

Summary
The catalytic conversion of lignin into N-containing chemicals is of great significance for the realization of value-added biorefinery concept. In this article, a one-pot strategy was designed for the transformation of lignin β-O-4 model compounds to imidazo[1,2-a]pyridines in yields up to 95% using 2-aminopyridine as a nitrogen source. This transformation involves highly coupled cleavage of C-O bonds, sp3C-H bond oxidative activation, and intramolecular dehydrative coupling reaction to construction of N-heterobicyclic ring. With this protocol, a wide range of functionalized imidazo[1,2-a]pyridines sharing the same structure skeleton as those commercial drug molecules, such as Zolimidine, Alpidem, Saripidem, etc., were synthesized from different lignin β-O-4 model compounds and one β-O-4 polymer, emphasizing the application feasibility of lignin derivatives in N-heterobicyclic pharmaceutical synthesis.
Subject areas: Organic chemistry, Applied chemistry
Graphical abstract

Highlights
-
•
The conversion of β-O-4 model compounds to imidazo[1,2-a]pyridines
-
•
Highly coupled cascade reaction
-
•
Drug skeletons
Organic chemistry; Applied chemistry
Introduction
With accelerating consumption of fossil resources and the increasing environmental concerns, lignocellulosic biomass as a renewable and abundant source of chemicals and fuels has recently attracted substantial attention.1,2,3,4,5 Lignin, a major component of lignocellulose and a complex plant-derived heteropolymer, is a unique precursor for aromatic compounds.6,7,8 However, most lignin in the paper and pulp industry has been discarded or burnt as a low-value fuel,9 which not only wastes renewable resources, but also poses different environmental concerns. Lignin valorization has therefore become a major topic in biomass conversion field and would play an important role in improving the economics of the overall biorefinery associated with addressing the problem of environmental impacts. So far, great efforts have been devoted to selective cleavage of C-O and C-C linkages to obtain phenols,2,10,11,12,13,14 arenes,15 cycloalkanes,16,17 aromatic aldehydes,18 ketones,19,20,21 acids,22 and benzoquinones.23 The above mentioned products are limited to C, H, O-containing compound.
Nitrogen-containing aryl compounds are typically value-added chemicals in the synthesis of biologically active pharmaceuticals and valuable materials.24,25,26,27 Recently, the conversion of lignin and lignin-derived compounds into N-containing products appears as a direction for lignin valorization.28,29,30 Li et al. described an efficient palladium-catalyzed formal cross-coupling of phenols with various amines and anilines to form aryl amines.31 Wang et al. converted lignin models and preoxidized lignin to isoxazole and aromatic nitrile.32 Maes et al. depicted a method that coverts lignin-derived 4-propylguaiacol to 3,4-dialkoxyanilines via a Beckmann rearrangement.33 Han et al. reported Pd-catalyzed coupling of aryl ethers and morpholines to 4-cyclohexylmorpholines.34 In our recent paper, we have accomplished the direct conversion of lignin β-O-4 dimers into benzylamines in the presence of organic amines overPd/C, and the potential of “lignin to benzylamines” was demonstrated by a two-step process.24 More recently, heterocyclic aromatic compounds, such as pyrimidines,26 functionalized quinolines,25 and quinoxalines27 have been synthesis from lignin β-O-4 segments via a one-pot multicomponent reaction. Of which, important marine alkaloid meridianin derivatives26 and an important drug compound AG129527 were obtained, showcasing good examples for the valorization of lignin to pharmaceutical molecules.
Imidazo[1,2-a]pyridine derivatives are essential ingredients for marketed medicines such as anti-ulcer Soraprazan and Zolimidine,35 vasodilator Olperinonr,36 anti-anxiety Alpidem and Saripidem,37 and sleeping Zolpidem (Scheme 1A). In addition, the heterobicyclic skeleton of imidazo[1,2-a]pyridine has high fluorescence activity, which can be used in the research of genetic fluorescent markers.38 Normally, The traditional synthesis methods of imidazo[1,2-a]pyridine derivatives are based on the construction of imidazole ring, which typically uses pyridines and olefins/alkynes as the starting materials via halogenation followed by in-situ generation of imidazole ring.39,40 In another route, imidazo[1,2-a]pyridines can also be constructed based on the formation of pyridine ring through halogenation of alkynes coupled with nucleophilic addition of imidazole41,42,43 (Scheme 1B). The above methods for the synthesis of imidazo[1,2-a]pyridine derivatives prevailingly rely on fossil-based feedstock and transition metal catalysts. Moreover, a great number of these progresses require multi-step synthetic routes, complicated ligands which suffer from high processing cost, low overall efficiency, and excessive waste emission. Hence, efficient strategy for the selective conversion of renewable feedstock such as lignin into such kind of products is highly desirable for cost-effective biorefinery conception and the sake of sustainable chemical industry.
Scheme 1.

Synthetic strategies to imidazo[1,2-a]pyridines
(A) Representative bioactive compounds derived from imidazo[1,2-a]pyridines.
(B) Current petroleum-based synthetic routes to imidazo[1,2-a]pyridines.
(C) The proposed renewable route in this work.
Ketones are important type of substrates for the construction of imidazole skeleton.44,45,46 Previous literature47,48,49 including our own19,50 have shown the possibility from lignin to aromatic ketones. It therefore provides us a potential choice using lignin β-O-4 segments as a renewable substrate for imidazo[1,2-a]pyridine production. Enlightened by the above cases, in this work, a one-pot two-step synthesis method of imidazo[1,2-a]pyridine derivatives from β-O-4 model compounds, by far the most frequent linkage in almost all types of lignin, using 2-aminopyridine as nitrogen source, commercial available Pd/C as catalyst and iodine as a promoter was developed (Scheme 1C). Such a strategy involves highly coupled cleavage of C-O bonds, dehydrative condensation, sp3C-H bond oxidative activation, and intramolecular dehydrative coupling reaction. Moreover, a β-O-4 polymer that mimicking natural lignin, was successfully converted to imidazo[1,2-a]pyridine derivative based on this catalytic route.
Results and discussion
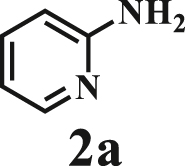
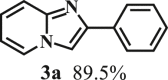
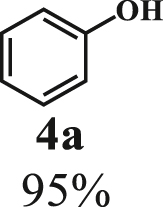
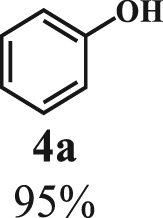
Our exploration started from the reaction of typical lignin β-O-4 model compound 2-(2-methoxyphenoxy)-1-phenylethanol 1a with 2-aminopyridine 2a to afford 2-phenylimidazo[1,2-a]pyridine 3a. This transformation is anticipated to have two major steps, namely cleavage of 1a to afford ketone intermediate acetophenone 5a, which then reacts with 2a to provide targeted product 3a. As Pd/C is a typical catalyst for C-O bond cleavage,49,51,52 it is used in this work for dissembling of 1a. According to Samec et al. research, addition of catalytic amount of hydrogen source could activate Pd/C catalyst through a low energy barrier pathway.47 Hence, 10 mol% NaBH4 (based on 1a) was employed as hydrogen source to activate Pd/C. Because water is a good solvent for NaBH4, it was selected as an integral solvent in the reaction. On the other hand, organic solvent is required to dissolve β-O-4 model compound. Therefore, a mixed solvent system containing water and organic solvent was adopted for this one-pot transformation. Our initial experiment was conducted without iodine source, it was found that no target product was generated (Table 1, entry 1), instead, C-O bond cleavage products 4a and 5a were obtained in excellent yields of 95.3% and 92.6%, respectively. In organic synthesis, molecular iodine has been used extensively in various reactions.53,54 It can act as a mild oxidizing reagent,55,56,57 and has mild Lewis acidic properties.58,59 Molecular iodine has the lowest homolytic dissociation energy among the nonradioactive halogens (151 kJ/mol). After astatine, iodine has the lowest electronegativity among the halogens. Therefore it is much easier to generate the monocationic iodine species (‘I+’) in situ than monocationic bromine or chlorine derivatives.60 Das et al.61 have reported that iodine source can promote the C-N bond formation between 2-aminobenzyl alcohol/2-aminobenzamide and different coupling partners (such as nitriles, aldehydes and ketones) to construct imidazole ring. Enlightened by this finding, we therefore added iodine in the reaction system, and obtained 40.0% of 3a yield (Table 1, entry 8). This encourages us to further screen different iodine sources. As shown in Table 1, this reaction is very sensitive to iodine species. In detail, iodine anions cannot promote the generation of 3a, the reactions in the presence of KI and CuI gave similar results to that without iodine source (Table 1, Entries 1–3), whereas it proceeded well in the presence of I2, KIO3, NaIO3, KIO4 and NaIO4promotors (Table 1, Entries 4–8), indicating that the promotors containing iodine cations could also catalyze the reaction, albeilt they showed lower activity than iodine (Table 1, entry 8 vs. entries 4–7), which might due that these promotors have weaker electrophilicity than I2. Therefore, I2 was employed as a promotor in the subsequent reactions. To further improve the yield of 3a, the solvent system was then screened. Under the same conditions, the solvent mixture containing polar-aprotic solvents such as CH2Cl2, CH3CN, tetrahydrofuran (THF) and ethyl acetate (EtOAc) gave 3a yield no more than 40% (Table 1, entries 8–12). Polar-protic solvent tert-amyl alcohol (t-AmOH) also provided 3a yield of 22%. Specifically, much higher 3a yield (60.5%), with 96.8% yield of phenol 4a (Table 1, entry 13) were obtained in toluene/water mixture (See Tables S1 and S2 for the screening of temperature and different solvents). It should be noted that the yield of 4a was always higher than 3a in all reactions, probably because 4a from depolymerization of lignin does not participate in downstream transformation, while the formation of 3a requires more steps from 1a. To sum up, the above experiments demonstrated the possibility for the conversion of lignin β-O-4 model compound into imidazo[1,2-a]pyridine derivatives. Yang62 and Kapoor63 disclosed that both amines and inorganic ammonium additives can promote the reaction.62 Accordingly, several organic amines and inorganic ammoniums were added in the reaction system with the aim to further improve the conversion efficiency. It is gratifyingly to note that most of these additives gave better conversion results except for ammonium chloride and aniline (Table 2). Amongst all these additives, ammonium bicarbonate (NH4HCO3) exhibited the greatest acceleration effect, providing 3a in yield of 71.6% (Table 2, entry 4). We speculate that ammonium bicarbonate additive can promote the cyclization reaction63 of acetophenone and aminopyridine in the presence of I2, by releasing NH3 to capture HI, a byproduct generated from the iodination of Cα in acetophenone, which will be further discussed in the following mechanism study. After optimizing the molar ratio of 1a and 2a (Tables S3 and S4), 3a yield up to 89.5% could be achieved (Table 2, entry 8).
Table 1.
Conversion of β-O-4 model compound 1a with 2-aminopyridine 2a at different conditions
 | |||||
|---|---|---|---|---|---|
| Entrya | Iodine Source | Solvent | 3a Yield (%) | 4a Yield (%) | 5a Yield (%) |
| 1 | – | EtOAc/H2O | 0.0 | 95.3 | 92.6 |
| 2 | KI | EtOAc/H2O | 0.0 | 94.8 | 85.1 |
| 3 | CuI | EtOAc/H2O | 0.0 | 95.7 | 89.7 |
| 4 | KIO3 | EtOAc/H2O | 31.5 | 96.1 | trace |
| 5 | NaIO3 | EtOAc/H2O | 23.4 | 94.7 | trace |
| 6 | KIO4 | EtOAc/H2O | 28.8 | 96.4 | trace |
| 7 | NaIO4 | EtoAc/H2O | 26.6 | 96.0 | trace |
| 8 | I2 | EtOAc/H2O | 40.0 | 95.3 | – |
| 9 | I2 | CH2Cl2/H2O | 2.3 | 45.6 | 14.3 |
| 10 | I2 | t-AmOH/H2O | 10.8 | 38.1 | 16.5 |
| 11 | I2 | CH3CN/H2O | 22.0 | 60.1 | 36.0 |
| 12 | I2 | THF/H2O | 33.6 | 65.0 | 38.5 |
| 13 | I2 | Toluene/H2O | 60.5 | 96.8 | – |
Reaction conditions: Step 1: 1a (0.2 mmol), 5 wt% Pd/C (5 mol%), NaBH4 (10 mol%) in water (0.5 mL), toluene (1.5 mL), 140°C, t = 2 h. Step 2: 2a (0.4 mmol), iodine source (0.35 mmol), 140°C, t = 15 h. Yield was determined by GC-FID using dodecane as an internal standard.
Table 2.
Screening of Amine additives
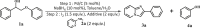 | |||
|---|---|---|---|
| Entrya | Additive | 3a Yield (%) | 4a Yield (%) |
| 1 | – | 60.5 | 96.8 |
| 2 | NH4Cl | 47.3 | 96.5 |
| 3 | NH4OAc | 62.5 | 95.9 |
| 4 | NH4HCO3 | 71.6 | 96.3 |
| 5 | 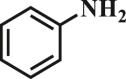 |
43.5 | 95.2 |
| 6 |  |
61.5 | 94.1 |
| 7 | 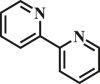 |
68.1 | 93.4 |
| 8b | NH4HCO3 | 89.5 | 96.8 |
Unless otherwise specified, the reaction conditions are: Step 1: 1a (0.2 mmol),5 wt% Pd/C (5 mol%), NaBH4 (10 mol%) in water (0.5 mL), toluene (1.5 mL), 140°C, t = 2 h. Step 2: 2a (0.4 mmol), I2 (0.35 mmol), additive (0.4 mmol), 140°C, t = 15 h. Yield was determined by GC-FID using dodecane as an internal standard.
1a (0.2 mmol), 2a (0.8 mmol), NH4HCO3 (0.4 mmol).
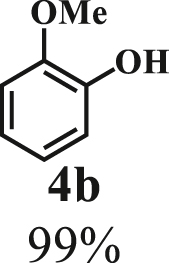
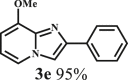
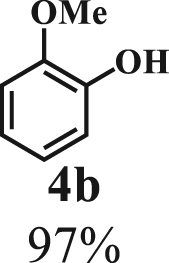
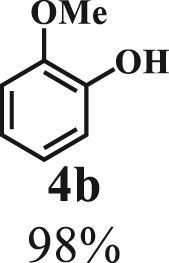
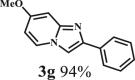
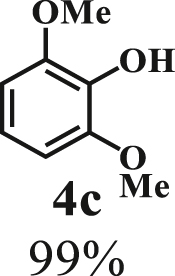
To examine the generality of this route, the activity of various lignin β-O-4 model compounds was explored. Substrates 1 with different functional groups on the aryl ring (Table 3) are found as fragments in different types of lignin.26 Under optimized reaction condition, all β-O-4 model compounds were completely consumed to afford the corresponding imidazo[1,2-a]pyridines and monophenols, whereas the yields and selectivity showed some diversity. In detail, when β-O-4 model compounds 1a-1g containing different methoxy groups reacted with 2a, moderate to good isolated yields (64–89.5%) of imidazo[1,2-a]pyridines 3a-3c, with excellent yields (95–99%) of phenol derivatives 4a-4c were achieved (Table 3, entries 1–7), indicating that the formation of imidazo[1,2-a]pyridine heterobicycles occurred associated with selective C-O bond breaking and C-N bond construction. Compared with the β-O-4 model compound bearing no functional group, methoxyl substitution on the O-terminal aryl ring had slight negative impact on the product yield (Table 3, entry 1 versus entries 4–5), whereas methoxyl group on the C-terminus aryl ring exhibited greater negative influence on the reaction efficiency (74–80%) (Table 3, entry1 vs. entries 6–7). It is also found that, the yields of heterocycle products were 89.5%, 86%, 74% and 64%, respectively, when the methoxyl groups of β-O-4 model compounds gradually increased from 0 to 4 (Table 3, entries 1, 4, 2, and 3). Overall, the more methoxyl groups on the aryl rings, the greater the negative impact on the reaction performance. Notwithstanding, this route leads to the successful cleavage of various β-O-4 model compounds and to the synthesis of imidazo[1,2-a]pyridines in a one-pot manner with fairly good yields. It is worth noting that all the prepared imidazo[1,2-a]pyridine molecules share the same structure skeleton as those commercial drug molecules (Scheme 1A), such as Zolimidine,35 Alpidem,61 Saripidem,37 etc. It therefore can be expected that themselves, or after proper modification,61 could have similar biological activity to above-mentioned drug molecules.
Table 3.
Scope of lignin β-O-4 model compounds and aminopyridine for the synthesis of imidazo[1,2-a]pyridines
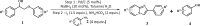 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entrya | Sub.1 | Sub.2 | 3 Yield (%) | 4 Yield (%) | Entry | Sub.1 | Sub.2 | 3 Yield (%) | 4 Yield (%) | |
| 1 | 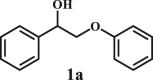 |
 |
 |
 |
8 | 1a |  |
 |
4a 96% |
|
| 2 | 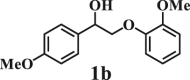 |
2a | 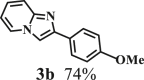 |
 |
9 | 1a | 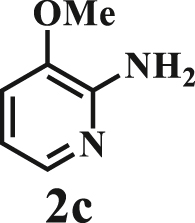 |
 |
4a 96% |
|
| 3 | 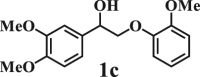 |
2a |  |
 |
10 | 1a |  |
 |
4a 97% |
|
| 4 |  |
2a | 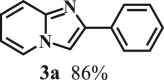 |
 |
11 | 1a | 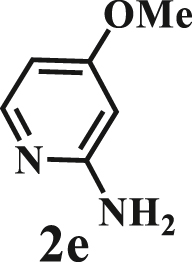 |
 |
4a 96% |
|
| 5 | 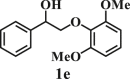 |
2a |  |
 |
12 | 1a |  |
 |
4a 95% |
|
| 6 | 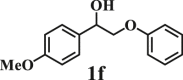 |
2a |  |
 |
13 | 1a | 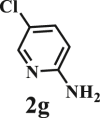 |
 |
4a 96% |
|
| 7 | 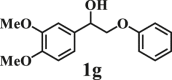 |
2a | 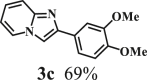 |
 |
14 | 1a |  |
 |
4a 95% |
|
Reaction conditions: for step 1: 1a (0.2 mmol), 5 wt% Pd/C (5 mol%), NaBH4 (10 mol%) in water (0.5 mL), toluene (1.5 mL), 140°C, t = 2 h. Forstep 2: 2a (0.8 mmol), I2 (0.35 mmol), NH4HCO3 (0.4 mmol), 140°C, t = 15 h. Yield refers to isolated yield.
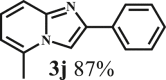
This reaction system has also proven to be effective for a broad range of aminopyridines. As shown in Table 3, seven aminopyridines have been successfully employed in the transformation, providing corresponding imidazo[1,2-a]pyridine products 3d-3j in high yields of 87%–95% (Table 3, entries 8–14). Aminopyridines possessing either electron-donating group (Table 3, entries 8–12,14) or electron-withdrawing group (Table 3, entry 13) on pyridine ring can react well with β-O-4 model compound to obtain excellent yield of imidazo[1,2-a]pyridines. Taking together, this one-pot catalytic system has proven to be effective not only for a variety of β-O-4 model compounds, but also for a broad range of aminopyridines. Lignin model compounds and aminopyridines with different functional groups are compatible to afford good to excellent yields of imidazo[1,2-a]pyridines, which thus offers an interesting opportunity for the valorization of lignin major segments to value-added N-heterobicyclic compounds.
To further determine the compatibility of the developed route, a β-O-4 polymer 1h (number-average molecular weight Mn = 23494, weight-average molecular weight Mw = 31680) that mimicking natural lignin was prepared and was then used as a substrate for the synthesis of imidazo[1,2-a]pyridine derivative. Under the same treatment condition as that in Table 3, 1h first underwent Pd/C-catalyzed C-O bond cleavage to afford p-hydroxyacetophenone 5b.
Subsequently, 5b reacted with 2-aminopyridine 2a in the presence of NH4HCO3/I2, and the corresponding target product 3k was obtained in 68% overall yield (isolated) based on β-O-4 polymer (Scheme 2). Product 3k is very similar in structure to Zolimidine,37 a commercially marketed anti-ulcer drug. Therefore, the above results further manifest the potential of this route in the synthesis of value-added N-heterobicyclic drug molecular from lignin β-O-4 polymer.
Scheme 2.

Conversion of lignin β-O-4 polymer to imidazo[1,2-a]pyridine derivative 3k
To understand the whole reaction scheme in depth, and to clarify the roles of different functional groups in β-O-4 aryl ethers, several comparative experiments were carried out to determine the possible reaction intermediates. For the first step, viz. the depolymerization of C-O bonds in β-O-4 model compounds, substrate 1i without substituted functional group in side chain cannot initiate any reaction over Pd/C in the presence of NaBH4 at 140°C, which is in sharp contrast to the reaction of 1a (Scheme 3, Reaction 1 versus Table 3, entry 1), implying that the hydroxyl group at Cα position should play a key role. Substrate 1j, in which the Cα position is blocked by a methyl group, also did not react under the same condition (Scheme 3 and Reaction 2), suggesting the hydrogen at Cα position is involved in the reaction. The same result was obtained on compound 1k (Scheme 3 and Reaction 3), which contains a methoxyl group in the Cα position, indicating that the proton of hydroxyl group at the Cα position also plays an important role in the reaction. The above three experiments show that both the proton of hydroxyl group and the hydrogen at Cα position are involved in C-O bond breaking. According to the above cases and literature,47 a plausible reaction pathway for the cleavage of C-O bonds of β-O-4 model compounds (taking 1a as an example) is proposed: the reaction begins with the dehydrogenation of 1a to produce intermediate 2-phenoxy-1-phenylethanone 6 and Pd/C-adsorbed hydrogen.47 This hydrogen participates in the subsequent cleavage of C-O bonds via an intramolecular hydrogenolysis step to generate another intermediate acetophenone 5a, with phenol 4a as a final product. The following time course experiment using 1a as the substrate gives direct evidence on the proposed pathway. As shown in Scheme 4A, at the very beginning of the reaction (10 min), the conversion of 1a only provided 6, and the yield of 6 increased fast at the initial stage but then quickly decreased after 20 min. In parallel, 5a and 4a appeared after 10 min and the corresponding yields increased very slow at the initial stage (from 0 to 20 min) with increasing of the reaction time. Of interest, after 20 min, with the consuming of 1a, the yield of 6 showed a decline trend whereas the yields of both 4a and 5a increased faster than before. Apparently, 2-phenoxy-1-phenylethanone 6 would be a plausible intermediate for the generation of 4a and 5a.
Scheme 3.

Control experiments
Scheme 4.

Time course reactions
(A) Time course profile of compound 1a conversion overPd/C.
(B) Time course profile of compound 5a conversion.
For the second step, to produce imidazo[1,2-a]pyridine compounds, we also carried out several comparative experiments to catch the possible reaction intermediates. First, treatment of acetophenone 5a alone under otherwise identical conditions yielded 68.2% of 2-iodo-1-phenylethanone 5c within 5 h (Scheme 3 and Reaction 4), indicating that the reaction of 5a with iodine to form 5c may be one step of the reaction. This assumption was confirmed by the reaction using 5c and 2a as the substrates, which afforded 95.7% of 3a, a slightly higher yield than that in Table 2, entry 8 using 1a as the substrate. According to the above facts, a plausible reaction pathway for the conversion of 5a to 3a is proposed: the reaction begins with the electrophilic substitution of α-H on acetophenone by I2 to produce intermediate 5c. This intermediate then reacts with 2-aminopyridine 2a via a cascade cyclization-dehydration step to generate 3a as a final product. Aforementioned experiments in Table 2 have demonstrated the acceleration effect of NH4HCO3 on the whole transformation. Two comparative experiments in Scheme 3 Reactions 4 and 6) further showed that NH4HCO3 additive can promote the formation of intermediate 5c, namely the electrophilic substitution of acetophenone by I2, because the reaction in Reaction 6 without NH4HCO3 gave lower yield of 5c (56.8%) than Reaction 4 (68.2%) under the same conditions. The time course profile of 5a conversion (Scheme 4B) showed that 5a was almost quantitatively converted to 3a in the end, and 5c yield appeared a volcanic trend with the increase of time, indicating it is an intermediate during the transformation. Noting that only a very low yield of 5c was observed, one can expect 5c is rapidly converted into 3a once it is generated. Therefore, 5c formation might be a rate-determining step in the reaction.
Based on all above results, the possible mechanism of the whole transformation is proposed in Scheme 5. The dehydrogenation of 1a overPd/C forms intermediate 6 and [Pd-H] species,47 then the in-situ generated [Pd-H] species participates the cleavage of the C-O bond of intermediate 6 to afford 5a and 4a. After adding I2 in the reaction mixture, intermediate 5a reacts with I2 via sp3C-H bond oxidative activation to form another intermediate 2-iodo-1-phenylethanone 5c and HI. In this step, NH4HCO3 can promote the reaction via neutralization of HI through releasing NH3. Once 5c is generated, a fast intermolecular nucleophilic substitution of 5c by 2-aminopyridine 2a occurs to obtain compound 7, which then releases intermediate 8 and HI via an intramolecular electron transfer. NH4HCO3further consumes HI and intermediate 8 undergoes an intramolecular cyclization to afford 9, which finally is dehydrated to targeted product 3a.
Scheme 5.

The plausible mechanism of the transformation of 1a to 3a
In conclusion, we have developed a route for the effective synthesis of imidazo[1,2-a]pyridines from lignin β-O-4 model compounds via a one-pot multicomponent transformation in the presence of pyridine, I2 and Pd/C. The developed method shows versatility in production of various imidazo[1,2-a]pyridine derivatives by varying β-O-4 model compounds and the substituted aminopyridines. The feasibility for the production of imidazo[1,2-a]pyridine derivatives from lignin-related polymer has also been demonstrated. A highly coupled cascade pathway, including cleavage of C-O bonds, sp3C-H bond oxidative activation and dehydration aromatization reaction has been established. The methodology described here makes it possible to build a bridge between renewable lignin and heterobicyclic aromatic compounds, thus providing a petroleum-independent choice for value-added fine chemicals and pharmaceutical molecules.
Limitations of the study
Unless otherwise specified, there are no limitations in this study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Pd/C | Cat. No. 75992 | Sigma-Aldrich |
| Iodine | CAS: 12190-71-5 | Mackun |
| 2-phenylimidazo[1,2-a]pyridine | CAS: 4249-72-3 | Mackun |
| NH4HCO3 | CAS: 1066-33-7 | Aladdin |
| NaBH4 | CAS: 1066-33-7 | Aladdin |
| 2-aminopyridine | CAS: 504-29-0 | Mackun |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Changzhi Li (licz@dicp.ac.cn).
Materials availability
This study did not generate new unique reagents.
All reagents were purchased from commercial sources (Mackun, Aladdin, Sigma-Aldrich) and used without purification unless otherwise mentioned. Lignin model compounds 1a-1g were synthesized according to the literature.19 2-Phenoxyphenylethane was extracted according to the literature.2 2-methoxy-1-phenoxy-1-phenylethane was extracted according to the literature.47 2,3 A lignin-related polymer was synthesized according to the literature.47 The products were purified by column chromatography over silica gel (200–400 size). Thin layer chromatography (TLC) analyses were conducted with glass-backed silica gel plates and visualized by UV light (254 nm). Flash column chromatography was performed using Qingdao Haiyang silica gel (200–300 mesh) with freshly distilled solvents. Gas chromatography (GC) analysis was performed on an Agilent 7890B gas chromatograph with a HP-5 MS column (quartz capillary column, 30 m × 0.25 mm × 0.25 μm). GC-MS analysis was carried out on a SHIMADZU GC-MS QP2020 NX with a HP-5 MS column (quartz capillary column, 30 m × 0.25 mm × 0.25 μm). NMR spectra were recorded on a Bruker 400 MHz or Bruker 700 MHz NMR spectrometer with TMS as the internal standard. Multiplicities are given as: s (singlet), d (doublet), t (triplet), q (quartet), brs (broad singlet), dd (doublets of doublet), dt (doublets of triplet), td (triplets of doublet) or m (multiplet).
Method details
Typical procedure for the Imidazo[1,2-a]pyridine synthesis from lignin β-O-4 model compound
Lignin model compound (0.2 mmol), Pd/C (22 mg, 0.01 mmol), magnetic stir bar, and Toluene (1.5 mL) were placed in the pressure tube (10 mL), next to the mixture freshly prepared NaBH4 water solution (0.5 mL, 0.092M) was added. The mixture was sealed and heated to 140°C (oil bath temperature) for 2h. After the reaction, the solution was cooled to room temperature, Aminopyridine (4 equiv.), I2 (89 mg, 0.35 mmol) and NH4HCO3(31.6 mg, 0.4 mmol) was added to the mixture. The mixture was sealed and heated to 140°C (oil bath temperature) for 15h. After the reaction, the solution was cooled to room temperature. The reaction mixture was quenched with NaS2O3 and extracted with ethyl acetate (3 × 15 mL) and dried over anhydrous Na2SO4. The organic phase was analyzed by GC to determine the yield of imidazo[1,2-a]pyridines. Then the solvent was evaporated under reduced pressure, and the crude products were purified by column chromatography using petroleum ether/ethyl acetate (8:1) to obtain the desired products.
Procedure for cleavage of a lignin-related polymer
A lignin-related polymer was synthesized according to the literature.47 Procedure for cleavage of the polymer: the lignin related polymer 1h (27.4 mg), Pd/C (22 mg), magnetic stir bar, and toluene (1.5 mL) were placed in the pressure tube (10 mL), then freshly prepared NaBH4 water solution (0.5 mL, 0.092 M) was added. The mixture was then heated at 140°C for 2 h. After cooling the reaction mixture to room temperature, 1 mL dodecane EtOAc solution (10.8 mg/mL), and 5 mL EtOAc were added in the recation. Gas chromatography was employed to determine the yield of 5b using dodecane as internal standard.
Procedure for conversion of lignin-related polymer to 3k
The lignin related polymer (27.4 mg), Pd/C (22 mg), magnetic stir bar, and toluene (1.5 mL) were placed in the pressure tube (10 mL), next to the mixture freshly prepared NaBH4 water solution (0.5 mL, 0.092M) was added. The mixture was then heated at 140°C for 2 h. After that, the solution was cooled to room temperature, 2a (75.2 mg), I2 (89 mg) and NH4HCO3(31.6 mg) was added in. The mixture was sealed and heated to 140°C for another 15 h. After reaction, the solution was cooled to room temperature. The reaction mixture was quenched with NaS2O3 and extracted with ethyl acetate (3 × 15 mL) and dried over anhydrous Na2SO4. Then the solvent was evaporated under reduced pressure, and the crude products were purified by column chromatography using petroleum ether/ethyl acetate to obtain the desired product 3k in isolated yield of 68%.
Characterization data for the products

2-phenylimidazo[1,2-a]pyridine (3a)
White solid; 1H NMR (CDCl3, 400 MHz) δ (ppm) 8.10 (d, 1H, J = 6.8 Hz), 7.95 (d, 2H, J = 7.6 Hz), 7.85 (s, 1H), 7.64 (d, 1H, J = 8.8 Hz), 7.43 (t, 2H, J = 7.6 Hz), 7.33 (t, 1H, J = 7.6 Hz), 7.17 (t, 1H, J = 8.0 Hz), 6.77 (t, 1H, J = 6.8 Hz). 13C NMR (CDCl3, 100 MHz) δ (ppm) 145.8, 145.5, 133.5, 129.9, 128.7, 128.4, 128.0, 126.0, 125.6, 124.8, 117.4, 112.5, 108.1.

2-(4-methoxyphenyl)imidazo[1,2-a]pyridine (3b)
Dark yellow solid; 1H NMR (CDCl3, 400 MHz) δ (ppm) 8.07 (m, 1H), 7.87 (dd, 2H, J1= 8.4 Hz, J2 = 1.2 Hz), 7.74 (d, 1H, J = 4.4 Hz), 7.60 (d, 1H, J = 9.2 Hz), 7.13 (m, 1H), 6.96 (dd, 1H, J1 = 8.4 Hz, J2 = 1.2 Hz), 6.74 (m, 1H), 3.84 (s, 3H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 159.5, 145.6, 145.5, 127.2, 126.3, 125.4, 124.5, 117.2, 114.1, 112.2, 107.2, 55.3.

2-(3,4-Dimethoxyphenyl)imidazo[1,2-a]pyridine (3c)
Yellow solid; 1H NMR (CDCl3, 400 MHz) δ (ppm) 7.84 (s, 1H), 7.74-7,72 (m, 1H), 7.61-7.54 (m, 1H), 7.48 (dd, J = 8.3, 1.6 Hz, 1H), 7.23 (t, J = 8.1 Hz, 2H), 6.96 (d, J = 8.3 Hz, 1H), 6.84 (t, J = 8.1 Hz, 1H), 4.03 (s, 3H), 3.95 (s, 3H); 3C NMR (CDCl3, 100 MHz) δ (ppm) 150.8, 149.0, 145.4, 144.2, 126.6, 125.0, 123.3, 118.6, 116.2, 112.5, 111.6, 110.0, 109.9, 56.0, 55.9.

8-methyl-2-phenylimidazo[1,2-a]pyridine (3d)
Yellow solid; 1H NMR (CDCl3, 400 MHz) δ (ppm) 7.97 (m, 3H), 7.82 (s, 1H), 7.44 (td, 2H, J1 = 7.6 Hz, J2 = 1.6 Hz), 7.32 (m, 1H), 6.93 (dt, 1H, J1 = 6.8 Hz, J2 = 0.8 Hz), 6.66 (t, 1H, J = 6.8 Hz), 2.67 (s, 3H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 146.2, 145.1, 134.0, 128.6, 127.7, 127.5, 126.1, 123.3, 123.2, 112.3, 108.5, 17.1.

8-Methoxy-2-phenylimidazo[1,2-a]pyridine (3e)
Yellow viscous liquid; 1H NMR (CDCl3, 400 MHz) δ (ppm) 4.01 (s, 3H), 6.41 (d, J = 7.5 Hz, 1H), 6.64 (t, J = 7 Hz,1H), 7.30 (t, J = 7 Hz, 1H), 7.40 (t, J = 8 Hz, 2H), 7.71 (d, J = 6.5 Hz,1H), 7.80 (s, 1H), 8.00 (d, J = 7.5 Hz, 2H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 54.7, 99.6, 108.1, 111.2, 117.4, 125.1, 126.7, 127.5, 132.5, 139.0, 143.8, 147.9.

7-methyl-2-phenylimidazo[1,2-a]pyridine (3f)
Light yellow solid; 1H NMR (CDCl3, 400 MHz) δ (ppm) 7.98 (d, 1H, J = 6.8 Hz), 7.94 (m, 2H), 7.77 (s, 1H), 7.42 (m, 3H), 7.32 (m, 1H), 6.60 (dd, 1H, J1 = 6.8 Hz, J2 = 1.2 Hz), 2.39 (s, 3H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 146.0, 145.3, 135.7, 133.7, 128.7, 127.8, 125.9, 124.7, 115.8, 115.1, 107.5, 21.4.

7-Methoxy-2-phenylimidazo[1,2-a]pyridine (3g)
White solid;.1H NMR (CDCl3, 400 MHz) δ (ppm) 7.93−7.88 (m, 3H), 7.67 (s, 1H), 7.42 (t, J = 7.8 Hz, 2H),7.30 (t, J = 7.4 Hz, 1H), 6.90 (d, J = 2.4 Hz, 1H), 6.48 (dd, J =7.4, 2.5 Hz, 1H), 3.85 (s, 3H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 157.9, 147.1, 145.5, 133.9, 128.6, 127.6, 125.9, 125.7, 107.4,106.8, 94.7, 55.4.

6-methyl-2-phenylimidazo[1,2-a]pyridine (3h)
White solid; 1H NMR (CDCl3, 400 MHz) δ (ppm) 7.93 (m, 2H), 7.85 (m, 1H), 7.74 (d, 1H, J = 0.4 Hz), 7.52 (d, 1H, J = 9.6 Hz), 7.42 (m, 2H), 7.30 (m, 1H), 7.00 (dd, 1H, J1 = 9.6 Hz, J2 = 2.0 Hz), 2.29 (s, 3H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 145.3, 144.6, 133.8, 128.6, 127.8, 127.7, 125.9, 123.3, 122.0, 116.7, 107.8, 18.0.

6-chloro-2-phenylimidazo[1,2-a]pyridine (3i)
White solid; 1H NMR (CDCl3, 400 MHz) δ (ppm) 8.12 (d, 1H, J = 1.2 Hz), 7.92 (m, 2H), 7.79 (s, 1H), 7.56 (d, 1H, J = 9.6 Hz), 7.43 (t, 2H, J = 7.6 Hz), 7.34 (m, 1H), 7.11 (dd, 1H, J1 = 9.6 Hz, J2 = 1.6 Hz). 13C NMR (CDCl3, 100 MHz) δ (ppm) 146.7, 144.0, 133.2, 128.7, 128.2, 126.0, 123.3, 120.5, 117.8, 108.4.

5-methyl-2-phenylimidazo[1,2-a]pyridine (3j)
Clear liquid; 1H NMR (CDCl3, 400 MHz) δ (ppm) 7.98 (d, J = 7.7 Hz, 2H), 7.73 (s, 1H), 7.54 (d, J = 9.1 Hz, 1H), 7.42 (t, J = 7.6 Hz, 2H), 7.34 – 7.29 (m, 1H), 7.13 (dd, J = 9.1, 6.8 Hz, 1H), 6.60 (d, J = 6.8 Hz, 1H), 2.60 (s, 3H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 145.7, 134.3, 133.9, 128.7, 127.9, 126.0, 124.9, 114.9, 111.7, 105.3, 18.8.

4-(imidazo[1,2-a]pyridin-2-yl)phenol (3k)
Brown solid; 1H NMR (d6-DMSO, 400 MHz) δ (ppm) 9.65 (s, 1H), 8.48 (d, 1H, J = 6.4Hz), 8.21 (s, 1H), 7.78 (d, 2H, J = 8.8 Hz), 7.53 (d, 1H, J = 8.8 Hz), 7.20 (m, 1H), 6.85 (m, 3H). 13C NMR (d6-DMSO, 100 MHz) δ (ppm) 157.4, 144.9, 144.6, 127.0, 126.7, 124.9, 124.5, 116.3, 115.5, 112.0, 107.6.
Acknowledgments
Support from the National Key R&D Program of China (2019YFC1905300, 2019YFC1905302), the National Natural Science Foundation of China (21878288, 21721004), the science and technology bureau of Dalian city (No. 2021RT04), and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB17020100) is gratefully acknowledged.
Author contributions
C.L. conceived the project. L.G. and Y.D. contributed to the experimental studies and analyzed the data. H.W., Y.L., Q.Q., Q.L., and F.S. synthesized some of the intermediates. C.L. supervised the research. C.L. and L.G. contributed to the original draft of the manuscript, which was revised by all authors.
Declaration of interests
There are no conflicts to declare.
Published: May 9, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106834.
Supplemental information
Data and code availability
-
•
This study does not generate new unique reagent.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper can be obtained from the lead contact upon request.
References
- 1.Zhou C.H., Xia X., Lin C.X., Tong D.S., Beltramini J. Catalytic conversion of lignocellulosic biomass to fine chemicals and fuels. Chem. Soc. Rev. 2011;40:5588–5617. doi: 10.1039/c1cs15124j. [DOI] [PubMed] [Google Scholar]
- 2.Li C., Zhao X., Wang A., Huber G.W., Zhang T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015;115:11559–11624. doi: 10.1021/acs.chemrev.5b00155. [DOI] [PubMed] [Google Scholar]
- 3.Zakzeski J., Bruijnincx P.C.A., Jongerius A.L., Weckhuysen B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010;110:3552–3599. doi: 10.1021/cr900354u. [DOI] [PubMed] [Google Scholar]
- 4.Zhou J.F.J., Xiao Y., Fung Kin Yuen V., Gözaydın G., Ma X., Panda S., Pham T.T., Yan N., Zhou K. An integrated process for l-tyrosine production from sugarcane bagasse. ACS Sustain. Chem. Eng. 2021;9:11758–11768. doi: 10.1021/acssuschemeng.1c03098. [DOI] [Google Scholar]
- 5.Yang See J., Song S., Xiao Y., Trang Pham T., Zhao Y., Lapkin A., Yan N. Transformation of corn lignin into sun cream ingredients. ChemSusChem. 2021;14:1586–1594. doi: 10.1002/cssc.202002739. [DOI] [PubMed] [Google Scholar]
- 6.Sun Z., Fridrich B., de Santi A., Elangovan S., Barta K. Bright side of lignin depolymerization: toward new platform chemicals. Chem. Rev. 2018;118:614–678. doi: 10.1021/acs.chemrev.7b00588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rinaldi R., Jastrzebski R., Clough M.T., Ralph J., Kennema M., Bruijnincx P.C.A., Weckhuysen B.M. Paving the way for lignin valorisation: recent advances in bioengineering, biorefining and catalysis. Angew. Chem. Int. Ed. Engl. 2016;55:8164–8215. doi: 10.1002/anie.201510351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van den Bosch S., Schutyser W., Vanholme R., Driessen T., Koelewijn S.F., Renders T., De Meester B., Huijgen W.J.J., Dehaen W., Courtin C.M., et al. Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps. Energy Environ. Sci. 2015;8:1748–1763. doi: 10.1039/c5ee00204d. [DOI] [Google Scholar]
- 9.Schutyser W., Renders T., Van den Bosch S., Koelewijn S.F., Beckham G.T., Sels B.F. Chemicals from lignin: an interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018;47:852–908. doi: 10.1039/c7cs00566k. [DOI] [PubMed] [Google Scholar]
- 10.Shuai L., Amiri M.T., Questell-Santiago Y.M., Héroguel F., Li Y., Kim H., Meilan R., Chapple C., Ralph J., Luterbacher J.S. Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization. Science. 2016;354:329–333. doi: 10.1126/science.aaf7810. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J., Teo J., Chen X., Asakura H., Tanaka T., Teramura K., Yan N. A series of NiM (M = Ru, Rh, and Pd) bimetallic catalysts for effective lignin hydrogenolysis in water. ACS Catal. 2014;4:1574–1583. doi: 10.1021/cs401199f. [DOI] [Google Scholar]
- 12.Wang B., Zhou P., Yan X., Li H., Wu H., Zhang Z. Cooperative catalysis of Co single atoms and nanoparticles enables selective CAr − OCH3 cleavage for sustainable production of lignin-based cyclohexanols. J. Energy Chem. 2023;79:535–549. doi: 10.1016/j.jechem.2022.12.020. [DOI] [Google Scholar]
- 13.Zhang B., Qi Z., Li X., Ji J., Luo W., Li C., Wang A., Zhang T. ReOx/AC-catalyzed cleavage of C–O bonds in lignin model compounds and alkaline lignins. ACS Sustain. Chem. Eng. 2019;7:208–215. doi: 10.1021/acssuschemeng.8b02929. [DOI] [Google Scholar]
- 14.Li X., Ding Y., Pan X., Xing Y., Zhang B., Liu X., Tan Y., Wang H., Li C. Scission of C–O and C–C linkages in lignin over RuRe alloy catalyst. J. Energy Chem. 2022;67:492–499. doi: 10.1016/j.jechem.2021.10.040. [DOI] [Google Scholar]
- 15.Shao Y., Xia Q., Dong L., Liu X., Han X., Parker S.F., Cheng Y., Daemen L.L., Ramirez-Cuesta A.J., Yang S., Wang Y. Selective production of arenes via direct lignin upgrading over a niobium-based catalyst. Nat. Commun. 2017;8:16104. doi: 10.1038/ncomms16104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X., Zhang B., Pan X., Ji J., Ren Y., Wang H., Ji N., Liu Q., Li C. One-pot conversion of lignin into naphthenes catalyzed by a heterogeneous rhenium oxide-modified iridium compound. ChemSusChem. 2020;13:4409–4419. doi: 10.1002/cssc.201903286. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X., Song M., Liu J., Zhang Q., Chen L., Ma L. Synthesis of high density and low freezing point jet fuels range cycloalkanes with cyclopentanone and lignin-derived vanillins. J. Energy Chem. 2023;79:22–30. doi: 10.1016/j.jechem.2022.12.017. [DOI] [Google Scholar]
- 18.Lv S., Liu H., Zhang J., Wu Q., Wang F. Water promoted photocatalytic transfer hydrogenation of furfural to furfural alcohol over ultralow loading metal supported on TiO2. J. Energy Chem. 2022;73:259–267. doi: 10.1016/j.jechem.2022.06.012. [DOI] [Google Scholar]
- 19.Liu Y., Li C., Miao W., Tang W., Xue D., Li C., Zhang B., Xiao J., Wang A., Zhang T., Wang C. Mild redox-neutral depolymerization of lignin with a binuclear Rh complex in water. ACS Catal. 2019;9:4441–4447. doi: 10.1021/acscatal.9b00669. [DOI] [Google Scholar]
- 20.Liu Y., Li C., Miao W., Tang W., Xue D., Xiao J., Zhang T., Wang C. Rhodium-terpyridine catalyzed redox-neutral depolymerization of lignin in water. Green Chem. 2020;22:33–38. doi: 10.1039/c9gc03057c. [DOI] [Google Scholar]
- 21.Rahimi A., Ulbrich A., Coon J.J., Stahl S.S. Formic-acid-induced depolymerization of oxidized lignin to aromatics. Nature. 2014;515:249–252. doi: 10.1038/nature13867. [DOI] [PubMed] [Google Scholar]
- 22.Chu D., Luo Z., Zhao C. Selective production of acetol or methyl lactate from cellulose over RuSn catalysts. J. Energy Chem. 2022;73:607–614. doi: 10.1016/j.jechem.2022.06.043. [DOI] [Google Scholar]
- 23.Cooper C.J., Alam S., Nziko V.d.P.N., Johnston R.C., Ivanov A.S., Mou Z., Turpin D.B., Rudie A.W., Elder T.J., Bozell J.J., Parks J.M. Co(salen)-Catalyzed oxidation of lignin models to form benzoquinones and benzaldehydes: a computational and experimental study. ACS Sustain. Chem. Eng. 2020;8:7225–7234. doi: 10.1021/acssuschemeng.0c01970. [DOI] [Google Scholar]
- 24.Zhang B., Guo T., Liu Y., Kühn F.E., Wang C., Zhao Z.K., Xiao J., Li C., Zhang T. Sustainable production of benzylamines from lignin. Angew. Chem. Int. Ed. Engl. 2021;60:20666–20671. doi: 10.1002/anie.202105973. [DOI] [PubMed] [Google Scholar]
- 25.Ding Y., Guo T., Li Z., Zhang B., Kühn F.E., Liu C., Zhang J., Xu D., Lei M., Zhang T., Li C. Transition-metal-Free synthesis of functionalized quinolines by direct conversion of beta-O-4 model compounds. Angew. Chem. Int. Ed. Engl. 2022;61:e202206284. doi: 10.1002/anie.202206284. [DOI] [PubMed] [Google Scholar]
- 26.Zhang B., Guo T., Li Z., Kühn F.E., Lei M., Zhao Z.K., Xiao J., Zhang J., Xu D., Zhang T., Li C. Transition-metal-free synthesis of pyrimidines from lignin beta-O-4 segments via a one-pot multi-component reaction. Nat. Commun. 2022;13:3365. doi: 10.1038/s41467-022-30815-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y., Luo Q., Qiang Q., Wang H., Ding Y., Wang C., Xiao J., Li C., Zhang T. Successive cleavage and reconstruction of lignin beta-O-4 models and polymer to access quinoxalines. ChemSusChem. 2022;15:e202201401. doi: 10.1002/cssc.202201401. [DOI] [PubMed] [Google Scholar]
- 28.Li H., Bunrit A., Li N., Wang F. Heteroatom-participated lignin cleavage to functionalized aromatics. Chem. Soc. Rev. 2020;49:3748–3763. doi: 10.1039/d0cs00078g. [DOI] [PubMed] [Google Scholar]
- 29.Upton B.M., Kasko A.M. Strategies for the conversion of lignin to high-value polymeric materials: review and perspective. Chem. Rev. 2016;116:2275–2306. doi: 10.1021/acs.chemrev.5b00345. [DOI] [PubMed] [Google Scholar]
- 30.Rong Y., Ji N., Yu Z., Diao X., Li H., Lei Y., Lu X., Fukuoka A. Lignin amination valorization: heterogeneous catalytic synthesis of aniline and benzylamine from lignin-derived chemicals. Green Chem. 2021;23:6761–6788. doi: 10.1039/d1gc02741g. [DOI] [Google Scholar]
- 31.Chen Z., Zeng H., Girard S.A., Wang F., Chen N., Li C.J. Formal direct cross-coupling of phenols with amines. Angew. Chem. Int. Ed. Engl. 2015;54:14487–14491. doi: 10.1002/anie.201506751. [DOI] [PubMed] [Google Scholar]
- 32.Li H., Wang M., Liu H., Luo N., Lu J., Zhang C., Wang F. NH2OH–Mediated lignin conversion to isoxazole and nitrile. ACS Sustain. Chem. Eng. 2018;6:3748–3753. doi: 10.1021/acssuschemeng.7b04114. [DOI] [Google Scholar]
- 33.Blondiaux E., Bomon J., Smoleń M., Kaval N., Lemière F., Sergeyev S., Diels L., Sels B., Maes B.U.W. Bio-based aromatic amines from lignin-derived monomers. ACS Sustain. Chem. Eng. 2019;7:6906–6916. doi: 10.1021/acssuschemeng.8b06467. [DOI] [Google Scholar]
- 34.Zheng B., Song J., Wu H., Han S., Zhai J., Zhang K., Wu W., Xu C., He M., Han B. Palladium-catalyzed synthesis of 4-cyclohexylmorpholines from reductive coupling of aryl ethers and lignin model compounds with morpholines. Green Chem. 2021;23:268–273. doi: 10.1039/d0gc03188g. [DOI] [Google Scholar]
- 35.Katritzky A.R., Xu Y.J., Tu H. Regiospecific synthesis of 3-substituted imidazo[1,2-a]pyridines, imidazo[1,2-a]pyrimidines, and imidazo[1,2-c]pyrimidine. J. Org. Chem. 2003;68:4935–4937. doi: 10.1021/jo026797p. [DOI] [PubMed] [Google Scholar]
- 36.Enguehard-Gueiffier C., Gueiffier A. Recent progress in the pharmacology of imidazo[1,2-a]pyridines. Mini Rev. Med. Chem. 2007;7:888–899. doi: 10.2174/138955707781662645. [DOI] [PubMed] [Google Scholar]
- 37.Hsu N., Jha S.K., Coleman T., Frank M.G. Paradoxical effects of the hypnotic Zolpidem in the neonatal ferret. Behav. Brain Res. 2009;201:233–236. doi: 10.1016/j.bbr.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 38.Srivastava S., Thakur N., Singh A., Shukla P., Maikhuri V.K., Garg N., Prasad A., Pandey R. Development of a fused imidazo[1,2-a]pyridine based fluorescent probe for Fe(3+) and Hg(2+) in aqueous media and HeLa cells. RSC Adv. 2019;9:29856–29863. doi: 10.1039/c9ra04743c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He C., Hao J., Xu H., Mo Y., Liu H., Han J., Lei A. Heteroaromatic imidazo[1,2-a]pyridines synthesis from C-H/N-H oxidative cross-coupling/cyclization. Chem. Commun. 2012;48:11073–11075. doi: 10.1039/c2cc35927h. [DOI] [PubMed] [Google Scholar]
- 40.Cao H., Liu X., Liao J., Huang J., Qiu H., Chen Q., Chen Y. Transition metal-mediated C horizontal lineO and C horizontal lineC bond-forming reactions: a regioselective strategy for the synthesis of imidazo[1,2-a]pyridines and imidazo[1,2-a]pyrazines. J. Org. Chem. 2014;79:11209–11214. doi: 10.1021/jo501671x. [DOI] [PubMed] [Google Scholar]
- 41.Zheng L., Hua R. Modular assembly of ring-fused and pi-extended phenanthroimidazoles via C-H activation and alkyne annulation. J. Org. Chem. 2014;79:3930–3936. doi: 10.1021/jo500401n. [DOI] [PubMed] [Google Scholar]
- 42.Knölker H., Hitzemann R., Boese R. Imidazole derivatives, III. Regiospecific Synthesis,Structure, and fluorescence properties of highly substituted imidazo[1,2-a ]pyridines and pyrido[1,2-a]benzimidazoles. Chem. Ber. 2006;123:327–339. doi: 10.1002/cber.19901230218. [DOI] [Google Scholar]
- 43.Peng J., Shang G., Chen C., Miao Z., Li B. Nucleophilic addition of benzimidazoles to alkynyl bromides/palladium-catalyzed intramolecular C-H vinylation: synthesis of benzo[4,5]imidazo[2,1-a]isoquinolines. J. Org. Chem. 2013;78:1242–1248. doi: 10.1021/jo302471z. [DOI] [PubMed] [Google Scholar]
- 44.Ujwaldev S.M., Rohit K.R., Harry N.A., Anilkumar G. Novel one step synthesis of imidazo[1,2-a]pyridines and Zolimidine via iron/iodine-catalyzed Ortoleva-King type protocol. Tetrahedron Lett. 2019;60:150950. doi: 10.1016/j.tetlet.2019.150950. [DOI] [Google Scholar]
- 45.Cai Q., Liu M.-C., Mao B.-M., Xie X., Jia F.-C., Zhu Y.-P., Wu A.-X. Direct one-pot synthesis of zolimidine pharmaceutical drug and imidazo[1,2-a]pyridine derivatives via I2/CuO-promoted tandem strategy. Chin. Chem. Lett. 2015;26:881–884. doi: 10.1016/j.cclet.2014.12.016. [DOI] [Google Scholar]
- 46.Wen Q., Lu P., Wang Y. Copper-mediated three-component synthesis of 3-cyanoimidazo[1,2-a]pyridines. Chem. Commun. 2015;51:15378–15381. doi: 10.1039/c5cc05821j. [DOI] [PubMed] [Google Scholar]
- 47.Galkin M.V., Dahlstrand C., Samec J.S.M. Mild and robust redox-neutral Pd/C-catalyzed lignol β-O-4′ bond cleavage through a low-energy-barrier pathway. ChemSusChem. 2015;8:2187–2192. doi: 10.1002/cssc.201500117. [DOI] [PubMed] [Google Scholar]
- 48.Galkin M.V., Sawadjoon S., Rohde V., Dawange M., Samec J.S.M. Mild heterogeneous palladium-catalyzed cleavage of β-O-4′-Ether linkages of lignin model compounds and native lignin in air. ChemCatChem. 2014;6:179–184. doi: 10.1002/cctc.201300540. [DOI] [Google Scholar]
- 49.Zhou X., Mitra J., Rauchfuss T.B. Lignol cleavage by Pd/C under mild conditions and without hydrogen: a role for benzylic C-H activation? ChemSusChem. 2014;7:1623–1626. doi: 10.1002/cssc.201301253. [DOI] [PubMed] [Google Scholar]
- 50.Guo H., Miles-Barrett D.M., Zhang B., Wang A., Zhang T., Westwood N.J., Li C. Is oxidation–reduction a real robust strategy for lignin conversion? A comparative study on lignin and model compounds. Green Chem. 2019;21:803–811. doi: 10.1039/C8GC02670J. [DOI] [Google Scholar]
- 51.Gao F., Webb J.D., Sorek H., Wemmer D.E., Hartwig J.F. Fragmentation of lignin samples with commercial Pd/C under ambient pressure of hydrogen. ACS Catal. 2016;6:7385–7392. doi: 10.1021/acscatal.6b02028. [DOI] [Google Scholar]
- 52.Zhang J., Zhou J., Xiao S., Wang Y. Hydrodeoxygenation of lignin-derived aromatic oxygenates over Pd-Fe bimetallic catalyst: a mechanistic study of direct C–O bond cleavage and direct ring hydrogenation. Catal. Lett. 2020;31:932–941. doi: 10.1007/s10562-020-03352-3. [DOI] [Google Scholar]
- 53.Parvatkar P.T., Parameswaran P.S., Tilve S.G. Recent developments in the synthesis of five- and six-membered heterocycles using molecular iodine. Chem. Eur J. 2012;18:5460–5489. doi: 10.1002/chem.201100324. [DOI] [PubMed] [Google Scholar]
- 54.Küpper F.C., Feiters M.C., Olofsson B., Kaiho T., Yanagida S., Zimmermann M.B., Carpenter L.J., Luther G.W., Iii, Lu Z., Jonsson M., Kloo L. Commemorating two centuries of iodine research: an interdisciplinary overview of current research. Angew. Chem. Int. Ed. Engl. 2011;50:11598–11620. doi: 10.1002/anie.201100028. [DOI] [PubMed] [Google Scholar]
- 55.Miller R.A., Hoerrner R.S. Iodine as a chemoselective reoxidant of TEMPO: application to the oxidation of alcohols to aldehydes and ketones. Org. Lett. 2003;5:285–287. doi: 10.1021/ol0272444. [DOI] [PubMed] [Google Scholar]
- 56.Mori N., Togo H. Facile oxidative conversion of alcohols to esters using molecular iodine. Tetrahedron. 2005;61:5915–5925. doi: 10.1016/j.tet.2005.03.097. [DOI] [Google Scholar]
- 57.Gogoi P., Konwar D. Transition-metal- and organic-solvent-free: a highly efficient anaerobic process for selective oxidation of alcohols to aldehydes and ketones in water. Org. Biomol. Chem. 2005;3:3473–3475. doi: 10.1039/B509527A. [DOI] [PubMed] [Google Scholar]
- 58.Deka N., Sarma J.C. Microwave-Mediated selective monotetrahydropyranylation of symmetrical diols catalyzed by iodine. J. Org. Chem. 2001;66:1947–1948. doi: 10.1021/jo000863a. [DOI] [PubMed] [Google Scholar]
- 59.Mukhopadhyay B., Kartha K.P.R., Russell D.A., Field R.A. Streamlined synthesis of per-O-acetylated sugars, glycosyl iodides, or thioglycosides from unprotected reducing Sugars1. J. Org. Chem. 2004;69:7758–7760. doi: 10.1021/jo048890e. [DOI] [PubMed] [Google Scholar]
- 60.Nachtsheim B., Finkbeiner P. Iodine in modern oxidation catalysis. Synthesis. 2013;45:979–999. doi: 10.1055/s-0032-1318330. [DOI] [Google Scholar]
- 61.Saha M., Das A.R. I2/TBHP promoted oxidative C–N bond formation at room temperature: divergent access of 2-substituted benzimidazoles involving ring distortion. Tetrahedron Lett. 2018;59:2520–2525. doi: 10.1016/j.tetlet.2018.05.028. [DOI] [Google Scholar]
- 62.Zhu X., Li X., Li X., Lv J., Sun K., Song X., Yang D. Decarboxylative C–H alkylation of heteroarenes by copper catalysis. Org. Chem. Front. 2021;8:3128–3136. doi: 10.1039/d1qo00210d. [DOI] [Google Scholar]
- 63.Kour D., Khajuria R., Kapoor K.K. Iodine–ammonium acetate promoted reaction between 2-aminopyridine and aryl methyl ketones: a novel approach towards the synthesis of 2-arylimidazo[1,2-a]pyridines. Tetrahedron Lett. 2016;57:4464–4467. doi: 10.1016/j.tetlet.2016.08.058. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This study does not generate new unique reagent.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper can be obtained from the lead contact upon request.