

Summary
Uncertainty persists whether anaerobic bacteria represent important pathogens in aspiration pneumonia. In a nested case-control study of mechanically ventilated patients classified as macro-aspiration pneumonia (MAsP, n = 56), non-macro-aspiration pneumonia (NonMAsP, n = 91), and uninfected controls (n = 11), we profiled upper (URT) and lower respiratory tract (LRT) microbiota with bacterial 16S rRNA gene sequencing, measured plasma host-response biomarkers, analyzed bacterial communities by diversity and oxygen requirements, and performed unsupervised clustering with Dirichlet Multinomial Models (DMM). MAsP and NonMAsP patients had indistinguishable microbiota profiles by alpha diversity and oxygen requirements with similar host-response profiles and 60-day survival. Unsupervised DMM clusters revealed distinct bacterial clusters in the URT and LRT, with low-diversity clusters enriched for facultative anaerobes and typical pathogens, associated with higher plasma levels of SPD and sCD14 and worse 60-day survival. The predictive inter-patient variability in these bacterial profiles highlights the importance of microbiome study in patient sub-phenotyping and precision medicine approaches for severe pneumonia.
Subject areas: Clinical finding, Microbiome, Respiratory medicine
Graphical abstract
Highlights
-
•
Aspiration pneumonia is not enriched for obligate anaerobic bacteria
-
•
Anaerobic spectrum antibiotics deplete obligate anaerobes in the respiratory tract
-
•
Low diversity and high pathogen abundance predicts adverse outcome in pneumonia
-
•
Microbiota profiling can sub-phenotype patients with severe pneumonia
Clinical finding; Microbiome; Respiratory medicine
Introduction
Aspiration is a common phenomenon in the human respiratory tract, occurring on a spectrum from routine physiologic micro-aspiration in the awake or asleep person, to large volume influx of gastric or oropharyngeal secretions through the glottis (i.e., macro-aspiration) leading to lung tissue injury and pneumonia.1,2 Although the pathogenesis of most bacterial pneumonias involves micro-aspiration or inhalation of pathogenic organisms from the upper to the lower respiratory tract (URT to LRT), clinicians have classically ascribed the term aspiration pneumonia to the subset of infections caused by macro-aspiration of gastric or upper respiratory secretions, often in vulnerable patients with stroke, altered mental status, or dysphagia.3,4
Early studies using invasive transtracheal sampling or thoracentesis first attributed the etiology of aspiration pneumonia to anaerobic bacteria such as Bacteroides, Fusobacterium, and Peptostreptococcus.5 Later studies isolated more typical pathogens of community-acquired pneumonia from sputum or bronchoalveolar lavage (BAL) cultures from patients with clinical diagnosis of aspiration.4,6 Notably, identification of anaerobes through routine culture-based study of the LRT is difficult, requiring special processing of specimens and anaerobic growth conditions. In recent years, culture-independent studies with DNA sequencing have enabled comprehensive profiling of microbial communities, allowing for the detection of fastidious organisms and anaerobes without the need for ex vivo organismal growth. From BAL examinations of patients with community- or healthcare-acquired pneumonia via 16S rRNA gene sequencing (16S-Seq), patients with aspiration risk factors had higher detection of oral Streptococci (31% versus 15%, p = 0.009) and lower detection of anaerobic bacteria compared to patients without risk factors (6% versus 18%, p = 0.02, respectively).7,8 Conversely, in patients with aspiration pneumonia complicated by lung abscess formation, 16S-Seq revealed high abundance of anaerobes not isolated by cultures.9 Among mechanically ventilated patients with aspiration pneumonia, abundance of anaerobes in the LRT was reduced from 12% after intubation to <3% by 72 h.10 The available evidence from small-scale culture-independent studies suggests that obligate anaerobic bacteria are implicated in the minority of aspiration pneumonias.
Clinical practice guidelines advise against routinely adding anaerobic coverage for suspected aspiration pneumonia beyond standard empiric treatment for community-acquired pneumonia, unless empyema or abscess is suspected.11 Antibiotic prescription data before the publication of these guidelines showed that the majority of patients with aspiration pneumonia (65–90%) received antibiotics with anaerobic coverage, either in the form of broad-spectrum agents or with specifically selected narrow-spectrum antibiotics (e.g., metronidazole).4,12 With the supportive evidence for the guideline recommendations relying on earlier clinical studies using conventional microbiology, there is remaining uncertainty on the epidemiology of causal microbes in aspiration pneumonia.
To understand the microbiome of aspiration pneumonia, we conducted a nested case-control study of patients with and without pneumonia, selected from a prospective cohort of adult patients with acute respiratory failure requiring mechanical ventilation in intensive care units (ICUs) at UPMC. Through in-depth review of available clinical and radiological data providing evidence for or against a macro-aspiration event leading to pneumonia, we classified patients into the following groups: (1) Macro-aspiration pneumonia (MAsP – cases), (2) Non-aspiration pneumonia (NonMAsP – diseased controls), and (3) Intubated Controls (patients intubated for airway protection without evidence of infection). We performed culture-independent sequencing of URT and LRT samples (16S-Seq and metagenomics) and measured biomarkers of host response in simultaneously collected plasma samples. We incorporated previously generated 16S-Seq data from URT and LRT samples collected from healthy volunteers (Healthy Controls) for comparing microbiota from ICU patients versus the healthy URT and LRT. We systematically quantified the abundance of bacteria by oxygen requirements for growth; we agnostically interrogated microbial community composition for prognostic features on patient outcomes, and integrated microbiome profiles with systemic host-response biomarkers.
Results
Cohort description and aspiration risk factor assessments
We included 158 mechanically ventilated patients, classified as MAsP (n = 56), NonMAsP (n = 91) and uninfected, intubated controls (n = 11). Compared to intubated controls, patients with pneumonia (both MAsP and NonMAsP) had higher prevalence of comorbid conditions, worse ventilatory mechanics and hypoxemia, higher organ dysfunction and lung injury risk factor burden, and worse clinical outcomes (Table 1). However, there were no significant differences between MAsP and NonMAsP subjects (Table 1), who had similar 60-day survival and liberation from mechanical ventilation (Figure S1). Patients with pneumonia were exposed to antibiotics of similar spectrum and duration independent of macro-aspiration diagnosis (Figure S2).
Table 1.
Baseline characteristics of enrolled mechanically ventilated subjects, stratified by clinical groups of macro-aspiration diagnosis
| Macro-aspiration Pneumonia | Non-macro-aspiration Pneumonia | Intubated Controls | p | |
|---|---|---|---|---|
| N | 56 | 91 | 11 | |
| Age, years (median [IQR]) | 55.3 [36.4, 67.4] | 59.1 [48.2, 69.2] | 46.8 [39.8, 56.2] | 0.07 |
| Men, n (%) | 31 (55.4) | 52 (57.1) | 8 (72.7) | 0.56 |
| Caucasians, n (%) | 50 (89.3) | 85 (93.4) | 10 (90.9) | 0.67 |
| BMI (median [IQR]) | 27.8 [25.3, 35.3] | 29.5 [24.2, 35.0] | 27.2 [23.1, 27.8] | 0.21 |
| COPD, n (%) | 8 (14.3) | 32 (35.2) | 0 (0.0) | <0.01 |
| Diabetes, n (%) | 23 (41.1) | 26 (28.6) | 0 (0.0) | 0.02 |
| Alcohol use, n (%) | 10 (17.9) | 12 (13.2) | 7 (63.6) | <0.01 |
| Immunosuppression, n (%) | 9 (16.1) | 17 (18.7) | 0 (0.0) | 0.29 |
| ARDS, n (%) | 12 (21.4) | 35 (38.5) | 0 (0.0) | 0.01 |
| WBC (median [IQR]) | 14.0 [10.2, 18.3] | 12.4 [9.6, 17.1] | 8.9 [6.8, 10.7] | 0.02 |
| Creatinine (median [IQR]) | 1.0 [0.7, 1.6] | 1.5 [0.8, 2.3] | 0.9 [0.6, 1.2] | 0.04 |
| Plateau Pressure (median [IQR]) | 20.0 [16.0, 23.0] | 20.0 [17.0, 25.0] | 13.0 [12.0, 16.0] | <0.01 |
| PaO2:FiO2 ratio (median [IQR]) | 156.5 [117.0, 205.0] | 148.0 [108.0, 205.0] | 273.0 [205.0, 311.0] | <0.01 |
| SOFA scores (median [IQR]) | 6.0 [4.0, 8.0] | 7.0 [5.0, 9.0] | 4.0 [3.0, 6.0] | 0.04 |
| LIPS score (median [IQR]) | 6.8 [5.5, 8.1] | 5.5 [4.5, 7.5] | 3.0 [1.5, 5.8] | <0.01 |
| Hypoinflammatory subphenotype, n (%) | 43 (86.0) | 64 (77.1) | 10 (90.9) | 0.31 |
| Culture-Positive, n (%) | 16 (28.6) | 38 (41.8) | 0 (0.0) | 0.16a |
| Anaerobic spectrum antibiotics on day of sampling, n (%) | 30 (53.6) | 51 (56.0) | 1 (9.1) | 0.01 |
| VFD (median [IQR]) | 22.0 [15.8, 25.0] | 20.5 [12.2, 24.0] | 26.0 [24.5, 27.0] | <0.01 |
| 14-day mortality, n (%) | 8 (14.3) | 13 (14.3) | 0 (0.0) | 0.40 |
| 30-day mortality, n (%) | 10 (17.9) | 20 (22.0) | 0 (0.0) | 0.21 |
| 60-day mortality, n (%) | 13 (23.2) | 22 (24.2) | 0 (0.0) | 0.18 |
Abbreviations: IQR: Interquartile Range; BMI: body mass index; COPD: chronic obstructive pulmonary disease, LIPS: lung injury prediction score; WBC: white blood cell count; PaO2: partial pressure of arterial oxygen; FiO2: Fractional inhaled concentration of oxygen; SOFA: sequential organ failure assessment; VFD: ventilator free days; ARDS: acute respiratory distress syndrome.
We compared continuous variables with non-parametric Wilcoxon tests and categorical variables with Fisher’s exact tests between the three groups. Statistically significant differences (p<0.05) are highlighted in bold.
Comparison between MAsP and NonMAsP subjects only because culture results were by definition negative in the intubated controls group.
Owing to the inherent subjectivity of macro-aspiration diagnosis, we further reviewed subjects for presence of risk factors for aspiration (both historical and related to acute presentation)4 and aimed to capture the clinicians’ suspicion of aspiration at the time of the clinical encounter. With regards to historic risk factors for aspiration, NonMAsP subjects had higher baseline use of chronic oxygen supplementation (p = 0.03), but otherwise no significant differences in factors such as history of stroke, dementia, or bulbar dysfunction (Table S1). With regards to acute factors, MAsP subjects were more commonly intubated for drug overdose compared to NonMAsP, whereas intubated controls were more frequently intubated for seizures compared to pneumonia subjects (p≤0.01) (Table S1). By examining the distribution of acute versus chronic risk factors for aspiration, we classified subjects into those with high burden (≥2) of acute risk factors, (26.5%), high burden (≥2) of chronic risk factors (22.8%) and those with low burden of acute or chronic risk factors (50.6%) (Figure S3). Among MAsP subjects, patients with high burden of acute risk factors had better 60-day survival and liberation compared to those with chronic risk factor burden (Figures S1B and S1E), whereas among NonMAsP subjects, there were no outcome differences between those with acute or chronic risk factor burden (Figures S1C and S1F).
Notably, for patients with no documentation of a macro-aspiration event (classified as NonMAsP), clinicians used the term “aspiration” in documentation in 30% of them, whereas radiographic reports indicated possibility of aspiration in 84% (Table S1).
We measured 13 biomarkers of host injury and inflammation in plasma samples collected at baseline (within the first 48 h of intubation). Both MAsP and NonMAsP subjects had higher plasma levels of sTNFR1, sST2, fractalkine, sRAGE, Ang-2, procalcitonin, pentraxin-3 and LBP compared to intubated controls (all p<0.05, Table S2).
Bacterial community profiles by clinical group
At baseline, we collected a posterior oropharyngeal swab (OPS) and an endotracheal aspirate (ETA) within 48 h of intubation. For patients who remained intubated in the ICU, we collected follow-up samples at a middle time-point (days 3–6) and a late follow-up interval (days 7–11 after intubation). For contextualization, we also analyzed available 16S-Seq data from a cross-sectional oral wash and BAL sample obtained from 24 Healthy Controls.
In a quality control step, we verified that clinical samples had significantly higher 16S-Seq yield (number of quality reads) compared to experimental negative controls (Figure S4). In clinical sample comparisons for bacterial burden in the URT and LRT at baseline, MAsP subjects had higher number of 16S rRNA gene copies in OPS samples compared to NonMAsP (p = 0.009), but otherwise no significant differences between groups (Figures 1A and 1B). For both URT and LRT samples, the three clinical groups of ICU patients (MAsP, NonMAsP and intubated controls) had similar alpha diversity (Shannon index) in OPS and ETA samples respectively, but overall, significantly lower Shannon indices compared to Healthy Controls (p<0.0001) (Figures 1C and 1D). There were significant differences in beta diversity comparisons between clinical groups (Figures 1E and 1F, p<0.0001), with evidence of compositional overlap for both OPS and ETA samples among MAsP and NonMAsP subjects (Figure S5). By examining the individual risk factors for aspiration among all three clinical groups, alcohol use disorder was associated with higher bacterial burden in both OPS and ETA samples, whereas seizures and radiographic suspicion of aspiration with higher bacterial burden in ETA samples (Figures S6–S9). Higher burden of acute risk factors for aspiration was associated with higher alpha diversity in OPS samples and bacterial burden in ETA samples (Figure S10). Culture-positive MAsP and NonMAsP subjects had higher bacterial burden and lower Shannon index in ETA samples compared to culture-negative subjects (Figure S11).
Figure 1.

Macro-aspiration pneumonia subjects had similar bacterial community profiles to non-aspiration pneumonia subjects and intubated controls in both upper and lower respiratory tract specimens
(A and B): Comparisons of bacterial burden (number of 16S rRNA gene copies by quantitative PCR) in oropharyngeal swabs (OPS) and endotracheal aspirate (ETA) samples for macro-aspiration pneumonia (MAsP), non-macro-aspiration pneumonia (NonMAsP) and intubated control (Ctrl) subjects. MAsP patients had significantly higher bacterial burden compared to NonMAsP patients in OPS samples.
(C and D): Shannon index comparisons between MAsP, NonMAsP, Ctrl (ICU patients) and healthy control (healthy volunteers) specimens revealed significantly lower Shannon index in MAsP subjects compared to healthy controls in both OPS and ETA samples.
(E and F): Principal coordinates analyses for beta diversity comparisons (Manhattan distances) between MAsP, NonMAsP, Ctrl (ICU patients) and healthy controls in OPS and ETA samples. We found significant beta diversity differences by permutational analysis of variance (p<0.0001) for both OPS and ETA samples, but no significant differences between MAsP and NonMAsP samples (black and orange ellipses). Data in boxplots are represented as individual values with median values and interquartile range depicted by the boxplots.
By summary relative abundance, Streptococcus, Veillonella, and Prevotellaceae_unclassified were the most common genera in OPS samples, whereas Streptococcus, Prevotellaceae_unclassified and Staphylococcus were the most common ones in ETA samples (Figure S12). Following adjustment for multiple comparisons, we found no differences in individual genera abundance between MAsP subjects versus NonMAsP or intubated controls (Data S1 and S2, related to Figure 1), whereas 18% and 32% of genera in URT and LRT samples, respectively, were differentially abundant between MAsP and Healthy Controls (Figure S13), highlighting the deviation of bacterial composition in MAsP subjects from the healthy respiratory microbiome.
We classified the 16S-Seq identified genera into categories defined by oxygen requirements (aerobes, anaerobes, facultative anaerobes, microaerophiles, variable) and membership in the typical lung microbiome in health (i.e., oral-origin commensals) versus organisms typically involved in LRT infections (i.e., common respiratory pathogens). By oxygen requirement, obligate anaerobes represented a median summary relative abundance of 18% (interquartile range: 5–50%) in MAsP ETA samples, with no significant difference from NonMAsP (21% [3–57]) and intubated controls (6% [0–29]), but significantly lower abundance compared to healthy controls (45% [23–62], p = 0.024) (Figure 2). A quarter of MAsP subjects had >50% relative abundance of obligate anaerobes. Conversely, MAsP and NonMAsP ETA samples were significantly enriched for facultative anaerobe abundance compared to healthy controls (p<0.001) (Figure 2). By classifying bacteria into oral-origin commensals versus plausible pathogens, we found no significant differences in ETA samples between MAsP versus NonMAsP subjects, whereas both MAsP and NonMAsP subjects had lower abundance of oral commensals and higher abundance of plausible pathogens compared to intubated, uninfected controls (Figure S14).
Figure 2.

Macro-aspiration subjects had similar abundance of obligate anaerobes in the upper and lower respiratory tract compared to non-macro-aspiration pneumonia subjects
We classified all component bacteria genera by oxygen requirement into obligate anaerobes, aerobes, facultative anaerobes, microaerophiles, genera of variable oxygen requirement and unclassifiable.
(A and B): Relative abundance barplots for all oropharyngeal swab (OPS-top) and endotracheal aspirate (ETA-bottom) samples, stratified in facets by clinical group categories: macro-aspiration pneumonia (MAsP), non-macro-aspiration pneumonia (NonMAsP), intubated controls and healthy control subjects.
(C–H): Comparisons of relative abundance for the three main categories of bacteria (obligate anaerobes, aerobes and facultative anaerobes) by clinical groups in OPS (top) and ETA (bottom) samples. MAsP subjects had similar abundance of obligate anaerobes to NonMAsP subjects in ETA samples, but significantly lower compared to healthy controls (p = 0.024). Conversely, MAsP subjects were enriched for facultative anaerobes compared to healthy controls in ETA samples (p = 0.0005). Data in boxplots are represented as individual values with median values and interquartile range depicted by the boxplots.
Metagenomic profiles in LRT specimens
We conducted Nanopore metagenomic sequencing of all microbial DNA molecules in a random subset of 64 available ETA samples (n = 24 MAsP, n = 29 NonMAsP and n = 11 Controls). There was no difference between clinical groups for DNA reads assigned to bacteria, fungi or viruses (Figure S15). Among bacteria, the most abundant genus Streptococcus (31% of all bacterial reads), with oral commensal Streptococcus spp., such as Streptococcus mitis, Streptococcus oralis, and Streptococcus salivarius, accounting for 76% of Streptococcus genus reads, and the remaining 24% of reads mapped to Streptococcus pneumoniae (Figure S15). There were no significant differences in individual Streptococcus spp. abundance between clinical groups. Among fungi, Candida spp. accounted for 95% of all fungal reads, without any significant difference in abundance between clinical groups for each Candida spp. (Figure S15).
Unsupervised clusters of bacterial composition
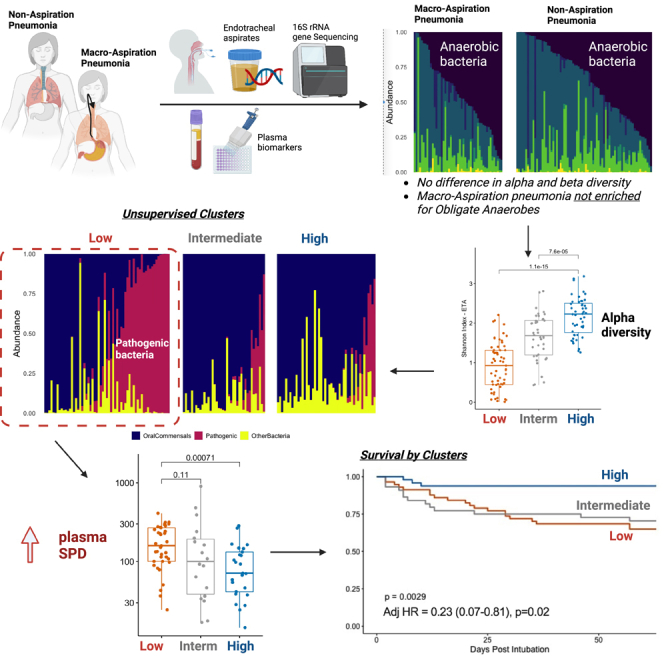
Because there were no large differences in microbial communities between MAsP and NonMAsP subjects, we then agnostically examined their 16S-Seq communities for any distinct clusters of microbial composition (“metacommunities”) with unsupervised Dirichlet Multinomial Models (DMMs). DMMs showed optimal fit for two clusters in OPS and three clusters in ETA samples (Figure S16). We found striking differences between clusters by Shannon index (Figure 3); thus, we refer to OPS clusters as high- versus low-diversity and to ETA clusters as high-, intermediate-, and low-diversity. DMM clusters in both compartments differed significantly by bacterial burden and beta diversity (Figure 3). The low diversity OPS cluster was enriched for Staphylococcus, Enterococcus, and Enterobacteriaceae genera whereas the low diversity ETA clusters for Pseudomonas (Figures S17 and S18). By bacterial oxygen requirement categories, the low diversity clusters were depleted for obligate anaerobes and enriched for facultative anaerobes compared to high diversity clusters (Figure S19). Similarly, the low diversity ETA cluster was depleted for oral commensals and markedly enriched for plausible pathogens (Figure S20).
Figure 3.

Unsupervised clustering by Dirichlet Multinomial Mixture (DMM) models revealed distinct clusters in upper and lower respiratory tract specimens in pneumonia subjects
(A–C): DMM in oropharyngeal swabs (OPS) showed optimal fit with two clusters (low- and high-diversity), which had significant differences in bacterial burden (by 16S rRNA gene qPCR), Shannon index and beta-diversity (Manhattan distances – principal coordinates analyses).
(D–F): DMM in endotracheal aspirates (ETA) showed optimal fit for three clusters (termed low-, intermediate- and high-diversity) with significant differences in bacterial burden, Shannon index and beta diversity. Data in boxplots are represented as individual values with median values and interquartile range depicted by the boxplots.
Patients in the low diversity ETA cluster had higher prevalence of COPD and use of chronic oxygen therapy, were more likely to have been diagnosed with a drug overdose or have a radiology report suspicious for aspiration compared to patients in the intermediate or high diversity cluster (p<0.01) (Tables S3 and S4).
Integration of host-response plasma biomarkers with microbial profiles
Cross-correlations between the 20 most abundant taxa in OPS and ETA samples and plasma biomarkers showed statistical significance for individual comparisons (e.g., Staphylococcus with Surfactant Protein D [SPD] levels, Figure S21), but comparisons were not significant following adjustment for multiple testing. By oxygen requirement classifications, we found significant correlations between facultative anaerobes abundance in ETA samples with IL-8 and IL-10 levels (Figure S22). By DMM clustering, we found significant differences for SPD and soluble CD14 (sCD14) for both OPS and ETA clusters, with the low diversity cluster in each compartment associated with higher plasma levels for both SPD and sCD14 (Figure 4,Tables S3, and S4).
Figure 4.

Pneumonia subjects classified in the low diversity cluster of bacterial composition in the upper and the lower respiratory tract had significantly higher plasma levels of SPD and sCD14 compared to high diversity cluster subjects
Data in boxplots are represented as individual values with median values and interquartile range depicted by the boxplots.
DMM clusters are predictive of outcome in patients of pneumonia
Among all subjects with pneumonia, non-survivors had lower Shannon index and differential composition by beta diversity for ETA samples (Figure 5), a difference that was significant also within MAsP and NonMAsP groups. Non-survivors had lower abundance of obligate anaerobes (p<0.0001), higher abundance of facultative anaerobes (p<0.0001), also reflected by lower abundance of oral commensals (p = 0.003) and higher abundance of plausible pathogens (p<0.0001, Figure S23). DMM clusters were strongly predictive of 60-day survival (Figure 5), with membership in the high diversity cluster in ETA samples being protective in Cox proportional hazards models adjusted for age, sex, history of COPD, chronic oxygen use and total antibiotic exposure by the day of baseline sampling (adjusted hazard ratio [95% confidence interval]: 0.23 [0.07–0.81]). When we combined cluster classifications for OPS and ETA samples, we found that subjects in the low diversity cluster in both sample types had worse 60-day survival than subjects in the high diversity cluster in both compartments (logrank p = 0.02), highlighting the impact of combined dysbiosis in URT and LRT (Figure S24).
Figure 5.

Patients with pneumonia in the high diversity cluster in either oropharyngeal swab (OPS) samples or endotracheal aspirates (ETA) had improved 60-day survival compared to patients into low or intermediate-diversity clusters of bacterial composition
(A and B): Shannon index comparisons between survivors and non-survivors showed significantly higher alpha diversity in ETA samples of survivors.
(C and D): Principal coordinates analyses of beta diversity (Manhattan distances) showed significant differences (by permutational analysis of variance) in ETA samples between survivors and non-survivors.
(E and F): Kaplan-Meier curves of 60-day survival between Dirichlet Multinomial Mixture model clusters in OPS and ETA samples demonstrate improved survival for patients in the high diversity cluster in both OPS and ETA samples. Adjusted hazard ratios (with 95% confidence intervals) for membership in the high diversity cluster are shown, as derived from Cox proportional hazards models adjusted for age, sex, history of COPD, chronic oxygen use, and antibiotic exposure by the day of sampling. Data in boxplots are represented as individual values with median values and interquartile range depicted by the boxplots.
Longitudinal microbiome evolution in the URT and LRT
We analyzed ecological metrics of bacterial burden, alpha diversity and composition in available longitudinal samples for OPS (n = 146 baseline, n= 65 middle and n = 25 late follow-up intervals) and ETA samples (n = 158 baseline, n= 46 middle and n = 12 late follow-up intervals). Despite global differences between samples at different follow-up intervals, we found no significant differences between MAsP and NonMAsP subjects within each follow-up interval (Figures S25 and S26). With mixed regression models, we found that time spent on mechanical ventilation was significantly associated with lower Shannon index at time of sampling in both OPS (p = 0.003) and ETA samples (p = 0.002, Figures S25 and S26). We then examined for time-dependent change in the relative abundance of bacteria by oxygen requirements, when subjects were stratified by exposure to antibiotics with anaerobic coverage. We found a significant reduction in OPS abundance of obligate anaerobes with enrichment for facultative anaerobes over time only for subjects who were exposed to antibiotics with anaerobic coverage (Figure S27).
Discussion
We conducted detailed profiling of URT and LRT microbiota in a cohort of mechanically-ventilated patients who were carefully phenotyped for aspiration and associated risk factors. We demonstrated that clinical diagnosis of macro-aspiration is not associated with a distinct microbiome signature, either at baseline post-intubation or in follow-up samples. Instead, we highlight that microbial communities in critically ill patients have systematically deviated from their expected profiles of the healthy respiratory tract, and such deviations have resulted in substantial heterogeneity in bacterial burden, diversity and composition. Overall, patients with pneumonia had depletion of obligate anaerobe bacteria in both the URT and LRT, matched by enrichment with facultative anaerobes, corresponding to a shift from typical oral-origin commensals to bacteria implicated in LRT infections. However, individual taxonomic features or ecologic parameters had limited predictive value; instead, unsupervised clusters emerged as informative, predictive summaries of microbiota heterogeneity. A high diversity cluster in both the URT and LRT, resembling the microbiome in health, was strongly associated with systemic host responses and predicted improved survival.
Diagnosis of aspiration from available clinical documentation is challenging, despite our protocolized definitions and approach for determining the diagnosis. Macro- or micro-aspiration is involved in the pathogenesis of pneumonia and is often indicted as a diagnostic possibility in clinical practice. However, the term “aspiration” is likely used liberally by bedside providers and radiologists. In subjects for whom we could not find any evidence of a macro-aspiration event (NonMAsP), clinicians had invoked aspiration as cause of pneumonia in 30%, and radiologists reported radiographic consolidations consistent with aspiration in a remarkable 84%. As the index macro-aspiration event may not be possible to be reported by patients or witnessed by providers, we paid close attention to observable risk factors for aspiration, relating to comorbid conditions (e.g., stroke or dementia), treatments (such as antacid use or chronic oxygen) and peri-intubation events (e.g., seizures or intoxication). Such factors were easier to capture compared to a witnessed macro-aspiration event and offer a reproducible framework for epidemiologic study of aspiration pneumonia.4
Our systematic assessment of URT and LRT microbiota did not reveal a distinct microbiome signature for MAsP, which was largely indistinguishable from NonMAsP. Although obligate anaerobes from the oropharynx or gastrointestinal tract have been historically implicated in the causal microbiology of MAsP, our comparisons with other critically ill groups and healthy volunteers showed a consistent pattern: MAsP patients had lower abundance of obligate anaerobes compared to healthy controls in both the URT and LRT tract, coupled by enrichment for facultative anaerobes. Consistent with published trends on anaerobic spectrum antibiotic prescriptions,12 56% of our MAsP patients were receiving anaerobic coverage on the day of sampling, yet only 24% had high (>50%) relative abundance of obligate anaerobes in the LRT. Detection of such anaerobic bacteria in the LRT does not necessarily mean that these were causing pneumonia. Furthermore, we found that anaerobic coverage antibiotics were associated with a progressive decline in abundance of obligate anaerobes in the URT. Recent research in critically ill patients has implicated anti-anaerobic antibiotics with enrichment of the gut microbiome with respiratory pathogens and increased risk for ventilator-associated pneumonia (VAP).13 Our study found that anaerobic coverage antibiotics were associated with a progressive decline in abundance of obligate anaerobes with a corresponding increase in abundance of facultative anaerobes in the URT. As such, treatment with anti-anaerobic antibiotics may have “off-target” effects beyond the gut, perhaps accounting for an increased risk for VAP because of loss of commensal obligative anaerobes in the respiratory tract.14 Therefore, the guideline recommendations to avoid anaerobic coverage in suspected aspiration pneumonia (unless abscess or empyema have been diagnosed)11 appear well justified.
Similar to earlier findings from our group,15 there is a wide spectrum of alpha/beta diversity in communities of patients diagnosed with pneumonia, and only a subset of subjects have enrichment for typical respiratory pathogens. Most patients diagnosed with pneumonia and treated empirically with antibiotics have LRT communities with high abundance of oral-origin commensal bacteria, with Streptococcus genera being the most abundant. Whereas 16S-Seq can be performed reproducibly in clinical samples and provide genus-level bacterial identification, more detailed taxonomic assessments require untargeted metagenomic screenings, which are nonetheless challenging to perform because of high amounts of contaminating human DNA in ETA specimens. We have overcome this signal/noise ratio issue in metagenomics by implementing a human DNA depletion step during sample processing, which allows for substantial enrichment of microbial DNA for metagenomics.16,17 Thus, by Nanopore metagenomics, we showed that non-pneumoniae Streptococcus spp. accounted for the majority of the Streptococcus 16S-Seq signal. These findings further support the theory of respiratory tract biogeography, according to which a high biomass URT determines the composition of the lower biomass LRT via downward microbial immigration, despite (or perhaps because of) the presence of an endotracheal tube.14,18 Nonetheless, microbial pathogenicity is a context-dependent variable, and oral-origin commensals are likely perfectly capable of causing an LRT infection in the right combination of bacterial burden and host defense circumstances,19 or may regulate the immune tone of the LRT to mitigate host susceptibility against invasive pathogens.20 Our LRT profiling highlights opportunities for improved antibiotic targeting and stewardship, as well as the need to integrate host responses when making the diagnosis of pneumonia.21,22
Agnostic clusters in both URT and LRT microbiota were predictive of outcomes, but also consistently correlated with plasma-measured biomarkers of host defenses. The low diversity cluster, enriched for facultative anaerobes and plausible pathogens, was associated with higher SPD and sCD14 levels. SPD plays a key role in lung innate immunity as a pattern recognition receptor involved in binding of microbes for phagocyte recognition and clearance.23 Plasma SPD levels have been associated with worse outcome in patients with acute lung injury.24 Similarly, membrane-bound CD14 is an accessory molecule that enables the transfer of LPS to the TLR4-MD2 complex involved in canonical pathogen sensing of Gram-negative bacteria by host cells.25 Plasma levels of sCD14 have been associated with clinical outcomes in patients with acute respiratory failure or pneumonia,26 although the triggers of systemic levels remain elusive. Our microbiota cluster analyses revealed consistent associations for both URT and LRT clusters with SPD and sCD14 levels, suggesting that lung dysbiosis or overt LRT infection may account for the observed plasma biomarker elevations levels and their associations with adverse outcomes.
In summary, we demonstrate that clinically diagnosed macro-aspiration does not result in enrichment of obligate anaerobes in the LRT of mechanically ventilated patients. On the contrary, critically ill patients are depleted of oral-origin anaerobic commensals. Thus, aspiration diagnosis in a ventilated patient should not lead to indiscriminate use of antibiotics with anaerobic coverage. The clinical label of aspiration is inadequate to capture the wide variability of microbial communities in URT and LRT of critically-ill patients. Detailed profiling with amplicon sequencing and metagenomics reveals predictive clusters of microbiota, with implications in etiologic pathogen diagnosis, guidance of antimicrobials, study of host responses and outcome prediction.27,28,29 With improvements in timeliness of experimental and analytical pipelines, portability of sequencing platforms, and empirical evidence on microbiome data synthesis for clinical diagnosis of pneumonias,30 prospective, real-time study is needed to define the clinical impact of microbiome science at bedside in the ICU.
Limitations of the study
Our study is limited by the single-center size design and the available sample size, which nonetheless allowed us to identify informative variability in microbiota profiles. Our assessment of macro-aspiration and related risk factors was retrospective and based on available documentation. We lacked systematic, objective assessments of swallowing function with dedicated testing. Our diagnostic approach is reflective of clinical practices on aspiration diagnosis in a tertiary academic center, and highlighted the challenges of epidemiologic studies on aspiration pneumonia. For patient safety and practical purposes of subject participation in our observational research study, we relied on study of non-invasive biospecimens (OPS and ETA) for microbiota profiling, as opposed to more invasive biospecimens (e.g., BAL or pleural fluid). Non-invasive ETA specimens are routinely used etiologic diagnosis in pneumonia by culture-based studies,31 but we cannot draw any inferences on role of anaerobes in closed space infections (i.e., empyemas or lung abscesses), which were not within the reach of our sampling strategy. Nanopore sequencing data may also be prone to sequencing errors, which can be impacted by the GC content of the component bacteria DNA. Nonetheless, overall error rates are declining with evolving sequencing chemistries and software.32 Timings of sample acquisition may have impacted our results, given that microbiota can be rapidly influenced by high oxygen concentrations on mechanical ventilation and systemic antibiotics in critically ill patients. We obtained non-invasive LRT samples at pre-defined feasibility time-intervals under a protocol with no greater than minimal risk. Nonetheless, our longitudinal analyses with mixed regression models revealed time-dependent changes in anaerobic bacteria abundance in subjects exposed to antibiotics with anaerobic spectrum, demonstrating the feasibility of detecting clinically relevant associations with our sampling strategy.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Oropharyngeal swabs, Endotracheal aspirates, Plasma samples, Oral washes, Bronchoalveolar lavage samples | University of Pittsburgh, Division of Pulmonary, Allergy and Critical Care Medicine | Not applicable |
| Chemicals, peptides, and recombinant proteins | ||
| HL-SAN | ArcticZymes | #70910-202 |
| Sputasol | Thermo Scientific | SR0233A |
| Saponin | Millipore Sigma | S4521 |
| ZymoBIOMICS Microbial Community DNA Standard | Zymo Research | D6305 |
| Critical commercial assays | ||
| Dneasy Powersoil DNA Kit | QIAGEN | #12888-100 |
| MiNION Flow Cells R9.4.1 | Oxford Nanopore Technologies | FLO-MIN106D |
| Rapid PCR Barcoding Kit | Oxford Nanopore Technologies | SQK-RPB004 |
| Luminex 10-plex (IL6, IL8, IL10, sRAGE, Ang-2, ST2, sTNFR1, fractalkine, procalcitonin, pentraxin3) | R&D Systems | Custom made |
| Luminex 2-plex (LBP, sCD14) | R&D Systems | Custom made |
| ELISA – SPD | Biovendor, LLC | RD194059101 |
| MiSeq Reagent Kit v2 (300-cycles) | Illumina | MS-102-2002 |
| Deposited data | ||
| 16S rRNA gene sequences | Sequence Resource Archive | PRJNA595346 for biospecimens from ICU patients; PRJNA940725 for Healthy Controls |
| Software and algorithms | ||
| R version v4.2.0 | The R Foundation for Statistical Computing | Not applicable |
| Prism 9 for macOS | GraphPad | Not applicable |
| ChatGPT | OpenAI.com | Not applicable |
| EPI2ME – What’s In My Pot (WIMP) | Oxford Nanopore Technologies | Not applicable |
| Mothur | Mothur.org | Not applicable |
| Other | ||
| Primary Code in R language | GitHub - Zenodo |
https://github.com/MicrobiomeALIR/AspirationPneumonia https://doi.org/10.5281/zenodo.7872061 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Kitsios.
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Method details
We conducted a case-control study of adult male and female patients with and without pneumonia, nested within a prospective observational cohort (the Acute Lung Injury Registry) at the University of Pittsburgh Medical Center (UPMC).
From April 2015 to February 2020, we prospectively enrolled a convenience sample of adult patients with acute respiratory failure requiring mechanical ventilation, who were hospitalized in intensive care units (ICUs) at UPMC (see flowchart, Figure S28).15,16,29,33,34 We obtained informed consent from the patients or their legally authorized representatives under the study protocol STUDY19050099 approved by the University of Pittsburgh Institutional Review Board (IRB). We enrolled both men and women aged 18 yearsold to 90 years old (age and sex information of enrolled subjects provided in Table 1). Exclusion criteria were inability to obtain informed consent, the presence of tracheostomy, and mechanical ventilation for more than 72 hprior to enrollment. For this analysis, we considered patients with clinical diagnosis of pneumonia and patients intubated for airway protection and no signs of infection. We diagnosed pneumonia based on established clinical criteria (2 out of 3: i) new radiographic infiltrate, ii) abnormal temperature or white blood cell count, and iii) positive microbiologic culture).35 Uninfected controls were subjects who were intubated for airway protection without any signs of lower respiratory tract or other infection. All available clinical data were reviewed by a consensus phenotyping committee of physician-scientists who were unaware of research biospecimen analyses (e.g. microbiome sequencing) at the time of the clinical phenotyping.
Study group definitions
Following selection of subjects with pneumonia and uninfected controls and without any knowledge of research assay data, we then performed further in-depth review of available clinical and radiological data with specific attention to factors/events related to aspiration to subclassify patients into the following three clinical groups.
-
a.
Macro-aspiration pneumonia (MAsP – cases): Clinical diagnosis of pneumonia following a macro-aspiration event (witnessed or documented aspiration event, such as vomiting with altered mental status followed by immediate hypoxemia, or visualization/retrieval of gastric/bilious contents from oropharynx, glottis, endotracheal tube or bronchial tree during intubation or bronchoscopy).36
-
b.
Non-macro-aspiration pneumonia (NonMAsP – diseased controls): clinical diagnosis of pneumonia without evidence of a macro-aspiration event as defined above.36
-
c.
Patients intubated for airway protection (Intubated Controls – uninfected diseased controls): Patients with acute respiratory failure and altered mental status, intubated for airway protection without clinical evidence of infection or macro-aspiration prior to or during intubation.
To contextualize the findings on microbiota from critically-ill patients with what is expected for the healthy respiratory tract, we also included data from 24 healthy volunteers who had contributed URT and LRT microbiome data in a previously published cohort (Lung HIV Microbiome Project - STUDY19060243).37
This group of healthy volunteers had been included in the Lung HIV Microbiome Project as subjects who were HIV negative and did not have obstructive airways disease. We designated these healthy volunteers as Healthy Controls.
Main analyses involved comparisons between cases (MAsP), the two diseased control groups (NonMAsP and Intubated Controls) and the Healthy Controls. Given the challenges and inherent subjectivity in establishing a clinical diagnosis of macro-aspiration, we further reviewed subjects for the presence of risk factors for aspiration (both historical and related to acute presentation), as adapted from a secondary analysis of the Global Initiative for Methicillin-resistant Staphylococcus aureus Pneumonia (GLIMP) study:4
-
a.
Chronic risk factors: history of stroke, bulbar dysfunction, dementia, esophageal pathology, chronic antiacid use, home oxygen use, residence at a skilled nursing facility.
-
b.
Acute presentation risk factors (during or before intubation): altered mental status with or without seizure, drug overdose, alcohol intoxication or withdrawal, vomiting.
Beyond our systematic assessment of macro-aspiration and associated risk factors, we also aimed to capture the clinicians’ suspicion of aspiration at the time of the clinical encounter. For that purpose, we performed a detailed review of the electronic medical record by searching for the term “aspiration” in physician documentation (progress notes and discharge documentation), as well as in radiology reports of available chest imaging studies. We also recorded clinical microbiological results from respiratory specimen and blood cultures, as obtained by the treating physicians. We considered microbiologic cultures as positive when pathogenic bacterial species were isolated and reported by the clinical microbiology for clinical specimens obtained within 48 h of research samples acquisition, i.e. not normal respiratory flora in respiratory cultures and not skin contaminants in blood cultures.
We systematically reviewed administered antibiotic therapies since hospital admission and recorded the antibiotic exposure for each subject according to the following three metrics.
-
1.
Anaerobic coverage (yes/no): whether antibiotics with anaerobic coverage were given on the day of sampling.
-
2.
The Antibiotic Exposure score by Zhao et al.38: a numerical scale with antibiotic weighting based on dosing duration, timing of administration relative to sample collection and antibiotic type and route of administration. We utilized the convex increasing weighting scheme and modeled the antibiotic exposure from hospital admission until day of sampling.
-
3.
The Narrow Antibiotic Treatment (NAT) score developed for community-acquired pneumonia treatment studies.39,40 We calculated the daily NAT score from −5 days from sampling to post 10 days after sampling on day 1.
Research sample collection
Within the first 48 h of intubation (baseline time-point), we collected a posterior oropharyngeal (oral) swab (OPS) via gentle swabbing the posterior oropharynx next to the endotracheal tube with a cotton tip swab for 5 s, and an endotracheal aspirate (ETA) via suctioning secretions from the endotracheal tube with the in-line suction catheter and without breaking seal in the ventilatory circuit.1,4
In ICUs at our institution, as part of a ventilator-associated pneumonia prevention bundle, a standard protocol has been implemented involving chlorhexidine oral rinses twice daily. Therefore, all subjects enrolled in our study underwent OPS sampling were exposed to the chlorhexidine oral rinse protocol. For ETA sampling, no clinical protocol was in place to interfere with research sample acquisition, which involved instillation of 5 mL of sterile 0.9% saline and retrieval of airway secretions in a closed specimen system via advancement of the in-line suction catheter.
We also collected simultaneous blood samples for centrifugation and separation of plasma, which was stored in −80C until conduct of experiments. For patients who remained intubated in the ICU, we collected follow-up samples at a middle time-point (days 3–6) and a late follow-up interval (days 7–11 post-intubation).
For Healthy Controls, an oral wash and BAL sample were collected with a standardized protocol.37Subjects were asked to fast and refrain from smoking for at least 12hrs before sample collection. Oral washes were performed by having participants gargle with 10 mL sterile 0.9% saline immediately before bronchoscopy. BAL was performed according to standardized procedures developed to minimize oral contamination. Participants gargled with an antiseptic mouthwash (Listerine) immediately before topical anesthesia. The bronchoscope was then inserted through the mouth and advanced to a wedge position quickly and without use of suction. BAL was performed in the right middle lobe or lingula up to a maximum of 300 mL 0.9% saline.
Laboratory analyses
Microbiome assays
From OPS and ETAs, we extracted genomic DNA and performed quantitative PCR (qPCR) of the V3-V4 region of the 16S rRNA gene to obtain the number of gene copies per sample, as a surrogate for bacterial load. From a separate aliquot of extracted DNA from OPS, ETA, oral washes and BAL samples, we performed amplicon sequencing for bacterial DNA (16S-Seq of the V4 hypervariable region) on the Illumina MiSeq platform.15,29 We used extensive experimental negative controls in all processing steps to rule out contamination, as well as mock microbial community positive controls (Zymo) to ensure target amplification success. We processed derived 16S sequences with a custom Mothur-based pipeline and performed analyses at genus level. From a random subset of 64 available ETA samples, we performed metagenomic Nanopore sequencing (following human DNA depletion) with a rapid PCR barcoding kit (SQK-RPB004) on the MinION device (Oxford Nanopore Technologies-ONT, Oxford, UK) for 5 h.16,17 We analyzed microbial metagenomic sequences with the EPI2ME platform (ONT) and the “What’s In My Pot” [WIMP] workflow to quantify abundance of microbial species.41 WIMP is an EPI2ME workflow for taxonomic classification of nanopore sequencing reads, classifying each sequence present in FASTQ files. We filtered FASTQ files with a mean quality (q-score) below a minimum threshold of 7. For reads above the quality threshold, WIMP uses the Centrifuge classification engine to assign each read to a taxon in the NCBI taxonomy.42 The Centrifuge classification results are then filtered and aggregated to calculate and report counts of reads at the species rank. For reads without a reliable assignment at the species rank, higher ranks of the taxonomy tree are used for the assignment. If no placement is reliable enough (below a scoring threshold), the sequence is labeled as "Unclassified".
Host-response assays
We measured 13 plasma biomarkers of tissue injury and inflammation with custom Luminex multi-analyte panels and ELISA from plasma samples stored in EDTA tubes in −80C. Specifically, we used a 10-plex Luminex panel (R&D Systems, Minneapolis, MI, United States) with 1:2 plasma dilution to measure interleukin(IL)-6, IL-8, IL-10, soluble tumor necrosis factor receptor 1 (sTNFR1), suppressor of tumorigenicity-2 (ST2), fractalkine, soluble receptor of advanced glycation end-products (sRAGE), angiopoietin-2, procalcitonin and pentraxin-3.43 We used a 2-plex Luminex panel (R&D Systems, Minneapolis, MI, United States) with 1:100 plasma dilution to measure lipopolysaccharide binding protein (LBP) and soluble cluster of differentiation 14 (sCD14). We used an ELISA panel to measure surfactant protein D (SPD) according to the manufacturer’s instructions (Biovendor, LLC, Asheville, NC, United States).23
Quantification and statistical analysis
We performed non-parametric comparisons for continuous (described as median and interquartile range – IQR) and categorical variables between clinical groups (Wilcoxon and Fisher’s exact tests, respectively). For microbial community profiling, we included samples that produced >300 high quality microbial reads for both 16S-Seq and Nanopore sequencing. 16S-Seq data from 47% baseline OPS and ETA had been previously used in a different analysis of patients with acute respiratory failure.29 We performed alpha diversity (Shannon index) calculations for each available sample, and then conducted between group comparisons of alpha diversity with non-parametric tests to draw inferences on systematic differences of alpha diversity between groups as a measure of relative community fitness.44 We conducted beta diversity analyses (Manhattan distances, analyzed via permutation analysis of variance and visualized via principal coordinates analyses) with the R vegan and mia packages.45 We examined for differentially abundant taxa between groups following centered log-ratio (CLR) transformations with the limma package to fit weighted linear regression models, perform tests based on an empirical Bayes moderated t-statistic and obtain False Discovery Ratio corrected pvalues.
We then examined the discovered bacterial taxa at genus level (n = 136) and classified them by two different classification schemes with clinical relevance:
A. By oxygen requirements for bacterial metabolism.
-
1.
Obligate aerobes (referred to throughout as aerobes) [14.0% of total 136 taxa]: bacteria that require oxygen to grow and survive, as they use oxygen as final electron acceptor in their respiratory chain.
-
2.
Obligative anaerobes (referred to throughout as anaerobes) [41.2%]: bacteria that are unable to grow in the presence of oxygen, as they are unable to use oxygen as a final electron acceptor and are killed in the presence of oxygen.
-
3.
Facultative anaerobes [17.6%]: bacteria that can grow in the presence or absence of oxygen. They are able to use both aerobic and anaerobic respiration, depending on the availability of oxygen in their environment, switching from aerobic to anaerobic metabolism.
-
4.
Microaerophiles [2.2%]: bacteria that require a low level of oxygen to grow and survive, as they are able to grow at oxygen concentrations lower than those required by obligate aerobes but higher than those tolerated by obligate anaerobes.
-
5.
Variable [14.7%]: genera that included both aerobes and anaerobes and could not be classified further with confidence.
-
6.
Unclassifiable [9.6%]: taxa that were not classified at the genus or family level with confidence to allow assessment of their metabolic needs.
B. By pathogenicity for LRT infections:
-
1.
Common respiratory pathogens [5.1% of total 136 taxa]: bacteria considered to be typical pathogens when isolated in LRT microbiologic cultures.
-
2.
Oral-origin commensal bacteria [10.3%]: bacterial taxa that have been characterized as typical members of the lung microbiome in health and originate from the oral cavity.
-
3.
Other [84.6%]: taxa with unclear clinical significance that do not fall into categories B1 or B2 above.
We performed these classifications following extensive literature review for each taxon, supplemented by chatGPT searches. For the case of Steptococcus, which was the most abundant taxon in both URT and LRT specimens, our literature searches concluded that this genus contains both obligate and facultative anaerobes, and would then have to be classified as variable. We reviewed our metagenomic data from Nanopore sequencing and identified that more than 90% of the reads assigned to Streptococcus spp. belonged to organisms that were facultative anaerobes. Therefore, to avoid any bias against the null (by erroneously assigning Steptococcus taxa as variable and artificially decreasing the number of bacterial reads belonging to the facultative anaerobe category), we operationally analyzed all Steptococcus reads as facultative anaerobes.
All genus-level classifications by oxygen requirement and plausible pathogenicity are provided in the Supplement (Datas S3 and S4, related to STAR Methods).
To agnostically examine our samples for distinct clusters of microbial composition (“metacommunities”), we applied unsupervised Dirichlet multinomial models (DMMs) with Laplace approximations46 to define the optimal number of clusters in our dataset, and then examined for associations with clinical parameters and outcomes. We followed patients prospectively and constructed Kaplan-Meier curves and Cox-proportional hazard models for 60-day survival, adjusted for the predictors of age and sex, as well as plausible confounders of microbiome associations diagnosis based on our findings (history of COPD, chronic oxygen use and antibiotic exposure score by the day of sampling). To examine for the impact of mechanical ventilation and antibiotics pressure on longitudinal microbiota profiles, we constructed mixed regression models with random patient intercepts and adjusted for the number of days post-intubation that each sample was taken (as a proxy for the exposure to the hyperoxic environment of the ventilator) and the antibiotic exposure score (convex model) by the day of sampling. We performed all analyses in R v.4.2.0.47
Power analysis
We did not perform a priori sample size calculations as the primary objective of this study was descriptive and we could not have reliable estimates of microbial composition differences between MAsP and NonMAsP subjects. However, we note that our study population represents the largest sample size analyzed to date on this topic. In post-hoc power analysis and given the observed distributions in healthy controls, MAsP and NonMAsP subjects, we conclude that our sample size of pneumonia cases provided >90% statistical power to detect a 20% relative abundance difference of obligate anaerobes between clinical groups.
Acknowledgments
The authors thank all members of the research team of the Acute Lung Injury Registry at the University of Pittsburgh, the medical and nursing staff in the ICUs at the University of Pittsburgh Medical Center, and all patients and their families for participating in this research project.
Funding Sources:National Institutes of Health (P01 HL114453 [BJM], K23 HL139987 [GDK], R03 HL162655[GDK], R01 HL142093-01 [RMB], R01HL159805 [PVB], R01AA028436 [PVB]); Veterans Affairs (IK2BX004886 [WB]).
Role of funding source: The funders of the study had no role in study design, data collection, data analysis, data interpretation, writing of the report or decision to submit the manuscript for publication.
Author contributions
Conceptualization, G.D.K. and V.D.N.; Methodology, G.D.K., K.L., B.M., K.S., and P.V.B.; Formal Analysis, G.D.K., V.D.N., K.L., and K.S.; Investigation, G.D.K., V.D.N., K.S., N.A., C.S., F.S., H.Y., A.F., K.L., X.W., S.Q., W.B., H.G., Y.Z., J.V., A.A., J.A.E., R.M.B., J.S.L., B.M., P.V.B., A.M., and B.J.M.; Resources, G.D.K., B.J.M., and A.M.; Data Curation, G.D.K., V.D.N., K.S., and C.S.; Writing – Original Draft, G.D.K. and V.D.N.; Writing – Review and Editing, G.D.K, V.D.N., K.S., N.A., C.S., F.S., H.Y., A.F., K.L., X.W., S.Q., W.B., H.G., Y.Z., J.V., A.A., J.A.E., R.B.M., J.S.L., B.M., P.V.B., A.M., and B.J.M.; Visualization, G.D.K., V.D.N., and K.S.; Supervision, G.D.K.; Project Administration, G.D.K., B.J.M., and A.M.; Funding Acquisition, G.D.K., B.J.M., R.M.B., A.M., W.B., and P.V.B.
Declaration of interests
G.D.K. has received research funding from Karius, Inc and Pfizer, Inc. A.M. has received research funding from Pfizer, Inc. B.J.M. has received research funding from Bayer Pharmaceuticals, Inc., consulting fees from Boehringer Ingelheim, BioAegis and Synairgen, and payments from expert testimony from VeraMedica, LLC. The other authors have no conflicts of interest to disclose.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: May 6, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106832.
Supplemental information
We examined for differentially abundant taxa between groups following centered log-ratio (CLR) transformations with the limma package to fit weighted linear regression models, perform tests based on an empirical Bayes moderated t-statistic and obtain False Discovery Ratio corrected p-values related to Figure 1
We examined for differentially abundant taxa between groups following centered log-ratio (CLR) transformations with the limma package to fit weighted linear regression models, perform tests based on an empirical Bayes moderated t-statistic and obtain False Discovery Ratio corrected p-values related to Figure 1
We classified the 16S-Seq identified genera into categories defined by oxygen requirements (aerobes, anaerobes, facultative anaerobes, microaerophiles, variable) and membership in the typical lung microbiome in health (i.e., oral-origin commensals) versus organisms typically involved in LRT infections (i.e., common respiratory pathogens) related to STAR Methods
We classified the 16S-Seq identified genera into categories defined by oxygen requirements (aerobes, anaerobes, facultative anaerobes, microaerophiles, variable) and membership in the typical lung microbiome in health (i.e., oral-origin commensals) versus organisms typically involved in LRT infections (i.e., common respiratory pathogens) related to STAR Methods
Data and code availability
-
•
Sequencing data collected for the study are publicly available as of the date of publication through the Sequencing Resource Archive, through the Accession numbers PRJNA595346 for the ICU patients and PRJNA940725 for the Healthy Controls.
-
•
Primary code and de-identified data for replication of analyses are available on the github repository (https://github.com/MicrobiomeALIR/AspirationPneumonia). DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contactupon request.
References
- 1.Mac Aogáin M., Baker J.M., Dickson R.P. On bugs and blowholes: why is aspiration the rule, not the exception? Am. J. Respir. Crit. Care Med. 2021;203:1049–1051. doi: 10.1164/rccm.202011-4257ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butler S.G., Stuart A., Markley L., Feng X., Kritchevsky S.B. Aspiration as a function of age, sex, liquid type, bolus volume, and bolus delivery across the healthy adult life span. Ann. Otol. Rhinol. Laryngol. 2018;127:21–32. doi: 10.1177/0003489417742161. [DOI] [PubMed] [Google Scholar]
- 3.Mandell L.A., Niederman M.S. Aspiration pneumonia. N. Engl. J. Med. 2019;380:651–663. doi: 10.1056/NEJMra1714562. [DOI] [PubMed] [Google Scholar]
- 4.Marin-Corral J., Pascual-Guardia S., Amati F., Aliberti S., Masclans J.R., Soni N., Rodriguez A., Sibila O., Sanz F., Sotgiu G., et al. Aspiration risk factors, microbiology, and empiric antibiotics for patients hospitalized with community-acquired pneumonia. Chest. 2021;159:58–72. doi: 10.1016/j.chest.2020.06.079. [DOI] [PubMed] [Google Scholar]
- 5.Cesar L., Gonzalez C., Calia F.M. Bacteriologic flora of aspiration-induced pulmonary infections. Arch. Intern. Med. 1975;135:711–714. doi: 10.1001/archinte.135.5.711. [DOI] [PubMed] [Google Scholar]
- 6.Lauterbach E., Voss F., Gerigk R., Lauterbach M. Bacteriology of aspiration pneumonia in patients with acute coma. Intern. Emerg. Med. 2014;9:879–885. doi: 10.1007/s11739-014-1120-5. [DOI] [PubMed] [Google Scholar]
- 7.Akata K., Noguchi S., Kawanami T., Hata R., Naito K., Mukae H., Yatera K. [microbiology of aspiration pneumonia] J. UOEH. 2019;41:185–192. doi: 10.7888/juoeh.41.185. [DOI] [PubMed] [Google Scholar]
- 8.Akata K., Yatera K., Yamasaki K., Kawanami T., Naito K., Noguchi S., Fukuda K., Ishimoto H., Taniguchi H., Mukae H. The significance of oral streptococci in patients with pneumonia with risk factors for aspiration: the bacterial floral analysis of 16S ribosomal RNA gene using bronchoalveolar lavage fluid. BMC Pulm. Med. 2016;16:79. doi: 10.1186/s12890-016-0235-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aoki K., Ishii Y., Tateda K. Detection of associated bacteria in aspiration pneumonia and lung abscesses using partial 16S rRNA gene amplicon sequencing. Anaerobe. 2021;69 doi: 10.1016/j.anaerobe.2021.102325. [DOI] [PubMed] [Google Scholar]
- 10.Otsuji K., Fukuda K., Ogawa M., Fujino Y., Kamochi M., Saito M. Dynamics of microbiota during mechanical ventilation in aspiration pneumonia. BMC Pulm. Med. 2019;19:260. doi: 10.1186/s12890-019-1021-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Metlay J.P., Waterer G.W., Long A.C., Anzueto A., Brozek J., Cr K., Cooley L.A., Dean N.C., Fine M.J., Flanders S.A., et al. Diagnosis and treatment of adults with community-acquired pneumonia. Am. J. Respir. Crit. Care Med. 2019;200:e45–e67. doi: 10.1164/rccm.201908-1581ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lanspa M.J., Peyrani P., Wiemken T., Wilson E.L., Ramirez J.A., Dean N.C. Characteristics associated with clinician diagnosis of aspiration pneumonia: a descriptive study of afflicted patients and their outcomes. J. Hosp. Med. 2015;10:90–96. doi: 10.1002/jhm.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chanderraj R., Baker J.M., Kay S.G., Brown C.A., Hinkle K.J., Fergle D.J., McDonald R.A., Falkowski N.R., Metcalf J.D., Kaye K.S., et al. In critically ill patients, anti-anaerobic antibiotics increase risk of adverse clinical outcomes. Eur. Respir. J. 2023;61 doi: 10.1183/13993003.00910-2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitsios G.D., McVerry B.J. Host-microbiome interactions in the subglottic space. Bacteria ante portas. Am. J. Respir. Crit. Care Med. 2018;198:294–297. doi: 10.1164/rccm.201802-0276ED. [DOI] [PubMed] [Google Scholar]
- 15.Kitsios G.D., Fitch A., Manatakis D.V., Rapport S.F., Li K., Qin S., Huwe J., Zhang Y., Doi Y., Evankovich J., et al. Respiratory microbiome profiling for etiologic diagnosis of pneumonia in mechanically ventilated patients. Front. Microbiol. 2018;9:1413. doi: 10.3389/fmicb.2018.01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang L., Haidar G., Zia H., Nettles R., Qin S., Wang X., Shah F., Rapport S.F., Charalampous T., Methé B., et al. Metagenomic identification of severe pneumonia pathogens in mechanically-ventilated patients: a feasibility and clinical validity study. Respir. Res. 2019;20:265. doi: 10.1186/s12931-019-1218-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Charalampous T., Kay G.L., Richardson H., Aydin A., Baldan R., Jeanes C., Rae D., Grundy S., Turner D.J., Wain J., et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat. Biotechnol. 2019;37:783–792. doi: 10.1038/s41587-019-0156-5. [DOI] [PubMed] [Google Scholar]
- 18.Dickson R.P., Erb-Downward J.R., Freeman C.M., McCloskey L., Falkowski N.R., Huffnagle G.B., Curtis J.L. Bacterial topography of the healthy human lower respiratory tract. mBio. 2017;8 doi: 10.1128/mBio.02287-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Musher D.M., Jesudasen S.S., Barwatt J.W., Cohen D.N., Moss B.J., Rodriguez-Barradas M.C. Normal respiratory flora as a cause of community-acquired pneumonia. Open Forum Infect. Dis. 2020;7:ofaa307. doi: 10.1093/ofid/ofaa307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu B.G., Sulaiman I., Tsay J.-C.J., Perez L., Franca B., Li Y., Wang J., Gonzalez A.N., El-Ashmawy M., Carpenito J., et al. Episodic aspiration with oral commensals induces a MyD88-dependent, pulmonary T-helper cell type 17 response that mitigates susceptibility to Streptococcus pneumoniae. Am. J. Respir. Crit. Care Med. 2021;203:1099–1111. doi: 10.1164/rccm.202005-1596OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitsios G.D. Translating lung microbiome profiles into the next-generation diagnostic gold standard for pneumonia: a clinical investigator’s perspective. mSystems. 2018;3 doi: 10.1128/mSystems.00153-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langelier C., Kalantar K.L., Moazed F., Wilson M.R., Crawford E.D., Deiss T., Belzer A., Bolourchi S., Caldera S., Fung M., et al. Integrating host response and unbiased microbe detection for lower respiratory tract infection diagnosis in critically ill adults. Proc. Natl. Acad. Sci. USA. 2018;115:E12353–E12362. doi: 10.1073/pnas.1809700115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varon J., Arciniegas Rubio A., Amador-Munoz D., Corcoran A., DeCorte J.A., Isabelle C., Pinilla Vera M., Walker K., Brown L., Cernadas M., et al. Surfactant protein D influences mortality during abdominal sepsis by facilitating Escherichia coli colonization in the gut. Crit. Care Explor. 2022;4:e0699. doi: 10.1097/CCE.0000000000000699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ware L.B., Koyama T., Zhao Z., Janz D.R., Wickersham N., Bernard G.R., May A.K., Calfee C.S., Matthay M.A. Biomarkers of lung epithelial injury and inflammation distinguish severe sepsis patients with acute respiratory distress syndrome. Crit. Care. 2013;17:R253. doi: 10.1186/cc13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anas A., van der Poll T., de Vos A.F. Role of CD14 in lung inflammation and infection. Crit. Care. 2010;14:209. doi: 10.1186/cc8850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mabrey F.L., Morrell E.D., Bhatraju P.K., Sathe N.A., Sakr S.S., Sahi S.K., West T.E., Mikacenic C., Wurfel M.M. Plasma soluble CD14 subtype levels are associated with clinical outcomes in critically ill subjects with coronavirus disease 2019. Crit. Care Explor. 2021;3 doi: 10.1097/CCE.0000000000000591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dickson R.P., Schultz M.J., van der Poll T., Schouten L.R., Falkowski N.R., Luth J.E., Sjoding M.W., Brown C.A., Chanderraj R., Huffnagle G.B., et al. Lung microbiota predict clinical outcomes in critically ill patients. Am. J. Respir. Crit. Care Med. 2020;201:555–563. doi: 10.1164/rccm.201907-1487OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fenn D., Abdel-Aziz M.I., van Oort P.M.P., Brinkman P., Ahmed W.M., Felton T., Artigas A., Póvoa P., Martin-Loeches I., Schultz M.J., et al. Composition and diversity analysis of the lung microbiome in patients with suspected ventilator-associated pneumonia. Crit. Care. 2022;26:203. doi: 10.1186/s13054-022-04068-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kitsios G.D., Yang H., Yang L., Qin S., Fitch A., Wang X.H., Fair K., Evankovich J., Bain W., Shah F., et al. Respiratory tract dysbiosis is associated with worse outcomes in mechanically ventilated patients. Am. J. Respir. Crit. Care Med. 2020;202:1666–1677. doi: 10.1164/rccm.201912-2441OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Charalampous T., Alcolea-Medina A., Snell L.B., Williams T.G.S., Batra R., Alder C., Telatin A., Camporota L., Meadows C.I.S., Wyncoll D., et al. Evaluating the potential for respiratory metagenomics to improve treatment of secondary infection and detection of nosocomial transmission on expanded COVID-19 intensive care units. Genome Med. 2021;13:182. doi: 10.1186/s13073-021-00991-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalil A.C., Metersky M.L., Klompas M., Muscedere J., Sweeney D.A., Palmer L.B., Napolitano L.M., O’Grady N.P., Bartlett J.G., Carratalà J., et al. Management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the infectious diseases society of America and the American thoracic society. Clin. Infect. Dis. 2016;63:e61–e111. doi: 10.1093/cid/ciw353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delahaye C., Nicolas J. Sequencing DNA with nanopores: troubles and biases. PLoS One. 2021;16 doi: 10.1371/journal.pone.0257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drohan, C.M., Nouraie, S.M., Bain, W., Shah, F.A., Evankovich, J., Zhang, Y., Morris, A., McVerry, B.J., and Kitsios, G.D. (2020). Host-response subphenotypic classification with A parsimonious model offers prognostic information in patients with acute respiratory failure: a prospective cohort study. 10.21203/rs.3.rs-57907/v1. [DOI]
- 34.Yang H., Haidar G., Al-Yousif N.S., Zia H., Kotok D., Ahmed A.A., Blair L., Dalai S., Bercovici S., Ho C., et al. Circulating microbial cell-free DNA is associated with inflammatory host-responses in severe pneumonia. Thorax. 2021;76:1231–1235. doi: 10.1136/thoraxjnl-2020-216013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gong M.N., Thompson B.T., Williams P., Pothier L., Boyce P.D., Christiani D.C. Clinical predictors of and mortality in acute respiratory distress syndrome: potential role of red cell transfusion. Crit. Care Med. 2005;33:1191–1198. doi: 10.1097/01.ccm.0000165566.82925.14. [DOI] [PubMed] [Google Scholar]
- 36.Bajwa E.K., Malhotra C.K., Thompson B.T., Christiani D.C., Gong M.N. Statin therapy as prevention against development of acute respiratory distress syndrome: an observational study. Crit. Care Med. 2012;40:1470–1477. doi: 10.1097/CCM.0b013e3182416d7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morris A., Beck J.M., Schloss P.D., Campbell T.B., Cr K., Curtis J.L., Flores S.C., Fontenot A.P., Ghedin E., Huang L., et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am. J. Respir. Crit. Care Med. 2013;187:1067–1075. doi: 10.1164/rccm.201210-1913OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao J., Murray S., Lipuma J.J. Modeling the impact of antibiotic exposure on human microbiota. Sci. Rep. 2014;4:4345. doi: 10.1038/srep04345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang A.A., Pickens C.O., He H., Scholtens D.M., Pawlowski A.E., Singer B.D., Kruser J.M., Walter J.M., Donnelly H., Wunderink R.G. B28. HOST AND MICROBIAL CLINICAL STUDIES IN LUNG INFECTIONS AND LUNG DISEASES. American Thoracic Society); 2020. The narrow-spectrum antibiotic treatment score: a novel quantitative tool for assessing broad- and narrow-spectrum antibiotic use in severe community-acquired pneumonia; p. A2929. [DOI] [Google Scholar]
- 40.Pickens C.O., Gao C.A., Cuttica M.J., Smith S.B., Pesce L.L., Grant R.A., Kang M., Morales-Nebreda L., Bavishi A.A., Arnold J.M., et al. Bacterial superinfection pneumonia in patients mechanically ventilated for COVID-19 pneumonia. Am. J. Respir. Crit. Care Med. 2021;204:921–932. doi: 10.1164/rccm.202106-1354OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Juul S., Izquierdo F., Hurst A., Dai X., Wright A., Kulesha E., Pettett R., Turner D.J. What’s in my pot? Real-time species identification on the MinION. bioRxiv. 2015 doi: 10.1101/030742. Preprint at. [DOI] [Google Scholar]
- 42.Kim D., Song L., Breitwieser F.P., Salzberg S.L. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016;26:1721–1729. doi: 10.1101/gr.210641.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kitsios G.D., Yang L., Manatakis D.V., Nouraie M., Evankovich J., Bain W., Dunlap D.G., Shah F., Barbash I.J., Rapport S.F., et al. Host-response subphenotypes offer prognostic enrichment in patients with or at risk for acute respiratory distress syndrome. Crit. Care Med. 2019;47:1724–1734. doi: 10.1097/CCM.0000000000004018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitsios G.D., Morowitz M.J., Dickson R.P., Huffnagle G.B., McVerry B.J., Morris A. Dysbiosis in the intensive care unit: microbiome science coming to the bedside. J. Crit. Care. 2017;38:84–91. doi: 10.1016/j.jcrc.2016.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huber W., Carey V.J., Gentleman R., Anders S., Carlson M., Carvalho B.S., Bravo H.C., Davis S., Gatto L., Girke T., et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods. 2015;12:115–121. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holmes I., Harris K., Quince C. Dirichlet multinomial mixtures: generative models for microbial metagenomics. PLoS One. 2012;7 doi: 10.1371/journal.pone.0030126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.R Foundation for Statistical Computing . CRAN; 2016. R: A Language and Environment for Statistical Computing. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
We examined for differentially abundant taxa between groups following centered log-ratio (CLR) transformations with the limma package to fit weighted linear regression models, perform tests based on an empirical Bayes moderated t-statistic and obtain False Discovery Ratio corrected p-values related to Figure 1
We examined for differentially abundant taxa between groups following centered log-ratio (CLR) transformations with the limma package to fit weighted linear regression models, perform tests based on an empirical Bayes moderated t-statistic and obtain False Discovery Ratio corrected p-values related to Figure 1
We classified the 16S-Seq identified genera into categories defined by oxygen requirements (aerobes, anaerobes, facultative anaerobes, microaerophiles, variable) and membership in the typical lung microbiome in health (i.e., oral-origin commensals) versus organisms typically involved in LRT infections (i.e., common respiratory pathogens) related to STAR Methods
We classified the 16S-Seq identified genera into categories defined by oxygen requirements (aerobes, anaerobes, facultative anaerobes, microaerophiles, variable) and membership in the typical lung microbiome in health (i.e., oral-origin commensals) versus organisms typically involved in LRT infections (i.e., common respiratory pathogens) related to STAR Methods
Data Availability Statement
-
•
Sequencing data collected for the study are publicly available as of the date of publication through the Sequencing Resource Archive, through the Accession numbers PRJNA595346 for the ICU patients and PRJNA940725 for the Healthy Controls.
-
•
Primary code and de-identified data for replication of analyses are available on the github repository (https://github.com/MicrobiomeALIR/AspirationPneumonia). DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contactupon request.