

Summary
Seven in absentia homolog 1 (SIAH1) was reported to be downregulated in hepatocellular carcinoma (HCC) and played an important role in HCC progression; however, the underlying reason remains unknown. Here, we found that Cathepsin K (CTSK), a protein potentially interacting with SIAH1, inhibits SIAH1 protein level. CTSK was highly expressed in HCC tissues. CTSK inhibition or downregulation suppressed HCC cell proliferation, whereas CTSK overexpression had the opposite effect; it promotes HCC cell proliferation by regulating the SIAH1/protein kinase B (AKT) pathway, wherein promotes SIAH1 ubiquitination. Neural precursor cells expressing developmentally downregulated 4 (NEDD4) was found to be a potential upstream ubiquitin ligase of SIAH1. Further, CTSK could mediate SIAH1 ubiquitination and degradation by increasing SIAH1 autoubiquitination and recruiting NEDD4 to ubiquitinate SIAH1. Finally, the roles of CTSK were confirmed in a xenograft mouse model. In conclusion, oncogenic CTSK was upregulated in human HCC tissues and accelerated HCC cell proliferation by downregulating SIAH1.
Subject areas: Molecular biology, Cell biology, Cancer
Graphical abstract
Highlights
-
•
SIAH1 expression is downregulated in HCC cells
-
•
CTSK interacts with SIAH1, and its expression is upregulated in HCC
-
•
CTSK and NEDD4 form complexes with SIAH1 to promote its ubiquitination
-
•
CSTK regulates SIAH1 expression and promotes HCC development and progression
Molecular biology; Cell biology; Cancer
Introduction
Hepatocellular carcinoma (HCC) is a common malignant tumor of the digestive system. The annual number of new cases and deaths due to HCC in China accounts for more than half of those in the world.1 Despite the rapid development of comprehensive treatment strategies for HCC, the prognosis of HCC patients remains unsatisfactory. The 5-year survival rate of HCC patients in China from 2000 to 2014 was only 11.9–13.1%.2 Uncontrolled proliferation of HCC cells is an important mechanism underlying the development of malignant HCC; however, the molecular mechanism remains unclear. Therefore, exploring the molecular mechanism underlying HCC cell proliferation may provide a new approach for the targeted treatment of HCC.
Seven in absentia homolog 1 (SIAH1), a member of the SIAH family, is an evolutionarily conserved RING-type E3 ubiquitin ligase. It has been demonstrated that SIAH1 is involved in a range of cellular activities, such as DNA damage response, hypoxia adaptation, apoptosis, angiogenesis, and cell proliferation.3,4 In addition, studies have reported that SIAH1 expression is closely associated with the occurrence and development of human cancers, such as breast cancer, colorectal cancer, stomach cancer, glioma, and HCC. It has been reported that SIAH1 is downregulated in HCC, and its inactivation plays an important role in HCC progression.5,6,7,8,9 SIAH1 expression can lead to growth stagnation and apoptosis in HCC cells through ubiquitination of β-catenin10; SIAH1 has also been reported to promote the ubiquitination and degradation of hepatitis B virus X (HBX), which in turn affects HCC progression.11 However, the reason underlying the downregulation of SIAH1 in HCC remains unknown and requires further study. For this purpose, we screened proteins interacting with SIAH1 using the yeast two-hybrid system.
Cathepsin K (CTSK), a lysosomal cysteine protease belonging to the papain superfamily,12 was identified as a potential interacting protein of SIAH1 in the yeast two-hybrid screen. Studies have shown that CTSK is an important extracellular matrix degrading enzyme that is necessary for normal bone resorption and is mainly involved in the degradation of the bone matrix.12 The decreased or increased expression of CTSK can result in the development of various bone diseases, such as osteogenesis imperfecta dense bone, osteoporosis, and multiple myeloma.13,14 Recently, it was reported that CTSK is specifically expressed in many human cancers and is closely related to the occurrence and malignant progression of tumors. For example, increased CTSK expression potentiates the metastasis of epithelial ovarian cancer,15 gut microbiota-stimulated CTSK secretion promotes tumor metastasis in colorectal cancer,16 and CTSK inhibition-induced mitochondrial reactive oxygen species (ROS) enhance the sensitivity of cancer cells to anti-cancer drugs.17 Therefore, CTSK can be considered a potential biomarker for several human cancers.
In this study, we investigated the precise mechanisms underlying the downregulation of SIAH1 in HCC and identified a connection between SIAH1 and CTSK using a yeast two-hybrid screen. We aimed to explore the role of CTSK in HCC cells and investigate the interaction and regulatory relationship between SIAH1 and CTSK.
Results
CTSK is an interacting protein of SIAH1
It has been reported that SIAH1, an E3 ubiquitin ligase, plays an important role in inhibiting the occurrence and development of HCC, and its protein level is frequently downregulated in human HCC patients. To explore the reasons for the low expression of SIAH1 in HCC, we screened SIAH1-interacting proteins in a human cDNA library using a yeast two-hybrid assay with SIAH1 as bait. CTSK was identified as a potential SIAH1-interacting protein by subjecting the results of the yeast two-hybrid experiment to BLAST in the NCBI database (Figure 1A). The interaction between SIAH1 and CTSK was further confirmed using a co-immunoprecipitation (coIP) assay in HEK293T cells. After ectopic expression of HIS-SIAH1 and 3×FLAG-CTSK in HEK293T cells, we found that 3×FLAG-CTSK co-immunoprecipitated with HIS-SIAH1 (Figure 1B), as well as endogenous CTSK, also interacted with SIAH1 (Figure 1C). To elucidate the regulatory relationship between SIAH1 and CTSK, we overexpressed SIAH1 and CTSK in HEK293T cells. The protein levels of SIAH1 decreased after CTSK overexpression (Figure 1D). Conversely, the expression level of CTSK did not change after the overexpression of SIAH1 (Figure 1E). These results suggested that CTSK is an upstream molecule of SIAH1 and can downregulate the protein level of SIAH1.
Figure 1.

CTSK is an interacting protein of SIAH1
(A) Yeast two-hybrid experiment was conducted with SIAH1 bait, and the obtained sequence was subjected to BLAST with NCBI to obtain CTSK.
(B) 3×FLAG-CTSK and HIS-SIAH1 plasmids were co-transfected in HEK293T cells and subjected to coIP assay to detect whether the two exist in the same complex.
(C) Antibody against SIAH1 was used to detect whether endogenous SIAH1 interacted with CTSK.
(D) CTSK was overexpressed in HEK293T cells, and the protein level of SIAH1 was assessed using western blotting.
(E) SIAH1 was overexpressed in HEK293T cells, and the protein level of CTSK was assessed using western blotting.
CTSK is highly expressed in HCC patients
To determine the clinical significance of CTSK, we first analyzed the effect of CTSK on the prognosis of HCC patients in The Cancer Genome Atlas (TCGA) database and found that high expression of CTSK significantly decreased the survival rate of HCC patients (Figure 2A). Then, 14 HCC tissues and 14 normal liver tissues were randomly chosen for CTSK protein measurement. As shown in Figure 2B, the protein level of CTSK in HCC tissues was significantly higher than that in the normal liver tissues. These results indicate that CTSK is highly expressed in HCC tissues and may influence the development of HCC.
Figure 2.

CTSK is highly expressed in HCC patients
(A) TCGA database analysis of the effect of CTSK on the prognosis of patients with liver cancer.
(B) Protein expression level and statistical plot of expression levels of CTSK in 14 HCC tissues and 14 normal liver tissues. All quantitative data are means ± SEM for 14 human samples per group (n = 14). ∗∗p < 0.01, as evaluated by unpaired two-tailed Student’s t test.
CTSK promotes the proliferation of HCC cells
Since the above results indicated that CTSK was highly expressed in HCC cells, we considered whether inhibition of CTSK suppressed the proliferation of HCC cells. We treated cells with a CTSK-specific inhibitor (Balicatib) to achieve loss of function. The efficiency of CTSK expression was assessed using western blotting (Figure 3A). After 24 h of treatment, the cells were used to detect cell growth. 5-Ethynyl-2ʹ-deoxyuridine (EdU) incorporation assays revealed that inhibition of CTSK expression significantly decreased the proportion of EdU-positive Huh7 and HepG2 cells (Figures 3B and 3C). The Cell Counting Kit-8 (CCK-8) assay also showed that CTSK inhibition exerted critical suppressive effects on the proliferation of HCC cells (Figure 3D). In order to further confirm the effect of CTSK on the proliferation of HCC cells, we used a short hairpin RNA (shRNA) strategy to downregulate CTSK and detected the proliferation ability of HCC cells upon downregulating CTSK. We designed three sets of shRNA for CTSK (shCTSK#1, shCTSK#2, and shCTSK#3), and a non-targeting shRNA served as negative control (shCtrl). The efficiency of shCTSKs was analyzed by western blotting. It was found that shCTSK#1 displayed the highest silencing efficiency (Figure S1A). Then, the shCTSK#1 and shCtrl were developed stable cell lines with CTSK knockdown through the strategy of lentivirus infection. Western blotting was used to detect the CTSK downregulation efficiency (Figure S1B). Next, we assessed the proliferation in Huh7 and HepG2 cells silencing CTSK. Briefly, silencing of CTSK significantly decreased the EdU-positive cells and cell activity, which showed similar effects with CTSK inhibitor Balicatib (Figures S1C–S1E).
Figure 3.

CTSK promotes the proliferation of HCC cells, related to Figure S1
(A) Western blotting revealed the inhibitory effect of Balicatib on CTSK protein levels in Huh7 and HepG2 cells.
(B) Representative images of the EdU experiment. Scale bar: 200 μm.
(C) Statistical analysis of EdU experiments performed in HuH7 and HepG2 cells after treatment with Balicatib.
(D) Results of the CCK-8 assay.
(E) Western blotting revealed the overexpression efficiency of CTSK.
(F) Representative images of the EdU experiment. Scale bar: 200 μm.
(G) Statistical analysis of EdU experiments performed in Huh7 and HepG2 cells after the overexpression of CTSK. (H) Results of the CCK-8 assay. All quantitative data are means ± SEM from three independent experiments (n = 3). The value of first day in CCK-8 assay was labeled “1”. ∗p < 0.05, ∗∗p < 0.01, as evaluated by paired two-tailed Student’s t test.
To further clarify the role of CTSK, we transiently transfected 3×FLAG-tagged CTSK cDNA into Huh7 and HepG2 cells to achieve functional gain, and the expression efficiency of CTSK was tested using western blotting (Figure 3E). EdU incorporation assays showed that overexpression of CTSK significantly increased the proportion of EdU-positive Huh7 and HepG2 cells compared to the control group (Figures 3F and 3G). Similarly, the CCK-8 assay showed a significant increase in cell viability in the 3×FLAG-CTSK group (Figure 3H).
CTSK promotes the proliferation of HCC cells by regulating SIAH1/AKT pathway
To explore the signaling pathway in which CTSK is involved, we used transcriptome sequencing for the data from human HCC patients obtained from TCGA database to perform gene set enrichment analysis (GSEA) enrichment analysis. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis showed that the genes of the CTSK-high-expression group in the PI3K-AKT-mTOR pathway were highly enriched (Figure 4A). It has been reported that SIAH1 inhibits cell proliferation and invasion in tumor cells in part by downregulating AKT expression. In contrast, SIAH1 plays an important role in inhibiting HCC cell proliferation. Thus, we speculated that CTSK might promote the proliferation of HCC cells by regulating the SIAH1/AKT pathway. As expected, inhibition or knockdown of CTSK significantly elevated the protein level of SIAH1, thereby decreasing the phosphorylation of AKT (S473) (Figure 4B). Conversely, overexpression of CTSK had the opposite effect (Figure 4C). More importantly, to confirm that CTSK-induced changes in the phosphorylation of AKT were attributed to SIAH, silencing of SIAH1 was used to perform the rescue experiment. Inhibition or knockdown of CTSK in Huh7 and HepG2 cells significantly upregulated the levels of SIAH1, thereby decreasing the levels of p-AKT (S473), and this regulation was significantly blocked by silencing of SIAH1 (Figure 4D). These results indicate that the downregulation of p-AKT (S473) induced by CTSK inhibition was largely attributed to the upregulation of SIAH1 expression. CCK-8 assays were conducted to confirm these results. Silencing of SIAH1 significantly attenuated CTSK inhibition or knockdown-induced suppression of cell proliferation (Figures 4E and 4F). Taken together, these results revealed that the promoting effects of CTSK upregulation on HCC proliferation were mediated through the SIAH1/AKT pathway.
Figure 4.

CTSK promotes the proliferation of HCC cells by regulating SIAH1/AKT pathway
(A) KEGG enrichment analysis of high-CTSK-expression group in TCGA database.
(B) Inhibition or knockdown of CTSK increased SIAH1 protein level and decreased the level of p-AKT.
(C) Overexpression of CTSK decreased SIAH1 protein level and increased the level of p-AKT.
(D) Silencing of SIAH1 significantly rescued the downregulation of p-AKT induced via CTSK inhibition or silencing in Huh7 and HepG2 cells.
(E and F) CCK-8 assay showed silencing of SIAH1 rescued the restrain of growth induced by CTSK inhibition or silencing in Huh7 and HepG2 cells. All quantitative data are means ± SEM from three independent experiments (n = 3). The value of first day in CCK-8 assay was labeled “1”. ∗ (down): Balicatib vs. DMSO, shCTSK#1 vs. shCtrl; ∗ (up): Balicatib+shSIAH1#1 vs. Balicatib+shCtrl, shCTSK#1+ shSIAH1#1 vs. shCTSK#1+ shCtrl. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, as evaluated by paired two-tailed Student’s t test.
CTSK recruits NEDD4 to promote the ubiquitination and degradation of SIAH1
Based on the previously reported studies and the above results, we speculated that CTSK might regulate the ubiquitination and degradation of SIAH1. To answer this question, we first examined the degradation pathway and ubiquitination level of SIAH1 after CTSK overexpression. MG-132 blocked the degradation of SIAH1 in cells with the expression of CTSK upregulated (Figure 5A). Furthermore, compared to the control group, overexpression of CTSK promoted the ubiquitination of SIAH1 (Figure 5B). Since it has been reported that SIAH1 is a ubiquitin ligase that can perform autoubiquitination, we hypothesized that CTSK is involved in regulating SIAH1 autoubiquitination. To address this, we generated an SIAH1-RING mutant in which Cys44 in the RING domain was converted to serine (C44S). Previous studies have shown that this mutant has no E3 ligase activity. As shown in Figure 5C, mutation of the ubiquitination domain of SIAH1 could not fully block the promoting effect of CTSK on SIAH1 ubiquitination, suggesting that CTSK induced the ubiquitination of SIAH1 not only by autoubiquitination but also by other E3s. To screen for the related upstream ubiquitin ligase of SIAH1, we used the Ubibrowser database with SIAH1 as a substrate and found that NEDD4 was the potential upstream E3 ligase with the highest score (Figure 5D). Studies have also reported that NEDD4 plays an important role in HCC proliferation and progression. Thus, we investigated whether NEDD4 is involved in CTSK-induced ubiquitination of SIAH1. It was found that NEDD4 promoted the ubiquitination and degradation of SIAH1 by the proteasome (Figures 5E and 5F). In addition, MYC-NEDD4 interacted with HIS-SIAH1 and 3×FLAG-CTSK (Figures 5G and 5H, upper panels); endogenous NEDD4 co-immunoprecipitated with SIAH1 and CTSK (Figures 5G and 5H, lower panels), suggesting that NEDD4, SIAH1, and CTSK formed a complex. Next, to explore whether CTSK promotes the ubiquitination and degradation of SIAH1 by recruiting NEDD4, we examined the ubiquitination of SIAH1 in the presence of NEDD4 and different doses of the CTSK inhibitor. It was revealed that as the dose of Balicatib was increased, the protein level of CTSK decreased, and the ubiquitination of SIAH1 showed a dose-dependent effect of CTSK (Figure 5I). More importantly, it was shown that compared with the corresponding control group, overexpression of NEDD4 effectively aggravated the ubiquitination levels of SIAH1, which could be blocked by CTSK inhibition (Figure 5J). Taken together, these results indicate that CTSK promotes SIAH1 degradation, not only by intensifying the autoubiquitination of SIAH1 but also by recruiting NEDD4 to ubiquitinate SIAH1.
Figure 5.

CTSK recruits NEDD4 to promote the ubiquitination and degradation of SIAH1
(A) Proteasome inhibitor MG132 rescued the downregulation of SIAH1 expression induced by CTSK overexpression.
(B) Overexpression of CTSK increased the ubiquitination level of SIAH1.
(C) Mutant SIAH1 with E3 activity deletion could not fully block the promoting effect of CTSK on SIAH1 ubiquitination.
(D) Ubibrowser database used SIAH1 as substrate, analyzed its upstream E3 ubiquitin ligase, and selected NEDD4 with the highest score.
(E) NEDD4 overexpression promoted the degradation of SIAH1 from proteasome.
(F) NEDD4 overexpression promoted the ubiquitination level of SIAH1.
(G and H) CoIP assays showed NEDD4, SIAH1, and CTSK formed a complex.
(I) Ubiquitination level of SIAH1 revealed the dose dependence of Balicatib. (J) Overexpression of NEDD4 induced ubiquitination of SIAH1 could be blocked via CTSK inhibition.
CTSK promotes HCC cell growth by regulating SIAH1 in vivo
Finally, we examined the above mechanism in an HCC tumor model. In this experiment, CTSK-silenced Huh7 cells or shCtrl Huh7 cells were transplanted subcutaneously inoculated into Balb/C nude mice. Consistent with the in vitro observations, CTSK knockdown markedly inhibited tumor growth (Figures 6A–6C). Moreover, western blotting revealed that, compared to the shCtrl group, SIAH1 protein was significantly increased in tumors with downregulation of CTSK (Figure 6D). These findings suggested that CTSK acts as a tumor promoter in HCC by regulating SIAH1 expression.
Figure 6.

CTSK promotes HCC cell growth by regulating SIAH1 in vivo
(A) Tumor tissues isolated from tumors initiated with cells infected with shCtrl or shCTSK#1 vectors.
(B) Tumor mass.
(C) Growth curve obtained by measuring tumor size on the indicated days.
(D) Representative blots of CTSK and SIAH1 levels in tumor tissues.
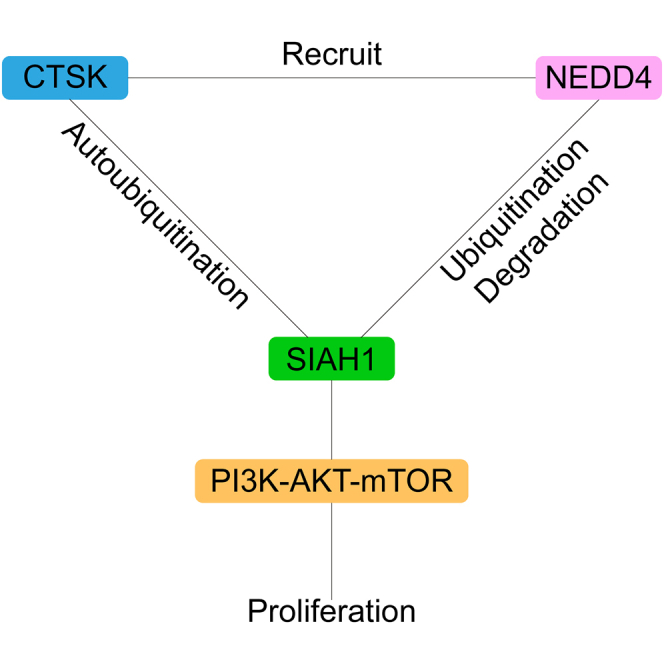
(E) Working model: CTSK promotes SIAH1 degradation, which not only intensifies the autoubiquitination of SIAH1 but also recruits NEDD4 to ubiquitinate SIAH1. All quantitative data are means ± SEM for 5 mice per group (n = 5). ∗p < 0.05, ∗∗∗p < 0.001, as evaluated by unpaired two-tailed Student’s t test.
Discussion
The SIAH family, a mammalian homolog of the Drosophila Sina protein, is a family of ubiquitin ligases. It has been reported that SIAH1 is involved in DNA damage response, hypoxia adaptation, apoptosis, angiogenesis, and cell proliferation.3,4 In addition, studies have shown that SIAH1 expression is closely associated with the occurrence and malignant progression of multiple human cancers such as breast cancer, colorectal cancer, stomach cancer, glioma, and HCC.5,6,7,8,9 SIAH1 is expressed at low levels in HCC and plays an important role in HCC.9 In the present study, we investigated the downregulation of SIAH1 expression in HCC and demonstrated that CTSK is involved in the regulation of SIAH1 expression. We found that CTSK and SIAH1 form a complex and that CTSK can accelerate SIAH1 ubiquitination and degradation, thereby promoting the proliferation of HCC cells by regulating the phosphorylation of AKT. These findings explain the reasons for the decreased expression of SIAH1 in HCC tissues and provide a theoretical basis for exploring the factors that regulate SIAH1 expression during the occurrence and development of HCC.
CTSK was first observed in osteoclasts and is a cysteine protease member of the cathepsin lysosomal protease family. It has been identified as an osteolytic enzyme and protease that can degrade bone matrix proteins including type I collagen, osteopontin, and osteonectin.18,19 Recent studies have shown that CTSK expression is closely associated with the occurrence and development of human cancers. For example, CTSK commonly causes bone metastasis in breast cancer by promoting the differentiation of fat cells20; increased expression of CTSK can enhance the metastasis of epithelial ovarian cancer21; and CTSK secretion can mediate TLR4-dependent M2 macrophage polarization and promote tumor metastasis of colorectal cancer.22 These findings indicate that CTSK might be a potential biomarker of human cancers. In this study, we assessed CTSK expression in human HCC and non-tumor liver tissues. We confirmed that the protein levels of CTSK were dramatically increased in tissues obtained from patients with HCC. We also examined the effect of CTSK on HCC cell proliferation and found that CTSK promoted HCC cell proliferation by promoting ubiquitination and degradation of SIAH1, in turn increasing the phosphorylation of AKT. These results indicate that CTSK plays a carcinogenic role in HCC and may be a potential biomarker for HCC. This study expounds the specific mechanisms by which CTSK promotes the occurrence and development of HCC and provides a theoretical basis for future investigations into the effect of CTSK in tumors.
NEDD4, a ubiquitin ligase belonging to the homologous to E6-AP carboxyl terminus (HECT) subfamily, is involved in processes such as ubiquitin modification, virus budding, transcriptional regulation, and neural development.23 NEDD4 plays an important role in the occurrence and development of various tumors; for example, NEDD4 can disrupt programmed cell death 1 ligand 1 (PD-L1) to control T cell-mediated immune surveillance in bladder cancer24; Nedd4 inhibits erastin-induced melanoma by ubiquitinating voltage-dependent anion selective channel 2/3 (VDAC2/3) ferroptosis25; NEDD4 can regulate the migration of lung cancer cells26; and NEDD4 ubiquitin ligase activates insulin-like growth factor 1 receptor (IGF-1R)/phosphoinositide-3 kinase (PI3K)/Akt signaling to mediate the development of endometrial cancer.27 It has been reported that NEDD4 is associated with the development of hepatitis B virus (HBV)-associated/disassociated HCC. For example, NEDD4 can promote the growth and motility of HCC cells by regulating large tumor suppressor gene 1 (LATS1) and phosphate and tension homology deleted on chromosome ten (PTEN)/PI3K/AKT7,28,29; NEDD4 induces degradative ubiquitination of the hepatitis B virus X protein and inhibits HBV-associated HCC progression.30 In the present study, we found that NEDD4 and SIAH1 form a complex and that NEDD4 might induce the degradative ubiquitination of SIAH1. We verified that CTSK could also form complexes with NEDD4. Furthermore, CTSK promoted the ubiquitination of SIAH1 through the recruitment of NEDD4. This is the first report of NEDD4 acting as an E3 ubiquitination ligase of SIAH1 and degrading it through ubiquitination. These results may provide a foundation for future exploration of the role of SIAH1 in tumors.
In conclusion, existing research on the role of SIAH1 in the occurrence and development of HCC is incomplete, and the reasons for its low expression in HCC tissues are unknown. In this study, we provide the first evidence that CTSK interacts with SIAH1 and mediates the proliferation of human HCC through its regulation. We found that CTSK expression is upregulated in human HCC tissues and promotes HCC cell proliferation by promoting AKT phosphorylation. CTSK promotes SIAH1 degradation by promoting SIAH1 ubiquitination and recruiting NEDD4 (Figure 6E). These findings provide a basis for further studies on the roles of CTSK and SIAH1 in HCC progression and the underlying molecular mechanisms.
Limitations of the study
Although we confirmed that CTSK regulation of SIAH1 can promote the proliferation of HCC cells, whether the regulation of SIAH1 by CTSK is also affected by other factors and the mechanism underlying the abnormally high expression of CTSK in HCC remain unclear. These issues require further investigation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CTSK | Proteintech | Cat# 11239-1-AP; RRID: AB_2245581 |
| SIAH1 | Proteintech | Cat# 13886-1-AP; RRID: AB_10643374 |
| NEDD4 | Proteintech | Cat# 67865-1-Ig; RRID: AB_2918623 |
| AKT | Proteintech | Cat# 60203-2-Ig; RRID: AB_10912803 |
| p-AKT (Ser473) | Proteintech | Cat# 80455-1-RR; RRID: AB_2918892 |
| HA | Proteintech | Cat# 51064-2-AP; RRID: AB_11042321 |
| FLAG | Proteintech | Cat# 66008-4-Ig; RRID: AB_2918475 |
| MYC | Proteintech | Cat# 60003-2-Ig; RRID: AB_2734122 |
| HIS | Proteintech | Cat# 66005-1-Ig; RRID: AB_11232599 |
| Ub | Proteintech | Cat# 10201-2-AP; RRID: AB_671515 |
| β-actin | Proteintech | Cat# 66009-1-Ig; RRID: AB_2687938 |
| HRP-conjugated Affinipure Goat Anti-Rabbit IgG(H+L) | Proteintech | Cat# SA00001-2; RRID: AB_2722564 |
| HRP-conjugated Affinipure Goat Anti-Mouse IgG(H+L) | Proteintech | Cat# SA00001-1; RRID: AB_2722565 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | Yuanpei | L110KJ |
| MEM | Yuanpei | L550KJ |
| FBS | Gibco | Cat#12664025C |
| MG132 | MCE | HY-13259 |
| DMSO | MCE | HY-Y0320 |
| Balicatib | MCE | HY-15100 |
| Yeast strain Y187 | Clontech | Cat#630486 |
| 0.45-μm nitrocellulose membrane | Millipore | ipvh00010 |
| BSA | Yeasen | 36101ES25 |
| Hieff Trans™ Liposomal Transfection Reagent | Yeasen | 40802ES02 |
| Chemiluminescent ECL Plus reagents | Thermo Fisher Scientific | 32106 |
| Protein A/G Magnetic Beads | MCE | HY-K0202 |
| Critical commercial assays | ||
| CCK-8 assay kit | Dojindo | CK04 |
| 5-Ethynyl-2ʹ-deoxyuridine assay kit | RiboBio | c10310-1 |
| Experimental models: Cell lines | ||
| HEK293T cell line | Stem Cell Bank, Chinese Academy of Sciences | N/A |
| HepG2 cell line | Stem Cell Bank, Chinese Academy of Sciences | N/A |
| Huh7 cell line | Stem Cell Bank, Chinese Academy of Sciences | N/A |
| Experimental models: Organisms/strains | ||
| Mouse, Balb/C nude | Charles River | https://www.criver.com/ |
| See Table S1 for information on human studies | N/A | |
| Oligonucleotides | ||
| shSIAH1#1 primers: | Youbio | N/A |
| F: 5′-GGATCCGGAAAGGCTACTCCAC CTTCTTTCAAGAGAAGAAGGTGGAGT AGCCTTTCCTTTTTTG-3′ |
||
| R: 5′-AATTCAAAAAAGGAAAGGCTACT CCACCTTCTTCTCTTGAAAGAAGGTGG AGTAGCCTTTCCG-3′ |
||
| shCTSK#1 primers: | Youbio | N/A |
| F: 5′- GATCCGTGAAATCTCTCGGCG TTTAATTTCAAGAGAATTAAACGCCG AGAGATTTCATTTTTTG-3′ |
||
| R: 5′- AATTCAAAAAATGAAATCTCTC GGCGTTTAATTCTCTTGAAATTAAAC GCCGAGAGATTTCACG-3′ |
||
| Recombinant DNA | ||
| pcDNA3.1-SIAH1-HIS | Youbio | N/A |
| pcDNA3.1-SIAH1 C44S-HIS | Youbio | N/A |
| pcDNA3.1-CTSK-3∗FLAG | Youbio | N/A |
| pcDNA3.1-NEDD4-MYC | Youbio | N/A |
| pcDNA3.1-Ub-HA | Youbio | N/A |
| shCTSK pLVX-shRNA2-PURO | Youbio | N/A |
| shSIAH1 pLVX-shRNA2-PURO | Youbio | N/A |
| Software and algorithms | ||
| Adobe Photoshop CS6 | Adobe | https://www.adobe.com/products/photoshop.html |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| SPSS 22.0 | SPSS | http://www.spss.com.cn |
| Graphpad prime 8.0 | Graphpad | https://www.graphpad.com/ |
| Other | ||
| TCGA - the Cancer Genome Atlas | HOME for Researchers | https://www.aclbi.com/static/index.html#/tcga |
| NCBI | National Center for Biotechnology Information | https://www.ncbi.nlm.nih.gov |
| GEO - Gene Expression Omnibus | National Center for Biotechnology Information | https://www.ncbi.nlm.nih.gov/geo/GEO: GSE209547; GEO: GSE188256 |
| UbiBrowser | Beijing proteome research center | http://ubibrowser.bio-it.cn/ubibrowser/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hengliang Shi, Professor (shl@xzhmu.edu.cn).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed material transfer agreement.
Experimental model and subject details
Tissues
Normal human liver and HCC tissue samples were obtained from the Affiliated Hospital of Xuzhou Medical University (Xuzhou, China). HCC tissue specimens were obtained from patients with HCC who underwent surgical resection, and normal liver specimens were obtained from patients undergoing partial hepatectomy for non-tumor reasons, such as trauma and hemangioma. All tissue samples were immediately frozen in liquid nitrogen after excision and stored at –80°C. This study was approved by the Ethical Research Committee of the Affiliated Hospital of Xuzhou Medical University (approval number: XYFY2019-KL129-01) and all samples were approved by the patients.
Cell culture
The HEK293T, HepG2, and Huh7 HCC cell lines were purchased from the Cell Bank, Chinese Academy of Sciences (Shanghai, China), and were cultured in Minimum Essential medium or Dulbecco’s modified Eagle’s medium (Yuanpei, Shanghai, China) supplemented with 10% fetal bovine serum (FBS, Gibco, Shanghai, China) at 37°C in an atmosphere of 5% CO2.
Method details
Transient transfection
Cell transfection was performed using the Hieff Trans™ Liposomal Transfection Reagent (Yeasen, Shanghai, China) in accordance with the manufacturer's instructions. The plasmid and the transfection agent were mixed with an appropriate amount of serum-free medium, and then the two were combined and mixed. After incubating at room temperature for 20 min, the mixture was added to the cells to be transfected. The medium was replaced 6 h after transfection.
Establishment of the stable cell lines
To produce the lentiviruses, HEK293T cells were cotransfected with the corresponding plasmids (shCtrl or shCTSKs) and helper plasmids using Hieff Trans™ Liposomal Transfection Reagent (Yeasen, Shanghai, China). After 72 h, the lentiviruses were collected and subsequently used to infect Huh7 and HepG2 cells. Forty-eight hours after infection, the cells were continuously cultured in a medium containing 2.5 μg/mL puromycin (Beyotime). The surviving cells were cultured into cell lines stably expressing shCtrl and shCTSKs.
5-Ethynyl-2ʹ-deoxyuridine (EdU) assay
To examine the effect of CTSK on HCC cell proliferation, EdU incorporation assays were performed using an EdU assay kit (RiboBio, Guangzhou, China). Briefly, cells were seeded in 96-well plates at a density of 1.2×104 cells/well and cultured for 24 h. Then, the cells were incubated with 50 μM EdU at 37°C for 2 h and washed with phosphate-buffered saline (PBS; 5 min twice). After fixation with 4% paraformaldehyde prepared in PBS for 30 min, cells were permeabilized with 0.1% Triton X-100 for 10 min. Next, the cells were washed twice with PBS for 5 min and treated with a 1×Apollo® reaction cocktail at room temperature for 30 min in the dark. Finally, nuclei were stained with 5 μg/mL Hoechst 33342 for 30 min and imaged under a fluorescence microscope (IX73; Olympus, Tokyo, Japan).
Cell counting Kit-8 (CCK-8) assay
Cell viability was assessed using a cell counting kit-8 assay (CCK-8, Dojindo, Japan). Briefly, cells were seeded in a 96-well plate at a density of 5000 cells/well and cultured for 24 h. At the designated time point, 10 μL of CCK-8 reagent was added to the cells. After incubation at 37°C for 4 h, a SynergyMx MultiMode Microplate reader was used to detect the absorbance at 450 nm. The cell viability was calculated based on the absorbance values.
Western blotting
For western blotting, cells were lysed in RIPA buffer supplemented with a protease inhibitor cocktail and centrifuged at 12,000×g at 4°C for 10 min; equal amounts of protein were subjected to 8% SDS-PAGE and then transferred onto a 0.45-micrometer pore size PVDF membrane (Millipore, Billerica, MA, USA). After blocking with 3% bovine serum albumin (BSA; Yeasen), the membrane was incubated overnight with the primary antibodies at 4°C and then with secondary antibodies for 1 h at room temperature. After washing with 1×TBST, ECL Plus western blotting Substrate (Thermo Fisher Scientific, Waltham, MA, USA) was used to detect the protein bands, and a chemiluminescence detection system (Tanon, Shanghai, China) was used for visualization. ImageJ 1.8.0 (NIH, Bethesda, MD, USA) was used to quantify band density. Relative protein levels were determined by normalizing the optical density values of the target protein with those of the loading control.
Immunoprecipitation
The treated cells were lysed using a Triton X-100-based lysis buffer (1% Triton X-100; 150 mM NaCl; 20 mM 4-(2-hydroxyethyl)-1- piperazineethanesulfonic acid (HEPES), pH 7.4; 2 mM ethylenediaminete-traaceticacid; 5 mM MgCl2) supplemented with the protease inhibitor cocktail and then centrifuged at 12000×g at 4°C. Next, a 1 mg/mL protein aliquot was prepared, and incubated with 20 μL magnetic beads at 4°C for 1 h to eliminate non-specificity. Next, the supernatant was mixed with appropriate amount of primary antibodies (FLAG, HIS, MYC, etc.) and incubated overnight at 4°C. After adding 40 μL magnetic beads and continuing to incubate at 4°C for 4 h, the magnetic beads were washed 5 times with pre-cooled 1×TBS; the proteins were recovered by boiling the beads in SDS sample buffer. They were then analyzed by western blotting.
Ubiquitination assay
HA-Ub, HIS-SIAH1, and corresponding plasmids such as HIS-SIAH1(C44S), FLAG-CTSK, and MYC-NEDD4 were co-expressed in HEK293T cells, a proteasome inhibitor (MG132) was added, and the cells were incubated for 6 h before harvesting. The collected cells were lysed with Triton X-100-based lysis buffer (1% Triton X-100; 150 mM NaCl; 20 m 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.4; 2 mM EDTA; 5 mM MgCl2) supplemented with protease inhibitors, followed by centrifugation at 12,000×g at 4°C. A 1 mg/mL protein solution was prepared, 20 μL of magnetic beads were added, and the tube was incubated at 4°C for 1 h to remove nonspecific binding. Next, the supernatant was mixed with an appropriate amount of primary antibody (HIS, etc.) and incubated overnight at 4°C. After adding 40 μL of magnetic beads, the application was continued for 4 h at 4°C, the magnetic beads were washed 5 times with pre-chilled 1×TBS, and the magnetic beads were boiled in SDS sample buffer to recover the protein, which was then analyzed using western blotting.
KEGG database analysis
The mRNA expression matrix and clinical information of 50 normal tissue samples and 374 HCC tissue samples was downloaded from The Cancer Genome Atlas (TCGA) database (http://portal.gdc.cancer.gov/). The 424 samples were divided into high and low expression groups according to CTSK expression levels. The grouped expression matrix was used as the dataset file, and CTSK high- and low-expression information was used as the group file. Next, the data were uploaded to the GSEA4.1.0 reference gene set c2.cp.kegg.v7.4. symbols.gmt (Curated) for KEGG enrichment analysis, and the number of permutations was set to 1000. After screening and filtering out signal pathways unrelated to the tumors, the output results were enriched and analyzed.
Yeast two-hybrid screening
The SIAH1 cDNA was cloned into the pGBK-T7 vector to generate a bait plasmid. Recombinant pGBK-T7-SIAH1 was transformed into yeast strain Y2HGold. Yeast strain Y187 carrying the human cDNA library plasmid was purchased from Clontech (Cat No. 630486). Y2HGold/pGBK-T7-SIAH1 recombinant yeast was mated with theY187 library and screened on SD/–Ade/–His/–Leu/–Trp/X-a-Gal/AbA agar plates. The blue colonies that grew on Ade/–His/–Leu/–Trp/X-a-Gal/AbA agar plates were subjected to plasmid extraction. The isolated plasmids were identified by sequencing with primers specific to the T7 promoter. Library insertion was determined via Basic Local Alignment Search Tool (BLAST) analysis of the sequencing results with the NCBI database.
Establishment of the mouse xenograft model
The animal studies have been conducted in accordance with the Institutional Animal Care and Use Committee of Xuzhou Medical University. Ten male nude mice (Vital River Laboratory Animal Technology, Beijing, China) with no significant difference in body weight (at 4 weeks of age) were reared in a sterile environment for 1 week. The mice were then randomly divided into two groups (n = 5/group). After disinfecting their skin, 100 μL of cell suspension (1 × 106 /mL) was injected into the dorsal side of the right hind limb. The mice were then returned to their cages and housed under the same conditions. Tumor length and width were measured every other day using Vernier calipers, and tumor volume was calculated as follows: length × (width2/2). After 6 weeks, all mice were euthanized, and their subcutaneous tumors were removed and then all tumors were isolated for protein extraction.
Statistical analysis
Data represent the results of cell-related experiments repeated at least three times, and all quantitative data are presented as means ± s.e.m. and were analyzed with the unpaired or paired two-tailed Student’s t test. In CCK-8 assay, the value of first day was labeled “1”. Statistical analysis was performed using SPSS (version 22.0; IBM Corp., Armonk, NY, USA). Statistical significance was set at P < 0.05.
Acknowledgments
This research was supported by the Jiangsu Provincial Medical Key Discipline [ZDXK202224], Young Science and Technology Innovation Team of Xuzhou Medical University Key Research and Development Plan of Jiangsu Province [TD202006], Jiangsu Provincial Commission of Health and Family Planning [M2020082], Xuzhou Institute of Technology [KC20128], National Natural Science Foundation Cultivation Project of Xuzhou Medical University Affiliated Hospital [2020Z001, 2020Z006], Outstanding Talents Fund project of Xuzhou Medical University [XYFY2021006], and Jiangsu Postgraduate Research Innovation Program [KYCX21_2670, KYCX22_2892, KYCX22_2907].
Author contributions
Chengming Zhang, Zhiyi Liu, Xiaotian Wang, Bin Zhang, Licheng Cui, Kuan Cao, Qinhe Hu, and Bin Hu conducted the experiments; Chengming Zhang, Zhiyi Liu, Hengliang Shi, and Renhao Wang designed the experiments. Chengming Zhang and Zhiyi Liu wrote the manuscript. Chengming Zhang, Hengliang Shi, and Renhao Wang revised the manuscript. All authors participated in the study design, data analysis, interpretation of the findings, and review of the manuscript and agreed to the publication of the final version.
Declaration of interests
The authors declare no competing interests.
Published: May 9, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106852.
Contributor Information
Hengliang Shi, Email: shl@xzhmu.edu.cn.
Renhao Wang, Email: wangrenhao@xzhmu.edu.cn.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Hartke J., Johnson M., Ghabril M. The diagnosis and treatment of hepatocellular carcinoma. Semin. Diagn. Pathol. 2017;34:153–159. doi: 10.1053/j.semdp.2016.12.011. [DOI] [PubMed] [Google Scholar]
- 2.Allemani C., Matsuda T., Di Carlo V., Harewood R., Matz M., Nikšić M., Bonaventure A., Valkov M., Johnson C.J., Estève J., et al. Global surveillance of trends in cancer survival 2000-14 (CONCORD-3): analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet. 2018;391:1023–1075. doi: 10.1016/S0140-6736(17)33326-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu J., Xue Y., Gao X., Zhou Q. Host cell factors stimulate HIV-1 transcription by antagonizing substrate-binding function of Siah1 ubiquitin ligase to stabilize transcription elongation factor ELL2. Nucleic Acids Res. 2020;48:7321–7332. doi: 10.1093/nar/gkaa461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim S., Cho H.Y., Kim D.G., Roh Y., Son S.Y., Mushtaq A.U., Kim M., Bhattarai D., Sivaraman A., Lee Y., et al. Targeting the interaction of AIMP2-DX2 with HSP70 suppresses cancer development. Nat. Chem. Biol. 2020;16:31–41. doi: 10.1038/s41589-019-0415-2. [DOI] [PubMed] [Google Scholar]
- 5.He H.T., Fokas E., You A., Engenhart-Cabillic R., An H.X. Siah1 proteins enhance radiosensitivity of human breast cancer cells. BMC Cancer. 2010;10:403. doi: 10.1186/1471-2407-10-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naishiro Y., Adachi M., Okuda H., Yawata A., Mitaka T., Takayama S., Reed J.C., Hinoda Y., Imai K. BAG-1 accelerates cell motility of human gastric cancer cells. Oncogene. 1999;18:3244–3251. doi: 10.1038/sj.onc.1202661. [DOI] [PubMed] [Google Scholar]
- 7.Shi H., Zheng B., Wu Y., Tang Y., Wang L., Gao Y., Gong H., Du J., Yu R. Ubiquitin ligase Siah1 promotes the migration and invasion of human glioma cells by regulating HIF-1alpha signaling under hypoxia. Oncol. Rep. 2015;33:1185–1190. doi: 10.3892/or.2014.3695. [DOI] [PubMed] [Google Scholar]
- 8.Leung C.O.N., Deng W., Ye T.M., Ngan H.Y.S., Tsao S.W., Cheung A.N.Y., Pang R.T.K., Yeung W.S.B. miR-135a leads to cervical cancer cell transformation through regulation of beta-catenin via a SIAH1-dependent ubiquitin proteosomal pathway. Carcinogenesis. 2014;35:1931–1940. doi: 10.1093/carcin/bgu032. [DOI] [PubMed] [Google Scholar]
- 9.Matsuo K., Satoh S., Okabe H., Nomura A., Maeda T., Yamaoka Y., Ikai I. SIAH1 inactivation correlates with tumor progression in hepatocellular carcinomas. Genes Chromosomes Cancer. 2003;36:283–291. doi: 10.1002/gcc.10170. [DOI] [PubMed] [Google Scholar]
- 10.Yoshibayashi H., Okabe H., Satoh S., Hida K., Kawashima K., Hamasu S., Nomura A., Hasegawa S., Ikai I., Sakai Y. SIAH1 causes growth arrest and apoptosis in hepatoma cells through beta-catenin degradation-dependent and -independent mechanisms. Oncol. Rep. 2007;17:549–556. [PubMed] [Google Scholar]
- 11.Zhao J., Wang C., Wang J., Yang X., Diao N., Li Q., Wang W., Xian L., Fang Z., Yu L. E3 ubiquitin ligase Siah-1 facilitates poly-ubiquitylation and proteasomal degradation of the hepatitis B viral X protein. FEBS Lett. 2011;585:2943–2950. doi: 10.1016/j.febslet.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 12.Zaidi M., Troen B., Moonga B.S., Abe E. Cathepsin K, osteoclastic resorption, and osteoporosis therapy. J. Bone Miner. Res. 2001;16:1747–1749. doi: 10.1359/jbmr.2001.16.10.1747. [DOI] [PubMed] [Google Scholar]
- 13.Fratzl-Zelman N., Valenta A., Roschger P., Nader A., Gelb B.D., Fratzl P., Klaushofer K. Decreased bone turnover and deterioration of bone structure in two cases of pycnodysostosis. J. Clin. Endocrinol. Metab. 2004;89:1538–1547. doi: 10.1210/jc.2003-031055. [DOI] [PubMed] [Google Scholar]
- 14.Motyckova G., Fisher D.E. Pycnodysostosis: role and regulation of cathepsin K in osteoclast function and human disease. Curr. Mol. Med. 2002;2:407–421. doi: 10.2174/1566524023362401. [DOI] [PubMed] [Google Scholar]
- 15.Szumilo J., Burdan F., Zinkiewicz K., Dudka J., Klepacz R., Dabrowski A., Korobowicz E. Expression of syndecan-1 and cathepsins D and K in advanced esophageal squamous cell carcinoma. Folia Histochem. Cytobiol. 2009;47:571–578. doi: 10.2478/v10042-008-0012-8. [DOI] [PubMed] [Google Scholar]
- 16.Pin F., Prideaux M., Huot J.R., Essex A.L., Plotkin L.I., Bonetto A., Bonewald L.F. Non-bone metastatic cancers promote osteocyte-induced bone destruction. Cancer Lett. 2021;520:80–90. doi: 10.1016/j.canlet.2021.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seo S.U., Woo S.M., Kim M.W., Lee H.S., Kim S.H., Kang S.C., Lee E.W., Min K.J., Kwon T.K. Cathepsin K inhibition-induced mitochondrial ROS enhances sensitivity of cancer cells to anti-cancer drugs through USP27x-mediated Bim protein stabilization. Redox Biol. 2020;30:101422. doi: 10.1016/j.redox.2019.101422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borel O., Gineyts E., Bertholon C., Garnero P. Cathepsin K preferentially solubilizes matured bone matrix. Calcif. Tissue Int. 2012;91:32–39. doi: 10.1007/s00223-012-9604-7. [DOI] [PubMed] [Google Scholar]
- 19.Novinec M., Lenarčič B. Cathepsin K: a unique collagenolytic cysteine peptidase. Biol. Chem. 2013;394:1163–1179. doi: 10.1515/hsz-2013-0134. [DOI] [PubMed] [Google Scholar]
- 20.Vashum Y., Khashim Z. Obesity and cathepsin K: a complex pathophysiological relationship in breast cancer metastases. Endocr., Metab. Immune Disord.: Drug Targets. 2020;20:1227–1231. doi: 10.2174/1871530320666200505115132. [DOI] [PubMed] [Google Scholar]
- 21.Fan X., Wang C., Song X., Liu H., Li X., Zhang Y. Elevated Cathepsin K potentiates metastasis of epithelial ovarian cancer. Histol. Histopathol. 2018;33:673–680. doi: 10.14670/hh-11-960. [DOI] [PubMed] [Google Scholar]
- 22.Li R., Zhou R., Wang H., Li W., Pan M., Yao X., Zhan W., Yang S., Xu L., Ding Y., Zhao L. Gut microbiota-stimulated cathepsin K secretion mediates TLR4-dependent M2 macrophage polarization and promotes tumor metastasis in colorectal cancer. Cell Death Differ. 2019;26:2447–2463. doi: 10.1038/s41418-019-0312-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zou X., Levy-Cohen G., Blank M. Molecular functions of NEDD4 E3 ubiquitin ligases in cancer. Biochim. Biophys. Acta. 2015;1856:91–106. doi: 10.1016/j.bbcan.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 24.Jing W., Wang G., Cui Z., Xiong G., Jiang X., Li Y., Li W., Han B., Chen S., Shi B. FGFR3 destabilizes PD-L1 via NEDD4 to control T-cell-mediated bladder cancer immune surveillance. Cancer Res. 2022;82:114–129. doi: 10.1158/0008-5472.CAN-21-2362. [DOI] [PubMed] [Google Scholar]
- 25.Yang Y., Luo M., Zhang K., Zhang J., Gao T., Connell D.O., Yao F., Mu C., Cai B., Shang Y., Chen W. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat. Commun. 2020;11:433. doi: 10.1038/s41467-020-14324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun A., Zhu J., Xia S., Li Y., Wu T., Shao G., Yang W., Lin Q. MEKK5 interacts with and negatively regulates the E3 ubiquitin ligase NEDD4 for mediating lung cancer cell migration. Life. 2021;11:1153. doi: 10.3390/life11111153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y., Goodfellow R., Li Y., Yang S., Winters C.J., Thiel K.W., Leslie K.K., Yang B. NEDD4 ubiquitin ligase is a putative oncogene in endometrial cancer that activates IGF-1R/PI3K/Akt signaling. Gynecol. Oncol. 2015;139:127–133. doi: 10.1016/j.ygyno.2015.07.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X., Trotman L.C., Koppie T., Alimonti A., Chen Z., Gao Z., Wang J., Erdjument-Bromage H., Tempst P., Cordon-Cardo C., et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128:129–139. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salah Z., Cohen S., Itzhaki E., Aqeilan R.I. NEDD4 E3 ligase inhibits the activity of the Hippo pathway by targeting LATS1 for degradation. Cell Cycle. 2013;12:3817–3823. doi: 10.4161/cc.26672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wan T., Lei Z., Tu B., Wang T., Wang J., Huang F. NEDD4 induces K48-linked degradative ubiquitination of hepatitis B virus X protein and inhibits HBV-associated HCC progression. Front. Oncol. 2021;11:625169. doi: 10.3389/fonc.2021.625169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.