulation.
FIGURE 3.

DKO mice have more aberrant bile acid metabolism. (A–D) Male WT (n = 5), Tlr5KO (n = 5), FxrKO (n = 16), and DKO (n = 18) mice were aged for 16 months, and liver tissues were harvested for RNA extraction, cDNA synthesis and RT-qPCR for bile acid–related transcripts. (A) De novo synthesis genes i. Cyp7a1, ii. Cyp8b1, iii. Cyp27a1 and iv. Cyp7b1. (B) Bile acid metabolism regulator genes i. Hnf4a, ii. Shp, iii. Fgfr4, and iv. Klb. (C) Basolateral transporter genes i. Slc10a1 and ii. Slco1b2. (D) Canalicular exporter genes i. Abcb11 and ii. Abcb4. (E-G) WT (n = 5), Tlr5KO (n = 5), FxrKO (n = 16), and DKO (n = 18) mice were aged for 16 months, and ileal tissues were harvested for RNA extraction, cDNA synthesis and RT-qPCR for bile acid–related transcripts. (E) Bile acid metabolism regulator gene Fgf15. (F) Apical transporter genes i. Asbt and ii. Fabp6. (G) Basolateral export genes i. Ostα and ii. Ostβ. 36b4 was used as internal control for RT-qPCR. Results are expressed as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Abbreviations: DKO, double knockout; HNF4α, hepatocyte nuclear factor 4α; Klb, β-Klotho; SHP, small heterodimer partner; WT, wild type.
When analyzing mRNAs associated with enterohepatic circulation of bile acids, DKO mice had lower expression of mRNAs encoding sodium-dependent and independent transporters for basolateral uptake of bile acids into hepatocytes (Figure 3Ci, ii). Of note, we also determined bile acid excretion to be impaired as the bile salt exporter pump (BSEP, encoded by Abcb11) and the multidrug-resistant protein 2 (MDR2, encoded by Abcb4) were further significantly decreased in the DKO mice compared with FxrKO mice (Figure 3Di-ii). Compared with the observed changes in hepatic bile acid transporters, mRNAs for apical uptake into enterocytes and basolateral export from enterocytes were similarly suppressed between DKO and FxrKO mice (Figure 3Fi, ii and 3Gi, ii). This could be an adaptation to minimize the recirculation of bile acids from the intestine to the liver. Overall, it seems that the DKO mice have a more aberrant bile acid metabolism, and this could contribute to their exacerbated cholestatic and HCC pathologies.
HCC severity in DKO mice is associated with a bloom of opportunistic pathobionts
Plausible gut microbiota alterations in our study groups were profiled by 16S rRNA gene sequencing. The gut microbiota between WT and FxrKO mice were first compared to establish any possible link with liver cancer progression. There were no changes in α- or β-diversity between WT and FxrKO mice (Supplemental Figure S4A, B, http://links.lww.com/HC9/A304), but LEfSe determined a differential abundance of Verrucomicrobia, Tenericutes, and Cyanobacteria for the FxrKO mice (Supplemental Figure S4C, http://links.lww.com/HC9/A304). Analysis of the DKO microbiota identified an increase in total bacterial load (Figure 4A) and substantial β-diversity (Figure 4B). Of note, there was no enrichment in Bacteroidetes, whereas the Firmicutes phylum was an enriched taxon with a bloom in Lactobacillus spp. in the DKO mice (Figure 4C). When conducting LEfSe analysis, we hypothesized that DKO mice might exhibit 2 major microbiota shifts, that is, expansion in gut pathobionts like Proteobacteria and secondary bile acid producers like Clostridia spp. The rationale for our hypothesis was based on our prior report of γ-Proteobacteria and Clostridium cluster XIVa blooms in Tlr5KO mice with HCC induced by a purified diet.25 As expected, DKO mice had enrichment of the Proteobacteria phylum with an augmentation of γ-Proteobacteria species (Figure 4C). This is remarkable because most liver tumor microbiota are dominated by the Proteobacteria phylum.16–18 Unexpectedly, our findings demonstrated an expansion of Clostridia spp. in the FxrKO mice but not in DKO mice (Figure 4C). As such, it appears that microbiota-mediated bile acid synthesis might be a predominant factor for tumor progression in FxrKO mice but may not the reason for tumor aggravation in DKO mice. We also explored the possibility that differences in gut permeability between FxrKO and DKO mice might contribute to exacerbated HCC. However, the similar levels of immunoreactivity to lipopolysaccharide and flagellin, which can be used as a surrogate marker of permeability, in FxrKO and DKO mice argued against this notion (Supplemental Figure S5A, B, http://links.lww.com/HC9/A303). Overall, the worsened HCC severity in the DKO mice is associated with a bloom in gut pathobionts like γ-Proteobacteria and a reduction in beneficial microbes like Bacteroidetes.
FIGURE 4.
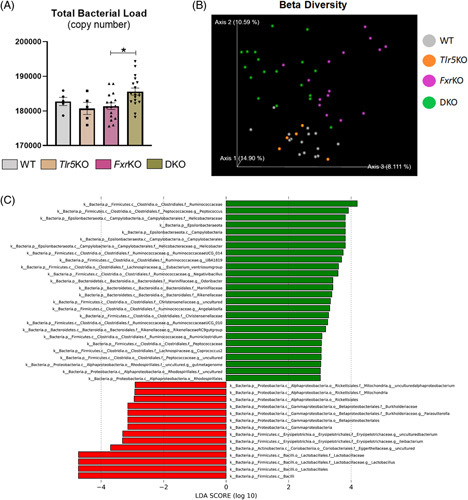
Gut microbiota for the DKO mice is enriched in HCC-associated Proteobacteria. (A–C) Male WT (n = 5 or 15), Tlr5KO (n = 5), FxrKO (n = 16), and DKO (n = 18) mice were aged for 16 months, fecal samples were snap frozen and bacterial DNA was extracted for RT-qPCR and 16S microbiota analysis. (A) Total bacterial load (copy number), (B) β-diversity through Bray-Curtis, and (C) Linear discriminant analysis Effect Size (LEfSe) comparing FxrKO and DKO groups. Results are expressed as means ± SEM. *p < 0.05. Raw data are deposited into the public database GenBank (BioProject ID: PRJNA941305, BioSample accessions: SAMN33601343, SAMN33601344, SAMN33601345, SAMN33601346). Abbreviations: DKO, double knockout; LDA, linear discriminant analysis; WT, wild type.
DISCUSSION
Perturbation in FXR signaling is a prominent feature of cholestasis and HCC progression in rodents and humans.8,10,12 In detail, mutations in the NR1H4 gene (encoding FXR) cause familial intrahepatic cholestasis, and this results in liver dysfunction denoted by impeded bile acid secretion, neonatal jaundice, and a high potential for hepatobiliary cancers.27,28 High bile acid levels from impaired FXR signaling are strongly suggestive to be a major risk factor for HCC because they can generate an inflammatory milieu, abnormal cell death, stimulate cellular proliferation, promote resistance to apoptosis, and activate oncogenic pathways.29 Although bile acids contribute to HCC pathogenesis, several other triggers are required for liver cancer progression. Herein, we investigated the tandem influence of the gut microbiota because studies have identified a potential panel of microbial dysbiosis changes associated with HCC,14 and microbiota are implicated in HCC pathogenesis.16–18 In this study, we found that gut microbiota dysbiosis worsens bile acid dysmetabolism, cholestatic liver disease, and HCC severity.
To explore this concept, we introduced gut microbiota dysbiosis in a murine HCC model (ie, FxrKO mice) by means of TLR5 deletion. On this intervention, we observed that gut microbiota dysbiosis worsened cholestatic liver injury as marked by the hyperbilirubinemia phenotype in DKO mice. Quantitative PCR demonstrated several attributes that could have contributed to the exacerbated cholestasis, which, in the future, should be supplemented with unbiased gene expression analysis like RNA sequencing. When compared with FxrKO mice, we found the mRNA expression levels of bile acid transporters (ie, BSEP and MDR2) to be significantly suppressed by an additional 45%–60% in DKO mice. Moreover, DKO mice had a 12% upregulation of the gene (ie, CYP7A1) that encodes the rate-limiting enzyme for bile acid biosynthesis. These findings relate to a recent study demonstrating that gut microbiota depletion consequently suppressed FXR-mediated inhibition of bile acid biosynthesis, resulting in fatal liver injury in a mouse model of primary sclerosing cholangitis.30
Tissue-specific FXR signaling differentially regulates bile acid metabolism and HCC risk. For instance, the intestinal FXR-FGF15-FGFR4 pathway can strongly suppress both CYP7A1 and CYP8B1, whereas liver FXR-SHP signaling predominately inhibits CYP8B1 and has a minor role in CYP7A1 repression.31 Alongside, hepatocyte-specific FxrKO mice have much lower HCC incidence, while intestinal-specific FxrKO mice still have robust liver cancer-like global knockouts.32 In our study, we found intestinal Fgf15 and hepatic Fgfr4 mRNA expressions to be further significantly suppressed by 90% and 25%, respectively, in DKO mice compared with FxrKO mice. Opposingly, FxrKO and DKO groups had a similar decrease in hepatic Shp levels. These results raise the possibility that microbiota changes in DKO mice affected intestinal FXR signaling more so than the hepatic FXR counterpart. To better determine whether gut microbiota-dependent HCC is mediated through dysregulation of hepatic and/or intestinal FXR signaling, HCC should be monitored in DKO mice with tissue-specific FXR deficiency.
The potent influence of the gut microbiota on FXR signaling is derived from certain bacterial species modifying the bile acid composition. One action involves the biotransformation of primary (ie, cholate vs. chenodeoxycholate) to secondary bile acids (ie, deoxycholate vs. lithocholate), where this gut microbiota effect can introduce a strong hydrophobic FXR antagonist (lithocholate) and lower levels of a less hydrophobic FXR agonist (chenodeoxycholate).33 Importantly, gut microbiota-derived bile acids are generally more hydrophobic and hepatotoxic, which may explain why HCC severity is more pronounced in DKO mice. Although this hypothesis requires further investigation, it is notable that our previous study demonstrated secondary bile acids to have a direct role in HCC progression for a subset of Tlr5KO mice fed a purified, inulin-containing diet.34 Moreover, we found vancomycin intervention lowered secondary bile acid production and abated tumor progression in these mice.34 It is possible that antibiotics could serve as a treatment for the DKO mice. It would also be important to experiment with fecal microbiota transplantation from DKO to germ-free mice to further establish that gut microbiota dysbiosis is pathological in HCC.
Therapies that restore the gut microbiota toward “eubiosis” or a balanced state could be of value in HCC prevention. Recent evidence supports this notion, as probiotic treatment was found to reduce HCC growth in mice inoculated with tumor cells and in hepatocyte-specific PTEN knockout mice.35,36 Similarly, treatment with antibiotics significantly attenuated tumor development in the streptozotocin and high-fat diet–induced NASH-HCC mouse model.37 These studies suggested gut microbiota dysbiosis as a contributor to HCC, and our results with the DKO mice further support this notion. Accordingly, we propose TLR5 as a potential target for HCC prevention. Of note, the TLR5 agonist entolimod (ie, CBLB502, a pharmacologically optimized flagellin derivative) was recently found to serve as an adjuvant by stimulating antitumor immunity38 and reducing side effects of anti-TNF cancer therapy39 in mouse hepatocellular and colorectal tumor models. Future studies are warranted to determine whether entolimod or recombinant flagellin can alleviate HCC by reversing gut microbiota dysbiosis to eubiosis.
Our study herein indicates that gut microbiota dysbiosis can be a causative factor in HCC progression, as seen in the DKO mice. It is impressive that out of the 14 enriched taxon signatures seen in the LEfSe plot for DKO mice, there were 3 dominant phyla that is, 50% by the Proteobacteria, 42.9% from Firmicutes, and 7.1% by Actinobacteria, whereas Bacteroidetes were not an enriched phylum. These results support the prior notion that the presence of TLR5 is essential for limiting the bloom of Proteobacteria.40 On the species level, the DKO microbiota was overwhelmed with the gut pathobiont γ-Proteobacteria that is known to be disease-promoting, including HCC.16–18 Comparatively, FxrKO microbiota was predominant in Firmicutes but had a minor proportion of Bacteroidetes and minimal Proteobacteria. FxrKO, but not DKO, mice also had an expansion of Clostridia spp., where these microbes are generally responsible for producing bile salt hydrolase and 7α-dehydroxylase in the 2-step process for hydrophobic secondary bile acid generation.41 As the DKO mice did not appear to have gut microbiota changes that would alter secondary bile acid production, it could be posited that the enhanced HCC severity is mediated by changes in primary bile acid biosynthesis and/or were instigated by the gut pathobiont γ-Proteobacteria. It would be important for future investigation to use fecal metatranscriptomics for identifying potential flagellin-positive bacteria that could also be potential opportunistic pathogens.
Collectively, this study highlights gut microbiota dysbiosis as an instigator of HCC pathogenesis primarily through bile acid dysmetabolism. Our findings complement prior studies that have found gut microbial metabolites such as LPS, lipoteichoic acid, and deoxycholate to be oncogenic in the liver, especially in the case of obesity-mediated HCC.42–44 However, our study suggests that flagellin-mediated activation of TLR5 may be tumor suppressive, similar to accounts of TLR2 signaling45,46 when compared with the many studies that demonstrate LPS-TLR4 signaling to promote HCC.42,47,48 Notably, a single-nucleotide polymorphism of the TLR5 gene in humans was recently uncovered that affected immune responses to flagellin and was associated with steatohepatitis-dependent HCC.49 Considering that regulation of the gut microbiota is predominately mediated by basolateral expression of TLR5 on intestinal epithelia,26,50 future studies should explore how tissue-specific disruption of Tlr5 (ie, liver vs. intestine) impacts the gut microbiota and HCC progression in FxrKO mice. Altogether, the current and prior reports support the concept of targeting TLR5 for stimulating immune responses, maintaining gut microbiota eubiosis, and better regulating bile acid metabolism to abate HCC.
Supplementary Material
FUNDING INFORMATION
Matam Vijay-Kumar is supported by R01 grant from the National Institutes of Health (NIH) (grant number CA219144). Rachel M. Golonka is supported by the National Cancer Institute of NIH under Award Number F31CA260842. Beng San Yeoh is supported by Postdoctoral Fellowship from American Heart Association (AHA) under Award ID 831112. Piu Saha is supported by Crohn’s and Colitis Foundation (CCF) and AHA Career Development Awards, grant numbers #854385 and #855256, respectively.
CONFLICTS OF INTEREST
The authors have no conflicts to report.
Footnotes
Abbreviations: AFP, α-fetoprotein; ALP, alkaline phosphatase; ALT, alanine aminotransferase; BSEP, bile salt exporter pump; CK-19, cytokeratin 19; CYP, cytochrome P450; DKO, double knockout; Fgfr4, FGF receptor 4; FXR, farnesoid X receptor; HNF4α, hepatocyte nuclear factor 4α; Klb, β-Klotho; KO, knockout; LEfSe, linear discriminant analysis effect size; LPS, lipopolysaccharides MDR2, multidrug-resistant protein 2; RT-PCR, real-time PCR; SHP, small heterodimer partner; TLR, toll-like receptor; WT, wild-type.
Supplemental Digital Content is available for this article. Direct URL citations are provided in the HTML and PDF versions of this article on the journal's website, www.hepcommjournal.com.
Contributor Information
Rachel M. Golonka, Email: rachel.golonka@rockets.utoledo.edu.
Beng San Yeoh, Email: bengsan.yeoh@utoledo.edu.
Piu Saha, Email: piu.saha@utoledo.edu.
Amira Gohara, Email: Amira.Gohara@utoledo.edu.
Ramakumar Tummala, Email: Ramakumar.Tummala@UToledo.Edu.
Stanislaw Stepkowski, Email: Stanislaw.Stepkowski@utoledo.edu.
Amit K. Tiwari, Email: Amit.Tiwari@UToledo.edu.
Bina Joe, Email: bina.joe@utoledo.edu.
Frank J. Gonzalez, Email: gonzalef@mail.nih.gov.
Andrew T. Gewirtz, Email: agewirtz@gsu.edu.
Matam Vijay-Kumar, Email: MatamVijay.Kumar@UToledo.Edu.
REFERENCES
- 1. Singal AG, Lampertico P, Nahon P. Epidemiology and surveillance for hepatocellular carcinoma: new trends. J Hepatol. 2020;72:250–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65:1038–48. [DOI] [PubMed] [Google Scholar]
- 3. Tang M, Zhao Y, Zhao J, Wei S, Liu M, Zheng N, et al. Liver cancer heterogeneity modeled by in situ genome editing of hepatocytes. Sci Adv. 2022;8:eabn5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lai ECH, Yee Lau W. Hepatocellular carcinoma presenting with obstructive jaundice. ANZ J Surg. 2006;76:631–66. [DOI] [PubMed] [Google Scholar]
- 5. Vinayagamoorthy V, Srivastava A, Sarma MS. Newer variants of progressive familial intrahepatic cholestasis. World J Hepatol. 2021;13:2024–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Colosimo S, Tomlinson JW. Bile acids as drivers and biomarkers of hepatocellular carcinoma. World J Hepatol. 2022;14:1730–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim I, Ahn SH, Inagaki T, Choi M, Ito S, Guo GL, et al. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res. 2007;48:2664–72. [DOI] [PubMed] [Google Scholar]
- 8. Kim I, Morimura K, Shah Y, Yang Q, Ward JM, Gonzalez FJ. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis. 2007;28:940–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–44. [DOI] [PubMed] [Google Scholar]
- 10. Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007;67:863–7. [DOI] [PubMed] [Google Scholar]
- 11. Su H, Ma C, Liu J, Li N, Gao M, Huang A, et al. Downregulation of nuclear receptor FXR is associated with multiple malignant clinicopathological characteristics in human hepatocellular carcinoma. Am J Physiol Gastrointest Liver Physiol. 2012;303:G1245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu N, Meng Z, Lou G, Zhou W, Wang X, Zhang Y, et al. Hepatocarcinogenesis in FXR-/- mice mimics human HCC progression that operates through HNF1alpha regulation of FXR expression. Mol Endocrinol. 2012;26:775–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Petersen C, Round JL. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol. 2014;16:1024–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu Q, Li F, Zhuang Y, Xu J, Wang J, Mao X, et al. Alteration in gut microbiota associated with hepatitis B and non-hepatitis virus related hepatocellular carcinoma. Gut Pathog. 2019;11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kang Y, Cai Y, Yang Y. The gut microbiome and hepatocellular carcinoma: implications for early diagnostic biomarkers and novel therapies. Liver Cancer. 2022;11:113–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Komiyama S, Yamada T, Takemura N, Kokudo N, Hase K, Kawamura YI. Profiling of tumour-associated microbiota in human hepatocellular carcinoma. Sci Rep. 2021;11:10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science. 2020;368:973–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang JH, Wang J, Chai XQ, Li ZC, Jiang YH, Li J, et al. The intratumoral bacterial metataxonomic signature of hepatocellular carcinoma. Microbiol Spectr. 2022;10:e0098322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, et al. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest. 2007;117:3909–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gewirtz AT, Simon PO, Jr, Schmitt CK, Taylor LJ, Hagedorn CH, O’Brien AD, et al. Salmonella typhimurium translocates flagellin across intestinal epithelia, inducing a proinflammatory response. J Clin Invest. 2001;107:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nadkarni MA, Martin FE, Jacques NA, Hunter N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology (Reading). 2002;148(Pt 1):257–66. [DOI] [PubMed] [Google Scholar]
- 25. Singh V, Yeoh BS, Chassaing B, Xiao X, Saha P, Aguilera Olvera R, et al. Dysregulated microbial fermentation of soluble fiber induces cholestatic liver cancer. Cell. 2018;175:679–94 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gomez-Ospina N, Potter CJ, Xiao R, Manickam K, Kim MS, Kim KH, et al. Mutations in the nuclear bile acid receptor FXR cause progressive familial intrahepatic cholestasis. Nat Commun. 2016;7:10713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vitale G, Mattiaccio A, Conti A, Turco L, Seri M, Piscaglia F, et al. Genetics in familial intrahepatic cholestasis: clinical patterns and development of liver and biliary cancers: a review of the literature. Cancers (Basel). 2022;14. doi: 10.3390/cancers14143421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li T, Apte U. Bile acid metabolism and signaling in cholestasis, inflammation, and cancer. Adv Pharmacol. 2015;74:263–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schneider KM, Candels LS, Hov JR, Myllys M, Hassan R, Schneider CV, et al. Gut microbiota depletion exacerbates cholestatic liver injury via loss of FXR signalling. Nat Metab. 2021;3:1228–41. [DOI] [PubMed] [Google Scholar]
- 31. Kong B, Wang L, Chiang JYL, Zhang Y, Klaassen CD, Guo GL. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology. 2012;56:1034–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takahashi S, Tanaka N, Fukami T, Xie C, Yagai T, Kim D, et al. Role of farnesoid X eceptor and bile acids in hepatic tumor development. Hepatol Commun. 2018;2:1567–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yu J, Lo JL, Huang L, Zhao A, Metzger E, Adams A, et al. Lithocholic acid decreases expression of bile salt export pump through farnesoid X receptor antagonist activity. J Biol Chem. 2002;277:31441–7. [DOI] [PubMed] [Google Scholar]
- 34. Singh V, Yeoh BS, Abokor AA, Golonka RM, Tian Y, Patterson AD, et al. Vancomycin prevents fermentable fiber-induced liver cancer in mice with dysbiotic gut microbiota. Gut Microbes. 2020;11:1077–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arai N, Miura K, Aizawa K, Sekiya M, Nagayama M, Sakamoto H, et al. Probiotics suppress nonalcoholic steatohepatitis and carcinogenesis progression in hepatocyte-specific PTEN knockout mice. Sci Rep. 2022;12:16206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li J, Sung CYJ, Lee N, Ni Y, Pihlajamäki J, Panagiotou G, et al. Probiotics modulated gut microbiota suppresses hepatocellular carcinoma growth in mice. Proc Natl Acad Sci USA. 2016;113:E1306–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yamada S, Takashina Y, Watanabe M, Nagamine R, Saito Y, Kamada N, et al. Bile acid metabolism regulated by the gut microbiota promotes non-alcoholic steatohepatitis-associated hepatocellular carcinoma in mice. Oncotarget. 2018;9:9925–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brackett CM, Kojouharov B, Veith J, Greene KF, Burdelya LG, Gollnick SO, et al. Toll-like receptor-5 agonist, entolimod, suppresses metastasis and induces immunity by stimulating an NK-dendritic-CD8 + T-cell axis. Proc Natl Acad Sci USA. 2016;113:E874–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Haderski GJ, Kandar BM, Brackett CM, Toshkov IM, Johnson CP, Paszkiewicz GM, et al. TLR5 agonist entolimod reduces the adverse toxicity of TNF while preserving its antitumor effects. PLoS One. 2020;15:e0227940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carvalho FA, Koren O, Goodrich JK, Johansson MEV, Nalbantoglu I, Aitken JD, et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe. 2012;12:139–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol. 2014;30:332–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21:504–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Loo TM, Kamachi F, Watanabe Y, Yoshimoto S, Kanda H, Arai Y, et al. Gut microbiota promotes obesity-associated liver cancer through PGE(2)-mediated suppression of antitumor immunity. Cancer Discov. 2017;7:522–38. [DOI] [PubMed] [Google Scholar]
- 44. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. [DOI] [PubMed] [Google Scholar]
- 45. Shiau DJ, Kuo WT, Davuluri GVN, Shieh CC, Tsai PJ, Chen CC, et al. Hepatocellular carcinoma-derived high mobility group box 1 triggers M2 macrophage polarization via a TLR2/NOX2/autophagy axis. Sci Rep. 2020;10:13582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li S, Li F, Xu L, Liu X, Zhu X, Gao W, et al. TLR2 agonist promotes myeloid-derived suppressor cell polarization via Runx1 in hepatocellular carcinoma. Int Immunopharmacol. 2022;111:109168. [DOI] [PubMed] [Google Scholar]
- 47. Liu Y, Zhang X, Chen S, Wang J, Yu S, Li Y, et al. Gut-derived lipopolysaccharide promotes alcoholic hepatosteatosis and subsequent hepatocellular carcinoma by stimulating neutrophil extracellular traps through toll-like receptor 4. Clin Mol Hepatol. 2022;28:522–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou S, Du R, Wang Z, Shen W, Gao R, Jiang S, et al. TLR4 increases the stemness and is highly expressed in relapsed human hepatocellular carcinoma. Cancer Med. 2019;8:2325–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nischalke HD, Fischer J, Klüners A, Matz‐Soja M, Krämer B, Langhans B, et al. A genetic variant in toll-like receptor 5 is linked to chemokine levels and hepatocellular carcinoma in steatohepatitis. Liver Int. 2021;41:2139–48. [DOI] [PubMed] [Google Scholar]
- 50. Chassaing B, Ley RE, Gewirtz AT. Intestinal epithelial cell toll-like receptor 5 regulates the intestinal microbiota to prevent low-grade inflammation and metabolic syndrome in mice. Gastroenterology. 2014;147:1363–77. e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available on request from the corresponding author.