

Abstract
Activation of Natural Killer (NK) cells depends on a balance between signals received from activation and inhibitory ligands expressed on the surface of target cells. Tumorigenic human adenovirus 12 (Ad12) transformed cells express low levels of the NK cell inhibitory ligand MHC I, but do not exhibit increased sensitivity to NK cell lysis compared to their non-tumorigenic counterparts. Analysis of the expression of activation ligands that bind to the NKG2D receptor revealed that RAE1β and H60 were reduced on the surface of Ad12 mouse cells as well as at the level of transcription. In accord with these results, RAE1 localization to the synapse and sensitivity to NK cell cytotoxicity were also diminished. The reduced transcription of the rat NKG2D ligands, RAEt1L and RRTL, in tumorigenic rat cells compared to non-tumorigenic counterparts implies that both mouse and rat cell lines share a common mechanism of NKG2D ligand activation subverted by Ad12.
Keywords: NKG2D ligand, adenovirus 12, tumorigenesis, immune evasion
1. INTRODUCTION
While all human adenovirus (Ad) serotypes are capable of transforming primary rodent cells in vitro, only a subset of these serotypes is able to form tumors in immune-competent rodents [1]. For example, injection of cells transformation by Ad12 into immune-competent rodents typically results in tumor formation where as Ad5 transformed cells do not. E1A and E1B, the first viral products synthesized after virus entry, are entirely responsible for the transformation phenotype; however, only E1A encoded by tumorigenic adenoviruses can promote tumorigenesis. Ad12 E1A renders the host cell less immunogenic by manipulating pathways involved in regulating both the innate and adaptive immune responses [2,3]. This supported by the fact that non-tumorigenic adenovirus serotypes are capable of forming tumors in immuno-suppressed rodents [4,5]. Investigation into the mechanisms by which tumorigenic Ad12 cells inhibit both the adaptive and innate arms of the immune system remains a key area of scientific interest.
Tumorigenic Ad12 cells avoid recognition by cytotoxic T lymphocytes (CTL), effector cells in the adaptive immune response, by repressing the expression of the Major Histocompatability Complex class I (MHC I) antigens on the cell surface of infected cells (reviewed in [6]). Without MHC I, Ad12 viral antigens cannot be presented on the cell surface for detection by CTLs. Ad12 E1A uses two mechanisms to repress the transcription of all MHC I genes. Infected cells alert the immune system of viral invasion by using transcription factors such as NF-κB, which increases the expression of MHC I. To suppress NF-κB-mediated activation, E1A inhibits pKAc phosphorylation of the NF-κB subunits, p50 and p65, which are essential for NF-κB DNA binding and transactivation, respectively [6–9]. E1A also orchestrates the recruitment of COUP-TF II, a nuclear hormone receptor, to a hormone response element in the MHC I enhancer. COUP-TF II represses transcription through the recruitment of histone deacetylases, which condense the chromatin structure of the MHC I gene locus. Combined, these two strategies provide a fail-safe mechanism by which Ad12 tumorigenic cells avoid detection by CTLs.
In addition to facilitating CTL-mediated recognition and defense, MHC I is also critical for the inhibition of Natural Killer (NK) cell activation, a key mediator of the innate immune response. The ligation of MHC I on target cells and inhibitory receptors on NK cells prevents the release of cytolytic enzymes, the production of cytokines and the inappropriate lysis of target cells [10]. By contrast, interaction between activation ligands on the surface of target cells and NK activation receptors triggers the directed release of these cytolytic enzymes and cytokines leading to cytotoxicity. The NK cell surface receptor, NKG2D, triggers the activation of NK cells upon interaction with several different ligands [11,12]. These ligands share structural homology with MHC I and are therefore termed MHC I-related molecules; however, unlike MHC I, they do not associate with t β2-microglobulin [12,13]. The murine NKG2D ligands include the retinoic acid early inducible gene 1 (RAE1) family, which has five isoforms: α, β, γ, δ, and ε; histocompatibility antigen 60 (H60), which has three isoforms: a, b, and c; and murine UL16-binding protein-like transcript 1 (MULT1) [14–18]. Two members of the RAE-1 family have been identified in rat, RAEt1L and RRTL [19,20]. When bound to its ligands, NKG2D initiates the activation of NK cells and the destruction of target cells. The expression of most NKG2D ligands is induced in response to a cellular stress such as transformation or viral infection [12]. The predominance of NKG2D ligands on the surface of target cells initiates the activation of NK cells and their eventual destruction. Access to NK cell-mediated lysis depends on this balance in favor of the engagement of activation receptors over inhibitory receptors.
Despite retaining MHC I surface expression, cells transformed with non-tumorigenic Ad2 or Ad5 are significantly more susceptible to lysis by NK cells than tumorigenic Ad12 cells, which have diminished MHC I expression [21,22]. Others have shown that the expression of Ad5 E1A can increase the expression of RAE-1 activation ligands in some cells correlating with their increased sensitivity to NK cell lysis [22,23]. Thus, in the case of non-tumorigenic Ad5 cells, activation of NK cells is the result of an excess in activation signaling that is essential for the elimination of non-tumorigenic Ad5 cells [24].
Unlike Ad2 or Ad5 cells, tumorigenic Ad12 cells are resistant to NK lysis, even though the expression of MHC I is greatly reduced [21,22,25,26]. Our findings reveal that the expression of NKG2D ligands is also reduced on tumorigenic Ad12 cells. This suggests that to counter an increase in susceptibility to NK cell lysis due to diminished MHC I levels, Ad12 co-evolved a mechanism to reduce NKG2D activation ligand expression as a means of subverting the innate immune response. This indicates that Ad12 evades both the adaptive (CTL) and the innate (NK) arms of the immune system by mediating a reduction of both MHC I inhibitory and NKG2D activation ligands.
2. MATERIALS AND METHODS
2.1 Cell Lines
Ad5 transformed BrAd5 cells and Ad12 transformed 12A1 cells were made by infection of brain cell suspensions with either Ad5 or Ad12 virus, respectively, and single foci were subsequently isolated as described previously [27]. DP5-2 (Ad5) and 12-1 (Ad12) cell lines were made by transformation of baby rat kidney (BRK) cells from Hooded Lister rats as previously described [28,29].
2.2 Flow Cytometry
Adenovirus transformed cell lines were detached from plates using 5mM EDTA in PBS containing 1% FBS (FACS Buffer) or Versene (8g NaCl; 0.2 g KCl; 1.15g Na2HPO4; 0.2mM EDTA; 0.1g Phenol Red; pH 7.34). Suspension cells were washed in FACS buffer. All cells were incubated with Anti-H60 (1:10) or Anti-Pan RAE1 (1:5) conjugated to PE or FITC, respectively (R&D Systems). Cells were incubated for 30 minutes to an hour and then washed 3 times with FACS buffer and fixed with 4% paraformaldehyde. Cells were analyzed using a FACSCalibur (Becton Dickinson, San Jose, Ca). All statistical information was obtained using PRISM software (Graphpad). Mean Fluorescence Intensity (MFI) obtained by flow cytometry for each cell line was compared by two-tailed paired t-tests.
2.3 Microscopy of the Immunological Synapse
Fixed confocal microscopy was performed on non-tumorigenic Ad5 transformed BrAd5 cells, tumorigenic Ad12 transformed 12A1 cells, and Mel1106 RAE1 negative control cells as previously described [30]. In brief, splenocytes harvested from BALB/c SCID mice were activated using 4000units/mL of recombinant human IL-2 (Novartis; aldesleukin/proleukin) for 6 days. The resulting cells were >90% DX5+ NK cells as measured by flow cytometry. Activated cells were harvested and incubated at a 2:1 ratio with targets for 15 minutes to allow conjugates to form. Conjugates were further adhered to slides for 20 minutes at 37°C. Slides were washed 3x with PBS, fixed and permeabilized with Cytofix/cytoperm, (BD Biosciences) then incubated with rabbit anti-Granzyme B antibody followed by goat anti-rabbit Alexa Fluor 488 (Invitrogen). Slides were washed again and incubated with rat anti-RAE1 then goat anti-rat Alexa Flour 568 (Invitrogen). Coverslips were mounted with Vectashield mounting media (Vector Labs) and slides were dried overnight. All samples were viewed on the Olympus IX-81 DSU spinning disc confocal microscope. Images were acquired and processed using Volocity software (Improvision). All samples were imaged and subsequently thresholded using the same settings to allow for quantitative comparison. Quantification of the accumulation of RAE1 at the interface between the lytic cells and target cells was determined using the following algorithm [((MFI at the contact site)/(MFI of the whole cell))-1]X100. The number of synapses analyzed was 19. Statistical difference was analyzed using an unpaired two-tailed student’s t-test. P-value 0.028.
2.4 Chromium (51Cr) Release Assay
The 51Chromium release cytotoxicity assay was carried out as previously described [31] Briefly, adherent NK cells prepared from Balb/c splenocytes activated by growth in IL-2 for six days (ALAKs) were used as effector cells (E) against 100μCi Na2 51CrO4 labeled Ad5 (BrAd5) and Ad12 (12A1) target cells (T) at various effector:target ratios. Preparation of ALAKs results in approximately 85% TCRβ+/DX5+ NK cells with the remaining cells consisting of non-specific, non-cytolytic T cells and NKT cells. Effector and target cells were incubated for 4 h at 37°C, centrifuged and then 100μL of supernatant was collected and analyzed for 51Cr release by gamma counting. Percent cytotoxicity was determined by measuring the amount 51Cr release after 4 hours of incubation using the following equation: Percent lysis = (E-S)/(M-S) X 100 in which E is the release from experimental samples, S is the spontaneous release, and M is the maximum release upon lysis with 10% SDS. Data are presented as the mean of triplicate samples ±SD and are pooled from two experiments.
2.5 Real Time qPCR
Quantitative (real time) PCR Analysis was carried out using the ABI®-PRISM 7000 Sequence Detection System (Applied Biosystems) or the 7300 Real Time PCR System (Applied Biosystems) and Taqman PCR assays were carried out according to the manufacturer’s instructions. Eukaryotic 18S rRNA (Hs99999901_sl), beta actin (Mm01205647_gl), Histocompatibility 60a (Mm01311160_ml), UL16 binding protein 1 (Mm01180648_ml), Retinoic Acid Early transcript 1 (RAEt 1) delta (Mm02394669_sH), RAEt 1 epsilon (Mm02530623_sl), Rat 18S (Rn03928990_gl), Rat Beta actin (Rn00667869_m1), Rat RAEt1L (Rn02395357_mH), and Rat RRTL (Rn04222874_mH) were purchased from the Applied Biosystems inventoried Taqman assays. Primer-probes sequences for RAEt 1 alpha, RAEt 1 beta, RAEt 1 gamma were previously published and purchased as custom Taqman assays from Applied Biosystems [32]. All probes were conjugated with FAM at the 5’ end and the non-fluorescent quencher MGB at the 3’ end. Total RNA was obtained using the RNeasy Minikit for total RNA extraction (Qiagen) and cDNA was synthesized using First-Strand cDNA Synthesis Kit and random hexamers (Invitrogen). The Cycling conditions for Real-time PCR were as follows: 50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 15 sec followed by 60°C for 1 min. Real-time data was analyzed using the Mann-Whitney test to compare both the expression of individual ligands between BrAd5 and 12A1 cells.
3. RESULTS
3.1 Ad12 transformed cells are less sensitive to NK cytotoxicity
Previous studies indicate that, despite diminished MHC I expression, tumorigenic Ad12 cells are significantly less susceptible to NK lysis than non-tumorigenic serotypes [21–23,25,26]. We compared the susceptibility to cell-mediated lysis of two previously described cell lines, the non-tumorigenic Ad5 cell line-BrAd5 and the tumorigenic Ad12 cell line-12A1 [27,33–39]. Adherent IL-2 activated NK cells (ALAKs) were prepared as previously described [31]. Consistent with previous reports, we show that 51Chromium-labeled tumorigenic Ad12 cells (12A1) are more resistant to lysis by ALAKs than their non-tumorigenic Ad5 (BrAd5) counterparts, especially at higher effector to target ratios (Figure 1).
Figure 1.

Tumorigenic 12A1 cells are more resistant to lysis by NK cells than non-tumorigenic BrAd5 cells. NK (ALAK) cells were used as effector cells (E) against 51Cr-labeled BrAd5 and 12A1 target cells (T) at the indicated effector:target ratios. ALAKs represent roughly 85% of cells after culture with IL-2 for six days. Cytotoxicity was determined by measurement of 51Cr release. BrAd5 cells were tested against six independent sources of ALAK cells (mice) in three experiments and 12A1 cells were tested against four ALAK sources (mice) in two experiments. Data are presented as the mean ±SD.
3.2 NKG2D ligands are diminished on the surface of Ad12 cells
One mechanism by which tumorigenic Ad12 cells could escape NK-mediated innate immunity despite diminished MHC I could be a concomitant reduction in the expression of NKG2D activating ligands on the cell surface. To address this possibility, the presence of NKG2D ligands on the surface of BrAd5 and 12A1 cells lines was assessed by flow cytometry. Using a pan-RAE1 antibody, which recognizes all of the RAE1 family isoforms, we found that the total amount of RAE1 on the surface of tumorigenic 12A1 cells was diminished compared to non-tumorigenic Ad5 cells (Figure 2A). In addition, the presence of H60 on the surface of tumorigenic Ad12 (12A1) cells is also reduced compared to non-tumorigenic Ad5 (BrAd5) cells (Figure 2B). The Mean Fluorescence Intensity, or MFI, for the total surface expression of RAE1 (Figure 2C) and H60 (Figure 2D) from four experiments was quantified. The results show that the MFI was 5.6 fold higher for RAE1 and 7.4 fold higher for H60 on non-tumorigenic Ad5 (BrAd5) cells than on tumorigenic Ad12 (12A1) cells. We were unable to detect Mult1 on the surface of either BrAd5 cells or 12A1 cells by flow cytometry (Data not shown).
Figure 2.

Surface expression of NKG2D ligands is reduced on tumorigenic Ad12 cells compared to non-tumorigenic Ad5 cells as shown by flow cytometry. Representative histograms measuring the presence of RAE1 (A) and H60 (B) on the surface of Ad5 (BrAd5) and Ad12 (12A1) stable cell lines from one experiment are shown (solid lines) as well as IgG control samples (dashed lines). The MFI for either RAE1 or H60 is indicated in the appropriate upper right corner. The average MFI ± SD from at least 4 experiments is calculated for RAE1 (C) and H60 (D). Statistical significance was determined using the Wilcox matched pairs test. p< 0.05 for RAE1 and p<.01 for H60. MFI: Mean fluorescence Intensity.
3.3 Tumorigenic Ad12 cells display decreased localization of RAE1 at the immune synapse
We next investigated whether NKG2D ligands differ at the synapse with lytic cells. Adenovirus transformed cells were stained for RAE1 and lytic cells were identified by intracellular staining for lytic effector granzyme B (figure 3A). BrAd5 and 12A1 cells were incubated with IL-2 activated lytic cells for 15 minutes to allow for conjugates to form. Significantly more RAE1 accumulated at the synapse formed between BrAd5 and lytic cells than between 12A1 and lytic cells (Figure 3B). The increased presence of RAE1 localized to the synapse between BrAd5 cells and lytic cells (Figure 3) coincides with the higher overall concentration of RAE-1 on the surface of BrAd5 cells (Figure 2). This provides a possible explanation for the increased susceptibility to lysis of BrAd5 cells demonstrated in Figure 1.
Figure 3.

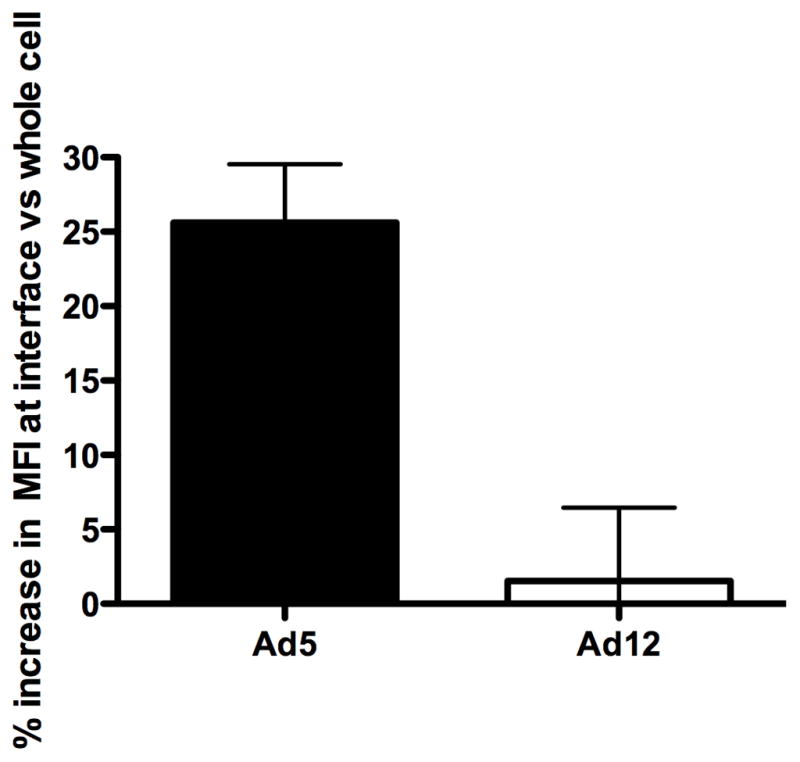
RAE1 accumulates at the interface between BrAd5 targets and NK effectors. (A) BrAd5 (top) or 12A1 (bottom) panels were incubated with IL-2 activated NK cells then fixed and stained with pan-RAE-1 antibody as described in Materials and Methods. Representative images from more than 20 images from two independent experiments are shown. (B) Quantification of fluorescence from images in (A) shows increased accumulation of RAE-1 in BrAd5 targets. Fluorescence intensity of RAE-1 staining was measured using Volocity software (Improvision). MFI of staining was measured at the interface and represented as a percentage of fluorescence of staining throughout the whole cell. p < 0.03.
3.4 Transcription of NKG2D ligands is lower in murine Ad12 cells
The lack of RAE1 and H60 on the surface of tumorigenic Ad12 cells could be due to one of several possibilities including increased cellular retention, increased protein degradation, miRNA silencing and repression of transcription. Since adenovirus is known to repress the transcription of MHC I, we investigated whether NKG2D ligands on the surface of tumorigenic Ad12 cells might be regulated in a similar fashion. We measured the mRNA expression levels of NKG2D ligands using quantitative real time PCR. As shown in Figure 4, expression of the negative control gene, β-actin (A), was nearly equivalent between the two cell lines while the positive control, MHC I gene H-2Dd (B), known to be repressed in Ad12 transformed cell lines, was significantly reduced in 12A1 cells compared to BrAd5 cells (Figure 4). Figure 4C shows that the mRNA expression of H60 is reduced on tumorigenic 12A1 cells compared to non-tumorigenic BrAd5 cells. The repression of MULT1 expression in tumorigenic Ad12 cells compared to BrAd5 cells (Figure 4D) was consistent with our results seen with other NKG2D ligands but was not statistically significant. The use of primers that distinguish between the different isoforms of RAE1 allowed us to identify which isoforms our pan-RAE1 antibody detected in the flow cytometry and immunoflourescence experiments mentioned above. Of all the RAE1 isoforms, only RAE1-β (F) is expressed by non-tumorigenic BrAd5 cells, however, the expression of RAE1-β was minimal in 12A1 cells. Negligible mRNA expression was detected for other members of the RAE1 family (RAE1-α (E), γ (G), δ (H), and ε (I)) in both cell lines. Taken together, the reduction in NKG2D ligand expression on the surface of tumorigenic Ad12 cells appears to be the consequence of decreased NKG2D mRNA levels resulting in escape from NK cell lysis.
Figure 4.

mRNA expression of NKG2D ligands is diminished in tumorigenic 12A1 cells compared to non-tumorigenic BrAd5 cells. TaqMan primers specific for β-actin (A), mouse MHC I gene H-2Dd (B), H60 (C), Mult1 (D), RAE1 Alpha (E), RAE1 Beta (F), RAE1 Gamma (G), RAE1 Delta (H), and RAE1 Epsilon (I) were used in RT-qPCR to analyze the relative quantities of RNA from BrAd5 cells and 12A1 cells. The data represents an average of at least 3 experiments. The p<0.05 for the murine MHC I gene, H-2Dd, RAE1-β and H60 using the Mann-Whitney t-test for statistical significance.
3.5 Transcription of NKG2D ligands is lower in rat Ad12 cells
To determine whether adenovirus is able to reduce the expression of NKG2D ligands in species other than mouse and in cells derived from a different tissue, we measured the expression of NKG2D ligands in Ad5 (DP5-2) and Ad12 (12-1) transformed BKR cells by realtime qPCR. Two members of the RAE-1 family have been identified in rats, RAEt1L and RRTL [19,20]. As shown in Figure 6, transformed rat DP5-2 cells showed significantly higher expression of both rat NKG2D ligands RAEt1L (B) and RRTL (C) compared to Ad12 transformed 12-1 cells. We also noted that RAEt1L appears to be expressed at a much higher level than RRTL as indicated by the scale on the Y-axis. No statistical difference was seen in the expression of the β-actin control gene (A). These results are consistent with our findings in transformed murine cells. This suggests adenovirus is capable of repressing the expression NKG2D ligands in multiple species, presumably by a common mechanism.
Figure 6.

Repression of NKG2D activation ligands protects tumorigenic Ad12 cells from immune surveillance. Non-tumorigenic Ad5 cells up-regulate NKG2D activation ligands resulting in their subsequent lysis by both CTLs and NK cells. Tumorigenic Ad12 cells lack NKG2D ligands on their surface allowing these cells to evade NK cells. NK cells: Natural Killer, Ag: Antigen.
4. DISCUSSION
Although all adenoviruses immortalize primary cells in tissue culture, adenovirus transformed cells capable of forming tumors in immune-competent hosts must evade the full arsenal of the immune system, including both the innate and adaptive immune responses. In this study, we observed a marked reduction in the presence of murine NKG2D activating ligands on the surface of tumorigenic adenovirus transformed cells using flow cytometry. Specifically, the murine NKG2D ligands, RAE1-β and H60, were significantly diminished on the surface of tumorigenic Ad12 cells (12A1) compared to non-tumorigenic Ad5 cells (BrAd5). RAE1-β and H60 mRNA transcripts were also reduced as measured by quantitative real-time PCR. These findings were further supported by our observation that the expression of rat NKG2D ligands, RAEt1L and RRTL, was diminished in Ad12 transformed BRK cells compared to their Ad5 transformed counterparts. Consistent with the reduction of NKG2D ligand expression on tumorigenic Ad12 cells, these cells were also less susceptible to NK lysis in cytotoxicity assays. The reduction of NKG2D activating ligands on tumorigenic Ad12 cells provides the first explanation for how tumorigenic Ad12 cells resist NK lysis despite their diminished MHC I expression [21,22,25,40].
The activation of an NK cell depends on a delicate balance between activation and inhibitory signals received from a target cell. Those signals are conveyed by activation and inhibitory ligands on the target cell surface that bind to NK cell receptors. MHC I antigens typically repress lysis by functioning as ligands for inhibitory receptors on the surface of NK Cells. NKG2D is one of several activation receptors on the surface of NK cells and has been shown to play an important role in the detection of adenovirus [24]. NKG2D recognizes several ligands that are structurally similar to MHC I. In the mouse, these activating ligands include the RAE1 family, the H60 family, and MULT1 [14–18]. The balance between the expression of NKG2D activating ligands and MHC I inhibitory ligands ultimately determines the fate of adenovirus-infected target cells. Uninfected cells do not elicit an immune response because they maintain normal expression of MHC I molecules and do not express NKG2D ligands. Infection by Ad5 causes an increase in both MHC I expression and NKG2D ligands alerting both CTLs and NK cells that a cell has been infected and these cells become targets for lysis [23,41]. Presumably, the balance between activating and inhibiting ligands on non-tumorigenic Ad5 cells favors activation leading to their increased susceptibility to NK cell lysis. By contrast, tumorigenic Ad12 cells avoid CTL lysis by repressing the transcription of MHC I. This loss of MHC I transcription should render tumorigenic Ad12 cells susceptible to NK cell lysis. however, tumorigenic Ad12 cells are resistant to lysis by NK cells. Our study reveals for the first time, a mechanism whereby protection of tumorigenic Ad12 cells from NK cell lysis can be explained by diminished surface expression of NKG2D ligands. Our model, as presented in Figure 6, illustrates how tumorigenic Ad12 cells evade NK cells by repressing the expression of NKG2D activating ligands. Although repression of MHC I and NKG2D activation ligands is not complete, we suspect that failure to expression these ligands above a critical threshold serves to protect Ad12 transformed cells from lysis by NK cells. The fact that the expression of NKG2D ligands was reduced in both mouse and rat transformed cells suggests that these species share a common mechanism of NKG2D ligand regulation manipulated by tumorigenic Ad12.
Manipulation of NK cell activation is not uncommon among viruses. Strategies used by other viruses include the expression of MHC I homologues that bind to NK inhibitory receptors (MCVM and HCMV), the prevention of NKG2D ligand transport to the cell surface (MCMV), the internalization of NKG2D ligands located on the cell surface (MCVM and KSHV), the shedding of NKG2D ligands form the surface of the cell (HIV), and the expression of viral miRNAs that target NKG2D ligand mRNAs for degradation (HCMV, KSHV, and EBV) [42–49]. Interestingly, during productive infection, adenovirus expresses two VA RNA genes, which are processed into functional miRNAs that target both cellular and viral genes for post transcriptional regulation [50]. While these miRNAs play a role in immune evasion during productive infection, their expression has not been detected in transformed cells making it unlikely that they are responsible for the differences seen in the expression of NKG2D ligands in our transformed cell lines [51]. Infection by other viruses has certainly been shown to increase the expression of NKG2D ligands; however, Ad12 appears to be unique in that it either fails to induce the expression of NKG2D ligands or actively represses their expression at the level of transcription.
As shown in this study, non-tumorigenic Ad5 cells express RAE1-β and H60 while the expression of these ligands is significantly reduced in tumorigenic Ad12 cells. This finding has an interesting parallel to MHC I down-regulation, which is mediated by Ad12 E1A. In tumorigenic Ad12 cells, E1A promotes the repression of MHC I expression by preventing the transcriptional activator NF-κB from binding the R1 element in the MHC I promoter and by recruiting the transcriptional repressor COUP-TF II to a nuclear hormone receptor recognition element, R2 [7,27,33,35–37,52–58]. While little is known about how NKG2D ligand gene expression is controlled, the activation of various NKG2D ligands by various stimuli suggests that their promoter regions can be differentially regulated, even within the same family such as the RAE1 family of ligands [45,59]. Inspection of all of the NKG2D ligand promoters, using the transcription factor binding site analysis program ConSite, suggests that there are common elements that could simultaneously govern all of these NKG2D promoters. We found putative NF-κB sites in all NKG2D ligand promoters as well as putative hormone response elements in the promoters of each NKG2D ligand except RAE1α. It remains to be determined whether COUP-TF and NF-κB regulate NKG2D ligands in the same way that they regulate MHC I. It is interesting to speculate that the E1A gene of tumorigenic Ad12 may mediate the binding of COUP-TF II and NK-κB to the promoters of NKG2D ligands in a similar manner to the mechanism responsible for the down-regulation of MHC I.
In summary, this study reveals a mechanism for the evasion of NK lysis by tumorigenic Ad12 cells. Unlike non-tumorigenic Ad5 cells, tumorigenic Ad12 cells express diminished levels of NKG2D activating ligands. The reduction of NKG2D ligand expression is significant because it provides the first explanation for how tumorigenic Ad12 cells evade NK lysis despite a reduction in MHC I expression. While loss of MHC I allows tumorigenic Ad12 cells to evade detection by CTLs, it should increase the susceptibility of these cells to NK lysis. Tumorigenic Ad12 cells compensate for the loss of MHC I by ensuring that NKG2D activation ligand genes are not transcribed and that the balance of NK regulatory signals favors inhibition. Viral mechanisms that inhibit the expression of NKG2D ligands described to date have primarily been at the post-transcriptional level. Reduced NKG2D ligand mRNA expression suggests that tumorigenic Ad12 cells fail to transcribe these genes resulting in protection from NK cell lysis. Future studies will determine whether the mechanism that down-regulates the transcription of NKG2D ligands is similar to that of MHC I, unifying viral control of both innate and adaptive immunity.
Figure 5.

Expression of NKG2D ligands is reduced in Ad12 (12-1) transformed rat cells compared to Ad5 (DP5-2) cells. Relative quantitation of mRNA expression for β-actin (A), RAEt1L (B), and RRTL (C) in DP5-2 and 12-1 cells. The data represents an average of at least 3 experiments. RAEt1L p < 0.05, RRTL p < 0.001.
Highlights.
Tumorigenic Ad12 cells are more resistant to NK lysis compared to non-tumorigenic Ad5 cells, despite their reduced MHC I expression.
The expression of NKG2D ligands on the surface of tumorigenic Ad12 cells is diminished compared to non-tumorigenic Ad5 cells.
Reduced surface expression of NKG2D ligands on tumorigenic Ad12 cells correlates with their reduced mRNA expression.
Acknowledgments
This paper is dedicated to the memory of John S. Doctor, Ph.D., an amazing mentor and friend. We wish to thank Sarah Passos, Sagie Wagage, Ari Glatman Zaretsky and Chris Hunter for providing balb/c splenocytes and their assistance in carrying out cytotoxcicty experiments. We would also like to thank Linda Shawver, Pinaki Banerjee and Keri Sanborn for their technical assistance and helpful scientific discussion.
This work was supported by grant CA29797 from the National Caner Institute (to R.P.R). and the NIH training grant in Virology 5-T32-AI-007324-16 (to C.Y.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Trentin JJ, Yabe Y, Taylor G. The quest for human cancer viruses. Science. 1962;137:835–841. doi: 10.1126/science.137.3533.835. [DOI] [PubMed] [Google Scholar]
- 2.Byrd PJ, Chia W, Rigby PWJ, Gallimore PH. Cloning of DNA fragments from the Left End of the Adenovirus Type 12 Genome: Transformation by Cloned Early Region I. Journal of General Virology. 1982;60:279–293. doi: 10.1099/0022-1317-60-2-279. [DOI] [PubMed] [Google Scholar]
- 3.Sawada Y, Urbanelli D, Raskova J, Shenk TE, Karel Raska J. Adenovirus tumor-specific transplantation antigen is a function of the E1A early region. Journal of Experimental Medicine. 1986;163:563–572. doi: 10.1084/jem.163.3.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernards R, Schrier PI, Houweling A, Bos JL, Van Der Eb AJ. Tumorigenicity of cells transformed by adenovirus type 12 by evasion of T-cell immunity. Nature. 1983;305:776–779. doi: 10.1038/305776a0. [DOI] [PubMed] [Google Scholar]
- 5.Gallimore P. Tumour Production in Immunosuppressed Rats with Cells transformed in vitro by Adenovirus type 2. Journal of General Virology. 1972;16:99–102. doi: 10.1099/0022-1317-16-1-99. [DOI] [PubMed] [Google Scholar]
- 6.Williams JF, Zhang Y, Williams MA, Hou S, Kushner D, Ricciardi R. E1A-based determinants of oncogenicity in human adenovirus groups A and C. Currents Topics Microbiology and Immunology. 2004;273:245–288. doi: 10.1007/978-3-662-05599-1_8. [DOI] [PubMed] [Google Scholar]
- 7.Guan H, Jiao J, Ricciardi RP. Tumorigenic adenovirus type 12 inhibits phosphorylation of NK-κB by PKAc, causing loss of DNA binding and transactivation. Journal of Virology. 2008;82:40–48. doi: 10.1128/JVI.01579-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiao J, Guan H, Lippa AM, Ricciardi RP. The N terminus of adenovirus type 12 E1A inhibits major histocompatibility complex class I expression by preventing phosphorylation of NK-kappaB p65 Ser276 through direct binding. Journal of Virology. 2010;84:7668–7674. doi: 10.1128/JVI.02317-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ricciardi RP, Zhao B, Guan H. Mechanism of tumorigenesis mediated by adenovirus-12 E1A. In: Tognon M, editor. Viral Oncogenesis. Research Signpost; Kerala: 2006. [Google Scholar]
- 10.Raulet DH, Vance ER. Self-tolerance of natural killer cells. Nature Immunology Reviews. 2006;6:520–531. doi: 10.1038/nri1863. [DOI] [PubMed] [Google Scholar]
- 11.Eagle RA, Trowsdale J. Promiscuity and the single receptor NKG2D. Nature Immunology Reviews. 2007;7:737–744. doi: 10.1038/nri2144. [DOI] [PubMed] [Google Scholar]
- 12.López-Larrea C, Suárez-Alvarez B, López-Soto A, López-Vázquez A, Gonzalez S. NKG2D receptor: sensing stressed cells. Trends in Molecular Medicine. 2008;14:179–189. doi: 10.1016/j.molmed.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Gomes AQ, Correia DV, Silva-Santos B. Non-classical major histocompatibility complex proteins as determinants of tumor immunosurveillance. EMBO. 2007;8:1024–1030. doi: 10.1038/sj.embor.7401090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carayannopoulos LN, Naidenko OV, Fremont DH, Yokoyama WM. Cutting Edge: murine UL16-binding protein-like transcript 1: a newly described transcript encoding a high-affinity ligands for murine NKG2D. Journal of Immunology. 2002;169:4079–4083. doi: 10.4049/jimmunol.169.8.4079. [DOI] [PubMed] [Google Scholar]
- 15.Malarkannan S, Shih PP, Eden PA, Horng T, Suberi AR. The molecular and functional characterization of a dominant minor H antigen, H60. Journal of Immunology. 1998;161:3501–3509. [PubMed] [Google Scholar]
- 16.Nomura M, Takihara Y, Shimada K. Isolation and characterization of retinoic acid-inducible cDNA clones in F9 cells: One of the early inducible clones encodes a novel protein sharing several highly homologous regions with a Drosophilia pholyhomeotic protein. Differeniation. 1994:57. doi: 10.1046/j.1432-0436.1994.5710039.x. [DOI] [PubMed] [Google Scholar]
- 17.Takada A, Yoshida S, Kajikawa M, Miyatake Y, Tamaru U, Sakai M, Chiba H, Maenaka K, Kohda D, Fugo K, Kasahara M. Two novel NKG2D ligands of the mouse H60 Family with differential expression patterns and binding affinities to NKG2D. Journal of Immunology. 2008;180:1678–1685. doi: 10.4049/jimmunol.180.3.1678. [DOI] [PubMed] [Google Scholar]
- 18.Zou Z, Nomura M, Takihara Y, Yasunaga T, Shimada K. Isolation and characterization of retinoic acid-inducible cDNA clones in F9 cells: a novel cDNA family encodes cell surface proteins sharing partial homology with MHC class I molecules. Journal of Biochemistry. 1996;19:319–328. doi: 10.1093/oxfordjournals.jbchem.a021242. [DOI] [PubMed] [Google Scholar]
- 19.Strausberg RL, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, Wagner L, Shenmen CM, Schuler GD, Altschul SF, Zeeberg B, Buetow KH, Schaefer CF, Bhat NK, Hopkins RF, Jordan H, Moore T, Max SI, Wang J, Hsieh F, Diatchenko L, Marusina K, Farmer AA, Rubin GM, Hong L, Stapleton M, Soares MB, Bonaldo MF, Casavant TL, Scheetz TE, Brownstein MJ, Usdin TB, Toshiyuki S, Carninci P, Prange C, Raha SS, Loquellano NA, Peters GJ, Abramson RD, Mullahy SJ, Bosak SA, McEwan PJ, McKernan KJ, Malek JA, Gunaratne PH, Richards S, Worley KC, Hale S, Garcia AM, Gay LJ, Hulyk SW, Villalon DK, Muzny DM, Sodergren EJ, Lu X, Gibbs RA, Fahey J, Helton E, Ketteman M, Madan A, Rodrigues S, Sanchez A, Whiting M, Madan A, Young AC, Shevchenko Y, Bouffard GG, Blakesley RW, Touchman JW, Green ED, Dickson MC, Rodriguez AC, Grimwood J, Schmutz J, Myers RM, Butterfield YS, Krzywinski MI, Skalska U, Smailus DE, Schnerch A, Schein JE, Jones SJ, Marra MA. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proceedings of the National Academy of Sciences, USA. 2002;26:16899–16903. doi: 10.1073/pnas.242603899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhuo M, Fujiki M, Wang M, Piard-Ruster K, Wai L-E, Wei L, Martiniez OM, Krams SM. Identification of the rat NKG2D ligands, RAEt1L and RRTL, and their role in allograft rejection. European Journal of Immunology. 2010;40:1748–1757. doi: 10.1002/eji.200939779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cook JL, Lewis AM. Differential NK cell and macrophage killing of hamster cells infected with non-oncogenic or oncogenic adenovirus. Science. 1984;224:612–615. doi: 10.1126/science.6710160. [DOI] [PubMed] [Google Scholar]
- 22.Cook JL, May DL, Lewis AMJ, Walker TA. Adenovirus E1A gene induction of susceptibility to Lysis by Natural Killer Cells and activated macrophages in Infected rodent cells. Journal of Virology. 1987;61:3510–3520. doi: 10.1128/jvi.61.11.3510-3520.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Routes JM, Ryan S, Morris K, Takaki R, Cerwenka A, Lanier LL. Adenovirus 5 sensitizes tumor cells to NKG2D-depended NK cell lysis and tumor rejection. Journal of Experimental Medicine. 2005:202. doi: 10.1084/jem.20050240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu J, Xiaopei H, Yang Y. NKG2D is required for NK cell activation and function in response to E1-deleted adenovirus. Journal of Immunology. 2010;185:7480–7486. doi: 10.4049/jimmunol.1002771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raska KJ, Gallimore PH. An Inverse Relation of the Oncogenic Potential of Adenovirus Transformed Cells and their Sensitivity to Killing By Syngeneic Natural Killer Cells. Virology. 1982;123:8–18. doi: 10.1016/0042-6822(82)90290-2. [DOI] [PubMed] [Google Scholar]
- 26.Sawada Y, Forhring B, Shenk TE, Raska KJ. Tumorigenicity of Adenovirus-transformed cells: Region E1A of Adenovirus 12 confers resistance to Natural Killer Cells. Virology. 1985;147:413–421. doi: 10.1016/0042-6822(85)90143-6. [DOI] [PubMed] [Google Scholar]
- 27.Eager KB, Williams J, Breiding D, Pan S, Knowles B, Appella E, Ricciardi RP. Expression of histocompatibility antigens H-2K, D, and L is reduced in adenovirus-12-transformed mouse cells and is restored by interferon-γ. Proceedings of the National Academy of Sciences, USA. 1985;82:5525–5529. doi: 10.1073/pnas.82.16.5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jelinek T, Pereira DS, Graham FL. Tumorigenicity of adenivirus-transformed rodent cells is influenced by at least two regions o adenovirus type 12 Early Region 1A. Journal of Virology. 1994;68:888–896. doi: 10.1128/jvi.68.2.888-896.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jenilek T, Graham FL. Recombinant human adenovirus containing hybrid adenovirus type 5 (Ad5)/Ad12 E1A genes: Characterization of hybrid E1A proteins and analysis of transforming activity and host range. Journal of Virology. 1992;66:4117–4125. doi: 10.1128/jvi.66.7.4117-4125.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanborn KB, Rak GD, Mentlik AN, Banerjee PP, Orange JS. Analysis of the NK cell immunological synapse. Methods in Molecular Biology. 1010;612:127–148. doi: 10.1007/978-1-60761-362-6_9. [DOI] [PubMed] [Google Scholar]
- 31.Makrigiannis AP, Rousselle E, Anderson SK. Independent control of Ly49g alleles: Implications for NK Cell repertoire selection and tumor cell killing. Journal of Immunology. 2004;172:1414–1425. doi: 10.4049/jimmunol.172.3.1414. [DOI] [PubMed] [Google Scholar]
- 32.Ogasawara K, Hammerman JA, Hsin H, Chikuma S, Bour-Jordan H, Chen T, Pertel T, Carnaud C, Bluestone JA, Lanier LL. Impairment of NK Cell Function by NKG2D Modulation in NOD Mice. Immunity. 2003;18:41–51. doi: 10.1016/s1074-7613(02)00505-8. [DOI] [PubMed] [Google Scholar]
- 33.Guan H, Smirnov DA, Ricciardi RP. Identification of genes associated with adenovirus 12 Tumorigenesis by microarray. Virology. 2003;309:114–124. doi: 10.1016/s0042-6822(02)00135-6. [DOI] [PubMed] [Google Scholar]
- 34.Khalli A, Ge R, Graeven U, Ricciardi RP. Negative Regulation of the Major Histocompatibility Complex Class I enhancer in Adenovirus Type 12-transformed cells via a Retinoic Acid Response Element. Journal of Virology. 1992;66:6979–6988. doi: 10.1128/jvi.66.12.6979-6988.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu X, Ge R, Westmoreland S, Cooney AJ, Tsai S, Tsai M-J, Ricciardi RP. Negative regulation by the R2 element of the MHC I enhancer in adenovirus-12 transformed cells correlats with high levels of COUP-TF binding. Oncogene. 1994;9:2183–2190. [PubMed] [Google Scholar]
- 36.Smirnov DA, Hou S, Liu X, Claudio E, Siebenlist UK, Ricciardi RP. COUP-TFII is Up-regulated in Adenovirus type 12 tumorigenic cells and is a repressor of MHC I transcription. Virology. 2001;284:13–19. doi: 10.1006/viro.2001.0913. [DOI] [PubMed] [Google Scholar]
- 37.Smirnov DA, Hou S, Ricciardi RP. Association of Histone Deacetylase with COUP-TF in Tumorigenic Ad12-transformed Cells and Its Potential Role in Shutoff of MHC class I transcription. Virology. 2000;268:319–328. doi: 10.1006/viro.1999.0181. [DOI] [PubMed] [Google Scholar]
- 38.Zhao B, Hou S, Ricciardi RP. Chromatin repression by COUP-TFII and HDAC dominates activation by NF-kB in regulating major histocompatibility complex class I transcription in adenovirus tumorigenic cells. Virology. 2003;306:68–76. doi: 10.1016/s0042-6822(02)00079-x. [DOI] [PubMed] [Google Scholar]
- 39.Zhao B, Ricciardi RP. E1A is the component of the MHC class I enhancer that mediates HDAC chromatin repression in adenovirus 12 tumorigenic cells. Virology. 2006;352:338–344. doi: 10.1016/j.virol.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 40.Huvent I, Cousin C, Kiss A, Baroni de Moraes MT, Bernard C, D'Halluin JC. Downregulation of Major Histocompatibility Complex Class I expression and susceptibility to Natural Killer cells in cells transformed with the oncogenic Adenovirus 12 are regulated by different E1A domains. Cancer Detection and Prevention. 1997;21:12–21. [PubMed] [Google Scholar]
- 41.Routes JM, Ryan S, Li H, Steinke J, Cook JL. Dissimilar immunogenicities of human papillomavirus E7 and Adenovirus E1A proteins influence primary tumor development. Virology. 2000;277:48–57. doi: 10.1006/viro.2000.0571. [DOI] [PubMed] [Google Scholar]
- 42.Hasan M, Krmpotic A, Ruzsics Z, Bubic I, Lenac T, Halenius A, Loewendorf A, Messerle M, Hengel H, Jonic S, Koszinowski UH. Selective down-regulation of the NKG2D ligand H60 by mouse cytomegalovirus m155 glycoprotein. Journal of Virology. 2005:79. doi: 10.1128/JVI.79.5.2920-2930.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jonjic S, Polic B, Krmpotic A. The role of NKG2D in immunoevasion by tumors and viruses: Viral inhibitors of NKG2D ligands: Friends or foes of immune surveillance. European Journal of Immunology. 2008;38:2952–2956. doi: 10.1002/eji.200838823. [DOI] [PubMed] [Google Scholar]
- 44.Lanier LL. Evolutionary struggles between NK cells and viruses. Nature Reviews: Immunology. 2008;8:259–268. doi: 10.1038/nri2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lodoen MB, Abenes G, Umamoto S, Houchins JP, Liu F, Lanier LL. The Cytomegalovirus m155 gene product subverts Natural Killer Cell antivirual protection by disruption of H60-NKG2D Interactions. Journal of Experimental Medicine. 2004;200:1075–1081. doi: 10.1084/jem.20040583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lodoen MB, Lanier LL. Viral modulation of NK cell immunity. Nature Reviews: Microbiology. 2005;3:59–69. doi: 10.1038/nrmicro1066. [DOI] [PubMed] [Google Scholar]
- 47.Nolting A, Dugast A-S, Rihn S, Luteijn R, Carrington MF, Kane K, Jost S, Toth I, Nagami E, Faetkenheuer G, Hartmann P, Altfeld M, Alter G. MHC Class I chain-related protein A shedding in chronic HIV-1 infection is associated with profound NK cell dysfunction. Virology. 2010;406:12–20. doi: 10.1016/j.virol.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas M, Boname JM, Field S, Nejentsev S, Salio M, Cerundolo V, Wills M, Lehner P. Down-regulation of NKG2D and NKp80 ligands by Kaposi's sarcoma-associated herpesvirus K5 protects against NK Cell cytotoxicity. Proceedings of the National Academy of Sciences, USA. 2005;105:1656–1661. doi: 10.1073/pnas.0707883105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas M, Wills M, Lehner P. Natural Killer cell evasion by an E3 ubiquitin ligase from Kaposi's sarcoma-associated herpesvirus. Biochemical Society Transactions. 2008;36:459–463. doi: 10.1042/BST0360459. [DOI] [PubMed] [Google Scholar]
- 50.Aparicio O, Carnero E, Abad X, Razquin N, Guruceaga E, Segura V, Fortes P. Adenovirus VA RNA-derived miRNAs target cellular genes involved in cell growth, gene expression and DNA repair. Nucleic Acids Research. 2010;38:750–763. doi: 10.1093/nar/gkp1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ortin J, Scheidtmann K-H, Greenberg R, Westphal M, Doerfler W. Transcription of the genes of Adenovirus type 12: Maps of stable RNA from productively infected human cells and abortively infected and transformed hamster cells. Journal of Virology. 1976;20:355–372. doi: 10.1128/jvi.20.2.355-372.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Friedman DJ, Ricciardi RP. Adenovirus type 12 E1A gene represses accumulation of MHC I mRNAs at the level of transcription. Virology. 1988;165:303–305. doi: 10.1016/0042-6822(88)90689-7. [DOI] [PubMed] [Google Scholar]
- 53.Hou S, Guan H, Ricciardi RP. In adenovirus type 12 tumorigenic cells, Major Histocompatibility Complex Class I transcription shutt of is overcome by induction of NF-κB and relief of COUP-TFII repression. Journal of Virology. 2002;76:3213–3220. doi: 10.1128/JVI.76.7.3212-3220.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hou S, Guan H, Ricciardi RP. Phosphorylaton of serine 337 of NK-κB is critical for DNA Binding. Journal of Biological Chemistry. 2003;278:45994–45998. doi: 10.1074/jbc.M307971200. [DOI] [PubMed] [Google Scholar]
- 55.Kralli A, Ge R, Graeven U, Ricciardi RP. Negative Regulation of the Major Histocompatibility Complex Class I enhancer in Adenovirus Type 12-transformed cells via a Retinoic Acid Response Element. Journal of Virology. 1992;66:6979–6988. doi: 10.1128/jvi.66.12.6979-6988.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kushner D, Ricciardi RP. Reduced phosphorylation of p50 is responsible for dinimished NF-κB binding to the Major Histocompatibility Complex Class I Enhancer in adenovirus type 12-transformed cells. Molecular and Cellular Biology. 1999;19:2169–2179. doi: 10.1128/mcb.19.3.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meijer I, Joecemsen AG, Witt CMd, Bos JL, Morello D, Eb AJVD. Adenovirus type 12 down regulates expression of a transgene under the control of a major histocompatibility complex class I promoter: Evidence for transcriptional control. Journal of Virology. 1989:63. doi: 10.1128/jvi.63.9.4039-4042.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schrier PI, Bernards R, Vaessen RTMJ, Houweling A, Eb AJVD. Expression on class I major histocompatibility antigens switched off by highly oncogenic adenovirus 12 in transformed rat cells. Nature. 1983;305:771–775. doi: 10.1038/305771a0. [DOI] [PubMed] [Google Scholar]
- 59.Gasser S, Orsulic S, Brown E, Raulet D. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–1190. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]