

Abstract
A proprietary library of novel N-aryl substituted amino acid derivatives bearing hydroxamate head group allowed the identification of compound 3a that possesses weak proadipogenic and PPARγ activating properties. The systematic optimization of 3a, in order to improve its PPARγ agonist activity, led to the synthesis of compound 7j (N-aryl substituted valine derivative) that possesses dual PPARγ / PPARα agonistic activity. Structural and kinetic analyses reveal that 7j occupies the typical ligand binding domain of the PPARγ agonists with, however, a unique high-affinity binding mode. Furthermore, 7j is highly effective in preventing CDK5-mediated phosphorylation of PPARγ serine 273. Although less proadipogenic than rosiglitazone, 7j significantly increases adipocyte insulin-stimulated glucose uptake and efficiently promotes white-to-brown adipocyte conversion. In addition, 7j prevents oleic acid-induced lipid accumulation in hepatoma cells. The unique biochemical properties and biological activities of compound 7j suggest that it would be a promising candidate for the development of compounds to reduce insulin resistance, obesity and non-alcoholic fatty liver disease.
Graphical Abstract
INTRODUCTION
Peroxisome proliferator-activated receptors (PPAR) are transcription factors belonging to the nuclear receptor superfamily and activated by ligands such as dietary fatty acids, particularly polyunsaturated fatty acids. The three PPAR subtypes: PPARα, γ, and δ (β), have different, yet overlapping, tissue expression patterns 1 and exert major roles in the regulation of specific physiological functions including glucose and lipid metabolism and energy homeostasis 2–4. These features make PPARs important molecular targets for the development of drugs for metabolic diseases.
PPARα is expressed in all metabolic tissues, but predominantly in the liver where it is involved in the regulation of the uptake and oxidation of fatty acids and lipoprotein metabolism 5. The fibrates family of drugs (clofibrate and fenofibrate) are pharmacological weak agonists of PPARα that are used to treat dyslipidemia as they lower plasma triglycerides and increase HDL cholesterol levels 6. The PPARα agonist fenofibrate has also beneficial effect in patient with non-alcoholic fatty liver disease (NAFLD) characterized by the accumulation of triglycerides in hepatocytes 7. However, fibrates increase markers of cardiovascular and renal disease and that of liver dysfunction, which underlines their ability to trigger adverse effects 8. Therefore, efforts are being made to develop PPARα agonists with improved clinical efficacy, Penafibrate being one of these new generation agonists 9.
PPARδ is involved in the regulation of fatty acid oxidation and mitochondrial respiration predominantly in skeletal muscle, liver and adipose tissue 10. Therefore, agonists targeting PPARδ may be considered as potential therapeutic agents for insulin-resistant related conditions. PPARδ agonists have been developed and used in research 11 but none are currently approved for clinical use.
PPARγ is considered the master regulator of adipogenesis via its promotion of lipid production and storage. Thiazolidinediones, including rosiglitazone (Rosi) and pioglitazone, are the most effective PPARγ activating drugs that were widely prescribed for the treatment of type 2 diabetes 12. However, their strong agonist activities are partly responsible for unwanted harmful side effects such as weight gain, fluid retention, osteoporosis, heart failure and cancer 13,14, which precipitated their withdrawal from the market.
The quest for antidiabetic compounds targeting PPAR with good therapeutic potential and reduced adverse effects has followed two main directions. The one based on the observation that a moderate, rather than full, activation of PPARγ dissociates the deleterious from the therapeutic effects of the agonist has led to the generation of selective PPARγ modulators (SPPARγMs) with higher therapeutic profiles than full agonists 15,16. The peculiar properties of SPPARγM being explained by the ability of PPARγ to adopt ligand-specific conformations with different transcriptional signatures. In addition, it has been shown that the clinical benefit of PPARγ partial agonists and SPPARγMs also involves their ability to inhibit the cyclin-dependent kinase 5 (CDK5)-mediated PPARγ phosphorylation at serine 273 17,18. The other concept for developing safe antidiabetic drugs targeting PPAR considers that beneficial effects of their activation could counteract their harmful effects. PPARα/γ dual agonists, so-called Glitazars, that combine the insulin sensitizing effect of PPARγ agonists with the beneficial effect of PPARα agonists on the lipid profile are representative of this class of drugs 19,20. Saroglitazar is approved in India for the treatment of peculiar type of diabetic dyslipidemia and hypertriglyceridemia 21,22. Moreover, dual α/δ and γ/δ PPAR agonists as well as “pan” agonists acting on all three isoforms are the subject of intense investigations 23–25 that could lead to the generation of molecules with potential additional therapeutic indications.
This work reports a mild two-step synthesis of a library of new N-aryl substituted amino acid derivatives from commercially easily available and inexpensive reagents. The effect of these compounds, in particular the influence of substitutions on their phenyl group, was evaluated on PPARs activity and led to the development of a new balanced and potent dual PPARα/γ agonist with unique ligand binding properties and singular biological activities that would be expected for a potential therapeutic candidate to reduce insulin resistance, obesity and NAFLD.
RESULTS AND DISCUSSION
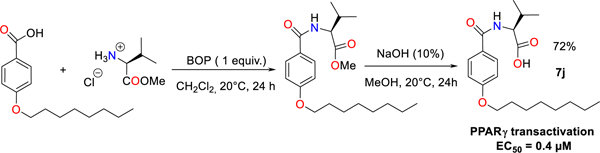
New N-aryl substituted amino acid derivatives bearing hydroxamate head group (head), initially designed to identify MMP inhibitors, were screened on their ability to induce spontaneous (in the absence of any other inductor) adipocyte differentiation of 3T3-L1 cells by measuring intracellular lipid accumulation. The molecule named 3a (4-Hexyloxy-N-((S)-1-hydroxycarbamoyl-2-methyl-propyl)-benzamide), that contains a L-Valine (core) and a 6 carbon atoms chain (capping group) (Figure 1A and Supplemental Figure S1), was identified as a weak activator of adipocyte differentiation as compared with Rosi (Supplemental Figure S2). We hypothesized that 3a increased adipocyte differentiation by activating the master regulator of adipogenesis: PPARγ. The PPARγ agonistic activity of 3a was confirmed by the use of a luminescence-based cell-based PPARγ transactivation assay, in which wild-type PPARγ ligand binding domain (PPARγ-LBD) is fused to the GAL4 DNA-binding domain (PPARγ-LBD-GAL4) and the Firefly luciferase reporter gene is under the control of GAL4 binding elements (Figure 1A). Importantly, 3a increased PPARγ activity by a factor of 2.8, while 1 μM of Rosi (a full PPARγ agonist) increased it by a factor of 25 ± 7. A thorough comparison of the PPARγ activation properties of the newly synthesized compounds with that of Rosi will be carried out at later stage. We initiated a process to identify the domains of 3a involved in PPARγ activation that would ideally lead to an optimization of its PPARγ agonist activity.
Figure 1.
Chemical structures and PPARγ agonist activity of 3a and its derivatives. The amino acid core (A), hydrophobic capping group (B) and binding head group (C) of 3a were modified (left panels) and PPARγ transactivation activity of each molecule (5 μM) was measured (right panels). Color fills accentuates the amino acid core (gray), hydrophobic capping group (green) and binding head group (yellow) of 3a. The percentage indicates the yield of the synthesis. PPARγ transactivation values are means ± SD expressed relative to the mean of control values, which was set to 1.
Effect of the amino acid core.
Analogues of the 3a hit were designed and synthesized through a reaction involving a two steps synthesis procedure in dichloromethane from amino acid methyl ester hydrochloride 1a-1h, easily prepared from amino acids. By using BOP reagent as an efficient and versatile reagent for the coupling of alkyloxybenzoic acid with 1a-1h, the synthesis of various substituted amino acid ester derivatives 2a-2h in high chemical yields of up to 90% was achieved. It is noteworthy that the transient ester species are successfully transformed into their corresponding hydroxamic acid parent derivatives 3a-3h by using hydroxylamine (40% H2O) in MeOH at reflux for 24 hours in yields varying from 36 to 72% (Figure 1A). Changing the L-Valine core by any other amino acid, decreased the PPARγ activation potency of the molecule (Figure 1A) perhaps due to the larger steric hindrance generated by the isopropyl moiety with respect to a benzyl or a hydroxy methyl group.
Effect of the carbon atoms chain capping group.
The different derivatives 4a-4h were synthesized according a similar procedure than for compounds 3a-3h in good-to-excellent yields, varying from 32 to 75% (Figure 1B). Altering the number of carbons in the polycarbon chain capping group of the aryl moiety showed that a linear 6 carbons chain was optimal for PPARγ activation (Figure 1B). This result suggests that the steric hindrance and/or the hydrophobic nature of the polycarbon chain are important factors to consider when designing most potent analogues.
Effect of the head group.
Substitution of the hydroxamate head moiety in 3a (Figure 1C) revealed that the preferred functions for PPARγ activation are OH > NHOH ≥ NH2 > COOMe (Figure 1C). The analog of 3a, with a carboxylic acid moiety in place of hydroxamate head group that optimally activated PPARγ was named 7a (Supplemental Figure S3).
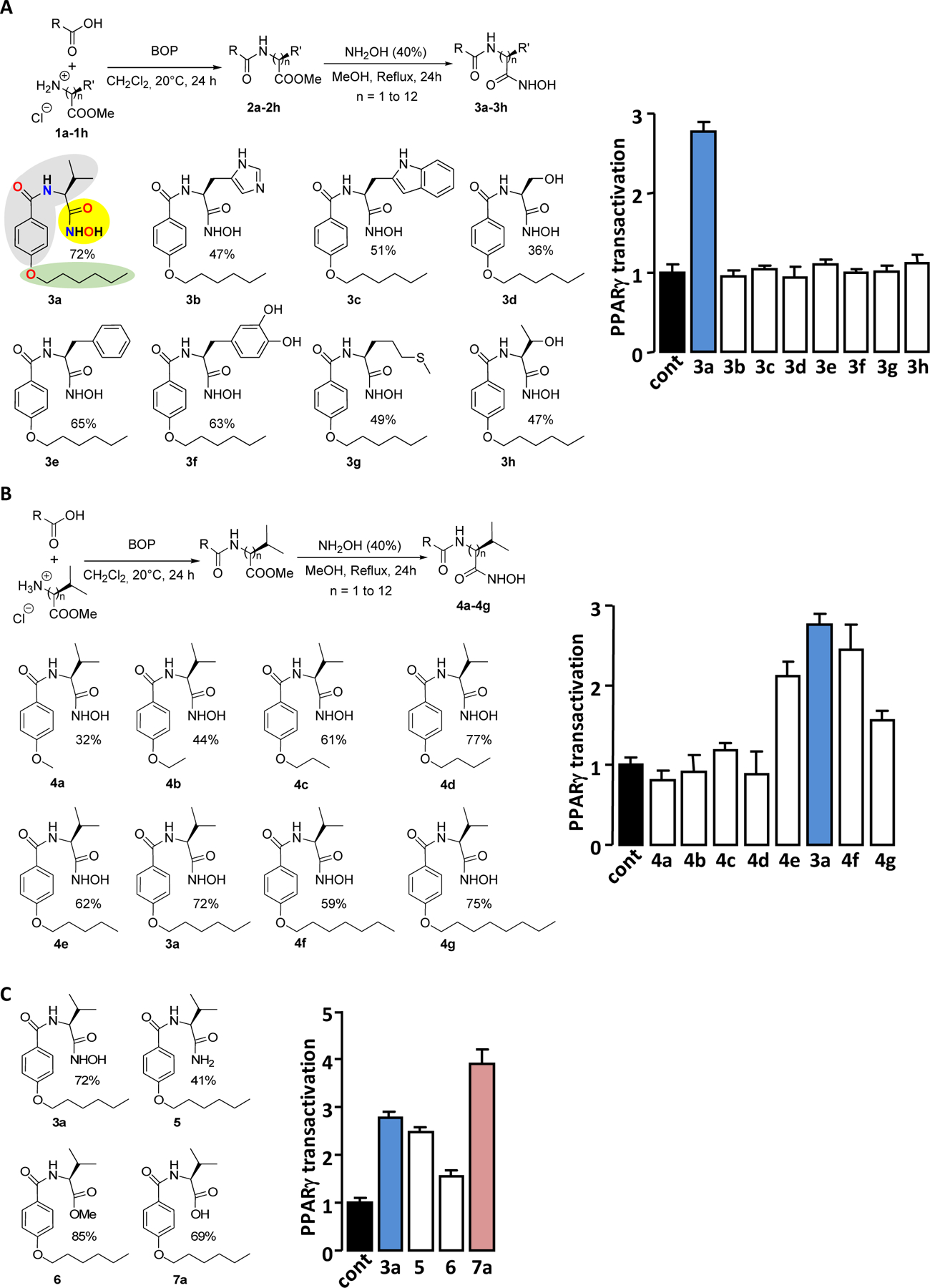
Optimization of PPARγ agonist activity of 7a.
Changing the absolute conformation of the core Valine from L to D abolished the PPARγ activation capacity of 7a (Figure 2A). Furthermore, substitution of the amino acid core (L-Valine) by other amino acid (L-form) and/or reducing the length of the polycarbon chain decreased the PPARγ activation efficiency of 7a (Figure 2B). The PPARγ transactivation activity of 7a was gradually enhanced by the extension of the polycarbon chain up to 8 carbon atoms (Figure 2C). The analog of 7a with the 8 carbon atoms chain in place of the 6 carbon atoms chain which optimally activates PPARγ was named 7j (Supplemental Figure S4). It should be noted that the presence of a chain with 10 carbon atoms in the molecule 7k did not further enhance its ability to activate PPARγ.
Figure 2.
Chemical structures and PPARγ agonist activity of 7a and its derivatives. The absolute conformation of the valine core (A), the amino acid core (B) or the hydrophobic capping group (B and C) of 7a were modified (left panels) and PPARγ transactivation activity of each molecule (5 μM) was measured (right panels). The percentage indicates the yield of the synthesis. PPARγ transactivations are expressed as in figure 1.
Typical PPAR agonists are known to consist of three parts: a polar head group (usually bearing a carboxylic acid functionality), a hydrophobic tail moiety and a linker which consists of flexible methylene units and an aromatic ring 26. Interestingly, 7a and 7j closely meet these elementary criteria.
Potency and efficacy of PPARγ activation.
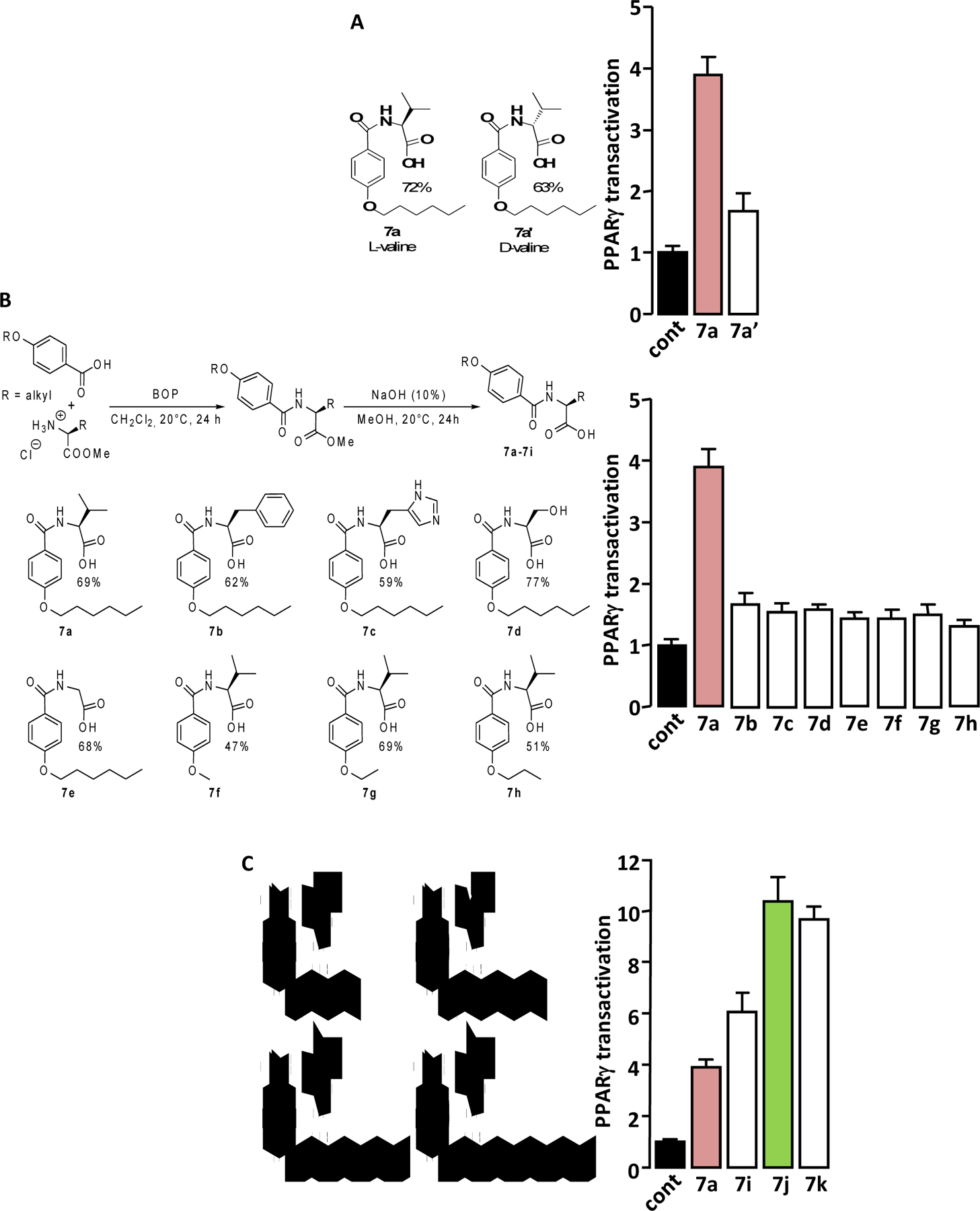
The concentration-dependent activation of PPARγ by the “hit” compound 3a and its two “lead” derivatives 7a and 7j were compared to that of the PPARγ full agonist Rosi using PPARγ-LBD-GAL4 chimera assay (Figure 3A).
Figure 3.
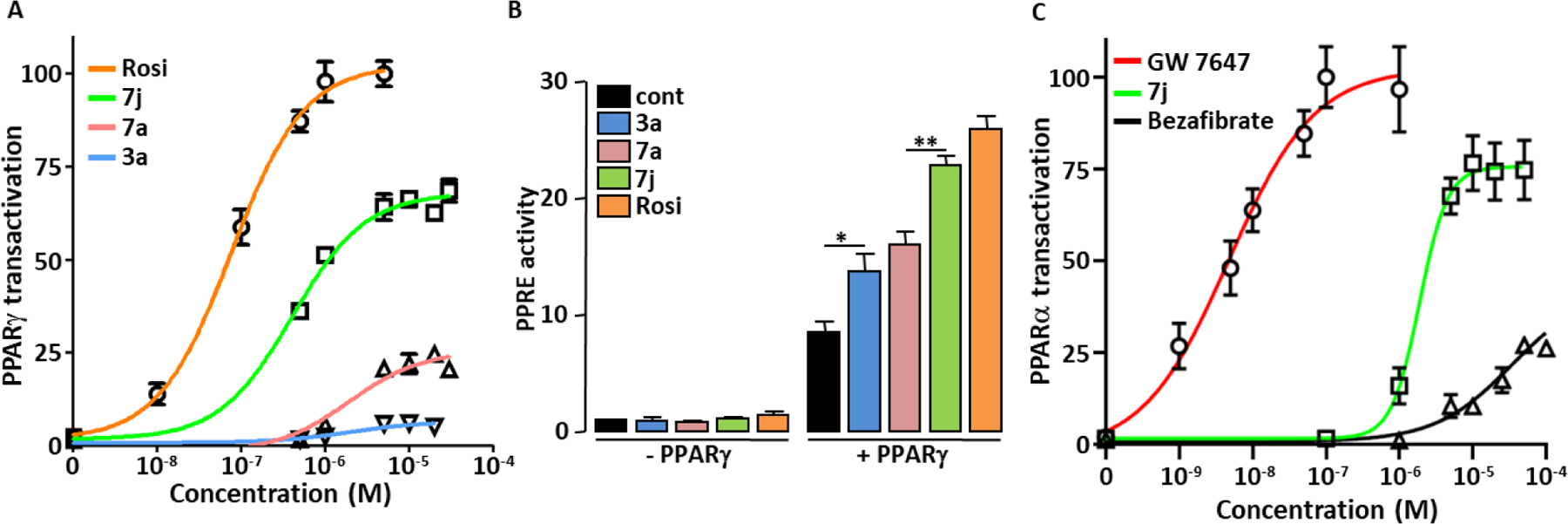
PPAR agonist activity of 3a, 7a and 7j. (A) Concentration-dependent PPARγ transactivation activities of 3a, 7a and 7j were compared to that of Rosi using PPARγ-LBD-GAL4 chimera assay. Values are means ± SD expressed as % of the maximal response measured with Rosi (5 μM). (B) HEK293 cells were transfected with PPRE-driven Firefly luciferase and SV40-driven Renilla luciferase coding vectors together with an empty plasmid (-PPARγ) or with the PPARγ expression vector (+PPARγ). 36 hours post transfection, cells were incubated for 17 hours with 3a, 7a, 7j (1μM) or Rosi (0.1μM). PPRE promoter activity was calculated as the ratio Firefly/Renilla luciferase. Values are means ± SD expressed relative to the control situation. *p < 0.05, **p < 0.01 (one-way ANOVA followed by Bonferroni’s post hoc test). (C) Concentration-dependent PPARα transactivation activities of 7j, GW 7647 and Bezafibrate were measured using PPARα-LBD-GAL4 chimera assay. Values are means ± SD expressed as % of the maximal response measured with GW 7647 (1 μM).
The potencies of the compounds were ranked as follow: 3a ≈ 7a < 7j < Rosi (Table 1). Regarding their efficacy (maximal PPARγ transactivation) compounds were ranked as follow: 3a < 7a < 7j ≤ Rosi (Table 1).
Table 1.
EC50 and maximal activation of PPARγ and PPAR
| Compound | PPAR | EC50 (nM) | Activity (%) |
|---|---|---|---|
| Rosi | γ | 76 ± 30 | 100 |
| 7j | γ | 400 ± 150 | 66.36 ± 3.41 |
| 7a | γ | 1940 ± 940 | 25.06 ± 2.03 |
| 3a | γ | 1840 ± 1020 | 6.33 ± 1.03 |
| GW 7647 | α | 5.4 ± 1 | 100 |
| 7j | α | 1882 ± 665 | 75.6 ± 3.83 |
| Bezafibrate | α | 31700 ± 5410 | 35.4 ± 3.81 |
Activities are expressed as % of the maximal response measured with 5 μM of Rosi (for PPARγ) or with 1 μM of GW 7647 (for PPARα). Values are means ± SD.
PPARγ activation elicited by 7j was around 66% of that obtained with Rosi. However, this value is certainly overestimated. Indeed, in this specific cell-based assay, we observed that the Renilla luciferase expression (Supplemental Figure S5A), which is used to normalize reporter gene values for variations inherent to transfection efficiency and sample handling, was more reduced by 7j concentrations that induce maximal PPARγ activity (> 10−6 M) than by Rosi. As a consequence, inclusion of Renilla luciferase expression values in the calculation of PPARγ activation by 7j leads to an “artificial rise” of the maximum response (Supplemental Figure S5B). Interestingly, the measurement of ATP cell content revealed that the number of viable HEK293 cells was no more reduced by 7j than by Rosi, ruling out that the 7j-dependent decrease in Renilla luciferase expression is due to a reduction in the number of viable cells. (Supplemental Figure S5C).
Taken together, these data allow us to define 3a and 7a as partial agonists of PPARγ and 7j as a strong partial agonist of PPARγ.
A PPAR Responsive Element (PPRE)-based luciferase assay was carried out to assess the ability of 3a, 7a and 7j to transactivate genes controlled by the binding of PPARγ to PPRE. The three molecules increased the expression of PPRE-driven luciferase only when full-length PPARγ was expressed (Figure 3B), showing that these molecules stimulate the actual transcriptional activity of PPARγ. Interestingly, the transactivation efficiencies were in agreement with those measured by the PPARγ-LBD-GAL4 chimera assay, i.e. 3a ≈ 7a < 7j. In addition, and as expected, 1 μM of 7j and 0.1 μM Rosi gave similar activation levels in both the PPARγ-LBD-GAL4 chimera assay and in the PPRE-based assay.
To attest that 3a, 7a and 7j are actual PPARγ ligands, their binding affinity (Kd) and rate constants (kon, koff) for PPARγ were compared to those of the reference ligand Rosi using surface plasmon resonance technology-based experiments (Table 2 and Supplemental Figure S6).
Table 2.
Affinity (Kd) and rate constants (kon, koff) for PPARγ/ligand and PPARα/ligand interactions
| Interaction | kon (M−1s−1) | koff (s−1) | Kd (nM) |
|---|---|---|---|
| PPARγ/Rosi | 3.45 (0.02) 105 | 0.0164 (0.0001) | 47.6 (0.3) |
| PPARγ/7j | 5.65 (0.06) 105 | 0.0170 (0.0001) | 30.1 (0.3) |
| PPARγ/7a | 1.52 (0.02) 105 | 0.0143 (0.0001) | 94 (1) |
| PPARγ/3a | 1.55 (0.02) 105 | 0.0190 (0.0001) | 123 (2) |
| PPARα/7j | 1.01 (0.03) 104 | 0.0312 (0.0005) | 3100 (50) |
Experimental error is reported in brackets.
The affinities of 3a and 7a for PPARγ were lower than those of 7j and Rosi, which is consistent with their low PPARγ transactivation capacity measured in cell-based assays. Remarkably, the partial agonist 7j shows an affinity for PPARγ similar or higher to that of the full agonist Rosi. It was shown that some compounds with moderate PPARγ agonist activity but with high binding affinity to PPARγ retain significant antidiabetic activity while having fewer and/or less severe adverse events than full PPARγ agonists 27. Therefore, 7j can reasonably be considered as scaffold molecule to develop new anti-diabetic drugs.
Specificity.
The concentration-dependent activations of PPARδ and PPARα by 3a, 7a and 7j were analyzed using the appropriate PPAR-LBD-GAL4 chimere assays. None of the molecules activated PPARδ (Supplemental Figure S7). Only 7j transactivated PPARα (Figure 3C and Table 1), with potency between that of Bezafibrate (a weak pan agonist for all three PPAR isoforms) and GW 7647 (a full PPARα agonist). 7j appeared to be a good PPARα agonist. In order to confirm that 7j is a PPARα ligand, their direct physical binding was measured using surface plasmon resonance technology-based experiment. We found that 7j concentration dependently bound to PPARα with a Kd value of 3.1 μM (Table 2).
PPAR form permissive heterodimers with RXR, thus allowing both the ligands of PPAR and RXR to regulate the transcription of PPAR target genes. Using mammalian-two hybrid system, we showed that 7j was less potent and less efficient than Rosi in inducing PPARγ / RXRα heterodimerization (Supplemental Figure 8A). Moreover, using RXRα-LBD-GAL4 chimera assay (Supplemental Figure 8B) and RXR Responsive Element (RXRE)-based luciferase assay (Supplemental Figure 8C) we demonstrated that 7j is not a direct RXR agonist.
Therefore, 7j is a dual PPARγ/α agonist (Glitazar) and as such, it could theoretically have beneficial synergistic activities on glucose and lipid homeostasis 19,20.
Binding of 7j, 7a and 3a to PPARγ.
The crystal structure of the complex of PPARγ LBD with the ligand 7j was solved collecting diffraction data from apo-crystals soaked for three days in the presence of the ligand at 0.5 mM. Initial difference Fourier maps revealed clear electron density for the ligand (Supplemental Figure S9) showing that it occupies the typical LBD region of the PPARγ agonists (Figure 4A and Supplemental Figure S10), similar to that of the full agonist Rosi (Figure 4B and Supplemental Figure S11), with its carboxylate group directly interacting through H-bonds with Y473 of helix 12 (H12), at the short distance of 2.3 Å, the two histidines (H323 and H449) of the canonical triad, and S289, (Figure 4A). The NH of 7j amide bond is also involved in a H-bond with S289 and the CO makes a H-bond with Y327. The terminal aliphatic chain of 7j is in equilibrium between two different conformations (occupation factors of 0.6 and 0.4, respectively) and makes vdW contacts with several residues of the internal strand of the β-sheet (Figure 4A and Supplemental S12) with a consequent effective stabilization of this domain. The isopropyl terminal group of 7j, makes vdW interactions in the hydrophobic pocket formed by the residues F282, C285, Q286, L453 and L469 (belonging to H3, H11 and H12), contributing in this way to a tighter binding of the ligand (Figure 4C and Supplemental Figure S13).
Figure 4.
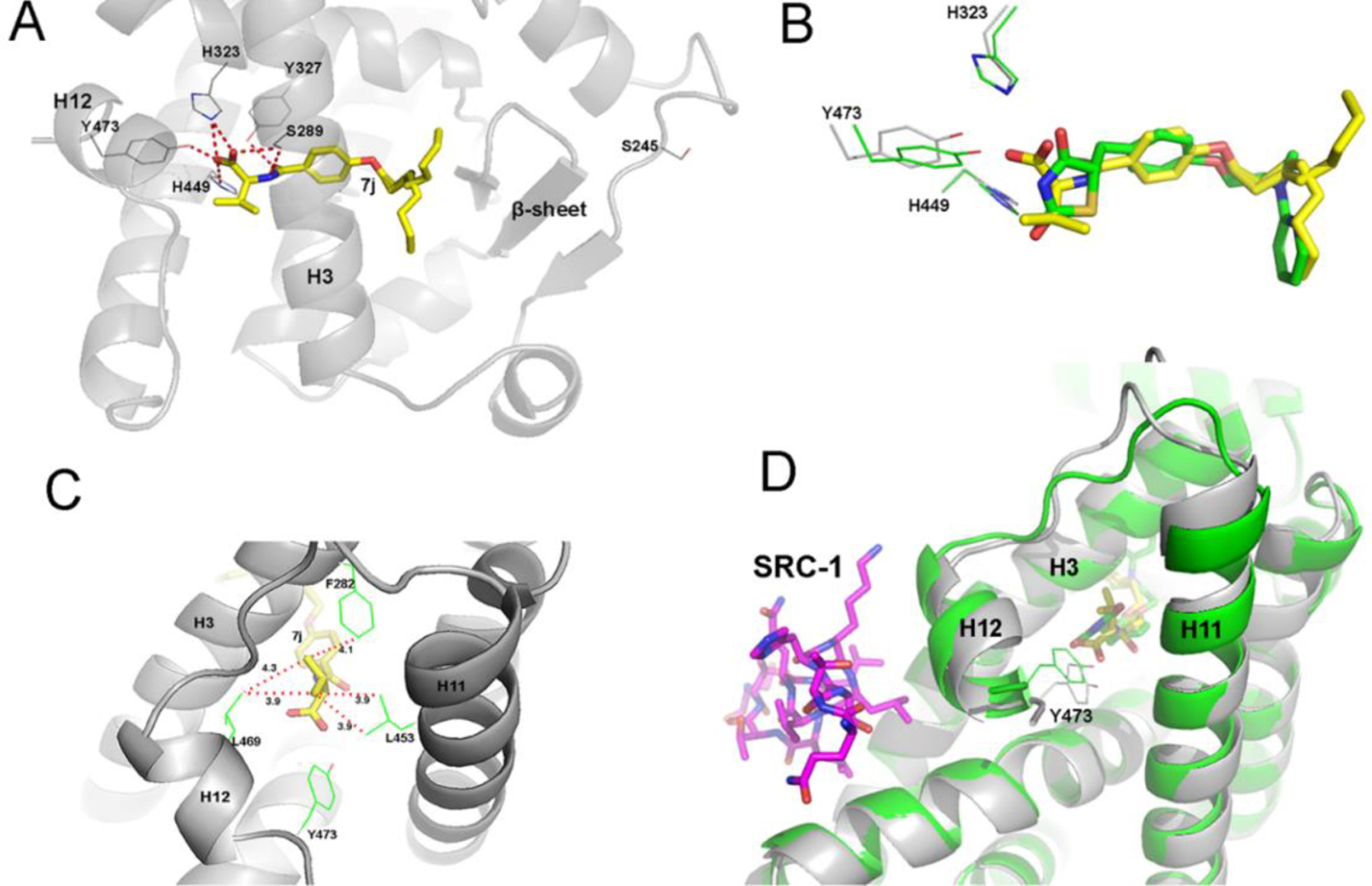
Binding of 7j to PPARγ. (A) Hydrogen-bond network of 7j in the PPARγ LBD. (B) Superimposition of 7j (yellow) and Rosi (green) structures. (C) vdW network of the 7j with residues belonging to H3, H11 and H12 of the PPARγ LBD. (D) Superimposition of 7j (yellow) and Rosi (green) with the co-activator SRC-1 (magenta); the PPARγ / Rosi structure is colored in green, PPARg / 7j in gray (PDB codes: 6QJ5 for PPARγ / 7j and 2PRG for PPARγ / Rosi).
The thorough analysis of the effective binding network of 7j into the PPARγ LBD indicates that the ligand is very tightly locked into the LBD, with its carboxylate group strongly interacting with Y473 of H12 (2.3 Å), but in a slightly distorted mode, with respect to Rosi. As a consequence, there is an adjustment of the conformation of helix 12 (H12), the loop 11/12 and the beginning of helix 11 (H11) (Figure 4D and Supplemental Figure S14). Importantly, H12 is a critical regulatory structural element in the Activation Function-2 (AF-2) co-regulator-binding surface, which determines the transcriptional output of PPARγ through differential recruitment of co-regulators.
Comparison of the crystal structures of PPARγ in complex with 7j, 7a or 3a 28 shows no difference in the binding mode of these ligands, regardless of the presence of a carboxylic or hydroxamic head (Supplemental Figure S15). However, the shorter aliphatic chains of 7a and 3a accommodate in a different part of the PPARγ LBD with respect to 7j, farther away from the β-sheet. (Supplemental Figure S15). Moreover, it is known that partial agonists of PPARγ preferentially stabilize the β-sheet region of the LBD. In the PPARγ/7j structure, the long aliphatic chain of the ligand strongly contributes to the stabilization of this region, through vdW interactions, with both its observed conformations facing residues of the innermost β-strand. The solvent entropic gain arising from a more efficacious displacement of water molecules, known to occupy the β-sheet pocket, could play an important role in lowering the free energy of the binding. The higher affinity of 7j for PPARγ with respect to those of 7a and 3a (Table 2) demonstrates that the affinity is largely affected by the length of the aliphatic chain, rather than the character of the acidic head.
Interestingly, a similar unique binding network was observed in the PPARγ crystal structure with the partial agonist SR2067 (Supplemental Figure S16) 29. Both ligands share a common amide group that forms two hydrogen bonds with Y327 and S289, interactions that are not possible with Rosi. However, unlike 7j, SR2067 does not interact with Y473.
Interaction of coregulators with PPARγ
Since Rosi and 7j stabilize different conformations of the AF2 co-regulator-binding surface of PPARγ, we analyzed how these molecules modulate the interaction of PPARγ with some of its coregulators. In mammalian two-hybrid system, 7j was less potent than Rosi in inducing displacement of the NCoR and SMRT co-repressors from PPARγ, as well as in inducing recruitment of TIF2 and MED1 co-activators to PPARγ (Figure 5).
Figure 5.
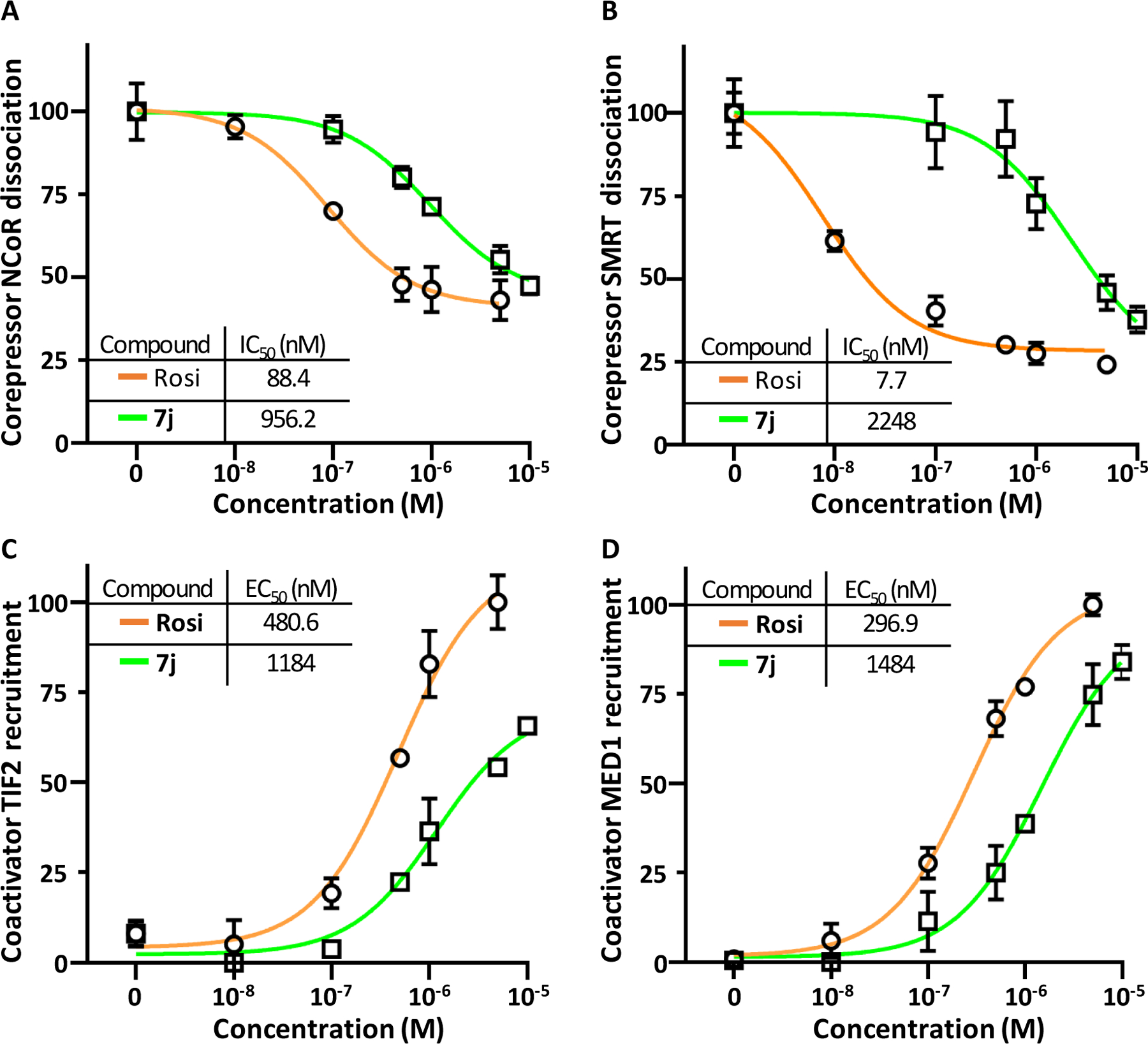
Ligand-specific co-regulator binding profiles. HEK293 cells were transfected with GAL4-NCoR (A), GAL4-SMRT (B), GAL4-TIF2 (C), GAL4-MED1 (D) and VP16-PPARγ-LBD expression vectors in a mammalian two-hybrid setting and treated with vehicle or various concentrations of 7j or Rosi. Values are means ± SD expressed as % of the maximal response measured.
The differences between the profiles of the concentration-response curves of 7j and Rosi to modulate the interaction of PPARγ with its partners (NCoR, SMRT, TIF2, MED1, Mediator and also RXRα) suggest that these two molecules induce distinct patterns of coregulatory protein recruitment.
CDK5-mediated phosphorylation of PPARγ.
Part of the anti-diabetic effects of PPARγ partial agonists have been associated with their ability to prevent the CDK5-mediated phosphorylation of PPARγ serine 273 residue 17,18. Therefore, we assessed the ability of 7j to inhibit such a phosphorylation. An ELISA protocol was optimized to quantify the phosphorylation of PPARγ triggered in vitro by recombinant CDK5. 7j prevented the phosphorylation of PPARγ serine 273 at least as efficiently as Rosi (Figure 6A). Furthermore, pretreatment of 3T3-L1 adipocytes with 7j or Rosi reduced the TNFα-stimulated phosphorylation of PPARγ serine 273 (Figure 6B). The involvement of CDK5 in this phosphorylation was attested by the fact that it was impeded by treatment of cells with Roscovitine, a CDK5 inhibitor. Moreover, out of ten genes known to be significantly controlled by CDK5-dependent PPARγ phosphorylation in fully differentiated adipocytes 17,18, seven were regulated in the same way by 7j and Rosi (Figure 6C). Two genes were only regulated by Rosi and the expression of one gene was not significantly modified by 7j and Rosi, although both molecules tended to alter this expression in the same direction.
Figure 6.
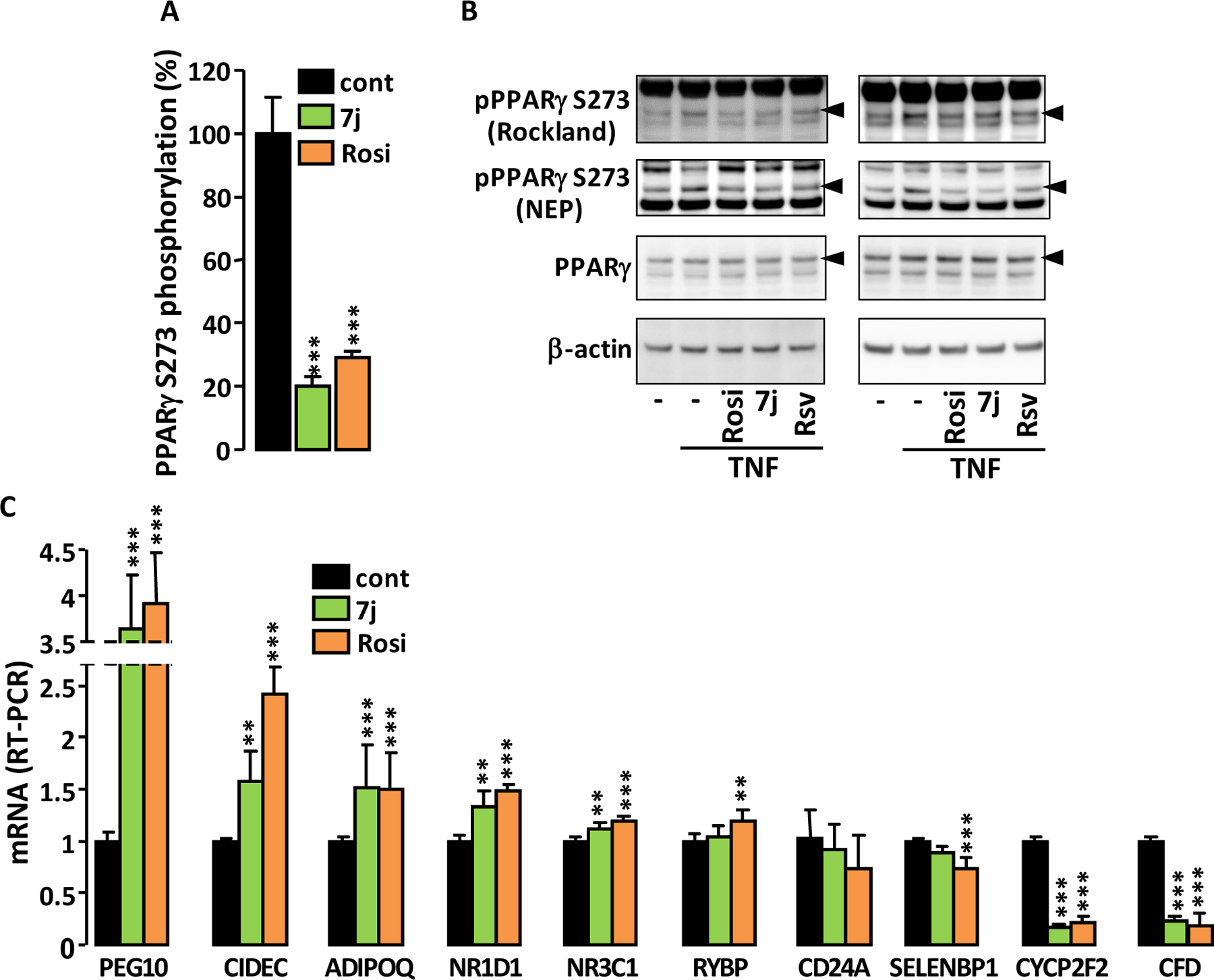
Effect of 7j on PPARγ phosphorylation. (A) Percentage of in vitro PPARγ Ser273 phosphorylation by CDK5 in the presence of 0.1 μM of 7j or Rosi. (B) Phosphorylation of PPARγ Ser273 in 3T3-L1 adipocyte incubated for 60 minutes with 5 μM of Rosi, 7j or Roscovitine (Rsv) before TNFα stimulation (50 ng/ml; 90 minutes). Two independent experiments are shown. Phosphospecific antibodies were from Rockland or New England Peptide (NEP). Arrow heads indicate phosphorylated PPARγ and PPARγ2. (C) RT-PCR analysis of the expression levels of a selection of genes known to be regulated by CDK5-dependent phosphorylation of PPARγ. Values are means ± SD (n = 5) expressed relative to the mean of control. **p < 0.01, ***p < 0.001 vs. control (one-way ANOVA followed by Dunnett’s post hoc test).
These results underline the ability of 7j to reduce CDK5-mediated phosphorylation of PPARγ serine 273 and to alter the expression of genes controlled by PPARγ phosphorylation. This feature supports a potential anti-diabetic effect of 7j.
Adipocyte differentiation and glucose uptake.
Adipogenesis-mediated weight gain is a major side effect of PPARγ full agonists 30. Partial PPARγ agonists are expected to have a reduced effect on lipid storage while maintaining a significant insulin sensitization effect. Therefore, the proadipogenic properties of 3a, 7a and 7j were compared to that of Rosi. 3T3-L1 fibroblast cells were incubated in the presence of PPARγ agonists as the sole inducer of adipocyte differentiation. Only 7j and Rosi (the strongest PPARγ agonists) significantly increased the intracellular lipid content (Figure 7A and Supplemental Figure S17A), showing that these two molecules stimulate adipocyte differentiation. However, for the same concentration, the proadipogenic property of 7j was significantly lower than that of Rosi. To ascertain that the proadipogenic effect of 7j involved PPARγ activation, 3T3-L1 cells were co-incubated with the specific PPARγ antagonist GW 9662 and with 7j, then intracellular lipid accumulation was measured. This approach has been successfully used to demonstrate the involvement of PPARγ activation in the proadipogenic effect of Rosi 31. Because of its short half-life, GW 9662 was added every day and to simplify the experiment the duration of the treatment was reduced to 4 days instead of 6 days, thus limiting the intracellular lipid accumulation. Antagonizing PPARγ activation significantly prevented 7j- and Rosi-induced adipogenesis (Figure 7B and Supplemental Figure S17B) showing that PPARγ activation is necessary for the proadipogenic effect of these molecules. When 3T3-L1 fibroblast cells were primed for adipocyte differentiation using the conventional adipogenic cocktail (Insulin, Dexamethasone, IBMX) and then treated for 7 days with the 3a, 7a, 7j or Rosi, mRNA levels of the markers of adipocyte differentiation were all increased in proportion to the ability of the molecules to activate PPARγ (Figure 7C).
Figure 7.
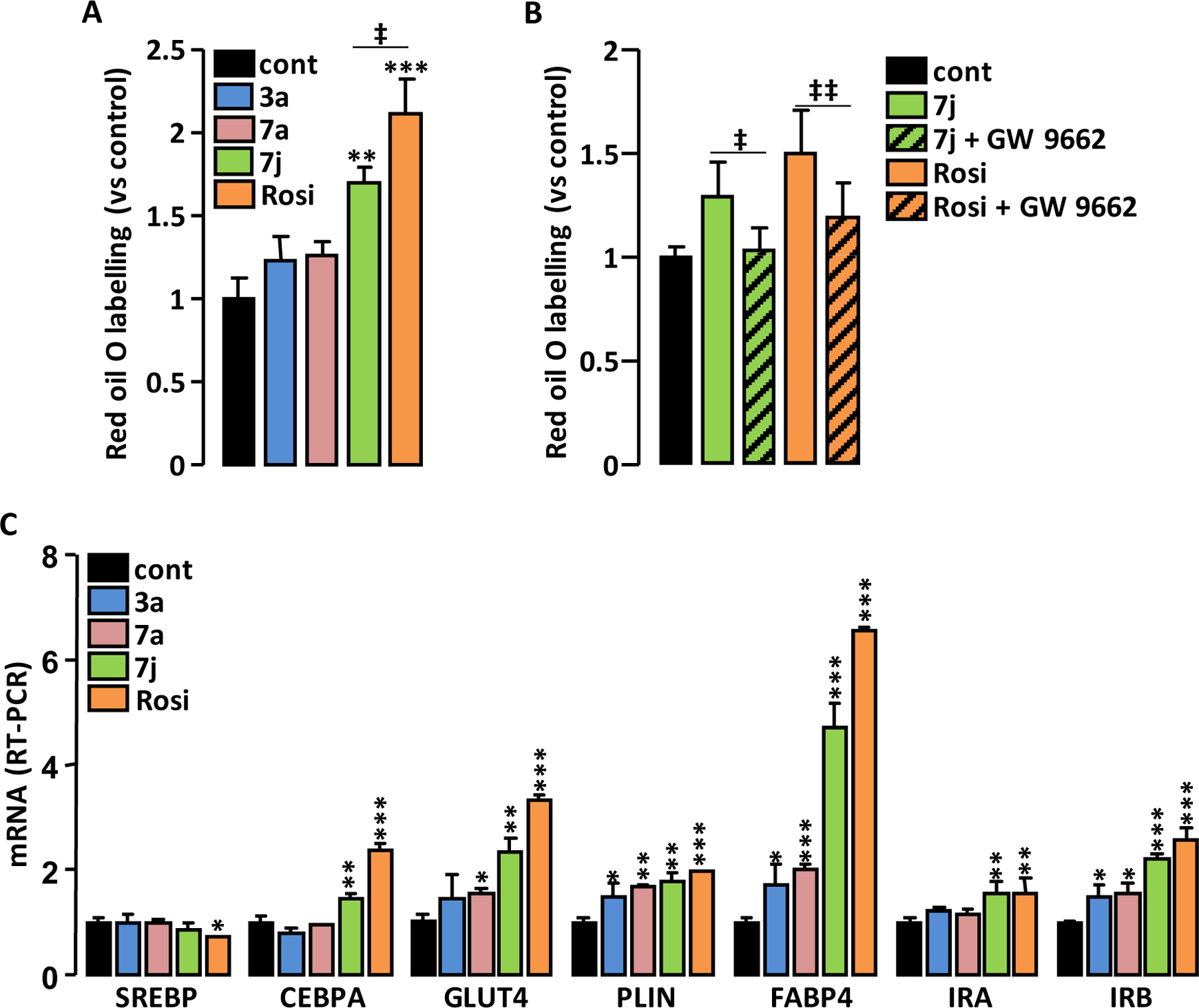
Adipogenic effect of the compounds. 3T3-L1 fibroblasts were incubated for 6 days (A) or for 4 days (B) with insulin (350 nM), the indicated PPARγ agonists (1 μM) and the PPARγ antagonist GW 9662 (5 μM). Intracellular lipids were stained with Red Oil O then colored lipids were quantified. (C) 3T3-L1 adipocyte differentiation was triggered by the standard mixture of inductors then cells were incubated for 7 days with 3a, 7a, 7j (1 μM) or Rosi (0.1 μM). The expression levels of adipogenesis-related genes were measured by RT-PCR. Values are means ± SD expressed as fold relative to untreated situation. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control (one-way ANOVA followed by Dunnett’s post hoc test); ‡p < 0.05, ‡‡p < 0.01 (t-test).
As expected for partial PPARγ agonists, 3a, 7a and 7j have reduced proadipogenic properties compared with the full agonist Rosi.
It has been shown that the ability of PPARγ ligands to increase cellular glucose uptake is not necessarily related to their transactivation activity or proadipogenic potential 32. Therefore, glucose uptake was measured in fully differentiated 3T3-L1 adipocytes after an acute treatment with PPARγ agonists. The insulin-dependent glucose uptake was increased by a short-term treatment with 7j or Rosi (Figure 8), denoting that part of their insulin sensitizing effect is independent of their ability to increase adipocyte differentiation.
Figure 8.
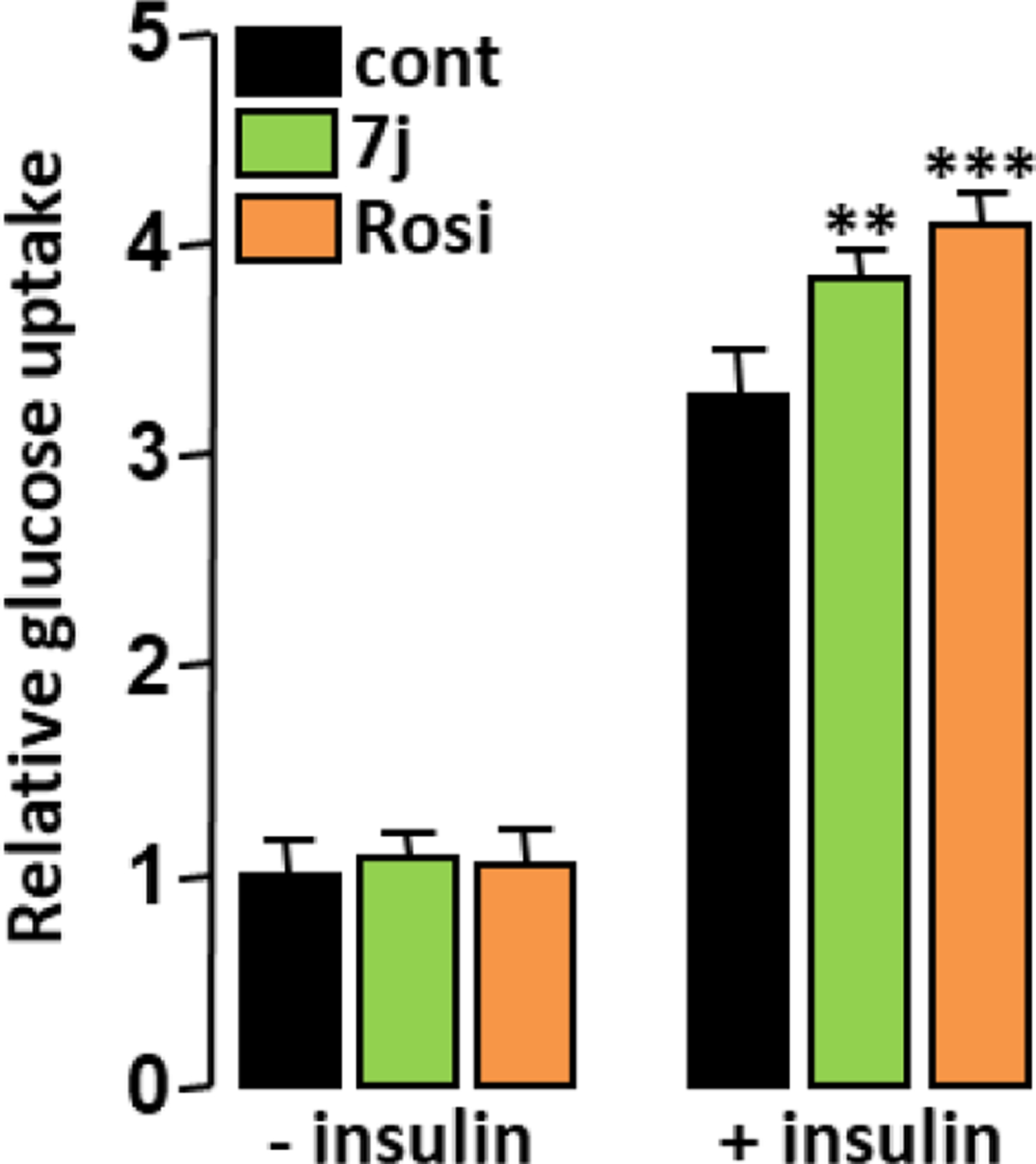
Adipocyte glucose uptake. 3T3-L1 adipocytes were treated with 7j (1 μM) or Rosi (0.1 μM) for 16 h prior insulin stimulation (50 nM; 10 min) and cellular glucose uptake was determined. Values are mean ± SD expressed as fold relative to the control situation. **p < 0.01, ***p < 0.001 vs. control (one-way ANOVA followed by Dunnett’s post hoc test).
Adipocyte browning.
Brown fat is a target for anti-obesity and anti-diabetes experimental therapies that aim to increase energy expenditure 33. Interestingly, strong PPARγ agonists activate the “browning” of white adipose tissues 34,35, suggesting that partial PPARγ agonists that retain significant white fat browning ability may also have therapeutic benefits in the treatment of obesity and diabetes. We therefore studied the ability of 3a, 7a and 7j to induce brite/brown-like adipocytes in 3T3-L1 cells.
Only 7j (and Rosi) upregulated mRNA levels of genes considered as brite/brown adipocyte-selective transcript (Figure 9A, B) 36. Immunoblotting analysis confirmed that 7j and Rosi increased the expression of Uncoupling Protein 1 (UCP1) protein, which is a functional marker of brown adipocytes (Figure 9B inset). Coherent with their adipocyte browning effect, Rosi and 7j increased mRNA levels of additional genes involved in (or associated with) mitochondria biogenesis (Figure 9C) 37 and in accordance these two molecules significantly increased the selective labeling of active mitochondria by the MitoTracker dye (Figure 9D). We have previously shown that Rosi induced the conversion of hMADS white adipocytes into brite adipocytes as evidenced by the strong expression of UCP1 38. In order to test whether compounds 3a, 7a and 7j were able to substitute for Rosi, hMADS cells first differentiated into white adipocytes were treated with these compounds between days 14 and 18. UCP1 expression was analyzed as an indicator of the degree of white-to-brown adipocyte conversion. UCP1 mRNA levels were increased in 3a, 7a and 7j-treated cells compared to untreated cells and 7j was found to be the most potent compound (Figure 9E), although less potent than Rosi (Figure 9F). The PPARγ agonists also increased the levels of UCP1 antigen, which is consistent with observations of its mRNA levels (Figure 9G).
Figure 9.
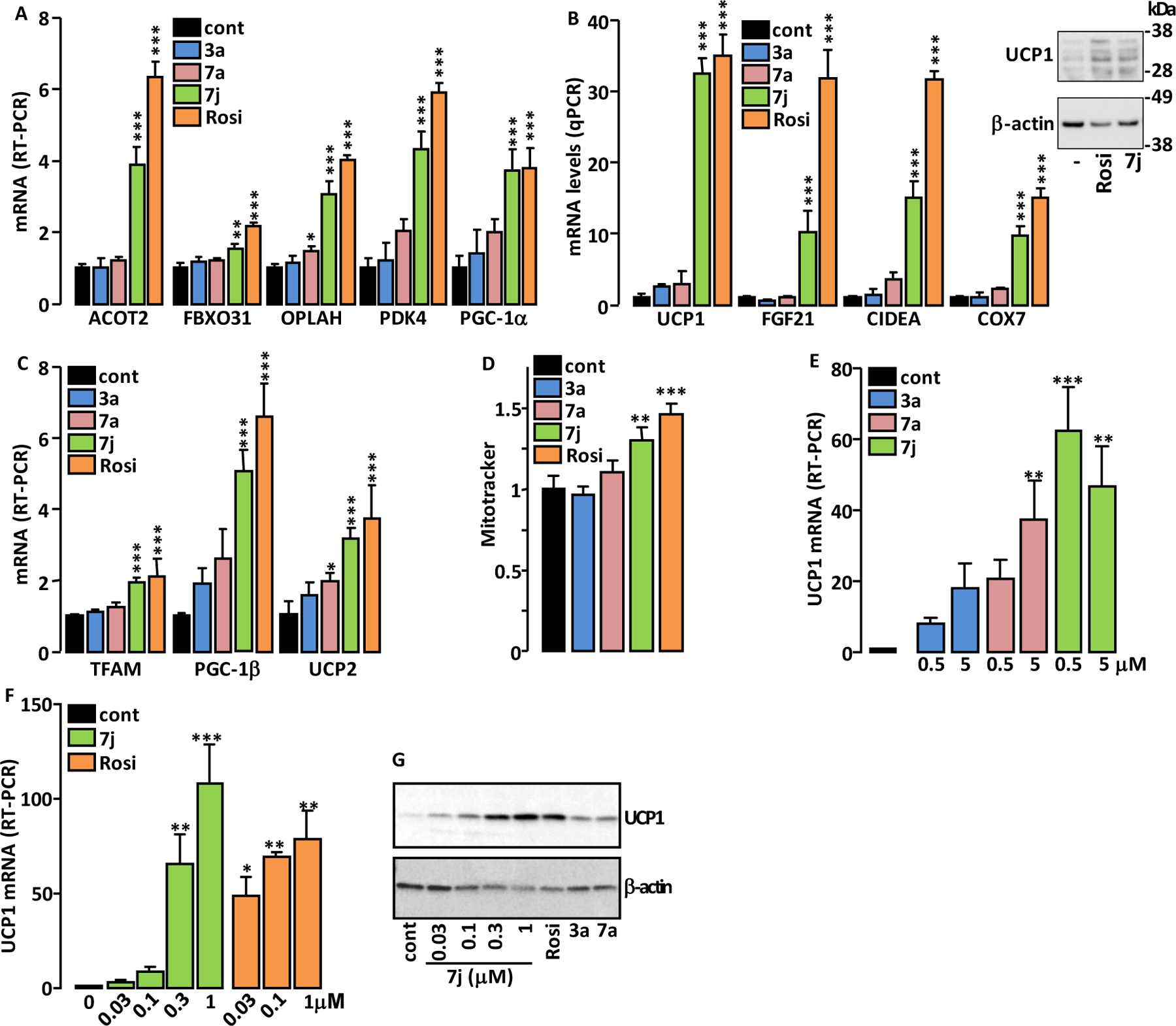
Adipocyte browning effect of the compounds. 3T3-L1 were treated as in Figure 7C then expression levels of beige (A) and brown (B) adipocyte markers as well as those of genes involved in mitochondrial biogenesis (C) were measured by RT-PCR. Inset in panel B: immunodetection of UCP1 and β-actin (loading control) in lysates of 3T3-L1 cells treated with 7j and Rosi. (D) Adipocyte mitochondria content was evaluated by flow cytometry after their selective labeling with MitoTracker dye. (E, F) hMADS white adipocytes were treated with the indicated compounds and UCP1 mRNA levels were measured by RT-PCR. (G) Immunodetection of UCP1 and β-actin (loading control) in lysates of hMADS treated with the indicated concentrations of 7j, Rosi (0.1 μM), 3a and 7a (1 μM). Values are mean ± SD expressed as fold relative to the control (cont) situation. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control (one-way ANOVA followed by Dunnett’s post hoc test).
The expression of the adipogenic marker Perilipin was not modified by Rosi or 7j (Supplemental Figure S18A), whereas the expression of Adiponectin, a PPARγ-responsive gene, was more efficiently increased by Rosi than by 7j (Supplemental Figure S18B), which confirms the that the PPARγ agonist activity of 7j is lower than that of Rosi.
These data suggest that 7j can efficiently promote the conversion from white to brown adipocytes.
Lipid accumulation in hepatocytes.
As PPARα agonists decrease hepatic steatosis5,7, we compared the effect of 7j (dual PPARα/γ agonist) to those of GW 7647 (PPARα agonist) and Rosi (PPARγ agonist) on the lipid accumulation into hepatocytes. In primary rat hepatocytes only GW 7647 prevented the basal intracellular lipid accumulation and oleic acid-induced lipid accumulation was prevented by Rosi, 7j and GW 7647 (Figure 10A). In human HuH7 hepatoma cells 7j and GW 7647 similarly prevented the oleic acid-stimulated lipid accumulation (Figure 10B). These results suggest that 7j is a promising compound to prevent hepatic steatosis.
Figure 10.
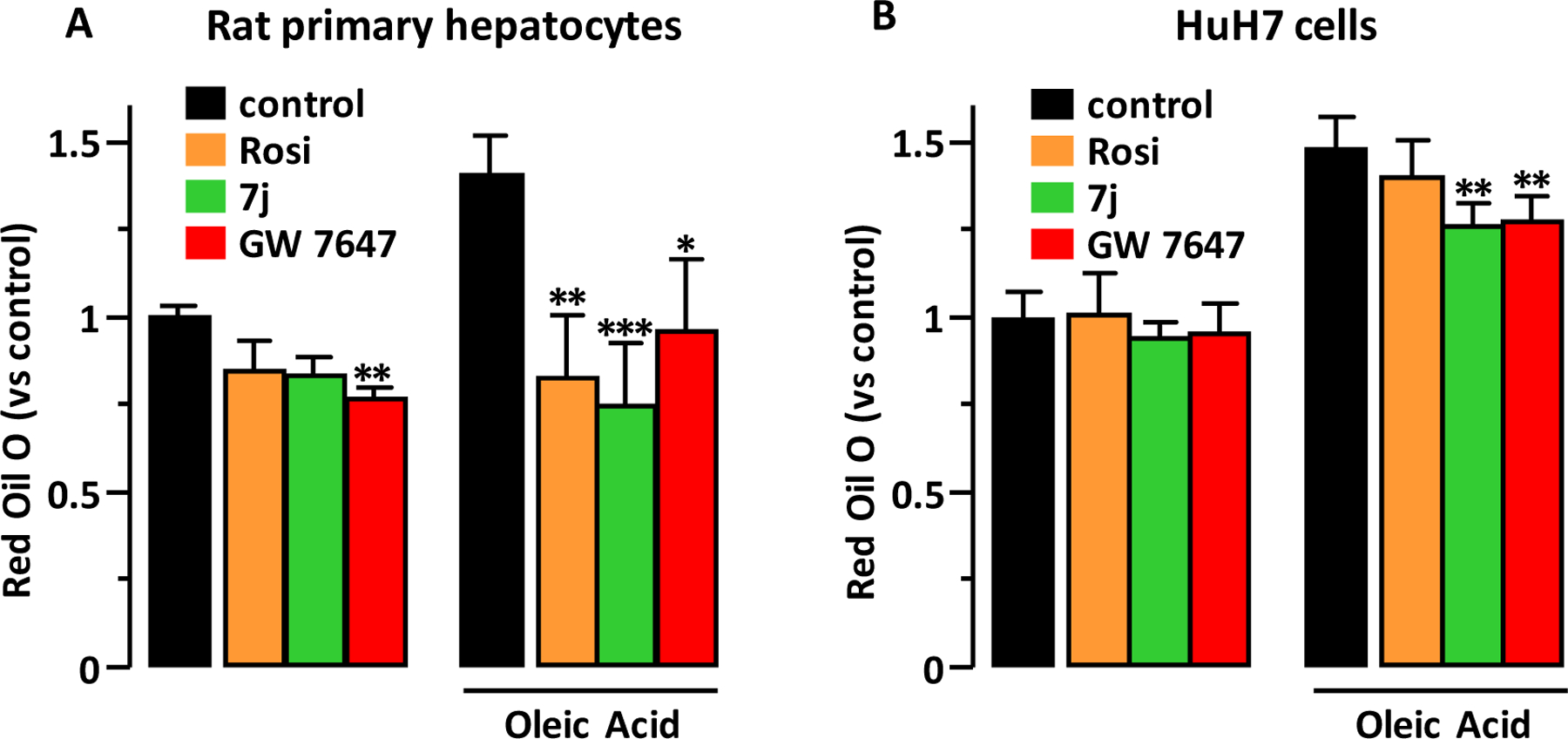
Lipid accumulation in hepatocytes. Primary rat hepatocytes (A) and human HuH7 hepatoma cells (B) were treated with 7j, Rosi or GW 7647 (1 μM) for 8 hours before addition of oleic acid (0.25 mM). 24 hours later, cellular lipid content was measured using Red Oil O staining. Values are mean ± SD expressed as fold relative to the control. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control (Kruskal-Wallis followed by Dunn’s post hoc test).
Cytotoxicity of 7j.
We confirmed, as previously reported39–41, that viability, membrane integrity and apoptosis of primary hepatocytes were not significantly altered by concentrations of Rosi up to 10 μM. These parameters were also not altered by the 7j compound (Supplemental Figure S19).
CONCLUSION
We have described the synthesis and optimization of compound 7j: a new N-aryl substituted valine derivative with a balanced agonist activity on PPAR α and γ. Compound 7j occupies the typical LBD region of the PPARγ agonists with a unique high-affinity binding mode and efficiently prevents CDK5-mediated phosphorylation of PPARγ. While poorly proadipogenic, compound 7j increases adipocyte insulin-stimulated glucose uptake and efficiently promotes white-to-brown adipocyte conversion. In addition, compound 7j prevents the oleic acid-induced lipid accumulation in hepatocytes. The unique biochemical properties of compound 7j, its specific biological activities and its low toxicity make it a promising candidate for the development of compounds to reduce insulin resistance, obesity and NAFLD.
EXPERIMENTAL SECTION
1. Chemistry
All solvents were purified according to reported procedures, and reagents were used as commercially available. Methanol, ethyl acetate, dichloromethane, ammonia and petroleum ether (35–60°C) were purchased from VWR and used without further purification. Column chromatography was performed on VWR silica gel (70–230 mesh). 1H NMR and 13C NMR spectra were recorded in CDCl3 or DMSO-d6 on a Bruker AC 300 spectrometer working at 300 MHz and 75 MHz, respectively (the usual abbreviations are used: s: singlet, d: doublet, t: triplet, q: quadruplet, m: multiplet). Tetramethylsilane was used as internal standard. All chemical shifts are given in ppm. Purity of all the new compounds evaluated by HPLC (Agilent 1100, C18) analysis was > 95%.
Typical procedure for the synthesis of amino acids methyl ester hydrochloride derivatives
Synthesis of L-Valine methyl ester hydrochloride
In a two necked round flask equipped with a condenser were placed at room temperature 3 g of L-Valine (2.56 10−2 mol) in 20 mL of methanol. The mixture was placed under stirring at 0°C and 3.4 mL of thionylchloride (4.7 10−3 mol) were slowly added. After removal of the solvents, diethylether was added and the product precipitate as a white solid. After filtration the product was dried under vacuum to afford the expected L-Valine methyl ester hydrochloride in 86% yield.
White solid; 1H NMR (D2O): δ = 4.14–4.17 (m, 1H), 3.80–3.85 (m, 3H), 2.50–2.56 (m, 1H), 1.12–1.15 (m, 6H). 13C (D2O): δ = 168.91, 58.57, 53.02, 29.95, 18.15.
(S)-2-(4-Hexyloxy-benzoylamino)-3-methyl-butyric acid methyl ester (6)
In a two necked round flask equipped with a condenser were placed at room temperature 2.34 g of benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate (BOP; 5.26 10−3 mol), 2.34 mL of diisopropylethylamine (1.8 10-2 mol), 1.2 g of 4-(Hexyloxy)benzoic acid (5.40 10−3 mol) and 0.88 g of L-Valine methyl ester hydrochloride (5.25 10−3 mol) in 15 mL of CH2Cl2. The mixture was placed under stirring at room temperature for 24 hours. Water was added to allow phase separation. The bottom phase layer was washed with NaHCO3 (10%) solution, dried over Na2SO4, filtered and concentrated in vacuo. After removal of the solvents, the crude residue was purified by chromatography on a silicagel column using CH2Cl2/Ethylacetate (1/1) eluent affording the expected product 6 in 85% yield.
White solid; 1H NMR (CDCl3): δ = 8.06 (m, 2H), 7.15 (m, 2H), 4.15–4.22 (m, 4H), 3.71–3.75 (m, 3H), 1.81–2.05 (m, 3H), 1.21–1.37 (m, 6H), 0.69–0.92 (m, 9H). 13C (CDCl3): δ = 172.90, 166.54, 160.52, 133.08, 130.21, 130.01, 114.55, 68.22, 57.56, 52.34, 33.49, 31.32, 29.34, 25.67, 22.61, 18.48, 14.03. MS (ESI) C19H29NO4 m/z 336.2146 (100%, (M+H+)).
4-Hexyloxy-N-((S)-1-hydroxycarbamoyl-2-methyl-propyl)-benzamide (3a)
In a 25 mL round flask were placed at room temperature 0.6 g of 6 (1.78 10−3 mol) in 15 mL of ethanol. 2 mL of a hydroxylamine solution (40%) were subsequently added and the mixture was allowed to stir at reflux for 24 hours. After removal of the solvents, the crude residue was purified by chromatography on a silicagel column using petroleum ether/ethylacetate (1/1) then methanol/ethylacetate (1/1) as eluents affording the expected product 3a in 72% yield.
White solid; 1H NMR (DMSO d6): δ = 8.01 (m, 2H), 7.09 (m, 2H), 3.95–3.98 (m, 3H), 1.78–1.82 (m, 2H), 1.01–1.31 (m, 6H), 0.71–0.93 (m, 9H). 13C (DMSO d6): δ = 167.20, 166.23, 161.1,133.41, 129.80, 113.98, 69.41, 57.31, 31.03, 29.42, 25.76, 22.43, 18.62, 13.89. MS (ESI) C18H28N2O4 m/z 337.2045 (100%, (M+H+)).
4-Hexyloxy-N-[(S)-1-hydroxycarbamoyl-2-(3H-imidazol-4-yl)-ethyl]-benzamide (3b)
Procedure similar to that applied for the preparation of 3a
Pale yellow solid; 1H NMR (DMSO d6): δ = 7.52–7.89 (m, 3H), 6.89–7.42 (m, 5H), 5.55 (s, 2H), 4.75–4.73 (m, 2H), 4.10–4.19 (m, 2H),1.22–1.92 (m, 9H), 0.89–0.96 (m, 3H). 13C (DMSO d6): δ = 174.19, 164.07, 162.03, 133.17, 129.03, 123.47, 118.22, 115.35, 67.56, 52.96, 30.99, 30.91, 29.05, 25.49, 22.54, 14.25. MS (ESI) C19H26N4O4 m/z 375.2014 (100%, (M+H+)).
4-Hexyloxy-N-[(S)-1-hydroxycarbamoyl-2-(1H-indol-2-yl)-ethyl]-benzamide (3c)
Procedure similar to that applied for the preparation of 3a
White solid; 1H NMR (DMSO d6): δ = 7.32–7.62 (m, 5H), 6.29–7.25 (m, 7H), 3.92–4.03 (m, 2H), 2.89–3.34 (m, 4H), 1.20–1.79 (m, 6H), 0.88–0.92 (m, 3H). 13C (DMSO d6): δ = 172.22, 168.98, 161.25, 139.48, 131.05, 129.14, 126.29, 120.15, 118.78, 113.69, 110.36, 104.25, 71.24, 53.32, 30.21, 28.28, 25.14, 22.04, 14.13. MS (ESI) C24H29N3O4 m/z 424.2236 (100%, (M+H+)).
4-Hexyloxy-N-((S)-2-hydroxy-1-hydroxycarbamoyl-ethyl)-benzamide (3d)
Procedure similar to that applied for the preparation of 3a
White solid; 1H NMR (DMSO d6): δ = 7.02–7.51 (m, 4H), 3.62–4.20 (m, 5H), 1.31–1.82 (m, 9H), 0.89–0.92 (m, 3H). 13C (DMSO d6): δ = 170.36, 168.24, 160.89, 130.14, 125.34, 113.47, 68.78, 61.24, 53.89, 31.01, 28.14, 25.98, 21.33, 13.88. MS (ESI) C16H24N2O5 m/z 325.1768 (100%, (M+H+)).
4-Hexyloxy-N-((S)-1-hydroxycarbamoyl-2-phenyl-ethyl)-benzamide (3e)
Procedure similar to that applied for the preparation of 3a
White solid; 1H NMR (DMSO d6): δ = 7.66–7.68 (m, 2H), 6.47–7.16 (m, 2H), 3.55–3.97 (m, 2H), 2.89–3.23 (m, 2H), 1.35–1.74 (m, 8H), 0.90–0.91 (m, 3H). 13C (DMSO d6): δ = 178.45, 168.32, 161.25, 137.47, 129.02, 125.68, 114.98, 60.01, 53.87, 40.36, 30.23, 28.14, 24.12, 22.98, 13.48. MS (ESI) C22H28N2O4 m/z 385.2057 (100%, (M+H+)).
N-[(S)-2-(3,4-Dihydroxy-phenyl)-1-hydroxycarbamoyl-ethyl]-4-hexyloxy-benzamide (3f)
Procedure similar to that applied for the preparation of 3a
White solid; 1H NMR (DMSO d6): δ = 7.71–7.73 (m, 2H), 6.59–6.93 (m, 4H), 6.32 (s, 1H), 5.87 (s, 1H), 3.99–4.02 (m, 2H), 2.95–2.99 (m, 2H), 1.17–2.02 (m, 9H), 0.87–0.89 (m, 3H). 13C (DMSO d6): δ = 171.25, 168.33, 160.58, 146.01, 143.69, 128.69, 126.47, 123.74, 116.36, 115.47, 69.71, 52.34, 38.38, 28.47, 22.14, 25.69, 22.47, 13.12. MS (ESI) C22H28N2O6 m/z 417.1934 (100%, (M+H+)).
4-Hexyloxy-N-((S)-1-hydroxycarbamoyl-4-methylsulfanyl-butyl)-benzamide (3g)
Procedure similar to that applied for the preparation of 3a
White solid; 1H NMR (DMSO d6): δ = 6.91–7.69 (m, 5H), 1.17–2.74 (m, 20H), 0.87–0.88 (m, 3H). 13C (DMSO d6): δ = 170.78, 168.65, 162.21, 128.12, 125.14, 117.77, 69.66, 51.45, 34.12, 31.78, 28.02, 25.63, 22.33, 15.15, 13.19. MS (ESI) C19H30N2O4S m/z 383.1935 (100%, (M+H+)).
4-(hexyloxy)-N-((2S,3S)-3-hydroxy-1-(hydroxyamino)-1-oxobutan-2-yl)benzamide (3h)
Procedure similar to that applied for the preparation of 3a
White solid; 1H NMR (DMSO d6): δ = 7.71–7.73 (m, 2H), 6.98–7.11 (m, 3H), 5.32 (s, 1H), 4.60–4.65 (m, 1H), 4.06–4.44 (m, 3H), 1.80–1.92 (m, 1H), 1.07–1.37 (m, 12H); 13C (DMSO d6): δ = 169.75, 167.63, 162.54, 132.53, 125.82, 114.51, 68.78, 67.63, 59.34, 31.88, 28.02, 24.89, 22.33, 15.12, 13.89. MS (ESI) C17H26N2O5 m/z 339.1879 (100%, (M+H+)).
(S)-N-(1-(hydroxyamino)-3-methyl-1-oxobutan-2-yl)-4-methoxybenzamide (4a)
White solid; 1H NMR (DMSO d6): δ = 0.91–0.96 (m, 6H), 2.16–2.23 (m, 1H), 3.84 (s, 3H), 4.33–4.45 (m, 1H), 6.98–7.53 (m, 2H), 7.83–7.89 (m, 2H), 8.79 (s, 2H); 13C (DMSO d6): δ = 174.44, 170.42, 164.51, 130.95, 127.71, 115.17, 56.40, 52.96, 32.19, 20.12, 19.65. MS (ESI) C13H18N2O4 m/z 267.1386 (100%, (M+H+)).
(S)-4-ethoxy-N-(1-(hydroxyamino)-3-methyl-1-oxobutan-2-yl)benzamide (4b)
White solid; 1H NMR (DMSO d6): δ = 0.91–0.95 (m, 6H), 1.49 (t, J = 7.0 Hz, 3H), 1.68–1.75 (m, 1H), 4.01–4.11 (m, 2H), 4.31–4.41 (m, 1H), 6.94–6.99 (m, 2H), 7.80–7.85 (m, 2H), 8.59 (s, 2H); 13C (DMSO d6): δ = 171.10, 166.47, 166.91, 132.01, 129.30, 114.59, 63.42, 56.95, 30.10, 18.64, 14.69. MS (ESI) C14H20N2O4 m/z 281.1412 (100%, (M+H+)).
(S)-N-(1-(hydroxyamino)-3-methyl-1-oxobutan-2-yl)-4-propoxybenzamide (4c)
White solid; 1H NMR (DMSO d6): δ = 0.92–1.12 (m, 9H), 1.69–1.89 (m, 2H), 3.27–3.38 (m, 2H), 3.90–4.02 (m, 2H), 6.90–7.07 (m, 2H), 7.76–7.89 (m, 2H); 13C (DMSO d6): δ = 171.14, 165.95, 162.22, 130.95, 128.03, 115.09, 70.35, 57.03, 30.75, 22.63, 18.64, 10.63. MS (ESI) C15H22N2O4 m/z 294.1634 (100%, (M+H+)).
(S)-4-butoxy-N-(1-(hydroxyamino)-3-methyl-1-oxobutan-2-yl)benzamide (4d)
White solid; 1H NMR (DMSO d6): δ = 0.90–0.98 (m, 9H), 1.52–1.62 (m, 2H), 1.77–1.83 (m, 2H), 2.16–2.22 (m, 2H), 3.97–4.02 (m, 2H), 4.37–4.42 (m, 1H), 7.75–7.77 (m, 2H), 7.98–8.02 (m, 2H), 8.53 (m, 1H); 13C (DMSO d6): δ = 171.49, 166.14, 166.16, 133.37, 130.18, 122.86, 69.16, 57.04, 30.74, 30.69, 18.48, 13.75. MS (ESI) C16H24N2O4 m/z 309.1701 (100%, (M+H+)).
(S)-N-(1-(hydroxyamino)-3-methyl-1-oxobutan-2-yl)-4-(pentyloxy)benzamide (4e)
White solid; 1H NMR (DMSO d6): δ = 0.90–0.98 (m, 9H), 1.52–1.62 (m, 2H), 1.77–1.83 (m, 2H), 2.16–2.22 (m, 2H), 3.97–4.02 (m, 2H), 4.37–4.42 (m, 1H), 7.75–7.77 (m, 2H), 7.98–8.02 (m, 2H), 8.53 (m, 1H); 13C (DMSO d6): δ = 169.71, 167.51, 162.83, 132.86, 126.86, 119.98, 68.72, 58.13, 31.14, 30.98, 29.34, 28.14, 18.54, 14.16. MS (ESI) C17H26N2O4 m/z 323.1934 (100%, (M+H+)).
(S)-4-(heptyloxy)-N-(1-(hydroxyamino)-3-methyl-1-oxobutan-2-yl)benzamide (4f)
White solid; 1H NMR (DMSO d6): δ = 0.90–0.96 (m, 9H), 1.28–1.74 (m, 10H), 2.02–2.16 (m, 1H), 3.97–4.07 (m, 2H), 4.37–4.42 (m, 1H), 7.03–7.08 (m, 2H), 8.02–8.09 (m, 2H), 8.53 (m, 1H); 13C (DMSO d6): δ = 171.09, 166.42, 162.02, 130.07, 129.03, 115.18, 68.15, 57.63, 31.80, 29.18, 26.04, 22.14, 18.74, 13.98. MS (ESI) C19H30N2O4 m/z 351.2245 (100%, (M+H+)).
(S)-N-(1-(hydroxyamino)-3-methyl-1-oxobutan-2-yl)-4-(octyloxy)benzamide (4g)
White solid; 1H NMR (DMSO d6): δ = 0.90–0.97 (m, 9H), 1.29–1.82 (m, 12H), 2.06–2.15 (m, 1H), 3.95–4.07 (m, 2H), 4.32–4.41 (m, 1H), 7.00–7.08 (m, 2H), 8.01–8.10 (m, 2H), 8.51 (m, 1H); 13C (DMSO d6): δ = 171.10, 166.35, 162.35, 131.37, 129.90, 115.11, 68.24, 57.52, 31.79, 30.95, 29.31, 26.82, 22.47, 18.63, 14.21. MS (ESI) C20H32N2O4 m/z 365.2423 (100%, (M+H+)).
(S)-2-(4-Hexyloxy-benzoylamino)-3-methyl-butyric acid (7a)
In a 25 mL round flask were placed at room temperature 0.6 g of 6 (1.78 10−3 mol) in 15 mL of ethanol. 2 mL of a sodium hydroxide solution (10%) were subsequently added and the mixture was allowed to stir at room temperature for 24 hours. The bottom phase layer was discarded, and the aqueous phase was acidified with HCl 1N. After extraction with ethylacetate, the organic phase was dried over Na2SO4, filtered and concentrated in vacuo. The crude residue was purified by chromatography on a silicagel column using petroleum ether/ethylacetate (1/1) as eluent affording the expected product 7a in 69% yield.
White solid; 1H NMR (CDCl3): δ = 7.80–7.84 (m, 2H), 6.95–7.04 (m, 2H), 4.44–4.46 (m, 1H), 4.02–4.05 (m, 2H), 2.19–2.35 (m, 1H), 1.73–1.84 (m, 2H), 1.43–1.57 (m, 2H), 1.26–1.35 (m, 5H), 1.04 (s, 3H), 1.03 (s, 3H), 0.91–0.94 (m, 3H). 13C (CDCl3): δ = 174.59, 168.63, 162.17, 128.97, 125.94, 113.84, 67.86, 58.88, 31.35, 30.54, 28.87, 25.42, 22.27, 18.43, 17.52, 12.98. MS (ESI) C18H27NO4 m/z 322.1932 (100%, (M+H+)).
(R)-2-(4-Hexyloxy-benzoylamino)-3-methyl-butyric acid (7a’)
Procedure similar to that applied for the preparation of 7a
White solid; 1H NMR (CDCl3): δ = 6.55–7.19 (m, 4H), 3.89–3.99 (m, 3H), 1.50–2.11 (m, 7H), 0.89–1.32 (m, 11H). 13C (CDCl3): δ = 177.56, 166.34, 159.95, 132.36, 130.12, 114.08, 72.30, 64.36, 31.32, 31.04, 29.14, 25.31, 22.14, 18.47, 13.96. MS (ESI) C18H27NO4 m/z 322.1932 (100%, (M+H+)).
(S)-2-(4-Hexyloxy-benzoylamino)-3-phenyl-propionic acid (7b)
Procedure similar to that applied for the preparation of 7a
White solid; 1H NMR (CDCl3): δ = 10.98 (s, 1H), 7.89–8.01 (m, 2H), 6.95–7.21 (m, 7H), 4.83–4.85 (m, 11H), 3.89–3.92 (m, 2H), 3.01–3.03 (m, 2H), 0.95–1.61 (m, 12H). 13C (CDCl3): δ = 176.15, 168.13, 161.14, 140.03, 129.18, 128.82, 123.52, 123.42, 115.22, 73.34, 61.56, 38.78, 33.14, 31.42, 26.14. MS (ESI) C23H30NO4 m/z 385.2221 (100%, (M+H+)).
(S)-2-(4-Hexyloxy-benzoylamino)-3-(3H-imidazol-4-yl)-propionic acid (7c)
Procedure similar to that applied for the preparation of 7a
Yellow solid; 1H NMR (CDCl3): δ = 7.93 (s, 1H), 7.63–7.65 (m, 2H), 6.75–6.92 (m, 4H), 4.69–4.72 (m, 2H), 4.12–3.92 (m, 2H), 2.67–2.78 (m, 2H), 0.76–1.52 (m, 11H). 13C (CDCl3): δ = 174.75, 169,70, 161.08, 132.04, 131.03, 129.92, 125.75, 121.56, 111.04, 68.13, 52.67, 31.14, 29.57, 29.15, 25.56, 22.59, 13.82. MS (ESI) C19H25N3O4 m/z 360.1834 (100%, (M+H+)).
(S)-2-(4-Hexyloxy-benzoylamino)-3-hydroxy-propionic acid (7d)
Procedure similar to that applied for the preparation of 7a
White solid; 1H NMR (DMSO d6): δ = 7.68 (s, 2H), 6.92–6.96 (m, 2H), 3.92–4.54 (m, 6H), 1.20–1.95 (m, 9H), 0.89–0.92 (m, 3H). 13C (DMSO d6): δ = 173.32, 166.74, 161.44, 128.45, 125.18, 111.14, 68.84, 62.30, 56.14, 31.12, 29.11, 24.13, 22.59, 14.13. MS (ESI) C16H23NO5 m/z 310.1667 (100%, (M+H+)).
(4-Hexyloxy-benzoylamino)-acetic acid (7e)
Procedure similar to that applied for the preparation of 7a
White solid; 1H NMR (DMSO d6): δ = 7.68 (s, 2H), 6.98 (s, 2H), 4.10–4.12 (m, 2H), 3.78 (m, 2H), 1.49–1.76 (m, 8H), 0.89–0.92 (m, 3H). 13C (DMSO d6): δ = 172.31, 168.28, 161.02, 130.06, 114.55, 114.53, 66.42, 42.85, 34.56, 32.12, 29.14, 23.12, 21.45, 14.14. MS (ESI) C15H21NO4 m/z 280.1511 (100%, (M+H+)).
(S)-2-(4-Methoxy-benzoylamino)-3-methyl-butyric acid (7f)
Procedure similar to that applied for the preparation of 7a
White solid; 1H NMR (DMSO d6): δ = 7.53–7.55 (m, 2H), 6.95–6.97 (m, 2H), 6.05 (s, 1H), 4.75–4.77 (m, 1H), 3.72 (s, 3H), 1.75–1.87 (m, 1H), 1.04–1.06 (m, 6H). 13C (DMSO d6): δ = 172.33, 167.28, 160.14, 131.12, 127.30, 110.15, 58.66, 55.65, 30.93, 19.03. MS (ESI) C13H17NO4 m/z 251.1232 (100%, (M+H+)).
(S)-2-(4-Ethoxy-benzoylamino)-3-methyl-butyric acid (7g)
Procedure similar to that applied for the preparation of 7a
White solid; 1H NMR (DMSO d6): δ = 7.59 (s, 2H), 7.02–7.05 (m, 2H), 4.16–4.40 (m, 3H), 1.87–1.92 (m, 1H), 1.32–1.36 (m, 3H), 0.98–1.03 (m, 6H). 13C (DMSO d6): δ = 173.15, 167.10, 160.83, 129.98, 127.04, 113.21, 61.89, 57.93, 29.98, 19.19, 14.31. MS (ESI) C14H19NO4 m/z 266.1342 (100%, (M+H+)).
(S)-3-Methyl-2-(4-propoxy-benzoylamino)-butyric acid (7h)
Procedure similar to that applied for the preparation of 7a
White solid; 1H NMR (DMSO d6): δ = 6.95–7.32 (m, 4H), 6.75 (s, 1H), 3.98–4.05 (m, 3H), 1.54–1.89 (m, 3H), 1.12–1.17 (m, 3H), 0.97–1.02 (m, 6H). 13C (DMSO d6): δ = 172.34, 167.42, 160.15, 128.64, 127.42, 112.45, 69.37, 58.62, 29.89, 21.12, 18.82, 10.29. MS (ESI) C15H21NO4 m/z 280.1534 (100%, (M+H+)).
(4-(heptyloxy)benzoyl)-L-valine (7i)
White solid; 1H NMR (DMSO d6): δ = 0.93–1.41 (m, 19H), 1.41–1.46 (m, 2H), 2.23–2.35 (m, 1H), 3.87–3.92 (t, J = 5Hz, 2H), 4.46–4.69 (m, 1H), 6.82–6.89 (m, 2H), 7.66–7.69 (m, 2H); 13C (DMSO d6): δ = 175.24, 167.12, 162.78, 129.02, 114.39, 68.28, 57.84,31.80, 31.27, 29.14, 29.07, 25.98, 22.64, 19.15, 17.92, 14.12. MS (ESI) C19H29NO4 m/z 336.2165 (100%, (M+H+)).
(4-(octyloxy)benzoyl)-L-valine (7j)
White solid; 1H NMR (DMSO d6): δ = 0.91–1.34 (m, 10H), 1.38–1.54 (m, 11H), 1.76–1.87 (m, 2H), 2.23–2.37 (m, 1H), 4.0.3–4.08 (t, J = 5Hz, 2H), 4.48–4.52 (dd, J = 5Hz, 1H), 6.98–7.01 (m, 2H), 7.83–8.86 (m, 2H); 13C (DMSO d6): δ = 175.31, 170.16, 163.59, 130.46, 127.28, 115.21, 69.24, 59.88, 33.04, 31.82, 30.53, 30.46, 30.34, 27.19, 23.77, 19.79, 18.97, 14.49. MS (ESI) C20H31NO4 m/z 350.2315 (100%, (M+H+)).
(4-(decyloxy)benzoyl)-L-valine (7k)
White solid; 1H NMR (DMSO d6): δ = 0.92–1.32 (m, 10H), 1.44–1.54 (m, 15H), 1.82–1.88 (m, 2H), 2.33–2.46 (m, 1H), 3.94–4.06 (m, 2H), 4.82–4.87 (m, 1H), 6.86–6.94 (m, 2H), 7.73–7.83 (m, 2H) 13C (DMSO d6): δ = 175.36, 167.71, 162.20, 129.31, 129.10, 125.71, 114.33, 114.15, 68.25, 57.62, 31.94, 31.43, 29.61, 29.43, 29.37, 29.16, 26.03, 22.73, 19.09, 17.91, 14.18. MS (ESI) C22H35NO4 m/z 378.2645 (100%, (M+H+)).
Chemicals.
Rosiglitazone, Bezafibrate and Roscovitine were from Sigma Aldrich (Saint-Quentin Fallavier, France). GW 7647 was from Axon Medchem (Groningen, The Netherlands). GW 9662 and SR 11235 were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Antibodies.
PPARγ antibody (E-8) was from Santa Cruz Biotechnology. PPARγ phospho Ser273 antibodies were from Rockland (Limerick, PA, USA) or custom produced by New England Peptide (Gardner, MA, USA). UCP1 antibodies (ab10983) were from Abcam (Cambridge, MA, USA). β-actin (13E5) was from Cell Signaling Technology (Danvers, MA, USA).
Cell culture.
Media and cell culture reagents were from Thermo Fisher Scientific (Illkirch-Graffenstaden, France). HEK293 cells (Griptite 293 MSR) were from Thermo Fisher Scientific. 3T3-L1 cells (from ATCC) were routinely cultured in DMEM with 4 mM l-glutamine, 4.5 g/liter glucose, 0.11 g/liter sodium pyruvate, and supplemented with 10% fetal bovine serum plus antibiotics. Two days after confluence, adipocytes differentiation was triggered by changing the adding the conventional induction mixture (0.1 μM dexamethazone, 500 μM 3-Isobutyl-1-methylxanthine, and 174.5 nM insulin from Sigma-Aldrich, L’Isle d’Abeau Chesnes, France). After 48 h, the medium was removed and replaced by a fresh medium containing only 174.5 nM insulin. HuH7 hepatoma cells from the Japanese Cancer Research Resources Bank were cultured in DMEM containing 10% fetal bovine serum (FBS). At the confluence, cells were treated for 24 hours with 0.25 mM oleic acid (Sigma-Aldrich) complexed with BSA. The establishment, characterization and culture protocols of human Multipotent Adipose-Derived Stem (hMADS) cells have been described previously 42. Briefly, confluent cells were submitted to differentiation medium (DMEM/Ham’s F12 media containing 10 µg/ml transferrin, 10 nM insulin, and 0.2 nM triiodothyronine from Sigma-Aldrich) supplemented with 1 µM dexamethasone and 500 µM isobutyl-methylxanthine. Two days later, the medium was changed, dexamethasone and isobutyl-methylxanthine were omitted and 100 nM Rosi (Sigma-Aldrich) were added for the indicated periods. Cells were treated between days 2 and 9 with Rosi to enable white adipocyte differentiation to take place. After 5 days in the absence of Rosi, brite adipocyte conversion was induced by adding compounds to be tested (day 14). Medium was changed every other day and cells were used at day 18. Pooled plateable cryopreserved primary rat hepatocytes (Male, Sprague Dawley) from Xenotech (Kansas city, KS, USA) were plated and cultured as described by the manufacturer.
Cell viability, Toxicity, apoptosis assay.
Cell viability was monitored using the CellTiter-Blue Cell Viability Assay (Promega, Madison, WI) based on the ability of living cells to convert resazurin into the fluorescent end product resorufin and by measuring cellular ATP levels, that declines rapidly when cells undergo necrosis or apoptosis, using ATPLite 1 step luminescence assay system (Perkin Elmer, Waltham, MA, USA) according to the manufacturer’s instructions. Cytotoxicity was determined by measuring the lactate dehydrogenase (LDH) released into the culture medium upon plasma membrane damage using the LDH-Glo Cytotoxicity Assay (Promega). Apoptosis was determined by measuring caspase 3/7-dependent cleavage of a luminogenic substrate using the Caspase-Glo 3/7 Assay (Promega). Experiments were performed 2 to 3 times in triplicate.
Cellular lipid content staining and measurement.
Oil Red O staining: 3T3-L1, HuH7 hepatoma cells or primary rat hepatocytes were washed with PBS and fixed with 4% formaldehyde solution for 20 minutes, then washed again and stained with 0.35% Oil Red O solution (Sigma-Aldrich) in 60% isopropanol for 20 minutes. Then, cells were washed with water, and photographs were taken. The stain from the cells was eluted using 100% isopropanol and the absorbance of the eluted stain was read at 490 nm. Experiments were performed 5 times in duplicate for cell lines and twice in triplicate for primary cells. AdipoRed assay: 3T3-L1 preadipocytes were cultured in black 96-well plate. Two days post confluency, cells were incubated with the test molecules (10 μM) for seven additional days. Cells were washed with PBS then a solution of AdipoRed reagent (1 / 40 in PBS) was added into each well. After ten minutes at room temperature and in the dark, fluorescence was measured at 485 nm and 572 nm for excitation and emission respectively on an EnSight Microplate Reader (Perkin Elmer) in the scanning mode. Since this technique was used for screening purposes, the experiment was performed once in duplicate.
2-deoxy-D-glucose uptake assay.
Glucose uptake activity of fully differentiated 3T3-L1 adipocytes was measured by the chemiluminescent assay 43 using Glucofax kit as described by the manufacturer (Yelen, Ensues la Redonne, France). Briefly, 3T3-L1 adipocytes (80% of the cells displaying the characteristic lipid-filled phenotype) were gently detached from the plate by Accutase (Thermo Fisher Scientific) treatment, seeded (3.5 104 cells/well) at confluence in 96-well culture dishes and cultured for two more days in DMEM supplemented with 10% FBS. Adipocytes were then incubated in glucose-free DMEM for 4 h and then washed twice with Krebs-Ringer-phosphate-Hepes (KRPH) buffer (20 mM Hepes, 5 mM KH2PO4, 1 mM MgSO4, 1 mM CaCl2, 136 mM NaCl, and 4.7 mM KCl at pH 7.4) containing 0.2% BSA and incubated 30 min in 100 μl of KRPH / BSA. KRPH buffer was removed and cells were incubated for 20 min with 170 μl of 100 nM insulin diluted in KRBH buffer, then 19 μl of 10 mM 2-deoxy-D-glucose (Sigma-Aldrich) was added and the cells were incubated for 20 min. Cells were then washed four times with cold PBS and lysed with 60 μl of reagent I of Glucofax kit. After 60 min incubation at 37 °C, 20 μL of cell lysates were collected and transferred into white 96-well plate. Then 100 μl of reagent II was added, and after 10 min incubation, the chemiluminescence was recorded on the EnSight multimode reader (Perkin Elmer). Experiments were performed 3 times in sextuplet.
Real Time PCR Analysis.
Total RNA was extracted using a Nucleospin RNA kit (Macherey-Nagel, Hoerdt, France), cDNA was synthesized from 0.5 μg of RNA using Moloney murine leukemia virus reverse transcriptase (Thermo Fisher Scientific) and used for PCR amplification. Real Time PCR (RT-PCR) were performed on the LightCycler 480 instrument (Roche Applied Science, Basel, Switzerland) using the Eva Green MasterMix (Euromedex, Souffelweyersheim, France). The comparative Ct method (2−(ΔΔCT)) was used to calculate the relative differences in mRNA expression. The acidic ribosomal phosphoprotein P0 (Rplp0) was used as housekeeping gene. Changes were normalized to the mean of control values, which was set to 1. Primer were synthesized by Eurogentec (Seraing, Belgium) their sequences were previously published 17,36,42. Experiments were performed 5 times in duplicate.
MitoTracker staining.
3T3-L1 adipocytes were trypsinized and centrifuged at 300 g at 4°C for 5 min. Cells were suspended in KRPH containing 0.5% BSA and incubated with 0.1 μM MitoTracker Green FM (Thermo Fisher Scientific) for 30 min at 37°C. Cells were spun at 300 g at 4°C for 5 min and suspended in 400 μl of fresh KRPH then 50 000 cells were analyzed using a BD Accuri C6 flow cytometer (BD Biosciences). Experiments were performed 3 times in triplicate.
Cell-based PPAR and RXRα transactivation assay.
PPARγ-LBD-Gal4 or PPARα-LBD-Gal4 expression vector (given by Dr. Teruo Kawada, Kyoto University, Japan) was transfected along with SV40-driven Renilla luciferase expression vector in HEK293 cells stably expressing the Gal4 response element driven Firefly luciferase reporter (pGL4.35[luc2P/9XGAL4UAS/Hygro] vector from Promega, Madison, WI, USA). 36 hours after transfection, cells were exposed to the tested compounds for additional 16 hours then Firefly and Renilla luciferase activities were measured in the cell lysates using the reagent Genofax A and C (Yelen) in an EnSight multimode reader (Perkin Elmer). PPAR transactivation activity of the compounds is calculated as ratio of Firefly to Renilla luciferase activity. Experiments were performed at least 3 times in duplicate (allowing the calculation of EC50 and maximal activation, Table 1), a representative experiment is shown in Figures 3 and S5. For the measure of PPARδ transactivation: HG5LN-GAL-PPARδ reporter cell line was previously described 44. Reporter cells were seeded at a density of 20,000 cells/well in 96-well white opaque tissue culture plates and maintained in phenol-red-free Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% dextran-coated, charcoal-treated fetal calf serum. 24 hours later, culture medium was replaced with DMEM containing tested compounds. 16 hours after exposure media was replaced with media containing 0.3 mM luciferin. Luminescence was measured in intact living cells for 2 sec in a Microbeta Wallac luminometer (PerkinElmer). Experiments were performed twice in duplicate. The measure of RXRα transactivation was performed as described for PPARγ except that RXRα-LBD-Gal4 expression vector was transfected.
Mammalian Two-Hybrid Assay.
HEK293 cells stably expressing the GAL4 response element driven Firefly luciferase reporter were transfected with SV40-driven Renilla luciferase expression vector together with expression vector of a fusion protein of the VP16 activation domain to PPARγ-LBD and with expression vector of a fusion protein of the GAL4-DNA binding domain to NCoR1, SMRT, TIF2, MED1 or RXRα. Then cells were treated as described for Cell-based PPAR transactivation assay. Experiments were performed 2 to 3 times in duplicate.
Immunoblot.
3T3-L1 adipocytes were incubated 24 hours with 1 % SVF then treated with PPARγ agonists for 60 min followed by 90 min of stimulation with murine TNF (50 ng/ml). Cells were lysed in the presence of a cocktail of protease and phosphatase inhibitor. Identical amounts of total protein were heat-denatured and reduced (70 °C; 10 min) then submitted to SDS-PAGE separation on 4–12% gradient (Thermo Fisher Scientific) and transferred to polyvinylidene fluoride membranes. Membranes were blocked for 1 h in 5% BSA solution and incubated with the appropriate primary and HRP-conjugated secondary antibodies (1:1000 and 1:10,000 dilutions, respectively). Immunodetections were performed using ECL reagent and image acquisition was performed by using a chemiluminescent CCD imager ImageQuant LAS 4000 (GE Healthcare, Velizy-Villacoublay, France).
Measure of in vitro PPARγ phosphorylation.
In vitro phosphorylation was performed on wild type PPARγ in the apo form and in the complex with 7j and Rosi. Stock solutions of ligands were prepared by diluting with 100% DMSO to a concentration of 50 mM. The stock solutions were further diluted with 50 mM Tris HCl pH 7.5 up to the final concentrations of 0.1 μM, 1 μM and 10 μM respectively, and pre-equilibrated overnight at 4°C with the protein. Phosphorylation was carried out at 30 °C for 3.5 hours in 300 μL of buffer containing 50 mM Tris HCl pH 7.5, 7.2 μg.mL−1 PPARγ, 0.1–1-10 μM ligand, 25 mM MgCl2, 50 μM DTT, 2 mM ATP, 0.66 ng mL−1 CDK5/p35 (Sigma Aldrich code n. SRP5011). Then, polystyrene micro well plates were coated overnight at 4°C with the reaction mixture, then washed three times with PBS + Tween 0.005% and left to block in PBS containing 1% bovine serum for 90 min at 37°C. The wells were washed three times and incubated for 60 min at 37°C with 100 μL of anti-phospho-Ser/The-Pro antibody (Sigma Aldrich code n. A05368) diluted 1:500 in PBS. After three washes, 100 μL of Anti-Mouse IgG-Peroxidase antibody produced in goat (Sigma Aldrich code n. A4416; 1:1000 in PBS) were added to the wells and incubated 60 min at 37°C. The wells were washed and 200 μL of o-phenylenediamine dihydrochloride (Sigmafast OPD code n. P9187) dissolved in water were added to the wells. Optical density was measured at 450nm using ApplyScan Thermofisher Reader and the data were processed using Excel.
Experiments were performed 3 times in triplicates.
Protein expression and purification.
PPARγ LBD was expressed as N-terminal His-tagged proteins using a pET28 vector and purified as previously described45. Briefly, freshly transformed E. coli BL21 DE3 were grown in LB medium with 30 μg of kanamycin/ml at 310 K to an OD of 0.6. The culture was then induced with 0.2 mM isopropyl-β-D-thio-galactopyranoside and further incubated at 291 K for 20 h. Cells were harvested and resuspended in a 20 ml/liter culture of Buffer A (20 mM Tris-HCl, 150 mM NaCl, 10% glycerol, 1mM Tris 2-carboxyethylphosphine HCL (TCEP), pH 8) in the presence of protease inhibitors (Complete Mini EDTA-free; Roche Applied Science). Cells were sonicated, and the soluble fraction was isolated by centrifugation (35,000 x g for 45 min). The supernatant was loaded onto a Ni2+-nitrilotriacetic acid column (GE Healthcare) and eluted with a gradient of imidazole 0–500 mM in Buffer A (20mM Tris-HCl, 20 mM NaCl, 10% glycerol, 1mM TCEP, pH 8) (batch method). The pure protein was identified by SDS PAGE. The protein was then dialyzed over Buffer A to remove imidazole, and it was cleaved with thrombin protease (Sigma-Aldrich Life Science) (10 units/mg) at room temperature for 2h. The digested mixture was reloaded onto a Ni2+-nitrilotriacetic acid column to remove the His tag and the undigested protein. The flow-through was dialyzed with Buffer B (20 mM Tris-HCl, 10% glycerol, 1mM TCEP, pH 8) to remove NaCl and loaded onto a Q-Sepharose HP column (GE Healthcare), and eluted with a gradient of NaCl 0–500 mM in Buffer B with a BioLogic DuoFlow FPLC system (Bio-Rad Laboratories, Italy). Finally, the protein was purified by gel-filtration chromatography on a HiLoad Superdex 75 column (GE Healthcare) and eluted with Buffer C (20 mM Tris-HCl, 1 mM TCEP, 0.5 mM EDTA, pH 8). The protein was then concentrated at 8 mg/ml using Amicon centrifugal concentrators with a 10 kDa cutoff membrane (Millipore, USA).
Crystallization and Data Collection.
Crystals of apo-PPARγ were obtained by vapor diffusion at 18°C using a sitting drop made by mixing 2 μL of protein solution with 2 μL of reservoir solution (0.8 M Na Citrate, 0.15M Tris, pH 8.0). The crystals were soaked for three days in a storage solution (1.2 M Na Citrate, 0.15 M Tris, pH 8.0) containing the ligand 7j or 7a (0.5 mM). The ligand dissolved in DMSO (50 mM) was diluted in the storage solution so that the final concentration of DMSO was 1%. The storage solution with glycerol 20% (v/v) was used as cryoprotectant. Crystals (0.15 × 0.15 mm) of PPARγ/7j and PPARγ/7a belong to the space group C2 with cell parameters shown in Supplemental Table S1.
Structure Determination and Refinement.
X-ray data set were collected at 100 K under a nitrogen stream using synchrotron radiation (beamline ID30B at ESRF, Grenoble, France). The collected data were processed using the programs Mosflm and Scala 46. Structure solution was performed with AMoRe 47, using the coordinates of PPARγ/LT175R (27) (PDB code 3D6D) as the starting model. The coordinates were then refined with CNS 48 and with PHENIX 49 including data between 58.2 and 2.0 Å for PPARγ/7j (57.4–1.8 Å for PPARγ/7a). The statistics of crystallographic data and refinement are summarized in Supplemental Table S1. The coordinates and structure factors described here have been deposited in the PDB under accession numbers 6QJ5 and 6ZLY for PPARγ/7j and PPARγ/7a, respectively.
Surface Plasmon Resonance.
Surface plasmon resonance analyses were performed by using Pioneer AE optical biosensor equipped with COOH5 chips (SensiQ). PPARγ surfaces were prepared by using standard amine-coupling procedures 50 and HBS (Hepes-buffered saline: 10 mM Hepes, 150 mM NaCl, 0.005% P20, DMSO 1%, pH 7.4) as the running buffer. Flow cells were activated for 7 min by injecting 140 μL of 50 mM N-hydroxysuccinimide (NHS):200 mM ethyl-3(3-dimethylamino) propylcarbodiimide (EDC). 150 μL of a 0.25 mg/mL PPARγ solution (in 10 mM NaOAc, pH 5.0) were injected for 15 min at 10 μL/min on channels 1 and 3 (channel 2 was used as reference, for a duplicate experiment), followed by a 70 μL injection of ethanolamine to block any remaining activated groups on the surface. 11,320 and 11,220 RU of protein were immobilized on channel 1 and 3, respectively. The screening of the analytes was performed using HBS, with 1% DMSO. To collect detailed a dilution protocol was used, injecting different concentrations of the analytes at a flow rate of 50 μL/min over the two channels at 20 °C (association phase of 60 s). A similar protocol was followed for the experiments with PPARα, using a PCH chip (Pall ForteBio).
Four buffer blanks were injected for double referencing. The regeneration of the surfaces between binding cycles was not necessary because all the analytes dissociate quickly in the 120 s dissociation phase. A DMSO calibration plot was constructed (buffer sample containing 0–2% (vol/vol) DMSO) to correct for bulk refractive index shifts. All sensorgrams were processed by using double referencing. To obtain kinetic rate constants and affinity constants the corrected response data were fit in the program QDAT. A kinetic analysis of each ligand/analyte interaction was obtained by fitting the response data to a 1:1 bimolecular interaction model. The equilibrium dissociation constant (Kd) was determined by the ratio koff/kon.
Statistical Analyses.
Statistical significance was estimated with one-way ANOVA followed by Bonferroni or Dunnett post hoc test, with two-tailed t-Test or with F-Test (statistical test is specified in figure legends) using Graph Pad Prism version 5.0 (GraphPad Software, San Diego, CA). Differences with p values of less than 0.05 were considered statistically significant.
Supplementary Material
AKNOWLEDGEMENTS
This work was supported by funds from Aix Marseille University, Inserm, CNRS, ANR and National Institutes of Health grant DK071900 to R.G.R.
ABBREVIATIONS
- BOP
benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate
- CDK5
cyclin-dependent kinase 5
- hMADS
human Multipotent Adipose-Derived Stem
- LBD
ligand binding domain
- NAFLD
non-alcoholic fatty liver disease
- PPRE
PPAR Responsive Element
- Rosi
Rosiglitazone
- SPPARγMs
selective PPARγ modulators
Footnotes
ASSOCIATED CONTENT
SUPPORTING INFORMATION
NMR and HPLC spectra of 3a, 7a, 7j; X-ray crystal structures of 3a, 7a, 7j, Rosiglitazone, SR2067 in PPARγ LBD; SPR sensorgrams of 3a, 7a, 7j; Adipogenic effect of the compounds; PPARγ, PPARδ and RXRα transactivation; PPARγ/RXRα heterodimerization; Toxicity of 7j; Statistics of crystallographic data and refinement.
Molecular formula string for 3a, 7a and 7j.
PDB ID
PDB 6QJ5, 6ZLY
Coordinates and structure factors of the PPARγ complexes with the compound 7j and 7a have been deposited in the Protein Data Bank under the accession code 6QJ5 and 6ZLY, respectively. Authors will release the atomic coordinates and experimental data upon article publication.
The authors declare no competing financial interest
Genes are identified by the symbols approved by the Human Genome Organization
REFERENCES
- (1).Braissant O; Foufelle F; Scotto C; Dauça M; Wahli W Differential Expression of Peroxisome Proliferator-Activated Receptors (PPARs): Tissue Distribution of PPAR-Alpha, -Beta, and -Gamma in the Adult Rat. Endocrinology 1996, 137 (1), 354–366. [DOI] [PubMed] [Google Scholar]
- (2).Botta M; Audano M; Sahebkar A; Sirtori CR; Mitro N; Ruscica M PPAR Agonists and Metabolic Syndrome: An Established Role? Int J Mol Sci 2018, 19 (4), 1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Derosa G; Sahebkar A; Maffioli P The Role of Various Peroxisome Proliferator-Activated Receptors and Their Ligands in Clinical Practice. J. Cell. Physiol 2018, 233 (1), 153–161. [DOI] [PubMed] [Google Scholar]
- (4).Silva AKS; Peixoto CA Role of Peroxisome Proliferator-Activated Receptors in Non-Alcoholic Fatty Liver Disease Inflammation. Cell. Mol. Life Sci 2018, 75 (16), 2951–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pawlak M; Lefebvre P; Staels B Molecular Mechanism of PPARα Action and Its Impact on Lipid Metabolism, Inflammation and Fibrosis in Non-Alcoholic Fatty Liver Disease. J. Hepatol 2015, 62 (3), 720–733. [DOI] [PubMed] [Google Scholar]
- (6).Tsunoda F; Asztalos IB; Horvath KV; Steiner G; Schaefer EJ; Asztalos BF Fenofibrate, HDL, and Cardiovascular Disease in Type-2 Diabetes: The DAIS Trial. Atherosclerosis 2016, 247, 35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yaghoubi M; Jafari S; Sajedi B; Gohari S; Akbarieh S; Heydari AH; Jameshoorani M Comparison of Fenofibrate and Pioglitazone Effects on Patients with Nonalcoholic Fatty Liver Disease. Eur J Gastroenterol Hepatol 2017, 29 (12), 1385–1388. [DOI] [PubMed] [Google Scholar]
- (8).Davidson MH; Armani A; McKenney JM; Jacobson TA Safety Considerations with Fibrate Therapy. Am. J. Cardiol 2007, 99 (6A), 3C–18C. [DOI] [PubMed] [Google Scholar]
- (9).Ida S; Kaneko R; Murata K Efficacy and Safety of Pemafibrate Administration in Patients with Dyslipidemia: A Systematic Review and Meta-Analysis. Cardiovasc Diabetol 2019, 18 (1), 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Liu Y; Colby JK; Zuo X; Jaoude J; Wei D; Shureiqi I The Role of PPAR-δ in Metabolism, Inflammation, and Cancer: Many Characters of a Critical Transcription Factor. Int J Mol Sci 2018, 19 (11), 3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wu C-C; Baiga TJ; Downes M; La Clair JJ; Atkins AR; Richard SB; Fan W; Stockley-Noel TA; Bowman ME; Noel JP; Evans RM Structural Basis for Specific Ligation of the Peroxisome Proliferator-Activated Receptor δ. Proc. Natl. Acad. Sci. U.S.A 2017, 114 (13), E2563–E2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Olefsky JM Treatment of Insulin Resistance with Peroxisome Proliferator-Activated Receptor Gamma Agonists. J. Clin. Invest 2000, 106 (4), 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Abbas A; Blandon J; Rude J; Elfar A; Mukherjee D PPAR- γ Agonist in Treatment of Diabetes: Cardiovascular Safety Considerations. Cardiovasc Hematol Agents Med Chem 2012, 10 (2), 124–134. [DOI] [PubMed] [Google Scholar]
- (14).Kung J; Henry RR Thiazolidinedione Safety. Expert Opin Drug Saf 2012, 11 (4), 565–579. [DOI] [PubMed] [Google Scholar]
- (15).Balint BL; Nagy L Selective Modulators of PPAR Activity as New Therapeutic Tools in Metabolic Diseases. Endocr Metab Immune Disord Drug Targets 2006, 6 (1), 33–43. [DOI] [PubMed] [Google Scholar]
- (16).Higgins LS; Depaoli AM Selective Peroxisome Proliferator-Activated Receptor Gamma (PPARgamma) Modulation as a Strategy for Safer Therapeutic PPARgamma Activation. Am. J. Clin. Nutr 2010, 91 (1), 267S–272S. [DOI] [PubMed] [Google Scholar]
- (17).Choi JH; Banks AS; Estall JL; Kajimura S; Boström P; Laznik D; Ruas JL; Chalmers MJ; Kamenecka TM; Blüher M; Griffin PR; Spiegelman BM Anti-Diabetic Drugs Inhibit Obesity-Linked Phosphorylation of PPARgamma by Cdk5. Nature 2010, 466 (7305), 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Choi JH; Banks AS; Kamenecka TM; Busby SA; Chalmers MJ; Kumar N; Kuruvilla DS; Shin Y; He Y; Bruning JB; Marciano DP; Cameron MD; Laznik D; Jurczak MJ; Schürer SC; Vidović D; Shulman GI; Spiegelman BM; Griffin PR Antidiabetic Actions of a Non-Agonist PPARγ Ligand Blocking Cdk5-Mediated Phosphorylation. Nature 2011, 477 (7365), 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Balakumar P; Mahadevan N; Sambathkumar R A Contemporary Overview of PPARα/γ Dual Agonists for the Management of Diabetic Dyslipidemia. Curr Mol Pharmacol 2019, 12 (3), 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Fiévet C; Fruchart J-C; Staels B PPARalpha and PPARgamma Dual Agonists for the Treatment of Type 2 Diabetes and the Metabolic Syndrome. Curr Opin Pharmacol 2006, 6 (6), 606–614. [DOI] [PubMed] [Google Scholar]
- (21).Jani RH; Pai V; Jha P; Jariwala G; Mukhopadhyay S; Bhansali A; Joshi S A Multicenter, Prospective, Randomized, Double-Blind Study to Evaluate the Safety and Efficacy of Saroglitazar 2 and 4 Mg Compared with Placebo in Type 2 Diabetes Mellitus Patients Having Hypertriglyceridemia Not Controlled with Atorvastatin Therapy (PRESS VI). Diabetes Technol. Ther 2014, 16 (2), 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Pai V; Paneerselvam A; Mukhopadhyay S; Bhansali A; Kamath D; Shankar V; Gambhire D; Jani RH; Joshi S; Patel P A Multicenter, Prospective, Randomized, Double-Blind Study to Evaluate the Safety and Efficacy of Saroglitazar 2 and 4 Mg Compared to Pioglitazone 45 Mg in Diabetic Dyslipidemia (PRESS V). J Diabetes Sci Technol 2014, 8 (1), 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Giampietro L; Laghezza A; Cerchia C; Florio R; Recinella L; Capone F; Ammazzalorso A; Bruno I; De Filippis B; Fantacuzzi M; Ferrante C; Maccallini C; Tortorella P; Verginelli F; Brunetti L; Cama A; Amoroso R; Loiodice F; Lavecchia A Novel Phenyldiazenyl Fibrate Analogues as PPAR α/γ/δ Pan-Agonists for the Amelioration of Metabolic Syndrome. ACS Med Chem Lett 2019, 10 (4), 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Laghezza A; Piemontese L; Cerchia C; Montanari R; Capelli D; Giudici M; Crestani M; Tortorella P; Peiretti F; Pochetti G; Lavecchia A; Loiodice F Identification of the First PPARα/γ Dual Agonist Able To Bind to Canonical and Alternative Sites of PPARγ and To Inhibit Its Cdk5-Mediated Phosphorylation. J. Med. Chem 2018, 61 (18), 8282–8298. [DOI] [PubMed] [Google Scholar]
- (25).Xu H-R; Zhang J-W; Chen W-L; Ning Z-Q; Li X-N Pharmacokinetics, Safety and Tolerability of Chiglitazar, A Novel Peroxisome Proliferator-Activated Receptor (PPAR) Pan-Agonist, in Healthy Chinese Volunteers: A Phase I Study. Clin Drug Investig 2019, 39 (6), 553–563. [DOI] [PubMed] [Google Scholar]
- (26).Gim HJ; Choi Y-S; Li H; Kim Y-J; Ryu J-H; Jeon R Identification of a Novel PPAR-γ Agonist through a Scaffold Tuning Approach. Int J Mol Sci 2018, 19 (10), 3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Guasch L; Sala E; Valls C; Blay M; Mulero M; Arola L; Pujadas G; Garcia-Vallvé S Structural Insights for the Design of New PPARgamma Partial Agonists with High Binding Affinity and Low Transactivation Activity. J. Comput. Aided Mol. Des 2011, 25 (8), 717–728. [DOI] [PubMed] [Google Scholar]
- (28).Montanari R; Capelli D; Yamamoto K; Awaishima H; Nishikata K; Barendregt A; Heck AJR; Loiodice F; Altieri F; Paiardini A; Grottesi A; Pirone L; Pedone E; Peiretti F; Brunel JM; Itoh T; Pochetti G Insights into PPARγ Phosphorylation and Its Inhibition Mechanism. J. Med. Chem 2020, 63 (9), 4811–4823. [DOI] [PubMed] [Google Scholar]
- (29).van Marrewijk LM; Polyak SW; Hijnen M; Kuruvilla D; Chang MR; Shin Y; Kamenecka TM; Griffin PR; Bruning JB SR2067 Reveals a Unique Kinetic and Structural Signature for PPARγ Partial Agonism. ACS Chem. Biol 2016, 11 (1), 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Eliasson B; Smith U; Mullen S; Cushman SW; Sherman AS; Yang J Amelioration of Insulin Resistance by Rosiglitazone Is Associated with Increased Adipose Cell Size in Obese Type 2 Diabetic Patients. Adipocyte 2014, 3 (4), 314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Liu C; Feng T; Zhu N; Liu P; Han X; Chen M; Wang X; Li N; Li Y; Xu Y; Si S Identification of a Novel Selective Agonist of PPARγ with No Promotion of Adipogenesis and Less Inhibition of Osteoblastogenesis. Scientific Reports 2015, 5 (1), 9530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Mukherjee R; Hoener PA; Jow L; Bilakovics J; Klausing K; Mais DE; Faulkner A; Croston GE; Paterniti JR A Selective Peroxisome Proliferator-Activated Receptor-Gamma (PPARgamma) Modulator Blocks Adipocyte Differentiation but Stimulates Glucose Uptake in 3T3-L1 Adipocytes. Mol. Endocrinol 2000, 14 (9), 1425–1433. [DOI] [PubMed] [Google Scholar]
- (33).Wu J; Cohen P; Spiegelman BM Adaptive Thermogenesis in Adipocytes: Is Beige the New Brown? Genes Dev 2013, 27 (3), 234–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ohno H; Shinoda K; Spiegelman BM; Kajimura S PPARγ Agonists Induce a White-to-Brown Fat Conversion through Stabilization of PRDM16 Protein. Cell Metab 2012, 15 (3), 395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Vernochet C; Peres SB; Davis KE; McDonald ME; Qiang L; Wang H; Scherer PE; Farmer SR C/EBPalpha and the Corepressors CtBP1 and CtBP2 Regulate Repression of Select Visceral White Adipose Genes during Induction of the Brown Phenotype in White Adipocytes by Peroxisome Proliferator-Activated Receptor Gamma Agonists. Mol. Cell. Biol 2009, 29 (17), 4714–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wu J; Boström P; Sparks LM; Ye L; Choi JH; Giang A-H; Khandekar M; Nuutila P; Schaart G; Huang K; Tu H; van Marken Lichtenbelt WD; Hoeks J; Enerbäck S; Schrauwen P; Spiegelman BM Beige Adipocytes Are a Distinct Type of Thermogenic Fat Cell in Mouse and Human. Cell 2012, 150 (2), 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Lin J; Handschin C; Spiegelman BM Metabolic Control through the PGC-1 Family of Transcription Coactivators. Cell Metabolism 2005, 1 (6), 361–370. [DOI] [PubMed] [Google Scholar]
- (38).Pisani DF; Djedaini M; Beranger GE; Elabd C; Scheideler M; Ailhaud G; Amri E-Z Differentiation of Human Adipose-Derived Stem Cells into “Brite” (Brown-in-White) Adipocytes. Front Endocrinol (Lausanne) 2011, 2, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Guo L; Zhang L; Sun Y; Muskhelishvili L; Blann E; Dial S; Shi L; Schroth G; Dragan YP Differences in Hepatotoxicity and Gene Expression Profiles by Anti-Diabetic PPAR Gamma Agonists on Rat Primary Hepatocytes and Human HepG2 Cells. Mol. Divers 2006, 10 (3), 349–360. [DOI] [PubMed] [Google Scholar]
- (40).Germano D; Uteng M; Pognan F; Chibout S-D; Wolf A Determination of Liver Specific Toxicities in Rat Hepatocytes by High Content Imaging during 2-Week Multiple Treatment. Toxicology in Vitro 2015, 30 (1, Part A), 79–94. [DOI] [PubMed] [Google Scholar]
- (41).Rachek LI; Yuzefovych LV; LeDoux SP; Julie NL; Wilson GL Troglitazone, but Not Rosiglitazone, Damages Mitochondrial DNA and Induces Mitochondrial Dysfunction and Cell Death in Human Hepatocytes. Toxicol Appl Pharmacol 2009, 240 (3), 348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Elabd C; Chiellini C; Carmona M; Galitzky J; Cochet O; Petersen R; Pénicaud L; Kristiansen K; Bouloumié A; Casteilla L; Dani C; Ailhaud G; Amri E-Z Human Multipotent Adipose-Derived Stem Cells Differentiate into Functional Brown Adipocytes. Stem Cells 2009, 27 (11), 2753–2760. [DOI] [PubMed] [Google Scholar]
- (43).Vidal N; Cavaillé JP; Poggi M; Peiretti F; Stocker P A Nonradioisotope Chemiluminescent Assay for Evaluation of 2-Deoxyglucose Uptake in 3T3-L1 Adipocytes. Effect of Various Carbonyls Species on Insulin Action. Biochimie 2012, 94 (12), 2569–2576. [DOI] [PubMed] [Google Scholar]
- (44).Seimandi M; Lemaire G; Pillon A; Perrin A; Carlavan I; Voegel JJ; Vignon F; Nicolas J-C; Balaguer P Differential Responses of PPARalpha, PPARdelta, and PPARgamma Reporter Cell Lines to Selective PPAR Synthetic Ligands. Anal. Biochem 2005, 344 (1), 8–15. [DOI] [PubMed] [Google Scholar]
- (45).Pochetti G; Godio C; Mitro N; Caruso D; Galmozzi A; Scurati S; Loiodice F; Fracchiolla G; Tortorella P; Laghezza A; Lavecchia A; Novellino E; Mazza F; Crestani M Insights into the Mechanism of Partial Agonism: Crystal Structures of the Peroxisome Proliferator-Activated Receptor Gamma Ligand-Binding Domain in the Complex with Two Enantiomeric Ligands. J. Biol. Chem 2007, 282 (23), 17314–17324. [DOI] [PubMed] [Google Scholar]
- (46).Kabsch W XDS. Acta Crystallogr. D Biol. Crystallogr 2010, 66 (Pt 2), 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Navaza J AMoRe: An Automated Package for Molecular Replacement. Acta Cryst A 1994, 50 (2), 157–163. [Google Scholar]
- (48).Brünger AT; Adams PD; Clore GM; DeLano WL; Gros P; Grosse-Kunstleve RW; Jiang JS; Kuszewski J; Nilges M; Pannu NS; Read RJ; Rice LM; Simonson T; Warren GL Crystallography & NMR System: A New Software Suite for Macromolecular Structure Determination. Acta Crystallogr. D Biol. Crystallogr 1998, 54 (Pt 5), 905–921. [DOI] [PubMed] [Google Scholar]
- (49).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L-W; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D Biol. Crystallogr 2010, 66 (Pt 2), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Jonsson U; Malmqvist M Real Time Biospecific Interaction Analysis. The Integration of Surface Plasmon Resonance Detection, General Biospecific Interface Chemistry and Microfluides into One Analytical System. In Advances in Biosensor, Turner A ed.; JAI Press, San Diego, 1992; pp 291–336. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.