

Summary
Background:
Components of the DNA damage checkpoint are essential for surviving exposure to DNA damaging agents. Checkpoint activation leads to cell cycle arrest, DNA repair, and apoptosis in eukaryotes. Cell cycle regulation and DNA repair appear essential for unicellular systems to survive DNA damage. The relative importance of these responses and apoptosis for surviving DNA damage in multicellular organisms remains unclear.
Results:
After exposure to ionizing radiation, wild-type Drosophila larvae regulate the cell cycle and repair DNA; grp (DmChk1) mutants cannot regulate the cell cycle but repair DNA; okra (DmRAD54) mutants regulate the cell cycle but are deficient in repair of double strand breaks (DSB); mei-41 (DmATR) mutants cannot regulate the cell cycle and are deficient in DSB repair. All undergo radiation-induced apoptosis. p53 mutants regulate the cell cycle but fail to undergo apoptosis. Of these, mutants deficient in DNA repair, mei-41 and okra, show progressive degeneration of imaginal discs and die as pupae, while other genotypes survive to adulthood after irradiation. Survival is accompanied by compensatory growth of imaginal discs via increased nutritional uptake and cell proliferation, presumably to replace dead cells.
Conclusions:
DNA repair is essential for surviving radiation as expected; surprisingly, cell cycle regulation and p53-dependent cell death are not. We propose that processes resembling regeneration of discs act to maintain tissues and ultimately determine survival after irradiation, thus distinguishing requirements between muticellular and unicellular eukaryotes.
Introduction
In eukaryotes, DNA damage checkpoints monitor the state of genomic DNA and delay the progress through the cell cycle as needed (reviewed in [1]). Central components of this checkpoint in mammals include four kinases: ATM, ATR, Chk1, and Chk2. Homologs of these exist in other eukaryotes and assume similar roles where examined. Human patients with ATM mutations, as well as their cells, show a dramatic sensitivity to killing by ionizing radiation [2, 3]. The importance of checkpoints in cellular survival to DNA damaging agents is presumed to be due to the role of checkpoints in cell cycle regulation. This is because mutants in the budding yeast gene rad9, the first checkpoint gene to be characterized, fail to arrest the cell cycle following damage and show increased radiation sensitivity; the latter phenotype was rescued by experimental induction of cell cycle delay. Consequently, cell cycle delay is thought to allow time for DNA repair and thereby ensure survival [4].
More recently, however, components of the DNA damage checkpoint are found to also activate DNA repair and to promote programmed cell death, which would cull cells with damaged DNA. For example, phosphorylation of NBS (a component of the Mre11 repair complex) by human ATM is of functional importance, while ATM knockout mice show a reduction in radiation-induced cell death in the CNS [5–7]. Therefore, the essential role of checkpoints in conferring survival to genotoxins may be due to DNA repair and cell death responses in addition to or instead of cell cycle regulation. Furthermore, what is important for survival at the cellular level may not be so in a multicellular context. For instance, the failure to arrest the cell cycle by checkpoints may be detrimental to individual cells, but removal of these by cell death and replacement via organ homeostasis may make cell cycle regulation inconsequential for survival of multicellular organs.
To address how DNA damage checkpoints operate in the context of multicellular organisms in vivo, we are studying the effect of ionizing radiation on Drosophila melanogaster. In Drosophila, mei-41 (ATR homolog) and grp (Chk1 homolog) are required to delay the entry into mitosis in larval imaginal discs after irradiation and to delay the entry into mitosis after incomplete DNA replication in the embryo [8–11]. Thus, mei-41 and grp play similar roles to their homologs in other systems. Moreover, mei-41 mutants are deficient in DNA repair [12]. The role of mei-41 and grp in radiation-induced cell death has not been tested, but mei-41 is dispensable for cell death after enzymatic induction of DNA double-strand breaks [13].
Here, we used mutants in mei-41, grp, p53, and okra, a homolog of budding yeast RAD54 that functions in repair of DNA double-strand breaks (DSB) to address the relative importance of cell cycle regulation, cell death, and DNA repair to the ability of a multicellular organism to survive ionizing radiation. The three responses are affected to different degrees in these mutants: wild-type larvae regulate S and M phases and repair DNA; grp mutants are unable to regulate the cell cycle ([10]; this study) but are able to repair DNA (this study); okra mutants are able to regulate the cell cycle (this study) but are deficient in DNA repair [14]; and mei-41 mutants are unable to regulate the cell cycle [10, 15] and are also deficient in DNA repair [12]. All genotypes with the exception of p53 mutants are proficient in radiation-induced cell death, suggesting that mei-41 and grp do not contribute to this response (in this study). Under these conditions, we find that while mei-41 and okra mutants are highly sensitive to killing by ionizing radiation, p53 mutants show reduced but significant survival and grp mutants resemble wild-type. These results suggest that cell death is neither sufficient nor absolutely necessary, DNA repair is essential, and optimal cell cycle regulation is dispensable for surviving ionizing radiation in Drosophila larvae.
Results
S Phase Checkpoint Is Compromised in mei-41 and grp Mutants
The CNS and imaginal discs of the Drosophila larva, which are precursors of adult tissues, proliferate by mitotic division during larval growth, while cells in the rest of the larva endoreplicate. In the CNS and eye imaginal discs of third instar larvae, cells undergo S phase in a stereotypical pattern (Figures 1A–1C show incorporation of a nucleotide analog, BrdU, in brain lobes). We find that brain lobes from irradiated larvae incorporate less BrdU than unirradiated controls, as detected by indirect immunofluoresence. The reduction in BrdU incorporation is seen as early as 1 hr after irradiation (1.5 hr time point is shown in Figure 1A). Estimates for the duration of S phase in third instar larvae range from <55 min in neuroblasts of the ventral nerve cord and 4.4 hr in wing imaginal disc in D. melanogaster to 11.9 hr in brain cells of D. virilis [16, 17]. Although the duration of S phase in D. melanogaster brain lobes is unknown, the average cell cycle duration in this tissue is 8–9 hr [18]. Taken together, S phase in the brain lobes could last one to several hours. Moreover, the distribution of S phase cells remains similar in irradiated and unirradiated brains. Therefore, we infer that irradiation led to inhibition of new S phase and/or slowing down of ongoing S phase. This is consistent with published reports that intra-S checkpoints in yeast and mammalian cell culture slow down S phase but do not completely block it [19, 20].
Figure 1.
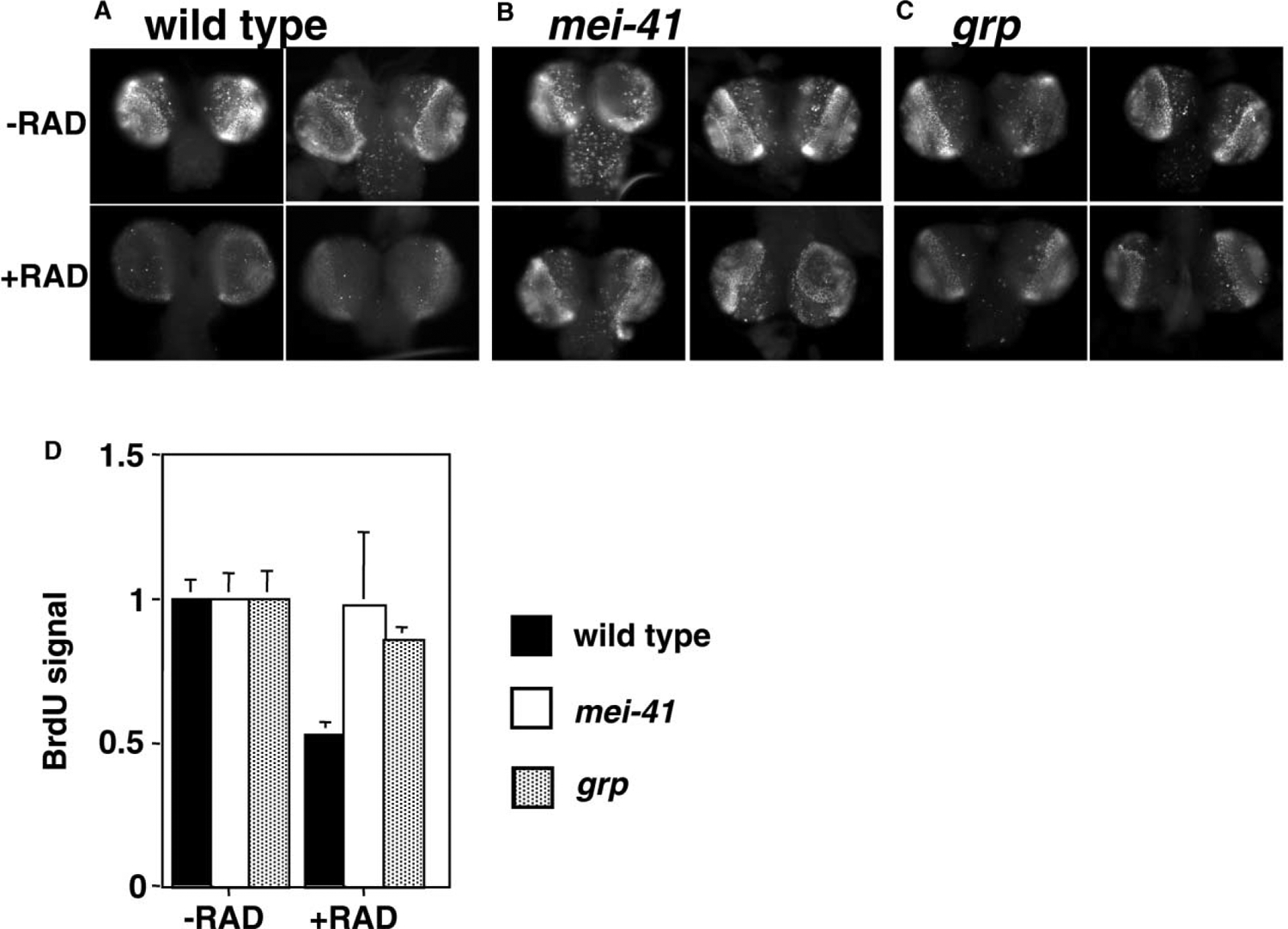
BrdU Incorporation in Larval Brains
Larvae were irradiated with 0R (−RAD) or 1600R (+RAD) of X-rays and allowed to recover for 1.5 hr before brains were extirpated and labeled with BrdU for 10 min. Incorporated BrdU was detected by immunofluoresence. Two representative brains from each genotype are shown for each treatment. All images were acquired and processed identically. BrdU incorporation is reduced after irradiation in wild-type (A), but not in mei-41 (B) or grp mutants (C). (D) shows quantification of anti-BrdU fluoresence. Data from four brains were averaged and shown for each genotype both with and without irradiation.
In contrast to wild-type, brains from mei-41 and grp homozygous mutant larvae (hereafter to be called mei-41 and grp larvae respectively; genotypes in Experimental Procedures) maintain robust BrdU incorporation after irradiation (Figures 1B and 1C; quantified in Figure 1D). Studies from yeast and mammalian cells have implicated ATM/ATR homologs in an intra-S checkpoint [21], and recent evidence from mammalian cell culture has implicated Chk1 in this checkpoint [22]. The requirement for mei-41 and grp in blocking mitosis after DNA damage in Drosophila larvae has been reported [10, 15]. Taken together, mei-41 and grp mutants are compromised for key cell cycle checkpoints that block S and M phases in response to DNA damage by ionizing radiation.
Radiation Sensitivity of mei-41 and grp Mutants
Given the above results and the notion that cell cycle checkpoints are of paramount importance, we were surprised to find that mei-41 and grp mutants exhibit vastly different sensitivity to killing by ionizing radiation. All wild-type larvae irradiated at 2000 or 4000Rads (R) of X-rays formed pupae, all of which eclosed. Those irradiated with 2000R eclosed into healthy viable adult flies, whereas those irradiated with 4000R eclosed into flies that were mostly unable to move and died by being stuck in food. In contrast to wild-type, mei-41 larvae irradiated with 2000R formed pupae but less than 1% eclosed (Table 1). The same mutants irradiated with 4000R formed pupae, none of which eclosed. These results are in agreement with previous reports on radiation sensitivity of mei-41 mutants [15, 23]. In contrast, survival of grp larvae was similar to that of wild-type at 2000R; all irradiated larvae formed pupae and more than 97% eclosed. Since grp larvae are defective in regulation of both M and S phases under these conditions, we conclude that regulation of these cell cycle phases by the DNA damage checkpoint is not absolutely essential for organismal survival following irradiation.
Table 1.
Quantification of Eclosion and Chromosome Breakage after Irradiation
| Eclosion after Irradiation | ||||
|---|---|---|---|---|
| Percent Eclosion (n/SD) | ||||
| Genotype | 0 R | 2000 R | 4000 R | |
| Wild-type | 98.1 (1659/0.9) | 97.9 (1305/2.1) | 87.0 (1215/10.5) | |
| grapes | 97.2 (415/1.2) | 97.3 (345/1.1) | 75.4 (336/7.9) | |
| mei-41 | 95.7 (695/1.8) | 0.9 (427/0.9) | 0.0 (329/0.0) | |
| okra | 97.8 (874/1.6) | 14.1 (747/10.9) | 0.9 (606/1.8) | |
| p53 | 83.8 (1672/3.3) | 71.0 (1632/10.3) | 46.1 (1479/16.6) | |
| Chromosome Breakage and Mitotic Index after Irradiation | ||||
| Percent Broken Metaphase Shromosomes (n/SD) | Mitotic Index (n/SD) | |||
| Genotype | 0 R | 220 R | 0 R | 220 R |
| Wild-type | 4.7 (366/4.6) | 6.1 (583/3.0) | 1.82 (2425/0.2) | 1.42 (3379/0.1) |
| grapes | 5.3 (581/5.9) | 8.3 (515/6.0) | 1.49 (3048/0.4) | 1.31 (3558/0.2) |
| mei-41 | 11.5 (477/6.3) | 45.9 (341/8.3) | ||
| okra | 5.3 (622/3.7) | 36.9 (769/15.3) | ||
(Top) Eclosion after irradiation. Larvae were irradiated during third larval instar with 0, 2000, or 4000 R of X-rays. Eclosed adults were counted for up to 10 days after irradiation and expressed as percent of total pupae. All larvae that were irradiated formed pupae under these conditions. Data are averaged from three to five experiments.
(Bottom) Chromosome breakage and mitotic index following irradiation. Larvae were irradiated during third larval instar with 0 or 220 R of X-rays. Such low doses are typically used for measurement of chromosome breakage, presumably for increased sensitivity [10]. Larval brains were extirpated at 3 hr after irradiation, fixed, squashed, and stained with a DNA dye. For breakage assay, the percent of metaphase cells showing at least one chromosome breakage is quantified. For mitotic index, the number of mitotic nuclei was calculated and expressed as a percent of total nuclei; at least 3 brains were quantified for each genotype and dose.
n = number of pupae counted (in the top section of the table), metaphase cells (for chromosome breakage), or nuclei (for mitotic index). SD, standard deviation; wt, wild-type. Genotypes of mutants are as indicated.
The Role of Cell Death in Survival
Of known consequences of DNA damage, cell cycle regulation is but one; the others include induction of cell death and DNA repair. Therefore, we asked if differences in the ability to cull damaged cells by cell death account for the differences in radiation sensitivity of wild-type, mei-41, and grp larvae.
Cell death, as visualized by staining with acridine orange (AO; a vital dye), is rare in discs from third instar larvae but increased dramatically following irradiation in wild-type discs ([24, 25]; Figures 2A and 2B). Irradiated mei-41 and grp mutants also show robust cell death comparable to that of wild-type (Figures 2D and 2F). We infer that gross differences in cell death cannot explain the differences in survival. In clonal analysis of third instar wing discs, 2000R of radiation was estimated to kill 57%–70% of cells [26]. We also estimate that dead cells constitute at least a third of irradiated wild-type eye discs in our experiments.
Figure 2.
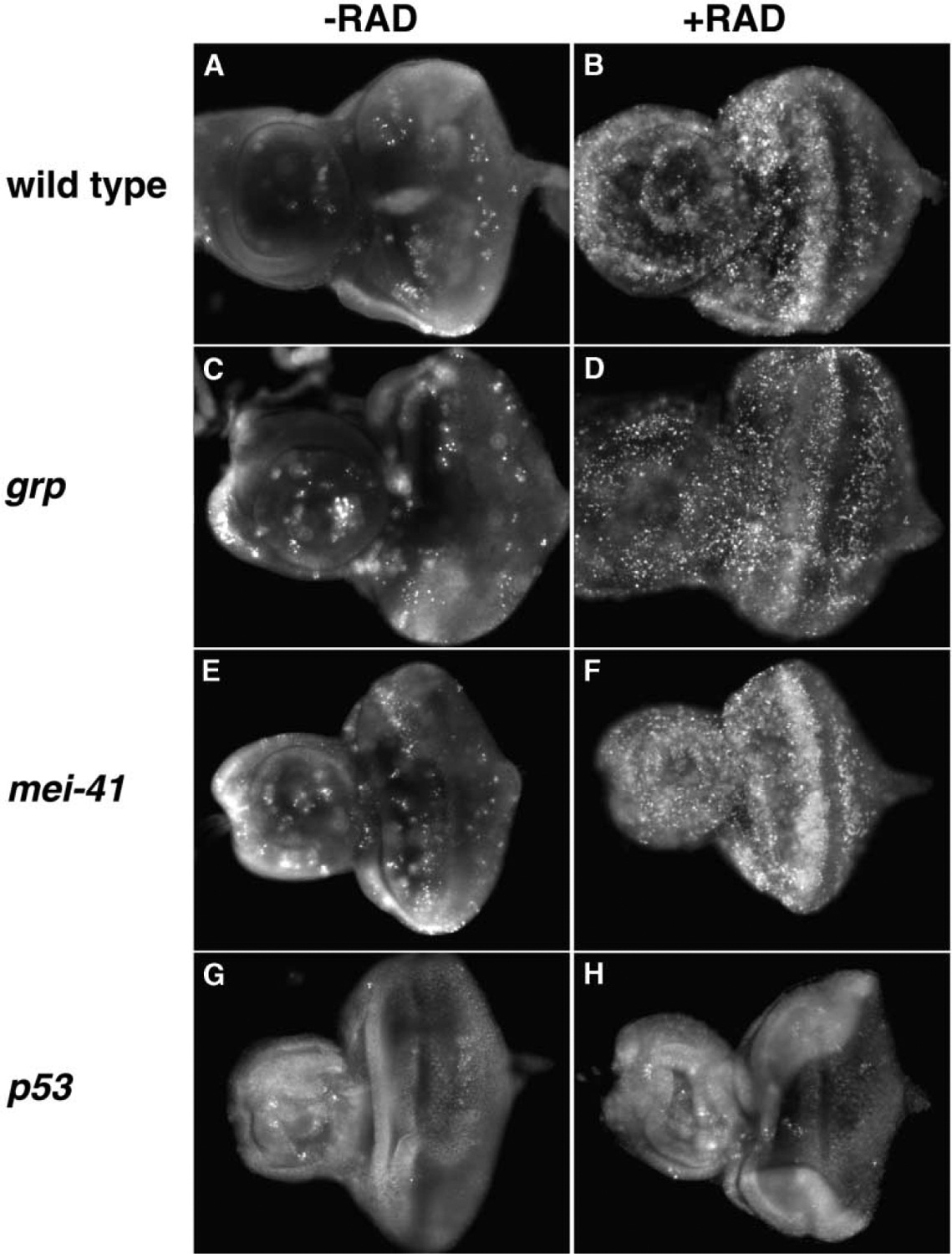
Cell Death after Irradiation
Larvae were irradiated with 0R (−RAD) or 2000R (+RAD) of X-rays and allowed to recover for 15–18 hr (B, D, F, and H). Eye-antennae discs were extirpated and stained with AO without fixing. Anterior is to the left. Genotypes are as indicated. Disc size should not be compared directly because unfixed tissues are squashed by the coverslip to different degrees during mounting.
mei-41 mutants demonstrate that culling of damaged cells by cell death is insufficient to ensure survival to radiation. We next addressed if it is necessary. Drosophila p53 is dispensable for viability but necessary for radiation-induced cell death. We find homozygous loss-of-function mutants in p53 to be compromised for viability, with 83.3% of third instar lavae eclosing into adults (compared to 98.1% in wild-type; Table 1). These mutants show a consistent, i.e., fully penetrant, lack of cell death by AO staining at times when it is readily detectable in other mutant backgrounds (Figures 2G and 2H), in agreement with previous reports [25, 27].Importantly, under these conditions, a significant portion of irradiated p53 mutant larvae eclosed into adults (71% at 2000R compared to 83.3% among unirradiated controls; Table 1). We conclude that p53-dependent cell death is largely dispensable for surviving ionizing radiation.
Maintenance of Cell Proliferation after Irradiation
Despite the death of a significant portion of cells in irradiated discs, both disc size and morphology remain well preserved in wild-type larvae (Figures 3G and 3H). In a previous study, quantification of mitotic index after irradiation suggested that this preservation is due to increased cell proliferation that may compensate for dead cells [26]. Here, we determine if cell proliferation and tissue preservation after irradiation requires components of the DNA damage checkpoint.
Figure 3.
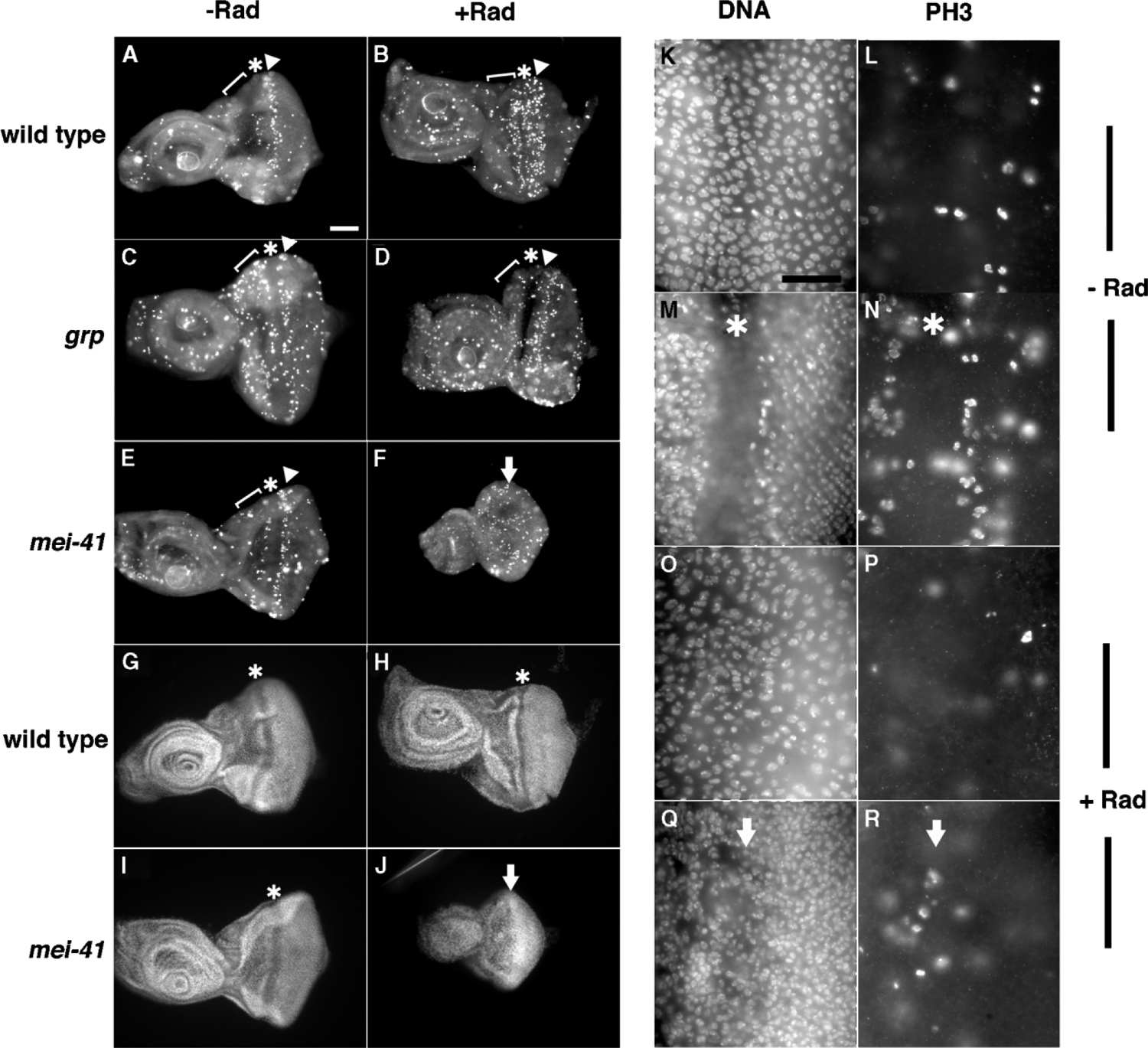
Mitosis, Disc Morphology, and Size after Irradiation
Third instar larvae were irradiated with 0 (−RAD) or 2000 Rads (+RAD) of X-rays. Discs were extirpated at 24 hr after irradiation, fixed, and stained for PH3, a marker for mitosis [42](A–F, L, N, P, and R) and DNA (G–J, K, M, O, and Q). The first (bracket) and second (arrowheads) mitotic waves, and MF (*) are indicated. The arrows indicate the presumptive location of the MF, which is absent in irradiated mei-41 discs (F, J, Q, and R). Anterior is to the left. Scale bar: 60 μm (A–J) or 15 μm (K–R). Genotypes are as indicated in (A)–(J); (K)–(R) are of mei-41 mutants.
(K–R) Higher magnification images show reduced proliferation in both cell layers of irradiated mei-41 discs, oriented as in (A)–(F); (K) and (M) are different focal planes of the same disc region that show peripodial (K) and columnar (M) layers. (L) and (N) show PH3 stain that corresponds to (K) and (M), respectively. (O) and (Q) are different focal planes of the same disc region that show peripodial (O) and columnar (Q) layers. (P) and (R) show PH3 stain that corresponds to (O) and (Q), respectively. An asterisk (*) indicates MF in (M) and (N). Arrow indicates presumptive MF in (Q) and (R). mei-41 controls (K–N) show mitoses in both peripodial (L) and columnar (N) cell layers. In contrast, irradiated mei-41 discs show few PH3 cells in both the peripodial (P) and columnar (R) cell layers; in particular, the second mitotic wave, which should reside just posterior to the MF, is absent in (R).
In control eye discs, asynchronous mitoses occur among undifferentiated cells anterior to the morphogenetic furrow (MF); this region is referred to as the “first mitotic wave”([28]; brackets in Figures 3A–3E). In addition, a second, more-confined region of mitoses is seen immediately posterior to the MF, corresponding to the “second mitotic wave” (arrowheads in Figures 3A–3E). In wild-type larvae, the number of mitotic cells decreased after irradiation, with maximal decrease seen at ~2 hr after irradiation, then recovered to control levels at ~6 hr after irradiation ([10, 15]; our unpublished data). At longer times after irradiation, we find that mitotic index increased further. This is most apparent at about 24 hr after irradiation when second mitotic waves are compared (Figures 3A and 3B, arrowheads). Closer examination revealed that the number of mitotic cells increased in both the peripodial and the columnar layers (data not shown; only the columnar layer contributes to the adult eye). The burst of mitoses seen in wild-type eye discs is in agreement with previous results from irradiated Drosophila wing discs in which mitotic index was found to increase above unirradiated controls after an initial checkpoint mediated decrease [26].
As stated above, grp mutants are defective in blocking mitosis after irradiation; as such, the mitotic index did not decrease to wild-type levels. At all times examined in our experiments, up to 24 hr after irradiation, the mitotic index remained similar in irradiated and unirradiated grp discs (Figures 3C and 3D). That is, we have not been able to detect a burst of mitoses in grp mutants, which nonetheless maintained organ size and morphology and therefore must be replacing dead cells. Possibly, the timing of compensatory mitoses in grp mutants is more heterogeneous. Importantly, grp mutants are able to sustain cell proliferation in an organized manner, i.e., undergo mitoses in two waves, after irradiation; further, mitoses were observed in both columnar and peripodial layers in irradiated grp eye discs (data not shown). This is in sharp contrast to mei-41 mutants.
mei-41 Mutant Discs Degenerate after Irradiation
In contrast to wild-type and grp mutant discs, mei-41 mutants are unable to sustain organized cell proliferation after irradiation; although mitotic cells are present, mitotic waves are no longer discernable at 24 hr after irradiation (Figures 3F and 3R). Furthermore, mei-41 mutant discs appear smaller at longer times (18–24 hr) after irradiation (Figure 3, compare 3J to 3I). Because cell size remains similar (compare Figures 3M and 3Q), we infer a reduction in cell number. The reduced size of mei-41 mutant discs is accompanied by the disappearance of the MF in the eye disc and tissue folds in the antennae disc, which is obvious by 24 hr postirradiation (Figure 3J), especially in comparison to discs at shorter times after irradiation (Figures 2F, 5D, and 5H). We infer that mei-41 larvae are unable to effectively replace cells lost to death following irradiation, resulting in degeneration of discs. Thus mei-41, but not grp, is required to preserve organ size and tissue morphology, as defined by the presence of a MF, after irradiation.
Figure 5.
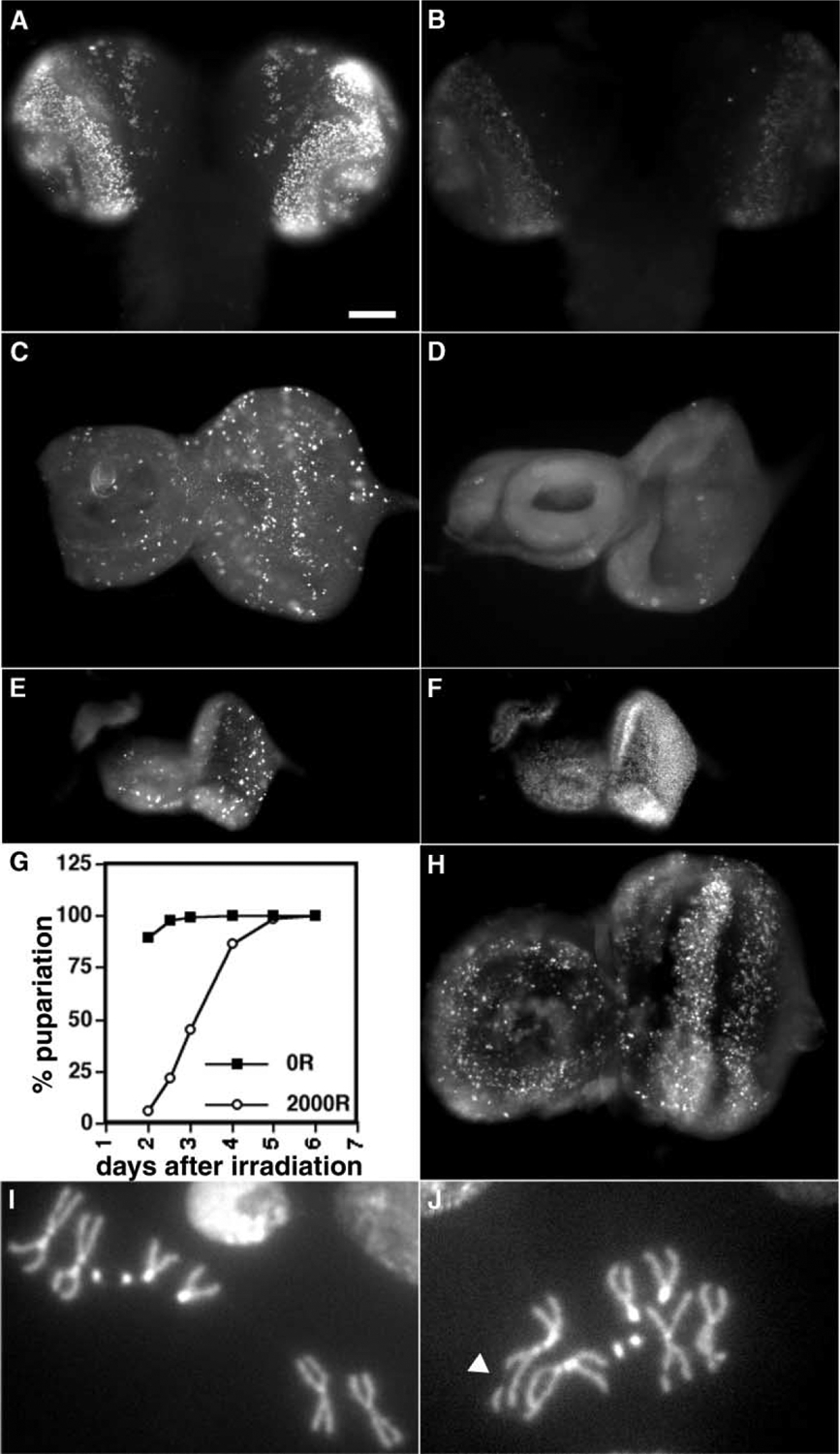
Radiation Responses of okra Mutants
(A and B) Larval brains were labeled with BrdU, as in Figure 1, 1.5 hr after irradiation with 0R (A) or 1600R (B) of X-rays. Samples were processed identically and the images were acquired and processed identically. (C and D) PH3 stain shows mitotic cells in eye-antennal discs from third instar larvae at 1 hr after exposure to 0R (C) or 4000R (D) of X-rays.
(E and F) An eye-antennae disc from a larva at 24 hr after irradiation with 2000R is shown; discs were fixed and stained for PH3 (E) and DNA (F).
(G) The onset of pupariation was determined as in Figure 4.
(H) Cell death in discs from irradiated third instar larvae was visualized by AO staining, exactly as in Figure 2.
(I and J) Representative larval metaphase cells from okra mutants to illustrate intact (I) and broken ([J]; arrowhead) chromosomes. The scale bar represents 60 μm.
Irradiation Delays the Onset of Pupariation
Experiments so far have been on early to mid third instar larvae, during the “feeding” stage. Next, larvae leave the food to crawl up the side of the container (the “wandering” stage). This is followed by pupariation. The larval-pupal transition is under hormonal control [29] and is believed to be sensitive to the extent of imaginal cell proliferation. For instance, mutations in a DNA replication factor, MCM2, slow down cell proliferation such that larval discs and brains are smaller in mcm2 mutants than in wild-type of similar age [30, 31]; mcm2 mutants delay the onset of pupariation (P.H. O’Farrell, personal communication). In another example, mutants in which cell proliferation continues unabated beyond normal levels (e.g., discs large) not only have larger than normal discs, but also delay the onset of pupariation [32]. Thus, either continued cell proliferation (in dlg and mcm2 mutants) or reduced disc size (in mcm2 mutants) can delay the onset of pupariation, although the exact nature of the link remains unclear.
If irradiation was inducing cell death and cell proliferation was being sustained to compensate for lost cells, we may expect to see a delay of pupariation. Indeed, irradiation delays pupariation in wild-type larvae in a dose-dependent manner (Figure 4A), in agreement with previous observations [26]. Since such a developmental delay is a novel response to irradiation, we addressed the requirement for DNA damage checkpoint components. grp mutants exposed to 2000R of X-rays also delay pupariation with wild-type kinetics (Figure 4B). This is consistent with the detection of sustained mitoses in irradiated grp mutants. Interestingly, mei-41 mutants irradiated with 2000R of X-rays also delay pupariation for even longer periods than wild-type and grp mutants. For instance, as of day two when most of the irradiated wild-type and grp larvae had formed pupae, only about 40% of irradiated mei-41 larvae had pupariated. We infer that continued cell proliferation (in wild-type and grp mutants) and smaller disc size and continued proliferation (in mei-41 mutants) contribute to delay pupariation after irradiation, apparently by mei-41-independent or grp-independent mechanisms.
Figure 4.
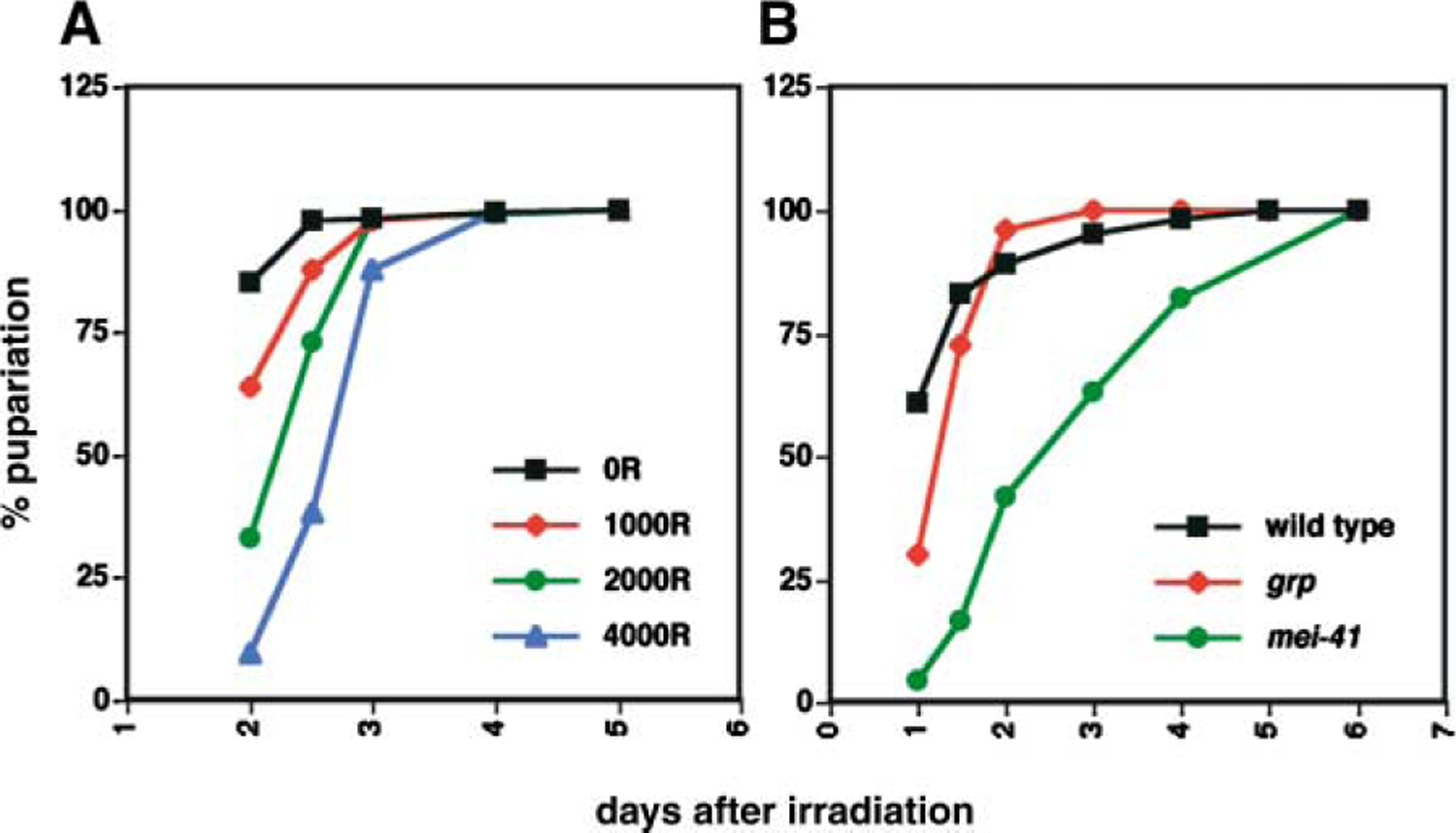
Irradiated Larvae Delay Pupariation
Feeding third instar larvae were irradiated and the number of pupae formed was counted on each subsequent day and expressed as a percentage of total pupae formed. All irradiated larvae pupariated under these conditions. (A) Wild-type, (B) 2000R. Unirradiated controls for all genotypes initiated pupariation with similar kinetics (not shown). These experiments were executed two to four times with similar trends in dose and genotype responses, although the exact numbers varied because of slight variations in larval culture.
Repair Mutants Have Intact Cell Cycle and Cell Death Responses but Are Radiation Sensitive
We document new mei-41-dependent responses to ionizing radiation: maintenance of organized cell proliferation, organ size, and tissue morphology. The ability to regulate S or M phases is dispensable for this response (as in grp mutants); cell death alone is insufficient (as in mei-41 mutants). We next addressed the role of DNA repair using mutants in okra, a homolog of yeast RAD54 that is needed for homologous recombinational repair (HR) of double-strand breaks (DSB) [14].
We find that irradiated okra mutants (genotype in Experimental Procedures) regulate both S and M phases (Figure 5A–5D) and undergo cell death (Figure 5H) as well as wild-type. Nonetheless, radiation sensitivity of okra mutants is closer to that of mei-41 mutants than wild-type and grp mutants (Table 1). okra mutants are also similar to mei-41 mutants with respect to the following phenotypes: the extent of delay of pupariation (Figure 5G), degeneration of discs (Figures 5E and 5F), and the inability to maintain organized cell proliferation (Figure 5E). In other words, mutational loss of efficient DSB repair reproduces all aspects of the mei-41 phenotype except for cell cycle regulation.
We see a small but significant difference in radiation sensitivity between okra and mei-41 mutants. There are three possible reasons. First, okra mutants may have residual okra activity, either because deposits from heterozygous mothers persist or because the putative null allele of okra is not a true null. Second, radiation sensitivity of mei-41 mutants may consist of a contribution by defective cell cycle regulation even though the latter is not absolutely required for survival. Finally, DSB may be repaired by HR, a process requiring okra, or by non-homologous end joining, and mei-41 may be needed for both while okra is needed for HR only.
Chromosome Breakage in mei-41 and okra Mutants
The similarity between radiation sensitivities of mei-41 and okra larvae suggests that DNA repair is the key determinant for survival. If so, we would predict that grp mutants that are resistant to radiation under our experimental conditions are able to repair DNA. One standard measure of DSB is the presence of breaks in metaphase chromosomes from squashed larval neuroblasts (e.g., [10]). In samples from mei-41 and okra larvae, which are known to be defective for repair of DSB, irradiation produced broken chromosomes in approximately 46% and 37% of metaphase cells respectively at 3 hr after irradiation (Figures 5I and 5J, and Table 1).The corresponding numbers are ~6% in wild-type and ~8% in grp mutants. At the low doses typically used in these assays (220R here), mitotic indices do not differ significantly between irradiated and unirradiated samples (Table 1, bottom). Therefore, lower break frequencies in grp mutants cannot be explained by arrest of cells in G2, which might have reduced the frequency of mitotic cells with broken chromosomes. We conclude that grp mutants are as capable as wild-type in the removal of X-ray-induced DSB.
Discussion
We examined the effects of DNA damage by ionizing radiation on the maintenance and survival of Drosophila larvae. Despite an extensive loss of cells to radiation-induced cell death, organ size and morphology are maintained remarkably well, and larvae survive to produce viable adults. Much to our surprise, optimal cell cycle regulation by checkpoints is neither necessary (as in grp mutants) nor sufficient (as in okra mutants) to ensure organ homeostasis and organismal survival. p53-dependent cell death is also largely dispensable in this regard. Instead, DNA repair appears to be of paramount importance as might be expected.
In mitotically proliferating cells of Drosophila larval imaginal discs and brains, the first responses to sublethal doses of irradiation (1000–4000R) are delays in cell cycle progression at 1–2 hr after irradiation (this study; [10]), followed by the induction of cell death at 4 hr after irradiation ([33]; this study). DNA synthesis resumes at ~5 hr after irradiation (our unpublished data), while mitotic index resumes at ~6 hr after [10, 15]. These relatively early responses are followed by an increase in proliferation that is detectable about a day after irradiation. Presumably, abundant cell death removes damaged cells, but sustained proliferation compensates to maintain proper organ size and morphology. Continued cell proliferation, we propose, delays the onset of the next major developmental transition, pupariation. The extent of delay correlates with radiation dose, presumably because more cells are lost at higher doses [26], requiring more compensatory proliferation.
Another response we monitored is DNA repair, a substantial portion of which must occur within 3 hr after 220R of irradiation because we see a significant difference in the incidence of chromosome breakage between wild-type and repair-deficient mutants by this time. However, cytologically visible chromosome breaks likely represent only a fraction of total DNA damage; for this reason, we cannot be certain if DNA repair is complete within this time frame.
Importance of Radiation Responses to Survival: DNA Repair Is Key
Having determined the sequence of responses to irradiation in wild-type larvae, we were able to document deviations from it in various mutants. mei-41 and grp mutants are unable to dampen DNA synthesis after irradiation. Previous work has shown that both mutants are unable to inhibit mitosis after irradiation, although grp mutants appear to retain a partial activity in this regard [10]. Thus, Drosophila ATR and Chk1 are needed for optimal regulation of both S and M phases after exposure to ionizing radiation. Induction of cell death, on the other hand, does not require mei-41 or grp. The most striking result we report is that grp mutants that are defective in regulation of both S and M phases are not sensitive to killing by 2000R of X-rays, doses that readily killed mei-41 and okra mutants. This finding strongly suggests that cell cycle regulation by checkpoints is not absolutely necessary for surviving irradiation under these conditions.
In determining what is necessary, the phenotype of okra mutants that can regulate both S and M phases and promote cell death is particularly informative because they are radiation sensitive. Thus, DNA repair is essential, suggesting that it is this defect in mei-41 mutants that renders them radiation sensitive (modeled in Figure 6). We speculate that irradiated mei-41 and okra larvae may attempt to increase proliferation, but the continual presence of unrepaired DNA likely channels these cells to death. This would lead to an eventual decline in cell number, which would undermine maintenance of cellular differentiation that is the basis of the MF. Signals from cells in the MF are thought be important for the generation of the second mitotic wave [34]. Loss of the MF could then explain the absence of the expected pattern of mitoses in mei-41 and okra discs.
Figure 6.
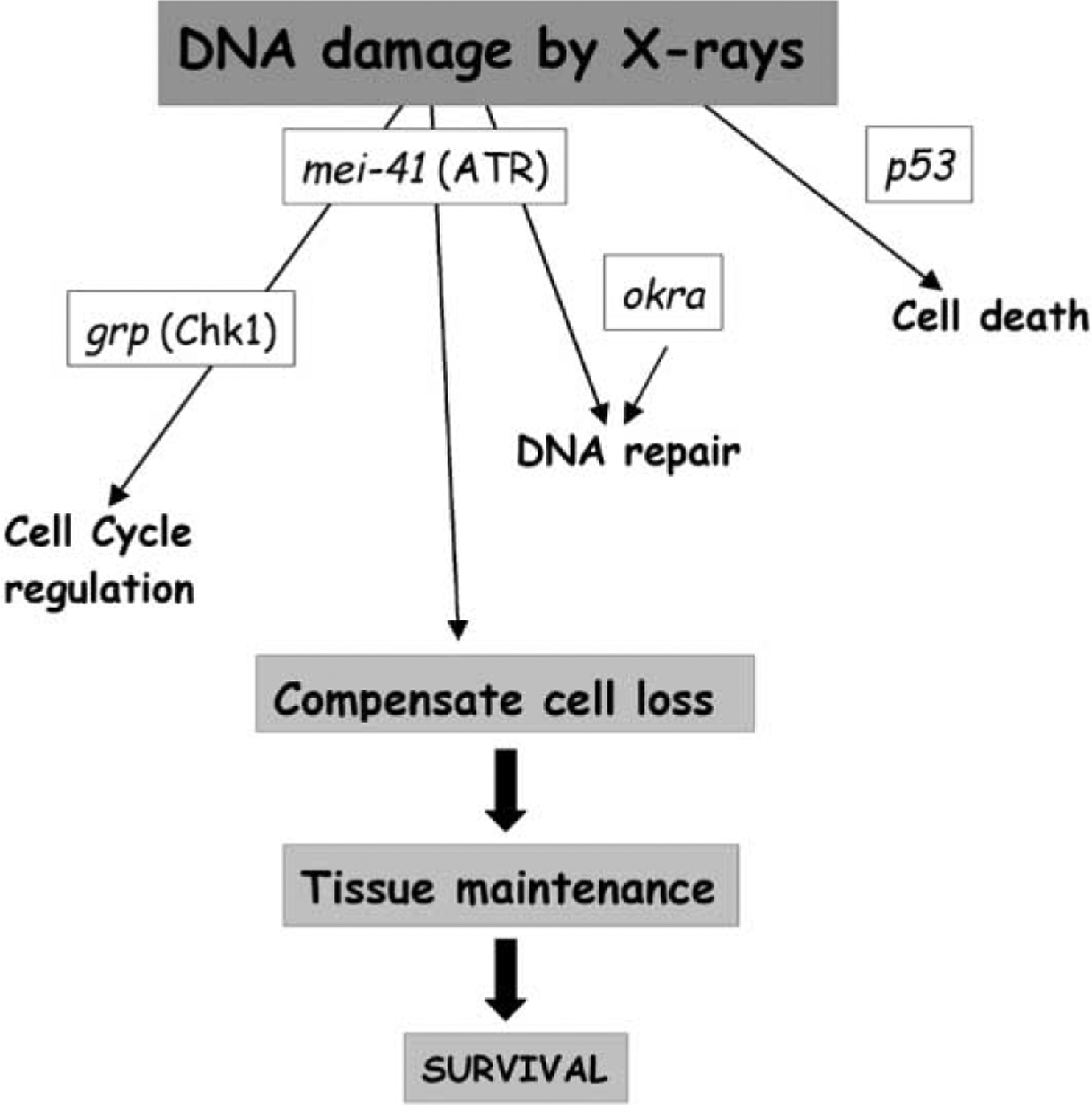
A Model for Surviving DNA Damage by Irradiation
mei-41 and grp function to regulate S and M phases after irradiation.This response is dispensable for survival. Radiation-induced cell death is p53 dependent but independent of mei-41 or grp; this response is largely dispensable. mei-41 and okra are needed for DNA repair, which is essential for survival to adulthood. mei-41 and okra act either in a common pathway to repair DNA or make independent contributions; only the latter is depicted.
Traditionally, checkpoints refer to the regulation of the cell cycle. Recent views propose the inclusion of the other responses among checkpoint responses, such as the preservation of DNA replication intermediates, transcriptional activation, and DNA repair [1]. Our data suggest that other responses may be more important in ensuring survival of multicellular organs and organisms. Interestingly, results from budding yeast also question the idea that cell cycle regulation by checkpoints is essential for surviving genotoxins even at the cellular level. For example, yeast Chk1 mutants show profoundly defective regulation of mitosis after irradiation and yet are only mildly radiation sensitive [35]. Another recent study indicates that stabilization of replication forks is crucial for surviving the alkylating agent MMS whereas the ability to inhibit mitosis is less important [43].
Relevance to Other Multicellular Systems
We emphasize that survival here refers to that of organs and organisms. At the cellular level, cell cycle regulation by checkpoints may well be crucial to allow time for DNA repair and for survival. In grp mutants that are defective for cell cycle checkpoints but are proficient for DNA repair, cells that progressed through S and M phases with damaged DNA may have been subject to cell death. Indeed, incidence of cell death appears higher in grp (and mei-41) mutants than in wild-type (our unpublished data). Loss of these cells, however, is clearly of little consequence to survival of imaginal discs and larvae. This could be because grp mutants are able to repair DNA in cells that are not in S and M phases, i.e., those in G1 or G2. These cells may then proliferate to compensate for lost cells. Numerous studies on tissue regeneration demonstrate the power of Drosophila larvae to restore not only cell number but also proper differentiation. In such a system, the failure of cell cycle checkpoints after irradiation may be of little consequence as long as damaged cells are replaced. We speculate that our findings may be particularly applicable to multicellular systems with similar regenerative powers such as the human liver [36].
Experimental Procedures
Fly Stocks
The A17-11 allele contains a G-to-A transition at the splice acceptor site of the second intron of the okra mRNA and at least another recessive lethal mutation on the same chromosome [37]. The mutation in okra is a putative null due to undetectable mRNA levels. JS17 contains a second chromosome deletion, which uncovers the okra gene. A17-11 and JS17 are balanced over CyO and are crossed to generate transheterozygous okra mutants, which were identified by the lack of GFP encoded by the balancer chromosome. mei-4129D is produced by imprecise excision of a P element in the coding region present in the mei-41RT allele. The resulting sequences are predicted to produce a 39 amino acid truncated protein (wild-type is 2347 aa) and is a putative null allele [38]. The grp1 allele has been described before [39]. The p535A-1-4 allele was generated by targeted deletion [40]. Homozygous or hemizygous grp and mei-41 mutants were identified by the lack of GFP encoded by the balancer.
Larvae Collection and Irradiation
Embryos were collected for 4 hr and aged at 25°C for 3 to 4 days to obtain third instar larvae. Larvae were irradiated using a TORREX X-ray generator, set at 115kV and 5 mA (producing 3.18 Rads/s).
BrdU Incorporation
Brains from irradiated and unirradiated larvae were extirpated in Scheider’s insect culture medium and incubated for 10 min in 0.8 ml of the same medium containing 0.5 mg/ml BromoDeoxyUridine (BrdU) on a nutator. Brains were fixed for 20 min in PBTx (phosphate buffered saline, 0.3% Triton X-100) containing 5% formaldehyde, washed three times with PBTx, denatured with 3.0 N HCl for 30 min, and neutralized with three washes of PBTx and blocked in PBTx + 10% Normal Goat Serum (blocking solution) before antibody staining.
Samples were incubated on the nutator for 5 hr with the monoclonal anti-BrdU antibody (1:50 in blocking solution; Beckton-Dickinson) at room temperature (rt), washed three times for 10 min each with PBTx, and incubated for 2 hr at rt with goat anti-mouse secondary antibody conjugated with Rhodamine (diluted 1:500 in blocking solution; Jackson). Samples were washed three times with PBTx, stained with 10 μg/ml Hoechst33258 in PBTx for 2 min, and washed three times with PBTx before mounting onto slides with Flourmount G (Southern Biotechnology Associates, Inc.).
AO Staining
Embryos were collected for 4 hr and aged at 25°C for 3 to 4 days to obtain third instar larvae. Larvae in food were irradiated with 2000R and allowed to recover at 25°C for 16 hr in bottles. Eye discs were extirpated in PBS, incubated for 5 min in PBS containing 0.5 mM acridine orange (Sigma) at rt, washed two times with PBS, mounted in PBS, and imaged immediately.
PH3 Antibody Staining
Third instar larvae in food were irradiated with 2000R of X-rays, transferred to fresh food, and recovered at 25°C. Larval eye-antennae discs were extirpated in PBS, fixed for 20 min in PBTx containing 5% formaldehyde, and washed three times with PBTx. Samples were incubated 2 hr at room temperature (rt) with a polyclonal anti-phospho histone 3 antibody (PH3) (diluted 1:1000 in blocking solution; Upstate Biotechnologies), washed three times with PBTx, and incubated 2–4 hr at rt with goat anti-rabbit secondary antibody conjugated with Rhodamine (diluted 1:500 in blocking solution; Jackson). Samples were washed three times with PBTx, stained with 10 μg/ml Hoechst33258 in PBTx for 2 min, and washed three times with PBTx before mounting onto slides with Flourmount G.
Viability Assays
Irradiated or control larvae were allowed to develop into pupae at 25°C. Homozygous mutant pupae were identified by the lack of GFP encoded by the balancer chromosome, transferred onto Whatmann 3MM paper, placed into food vials, and allowed to continue development at 25°C. Wild-type pupae and heterozygous pupae (GFP positive) were treated identically. The number of adults that eclosed and the number of empty pupae cases were scored for up to 10 days after irradiation. Percent eclosion is the number of empty pupae cases expressed as percent of total pupae formed. All irradiated larvae pupariated in these experiments.
Measurement of Pupariation
Irradiated and control larvae were allowed to recover at 25°C. Total number of pupae were counted at different times after irradiation for up to 7 days. Homozygous mutant pupae were identified by the lack of GFP.
Brain Squashes
Brains were dissected in saline and processed exactly as published before for Hoechst 33258 staining without colchicine treatment [41].
Image Acquisition and Analysis
Images were taken using a Leica DMR fluorescence compound microscope, a Sensicam CCD camera, and Slidebook software (Intelligent Imaging, Inc.). For quantification of BrdU intensities, larval brains were imaged at the same exposure time. Slidebook software was then used to quantify total signal in each image that is above the background level.
Acknowledgments
We thank Bob Duronio, Jeff Sekelsky, Kai Zinn, and Pat O’ Farrell for critical reading of the manuscript and members of the Su lab for helpful suggestions. This work was supported by grants to T.T.S. from the American Cancer Society (PRG-99-166-CCG) and the National Institute of Health (RO1 GM 66441). B.R.J. was supported by a National Institute of Health predoctoral training grant and by a scholarship from ARCS (Achievement Rewards for College Scientists).
References
- 1.Zhou BB, and Elledge SJ (2000). The DNA damage response: putting checkpoints in perspective. Nature 408, 433–439. [DOI] [PubMed] [Google Scholar]
- 2.Savitsky K, Bar-Shiva A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. (1995). A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268, 1749–1753. [DOI] [PubMed] [Google Scholar]
- 3.Jeggo PA, Carr AM, and Lehmann AR (1998). Splitting the ATM: distinct repair and checkpoint defects in ataxia-telangiectasia. Trends Genet. 14, 312–316. [DOI] [PubMed] [Google Scholar]
- 4.Weinert TA, and Hartwell LH (1988). The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science 241, 317–322. [DOI] [PubMed] [Google Scholar]
- 5.Gatei M, Young D, Cerosaletti KM, Desai-Mehta A, Spring K, Kozlov S, Lavin MF, Gatti RA, Concannon P, Khanna K, et al. (2000). ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat. Genet 25, 115–119. [DOI] [PubMed] [Google Scholar]
- 6.Petrini JH (2000). The Mre11 complex and ATM: collaborating to navigate S phase. Curr. Opin. Cell Biol 12, 293–296. [DOI] [PubMed] [Google Scholar]
- 7.Herzog KH, and Chong MJ, Kapsetaki M, Morgan JI, and McKinnon PJ (1998). Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science 280, 1089–1091. [DOI] [PubMed] [Google Scholar]
- 8.Sibon OC, Stevenson VA, and Theurkauf WE (1997). DNA-replication checkpoint control at the Drosophila midblastula transition. Nature 388, 93–97. [DOI] [PubMed] [Google Scholar]
- 9.Sibon OC, Laurencon A, Hawley R, and Theurkauf WE (1999). The Drosophila ATM homologue Mei-41 has an essential checkpoint function at the midblastula transition. Curr. Biol 9, 302–312. [DOI] [PubMed] [Google Scholar]
- 10.Brodsky MH, and Sekelsky JJ, Tsang G, Hawley RS, and Rubin GM (2000). mus304 encodes a novel DNA damage checkpoint protein required during Drosophila development. Genes Dev. 14, 666–678. [PMC free article] [PubMed] [Google Scholar]
- 11.Garner M, van Kreeveld S, and Su TT (2001). mei-41 and bub1 block mitosis at two distinct steps in response to incomplete DNA replication in Drosophila embryos. Curr. Biol 11, 1595–1599. [DOI] [PubMed] [Google Scholar]
- 12.Gatti M, Pimpinelli S, and Baker BS (1980). Relationships among chromatid interchanges, sister chromatid exchanges, and meiotic recombination in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 77, 1575–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahmad K, and Golic KG (1999). Telomere loss in somatic cells of Drosophila causes cell cycle arrest and apoptosis. Genetics 151, 1041–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kooistra R, Pastink A, Zonneveld JB, Lohman PH, and Eeken JC (1999). The Drosophila melanogaster DmRAD54 gene plays a crucial role in double-strand break repair after P-element excision and acts synergistically with Ku70 in the repair of X-ray damage. Mol. Cell. Biol 19, 6269– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hari KL, Santerre A, Sekelsky JJ, McKim KS, Boyd JB, Hawley RS (1995). The mei-41 gene of D. melanogaster is a structural and functional homolog of the human ataxia telangiectasia gene. Cell 82, 815–821. [DOI] [PubMed] [Google Scholar]
- 16.Spradling A, and Orr-Weaver T (1987). Regulation of DNA replication during Drosophila development. Annu. Rev. Genet 21, 373–403. [DOI] [PubMed] [Google Scholar]
- 17.Neufeld TP, de la Cruz AF, Johnston LA, and Edgar BA (1998). Coordination of growth and cell division in the Drosophila wing. Cell 93, 1183–1193. [DOI] [PubMed] [Google Scholar]
- 18.Meinertzhagen IA, and Hanson TE (1993). The development of the optic lobe. In The Development of Drosophila melanogaster, Bate M and Martinez Arias A, eds. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; ) pp. 1363–1491. [Google Scholar]
- 19.Paulovich AG, and Hartwell LH (1995). A checkpoint regulates the rate of progression through S phase in S. cerevisiae in response to DNA damage. Cell 82, 841–847. [DOI] [PubMed] [Google Scholar]
- 20.Rowley R, Phillips EN, and Schroeder AL (1999). The effects of ionizing radiation on DNA synthesis in eukaryotic cells. Int. J. Radiat. Biol 75, 267–283. [DOI] [PubMed] [Google Scholar]
- 21.Falck J, Mailand N, Syljuasen RG, Bartek J, and Lukas J (2001). The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 410, 842–847. [DOI] [PubMed] [Google Scholar]
- 22.Zhao H, and Watkins JL, and Piwnica-Worms H (2002). Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc. Natl. Acad. Sci. USA 99, 14795–14800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyd JB, Golino MD, Nguyen TD, and Green MM (1976). Isolation and characterization of X-linked mutants of Drosophila melanogaster which are sensitive to mutagens. Genetics 84, 485–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hay BA, Wolff T, and Rubin GM (1994). Expression of baculovirus P35 prevents cell death in Drosophila. Development 120, 2121–2129. [DOI] [PubMed] [Google Scholar]
- 25.Brodsky MH, Nordstrom W, Tsang G, Kwan E, Rubin GM, and Abrams JM (2000). Drosophila p53 binds a damage response element at the reaper locus. Cell 101, 103–113. [DOI] [PubMed] [Google Scholar]
- 26.Haynie JL, and Bryant PJ (1977). The Effects of X-rays on the proliferation dynamics of cells in the imaginal wing disc of Drosophila melanogaster. Wilhelm Roux’s Archives 183, 85–100. [DOI] [PubMed] [Google Scholar]
- 27.Sogame N, Kim M, and Abrams JM (2003). Drosophila p53 preserves genomic stability by regulating cell death. Proc. Natl. Acad. Sci. USA 100, 4696–4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolff T, and Ready DF (1993). Pattern formation in the Drosophila retina. In The Development of Drosophila melanogaster, Bate M and Martinez Arias A, eds. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; ) pp. 1277–1326. [Google Scholar]
- 29.Ashburner M (1989). Drosophila: A Laboratory Handbook (Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press; ). [Google Scholar]
- 30.Feger G, Vaessin H, Su TT, Wolff E, Jan LY, and Jan YN (1995). dpa, a member of the MCM family, is required for mitotic DNA replication but not endoreplication in Drosophila. EMBO J. 14, 5387–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Treisman JE, Follette PJ, O’Farrell PH, and Rubin GM (1995). Cell proliferation and DNA replication defects in a Drosophila MCM2 mutant. Genes Dev. 9, 1709–1715. [DOI] [PubMed] [Google Scholar]
- 32.Abbott LA, and Natzle JE (1992). Epithelial polarity and cell separation in the neoplastic l(1)dlg-1 mutant of Drosophila. Mech. Dev 37, 43–56. [DOI] [PubMed] [Google Scholar]
- 33.James AA, and Bryant PJ (1981). A quantitative study of cell death and mitotic inhibition in gamma-irradiated imaginal wing discs of Drosophila melanogaster. Radiat. Res 87, 552–564. [PubMed] [Google Scholar]
- 34.Baker NE, and Yu SY (2001). The EGF receptor defines domains of cell cycle progression and survival to regulate cell number in the developing Drosophila eye. Cell 104, 699–708. [DOI] [PubMed] [Google Scholar]
- 35.Sanchez Y, Bachant J, Wang H, Hu F, Liu D, Tetzlaff M, and Elledge SJ (1999). Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 286, 1166–1171. [DOI] [PubMed] [Google Scholar]
- 36.Michalopoulos GK, and DeFrances MC (1997). Liver regeneration. Science 276, 60–66. [DOI] [PubMed] [Google Scholar]
- 37.Kooistra R, Vreeken K, and Zonneveld JB, de Jong A, Eeken JC, Osgood CJ, Buerstedde JM, Lohman PH, and Pastink A (1997). The Drosophila melanogaster RAD54 homolog, DmRAD54, is involved in the repair of radiation damage and recombination. Mol. Cell. Biol 17, 6097–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laurençon A, Purdy A, Sekelsky J, Hawley RS, and Su TT (2003). Phenotypic analysis of separation-of-function alleles of MEI-41, a Drosophila ATR. Genetics 164, 589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fogarty P, and Campbell SD, Abu-Shumays R, Phalle BS, Yu KR, Uy GL, Goldberg ML, and Sullivan W (1997). The Drosophila grapes gene is related to checkpoint gene chk1/rad27 and is required for late syncytial division fidelity. Curr. Biol 7, 418–426. [DOI] [PubMed] [Google Scholar]
- 40.Rong YS, Titen SW, Xie HB, Golic NM, Bastiani M, Bandyopadhyay P, Olivera BM, Brodsky M, Rubin GM, and Golic KG, et al. (2002). Targeted mutagenesis by homologous recombination in D. melanogaster. Genes Dev. 16, 1568–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pimpinelli S, Bonaccorsi S, Fanti L, and Gatti M (2000). Preparation and analysis of Drosophila mitotic Chromosomes. In Drosophila Protocols, Sullivan W, Ashburner M, and Hawley RS, eds. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; ) pp. 3–23 [Google Scholar]
- 42.Su TT, Sprenger F, DiGregorio PJ, Campbell SD, and O’Farrell PH (1998). Exit from mitosis in Drosophila syncytial embryos requires proteolysis and cyclin degradation, and is associated with localized dephosphorylation. Genes Dev. 12, 1495–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tercero JA, Longhese MP, and Diffley JF (2003). A central role for DNA replication forks in checkpoint activation and response. Mol. Cell 11, 1323–1336. [DOI] [PubMed] [Google Scholar]