

Abstract
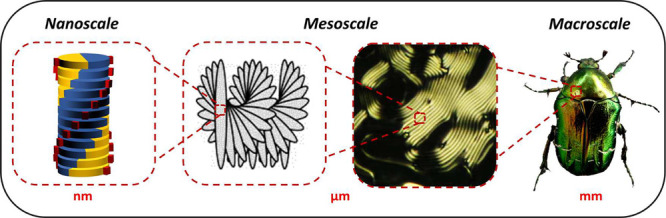
Several biomolecules can form dynamic aggregates in water, whose nanometric structures often reflect the chirality of the monomers in unexpected ways. Their twisted organization can be further propagated to the mesoscale, in chiral liquid crystalline phases, and even to the macroscale, where chiral, layered architectures contribute to the chromatic and mechanical properties of various plant, insect, and animal tissues. At all scales, the resulting organization is determined by a subtle balance among chiral and nonchiral interactions, whose understanding and fine-tuning is fundamental also for applications. We present recent advances in the chiral self-assembly and mesoscale ordering of biological and bioinspired molecules in water, focusing on systems based on nucleic acids or related aromatic molecules, oligopeptides, and their hybrid stuctures. We highlight the common features and key mechanisms governing this wide range of phenomena, together with novel characterization approaches.
Introduction
Transfer of chirality across length scales is ubiquitous in nature. Chiral biomolecules like nucleotides or amino acids give rise to helical and fibrillar structures. Cellulose nanocrystals and fibers, at high enough concentration, can spontaneously order into liquid crystalline (LC) phases, whose mesoscale chirality reflects the chiral morphology and interactions of the individual nanoscale components, although often in unexpected ways.1 On an even larger scale, layered, periodic architectures made of cellulose, chitin, or collagen contribute to the mechanical properties of biological tissues and are at the basis of the bright, pigment-free coloration observed in some plants and insects. Such structures are often similar despite the biochemical diversity of the underlying macromolecules.1,2 In general, predicting and controlling the propagation of chirality from nanoscale building blocks to aggregates to micro- and macroscopic structures constitutes a significant challenge.3 However, the same sensitivity to molecular details, which makes it difficult to track this process, is at the origin of a wide variety of applications, ranging from asymmetric catalysis to sensing and from tissue engineering and actuators to materials for photonics and electronics.4
Supramolecular polymers are based on the self-assembly of monomers into flexible and dynamic structures through weak, noncovalent interactions. As compared to their covalent counterparts, they are more sensitive to changes in concentration, temperature, or pH; moreover, kinetics often plays a role in the possible modes of their self-assembly.5 As such, they are a paradigmatic case of the variety of chiral behaviors, including inversion of the favored twist, of the mutual interplay between self-assembly and chiral interactions, and of the possible partitioning of enantiomers into homochiral aggregates, which bears relevance for the emergence of biomolecular homochirality within the context of life’s origin.6
In this mini-review, we will focus on the water-based self-assembly of chiral biomolecules and bioinspired moieties, including recent developments in their characterization. In water, hydrogen bonds, metal coordination, and electrostatic and hydrophobic interactions all contribute to the rich observed behavior, each with their own dependence on temperature and concentration.4,5a Moreover, we will describe how chiral molecular features are transferred to the mesoscale, with LC phases often competing with the bundling and formation of isotropic networks.
Before discussing the specific systems, we briefly recall a few recurring concepts and definitions for molecular self-assembly and the amplification of chiral asymmetry in supramolecular polymers. The two main polymerization modes are defined on the basis of the free energy gain upon aggregation of monomers with other monomers or with aggregates or between aggregates. In an isodesmic process (found, e.g., in chromonic molecules7), the energy per monomer is independent of the aggregation number; this results in a broad length distribution, with prevalence of free monomers. However, some degree of aggregation can be found even in dilute solutions. By contrast, other molecules, like surfactants and some peptides, are fully soluble and stable as monomers until a concentration or temperature threshold is reached. When nucleation of short aggregates occurs, elongation proceeds through an enhanced, cooperative incorporation of soluble monomers; i.e., the free energy of binding depends on the aggregation number. This results in narrower length distributions and, in the case of amphiphiles, in micelles with an optimal size, that is, number of molecules. Kinetics can clearly influence the growth process and lead to the formation of metastable structures.5b
Homochiral aggregates can easily emerge in solutions of a chiral nonracemic monomer but also with mixed enantiomers or when a small fraction of chiral molecules is mixed with compatible achiral molecules. Indeed, for flat, disc-shaped monomers, helical aggregates optimize intermolecular stacking interactions, as the rotational freedom of monomers is hampered. Often, the amplification of asymmetry can be quantified through optical techniques like circular dichroism (CD). When molecules with opposite chirality are mixed, within the so-called Majority-rules effect a small enantiomeric excess (ee) can sensitively affect the net helicity. Similarly, when some chiral, nonracemic molecule is used as a dopant in achiral molecules, within the so-called Sergeant-and-soldiers effect, the net helicity can be found to strongly depend on dopant concentration.3
For sake of brevity and based on our personal experience, in the next sections we will provide selected examples of such phenomena in systems based on—or related to—only two main, broadly defined classes of biomolecules: (i) nucleic acids and other flat, conjugated aromatic molecules, like porphyrins or dyes, whose self-assembly in water is typically dominated by stacking interactions; (ii) oligopeptides, for which electrostatics and hydrogen bonding often play a major role. Finally, we will discuss a few cases of hybrids of the two previous classes and highlight differences and similarities.
We are aware that we are not covering important areas of investigation, like, e.g., the emergence of chirality in lipid-based micelles and mesophases8 or the self-assembly of polysaccharides.1
Spiral Staircases: Self-Assembly and Ordering of Nucleic Acids and Disc-Like Molecules
Nucleotides, the building blocks of DNA and RNA polymers, are composed of nitrogen bases, sugars, and negatively charged phosphate groups. DNA is a paradigmatic case of how the chirality of the sugar is transferred to the whole macromolecule, mediated, and amplified by interactions and molecular constraints. Indeed, bases pair via specific Watson–Crick hydrogen bonds, and stacking forces hold neighboring, hydrophobic base pairs in close contact, excluding water molecules. When combined with the mutual repulsion of phosphate groups and with the length of the internucleotide covalent bonds, this yields the molecular features of the celebrated right-handed double helix.
A similar, subtle interplay of interactions also occurs in the formation of noncovalent, supramolecular assemblies of nucleic acids and other molecules with similar structure.
Aggregation of Nucleic Acid Bases and Aromatic Molecules
Self-Assembly of Guanosine Derivatives
Guanosine (G) monophosphate, one of the four nucleotides, besides taking part in canonical Watson–Crick base pairing with cytosine, can form Hoogsteen hydrogen bonds with other G bases, forming flat, hydrophobic quartets.9 Early studies in dilute basic solutions, with sodium counterions, have shown the coexistence of stacked monomers and stacked G-quartets, forming reversible, chiral aggregates.9b Aggregates of G-quartets are maximally stable at low temperatures and at pressures of up to 1 kbar.9c At even higher pressures, the G-quartet stacks dissociate laterally into monomer stacks without significantly changing the longitudinal dimension. Recently, it has been elucidated that the aggregation mode of quartets, either homopolar or heteropolar, can be tuned by different monovalent and divalent metal ions and determines the handedness of the resulting quadruplexes.10 Moreover, while Sr2+ ions favor thermodynamically stable right-handed aggregates, transient formation of left-handed quadruplexes is possible, as demonstrated by circularly polarized luminescence of intercalated dyes.11
At high enough concentration, quadruplexes of G monomers and G oligomers can align in an LC chiral nematic or cholesteric (N*) phase, i.e., a fluid with orientational order and an additional periodicity nested in the chiral interactions of the aggregates. Intriguingly, the molecular features of the quadruplexes composed by oligomers of different lengths result in LC phases of opposite handedness.9a
Base Analogs
Several nucleobase analogs have been investigated, either for their plausible prebiotic origin and their possible role as informational polymers preceding RNA and DNA6d or to fine-tune molecular self-assembly in aqueous solutions.5a Among these, mixtures of the achiral molecule 2,4,6-triaminopyrimidine (TAP) and cyanuric acid modified with a hexanoic acid tail (CyCo6) can form hexameric discs through hydrogen bonding, which on their turn stack into micrometer-sized fibrils (Figure 1a). Hydrogels generated by such fibrils show a strong CD signal, variable in sign and amplitude, a result of the simultaneous formation of macroscopic homochiral domains. This can be confirmed by 2D CD spectroscopy on a thin sample, which provides an inhomogeneous pattern of positive and negative CD signals (Figure 1b), and by scanning electron microscopy (SEM), which reveals domains of opposite handedness in different sample positions (Figure 1c). Figure 1d shows the structure of a TAP–CyCo6 reconstructed through a combination of X-ray diffraction and MD simulations. Both the addition of 1 chiral derivative of CyCo6 over 1000 achiral molecules and the usage of mixtures of opposite enantiomers of CyCo6 derivatives with ∼6% ee result in the appearance of fully homochiral structures in the whole sample, while other chiral dopants, unrelated to CyCo6, show a weaker effect. The strong amplification supports the hypothesis that nucleic acid homochirality originated from a symmetry breaking at the supramolecular polymer level, even in solutions of largely achiral molecules.6b
Figure 1.

Supramolecular fibers from stacked hexads of nucleobase analogs. (a) Chemical structures of the nucleobase analogs and proposed model of the supramolecular polymer. (b) A space-resolved CD of TAP–CyCo6 solution, obtained by moving a thin sample in the path of a beam with a submm diameter, reveals the coexistence of different homochiral domains. (c) SEM images of TAP–CyCo6 assemblies in two different locations of a solution containing 30 mM of each monomer. (d) Snapshot of MD simulation. CyCo6 and TAP are depicted in green and blue, respectively. The simulation shows that sodium ions and water molecules, represented respectively as orange and red/white spheres in the space-filling representation, can be captured within the hexad stacks. Adapted with permission from ref (6b) and ref (6c). Copyright 2019 Wiley and Copyright 2021 American Chemical Society.
Porphyrins
Another paradigmatic class of flat molecules is constituted by porphyrins, naturally occurring macrocyclic organic compounds. Because of their electronic properties, structure, and ability to host metal ions in their central pocket, porphyrins play important roles in nature for pigmentation, oxygen transport, and electron transfer and find applications in molecular electronics, photovoltaics, and biomimetics. Several water-soluble achiral porphyrins, like meso-tetrakis(4-sulfonatophenyl)porphyrin (TPPS), form stable supramolecular aggregates in aqueous solutions, often called J-aggregates.12 Spontaneous symmetry breaking can occur, e.g., as a result of hydrodynamic forces during stirring, with the prevalence of chiral aggregates of one handedness. By using the aggregates as catalysts in an enantioselective chemical Diels–Alder reaction, the supramolecular chirality can be transferred top-down to the molecular level, with enantiomeric excess in the reaction products.12a Stronger induction of aggregate chirality can be obtained through the action of chiral additives. In a biologically inspired system, water-soluble aggregates of a cationic porphyrin (Figure 2a) are obtained through the addition of ATP, which imparts chirality to the stacked porphyrin molecules.12b These further assemble into right-handed double helices, as shown by CD spectra, light-scattering measurements, transmission electron microscopy (TEM) imaging, and MD simulations (Figure 2b,c). Interestingly, enzymatic hydrolysis of ATP triggers disassembly of the aggregates, which can be reversed by addition of fresh ATP. Self-assembly of porphyrins has been investigated also in organic solvents; in methanol and chloroform mixtures, for example, TPPS forms long nanofibers,12c while the addition of a chiral diaminocyclohexane induces shorter nanorods with specific handedness, as demonstrated by correlated CD spectra and TEM imaging.
Figure 2.

Chiral porphyrin aggregates. (a) Chemical structure of the porphyrin 1 and adenosine triphosphate (ATP). (b) Scheme of the proposed self-assembly pathway, with the formation of helical multiscale aggregates of porphyrin 1 induced by ATP and disassembled by alkaline phosphatase (ALP). (c,d) CD spectra (c) and size distribution from dynamic light scattering (d) of solutions of porphyrin 1 alone or with different types of phosphates. (e) TEM images of 1 alone (blue frame) and the right-handed double helical 1-ATP assembly (red frames). The white arrow points to a double helix structure, while the yellow arrow points to a single helix structure. (f) Snapshot of a molecular dynamics trajectory with porphyrin 1 and ATP. The left panel represents side and top views of the aggregated structure, while the right panel shows a side view of the aggregate with porphyrin alkyl-ammonium side chains and ATP molecules hidden for clarity. Adapted with permission from ref (12b). Copyright 2019 American Chemical Society.
Ordering with a Twist: Liquid Crystalline Phases
DNA Oligomers
The propensity of concentrated solutions of DNA double helices longer than 100 base pairs to organize in (chiral) nematic LC phases has long been known and understood as the consequence of their shape anisotropy. However, although the double helical shape is symmetric and well characterized, describing how the molecular chirality propagates to mesoscale properties is challenging. Indeed, the repulsive interactions between the right-handed helical patterns of negative charges induce an overall left-handed chiral torque, which also depends on geometrical features and on adsorbed counterions; however, at high concentration, steric hindrance of the helical grooves also plays a role, inducing a right-handed torque. The overall balance, reflected in the observed micrometer-scale periodicity of the left-handed N* phase, thus depends on the mean interhelical distance.13
An even richer scenario emerges for shorter duplexes, down to a few base pairs, as in this case the LC formation is mediated by end-to-end aggregation into reversible aggregates, with implications on the prebiotic emergence of nucleic acid strands.14 Short duplexes display LC phases similar to those observed in longer strands but at higher concentrations. As a consequence, depending on oligonucleotide length and sequence, electrostatic and steric torques become comparable, and both right- and left-handed N* can be observed,13a as shown in Figure 3a. The correlation between self-assembly, LC ordering, and chiral interactions is also reflected in the temperature dependence of the N* pitch, with stronger chirality for shorter aggregates.13b
Figure 3.

Liquid crystalline phases in DNA oligomers and their mixtures with Sunset Yellow. (a) Dependence of the inverse N* periodicity on DNA concentration c (lower x axis) and on the corresponding interaxial distance dH (upper x axis) for different self-complementary oligomer sequences. Insets show POM textures. Positive and negative values correspond to right- and left-handed N*, respectively. (b) POM images of pure SSY solutions at room temperature, with increasing concentration from left to right, showing the isotropic I, nematic N, and columnar M mesophases. (c) POM images of pure DNA solutions at room temperature, with increasing concentration from left to right, showing the chiral nematic N* and two columnar M and M2 mesophases. (d) POM images of a SSY-DNA mixture at molar ratio of 0.57 at room temperature with increasing concentration from left to right, displaying N*, M, and a biphasic MS mesophase. (e) POM images of the SSY-DNA in (d) at increasing temperatures, showing the transition from MS to M. (f) Phase diagrams of SSY-DNA mixtures as a function of concentration, molar ratio, and temperature. The lower panel shows the projection at 20 °C. (g) CD and visible absorbance spectra of isotropic SSY-DNA mixtures with fixed [SSY] = 0.5 mg/mL and [DNA] increasing from 0 to 150 mg/mL. Since only SSY absorbs in this region, the spectra show that achiral SSY molecules are influenced by the chiral DNA duplexes. Adapted with permission from ref (18) and ref (13a). Copyright 2018 American Physical Society and 2021 National Academy of Sciences.
Moreover, new opportunities to study the propagation of chirality come from the different modes of reversible aggregation in mixtures of natural d-DNA double helices and their mirror symmetrical l-DNA counterparts.15 Blunt ended duplexes self-assemble through stacking interactions, which are not enantioselective, and form mixed aggregates, while duplexes with dangling tails assemble through base-pairing, which is enantioselective and yields a continuous helical pattern, and form homochiral aggregates of both chiralities. The different composition of aggregates gives rise to a different dependence of the N* pitch on the ee.
Chromonics
Chromonics are flat molecules with aromatic, hydrophobic cores, stacking in water into linear reversible aggregates which in turn can align in LC phases.7 A few inherently chiral chromonic molecules have been reported, which form chiral aggregates, ordering in an N* phase.16 A mixture of enantiomers of a perylene-based chromonic system has been characterized as a function of chiral fraction, at fixed total concentration within the (chiral) nematic phase.16c The inverse N* pitch grows linearly with the chiral fraction across the whole range, suggesting that opposite enantiomers can fit equally well within the aggregates, without affecting the amount of order. Increasing temperature reduces the overall phase chirality at any given chiral fraction and also the helical twisting power (HTP) of the chiral fraction; this is interpreted as a result of the decreased average length of aggregates, opposite to that observed in DNA oligomers.13b
Chiral dopants, like sugars and amino acids, can induce an N* phase also in achiral chromonic molecules.17 The relatively low measured HTP values, i.e., the low effectiveness of dopants, may result from the very large length of the aggregates, as revealed by freeze fracture electron microscopy, and from the fact that dopants lie in between columnar aggregates, without directly affecting their structure.
A different situation occurs when mixing molecules capable of intercalation into each other and stronger interactions. This is the case of the chromonic sunset yellow (SSY) and a self-complementary DNA dodecamer, forming a double strand.18 Both molecules alone and their mixtures display isotropic, nematic, and columnar phases, as shown in polarized optical microscopy textures (POM, Figure 3b–e) and summarized in the phase diagram in Figure 3f. A uniform, mixed N* phase appears at all mixing fractions (Figure 3f). This is intriguing, as mixtures of SSY and disodium cromoglycate (DSCG), a different but structurally similar chromonic molecule, have been found to phase separate upon transition from the isotropic to nematic phase.19 The binding between SSY and DNA can be assessed through CD and absorption in the visible range, where DNA does not absorb. Upon increasing DNA concentration at fixed SSY fraction, the appearance of distinct CD peaks and the shift of an SSY absorption peak are indicative of intercalation and induction of a twisted arrangement (Figure 3g). At high concentration, two separate columnar phases of SSY and DNA coexist, which however become a homogeneous phase at high temperature (Figure 3e).
Braided Braids: Self-Assembly and Ordering of Peptides
Polypeptides are chains of chiral amino acids connected by peptide bonds, from few monomers up to long proteins. Even considering only natural, l-amino acids found in biomolecules, the larger “alphabet” of residues—as compared to nucleic acids—provides an exponentially larger number of accessible structures. This is reflected by a variety of secondary structures, including chiral α-helices and twisted β-sheets, and several types of fibrillar aggregates, whose shape is determined by electrostatic and hydrophobic interactions, hydrogen bonds, and steric hindrance of amino acid residues. On their turn, such anisotropic aggregates can merge into bundles and form structures on a larger scale, like disordered networks or liquid crystalline phases. Understanding and mastering the key design principles across length scales is a fascinating and challenging task, with countless opportunities for applications4,20 and, even more so, when different moieties are conjugated to each other and contribute to self-assembly and chiral transfer.
Twist Control through Sequence Patterns
The repetition, or alternation, of interaction patterns within oligopeptides can result in supramolecular self-assembly into common structures, despite the diversity of the amino acid sequences. Dipeptides, like, e.g., two aromatic phenylalanine (Phe) residues, are an extreme example of this, as they can form chiral structures with long-range order via π–π interactions and hydrogen bonding, whose features can be further modulated by electrostatic interactions.4 Different amyloid fibers share twisted β-structures connected to form helical filaments, whose periodicity is often controlled by electrostatic interactions.21 These in turn aggregate into larger twisted fibers and ribbons. On the other hand, subtle changes in the interaction patterns can have a profound impact on supramolecular chirality. Indeed, in short, truncated segments of the Serum Amyloid A protein and removal or mutation of single amino acids can induce the inversion of handedness in filaments and in the resulting amyloid fibers.20 In another system of short amphiphilic peptides, left-handed fibrillar intermediates order into right-handed nanotubes in a process guided by histidine side-chain interactions.22
Combining amino acids with opposite chirality within the same strand and comparing their self-assembly behavior to peptides of single chirality provide a convenient way to investigate the propagation mechanisms.23 In short amphiphilic peptides, this approach has allowed us to demonstrate that CD signals are dominated by the chirality of hydrophobic isoleucine residues. On the contrary, the handedness of supramolecular fibrils—determined through SEM and atomic force microscopy (AFM)—is dictated by the twist of β-sheets, which in turn depends on the chirality of a terminal lysine (Lys) residue.23a Interestingly, in Phe-Phe dipeptides capped with fluorenyl aromatic groups, an opposite chirality of the two residues enhances the formation of nanofibrils in water and other solvents, in conditions at which the homochiral dipeptide displays polymorphism, likely by favoring the orientation of aromatic groups in the molecule.23b
In heterochiral peptides, conformational constraints can also disclose access to self-assembly intermediates.23c Indeed, while homochiral strands formed by tryptophan residues and stabilized by hydrophilic segments (Figure 4a) readily form flat and rectangular nanostructures (Figure 4 c), heterochiral peptides with alternating d- and l-residues (W and w, respectively), while converging to the same final structures, show markedly slower assembly kinetics. The CD signal evolves over time (Figure 4b), and during the first hours, helices with opposite handedness appear, which in a couple of days unwind and associate into flat ribbons and crystals, a process reminiscent of the evolution of amyloid fibers (Figure 4d). In asymmetrically heterochiral peptides, further intermediates can form, including helices with variable pitch and helices with alternating twist and overtwisted, sausage-like structures. In such cases, the system remains kinetically trapped in an interlaced network, as summarized in the energy landscape in Figure 4e.
Figure 4.

Self-assembly intermediates in heterochiral peptides. (a) Chemical structures of the amphiphilic peptides T-W6, T-(wW)3, and T-W(wW)2. (b) Time evolution of CD spectra for T-(wW)3. (c) AFM images of solutions of the two enantiomeric peptides T-W6 and T-w6. (d) AFM images showing the time evolution of the heterochiral (racemic) peptide T-(wW)3, with the proposed structure of the chiral aggregates. (e) Energy landscape of self-assembly pathways for homochiral and heterochiral peptides. Adapted with permission from ref (23c). Copyright 2020 American Chemical Society.
Further control over supramolecular organization can arise in mixtures of opposite enantiomers. In a system of aromatic tripeptides,24 while the pure enantiomers form hollow, achiral nanofibers, in turn assembling into hydrogels of isotropic or aligned fibers, racemic mixtures form a hydrogel composed of chiral nanofibers at a fraction related to the ratio of the two enantiomers (Figure 5). This peculiar behavior is nested in the formation of heterodimeric building blocks, as revealed by NMR.
Figure 5.

Mixtures of tripeptide enantiomers. (a) Chemical structures of the LMW-L1 and LMW-D1 peptides. (b) Illustration of the self-assembly mechanisms for three peptide solutions. Pure LMW-L1 and LMW-D1 form different homodimers, which further assemble in isotropic and aligned structures, respectively. An equimolar mixture of the two creates heterodimers, assembling in chiral nanofibers. (c) Cryo-TEM images corresponding to the three situations. The scale bar represents 100 nm. Adapted with permission from ref (24). Copyright 2019 Springer.
Chiral Order Goes to Great Lengths
As in the case of nucleic acids, the supramolecular chirality can propagate to mesoscopic length scales, often with a subtle dependence on molecular features. In a family of 10-residue β-peptides, the sequence pattern and the presence of modified amino acids determine a helical conformation with well-defined lipophilic and hydrophilic surfaces.25 Depending on small variations in the net charge, in the spatial arrangement of charged residues and in peptide terminals, nanofibers can self-assemble in aqueous solutions, and a chiral nematic LC phase can emerge only from peptides with the right balance of side and end-to-end interactions.
A second interesting example, crossing into the colloidal scale, comes from β-lactoglobulin amyloid fibrils.26 The length distribution can be controlled through high shear forces, followed by dialysis, down to rod-like helical particles of a few hundreds of nm in length. Nucleation and growth of N and N* tactoids, as well as the bulk N* phase, are observed at increasing concentrations, with left-handed fibrils assembling in a right-handed phase as a result of the helical twist angle.26a Chiral ordering inside the droplets is only observed for sufficiently shortened helices, because of the reduced surface anchoring strength parallel to the interface.26b
Joining Different Worlds: Hybrid Twisted Structures
The self-assembly capability of peptides can be further expanded by their conjugation with different molecular moieties, which also allows regulation of chiral propagation toward fine-tuning of hierarchically structured biomaterials. A widely investigated approach is the addition of hydrophobic tails with various topologies, which promotes the emergence of a wealth of micellar, tubular, and lamellar structures27,28 with emphasis either on peptide interactions or on the amphiphilic self-assembly. Also in this case, the chiral interactions can propagate to the mesoscale.29
An impressive demonstration of how achiral electrostatic and van der Waals interactions can control shape selection in chiral nanoscale structures is provided by C12–16–K, a construct of a single Lys with alkyl chains of 12–16 carbons (Figure 6a).27 Screening of electrostatic interactions through salt addition directly enhances chiral interactions. More subtly, variations of pH determine the degree of Lys ionization and thus the strength of electrostatic interactions, which however remain long-ranged. This yields helical bilayers and helicoidal rolls (called cochleates), as reconstructed from experimental techniques over Å to μm length scales (Figure 6b–e) and molecular simulations including electrostatic and surface tension terms. At low pH, with fully ionized headgroups, the amphiphiles display high aspect ratio, planar crystalline bilayers, which fold into helicoidal rolls at increasing salt concentrations. At higher pH, helices are instead observed, whose radius monotonically increases with the decreasing degree of ionization. Furthermore, the strength of chiral interactions can be tuned by varying the ratio of the two enantiomers, resulting in mixed, achiral assemblies with weakened CD signal (Figure 6f).
Figure 6.

Electrostatic control of chiral structures. (a) Chemical structure of Cn–K. (b) Sketches of different chiral structures: helical ribbon, closed helical tubule, scroll-like cochleate, twisted ribbon. (c) Cryo-TEM images of l-C16–K forming flat ribbons and cochleate structures. (d) Structural phase diagram for C16–K assemblies as a function of ionic strength and pH. The inset shows the characteristic small- and wide-angle X-ray scattering intensity profiles for planar bilayers, helices, and cochleates. (e) Ex situ AFM image of C16–K helical ribbon at pH ∼ 8.5, drop-casted and dried on a Si (1 0 0) substrate. (f) CD spectra for C16–K molecular assemblies in solutions at 10 mM NaCl, pH ≪ pKa (left) and pH ≈ pKa (right). Adapted with permission from ref (27). Copyright 2022 American Chemical Society.
A linear structure with two alternating Lys and C12 tails (Figure 7a), encoding chirality, amphiphilicity, and hydrogen bond formation, sequentially passes through various 1D intermediates, including fibrils, twisted ribbons, and coiled helical ribbons, to eventually assemble into closed nanotubes28a (Figure 7b,c). Temperature-induced breaking of the hydrogen bonds and disordering of C12 chains result in reversible disassembly into spherical micelles (Figure 7d).
Figure 7.

Self-assembly process of an amphiphilic peptide. (a) Chemical structure and schematic structure of C12–β12. Red spheres and green ribbons represent hydrophilic and hydrophobic portions, respectively. (b) Reconstructed pathway of structural evolution. (c) Cryo-TEM images display the time evolution of the peptide solution at T = 25 °C, from a few minutes to several months. Scale bar 100 nm. (d) Cryo-TEM image of spherical micelles of radius ∼4 nm at 40 °C. The inset represents the molecular organization within the micelle. Adapted with permission from ref (28a). Copyright 2011 American Chemical Society.
The conjugation of a long alkyl chain to a dendritic peptide results in a bilayer structure, which forms hydrogels over a wide pH range.28b Moreover, accompanying the pH change, which determines the ionization of carboxylic acids, it self-assembles into different chiral nanostructures, from helical nanotubes to coiled superhelices and dendrite nanostructures. The modification of peptides, containing β-sheet forming domains, with hydrophobic palmitic acid tails allows the assembly of micellar helical nanoribbons in water.28c The width, periodicity, and even handedness of such ribbons depend on peptide sequence through the interplay among twist, promoted by β-sheets, molecular packing, and repulsion between charged residues.
The incorporation of ferrocene moieties in Phe-Phe dipeptides favors the appearance of chiral nanostructures in the presence of several different counterions (Figure 8a,b).30 Temperature controls the kinetics of self-assembly of the twisted β-sheets at the basis of all structures and thus their diameter and helical pitch. The polarity of the solvent further controls the balance of interactions, even inducing handedness inversion in the nanostructures (Figure 8c,d). The interaction with chiral diamines introduces geometry frustration, which can propagate up to the μm-scale in confined films, with the formation of ordered helical arrays.30b
Figure 8.

Different self-assembled structures of a ferrocene-modified peptide. (a) Molecular structure of Fc-FF, with carbon in gray, oxygen in red, nitrogen in blue, and hydrogen in white. (b) Chemical structures of the counterions used to direct the chiral self-assembly of Fc-FF, as shown in the sketch. (c) SEM images of the various supramolecular structures obtained. From top to bottom: (1) nanoscrews (scale bar 1 μm), (2) nanohelices formed via the twist of two crossed ribbons (scale bar 300 nm), (3) big twists (scale bar 200 nm), (4 and 5) left- and right-handed twisted ribbons (scale bar 100 nm), (6) rigid nanosprings (scale bar 100 nm), (7) tube-like helical ribbons (scale bar 50 nm). (d) Generic pathway for the hierarchical chiral self-assembly of Fc-FF in which strands of a β-sheet are represented as yellow wires. The handedness of the structures can be inverted from right to left by slightly increasing the solvent polarity or temperature. Adapted with permission from ref (30a). Copyright 2015 American Chemical Society.
As a last example in our quick ride through different modes of chiral self-assembly, we cannot forget hybrids of our two main characters, nucleobases and peptides.31 Such a combination can have multiple facets, from PNA, a DNA analogue with covalent pseudopeptide backbone, to the evergreen Phe-Phe dipeptide conjugated with nucleic acids.31a However, a simple mixture of Fmoc-protected glutamic acid and different nucleobases is an emblematic case of their rich interplay and of the role of achiral molecules in mediating chirality transfer.31b While the amino acid alone, in both its pure enantiomers L or D, does not self-assemble at all, the addition of purines (guanine and adenine without the chiral sugar) promotes the formation of hydrogels composed by helical nanostructures. In contrast, pyrimidines thymine and cytosine do not trigger self-assembly, suggesting that stronger π–π stacking, enhanced by hydrogen bonds, stabilizes the nanostructures. The handedness of helical assemblies depends on the chirality of the amino acid and can be imparted to the achiral dye thioflavin T, leading to a strong circularly polarized fluorescence.
Future Outlook
Based on our brief overview of chirality transfer in supramolecular systems, it is quite evident that (i) recurring motifs, like nanohelices and ribbons, are observed in widely different macromolecules and (ii) upon little changes in molecular details or environmental conditions, dramatic variations can occur in aggregate structure and chirality, as well as in their liquid crystalline ordering. Although the lack of a simple and unified description, or of a universal model system, may be frustrating, the emergence of new modes of chiral propagation is a constant source of wonder and adds new pieces in our understanding of the critical patterns of interactions. Moreover, it provides an ever growing toolbox for the tailoring of materials with controlled optical and mechanical properties or for enantioselective molecular recognition, filtration, or catalysis. Awareness about novel space- and time-resolved characterization techniques and modeling tools, developed for particular systems, can be beneficial for the investigation of other molecules, within a positive cross-contamination loop. We see this mini-review as a small contribution in this direction.
Acknowledgments
The authors thank Tommaso Bellini, Tommaso Fraccia, and Marco Todisco for useful discussions.
Biographies

Federico Caimi obtained his MSc in Chemical Sciences from the University of Milano, Italy, in 2019, with a thesis on self-assembly, liquid crystal formation, and reactivity of phosphorimidazolides of nucleotides. He then earned a PhD in Physics from the same university in 2023, under the supervision of Professor Tommaso Bellini, performing research on ferroelectric nematic materials. His interests span through various aspects of soft matter and liquid crystals, from organic synthesis to surface treatment and phase characterization.

Giuliano Zanchetta obtained a PhD in Physics at the University of Milano, Italy, in 2007. After postdoctoral stays at University of Colorado Boulder (USA) and University of Fribourg (Switzerland), he is now associate professor of applied physics at the University of Milano. His main research interests include the self-assembly and liquid crystalline ordering of nucleic acids and their prebiotic implications, the quantitative measurement of biomolecular interactions through optical label-free techniques, and the investigation of yielding properties of soft materials through active microrheology.
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Omegavirtual special issue “Nucleic Acids: A 70th Anniversary Celebration of DNA”.
References
- a Parton T. G.; Parker R. M.; van de Kerkhof G. T.; Narkevicius A.; Haataja J. S.; Frka-Petesic B.; Vignolini S. Chiral self-assembly of cellulose nanocrystals is driven by crystallite bundles. Nat. Commun. 2022, 13, 2657. 10.1038/s41467-022-30226-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nyström G.; Arcari M.; Adamcik J.; Usov I.; Mezzenga R. Nanocellulose Fragmentation Mechanisms and Inversion of Chirality from the Single Particle to the Cholesteric Phase. ACS Nano 2018, 12, 5141–5148. 10.1021/acsnano.8b00512. [DOI] [PubMed] [Google Scholar]; c Frka-Petesic B.; Vignolini S. So much more than paper. Nat. Photonics 2019, 13, 365–367. 10.1038/s41566-019-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belamie E.; Mosser G.; Gobeaux F.; Giraud-Guille M. M. Possible transient liquid crystal phase during the laying out of connective tissues: α-chitin and collagen as models. J. Phys.: Condens. Matter 2006, 18, S115–S129. 10.1088/0953-8984/18/13/S08. [DOI] [Google Scholar]
- Morrow S. M.; Bissette A. J.; Fletcher S. P. Transmission of chirality through space and across length scales. Nat. Nanotechnol. 2017, 12, 410–419. 10.1038/nnano.2017.62. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Wang Y.; Hamley I. W.; Qi W.; Su R.; He Z. Chiral self-assembly of peptides: Toward the design of supramolecular polymers with enhanced chemical and biological functions. Prog. Polym. Sci. 2021, 123, 101469. 10.1016/j.progpolymsci.2021.101469. [DOI] [Google Scholar]
- a Krieg E.; Bastings M. M. C.; Besenius P.; Rybtchinski B. Supramolecular Polymers in Aqueous Media. Chem. Rev. 2016, 116, 2414–2477. 10.1021/acs.chemrev.5b00369. [DOI] [PubMed] [Google Scholar]; b Bruckner E. P.; Stupp S. I. Designing supramolecular polymers with nucleation and growth processes. Polym. Int. 2022, 71, 590–595. 10.1002/pi.6384. [DOI] [Google Scholar]
- a Tschierske C.; Dressel C. Mirror Symmetry Breaking in Liquids and Their Impact on the Development of Homochirality in Abiogenesis: Emerging Proto-RNA as Source of Biochirality?. Symmetry 2020, 12, 1098. 10.3390/sym12071098. [DOI] [Google Scholar]; b Karunakaran S. C.; Cafferty B. J.; Weigert-Muñoz A.; Schuster G. B.; Hud N. V. Spontaneous Symmetry Breaking in the Formation of Supramolecular Polymers: Implications for the Origin of Biological Homochirality. Angew. Chem., Int. Ed. 2019, 58, 1453–1457. 10.1002/anie.201812808. [DOI] [PubMed] [Google Scholar]; c Alenaizan A.; Borca C. H.; Karunakaran S. C.; Kendall A. K.; Stubbs G.; Schuster G. B.; Sherrill C. D.; Hud N. V. X-ray Fiber Diffraction and Computational Analyses of Stacked Hexads in Supramolecular Polymers: Insight into Self-Assembly in Water by Prospective Prebiotic Nucleobases. J. Am. Chem. Soc. 2021, 143, 6079–6094. 10.1021/jacs.0c12010. [DOI] [PubMed] [Google Scholar]; d Menor Salván C.; Bouza M.; Fialho D. M.; Burcar B. T.; Fernández F. M.; Hud N. V. Prebiotic Origin of Pre-RNA Building Blocks in a Urea “Warm Little Pond” Scenario. ChemBioChem. 2020, 21, 3504–3510. 10.1002/cbic.202000510. [DOI] [PubMed] [Google Scholar]
- Lydon J. Chromonic liquid crystalline phases. Liq. Cryst. 2011, 38, 1663–1681. 10.1080/02678292.2011.614720. [DOI] [Google Scholar]
- Ghosh S.; Ray A.; Pramanik N. Self-assembly of surfactants: An overview on general aspects of amphiphiles. Biophys. Chem. 2020, 265, 106429. 10.1016/j.bpc.2020.106429. [DOI] [PubMed] [Google Scholar]
- a Davis J. T.; Spada G. P. Supramolecular architectures generated by self-assembly of guanosine derivatives. Chem. Soc. Rev. 2007, 36, 296–313. 10.1039/B600282J. [DOI] [PubMed] [Google Scholar]; b Wong A.; Ida R.; Spindler L.; Wu G. Disodium Guanosine 5‘-Monophosphate Self-Associates into Nanoscale Cylinders at pH 8: A Combined Diffusion NMR Spectroscopy and Dynamic Light Scattering Study. J. Am. Chem. Soc. 2005, 127, 6990–6998. 10.1021/ja042794d. [DOI] [PubMed] [Google Scholar]; c Gao M.; Harish B.; Berghaus M.; Seymen R.; Arns L.; McCallum S. A.; Royer C. A.; Winter R. Temperature and pressure limits of guanosine monophosphate self-assemblies. Sci. Rep. 2017, 7, 9864. 10.1038/s41598-017-10689-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Sánchez-Ferrer A.; Bagnani M.; Adamcik J.; Azzari P.; Hao J.; Song A.; Liu H.; Mezzenga R. Metal ions confinement defines the architecture of G-quartet, G-quadruplex fibrils and their assembly into nematic tactoids. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 9832–9839. 10.1073/pnas.1919777117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Gao C.; Zhang Z.; Liu X.; Chen Y.; Feng L. Kinetic control of chirality and circularly polarized luminescence in G-quartet materials. J. Mater. Chem. B 2021, 9, 7140–7144. 10.1039/D1TB00683E. [DOI] [PubMed] [Google Scholar]
- a Arlegui A.; Soler B.; Galindo A.; Arteaga O.; Canillas A.; Ribó J. M.; El-Hachemi Z.; Crusats J.; Moyano A. Spontaneous mirror-symmetry breaking coupled to top-bottom chirality transfer: from porphyrin self-assembly to scalemic Diels–Alder adducts. Chem. Commun. 2019, 55, 12219–12222. 10.1039/C9CC05946F. [DOI] [PubMed] [Google Scholar]; b Li Z.; Zeman C. J.; Valandro S. R.; Bantang J. P. O.; Schanze K. S. Adenosine Triphosphate Templated Self-Assembly of Cationic Porphyrin into Chiral Double Superhelices and Enzyme-Mediated Disassembly. J. Am. Chem. Soc. 2019, 141, 12610–12618. 10.1021/jacs.9b04133. [DOI] [PubMed] [Google Scholar]; c Rong Y.; Chen P.; Liu M. Self-assembly of water-soluble TPPS in organic solvents: From nanofibers to mirror imaged chiral nanorods. Chem. Commun. 2013, 49, 10498–10500. 10.1039/c3cc46467a. [DOI] [PubMed] [Google Scholar]
- a Zanchetta G.; Giavazzi F.; Nakata M.; Buscaglia M.; Cerbino R.; Clark N. A.; Bellini T. Right-handed double-helix ultrashort DNA yields chiral nematic phases with both right- and left-handed director twist. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 17497–17502. 10.1073/pnas.1011199107. [DOI] [PMC free article] [PubMed] [Google Scholar]; b De Michele C.; Zanchetta G.; Bellini T.; Frezza E.; Ferrarini A. Hierarchical Propagation of Chirality through Reversible Polymerization: The Cholesteric Phase of DNA Oligomers. ACS Macro Lett. 2016, 5, 208–212. 10.1021/acsmacrolett.5b00579. [DOI] [PubMed] [Google Scholar]
- a Nakata M.; Zanchetta G.; Chapman B. D.; Jones C. D.; Cross J. O.; Pindak R.; Bellini T.; Clark N. A. End-to-End Stacking and Liquid Crystal Condensation of 6– to 20–Base Pair DNA Duplexes. Science 2007, 318, 1276–1279. 10.1126/science.1143826. [DOI] [PubMed] [Google Scholar]; b Smith G. P.; Fraccia T. P.; Todisco M.; Zanchetta G.; Zhu C.; Hayden E.; Bellini T.; Clark N. A.. Backbone-free duplex-stacked monomer nucleic acids exhibiting Watson–Crick selectivity. Proceedings of the National Academy of Sciences of the United States of America 2018; Vol. 115. [DOI] [PMC free article] [PubMed]
- Rossi M.; Zanchetta G.; Klussmann S.; Clark N. A.; Bellini T. Propagation of Chirality in Mixtures of Natural and Enantiomeric DNA Oligomers. Phys. Rev. Lett. 2013, 110, 107801. 10.1103/PhysRevLett.110.107801. [DOI] [PubMed] [Google Scholar]
- a Naidu J. J.; Bae Y.-J.; Jeong K.-U.; Lee S.-H.; Lee M.-H. Synthesis of New Chiral Chromonic Lyotropic Liquid Crystal Based on Perylenebis(dicarboximide). Bulletin of the Korean Chemical Society 2009, 30, 935–937. 10.5012/bkcs.2009.30.4.935. [DOI] [Google Scholar]; b Yang S.; Wang B.; Cui D.; Kerwood D.; Wilkens S.; Han J.; Luk Y.-Y. Stereochemical Control of Nonamphiphilic Lyotropic Liquid Crystals: Chiral Nematic Phase of Assemblies Separated by Six Nanometers of Aqueous Solvents. J. Phys. Chem. B 2013, 117, 7133–7143. 10.1021/jp401382h. [DOI] [PubMed] [Google Scholar]; c Ando J. K.; Collings P. J. A chiral–racemic lyotropic chromonic liquid crystal system. Soft Matter 2021, 17, 1409–1414. 10.1039/D0SM02013C. [DOI] [PubMed] [Google Scholar]
- Shirai T.; Shuai M.; Nakamura K.; Yamaguchi A.; Naka Y.; Sasaki T.; Clark N. A.; Le K. V. Chiral lyotropic chromonic liquid crystals composed of disodium cromoglycate doped with water-soluble chiral additives. Soft Matter 2018, 14, 1511–1516. 10.1039/C7SM02262J. [DOI] [PubMed] [Google Scholar]
- Theis J. G.; Smith G. P.; Yi Y.; Walba D. M.; Clark N. A. Liquid crystal phase behavior of a DNA dodecamer and the chromonic dye Sunset Yellow. Phys. Rev. E 2018, 98, 042701. 10.1103/PhysRevE.98.042701. [DOI] [Google Scholar]
- Yamaguchi A.; Smith G. P.; Yi Y.; Xu C.; Biffi S.; Serra F.; Bellini T.; Zhu C.; Clark N. A. Phases and structures of sunset yellow and disodium cromoglycate mixtures in water. Phys. Rev. E 2016, 93, 012704. 10.1103/PhysRevE.93.012704. [DOI] [PubMed] [Google Scholar]
- Rubin N.; Perugia E.; Wolf S. G.; Klein E.; Fridkin M.; Addadi L. Relation between Serum Amyloid A Truncated Peptides and Their Suprastructure Chirality. J. Am. Chem. Soc. 2010, 132, 4242–4248. 10.1021/ja909345p. [DOI] [PubMed] [Google Scholar]
- a Adamcik J.; Mezzenga R. Adjustable twisting periodic pitch of amyloid fibrils. Soft Matter 2011, 7, 5437. 10.1039/c1sm05382e. [DOI] [Google Scholar]; b Assenza S.; Adamcik J.; Mezzenga R.; De Los Rios P. Universal Behavior in the Mesoscale Properties of Amyloid Fibrils. Phys. Rev. Lett. 2014, 113, 268103. 10.1103/PhysRevLett.113.268103. [DOI] [PubMed] [Google Scholar]
- Ma X.; Zhao Y.; He C.; Zhou X.; Qi H.; Wang Y.; Chen C.; Wang D.; Li J.; Ke Y.; Wang J.; Xu H. Ordered Packing of β-Sheet Nanofibrils into Nanotubes: Multi-hierarchical Assembly of Designed Short Peptides. Nano Lett. 2021, 21, 10199–10207. 10.1021/acs.nanolett.1c02944. [DOI] [PubMed] [Google Scholar]
- a Wang M.; Zhou P.; Wang J.; Zhao Y.; Ma H.; Lu J. R.; Xu H. Left or Right: How Does Amino Acid Chirality Affect the Handedness of Nanostructures Self-Assembled from Short Amphiphilic Peptides?. J. Am. Chem. Soc. 2017, 139, 4185–4194. 10.1021/jacs.7b00847. [DOI] [PubMed] [Google Scholar]; b Gil A. M.; Casanovas J.; Mayans E.; Jiménez A. I.; Puiggalí J.; Alemán C. Heterochirality Restricts the Self-Assembly of Phenylalanine Dipeptides Capped with Highly Aromatic Groups. J. Phys. Chem. B 2020, 124, 5913–5918. 10.1021/acs.jpcb.0c04513. [DOI] [PubMed] [Google Scholar]; c Lee H.-s.; Lim Y.-b. Slow-Motion Self-Assembly: Access to Intermediates with Heterochiral Peptides to Gain Control over Alignment Media Development. ACS Nano 2020, 14, 3344–3352. 10.1021/acsnano.9b09070. [DOI] [PubMed] [Google Scholar]
- Yang X.; Lu H.; Tao Y.; Zhang H.; Wang H. Controlling supramolecular filament chirality of hydrogel by co-assembly of enantiomeric aromatic peptides. J. Nanobiotechnol. 2022, 20, 77. 10.1186/s12951-022-01285-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz W. C.; Yuwono V. M.; Drake R.; Hartgerink J. D.; Abbott N. L.; Gellman S. H. Lyotropic Liquid Crystals Formed from ACHC-Rich β-Peptides. J. Am. Chem. Soc. 2011, 133, 13604–13613. 10.1021/ja204874h. [DOI] [PubMed] [Google Scholar]
- a Nyström G.; Arcari M.; Mezzenga R. Confinement-induced liquid crystalline transitions in amyloid fibril cholesteric tactoids. Nat. Nanotechnol. 2018, 13, 330–336. 10.1038/s41565-018-0071-9. [DOI] [PubMed] [Google Scholar]; b Bagnani M.; Nyström G.; De Michele C.; Mezzenga R. Amyloid Fibrils Length Controls Shape and Structure of Nematic and Cholesteric Tactoids. ACS Nano 2019, 13, 591–600. 10.1021/acsnano.8b07557. [DOI] [PubMed] [Google Scholar]
- McCourt J. M.; Kewalramani S.; Gao C.; Roth E. W.; Weigand S. J.; Olvera de la Cruz M.; Bedzyk M. J. Electrostatic Control of Shape Selection and Nanoscale Structure in Chiral Molecular Assemblies. ACS Central Science 2022, 8, 1169. 10.1021/acscentsci.2c00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ziserman L.; Lee H.-Y.; Raghavan S. R.; Mor A.; Danino D. Unraveling the Mechanism of Nanotube Formation by Chiral Self-Assembly of Amphiphiles. J. Am. Chem. Soc. 2011, 133, 2511–2517. 10.1021/ja107069f. [DOI] [PubMed] [Google Scholar]; b Duan P.; Qin L.; Zhu X.; Liu M. Hierarchical Self-Assembly of Amphiphilic Peptide Dendrons: Evolution of Diverse Chiral Nanostructures Through Hydrogel Formation Over a Wide pH Range. Chemistry – A European Journal 2011, 17, 6389–6395. 10.1002/chem.201003049. [DOI] [PubMed] [Google Scholar]; c Sangji M. H.; Sai H.; Chin S. M.; Lee S. R.; R Sasselli I.; Palmer L. C.; Stupp S. I. Supramolecular Interactions and Morphology of Self-Assembling Peptide Amphiphile Nanostructures. Nano Lett. 2021, 21, 6146–6155. 10.1021/acs.nanolett.1c01737. [DOI] [PubMed] [Google Scholar]
- Jin H.-E.; Jang J.; Chung J.; Lee H. J.; Wang E.; Lee S.-W.; Chung W.-J. Biomimetic Self-Templated Hierarchical Structures of Collagen-Like Peptide Amphiphiles. Nano Lett. 2015, 15, 7138–7145. 10.1021/acs.nanolett.5b03313. [DOI] [PubMed] [Google Scholar]
- a Wang Y.; Qi W.; Huang R.; Yang X.; Wang M.; Su R.; He Z. Rational Design of Chiral Nanostructures from Self-Assembly of a Ferrocene-Modified Dipeptide. J. Am. Chem. Soc. 2015, 137, 7869–7880. 10.1021/jacs.5b03925. [DOI] [PubMed] [Google Scholar]; b Wang Y.; Li Q.; Zhang J.; Qi W.; You S.; Su R.; He Z. Self-Templated, Enantioselective Assembly of an Amyloid-like Dipeptide into Multifunctional Hierarchical Helical Arrays. ACS Nano 2021, 15, 9827–9840. 10.1021/acsnano.1c00746. [DOI] [PubMed] [Google Scholar]
- a Datta D.; Tiwari O.; Gupta M. K. Self-Assembly of Diphenylalanine–Peptide Nucleic Acid Conjugates. ACS Omega 2019, 4, 10715–10728. 10.1021/acsomega.9b00047. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Deng M.; Zhang L.; Jiang Y.; Liu M. Role of Achiral Nucleobases in Multicomponent Chiral Self-Assembly: Purine-Triggered Helix and Chirality Transfer. Angew. Chem., Int. Ed. 2016, 55, 15062–15066. 10.1002/anie.201608638. [DOI] [PubMed] [Google Scholar]