

Abstract
In recent years, Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system has become the most popular one for genome editing. When the nuclease domains of Cas9 protein are mutated into deactivated form (dCas9), CRISPR/dCas9 still retains the ability to bind the targeted DNA sequence, but loses the endonuclease cleavage activity. Taking advantage of the characteristics of this engineered nuclease inactive Cas9, the CRISPR/dCas system has been repurposed into versatile RNA-guided, DNA-targeting platforms, such as genome imaging, gene regulation, and epigenetic modification. Specifically, fusion of dCas9 with activation domains allows specific and efficient transcriptional activation on a genome-wide scale among diverse organisms. The purpose of this chapter is to review most important the recently published literature on CRISPR/dCas9-based transcriptional activation systems. Compared with the conventional approaches for enhancement of the expression of specific genes of interest, CRISPR/Cas9-based system has emerged as a promising technology for genome regulation, allowing specificity, convenience, robustness, and scalability for endogenous gene activation.
Keywords: CRISPR/Cas, Gene regulation, Transcriptional activation, Endogenous gene activation
10.1. Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) protein systems provide adaptive immunity against viruses and plasmids in bacteria and archaea. In contrast to type I and III CRISPR/Cas systems which employ a set of Cas proteins for RNA-guided immune surveillance, the type II bacterial CRISPR/Cas system uses only a single Cas protein, known as Cas9, to mediate foreign DNA recognition and cleavage [16]. In the process, CRISPR RNA (crRNA) hybridizes with cognate trans-activating crRNA (tracrRNA) to form a unique dual-RNA structure that directs Cas9 to specific DNA target site that is complementary to the 20-nucleotide (nt), guide-RNA sequence and further introduces site-specific double-stranded breaks (DSBs) in target DNA upon recognition of the protospacer adjacent motif (PAM) sequence [12, 17]. Notably, the single chimeric guide RNA (sgRNA) mimicking the natural dual RNA by fusing crRNA with tracrRNA via a tetraloop is sufficient to guide the endonuclease Cas9 to specific DNA target site for DNA degradation (Fig. 10.1a). By changing the 20-nucleotide guide-RNA sequence located on the 5′ end of sgRNA, this simplified two-component CRISPR–Cas9 system can be easily programmed to target virtually any DNA sequence of interest in the genome. In the cells, the generated site-specific DSBs by CRISPR–Cas9 can be further repaired, either by the error-prone nonhomologous end joining (NHEJ) pathway or by high-fidelity-homology-directed repair (HDR) pathway when a repair DNA template is present [11] (Fig. 10.1a). Since the first demonstration of its power for genome editing in mammalian cells [9, 22], CRISPR RNA-guided Cas9 system has drawn worldwide attention due to its simplicity and robustness and quickly become the most common tool for genome engineering in a variety of organisms [11].
Fig. 10.1. Schematic diagram showing how CRISPR/Cas9 system is used for genome engineering.
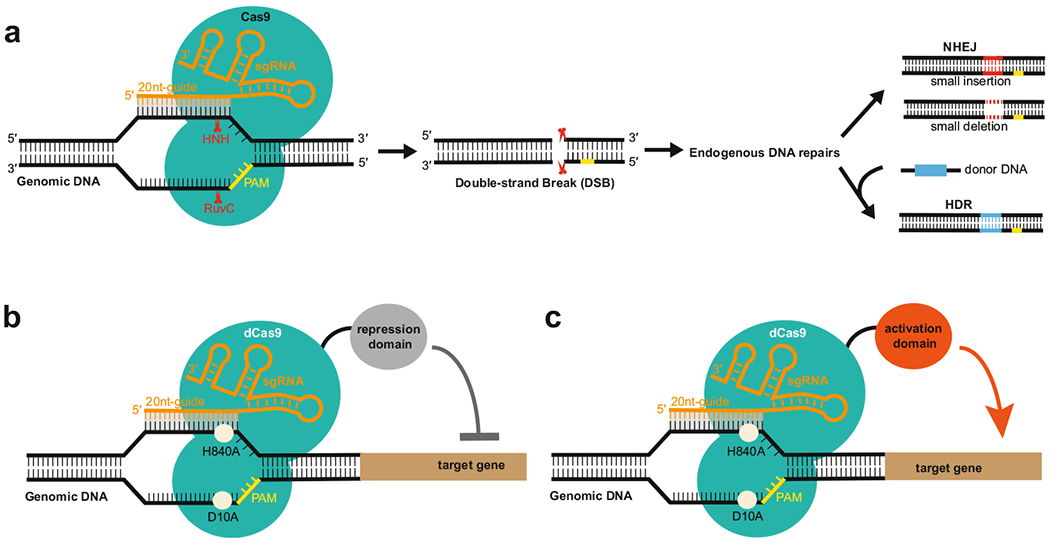
(a) Cas9 is guided by an sgRNA to a specific DNA locus, where HNH and RuvC nuclease domains cut the double-strand DNA to form a double-stranded break (DSB). The generated DSB is further repaired, either by the error-prone nonhomologous end joining (NHEJ) pathway or by high-fidelity homology-directed repair (HDR) pathway when a repair DNA template is present. (b) Activation or repression domain is fused to the catalytically inactive Cas9 variant for gene regulation
Cas9 is a multidomain and multifunctional DNA endonuclease [16]. It contains two distinct nuclease domains responsible for double-stranded (ds) DNA cleavage: the HNH domain of Cas9 cleaves the target DNA strand, while the RuvC-like domain of Cas9 cleaves the nontarget DNA strand [12, 17]. Mutations in both nuclease domains (D10A: Asp10 → Ala, H840A: His840 → Ala) result in an RNA-guided DNA-binding protein without endonuclease activity [17, 24]. This engineered nuclease-deficient Cas9, termed dCas9, when fused to effector domains with distinct regulatory functions, enables the repurposing of the CRISPR-Cas9 system to a general platform for RNA-guided DNA targeting without cleavage activity (Fig. 10.1b), thereby allowing versatile genome modification beyond permanent genome editing [10], such as gene regulation [13, 24], epigenetic modulation [19], and live-cell imaging [7]. Specifically, fusion of dCas9 to activation effector domains allows specific and efficient transcriptional activation on a genome-wide scale in diverse organisms [14].
10.2. CRISPRa Activation Systems
CRISPR/dCas9-mediated gene activation (hereafter referred to as CRISPRa) systems, consisting of dCas9-activation domain fusion proteins and sgRNA, can target the specific promoter or enhancer region of the gene of interest. The activation domain can be one or several activation domains, such as VP64 acidic transactivation domain (four copies of Herpes simplex virus protein 16), or can be full length or part of an epigenetic modifier, such as the core of histone acetyltransferase p300. Therefore, CRISPRa can function by either directly activating transcription, or modifying the chromatin conformation, or function through recruiting additional transcriptional and/or epigenetic activators to the targeted region. Basically, depending on which component of CRISPR/dCas9 is fused with the activation domain, there are at least three categories of CRISPRa systems: dCas9-activation domain fusion protein, sgRNA-activation domain fusion protein, or combined CRISPRa system with both dCas9- and sgRNA-conjugated with activation domains. For each category, they can also be further classified depending on what the activation domain is. These CRISPRa platforms will be discussed in detail in the following sections (Fig. 10.2).
Fig. 10.2. Different types of CRISPRa activation systems.
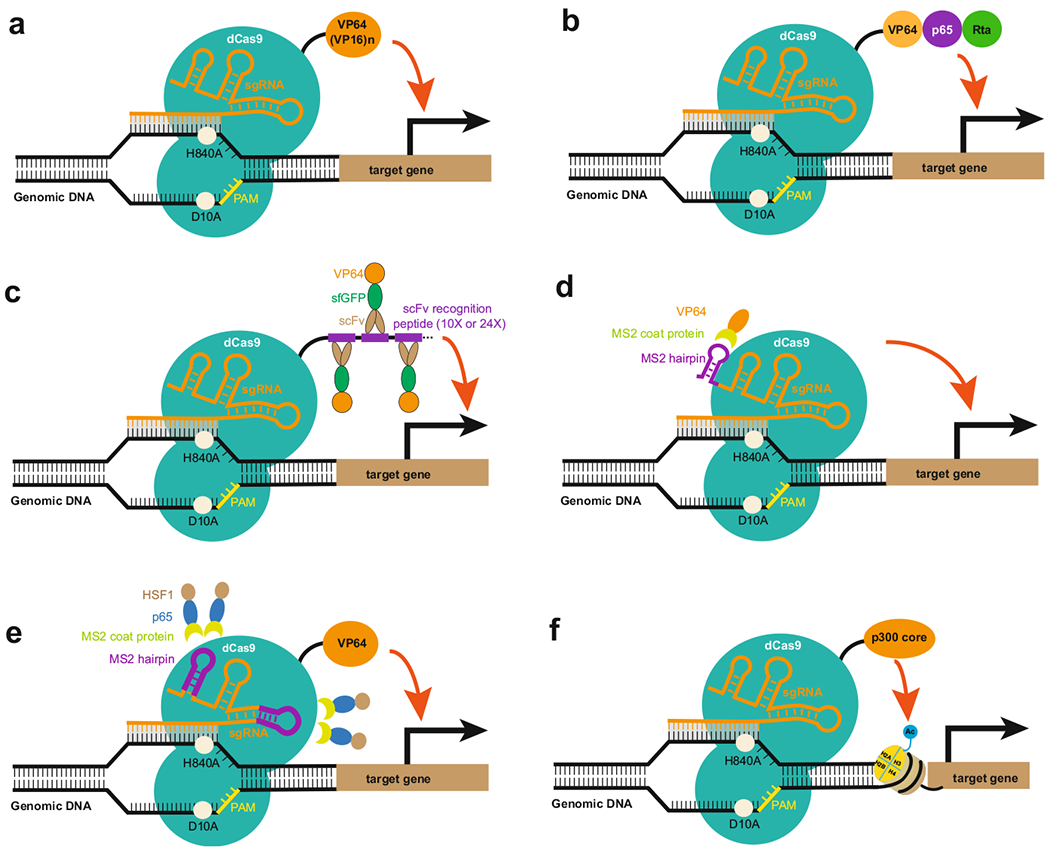
(a) The dCas9–VP64 (or multiple copies of VP16) system (b) The dCas9–VPR system (c) The dCas9–SunTag system (d) The sgRNA-activation domain system (e) The SAM system (f) The dCas9–p300 core system
10.2.1. The dCas9–VP64 CRISPRa System
The dCas9–VP64 CRISPRa system, first reported in 2013 as being able to activate targeted endogenous genes, represents the first-generation CRISPRa system, while all the other further improved versions are generally considered as second-generation activation systems. When dCas9 is genetically fused with a C-terminal VP64 acidic transactivation domain (four copies of Herpes simplex virus protein 16), it can activate both reporter gene and endogenous genes with a single sgRNA by transient delivery into mammalian cells (Fig. 10.2a). In addition, the use of multiple sgRNAs was able to achieve synergistic activation of a broad range of selected genes (interleukin 1 receptor antagonist, IL1RN), achaete-scute family bHLH transcription factor 1 (ASCL1), nanog homeobox (NANOG), myogenic differentiation 1 (MYOD1), hemoglobin subunit gamma ½ (HBG1/2), vascular endothelial growth factor A (VEGFA), and neurotrophin 3 (NTF3). Furthermore, RNA sequencing demonstrated that targeted gene activation was quite specific with no detectable off-target gene activation [21, 23].
As expected, increasing the number of VP16 repeat domains, such as dCas9–VP96, VP64–dCas9–VP64, dCas9–VP160, and dCas9–VP192 (Fig. 10.2a), has been shown to more efficiently upregulate the expression of endogenous genes, such as interleukin 1 receptor antagonist (IL1RN), SRY-Box2 (SOX2), POU class 5 homeobox 1 (POU5F1 or OCT4), both at mRNA and/or protein levels [1, 4, 8, 18]. Among them, the dCas9–VP192 leads to the highest increase in OCT4 expression levels, up to about 70-fold. Furthermore, human skin fibroblasts can be reprogrammed into inducing pluripotent stem cells (iPSCs) by replacing OCT4 overexpression with dCas9–VP192-mediated activation of endogenous OCT4. The epigenetic changes at OCT4 distal enhancer induced by CRISPRa were investigated and shown to have more active histone mark H3K27Ac, consistent with the previous report that the VP64 transactivation domain recruits the activating complex component p300 and facilitates histone acetylation [1]. In another study, Black et al. found that VP64–dCas9–VP64-mediated endogenous gene activation of mouse neuronal transcription factors Brn2, Ascl1, and Myt1l (BAM factors) directly reprogrammed cultured primary mouse embryonic fibroblasts (PMEFs) to functional induced neuronal cells[3]. Mechanistically, they found that the rapid and sustained elevated levels of endogenous gene expression corresponded to an increase of the epigenetically active markers H3K27ac and H3K4me3 at the target loci. Similar to dCas9–VP64, the efficient activation of endogenous genes also required multiple sgRNAs. In addition, the enhancement of gene activation was also observed with multiple sgRNAs tiling the promoter region, which suggested that recruitment of more activators could be helpful for increasing activation efficiency. This strategy was applied for the development of the second generation of CRISPR activation systems.
10.2.2. The dCas9–SunTag CRISPRa System
SunTag, a protein scaffold with repeating peptide epitope array, that can recruit multiple copies of antibody–activator fusion protein, has been initially developed for imaging of single molecule in living cells. When antibody–VP64 fusion protein was delivered with dCas9-SunTag fusion protein, the system demonstrated strong activation of endogenous gene expression. In one study, Marvin et al. used dCas9 fused with a carboxy-terminal SunTag array consisting of ten copies of a small peptide epitope, and recruited theoretically ten copies of single-chain variable fragment (scFV)–superfolded GFP (sfGFP)–VP64 (scFV–sfGFP–VP64) antibody–activator fusion proteins to a single dCas9 (Fig. 10.2c). Using the dCas9–SunTag–10x (scFV–sfGFP–VP64) system, 10–40-fold activation of the C-X-C Motif Chemokine Receptor 4 (CXCR4) gene was achieved with only one sgRNA, which led to the manipulation of cell migration. Using the SunTag system, Gilbert et al. performed a saturating screen in which they tested the activity of every unique sgRNA broadly tiling around the transcription start sites (TSSs) of 49 genes known to modulate cellular susceptibility to ricin, and observed a peak of active sgRNAs for SunTag CRISPRa system at −400 to −50 bp upstream from the transcription start site (TSS) [26].
10.2.3. The dCas9–VPR CRISPRa System
The tripartite activator domain that consists of VP64, Nuclear Factor NF-κ-B P65 subunit activation domain (p65AD) and Epstein–Barr virus R transactivator (Rta) (VPR) was developed to enhance the CRISPR/dCas9-based activation of endogenous genes (Fig. 10.2b). A set of genes related to cellular reprogramming, development, and gene therapy were activated with three to four gRNAs delivered in concert. When compared to the dCas9–VP64 activator, dCas9–VPR showed significantly (22–320-fold) greater activation of endogenous targets than dCas9–VP64. Furthermore, in accordance with previous studies, we noted an inverse correlation between basal expression level and relative expression gain induced by CRISPR activation systems [5].
10.2.4. The sgRNA-Activation Domain of CRISPRa System
In addition to fusing different transactivation domains to either the amino or carboxy terminus of the dCas9 protein, sgRNA can also be engineered to gain more robust activation. Zalatan et al. first introduced a single-RNA hairpin domain to the end of the sgRNA, connected by a two-base linker. For the recruitment RNA modules, they used the well-characterized viral RNA sequences MS2, PP7, and Com, which are recognized by the MCP, PCP, and Com RNA-binding proteins, respectively. Then they fused the transcriptional activation domain VP64 to each of the corresponding RNA-binding proteins for the purpose of the activation of targeted genes (Fig. 10.2d). On the other hand, when a repression domain KRAB is engineered into RNA-binding proteins, the system is good for transcriptional inhibition. Overall, the successful application of scaffold RNA-mediated transcriptional control in human and yeast cells paves the way for simultaneous ON/OFF gene regulatory switches mediated by orthogonal RNA-binding proteins fused to transcriptional activators (VP64) or repressors (KRAB) [29].
10.2.5. The Combined CRISPRa System
Based on the crystal structure of the Streptococcus pyogenes dCas9 (D10A/H840A) in complex with a single-guide RNA (sgRNA) and complementary target DNA, Konermann et al. developed synergistic activation mediator (SAM) system [20]. They selected a minimal hairpin aptamer, which selectively binds dimerized MS2 bacteriophage coat proteins (MCP), and appended it to the sgRNA tetraloop and stem loop 2 (Fig. 10.2e). Together with MS2-mediated transactivation factors MCP-p65AD-heat shock factor 1 (HSF1), dCas9–VP64 significantly enhanced the efficiency of activation of protein-coding genes and long noncoding RNAs (lincRNA) with one single-guide RNA, and enabled multiplexed activation of ten genes simultaneously. The ability to activate target genes using individual sgRNAs greatly facilitates the development of pooled, genome-wide transcriptional activation screening. Based on the SAM system, they successfully performed a screening for genes that confer resistance to a BRAF inhibitor in melanoma cells [20].
10.2.6. The dCas9–Epigenetic Modifier CRISPRa System
The dCas9 can also be fused with an epigenetic modifier to directly manipulate the epigenetic states at the enhancer region, thereby activating the targeted genes. This system uses different mechanism of action from the dCas9-activating transcription factor fusion protein systems mentioned as above. While the activator domains used in the previous engineered transcriptional factors such as VP64 act as scaffolds for recruiting multiple components of the preinitiation complex including transcriptional and epigenetic factors, and do not enzymatically modulate the chromatin state directly, the dCas9–epigenetic modifier fusion protein directly alter the specific epigenetic marks at specific location.
In one study, fusion of dCas9 to the catalytic core of the transcription activator acetyltransferase p300 (dCas9–p300core), a highly conserved acetyltransferase involved in a wide range of cellular processes, has been demonstrated to activate genes in human cells (Fig. 10.2f). The fusion protein catalyzes acetylation of histone H3 lysine 27 at its target sites, leading to specific and robust transcriptional activation of target genes including IL1RN, Myogenic Differentiation 1 (MYOD) and OCT4 from both promoters and enhancers with an individual guide RNA [15].
With the expansion of the CRISPRa toolbox, it will be necessary to compare the activation by these different systems across many endogenous genes in a variety of cell types, in order to determine which tool is best suited for specific genes and cell types. Recently, Chavez et al. performed a series of experiments in human embryonic kidney (HEK) 293T cells to compare the activation efficiency of many published CRISPRa activation systems, and three from the second generation in particular – VPR, SAM, and SunTag – appeared to be the most potent [6]. For nine selected coding genes and noncoding genes, the activation levels can reach up to several orders of magnitude higher than those of the first-generation dCas9–VP64 activator. Among the three, SAM seems to deliver high levels of gene induction most consistently, although none of the three was obviously superior to the other.
In addition to the application of CRISPRa in mammalian cells, CRISPR/Cas9-based activation system was also tested in the bacteria, Saccharomyces cerevisiae and Drosophila melanogaster cells for activating endogenous loci [6]. For example, a fusion of dCas9 with the ω-subunit of the E. coli polymerase allowed assembly of the holoenzyme for reporter gene activation in E. coli. Activation levels depended on the distance between the dCas9 binding sequence and the promoter element. It is possible that activation can be further optimized by changing the protein linker between dCas9 and the activation domain and/or by using different activation domains [2].
10.3. Advantages of CRISPRa System
The dCas9-guide RNA-mediated DNA target recognition requires both the PAM sequence in target DNA and Watson–Crick base pairing between the 20-nt guide RNA sequence and the complementary target DNA sequence. It has been shown that the sequences fully complementary to the guide RNA but lacking a nearby PAM are ignored by CRISPR/Cas9 system [25]. Compared with small activating RNA-mediated gene activation which only depend on Watson–Crick base pairing between mRNAs and saRNAs, the two-component requirement of CRISPR/Cas9 recognition renders more specificity with minimal off-target effects and we need only consider off-targets adjacent to a PAM, because potential targets lacking a PAM are unlikely to be interrogated.
In addition to small activating RNA, customized DNA-binding proteins such as zinc-finger proteins or transcription activator-like effectors (TALEs) have been used as tools for sequence-specific DNA targeting and gene regulation. These proteins robustly target DNA through programmable DNA-binding domains and can recruit effectors for transcription activation in a modular way. However, because each DNA-binding protein needs to be individually designed, the construction and delivery for the purpose of simultaneously regulating multiple loci is technically challenging. In contrast, one of the benefits of dCas9-based transcription effectors over the customized DNA-binding proteins is the ease with which multiple loci can be regulated, with only single-guide RNA (sgRNA) for each additional locus one desires to activate.
The conventional methods for gene overexpression include the use of cDNA overexpression vectors or cDNA libraries. However, cloning large cDNA sequences into viral vectors and manipulating several gene isoforms simultaneously are difficult. Also, the cDNA constructs often do not capture the full complexity of transcript isoforms, and they are independent of the endogenous regulatory context. Additionally, synthesizing large-scale libraries for genome-wide screening is not cost effective. Therefore, CRISPRa system has emerged as an ideal technology for genome regulation, providing specificity, convenience, robustness, and scalability for gene activation.
10.4. Applications and Limitations of CRISPRa System
CRISPR-based activation system could be applied to regulate gene expression in a variety of biological processes, including stem cell differentiation, silenced gene activation, genetic defect compensation, cell fate engineering, and genome-wide screening. To study whether CRISPRa could be used for direct cell reprogramming, Black et al. used a dCas9 with both N-terminal and C-terminal VP64 transactivation domains (VP64–dCas9–VP64) to achieve multiplex activation of the neurogenic factors Brn2, Ascl1, and Myt1l (BAM factors) and demonstrated direct cellular reprogramming from fibroblast to induced neuronal cells through targeted activation of endogenous genes [3].
Another example is for HIV treatment. Although the combined antiretroviral therapies (cARTs) have had a marked impact on the treatment and progression of HIV/AIDS, the most significant limitation of currently available cARTs is the inability to extinguish the integrated latent HIV reservoirs, resulting in a persistent infection even under lifelong treatment. A promising strategy to eradicate latent HIV reservoirs is to reactivate the dormant virus in the presence of combined antiretroviral therapies (cARTs). Recently, several groups simultaneously reported that CRISPR-based activation is highly effective at inducing transcriptional activation of latent HIV-1 infection specifically in human T cells, providing an exciting new avenue towards latent HIV therapy (Saayman et al. 2016).
Whereas loss-of-function screens can be conducted using RNAi or Cas9-based tools, gain-of-function screens have been confined to cDNA overexpression libraries. Compared with all the limitations with the available cDNA libraries, CRISPRa-based targeted gene regulation on a genome-wide scale is a powerful strategy for interrogating, perturbing, and engineering biological systems. Taking advantage of the robust SAM system, Konermann et al. performed a genome-wide screening for genes that, upon activation, confer resistance to a BRAF inhibitor, using a library consisting of 70,290 guides targeting all human coding isoforms. The screens exhibited a high degree of consistency with 100% validation of the top ten hits. The top hits included genes previously shown to be able to confer resistance, and novel candidates were validated using individual sgRNA and complementary DNA overexpression. Furthermore, gene expression signature based on the top screening hits correlated with markers of BRAF inhibitor resistance in cell lines and patient-derived samples, proving the potential of Cas9-based activators as a powerful genetic perturbation technology [20].
The discovery of the RNA-mediated programmable CRISPR/Cas9 technology, has transformed the field of biology. While CRISPR/dCas9-mediated gene activation represents dramatic advantages over conventional approaches, there are several concerns with its broad application. In addition to the general problems with CRISPR system, such as off-target effects, delivery issue, and potential immuno–genicity [28], the major concern is that the activation capability of endogenous genes by CRISPRa system is not as robust as that of cDNA overexpression approach and is heavily dependent on the selection of sgRNAs. This could be a potential concern for biological processes, such as direct reprogramming or transdifferentiation from one mature cell type to another, which might require a large amount of factors in order to overcome the force of gravity on the famous Waddington’s epigenetic landscape [27]. Thus how to effectively design the most robust and specific guide RNAs for transcriptional activation needs further exploration.
Contributor Information
Meng Chen, Department of Bioengineering, Stanford University, Stanford, CA 94305, USA; Gladstone Institute of Cardiovascular Disease, San Francisco, CA 94158, USA.
Lei Stanley Qi, Department of Bioengineering, Stanford University, Stanford, CA 94305, USA; Department of Chemical and Systems Biology, Stanford University, Stanford, CA 94305, USA; ChEM-H, Stanford University, Stanford, CA 94305, USA.
References
- 1.Balboa D, Weltner J, Eurola S et al. (2015) Conditionally stabilized dCas9 activator for controlling gene expression in human cell reprogramming and differentiation. Stem Cell Rep 5:448–459. doi: 10.1016/j.stemcr.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bikard D, Jiang W, Samai P et al. (2013) Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res 41:7429–7437. doi: 10.1093/nar/gkt520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Black JB, Adler AF, Wang H-G et al. (2016) Targeted epigenetic remodeling of endogenous loci by CRISPR/Cas9-based transcriptional activators directly converts fibroblasts to neuronal cells. Cell Stem Cell 19:406–414. doi: 10.1016/j.stem.2016.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chakraborty S, Ji H, Kabadi AM et al. (2014) A CRISPR/Cas9-based system for reprogramming cell lineage specification. Stem Cell Rep 3:940–947. doi: 10.1016/j.stemcr.2014.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chavez A, Scheiman J, Vora S et al. (2015) Highly efficient Cas9-mediated transcriptional programming. Nat Methods 12:326–328. doi: 10.1038/nmeth.3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chavez A, Tuttle M, Pruitt BW et al. (2016) Comparison of Cas9 activators in multiple species. Nat Methods 13:563–567. doi: 10.1038/nmeth.3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen B, Gilbert LA, Cimini BA et al. (2013) Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155:1479–1493. doi: 10.1016/j.cell.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng AW, Wang H, Yang H et al. (2013) Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res 23:1163–1171. doi: 10.1038/cr.2013.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cong L, Ran FA, Cox D et al. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. doi: 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dominguez AA, Lim WA, Qi LS (2016) Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol 17:5–15. doi: 10.1038/nrm.2015.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doudna JA, Charpentier E (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096. doi: 10.1126/science.1258096 [DOI] [PubMed] [Google Scholar]
- 12.Gasiunas G, Barrangou R, Horvath P, Siksnys V (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A 109:E2579–E2586. doi: 10.1073/pnas.1208507109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilbert LA, Larson MH, Morsut L et al. (2013) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154:442–451. doi: 10.1016/j.cell.2013.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gilbert LA, Horlbeck MA, Adamson B et al. (2014) Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159:647–661. doi: 10.1016/j.cell.2014.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hilton IB, D’Ippolito AM, Vockley CM et al. (2015) Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33:510–517. doi: 10.1038/nbt.3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang F, Doudna JA (2015) The structural biology of CRISPR-Cas systems. Curr Opin Struct Biol 30:100–111. doi: 10.1016/j.sbi.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jinek M, Chylinski K, Fonfara I et al. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. doi: 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kearns NA, Genga RMJ, Enuameh MS et al. (2014) Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells. Development 141:219–223. doi: 10.1242/dev.103341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kearns NA, Pham H, Tabak B et al. (2015) Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods 12:401–403. doi: 10.1038/nmeth.3325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konermann S, Brigham MD, Trevino AE et al. (2015) Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517:583–588. doi: 10.1038/nature14136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maeder ML, Linder SJ, Cascio VM et al. (2013) CRISPR RNA-guided activation of endogenous human genes. Nat Methods 10:977–979. doi: 10.1038/nmeth.2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mali P, Yang L, Esvelt KM et al. (2013) RNA-guided human genome engineering via Cas9. Science 339:823–826. doi: 10.1126/science.1232033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perez-Pinera P, Kocak DD, Vockley CM et al. (2013) RNA-guided gene activation by CRISPR–Cas9-based transcription factors. Nat Methods 10:973–976. doi: 10.1038/nmeth.2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qi LS, Larson MH, Gilbert LA et al. (2013) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183. doi: 10.1016/j.cell.2013.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sternberg SH, Redding S, Jinek M et al. (2014) DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507:62–67. doi: 10.1038/nature13011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanenbaum ME, Gilbert LA, Qi LS et al. (2014) A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159:635–646. doi: 10.1016/j.cell.2014.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waddington CH (1957) The strategy ofthe genes. Allen, London [Google Scholar]
- 28.Xiong X, Chen M, Lim WA et al. (2016) CRISPR/Cas9 for human genome engineering and disease research. Annu Rev Genomics Hum Genet 17:131–154. doi: 10.1146/annurev-genom-083115-022258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zalatan JG, Lee ME, Almeida R et al. (2015) Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 160:339–350. doi: 10.1016/j.cell.2014.11.052 [DOI] [PMC free article] [PubMed] [Google Scholar]