

Abstract
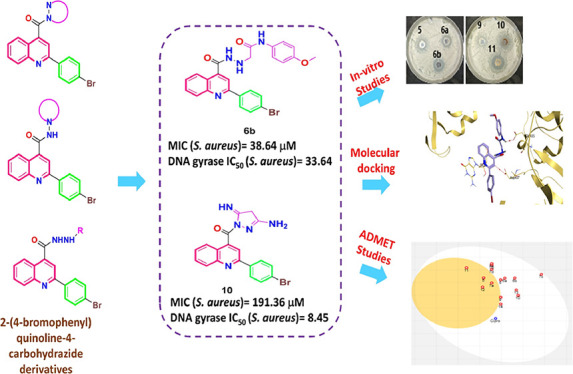
Microbial DNA gyrase is regarded as an outstanding microbial target. Hence, 15 new quinoline derivatives (5–14) were designed and synthesized. The antimicrobial activity of the afforded compounds was pursued via in vitro approaches. The investigated compounds displayed eligible MIC values, particularly against G-positive Staphylococcus aureus species. Consequently, an S. aureus DNA gyrase supercoiling assay was performed, using ciprofloxacin as a reference control. Obviously, compounds 6b and 10 unveiled IC50 values of 33.64 and 8.45 μM, respectively. Alongside, ciprofloxacin exhibited an IC50 value of 3.80 μM. Furthermore, a significant docking binding score was encountered by compound 6b (−7.73 kcal/mol), surpassing ciprofloxacin (−7.29 kcal/mol). Additionally, both compounds 6b and 10 revealed high GIT absorption without passing the blood brain barrier. Finally, the conducted structure−activity relationship study assured the usefulness of the hydrazine moiety as a molecular hybrid for activity either in cyclic or opened form.
1. Introduction
Bacterial infections are one of the main causes of the overwhelming global morbidity and mortality, even in hospitalized patients.1,2 By 2050, it is anticipated that 10 million people will die from these infectious diseases annually across the globe in the absence of improved medical interventions.3,4
The overwhelming surge of multidrug resistance exhibited by some microorganisms such as Staphylococcus aureus,5Escherichia coli,6S. epidermidis,7Pseudomonas aeruginosa,8Scedosporium apiospernum,9 and Enterococcus faecium(10) is regarded as a major threat to human health. It is still so challenging to get new antimicrobial lead compounds despite extensive studies being carried out to seek for structural modification of known antibacterial scaffolds over many years.11 Hence, the discovery of novel antibacterial agents with promising activity against both drug-sensitive and drug-resistant microbes is urgently required to combat bacterial infections, and these agents represent an attractive and appealing point of research in the medicinal chemistry field.12,13
On the other hand, bacterial DNA gyrase is a topoisomerase of type II, displaying a crucial and distinct role within bacteria. Gyrase is largely involved in chain elongation during chromosome replication and controlling DNA topological transitions.11,14 DNA gyrase inhibitors could hinder bacterial growth by two different mechanisms: either inhibiting the gyrase ATPase activity, leading to blocking of negative supercoil introduction in DNA (e.g., amino coumarin), or directly inhibiting DNA gyrase, called “gyrase poisoning” with a direct impact on cell physiology and division (e.g., ciprofloxacin).15 Consequently, DNA gyrase could be considered an attractive target for designing and affording new antimicrobial agents.
Quinolines are DNA gyrase inhibitors that have a crucial impact on bacterial DNA replication and recombination.16 The literature revealed that many of the reported quinoline derivatives were utilized as bacterial DNA gyrase inhibitors.16−24 For example, some phenylquinoline-oxadiazole derivatives were investigated for their DNA-gyrase inhibitory potential, with significant MIC values of 62, 10, and 35 ng/mL for compounds I, II, and III, respectively, against S. aureus, as depicted in Figure 1.16 Additionally, some (2-methylquinolin-4-yl)piperidine derivatives were assessed for their Mycobacterium tuberculosis DNA gyrase B inhibitory potential, with prominent MIC values of 3.81, 3.47, and 1.72 μM for compounds IV, V, and VI, respectively, as shown in Figure 1.17 Furthermore, some benzylidene hydrazineyl quinoline derivatives were tested for their DNA-gyrase inhibitory potential, with outstanding MIC values of 50, 12.5, and 12.5 μg/mL for compounds VII, VIII, and IX, respectively, against S. pneumonia, as shown in Figure 1.19
Figure 1.
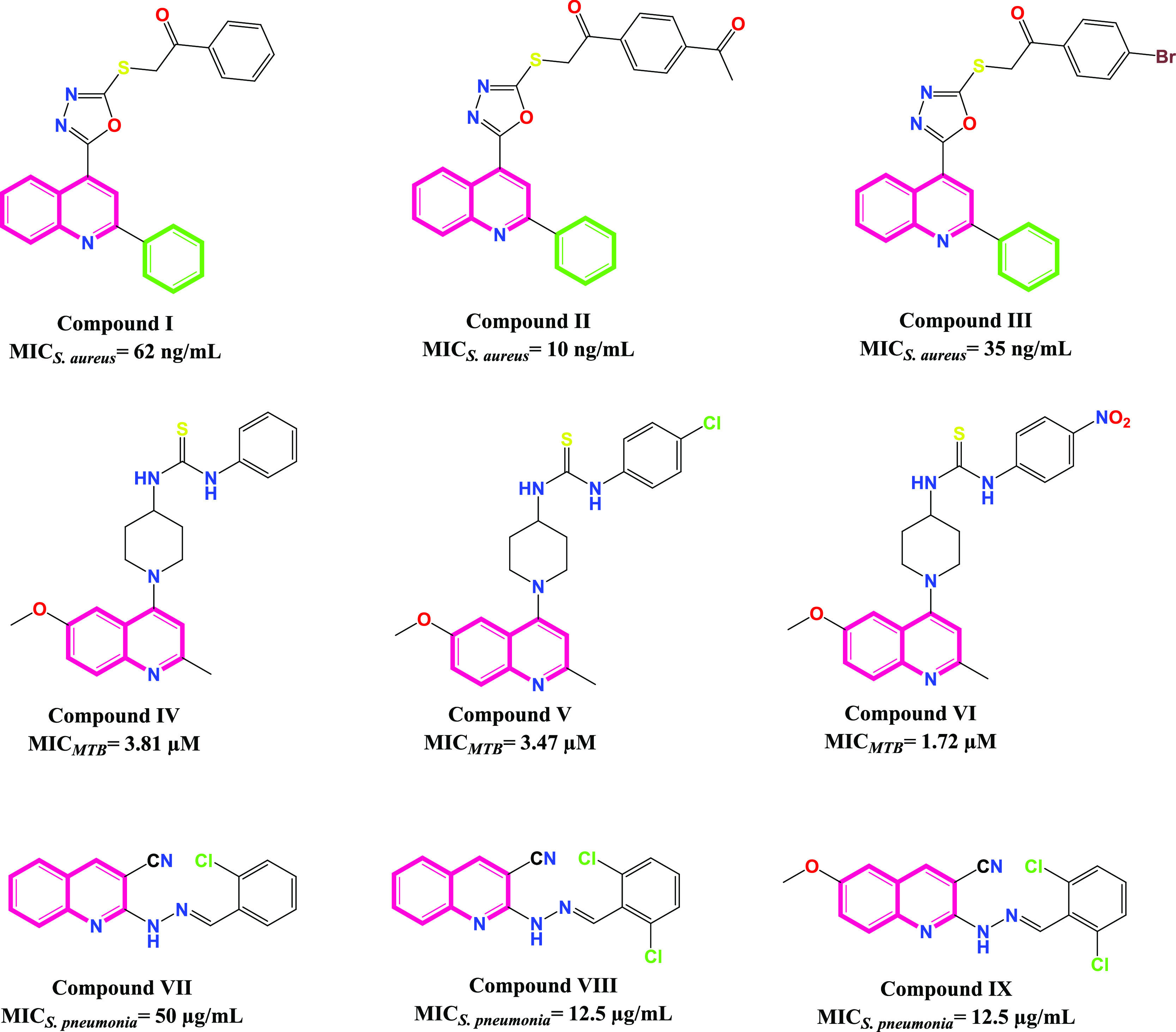
Some reported quinoline derivatives and their MIC values against different bacteria.
Compounds incorporating quinoline-based bioactive heterocycles such as pyrrole, pyrazoline, or benzopyrrole rings have drawn considerable attention with pronounced antimicrobial effect.25−27 These compounds were utilized as broad spectrum antibiotic drugs that work on both Gram-positive and Gram-negative pathogens as well as many fungal strains.28−30 The antimicrobial effect of these classes of compounds is mediated through different mechanisms. They have reported to act as inhibitors of DNA gyrase, topoisomerase, and as urease inhibitors.2,28,31
1.1. Rational of the Design
The molecular hybridization is a type of rational drug design strategy based on the recognition of two or more basic pharmacophoric groups and utilizing them for molecular hybrid formation to develop new chemical entities with enhanced potency and selectivity to the target than the parent lead compound.17 The literature revealed that phenyl quinoline derivatives could display significant antimicrobial potential as DNA gyrase inhibitors.16 In addition, the literature revealed as well that by using molecular hybridization of quinoline scaffolds with hydrazine, moieties could give new chemical hybrid entities with broad spectrum and promising antimicrobial activities,14 as depicted in Figure 2. Accordingly, in this current work, we aimed to design and synthesize novel 2-phenyl quinoline hydrazide derivatives as bacterial DNA gyrase inhibitors with estimating their antimicrobial potential via in silico and in vitro approaches. Diverse 2-phenyl quinoline hydrazide derivatives were designed to allow conducting SAR studies. This was approached by substituting one nitrogen of the hydrazine moiety with different substituents without including it in a ring system or by incorporating one or two nitrogen of the hydrazine moiety in a five-membered ring system, as depicted in Figure 3.
Figure 2.
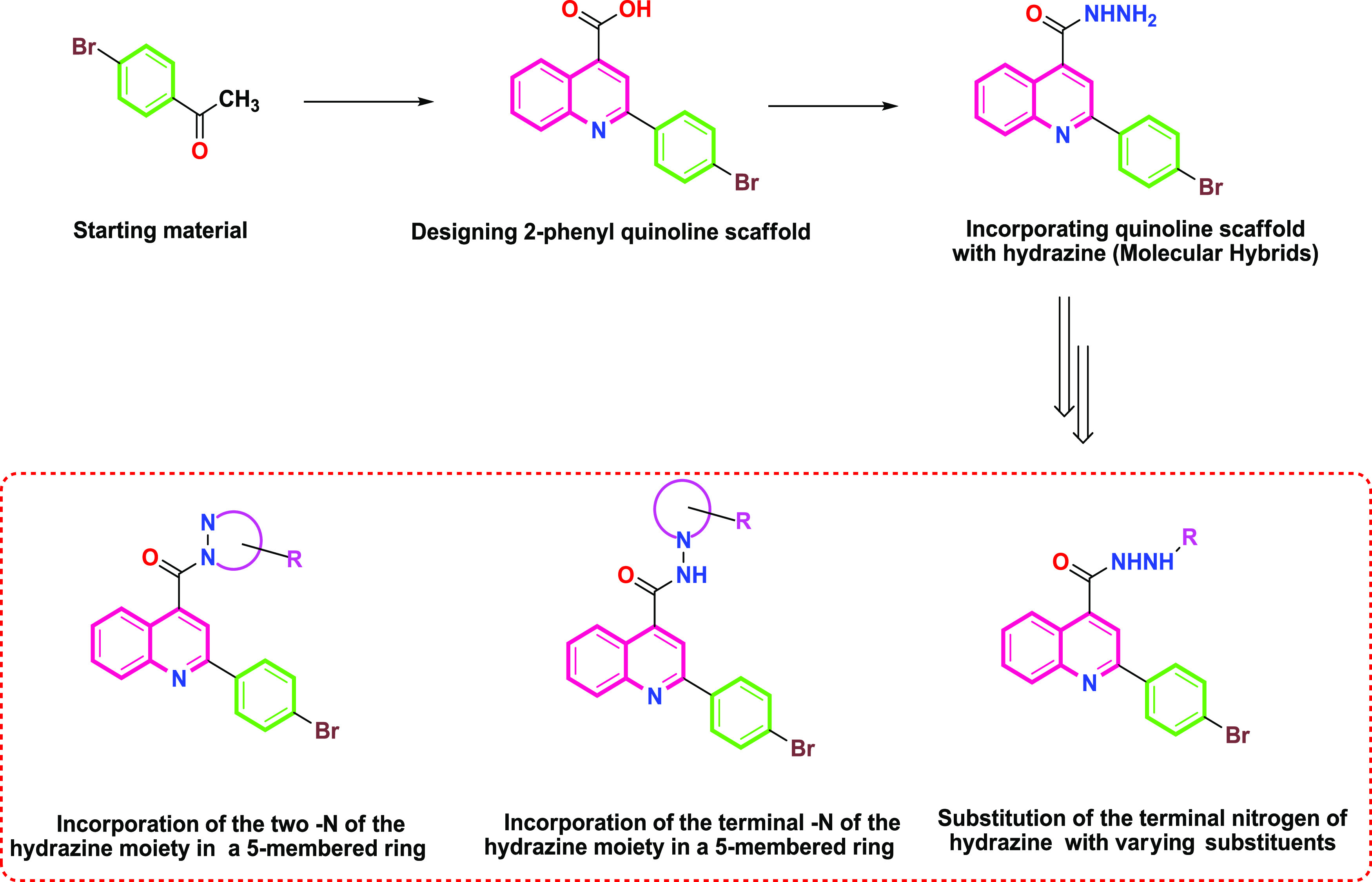
Rational drug design for the afforded compounds based on molecular hybridization.
Figure 3.
Designed compounds (5–14) and their basic pharmacophores.
2. Results and Discussion
2.1. Chemistry
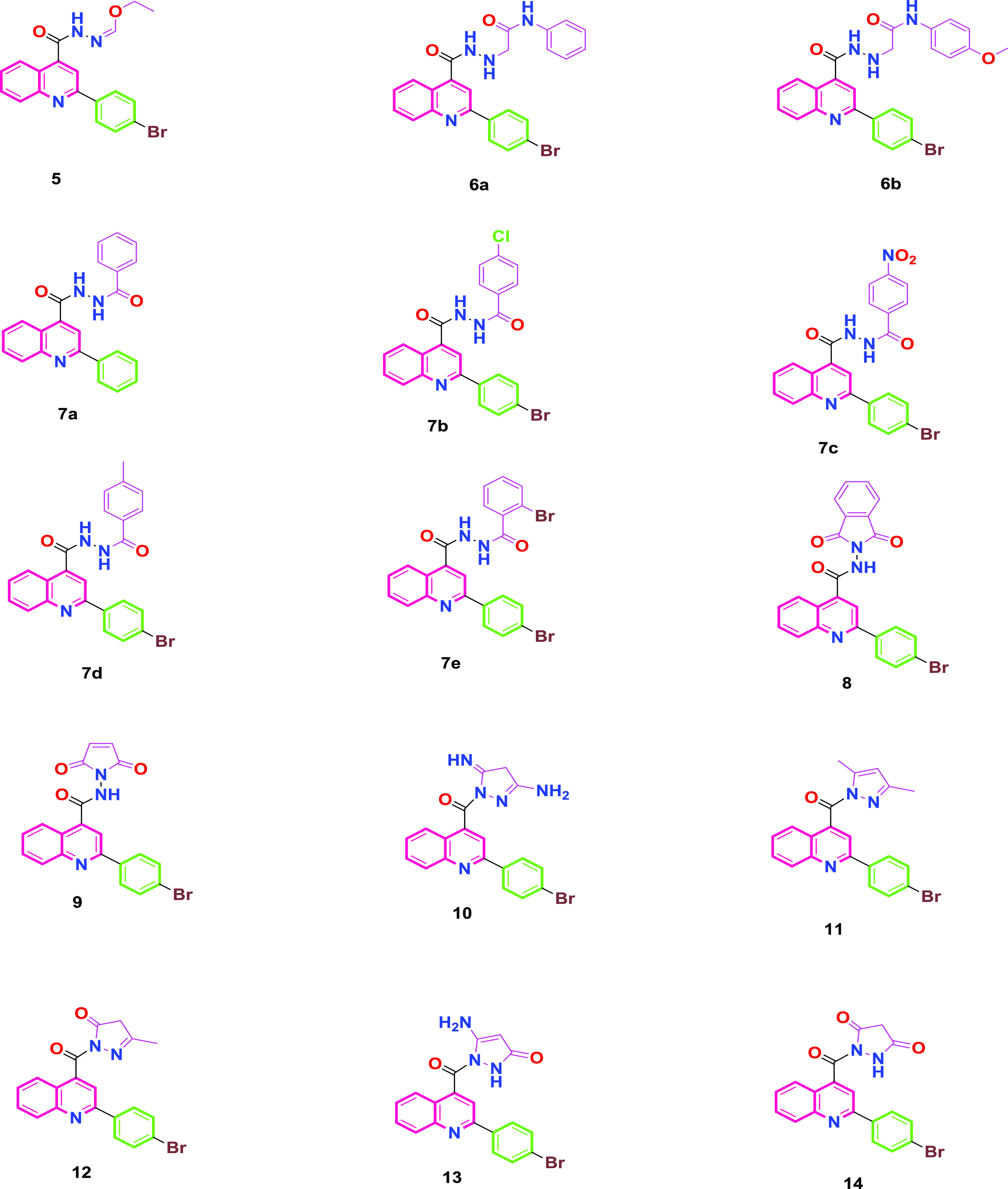
The synthetic route to the target compounds is described in Schemes 2 and 3, while Scheme 1 shows the routes adopted for synthesis of the starting materials. The structure of the synthesized compounds was confirmed based on their elemental analysis and spectral data (IR, 1H NMR, 13C NMR, and MS). 2-(4-Bromophenyl)quinoline-4-carboxylic acid (1) was prepared by reacting isatin with 4-bromoacetophenone in refluxing ethanol under basic conditions employing the Pfitzinger reaction protocol.32 The acid derivative 1 was subsequently heated in absolute ethanol containing catalytic amounts of concentrated sulfuric acid as a dehydrating agent to give the corresponding ester (2) as reported,33,34 which was then treated with hydrazine hydrate in boiling ethanol to afford the key intermediate 2-(4-bromophenyl)quinoline-4-carbohydrazide (3) as reported.34 The ethoxyformaldehyde hydrazone structure 5 was prepared via reaction of the acid hydrazide derivative 3 with triethyl orthoformate (Scheme 2). The structure of the new compound was confirmed by the IR spectra, which revealed the disappearance of the forked peak of the −NH2 group and the appearance of an absorption band at 3431 cm–1, referring to the −NH group. 1H NMR spectra showed a triplet signal at δ 1.40–1.43 ppm, referring to OCH2CH3, a quartet signal at δ 3.36–3.44 ppm corresponding to OCH2CH3, and a singlet signal at δ 9.60 ppm corresponding to the (N=CH) proton. Further support of the structure of compound 5 was obtained from the 13C NMR spectrum, which showed two signals corresponding to the ethoxy group at δ 14.87 and 62.09 ppm. On the other hand, reaction of acetohydrazide derivative 3 with 2-chloro-N-aryl-acetamides (4a,b), which were prepared according to reported procedures,35 gave 2-(2-(2-(4-bromophenyl)quinoline-4-carbonyl)hydrazinyl)-N-(substituted phenyl)acetamide (6a,b). The IR spectra of compounds 6a,b revealed the disappearance of the forked peak of NH2 and the appearance of an absorption band around 1647–1660 cm–1 pointing to two carbonyl groups. 1H NMR spectra showed a singlet signal at δ 4.18–4.27 ppm, corresponding to CH2 protons of the side chain, in addition to the presence of three singlets at δ 9.27–11.91 ppm, corresponding to three exchangeable NH protons. On the other hand, the 13C NMR spectra showed two signals corresponding to two carbonyl groups at δ 163.95–165.69 and 167.34–168.01 ppm and a signal corresponding to the carbons of CH2 at δ 57.34–57.72 ppm. Stirring benzoyl chloride derivatives with acid hydrazide 3 in dioxane afforded the corresponding N-acyl derivatives 7a–e. The 1H NMR spectra of compounds 7a-e showed two singlets at δ 10.70–11.92 ppm, corresponding to the two exchangeable NH protons. 13C NMR spectra of compounds 7a–e showed two signals corresponding to the two carbonyl groups at δ 161.24–167.05 ppm. In addition, reacting acid hydrazide 3 with phthalic anhydride and maleic anhydride in dioxane in the presence of glacial acetic acid afforded 2-(4-bromophenyl)-N-(1,3-dioxoisoindolin-2-yl)quinoline-4-carboxamide (8) and 2-(4-bromophenyl)-N-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)quinoline-4-carboxamide (9), respectively. The 1H NMR spectra of compounds 8 and 9 illustrated the disappearance of the NH2 signal of the parent acid hydrazide 3 and the presence of only one signal at δ 11.76 and 11.14 ppm, corresponding to one exchangeable NH proton of compounds 8 and 9, respectively. Compound 8 showed a multiplet signal at δ 8.00–8.09 ppm referring to four protons of the dioxoisoindolinyl moiety. The 13C NMR spectra of compounds 8 and 9 revealed the presence of two signals corresponding to two carbonyl groups at δ 165.30 and 166.66 ppm, respectively. Pyrazole derivative 10 was synthesized via the reaction of acid hydrazide 3 with malononitrile in DMF containing pyridine (Scheme 3). The structure of the synthesized compound was elucidated by the 1H NMR spectrum, which showed a singlet signal at δ 4.37 ppm corresponding to the −CH2 proton of the pyrazole moiety and two singlet signals of exchangeable NH2 and NH at δ 1.06 and 11.14 ppm, respectively. The 13C NMR spectra of compound 10 revealed three signals at δ 31.50, 160.58, and 163.28 ppm, referring to carbons of the pyrazole moiety. Cyclocondensation of acid hydrazide 3 with appropriate β-dicarbonyl compounds, namely, acetylacetone and ethyl acetoacetate, produced the corresponding pyrazolyl derivative (2-(4-bromophenyl)quinolin-4-yl)(3,5-dimethyl-1H-pyrazol-1-yl)methanone (11) and 2-(2-(4-bromophenyl)quinoline-4-carbonyl)-5-methyl-2,4-dihydro-3H-pyrazol-3-one (12), respectively. The structures of the titled compounds were deduced from IR spectra, which showed the absence of absorption bands of the NH2 and NH groups. The 1H NMR spectra of compound 11 were devoid of the presence of two singlet signals of the exchangeable protons of the NH and NH2 groups, besides the presence of two singlet signals at δ 1.75 and 2.03 ppm corresponding to the two methyl groups and a singlet signal at δ 6.91 ppm corresponding to the −CH proton of the pyrazole ring. Meanwhile, the 13C NMR spectrum showed two signals at δ 15.96 and 26.03 ppm corresponding to the two methyl groups and three signals at δ 115.51, 142.36, and 155.41 ppm relating to the carbons of the pyrazole ring. On the other hand, the 1H NMR spectra of compound 12 showed singlet signals at δ 2.00 ppm, corresponding to the methyl group, and a singlet signal δ at 3.43 ppm, corresponding to the −CH2 proton of the pyrazolone ring. The 13C NMR spectra of compound 12 revealed a signal at δ 17.19 ppm, corresponding to the methyl group, and two signals at δ 48.34 and 159.80 ppm, referring to the carbons of the pyrazolone ring, in addition to a signal at 166.49 ppm, corresponding to the carbonyl group of the pyrazolone moiety. 5-Amino-1-(2-(4-bromophenyl)quinoline-4-carbonyl)-1,2-dihydro-3H-pyrazol-3-one (13) was obtained in a good yield through the reaction of acid hydrazide 3 with ethyl cyanoacetate in glacial acetic acid. From the 1H NMR spectra of compound 13, the -CH proton signal of the pyrazolone moiety exhibited resonance at δ 3.76 ppm, and singlet signals of exchangeable NH2 and NH protons were observed at δ 3.56 and 10.09 ppm, respectively. The 13C NMR spectra of compound 13 showed two signals at δ 66.75 and 154.86 ppm referring to carbons of the pyrazolone ring, along with a signal at 162.47 corresponding to the carbonyl group of the pyrazolone moiety. Finally, the reaction of key intermediate 3 with diethyl malonate in basic medium afforded 1-(2-(4-bromophenyl)quinoline-4-carbonyl)pyrazolidine-3,5-dione (14) in a reasonable yield. The 1H NMR spectra of compound 14 showed the appearance of a singlet signal at δ 2.88 ppm pointing to the CH2 protons of the pyrazolidine moiety and a singlet signal at δ 12.50 ppm corresponding to the exchangeable NH proton. The 13C NMR spectra of compound 14 disclosed the appearance of a signal of the CH2 carbon of the pyrazolidine moiety at δ 54.93 ppm and two signals at δ 168.03 and 170.73 ppm referring to two carbonyl groups. Further, molecular ion peaks corresponding to the molecular weights of the synthesized compounds confirm the structure of these final compounds. The full spectral data for all afforded compounds (5–14) are depicted in the Supporting Information SI1.
Scheme 2. Synthesis of Target Compounds 5–9.

Scheme 3. Synthesis of Target Compounds 10–14.

Scheme 1. Synthesis of Starting Materials 1–4a,b.

2.2. Biological Evaluation
2.2.1. In Vitro Antimicrobial Activity
The newly synthesized quinoline derivatives 5–14 were preliminary screened for their antibacterial activity against S. aureus (ATCC 6538-P) representing Gram-positive bacteria and E. coli (ATTC 25933) representing Gram-negative bacteria using the agar well diffusion method. They were also evaluated for their antifungal potency against C. albicans (ATTC 10231) and A. niger-NRRL fungal strains. The antimicrobial activity results were recorded for each test molecule as the average diameter of the inhibition zones of microbial growth around the well in millimeters (mm). Regarding the antibacterial activity, as shown in Table 1, all the tested quinoline compounds possessed moderate-to-good antibacterial activity against Gram-positive bacteria (S. aureus). However, none of them was found to be effective against Gram-negative bacteria (E. coli). On the basis of zone of inhibition against the tested bacteria, compounds 6a, 6b, 10, 11, 13, and 14 were found to be the most effective against S. aureus, showing the maximum zone of inhibition in the range of 21–30 mm. Additionally, compounds 5 and 7d showed moderate antibacterial activity against S. aureus with zone of inhibition values of 18 and 16 mm, respectively.
Table 1. Antimicrobial Inhibition Zone in mm of Compounds 5–14, Neomycin, and Cyclohexamide.
| Gram +bacteria | Gram −bacteria | fungi |
||
|---|---|---|---|---|
| compd no | S. aureus (ATCC 6538-P) | E. coli (ATTC 25933) | C. albicans (ATTC 10231) | A. niger NRRL |
| 5 | 18 | 0 | 18 | 14 |
| 6a | 22 | 0 | 20 | 14 |
| 6b | 26 | 0 | 15 | 0 |
| 7a | 0 | 0 | 12 | 0 |
| 7b | 0 | 0 | 13 | 0 |
| 7c | 14 | 0 | 0 | 0 |
| 7d | 16 | 0 | 0 | 0 |
| 7e | 0 | 0 | 0 | 0 |
| 8 | 15 | 0 | 14 | 0 |
| 9 | 14 | 0 | 0 | 0 |
| 10 | 24 | 0 | 22 | 0 |
| 11 | 28 | 0 | 29 | 0 |
| 12 | 0 | 0 | 0 | 0 |
| 13 | 30 | 0 | 31 | 0 |
| 14 | 21 | 0 | 26 | 0 |
| neomycin | 26 | 24 | 30 | 0 |
| cyclohexamide | 0 | 0 | 0 | 21 |
With respect to the antifungal activity, it was noticed that most of the investigated molecules showed antifungal activity against C. albicans. However, the assessed molecules showed no apparent activity against A. niger, with the exception of compounds 5 and 6a, which showed equal antifungal activity against A. niger with a zone of inhibition value of 14 mm. Compounds 6a, 10, 11, 13, and 14 demonstrated the maximum inhibition zone against C. albicans, with inhibition zones between 20 and 31 mm. In addition, compound 5 showed moderate antifungal activity against C. albicans with a zone of inhibition value of 18 mm.
Accordingly, quinoline derivatives 11 and 13 displayed considerable antibacterial and antifungal activities (inhibition zones 28 and 30 mm against S. aureus as well as 29 and 31 mm against C. albicans, respectively). Hence, it was clear that compound 13 was marked as the most potent against both S. aureus and C. albicans. Additionally, compound 6b possessed higher antibacterial potency against S. aureus (inhibition zone value of 26 mm) compared to its potency against C. albicans (inhibition zone value of 15 mm).
2.2.2. Evaluation of Minimum Inhibitory Concentration (MIC)
The two-fold dilution method was used to assess the values of MIC for quinoline derivatives: 5, 6a, 6b, 10, 11, 13, and 14 against S. aureus and C. albicans. The values of MIC for the tested quinoline derivatives are listed in Table 2. Regarding the S. aureus bacterial strain, both quinoline derivatives linked to ethyl formohydrazonate (compound 5) and 4-(4-methoxyphenyl)acetamidohydazinyl (compound 6b) exhibited more effective and equal antibacterial activities against S. aureus. Hence, compounds 5 and 6b showed MIC values of 49.04 and 38.64 μM, respectively, against S. aureus. Compounds 6a, 10, and 11 exhibited decreased antibacterial activity against S. aureus with MIC values of 164.35, 191.36, and 192.29 μM, respectively. Compounds 13 and 14 showed weak antibacterial activities against S. aureus with MIC values of 381.81 and 761.77 μM, respectively. Finally, with respect to the tested C. albicans fungal strain, both compounds 5 and 6a were found to be the most active compounds against the tested fungal strain. Compounds 5 and 6a displayed MIC values of 24.53 and 41.09 μM, respectively. However, the quinoline compounds 6b and 10 demonstrated decreased antifungal activities against C. albicans strain with MIC values of 77.29 and 191.36 μM, respectively.
Table 2. Minimum Inhibition Concentration (MIC) for Quinoline Derivatives 5, 6a, 6b, 10, 11, 13, and 14 against Different Test Microbes.
| compd no | S. aureus (ATCC 6538-P) | C. albicans (ATTC 10231) |
|---|---|---|
| 5 | 49.04 | 24.53 |
| 6a | 164.35 | 41.09 |
| 6b | 38.64 | 77.29 |
| 10 | 191.36 | 191.36 |
| 11 | 192.29 | 769.17 |
| 13 | 381.81 | 381.81 |
| 14 | 761.77 | 1523.54 |
| neomycin | 78.125 | 156.25 |
2.2.3. Evaluation of Minimum Bactericidal Concentration (MBC)
Quinoline derivatives 5, 6a, 6b, 10, 11, 13, and 14 were subjected to the MBC assay against each S. aureus, E. coli, and C. albicans. The MBC was taken as the concentration of plant extract that do not exhibit any bacterial growth on the freshly inoculated agar plates. Obviously, the investigated quinoline derivatives exerted good MBC values against the tested microbial strains. According to the data recorded in Table 3, quinoline compounds 5, 6b, and 10 were found to be the most potent agents against the S. aureus bacterial strain, with MBC values of 196.17, 77.29, and 191.36 μM, respectively. Regarding the C. albicans fungal strain, the quinoline compounds 5, 6a, and 10 exhibited more effective antifungal activities. Hence, 5, 6a, and 10 showed MBC values of 98.08, 82.17, and 191.36 μM, respectively. It is worth mentioning that compound 10 showed bactericidal and fungicidal properties as it displayed equal MIC and MBC values of 191.36 μM against S. aureus and C. albicans.
Table 3. Minimum Bactericidal Concentration (MBC) for Quinoline Derivatives 5, 6a, 6b, 10, 11, 13, and 14 against Different Test Microbes.
| compd no | S. aureus (ATCC 6538-P) | C. albicans (ATTC 10231) |
|---|---|---|
| 5 | 196.17 | 98.08 |
| 6a | 657.41 | 82.17 |
| 6b | 77.29 | 309.18 |
| 10 | 191.36 | 191.36 |
| 11 | 384.59 | 1538.35 |
| 13 | 1527.22 | 763.61 |
| 14 | 3047.07 | 3047.07 |
| neomycin | 312.5 | 625 |
2.2.4. Evaluation of Minimum Biofilm Inhibitory Concentration (MBIC)
The biofilm inhibitory activity of quinolone derivatives 5, 6a, 6b, 10, 11, 13, and 14 against each of S. aureus and C. albicans was assessed. Crystal violet staining was employed to investigate the antibiofilm effect of the evaluated quinoline molecules using a 96-well microplate assay method. The results in this regard are illustrated in Table 4. The quinoline compounds 6b and 11, with MIC values of 154.59 and 192.29 μM, respectively, could inhibit the S. aureus biofilm. Meanwhile, compounds 6a, 10, and 11, with MIC values of 41.09, 95.67, and 96.14 μM, respectively, were the most active in inhibiting the biofilm formation of C. albicans fungal strain. Notably, the biofilm inhibitory activity of compound 6a against C. albicans fungal strains was promising (41.09 μM).
Table 4. MIC of Biofilm Inhibition Noted for Quinoline Derivatives 5, 6a, 6b, 10, 11, 13, and 14 against Different Test Microbes.
| compd no | S. aureus (ATCC 6538-P) | C. albicans (ATTC 10231) |
|---|---|---|
| 5 | 392.33 | 784.66 |
| 6a | 657.41 | 41.09 |
| 6b | 154.59 | 618.36 |
| 10 | 765.44 | 95.67 |
| 11 | 192.29 | 96.14 |
| 13 | 381.81 | 190.90 |
| 14 | 380.88 | 380.88 |
| neomycin | 625 | 625 |
S. aureus DNA Gyrase Supercoiling Assay
The afforded compounds with prominent average MIC values (compounds 6b and 10) were chosen for further pursuing their inhibitory potential against S. aureus DNA gyrase using ciprofloxacin as a reference drug.36 Obviously, compounds 6b and 10 unveiled concerning IC50 values of 33.64 and 8.45 μM, respectively. Alongside, ciprofloxacin exhibited an IC50 value of 3.80 μM, as illustrated in Figure 4. Hence, the suggested mode of action for the afforded compounds in this current work was emphasized.
Figure 4.

IC50 values of microbial DNA Gyrase supercoiling assay for the compounds 6b and 10 using ciprofloxacin (cipro) as a reference drug.
2.3. In Silico Studies
2.3.1. Molecular Docking
Using the co-crystallized inhibitor (ciprofloxacin) as a reference standard, molecular docking was conducted for the newly afforded antimicrobial candidates against the target protein of the S. aureus gyrase-DNA complex, opening our eyes to the binding modes of the afforded compounds at the S. aureus gyrase-DNA complex. Pre-screening validation was carried out to assure the MOE program’s accuracy. Thus, the validation was established by re-docking the native inhibitor (ciprofloxacin), and a reasonable low RMSD value of 0.94 Å was obtained, emphasizing MOE validity,37−40 as shown in Figure 5. The 2D superimposition of the re-docked co-crystallized ciprofloxacin and the native co-crystallized ciprofloxacin is depicted in Figure SI1. Accordingly, the co-crystallized inhibitor, ciprofloxacin, and the afforded compounds (5–14) underwent a molecular docking analysis. Table 5 shows the binding free energy values along with the binding interactions of the investigated compounds.
Figure 5.
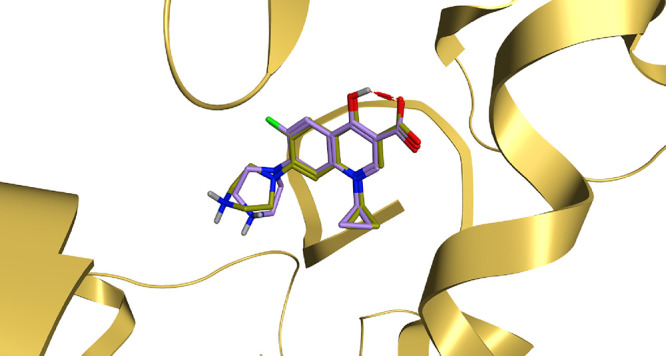
3D superimposition of the native co-crystallized ciprofloxacin (Olive green) and the docked co-crystallized ciprofloxacin (light violet) at S. aureus gyrase-DNA complex target protein.
Table 5. Binding Scores, Amino Acid Interactions, as Well as RMSD Values of the Afforded Compounds and the Co-crystallized Inhibitor (Ciprofloxacin) at S. aureus Gyrase-DNA Complex Target Protein.
| comp. no | S score (kcal/mol) | RMSD (Å) | interactions | distance (Å) |
|---|---|---|---|---|
| 5 | –6.52 | 2.06 | GLY1082/pi-H | 3.65 |
| 6a | –7.37 | 1.69 | ASP437/H-donor | 3.46 |
| DG9/H-donor | 3.39 | |||
| GLU435/H-donor | 3.15 | |||
| DG9/pi-pi | 4.00 | |||
| 6b | –7.73 | 1.67 | ASP437/H-donor | 3.46 |
| GLU435/H-donor | 3.11 | |||
| DG9/pi-pi | 3.90 | |||
| 7a | –6.81 | 1.25 | ASP437/H-donor | 3.45 |
| DG9/H-donor | 3.54 | |||
| DG9/pi-pi | 3.87 | |||
| 7b | –7.17 | 1.27 | GLU435/H-donor | 3.08 |
| GLU435/pi-H | 4.41 | |||
| DG9/pi-pi | 3.85 | |||
| 7c | –7.41 | 1.21 | ASP437/H-donor | 3.17 |
| GLU435/H-donor | 3.68 | |||
| DG9/pi-pi | 3.86 | |||
| 7d | –6.95 | 1.22 | DG9/pi-pi | 3.70 |
| 7e | –7.29 | 1.02 | ASP437/H-donor | 3.21 |
| ALA439/pi-H | 3.59 | |||
| DG9/pi-pi | 3.89 | |||
| 8 | –7.48 | 1.52 | DG9/H-donor | 3.35 |
| DG9/pi-pi | 3.78 | |||
| 9 | –6.80 | 1.68 | DG9/H-donor | 3.43 |
| ASP437/H-donor | 3.42 | |||
| GLY436/pi-H | 4.06 | |||
| DG9/pi-pi | 3.84 | |||
| 10 | –6.30 | 1.15 | SER1084/H-acceptor | 3.31 |
| 11 | –6.37 | 1.24 | ALA439/pi-H | 4.02 |
| 12 | –6.36 | 1.63 | ASP437/H-donor | 3.44 |
| 13 | –6.36 | 1.86 | ASP437/H-donor | 3.21 |
| DG9/pi-pi | 3.46 | |||
| DG9/pi-pi | 3.80 | |||
| 14 | –6.37 | 1.25 | DG9/H-donor | 3.01 |
| DT10/H-acceptor | 3.34 | |||
| DG9/pi-pi | 3.51 | |||
| DG9/pi-pi | 3.73 | |||
| cipro | –7.29 | 0.78 | GLU477/H-donor | 3.39 |
| SER1084/H-acceptor | 2.98 | |||
| SER1084/H-acceptor | 2.72 | |||
| ASP1083/H-acceptor | 2.96 | |||
| GLU477/ionic | 3.99 | |||
| DG9/pi-pi | 3.99 | |||
| DG9/pi-pi | 3.85 |
By interpreting the docking results attained, it was obviously revealed that the co-crystallized inhibitor (ciprofloxacin) could form multiple hydrogen bonds at the active site of the S. aureus gyrase-DNA complex target protein at an energy score of −7.29 kcal/mol with an RMSD value of 0.78 Å. The carboxylate moiety of ciprofloxacin could form one hydrogen bond with ASP1083 at a distance of 2.96 Å and two hydrogen bonds with SER1084 at distances of 2.72 and 2.98 Å. Additionally, the piperazine ring of ciprofloxacin could form one hydrogen bond as well as one ionic bond with GLU477 at a distance of 3.39 and 3.99 Å, respectively. Additionally, the quinolone nucleus could form two pi-pi interactions with the purine ring of DG9 at distances of 3.85 and 3.99 Å, as shown in Figure 6.
Figure 6.

3D protein positioning and binding interactions of ciprofloxacin at the active site of S. aureus gyrase-DNA complex target protein.
Moreover, regarding the prominent MIC values of compounds 6b and 10, it was revealed that compound 6b could bind at the active site of the S. aureus gyrase-DNA complex target protein with an energy score of −7.73 kcal/mol (surpassing the binding score of ciprofloxacin) with an RMSD value of 1.67 Å. The quinolone nucleus of compound 6b could form a hydrogen bond with ASP437 at a distance of 3.46 Å and a pi-pi interaction with the purine ring of DG9 at a distance of 3.90 Å. Additionally, the amide nitrogen of compound 6b could form a hydrogen bond with GLU435 at a distance of 3.11, as displayed in Figure 7.
Figure 7.

3D protein positioning and binding interactions of compound 6b at the active site of S. aureus gyrase-DNA complex target protein.
However, it was revealed that compound 10 could bind at the active site of the S. aureus gyrase-DNA complex target protein with an energy score of −6.30 kcal/mol and RMSD value of 1.15 Å. The imine group at position 5 of the pyrazole ring of compound 10 could form a hydrogen bond with SER1084 at a distance of 3.31 Å, as depicted in Figure 8. The 2D and 3D binding interactions for all the afforded compounds, along with their corresponding 3D protein positioning, are illustrated in Figure SI2.
Figure 8.

3D protein positioning and binding interactions of compound 10 at the active site of S. aureus gyrase-DNA complex target protein.
2.3.2. Physicochemical, ADME, and Pharmacokinetic Properties Prediction
The pharmacokinetic, physicochemical, and toxicity parameters prediction is a principal step after the synthesis of new molecular entities.41−43 Thus, the Swiss Institute of Bioinformatics’ (SIB) free Swiss ADME web application was employed to forecast the pharmacokinetic properties and ADME parameters of the newly afforded compounds as well as to estimate their physical and chemical properties. Accordingly, SMILES notations of the chemical structures of the synthesized compounds were put on the online server for more calculations.44 Additionally, the pkCSM descriptor algorithm protocol was utilized to forecast the toxicity parameters of the synthesized compounds.43
Hence, regarding their physicochemical features, except for compounds 7b, 7d, 7e, and 11, all the afforded compounds exhibited moderate water solubility. Hence, fewer problems may arise during pharmaceutical formulation since drugs have to be found in solution form at the absorption site to be easily absorbed.45 Additionally, considering their ADME properties, all the investigated compounds displayed high GIT absorption owing to their eligible lipophilicity. Hence, we could forecast reasonable bioavailabilities upon oral administration.46,47 Obviously, compounds 5, 7a, 11, and 12 can pass through the blood–brain barrier; consequently, these compounds could be employed for CNS microbial infections.48 Interestingly, all the synthesized compounds 6a, 6b, 6c, and 6e are not good substrates for P-glycoprotein (Pgp-); thus, they are not susceptible to the transporter efflux mechanism as shown in Figure 9. In addition, it is worth noting that all the afforded compounds could exhibit inhibition for all or most of the common hepatic metabolizing enzymes (CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4). Moreover, all of the synthesized derivatives may display feasible oral bioavailability according to Lipinski’s rule;49 hence, they could be incorporated in oral formulations. In addition, the bioavailability snapshot radars for the investigated compounds can be subjected to intuitive examination (Figure SI3). These unique snapshots for SwissADME (drug similarity graphs, which are expressed in a hexagonal shape with each vertex displaying a parameter that reflects a product’s bioavailability and the colored zone being the suitable physicochemical space for oral bioavailability) were illustrated in Figure SI3.
Figure 9.

Boiled-egg diagram for all the afforded compounds as well as ciprofloxacin as a reference control, where points located in the boiled-egg’s yolk are molecules anticipated to pass through the blood brain barrier, points located in the boiled-egg’s white are molecules anticipated to be passively absorbed from GIT, blue dots are molecules predicted to be effluated from CNS by P-glycoprotein, and red dots are molecules predicted not to be effluated from CNS by P-glycoprotein.
Furthermore, the toxicity parameters for the afforded compounds were anticipated using the pkCSM descriptor algorithm protocol. It was shown that, except for compounds 7a, 9, and 14, the assessed compounds could exhibit Ames toxicity; hence, they may be mutagenic.50 Additionally, all of the afforded compounds are fortunately non-inhibitors of hERG I, so they do not manifest a cardiotoxic effect on the human heart’s electrical activity.51 However, except for compounds 5, 9, 11, 12, and 14, the investigated compounds could be regarded as hERG II inhibitors and thus may provoke a cardiac arrhythmia threat.52 Notably, compound 11 is non-hepatotoxic. Last but not least, compounds 5, 7a, 7b, 7d, 7e, 8, and 11 could display eligible tolerability because of their smaller oral rat chronic toxicity values as depicted in Tables 6 and 7.
Table 6. Anticipated ADMET and Physicochemical Features of the Afforded Compounds (5–7e)a.
| investigated compounds |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| 5 | 6a | 6b | 7a | 7b | 7c | 7d | 7e | ||
| molecular properties | molar refractivity | 102.26 | 124.51 | 131.00 | 107.89 | 120.60 | 124.41 | 120.55 | 123.29 |
| TPSA (Az ) | 63.58 | 83.12 | 92.35 | 71.09 | 71.09 | 116.91 | 71.09 | 71.09 | |
| log P o/w (WLOGP) | 4.37 | 4.35 | 4.36 | 3.98 | 5.39 | 4.65 | 5.05 | 5.50 | |
| consensus log P o/w | 4.04 | 3.83 | 3.96 | 3.72 | 4.76 | 3.53 | 4.58 | 4.79 | |
| water solubility | MS | MS | MS | MS | PS | MS | PS | PS | |
| pharmacokinetics parameters | GI absorption | high | high | high | high | high | high | high | high |
| BBB permeant | yes | no | no | yes | no | no | no | no | |
| P-gp substrate | no | no | no | no | no | no | no | no | |
| CYP1A2 inhibitor | yes | yes | yes | yes | yes | yes | yes | yes | |
| CYP2C19 inhibitor | yes | yes | yes | yes | yes | yes | yes | yes | |
| CYP2C9 inhibitor | yes | yes | yes | yes | yes | yes | yes | yes | |
| CYP2D6 inhibitor | no | yes | yes | yes | yes | no | yes | yes | |
| CYP3A4 inhibitor | no | yes | yes | yes | yes | yes | yes | yes | |
| drug/lead likeness | drug likeness (Lipinski) | yes | yes | yes | yes | yes | yes | yes | no |
| lead likeness | no | no | no | no | no | no | no | no | |
| toxicity parameters | Ames toxicity | yes | yes | yes | no | yes | yes | yes | yes |
| max. tolerated dose (log mg/kg/day) | 0.423 | 0.799 | 0.753 | 0.825 | 0.604 | 0.356 | 0.602 | 0.6 | |
| hERG I inhibitor | no | no | no | no | no | no | no | no | |
| hERG II inhibitor | no | yes | yes | yes | yes | yes | yes | yes | |
| oral rat acute toxicity (LD50) (mol/kg) | 2.059 | 2.753 | 2.784 | 2.867 | 2.77 | 2.303 | 2.789 | 2.76 | |
| oral rat chronic toxicity (LOAEL) (log mg/kg_bw/day) | 1.104 | 2.305 | 2.166 | 1.149 | 1.14 | 1.779 | 1.093 | 1.127 | |
| hepatotoxicity | yes | yes | yes | yes | yes | yes | yes | yes | |
| minnow toxicity (log mM) | 0.85 | 0.293 | 0.658 | –2.506 | –0.841 | –0.305 | –0.623 | –1.581 | |
MS: moderately soluble, PS: poorly soluble.
Table 7. Anticipated ADMET and Physicochemical Features of the Afforded Compounds (8–14) as Well as Ciprofloxacina.
| investigated
compounds |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| 8 | 9 | 10 | 11 | 12 | 13 | 14 | ciprofloxacin | ||
| molecular properties | molar refractivity | 122.87 | 106.92 | 112.69 | 106.77 | 111.40 | 104.06 | 106.50 | 95.25 |
| TPSA (Az ) | 79.37 | 79.37 | 95.43 | 47.78 | 62.63 | 93.77 | 79.37 | 74.57 | |
| log P o/w (WLOGP) | 4.22 | 2.85 | 3.00 | 5.17 | 3.65 | 3.43 | 2.31 | 1.18 | |
| consensus log P o/w | 3.98 | 2.90 | 3.09 | 4.71 | 3.68 | 3.22 | 2.71 | 1.10 | |
| water solubility | MS | MS | MS | PS | MS | MS | MS | VS | |
| pharmacokinetics parameters | GI absorption | high | high | high | high | high | high | high | high |
| BBB permeant | no | no | no | yes | yes | no | no | no | |
| P-gp substrate | no | no | no | no | no | no | no | yes | |
| CYP1A2 inhibitor | yes | yes | yes | yes | yes | yes | yes | no | |
| CYP2C19 inhibitor | yes | yes | yes | yes | yes | no | yes | no | |
| CYP2C9 inhibitor | yes | yes | yes | yes | yes | yes | yes | no | |
| CYP2D6 inhibitor | no | no | no | no | no | no | no | no | |
| CYP3A4 inhibitor | no | no | no | no | no | no | no | no | |
| drug/lead likeness | drug likeness (Lipinski) | yes | yes | yes | yes | yes | yes | yes | yes |
| lead likeness | no | no | no | no | no | no | no | yes | |
| toxicity parameters | Ames toxicity | yes | no | yes | yes | yes | yes | no | no |
| max. tolerated dose (log mg/kg/day) | 0.021 | –0.212 | 0.166 | 0.075 | 0.072 | 0.567 | 0.206 | 0.924 | |
| hERG I inhibitor | no | no | no | no | no | no | no | no | |
| hERG II inhibitor | yes | no | yes | no | no | yes | no | no | |
| oral rat acute toxicity (LD50) (mol/kg) | 2.619 | 2.129 | 2.316 | 2.475 | 2.474 | 2.693 | 2.223 | 2.891 | |
| oral rat chronic toxicity (LOAEL) (log mg/kg_bw/day) | 0.853 | 1.627 | 2.774 | 1.077 | 1.267 | 1.612 | 1.719 | 1.036 | |
| hepatotoxicity | yes | yes | yes | no | yes | yes | yes | yes | |
| minnow toxicity (log mM) | –1.722 | –0.056 | 1.277 | 1.666 | 0.503 | 3.427 | 1.391 | 1.194 | |
MS: moderately soluble, PS: poorly soluble, VS: very soluble.
2.4. Structure–Antimicrobial Activity Relationship
A structure–antimicrobial activity relationship study was established to correlate the chemical structure of the diversely afforded compounds with their corresponding antimicrobial activity attained based on the zone of inhibition of these compounds utilizing S. aureus species. Interestingly, it was revealed that compound 13 displayed the best antimicrobial activity against S. aureus species by incorporating the hydrazine moiety in a five-membered ring with substitution of the ring with an amino group at position 5 and an oxo group at position 3 (1,2-dihydro-3H-pyrazol-3-one). Additionally, eligible antimicrobial activities were attained by incorporating the hydrazine moiety in a five-membered ring with substitution of the ring with a methyl group at positions 3 and 5 (compound 11), with an amino group at position 3 and an imino group at position 3 (compound 10), or with an oxo group at positions 3 and 5 (compound 14). However, the antimicrobial activity was obviously drained by incorporating the hydrazine moiety in a five-membered ring with substitution of the ring with a methyl group at position 3 and an oxo group at position 5 (compound 12), as shown in Figure 10. It is worth noting that eligible antimicrobial activities were attained as well without incorporating the hydrazine moiety in a cyclic form but rather by its substitution with the 4-methoxyphenyl acetamide moiety (compound 6b) or with the phenylacetamide moiety (compound 6a). However, lower antimicrobial activities were shown by incorporating the terminal nitrogen of the hydrazine moiety in a cyclic structure such as an isoindoline ring (compound 8) or pyrrole ring (compound 9) or by substituting the hydrazine moiety with ethyl formohydrazonate (compound 5) and 4-nitro benzoyl, 4-methyl benzoyl, or 2-bromo benzoyl derivatives (compounds 7c, 7d, and 7e, respectively). Notably, the antimicrobial activity was totally depleted by substituting the hydrazine moiety with benzoyl or 4-chloro benzoyl derivatives (compounds 7a and 7b, respectively), as shown in Figure 10.
Figure 10.
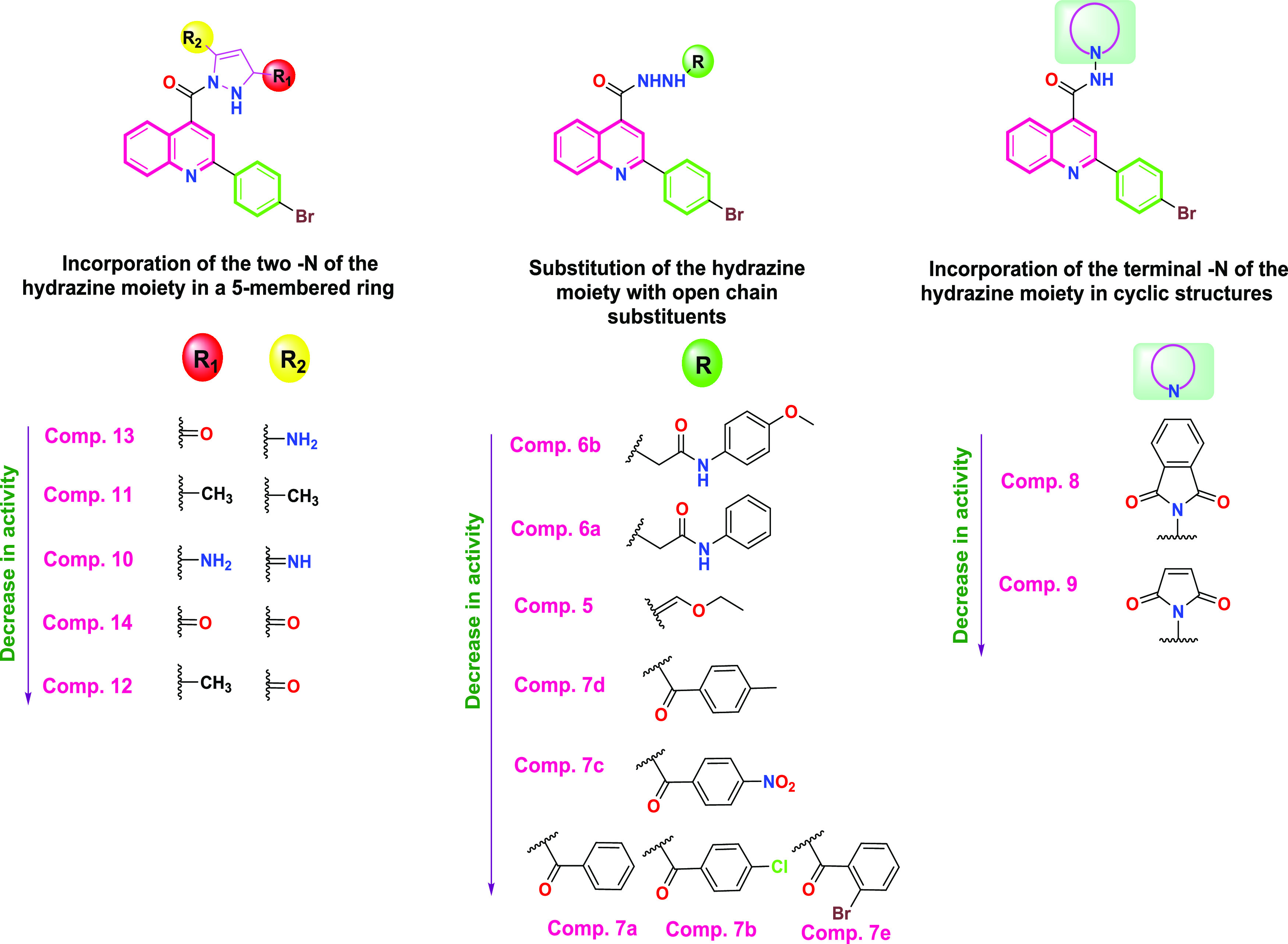
Structure–antimicrobial activity relationship of the afforded compounds (5–14) regarding S. aureus species.
3. Conclusions
Molecular hybridization is regarded as an important rational drug design strategy. Hence, 15 novel molecular hybrids of 2-phenyl quinoline hydrazine derivatives (5–14) were estimated as microbial DNA gyrase inhibitors against different microbial species using in vitro and in silico approaches. In particular, S. aureus species were clearly susceptible to most of the synthesized compounds. The most outstanding candidates with prominent average MIC values (6b and 10) were pursued for the S. aureus DNA supercoiling assay using ciprofloxacin as a reference control. The reasonable IC50 values exhibited by compounds 6b and 10 assure the proposed mechanism and the work perspective. Additionally, the conducted molecular docking studies assured the binding affinities of the afforded compounds to the microbial DNA gyrase, particularly compound 6b, which displayed a binding score exceeding that shown by ciprofloxacin. Furthermore, the established ADMET studies have put eyes on the eligible pharmacokinetics of the afforded compounds, particularly their oral bioavailability. The dedicated SAR elicited the usefulness of the hydrazine moiety for activity either in a cyclic or opened form and can pave the way for future structural modifications with anticipated activity.
4. Material and Methods
4.1. Chemistry
Solvents and reagents were obtained from Aldrich and were used without further purification unless otherwise indicated. Melting points were determined by the open capillary tube method using Stuart SMP-10 melting point apparatus and were uncorrected. The elemental analysis was carried out by Thermo Fisher Scientific FLASH 2000 CHNS/O analyzer, at The Regional Center for Mycology and Biotechnology, Al-Azhar University, Egypt. Infrared spectra were recorded on potassium bromide discs using a Bruker FT-IR spectrophotometer at MUST University and expressed in wave number υmax (cm–1). 1H NMR spectra were performed on a Bruker 400 MHz spectrophotometer using TMS as an internal standard, and the chemical shifts (δ) were recorded in ppm on the δ scale at Ain Shams University, Egypt. 13C NMR spectra were carried out using a Bruker 100 MHz instrument with TMS as an internal standard, and chemical shifts (δ) were recorded in ppm on the δ scale at Ain Shams University, Egypt. Mass spectra were run on a Hewlett Packard 5988 spectrometer or a Shimadzu QP-2010 Plus at The Regional Center for Mycology & Biotechnology, Al-Azhar University, Egypt. The progress of the reactions was monitored by TLC using pre-coated aluminum sheets silica gel (Merck 60 F254) with chloroform:methanol (9.5:0.5, v/v) as the eluting system and visualized by a UV lamp.
4.1.1. Procedure for Synthesis of 2-(4-Bromophenyl)quinoline-4-carboxylic Acid (1) as Reported32
A mixture of isatin (10 mmol, 1.47 g), 4-bromo acetophenone (10 mmol, 1.99 g), and 33% KOH (10 mL) in ethanol (10 mL) was heated under reflux for 12 h. The reaction mixture was left to cool and then acidified with HCl. The formed residue was washed with H2O, filtered, dried, and crystallized from ethanol to give compound 1.
Yellowish orange powder, yield 91%. mp 232–234 °C. IR (KBr, cm–1): 3446 (OH), 3178 (CH aromatic), 2980 (CH aliphatic), 1708 (C=O).
4.1.2. Procedure for Synthesis of Ethyl 2-(4-Bromophenyl)quinoline-4-carboxylate (2) as Reported33,34
2-(4-Bromophenyl)quinoline-4-carboxylic acid (1) (10 mmol, 3.28 g) in absolute ethanol (20 mL) containing 2 mL of conc H2SO4 was heated under reflux for 12 h. After cooling, the reaction mixture was rendered alkaline using an aqueous solution of NaHCO3. The formed residue was washed with H2O, filtered, dried, and crystallized from ethanol to give compound 2.
Buff crystals, yield 80%. mp 92–94 °C. IR (KBr, cm–1): 3138 (CH aromatic), 2993 (CH aliphatic), 1716 (C=O).
4.1.3. Procedure for Synthesis of 2-(4-Bromophenyl)quinoline-4-carbohydrazide (3) as Reported34
Ethyl 2-(4-bromophenyl)quinoline-4-carboxylate (2) (10 mmol, 3.56 g) was dissolved in absolute ethanol (20 mL), and 98% hydrazine hydrate (6 mL) was added. The reaction mixture was heated under reflux for 7 h. After cooling, the reaction mixture was poured into ice cooled water. The formed residue was, filtered, dried, and crystallized from ethanol to give the acid hydrazide 3.
White powder, yield 75%. mp 245–247 °C. IR (KBr, cm–1): 3263 (NH), 3305 (NH2), 3055 (CH aromatic), 2983 (CH aliphatic), 1645 (C=O).
4.1.4. Procedure for Synthesis of 2-Chloro-N-aryl-acetamides (4a,b) as Reported35
An excess amount of chloroacetyl chloride (15 mL) was added dropwise over 1 h to a solution of either aniline or 4-methoxyaniline (10 mmol) and sodium acetate (10 mmol, 0.82 g) in glacial acetic acid (10 mL). The reaction mixture was stirred at room temperature overnight. Then, it was poured onto ice-cold water. The solid obtained was filtered, washed with cold water, dried, and crystalized using 95% ethanol to obtain the desired products.
4.1.5. 2-Chloro-N-phenyl-acetamide (4a)
White powder, yield 90%. mp 135–137 °C. IR (KBr, cm–1): 3207 (NH), 3099 (CH aromatic), 2993 (CH aliphatic), 1674 (C=O).
4.1.6. 2-Chloro-N-(4-methoxy)phenyl-acetamide (4b)
Gray powder, yield 82%. mp 124–126 °C. IR (KBr, cm–1): 3294 (NH), 3072 (CH aromatic), 2956 (CH aliphatic), 1664 (C=O).
4.1.7. Procedure for Synthesis of Ethyl-N-(2-(4-bromophenyl)quinoline-4-carbonyl)formohydrazonate (5)
The acid hydrazide 3 (10 mmol, 3.41 g) and triethyl orthoformate (10 mL) was heated under reflux for 6 h, and then the reaction mixture was evaporated under reduced pressure. The obtained residue was collected, dried, and finally crystallized from ethanol to give compound 5.
White powder, yield 65% (2.6 g). mp 117–119 °C. IR (KBr, cm–1): 3431 (NH), 3089 (CH aromatic), 2980 (CH aliphatic), 1681 (C=O), 1587 (C=N). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.40–1.43 (t, 3H, CH2CH3), 3.36–3.44 (q, 2H, CH2CH3), 7.68–7.78 (m, 3H, Ar-H), 7.84 (t, 1H, Ar-H), 8.07–8.23 (m, 3H, Ar-H), 8.34–8.50 (m, 1H, Ar-H), 8.95 (d, 1H, J = 8.1 Hz. Ar-H), 9.60 (s, 1H, N=CH), 10.19 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 14.87, 62.09, 118.77, 122.52, 124.58, 125.55, 125.95, 129.01, 129.68 (2C), 130.38, 130.68, 132.04 (2C), 136.47, 141.22, 148.66, 155.83, 162. 60. MS m/z (%): 400.61 (M + 2, 13.90), 398.61 (M+, 14.66), 175.25 (100.00).). Anal. calcd. for C19H16BrN3O2 (398.26): C, 57.30; H, 4.05; N 10.55; Found: C, 57.54; H, 4.27; N, 10.78.
4.1.8. General Procedure for Synthesis of 2-(2-(2-(4-Bromophenyl)quinoline-4-carbonyl)hydrazinyl)-N-aryl Acetamide (6a,b)
To a suspension of acid hydrazide 3 (10 mmol, 3.41 g) in acetone (30 mL) containing anhydrous potassium carbonate (40 mmol, 5.52 g), the corresponding acetamide derivative (4a,b) (10 mmol) was added, and the reaction mixture was heated under reflux for 24 h and treated with water (10 mL), the solid obtained was filtered, dried, and crystallized from ethanol to afford compounds 6a,b.
4.1.8.1. 2-(2-(2-(4-Bromophenyl)quinoline-4-carbonyl)hydrazinyl)-N-phenylacetamide (6a)
Gray powder, yield 50% (2.4 g). mp 280–282 °C. IR (KBr, cm–1): 3419 (NH), 3064 (CH aromatic), 2914 (CH aliphatic), 1654, 1647 (2 C=O), 1627 (C=N). 1H NMR (400 MHz, CDCl3), δ ppm: 4.18 (s, 2H, CH2), 7.18 (t, 1H, Ar-H), 7.36 (t, 2H, Ar-H), 7.53–7.70 (m, 3H, Ar-H), 7.77 (t, 1H, Ar-H), 7.87 (d, 2H, J = 8.4 Hz. Ar-H), 8.01–8.09 (m, 2H, Ar-H), 8.18–8.27 (m, 3H, Ar-H), 9.27 (s, 1H, NH, D2O exchangeable), 11.88 (s, 1H, NH, D2O exchangeable), 11.91 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, CDCl3), δ ppm: 57.72, 123.52 (2C), 123.91, 124.88, 126.99, 127.96, 128.34 (2C), 128.64, 129.76, 130.38 (2C), 130.65, 132.04 (2C), 133.77, 136.46, 137.83, 143.25, 148.67, 155.44, 165.69, 168.01. MS m/z (%): 477.98 (M + 2, 16.81), 475.91 (M+, 18.14), 405.91 (100.00).). Anal. Calcd. for C24H19BrN4O2 (475.35): C, 60.64; H, 4.03; N 11.79; found: C, 60.85; H, 4.21; N, 12.06.
4.1.8.2. 2-(2-(2-(4-Bromophenyl)quinoline-4-carbonyl)hydrazinyl)-N-(4-methoxyphenyl)acetamide (6b)
White powder, yield 49% (2.5 g). mp > 350 °C. IR (KBr, cm–1): 3419 (NH), 3066 (CH aromatic), 2956 (CH aliphatic), 1660, 1647 (2 C=O), 1627 (C=N). 1H NMR (400 MHz, DMSO-d6), δ ppm: 3.72 (s, 3H, OCH3), 4.27 (s, 2H, CH2), 6.89 (d, 2H, J = 8.8 Hz. Ar-H), 7.55 (d, 2H, J = 8.8 Hz. Ar-H), 7.66 (t, 1H, Ar-H), 7.75 (d, 2H, J = 8.8 Hz. Ar-H), 7.84 (t, 1H, Ar-H), 8.07–8.15 (m, 2H, Ar-H), 8.24–8.29 (m, 3H, Ar-H), 10.59 (s, 1H, NH, D2O exchangeable), 10.95 (s, 1H, NH, D2O exchangeable), 11.14 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 55.67, 57.34, 118.08 (2C), 122.55, 123.89, 124.87, 125.93, 126.99, 127.63, 128.61 (2C), 129.33 (2C), 130.68, 131.75 (2C), 137.82, 142.88, 147.99, 149.73, 155.15, 156.79, 163.95, 167.34. MS m/z (%): 507.87 (M + 2, 34.49), 505.92 (M+, 35.02), 199.11 (100.00). Anal. calcd. for C25H21BrN4O3 (505.35): C, 59.42; H, 4.19; N 11.09; found: C, 59.70; H, 4.35; N, 11.27.
4.1.9. General Procedure for Synthesis of 2-(4-Bromophenyl)-N′-(substituted benzoyl)quinoline-4-carbohydrazide (7a–e)
To a well-stirred solution of acid hydrazide 3 (10 mmol, 3.41 g) in dioxane (20 mL), the appropriate benzoyl chloride derivative was added dropwise, and the reaction mixture was stirred overnight at room temperature. The reaction mixture was treated with sodium carbonate solution (10%, 25 mL), the solid obtained was filtered, dried, and crystallized from ethanol to give compounds 7a–e.
4.1.9.1. N′-Benzoyl-2-(4-bromophenyl)quinoline-4-carbohydrazide (7a)
White powder, yield 60% (2.7 g). mp 279–281 °C. IR (KBr, cm–1): 3284 (NH), 3066 (CH aromatic), 2929 (CH aliphatic), 1635 (2 C=O), 1604 (C=N). 1H NMR (400 MHz, DMSO-d6), δ ppm: 7.50–7.58 (m, 3H, Ar-H), 7.64 (t, 1H, Ar-H), 7.77–7.83 (m, 4H, Ar-H), 7.93 (d, 2H, J = 8.0 Hz. Ar-H), 8.11 (d, 1H, J = 8.4 Hz. Ar-H), 8.24–8.28 (m, 3H, Ar-H), 11.83 (s, 1H, NH, D2O exchangeable), 11.92 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 122.59, 123.14, 124.22, 124.76, 126.88, 127.67 (2C), 129.07 (2C), 129.82, 130.38 (2C), 130.96, 132.04 (2C), 132.54, 134.46, 138.22, 144.09, 148.40, 155.67, 164.56, 165.65. MS m/z (%): 448.66 (M + 2, 34.76), 446.43 (M+, 36.45), 210.50 (98.84). Anal. calcd. for C23H16BrN3O2 (446.30): C, 61.90; H, 3.61; N 9.42; found: C, 61.84; H, 3.74; N, 9.68.
4.1.9.2. 2-(4-Bromophenyl)-N′-(4-chlorobenzoyl)quinoline-4-carbohydrazide (7b)
White powder, yield 58%. (2.8 g). mp 305–307 °C. IR (KBr, cm–1): 3292 (NH), 3064 (CH aromatic), 2926 (CH aliphatic), 1655(2 C=O), 1611 (C=N). 1H NMR (400 MHz, DMSO-d6), δ ppm: 7.65 (d, 2H, J = 8.4 Hz. Ar-H), 7.73 (t, 1H, Ar-H), 7.81 (d, 2H, J = 8.4 Hz. Ar-H), 7.89 (t, 1H, Ar-H), 8.02 (d, 2H, J = 8.4 Hz. Ar-H), 8.17–8.21 (m, 2H, Ar-H), 8.28 (d, 2H, J = 8.4 Hz. Ar-H), 8.45 (d, 1H, J = 8.4 Hz. Ar-H), 10.87 (s, 1H, NH, D2O exchangeable), 10.89 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 116.90, 122.93, 123.60, 125.91, 127.60, 128.57, 129.23 (2C), 130.23, 130.90 (2C), 131.28 (2C), 131.59, 132.26 (2C), 134.29, 137.31, 141.29, 147.62, 154.32, 164.33, 166.38. MS m/z (%): 482.37 (M + 2, 18.46), 481.26 (M + 1, 32.92), 480.47 (M+, 94.62), 186 (100.00). Anal. calcd. for C23H15BrClN3O2 (480.75): C, 57.46; H, 3.15; N 8.74; found: C, 57.63; H, 3.23; N, 8.95.
4.1.9.3. 2-(4-Bromophenyl)-N′-(4-nitrobenzoyl)quinoline-4-carbohydrazide (7c)
Orange powder, yield 57% (2.8 g). mp charring 350 °C. IR (KBr, cm–1): 3462 (NH), 3064 (CH aromatic), 2970 (CH aliphatic), 1662, 1649 (2 C=O), 1612 (C=N). 1H NMR (400 MHz, DMSO-d6), δ ppm: 7.64 (t, 1H, Ar-H), 7.75–7.83 (m, 3H, Ar-H), 8.10–8.19 (m, 3H, Ar-H), 8.22–8.24 (m, 3H, Ar-H), 8.29 (d, 2H, J = 8.4 Hz. Ar-H), 8.49 (d, 1H, J = 8.4 Hz. Ar-H), 11.57 (s, 1H, NH, D2O exchangeable), 11.59 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 120.51, 123.89, 125.26, 125.57, 126.60 (2C), 127.32, 128.34 (2C), 129.69, 129.84 (2C), 130.00, 131.35, 132.39 (2C), 135.43, 138.50, 147.32, 149.72, 155.14, 161.24, 161.93. MS m/z (%): 493.74 (M + 2, 15.57), 492.83 (M + 1, 8.53), 491.41 (M+, 17.55), 368.88 (100.00). Anal. calcd. for C23H15BrN4O4 (491.30): C, 56.23; H, 3.08; N 11.40; found: C, 56.40; H, 3.27; N, 11.64.
4.1.9.4. 2-(4-Bromophenyl)-N′-(4-methylbenzoyl)quinoline-4-carbohydrazide (7d)
White powder, yield 71.6% (3.3 g). mp 278–280 °C IR (KBr, cm–1): 3224 (NH), 3032 (CH aromatic), 2991 (CH aliphatic), 16,365, 1635 (2 C=O), 1610 (C=N). 1H NMR (400 MHz, DMSO-d6), δ ppm: 2.40 (s, 3H, CH3), 7.37 (d, 2H, J = 7.6 Hz. Ar-H), 7.73 (t, 1H, Ar-H), 7.81 (d, 2H, J = 8.8 Hz. Ar-H), 7.87–7.91 (m, 3H, Ar-H), 8.18 (d, 1H, J = 7.2 Hz. Ar-H), 8.21 (s, 1H, Ar-H), 8.27 (d, 2H, J = 8.8 Hz. Ar-H), 8.46 (d, 1H, J = 7.2 Hz. Ar-H), 10.71 (s, 1H, NH, D2O exchangeable), 10.81 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 21.73, 117.12, 121.86, 123.90, 124.58, 125.56, 126.98, 127.29 (2C), 128.33 (2C), 128.65 (2C), 132.03 (2C), 133.39, 133.77, 137.15, 141.52, 142.18, 147.68, 155.44, 166.00, 167.05. MS m/z (%): 462.86 (M + 2, 12.13), 460.71 (M+, 13.70), 119.17 (100.00). Anal. calcd. for C24H18BrN3O2 (460.33): C, 62.62; H, 3.94; N 9.13; found: C, 62.88; H, 4.15; N, 9.41.
4.1.9.5. N′-(2-Bromobenzoyl)-2-(4-bromophenyl)quinoline-4-carbohydrazide (7e)
White powder, yield 64% (3.3 g). mp 296–298 °C. IR (KBr, cm–1): 3265 (NH), 3055 (CH aromatic), 2929 (CH aliphatic), 1669–1672 (2 C=O), 1612 (C=N). 1H NMR (400 MHz, DMSO-d6), δ ppm: 7.47 (t, 1H, Ar-H), 7.55 (t, 1H, Ar-H), 7.61 (d, 1H, J = 8 Hz. Ar-H), 7.71–7.76 (m, 2H, Ar-H), 7.81 (d, 2H, J = 8.4 Hz. Ar-H), 7.88 (t, 1H, Ar-H), 8.18 (d, 1H, J = 8.4 Hz. Ar-H), 8.22 (s, 1H, Ar-H), 8.30 (d, 2H, J = 8.4 Hz. Ar-H), 8.42 (d, 1H, J = 8.4 Hz. Ar-H), 10.70 (s, 1H, NH, D2O exchangeable), 10.98 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 117.29, 119.85, 123.53, 124.57, 125.89, 125.91, 128.20, 129.80 (2C), 129.99, 131.13, 131.74 (2C), 132.45, 133.60, 135.43, 136.96, 137.69, 141.22, 148.31, 154.77, 166.00, 167.01. MS m/z (%): 527.98 (M + 2, 26.83), 525.17 (M+, 22.60), 262.93 (100.00). Anal. calcd. for C23H15Br2N3O2 (525.20): C, 52.60; H, 2.88; N 8.00; found: C, 52.79; H, 3.06; N, 8.24.
4.1.10. Procedure for Synthesis of 2-(4-Bromophenyl)-N-(1,3-dioxoisoindolin-2-yl)quinoline-4-carboxamide (8)
A solution of acid hydrazide 3 (10 mmol, 3.41 g) and phthalic anhydride (10 mmol, 1.48 g) in dioxane (20 mL) containing glacial acetic acid (0.2 mL) was heated under reflux for 7 h. The mixture was poured onto ice cold water, and the solid obtained was filtered, dried, and crystallized from methanol to yield compound 8.
Buff crystals, yield 61% (2.9 g). mp 260–262 °C. IR (KBr, cm–1): 3292 (NH), 3080 (CH aromatic), 2972 (CH aliphatic), 1749 (C=O). 1H NMR (400 MHz, DMSO-d6), δ ppm: 7.76–7.80 (m, 1H, Ar-H), 7.82 (d, 2H, J = 8.4 Hz. Ar-H), 7.90–7.94 (m, 2H, Ar-H), 8.00–8.09 (m, 4H, Ar-H), 8.21 (d, 1H, J = 8.4 Hz. Ar-H), 8.32–8.36 (m, 3H, Ar-H), 11.76 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 123.22, 123.90, 124.58, 128.33, 128.65 (2C), 129.70, 130.68 (2C), 131.06 (2C), 131.39 (2C), 131.73, 132.45 (2C), 135.42, 136.78, 139.19, 148.36, 154.78, 165.30 (2C), 166.38. MS m/z (%): 474.01 (M + 2, 15.27), 473.89 (M + 1, 30.23), 472.85 (M+, 12.02), 141.42 (100.00). Anal. calcd. for C24H14BrN3O3 (472.30): C, 61.03; H, 2.99; N 8.90; found: C, 61.29; H, 3.15; N, 9.18.
4.1.11. Procedure for Synthesis of 2-(4-Bromophenyl)-N-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)quinoline-4-carboxamide (9)
A solution of acid hydrazide 3 (10 mmol, 3.41 g) and maleic anhydride (10 mmol, 0.98 g) in dioxane (20 mL) containing glacial acetic acid (0.2 mL) was heated under reflux for 10 h.
The mixture was poured onto ice cold water, and the solid obtained was filtered, dried, and crystallized from methanol to yield compound 9.
White powder, yield 52% (2.2 g). mp 325–327 °C. IR (KBr, cm–1): 3446 (NH), 3066 (CH aromatic), 2995 (CH aliphatic), 1658 (C=O), 1608 (C=N). 1H NMR (400 MHz, DMSO-d6), δ ppm: 7.75–7.83 (m, 5H, Ar-H, maleimide-H), 7.91 (t, 1H, Ar-H), 8.20 (d, 1H, J = 8 Hz. Ar-H), 8.30–8.38 (m, 3H, Ar-H), 8.50 (d, 1H, J = 8.8 Hz. Ar-H), 11.14 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 123.89, 124.57, 126.61, 128.34, 129.02 (2C), 129.99, 130.67 (2C), 131.73, 132.03, 134.06 (2C), 137.83, 141.91, 147.31, 155.13, 166.66 (2C), 168.72 MS m/z (%): 424.68 (M + 2, 29.28), 423.99 (M + 1, 12.24), 422.97 (M+, 27.84), 230.54 (100.00). Anal. calcd. for C20H12BrN3O3 (422.24): C, 56.89; H, 2.86; N 9.95; found: C, 57.03; H, 2.97; N, 10.24.
4.1.12. Procedure for Synthesis (3-Amino-5-imino-4,5-dihydro-1H-pyrazol-1-yl)(2-(4-bromophenyl)quinolin-4-yl)methanone (10)
A solution of acid hydrazide 3 (10 mmol, 3.41 g) and malononitrile (10 mmol, 0.66 g) in DMF (15 mL) containing 2 drops of pyridine was heated under reflux for 10 h. The mixture was poured onto ice cold water, and the solid obtained was filtered, dried, and crystallized from propanol to give compound 10.
Brown powder, yield 52% (2.12 g). mp 246–248 °C. IR (KBr, cm–1): 3444 (NH2) 3307 (NH), 3099 (CH aromatic), 2927 (CH aliphatic), 1685 (C=O), 1614 (C=N). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.06 (s, 2H, NH2, D2O exchangeable), 4.37 (s, 2H, CH2, pyrazole), 7.75–7.99 (m, 4H, Ar-H), 8.21 (d, 1H, J = 8.8 Hz. Ar-H), 8.29–8.32 (m, 3H, Ar-H), 8.49 (d, 1H, J = 8.8 Hz. Ar-H), 11.14 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 31.50, 123.21, 124.88, 125.55, 126.32, 127.97, 128.64 (2C), 129.99, 131.33 (2C), 132.41, 137.82, 141.23, 147.99, 155.14, 160.58, 163.28, 166.38. MS m/z (%): 410.95 (M + 2, 28.45), 409.03 (M + 1, 31.82), 408.02 (M+, 32.73), 88.56 (100.00). Anal. calcd. for C19H14BrN5O (408.26): C, 55.90; H, 3.46; N, 17.15; found: C, 56.12; H, 3.70; N, 17.39.
4.1.13. Procedure for Synthesis of (2-(4-Bromophenyl)quinolin-4-yl)(3,5-dimethyl-1H-pyrazol-1-yl)methanone (11)
A solution of acid hydrazide 3 (10 mmol, 3.41 g) and acetyl acetone (10 mmol, 1.00 mL) in dry DMF (10 mL) was heated under reflux for 7 h; then, the mixture was poured onto ice cold water. The aqueous layer was extracted with ethyl acetate (2 × 50 mL), and the combined organic extract was dried over anhydrous sodium sulfate and concentrated under vacuum. The remaining residue was crystallized from diethyl ether to give compound 11.
Brown powder, yield 49% (2 g). mp 220–222 °C. IR (KBr, cm–1): 3069 (CH aromatic), 2926 (CH aliphatic), 1631 (C=O), 1606 (C=N). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.75 (s, 3H, CH3), 2.03 (s, 3H, CH3), 6.91 (s, 1H, CH pyrazole), 7.63 (t, 1H, Ar-H), 7.75–7.83 (m, 4H, Ar-H), 8.04 (s, 1H, Ar-H), 8.12 (d, 1H, J = 8.8 Hz. Ar-H), 8.27 (d, 2H, J = 8.4 Hz. Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 15.96, 26.03, 115.51, 120.97, 123.46, 124.17, 125.67, 127.60, 130.05 (2C), 130.32, 130.82, 133.97 (2C), 137.47, 142.36, 145.54, 147.76, 155.41, 157.63, 165.20. MS m/z (%): 408.06 (M + 2, 17.77), 406.16 (M+, 18.44), 41.30 (100.00). Anal. calcd. for C21H16BrN3O (406.28): C, 62.08; H, 3.97; N 10.34; found: C, 61.91; H, 4.15; N, 10.60.
4.1.14. Procedure for Synthesis 2-(2-(4-Bromophenyl)quinoline-4-carbonyl)-5-methyl-2,4-dihydro-3H-pyrazol-3-one (12)
To a solution of acid hydrazide 3 (10 mmol, 3.41 g) in glacial acetic acid (20 mL), ethyl acetoacetate (20 mmol, 2.6 mL) was added, and the reaction mixture was heated under reflux for 8 h; then, the mixture was poured onto ice cold water. The solid obtained was filtered, dried, and crystallized from acetonitrile to give compound 12.
White powder, yield 50%. mp charring 350 °C. IR (KBr, cm–1): 3032 (CH aromatic), 2926 (CH aliphatic), 1675, 1633 (2 C=O), 1610 (C=N). 1H NMR (400 MHz, DMSO-d6), δ ppm: 2.00 (s, 3H, CH3), 3.43 (s, 2H, CH2 pyrazolone), 7.77 (t, 1H, Ar-H), 7.82 (d, 2H, J = 8.4 Hz. Ar-H), 7.91 (t, 1H, Ar-H), 8.21 (d, 1H, J = 8.8 Hz. Ar-H), 8.28–8.33 (m, 3H, Ar-H), 8.51 (d, 1H, J = 8.4 Hz. Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 17.19, 48.34, 117.28, 123.97, 124.33, 125.82, 128.25, 129.80 (2C), 130.07, 131.24, 132.48 (2C), 137.67, 141.65, 148.34, 155.14, 159.80, 166.49, 170.07. MS m/z (%): 410.05 (M + 2, 30.11), 408.17 (M+, 32.83), 407.08 (100.00). Anal. calcd. for C20H14BrN3O2 (408.26): C, 58.84; H, 3.46; N 10.29; Found: C, 59.12; H, 3.71; N, 10.52.
4.1.15. Procedure for Synthesis 5-Amino-1-(2-(4-bromophenyl)quinoline-4-carbonyl)-1,2-dihydro-3H-pyrazol-3-one (13)
To a solution of acid hydrazide 3 (10 mmol, 3.41 g) in glacial acetic acid (20 mL), ethyl cyanoacetate (10 mmol, 1.13 mL) was added and the reaction mixture was heated under reflux for 10 h; then, the mixture was poured onto ice cold water. The solid obtained was filtered, dried, and crystallized from acetonitrile to give compound 13.
Brown powder, yield 72% (2.9 g). mp 190–192 °C. IR (KBr, cm–1): 3444 (NH2), 3309 (NH), 3057 (CH aromatic), 2953 (CH aliphatic), 1643 (C=O), 1614 (C=N). 1H NMR (400 MHz, DMSO-d6), δ ppm: 3.56 (s, 2H, NH2, D2O exchangeable), 3.76 (s, 1H, CH pyrazolone), 7.66 (t, 1H, Ar-H), 7.77 (d, 2H, J = 8.0 Hz. Ar-H), 7.83 (t, 1H, Ar-H), 8.12–8.20 (m, 2H, Ar-H), 8.25–8.30 (m, 3H, Ar-H), 10.09 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 66.75, 121.97, 124.17, 126.03, 127.60, 128.47 (2C), 129.77, 130.04, 132.47 (2C), 132.76, 137.62, 140.29, 143.51, 148.90, 154.86, 162.47, 168.90. MS m/z (%):411.80 (M + 2, 25.31), 410.49 (M + 1, 56.30), 409.06 (M+, 28.61), 300.19 (100.00). Anal. calcd. for C19H13BrN4O2 (409.24): C, 55.76; H, 3.20; N 13.69; found: C, 55.93; H, 3.42; N, 13.95.
4.1.16. Procedure for Synthesis 1-(2-(4-Bromophenyl)quinoline-4-carbonyl)pyrazolidine-3,5-dione (14)
A mixture of acid hydrazide 3 (2 mmol, 0.68 g) and diethyl malonate (4 mmol, 0.64 mL) in sodium methoxide (4 mmol, 0.22 g, in methanol 20 mL) was heated under reflux for 16 h. The traction mixture was concentrated under vacuum, and the remaining residue was dissolved in water (15 mL) and then acidified with 2 N HCl (pH 3–4). The solid obtained was filtered, dried, and crystallized from ethanol to give compound 14.
White powder, yield 72% (0.59 g). mp 160–162 °C. IR (KBr, cm–1): 3419 (NH), 3032 (CH aromatic), 2954 (CH aliphatic), 1714 (C=O), 1608 (C=N). 1H NMR (400 MHz, DMSO-d6), δ ppm: 2.88 (s, 2H, CH2 pyrazolidine), 7.73–7.89 (m, 4H, Ar-H), 8.19 (d, 1H, J = 8.4 Hz. Ar-H), 8.24–8.33 (m, 2H, Ar-H), 8.47 (s, 1H, Ar-H), 8.64 (d, 1H, J = 8.8 Hz. Ar-H), 12.50 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 54.93, 119.44, 120.81, 122.86, 123.51, 125.94, 128.68 (2C), 129.31, 132.03 (2C), 137.14, 138.81, 149.03, 151.39, 155.15, 168.03, 170.73, 173.82. MS m/z (%): 412.39 (M + 2, 52.54), 410.62 (M+, 53.87), 322.66 (100.00). Anal. calcd. for C19H12BrN3O3 (410.23): C, 55.63; H, 2.95; N 10.24; found: C, 55.84; H, 3.17; N, 10.48.
4.2. Biological Evaluation
The antimicrobial assessment and the MIC, MBC, and MBIC assays were performed at Biotechnology Research Institute, National Research Centre, Cairo, Egypt.
4.2.1. Test Microorganism
Four representative test microbes used were Staphylococcus aureus ATCC 6538-P as Gram-positive bacteria, Escherichia coli ATCC 25933 as Gram-negative bacteria, and Candida albicans ATCC 10231 as yeast, whereas Aspergillus niger NRRL was used as a representative of filamentous fungi.
4.2.2. Agar Well Diffusion
The agar well diffusion method was employed to estimate the antimicrobial activity of the 15 quinolone derivatives (5–14) by measuring the zone of inhibition. (Supporting Information, Appendix A).
4.2.3. MIC, MBC, and MBIC Assay
The minimum inhibitory concentration (MIC), the minimum bactericidal concentration (MBC), and the minimum biofilm inhibitory concentration (MBIC) assays were utilized to pursue the antimicrobial activity for the prominently afforded compounds. S. aureus ATCC 6538 (Gram-positive bacteria) and C. albicans ATCC 10231 (yeast) were used and grown on Mueller–Hinton medium (Supporting Information, Appendix A).
S. aureus DNA Gyrase Supercoiling Assay
The DNA gyrase supercoiling assay was performed at the Tissue Culture Unit, VACSERA, Giza, Egypt. The S. aureus DNA gyrase supercoiling assay was conducted on the compounds with prominent average MIC values (compounds 6b and 10) along with the reference drug, ciprofloxacin, to assess inhibition % at the concentrations (0.1, 1, 10, 50, and 100 μM)53 using purified DNA Gyrase and relaxed DNA Kit (plasmid based).
4.3. In Silico Studies
4.3.1. Docking Studies
4.3.1.1. Molecular Docking
The binding interactions of the newly afforded 2-(4-bromophenyl)quinoline-4-carbohydrazide derivatives (5–14) at the S. aureus DNA gyrase active site were considered via molecular docking employing the MOE 2019 suite.54 The co-crystallized ligand (ciprofloxacin) at the target site was used as a reference control.
4.3.1.2. Preparation of the Investigated Derivatives
To get ready for the molecular docking process, the assessed compounds were drawn chemically utilizing PerkinElmer ChemOffice Suite 2017 and imported to one database file along with ciprofloxacin (MDB file) as previously described.39,55−62
4.3.1.3. Preparation of the S. aureus Gyrase-DNA Complex Target Protein
The PDB file of the S. aureus gyrase-DNA complex target protein was downloaded from the online protein data bank site (PDB entry: 2XCT).63 Subsequently, the target protein was prepared and energetically minimized to be thoroughly prepared for established docking, as previously discussed in detail.55−62
4.3.1.4. Docking of the Afforded Compounds (5–14) to S. aureus Gyrase-DNA Complex Target Protein
The general docking protocol was employed according to the default procedures discussed previously in detail,23,59,64−68 and the docking process was run for the selected compounds. For each docked compound, the pose with the best affinity energy scores, RMSD values, and prominent amino acid and nucleobase interactions was selected and saved for further visualization.
4.3.2. Physicochemical, ADMET, and Pharmacokinetic Properties Prediction
The Swiss Institute of Bioinformatics (SIB) offers the free Swiss ADME web tool, which can be employed for assessing the physicochemical characteristics, forecasting pharmacokinetic features, and anticipating the ADME parameters of the afforded compounds. Thus, SMILES notations regarding the synthesized compounds’ chemical structures were imported to the online server of the Swiss ADME web tool for more calculations processing.44 In addition, the investigated compounds’ toxicity features were examined employing the pkCSM descriptors algorithm tool.43
Acknowledgments
This work was supported through the Annual Funding track by the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia [GRANT 3104].
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c01156.
All the spectral analysis such as 1H-NMR, 13C-NMR, and MS spectra for all prepared compounds; and detailed descriptions for section 4.2. and 4.3 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ammar Y. A.; Farag A. A.; Ali A. M.; Hessein S. A.; Askar A. A.; Fayed E. A.; Elsisi D. M.; Ragab A. Antimicrobial evaluation of thiadiazino and thiazolo quinoxaline hybrids as potential DNA gyrase inhibitors; design, synthesis, characterization and morphological studies. Bioorg. Chem. 2020, 99, 103841–103853. 10.1016/j.bioorg.2020.103841. [DOI] [PubMed] [Google Scholar]
- Elbastawesy M. A. I.; Mohamed F. A. M.; Zaki I.; Alahmdi M. I.; Alzahrani S. S.; Alzahrani H. A.; Gomaa H. A. M.; Youssif B. G. M. Design, synthesis and antimicrobial activity of novel quinoline-2-one hybrids as promising DNA gyrase and topoisomerase IV inhibitors. J. Mol. Struct. 2023, 1278, 134902–134912. 10.1016/j.molstruc.2023.134902. [DOI] [Google Scholar]
- Blair J. M. A.; Webber M. A.; Baylay A. J.; Ogbolu D. O.; Piddock L. J. V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- Ragab A.; Abusaif M. S.; Gohar N. A.; Aboul-Magd D. S.; Fayed E. A.; Ammar Y. A. Development of new spiro[1,3]dithiine-4,11′-indeno[1,2-b]quinoxaline derivatives as S. aureus Sortase A inhibitors and radiosterilization with molecular modeling simulation. Bioorg. Chem. 2023, 131, 106307–106322. 10.1016/j.bioorg.2022.106307. [DOI] [PubMed] [Google Scholar]
- Jansen K. U.; Knirsch C.; Anderson A. S. The role of vaccines in preventing bacterial antimicrobial resistance. Nat. Med. 2018, 24, 10–19. 10.1038/nm.4465. [DOI] [PubMed] [Google Scholar]
- Kaatz G. W.; McAleese F.; Seo S. M. Multidrug resistance in Staphylococcus aureus due to overexpression of a novel multidrug and toxin extrusion (MATE) transport protein. Antimicrob. Agents Chemother. 2005, 49, 1857–1864. 10.1128/AAC.49.5.1857-1864.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edoh D.; Alomatu B. Comparison of antibiotic resistance patterns between laboratories in Accra East, Ghana. Afr. J. Sci. Technol. 2007, 8, 1–7. 10.4314/AJST.V8I1. [DOI] [Google Scholar]
- Nayak N.; Nag T.; Satpathy G.; Ray S. Ultrastructural analysis of slime positive & slime negative Staphylococcus epidermidis isolates in infectious keratitis. Indian J. Med. Res. 2007, 125, 767–771. [PubMed] [Google Scholar]
- Jensen L. B.; Baloda S.; Boye M.; Aarestrup F. M. Antimicrobial resistance among Pseudomonas spp. and the Bacillus cereus group isolated from Danish agricultural soil. Environ. Int. 2001, 26, 581–587. 10.1016/S0160-4120(01)00045-9. [DOI] [PubMed] [Google Scholar]
- Goldblatt D.; Thrasher A. Chronic granulomatous disease. Clin. Exp. Immunol. 2000, 122, 1–9. 10.1046/j.1365-2249.2000.01314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Kohlhoff S.; Valencia G.; Hammerschlag M. R.; Sharma R. Treatment of vancomycin-resistant Enterococcus faecium ventriculitis in a neonate. Int. J. Antimicrob. Agents 2007, 29, 740–741. 10.1016/j.ijantimicag.2006.11.025. [DOI] [PubMed] [Google Scholar]
- Fayed E. A.; Ebrahim M. A.; Fathy U.; Saeed H. S. E.; Khalaf W. S. Evaluation of quinoxaline derivatives as potential ergosterol biosynthesis inhibitors: design, synthesis, ADMET, molecular docking studies, and antifungal activities. J. Mol. Struct. 2022, 1267, 133578–133582. 10.1016/j.molstruc.2022.133578. [DOI] [Google Scholar]
- Fayed E. A.; Nosseir E. S.; Atef A.; El-Kalyoubi S. A. In vitro antimicrobial evaluation and in silico studies of coumarin derivatives tagged with pyrano-pyridine and pyrano-pyrimidine moieties as DNA gyrase inhibitors. Mol. Diversity 2022, 26, 341–363. 10.1007/s11030-021-10224-4. [DOI] [PubMed] [Google Scholar]
- Ammar Y. A.; Farag A. A.; Ali A. M.; Ragab A.; Askar A. A.; Elsisi D. M.; Belal A. Design, synthesis, antimicrobial activity and molecular docking studies of some novel di-substituted sulfonylquinoxaline derivatives. Bioorg. Chem. 2020, 104, 104164–104178. 10.1016/j.bioorg.2020.104164. [DOI] [PubMed] [Google Scholar]
- Puerto A. S.; Fernández J. G.; Del Castillo J. d. D. L.; Pino M. J. S.; Angulo G. P. In vitro activity of β-lactam and non−β-lactam antibiotics in extended-spectrum β-lactamase–producing clinical isolates of Escherichia coli. Diagn. Microbiol. Infect. Dis. 2006, 54, 135–139. 10.1016/j.diagmicrobio.2005.08.018. [DOI] [PubMed] [Google Scholar]
- Ragab A.; Elsisi D. M.; Abu Ali O. A.; Abusaif M. S.; Askar A. A.; Farag A. A.; Ammar Y. A. Design, synthesis of new novel quinoxalin-2 (1H)-one derivatives incorporating hydrazone, hydrazine, and pyrazole moieties as antimicrobial potential with in-silico ADME and molecular docking simulation. Arabian J. Chem. 2022, 15, 103497–103511. 10.1016/j.arabjc.2021.103497. [DOI] [Google Scholar]
- Collin F.; Karkare S.; Maxwell A. Exploiting bacterial DNA gyrase as a drug target: current state and perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. 10.1007/s00253-011-3557-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofny H. A.; Mohamed M. F. A.; Gomaa H. A. M.; Abdel-Aziz S. A.; Youssif B. G. M.; El-koussi N. A.; Aboraia A. S. Design, synthesis, and antibacterial evaluation of new quinoline-1, 3, 4-oxadiazole and quinoline-1, 2, 4-triazole hybrids as potential inhibitors of DNA gyrase and topoisomerase IV. Bioorg. Chem. 2021, 112, 104920–104932. 10.1016/j.bioorg.2021.104920. [DOI] [PubMed] [Google Scholar]
- Medapi B.; Renuka J.; Saxena S.; Sridevi J. P.; Medishetti R.; Kulkarni P.; Yogeeswari P.; Sriram D. Design and synthesis of novel quinoline–aminopiperidine hybrid analogues as Mycobacterium tuberculosis DNA gyraseB inhibitors. Bioorg. Med. Chem. 2015, 23, 2062–2078. 10.1016/j.bmc.2015.03.004. [DOI] [PubMed] [Google Scholar]
- Dighe S. N.; Collet T. A. Recent advances in DNA gyrase-targeted antimicrobial agents. Eur. J. Med. Chem. 2020, 199, 112326–112335. 10.1016/j.ejmech.2020.112326. [DOI] [PubMed] [Google Scholar]
- El-Gamal K. M.; El-Morsy A. M.; Saad A. M.; Eissa I. H.; Alswah M. Synthesis, docking, QSAR, ADMET and antimicrobial evaluation of new quinoline-3-carbonitrile derivatives as potential DNA-gyrase inhibitors. J. Mol. Struct. 2018, 1166, 15–33. 10.1016/j.molstruc.2018.04.010. [DOI] [Google Scholar]
- Sridhar P.; Alagumuthu M.; Arumugam S.; Reddy S. R. Synthesis of quinoline acetohydrazide-hydrazone derivatives evaluated as DNA gyrase inhibitors and potent antimicrobial agents. RSC Adv. 2016, 6, 64460–64468. 10.1039/C6RA09891F. [DOI] [Google Scholar]
- El-Shershaby M. H.; El-Gamal K. M.; Bayoumi A. H.; El-Adl K.; Alswah M.; Ahmed H. E. A.; Al-Karmalamy A. A.; Abulkhair H. S. The antimicrobial potential and pharmacokinetic profiles of novel quinoline-based scaffolds: synthesis and in silico mechanistic studies as dual DNA gyrase and DHFR inhibitors. New J. Chem. 2021, 45, 13986–14004. 10.1039/D1NJ02838C. [DOI] [Google Scholar]
- El-Shershaby M. H.; El-Gamal K. M.; Bayoumi A. H.; El-Adl K.; Ahmed H. E. A.; Abulkhair H. S. Synthesis, antimicrobial evaluation, DNA gyrase inhibition, and in silico pharmacokinetic studies of novel quinoline derivatives. Arch. Pharm. 2021, 354, 2000277–2000286. 10.1002/ardp.202000277. [DOI] [PubMed] [Google Scholar]
- Lakhrissi Y.; Rbaa M.; Tuzun B.; Hichar A.; Anouar E. H.; Ounine K.; Almalki F.; Hadda T. B.; Zarrouk A.; Lakhrissi B. Synthesis, structural confirmation, antibacterial properties and bio-informatics computational analyses of new pyrrole based on 8-hydroxyquinoline. J. Mol. Struct. 2022, 1259, 132683–132692. 10.1016/j.molstruc.2022.132683. [DOI] [Google Scholar]
- Pradeep M.; Vishnuvardhan M.; Thalari G. A simple and efficient microwave assisted synthesis of pyrrolidinyl-quinoline based pyrazoline derivatives and their antimicrobial activity. Chem. Data Collect. 2021, 32, 100666–100671. 10.1016/j.cdc.2021.100666. [DOI] [Google Scholar]
- Xu Z.; Zhao S.-J.; Lv Z.-S.; Gao F.; Wang Y.; Zhang F.; Bai L.; Deng J.-L. Fluoroquinolone-isatin hybrids and their biological activities. Eur. J. Med. Chem. 2019, 162, 396–406. 10.1016/j.ejmech.2018.11.032. [DOI] [PubMed] [Google Scholar]
- Gao F.; Wang P.; Yang H.; Miao Q.; Ma L.; Lu G. Recent developments of quinolone-based derivatives and their activities against Escherichia coli. Eur. J. Med. Chem. 2018, 157, 1223–1248. 10.1016/j.ejmech.2018.08.095. [DOI] [PubMed] [Google Scholar]
- Patel K. B.; Kumari P. A review: Structure-activity relationship and antibacterial activities of Quinoline based hybrids. J. Mol. Struct. 2022, 1268, 133634–133657. 10.1016/j.molstruc.2022.133634. [DOI] [Google Scholar]
- Liu B.; Jiang D.; Hu G. The antibacterial activity of isatin hybrids. Curr. Top. Med. Chem. 2022, 22, 25–40. 10.2174/1568026621666211116090456. [DOI] [PubMed] [Google Scholar]
- Elshaier Y. A. M. M.; Aly A. A.; Abdel-Aziz M.; Fathy H. M.; Brown A. B.; Bräse S.; Ramadan M. Synthesis and Identification of New N,N-Disubstituted Thiourea, and Thiazolidinone Scaffolds Based on Quinolone Moiety as Urease Inhibitor. Molecules 2022, 27, 7126–7143. 10.3390/molecules27207126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lübbers T.; Angehrn P.; Gmünder H.; Herzig S. Design, synthesis, and structure–activity relationship studies of new phenolic DNA gyrase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 4708–4714. 10.1016/j.bmcl.2006.12.065. [DOI] [PubMed] [Google Scholar]
- Jayashree B. S.; Thomas S.; Nayak Y. Design and synthesis of 2-quinolones as antioxidants and antimicrobials: a rational approach. Med. Chem. Res. 2010, 19, 193–209. 10.1007/s00044-009-9184-x. [DOI] [Google Scholar]
- Nicolaï E.; Güngör T.; Goyard J.; Cure G.; Fouquet A.; Teulon J.; Delchambre C.; Cloarec A. Synthesis and aldose reductase inhibitory activity of N-(quinolinyl thiocarbonyl) glycine derivatives. Eur. J. Med. Chem. 1992, 27, 977–984. 10.1016/0223-5234(92)90032-V. [DOI] [Google Scholar]
- Feist K.; Kuklinski M. Synthetische Versuche in der 2-Phenyl-chinolin-Reihe I. Synthese einiger brom-substituierter 2-Phenyl-chinolin-4-karbonsäuren, Untersuchungen über die Reaktionsfähigkeit des Bromatoms in ihnen sowie Abbau von 6- und 4′- Bromatophan nach Curtius. Arch. Pharm. 1936, 274, 244–255. 10.1002/ardp.19362740405. [DOI] [Google Scholar]
- Hamdy R.; Elseginy S.; Ziedan N.; Jones A.; Westwell A. New quinoline-based heterocycles as anticancer agents targeting bcl-2. Molecules 2019, 24, 1274–1284. 10.3390/molecules24071274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraldi P. G.; Preti D.; Tabrizi M. A.; Fruttarolo F.; Saponaro G.; Baraldi S.; Romagnoli R.; Moorman A. R.; Gessi S.; Varani K.; Borea P. A. N6-[(Hetero) aryl/(cyclo) alkyl-carbamoyl-methoxy-phenyl]-(2-chloro)-5′-N-ethylcarboxamido-adenosines: The first example of adenosine-related structures with potent agonist activity at the human A2B adenosine receptor. Bioorg. Med. Chem. 2007, 15, 2514–2527. 10.1016/j.bmc.2007.01.055. [DOI] [PubMed] [Google Scholar]
- Al-Wahaibi L. H.; Amer A. A.; Marzouk A. A.; Gomaa H. A. M.; Youssif B. G. M.; Abdelhamid A. A. Design, Synthesis, and Antibacterial Screening of Some Novel Heteroaryl-Based Ciprofloxacin Derivatives as DNA Gyrase and Topoisomerase IV Inhibitors. Pharmaceuticals 2021, 14, 399–412. 10.3390/ph14050399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Demerdash A.; Al-Karmalawy A. A.; Abdel-Aziz T. M.; Elhady S. S.; Darwish K. M.; Hassan A. H. E. Investigating the structure–activity relationship of marine natural polyketides as promising SARS-CoV-2 main protease inhibitors. RSC Adv. 2021, 11, 31339–31363. 10.1039/D1RA05817G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elebeedy D.; Elkhatib W. F.; Kandeil A.; Ghanem A.; Kutkat O.; Alnajjar R.; Saleh M. A.; Abd El Maksoud A. I.; Badawy I.; Al-Karmalawy A. A. Anti-SARS-CoV-2 activities of tanshinone IIA, carnosic acid, rosmarinic acid, salvianolic acid, baicalein, and glycyrrhetinic acid between computational and in vitro insights. RSC Adv. 2021, 11, 29267–29286. 10.1039/D1RA05268C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmaaty A. A.; Alnajjar R.; Hamed M. I. A.; Khattab M.; Khalifa M. M.; Al-Karmalawy A. A. Revisiting activity of some glucocorticoids as a potential inhibitor of SARS-CoV-2 main protease: theoretical study. RSC Adv. 2021, 11, 10027–10042. 10.1039/D0RA10674G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattab M.; Al-Karmalawy A. A. Computational repurposing of benzimidazole anthelmintic drugs as potential colchicine binding site inhibitors. Future Med. Chem. 2021, 13, 1623–1638. 10.4155/fmc-2020-0273. [DOI] [PubMed] [Google Scholar]
- Gaber A. A.; El-Morsy A. M.; Sherbiny F. F.; Bayoumi A. H.; El-Gamal K. M.; El-Adl K.; Al-Karmalawy A. A.; Ezz Eldin R. R.; Saleh M. A.; Abulkhair H. S. Pharmacophore-linked pyrazolo[3,4-d]pyrimidines as EGFR-TK inhibitors: Synthesis, anticancer evaluation, pharmacokinetics, and in silico mechanistic studies. Arch. Pharm. 2021, 8, e2100258. 10.1002/ardp.202100258. [DOI] [PubMed] [Google Scholar]
- El-Shershaby M. H.; Ghiaty A.; Bayoumi A. H.; Al-Karmalawy A. A.; Husseiny E. M.; El-Zoghbi M. S.; Abulkhair H. S. From triazolophthalazines to triazoloquinazolines: A bioisosterism-guided approach toward the identification of novel PCAF inhibitors with potential anticancer activity. Bioorg. Med. Chem. 2021, 42, 116266–116276. 10.1016/j.bmc.2021.116266. [DOI] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717–42729. 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires D. E. V.; Blundell T. L.; Ascher D. B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savjani K. T.; Gajjar A. K.; Savjani J. K. Drug solubility: importance and enhancement techniques. Int. Scholarly Res. Not. 2012, 2012, 1. 10.5402/2012/195727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalbert A.; Morand C.; Manach C.; Rémésy C. Absorption and metabolism of polyphenols in the gut and impact on health. Biomed. Pharmacother. 2002, 56, 276–282. 10.1016/S0753-3322(02)00205-6. [DOI] [PubMed] [Google Scholar]
- Thanou M.; Verhoef J. C.; Junginger H. E. Oral drug absorption enhancement by chitosan and its derivatives. Adv. Drug Delivery Rev. 2001, 52, 117–126. 10.1016/S0169-409X(01)00231-9. [DOI] [PubMed] [Google Scholar]
- Sullins A. K.; Abdel-Rahman S. M. Pharmacokinetics of Antibacterial Agents in the CSF of Children and Adolescents. Pediatr. Drugs 2013, 15, 93–117. 10.1007/s40272-013-0017-5. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2012, 64, 4–17. 10.1016/j.addr.2012.09.019. [DOI] [PubMed] [Google Scholar]
- Levy D. D.; Zeiger E.; Escobar P. A.; Hakura A.; van der Leede B.-j. M.; Kato M.; Moore M. M.; Sugiyama K.-i. Recommended criteria for the evaluation of bacterial mutagenicity data (Ames test). Mutat. Res., Genet. Toxicol. Environ. Mutagen. 2019, 848, 403074–403088. 10.1016/j.mrgentox.2019.07.004. [DOI] [PubMed] [Google Scholar]
- Roy S.; Mathew M. K. Fluid flow modulates electrical activity in cardiac hERG potassium channels. J. Biol. Chem. 2018, 293, 4289–4303. 10.1074/jbc.RA117.000432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti M. C. HERG1 channel agonists and cardiac arrhythmia. Curr. Opin. Pharmacol. 2014, 15, 22–27. 10.1016/j.coph.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezelarab H. A. A.; Abbas S. H.; Abourehab M. A. S.; Badr M.; Sureram S.; Hongmanee P.; Kittakoop P.; Abuo-Rahma G. E.-D. A.; Hassan H. A. Novel antimicrobial ciprofloxacin-pyridinium quaternary ammonium salts with improved physicochemical properties and DNA gyrase inhibitory activity. Med. Chem. Res. 2021, 30, 2168–2183. 10.1007/s00044-021-02798-3. [DOI] [Google Scholar]
- Inc C. C. G.Molecular operating environment (MOE); Chemical Computing Group Inc.: 1010 Sherbooke St. West, S., Montreal; 2016. [Google Scholar]
- Elmaaty A. A.; Darwish K. M.; Khattab M.; Elhady S. S.; Salah M.; Hamed M. I. A.; Al-Karmalawy A. A.; Saleh M. M. In a search for potential drug candidates for combating COVID-19: computational study revealed salvianolic acid B as a potential therapeutic targeting 3CLpro and spike proteins. J. Biomol. Struct. Dyn. 2022, 40, 8866–8893. 10.1080/07391102.2021.1918256. [DOI] [PubMed] [Google Scholar]
- Abo Elmaaty A.; Hamed M. I. A.; Ismail M. I.; Elkaeed E. B.; Abulkhair H. S.; Khattab M.; Al-Karmalawy A. A. Computational Insights on the Potential of Some NSAIDs for Treating COVID-19: Priority Set and Lead Optimization. Molecules 2021, 26, 3772–3782. 10.3390/molecules26123772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamed M. I. A.; Darwish K. M.; Soltane R.; Chrouda A.; Mostafa A.; Abo Shama N. M.; Elhady S. S.; Abulkhair H. S.; Khodir A. E.; Elmaaty A. A.; Al-karmalawy A. A. β-Blockers bearing hydroxyethylamine and hydroxyethylene as potential SARS-CoV-2 Mpro inhibitors: rational based design, in silico, in vitro, and SAR studies for lead optimization. RSC Adv. 2021, 11, 35536–35558. 10.1039/D1RA04820A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmaaty A. A.; Darwish K. M.; Chrouda A.; Boseila A. A.; Tantawy M. A.; Elhady S. S.; Shaik A. B.; Mustafa M.; Al-karmalawy A. A. In Silico and In Vitro Studies for Benzimidazole Anthelmintics Repurposing as VEGFR-2 Antagonists: Novel Mebendazole-Loaded Mixed Micelles with Enhanced Dissolution and Anticancer Activity. ACS Omega 2022, 7, 875–899. 10.1021/acsomega.1c05519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elebeedy D.; Badawy I.; Elmaaty A. A.; Saleh M. M.; Kandeil A.; Ghanem A.; Kutkat O.; Alnajjar R.; Abd El Maksoud A. I.; Al-karmalawy A. A. In vitro and computational insights revealing the potential inhibitory effect of Tanshinone IIA against influenza A virus. Comput. Biol. Med. 2022, 141, 105149. 10.1016/j.compbiomed.2021.105149. [DOI] [PubMed] [Google Scholar]
- Hammoud M. M.; Nageeb A. S.; Morsi M. A.; Gomaa E. A.; Elmaaty A. A.; Al-Karmalawy A. A. Design, synthesis, biological evaluation, and SAR studies of novel cyclopentaquinoline derivatives as DNA intercalators, topoisomerase II inhibitors, and apoptotic inducers. New J. Chem. 2022, 46, 11422–11436. 10.1039/D2NJ01646J. [DOI] [Google Scholar]
- Hammouda M. M.; Elmaaty A. A.; Nafie M. S.; Abdel-Motaal M.; Mohamed N. S.; Tantawy M. A.; Belal A.; Alnajjar R.; Eldehna W. M.; Al-Karmalawy A. A. Design and synthesis of novel benzoazoninone derivatives as potential CBSIs and apoptotic inducers: In Vitro, in Vivo, molecular docking, molecular dynamics, and SAR studies. Bioorg. Chem. 2022, 127, 105995–106006. 10.1016/j.bioorg.2022.105995. [DOI] [PubMed] [Google Scholar]
- Saleh M. A.; Elmaaty A. A.; El Saeed H. S.; Saleh M. M.; Salah M.; Ezz Eldin R. R. Structure based design and synthesis of 3-(7-nitro-3-oxo-3,4-dihydroquinoxalin-2-yl)propanehydrazide derivatives as novel bacterial DNA-gyrase inhibitors: In-vitro, In-vivo, In-silico and SAR studies. Bioorg. Chem. 2022, 129, 106186. 10.1016/j.bioorg.2022.106186. [DOI] [PubMed] [Google Scholar]
- Bax B. D.; Chan P. F.; Eggleston D. S.; Fosberry A.; Gentry D. R.; Gorrec F.; Giordano I.; Hann M. M.; Hennessy A.; Hibbs M.; et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935–940. 10.1038/nature09197. [DOI] [PubMed] [Google Scholar]
- Hazem R. M.; Antar S. A.; Nafea Y. K.; Al-Karmalawy A. A.; Saleh M. A.; El-Azab M. F. Pirfenidone and vitamin D mitigate renal fibrosis induced by doxorubicin in mice with Ehrlich solid tumor. Life Sci. 2022, 288, 120185–120194. 10.1016/j.lfs.2021.120185. [DOI] [PubMed] [Google Scholar]
- Kandeil A.; Mostafa A.; Kutkat O.; Moatasim Y.; Al-Karmalawy A. A.; Rashad A. A.; Kayed A. E.; Kayed A. E.; El-Shesheny R.; Kayali G.; Ali M. A. Bioactive Polyphenolic Compounds Showing Strong Antiviral Activities against Severe Acute Respiratory Syndrome Coronavirus 2. Pathogens 2021, 10, 758–775. 10.3390/pathogens10060758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud D. B.; Bakr M. M.; Al-karmalawy A. A.; Moatasim Y.; El Taweel A.; Mostafa A. Scrutinizing the Feasibility of Nonionic Surfactants to Form Isotropic Bicelles of Curcumin: a Potential Antiviral Candidate Against COVID-19. AAPS PharmSciTech 2022, 23, 1–12. 10.1208/s12249-021-02197-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.