

Abstract
Puromycin derivatives containing an emissive thieno[3,4-d]-pyrimidine core, modified with azetidine and 3,3-difluoroazetidine as Me2N surrogates, exhibit translation inhibition and bactericidal activity similar to the natural antibiotic. The analogues are capable of cellular puromycylation of nascent peptides, generating emissive products without any follow-up chemistry. The 3,3-difluoroazetidine-containing analogue is shown to fluorescently label newly translated peptides and be visualized in both live and fixed HEK293T cells and rat hippocampal neurons.
Keywords: fluorescence, antibiotics, labelling, modified nucleosides, heterocycles
Graphical Abstract
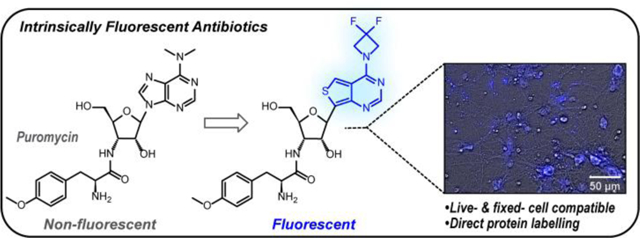
Inherently emissive puromycin analogues can fluorescently tag newly synthesized proteins in live-cells without the need for further chemical treatments.
Translation is a complex and tightly regulated process.[1–3] Diverse techniques have therefore been developed to monitor cellular protein synthesis in a spatiotemporal manner,[4] including metabolic labeling through the incorporation of modified amino acids (e.g., azidohomoalanine)[5] and translating ribosome affinity purification (TRAP) utilizing stalled ribosomes.[6,7] However, one of the simplest and most utilized experimental techniques relies on puromycin (1), a naturally occurring aminonucleoside antibiotic isolated from Streptomyces alboniger (Figure 1).[8,9] Acting as a charged tRNA mimic, it incorporates into the C-terminus of elongating nascent peptide chains, thus preventing elongation and prematurely terminating translation.[10–12] Puromycin is used as a selection marker for bacterial cell lines genetically engineered to express resistance, and as a probe for protein synthesis with purified ribosomes and cell-free translation kits as well as with cultured cells and animals.[13] Puromycin treatment also facilitates visualization of newly synthesized proteins following immunostaining of puromycin-terminated peptides.[14]
Figure 1.

Chemical structure of puromycin (1) and puromycin derivatives ThPuromycin(2), AzThPuromycin (3), and F2AzThPuromycin (4).
A diverse toolbox of puromycin-based reagents with supplementary functionalities has been developed, employing biotin for affinity purification or fluorophores for fluorescent microscopy applications.[13] Such analogues have been crucial for deconvoluting translation-dependent processes. Currently available analogues including “clickable” derivatives such as O-propargyl puromycin, by their nature, necessitate subsequent reactions that frequently preclude real-time and/or live-cell experiments. They also do not facilitate continual reagent administration and time-dependent puromycylation without altering the system with additional reagents and kinetic biases.[13,15] We thus aimed to develop intrinsically emissive puromycin surrogates. We opted to replace its nucleobase with minimally perturbing emissive adenine surrogates and create microenvironmentally responsive antibiotics for potential real-time assessment of cellular protein expression.
Puromycin’s N6,N6 dimethylated adenine is practically non emissive.[16,17] Fluorescent, isomorphic and isofunctional purine and pyrimidine analogues derived from thieno[3,4-d]-pyrimidine and isothiazolo[4,3-d]pyrimidine heterocyclic nuclei have been developed by replacing the imidazole ring with 5-membered aromatic heterocycles.[18–22] Both emissive RNA alphabets exhibit significantly improved photophysical properties compared to their native counterparts as evidenced by their employment for analyzing enzymatic transformations and protein/nucleic acids interactions.[23–28] We have also explored the effect of exocyclic amine dialkylation on the fluorescence of both adenosine analogues.[29] Intriguingly, replacing the N6,N6-dimethylamino with an azetidine dramatically increased the emission quantum yield of these purine mimics, particularly when strong electron-withdrawing groups were present at the 4-membered ring’s 3-position [e.g., 𝜙(H2O)=0.64 for the 3,3-difluoroazetidine].[29] Here we investigate inherently emissive puromycin analogues employing the thieno[3,4-d]-pyrimidine core (Figure 1) and report their synthesis and photophysical properties. We demonstrate their antibacterial features and cell entry as well as their cellular visualization by fluorescence microscopy, either on their own or via antibody staining of puromycylated translation products.
The synthesis of the puromycin analogues, utilizing nucleobase 8, ribonolactone 13, and the tyrosine analogue Fmoc-O-methyl-l-tyrosine, is shown in Scheme 1. Cyclization of 4-aminothiophene-3-carboxylate hydrochloride 5 followed by thionation with phosphorous pentasulfide produced compound 6, as reported.[18,29] Methylation of 6 with methyl iodide selectively produced 7, which was brominated with 1,3-Dibromo-5,5-dimethylhydantoin (DBDMH) to give 8. Subsequently, azidolactone 13 was synthesized from d-xylose 9, which was protected at the 1’ and 2’ positions via an isopropylidene and at the 5’ position with a benzyl group to give 10. Activation of the 3’-OH with triflic anhydride and subsequent azide displacement yielded the desired 3’ α-azido derivative 11 in 53% yield. Isopropylidene deprotection with acidic methanol and protection of the newly freed 2’-OH produced intermediate 12. Hydrolysis of the anomeric acetal with hydrochloric acid yielded the hemi-acetal, which was then oxidized to the desired lactone 13 with pyridinium chlorochromate. Lithium halogen exchange utilizing nucleobase 8 and addition to lactone 13 produced hemi-ketal 14, which was subjected to a BF3·Et2O mediated triethylsilane reduction to yield 15. The stereochemistry of this key precursor was confirmed by X-ray crystallography (Scheme 1, Table S1). Displacing the thiomethyl group of 15 with secondary amines in DMSO provided analogues 16, 17 and 18. Subsequent reduction of the 3’ azide with trimethylphosphine in THF followed by peptide coupling with Fmoc protected O-methyl-l-tyrosine yielded the fully protected antibiotic derivatives 19–21. Fmoc deprotection with piperidine in DMF followed by benzyl deprotection with boron trichloride produced the desired compounds 2–4, respectively, in good yield (Scheme 1).[31]
Scheme 1.

(a) Formamidine acetate, EtOH, reflux, ON, 78%. (b) i. P2S5, Pyridine, 110 °C, 2 h; ii. MeI, K2CO3, DMF, RT, 1 hr, 65%. (c) DBDMH, DMF, 0 °C, 2.5 h, 45%.(d) i. H2SO4, Acetone, RT, 2.5 hr; ii. TBAI, DIPEA, BnBr, 90 °C, 22h, 65%. (e) i. Tf2O, Pyridine, DCM, 0 °C, 2h; ii. NaN3, DMF, RT, 48 hr, 53%. (f) i. AcCl, MeOH, RT, ON; ii. NaH, BnBr, TBAI, DMF, 0 °C to RT, ON, 83%. (g) i. HCl, H2O, Dioxane, 60 °C, ON; ii. PCC, DCM, RT, ON, 78%. (h) n-BuLi, THF,−78°C, 27%. (i) BF3·OEt2, Et3SiH, DCM, −78°C to RT, 55%. (j) RH2Cl, DBU, DMSO, RT. (k) Fmoc-O-methyl-l-tyrosine, EDC, HOBt, (Me)3P, THF. (l) i. Piperidine, DMF, RT; ii. BCl3, DCM, HPLC. Bottom left: Crystal structure of 15.[30]
The absorption and emission spectra of 2, 3, and 4 are presented in figure 2 and their corresponding photophysical properties are summarized in Table 1 and Table S2.[32] All analogues exhibited absorbance maxima at approximately 354 nm and emission maxima between 417 and 424 nm. Notably, however, these puromycin analogues display emission quantum yields that are 104–105-fold higher than the corresponding adenine or N6,N6 dimethylated adenine.[16,17,33,34]
Figure 2.

Absorption (dashed line) and emission (solid line, excitation 350 nm) spectra of 2 (green), 3 (blue), and 4 (red) in PBS. The absorption and emission spectra were normalized to 0.1 at 350 nm.
Table 1.
Photophysical and Bactericidal properties of puromycin derivatives 2–4.
| Photophysical Properties |
Antibacterial Activity |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| (MIC) |
|||||||||
| Cpd. | Solvent | λabsa | λem (ϕ)a | Stokes Shifta |
S. aureus (29213)b |
B. cereus (14579) |
S. marcescens (8100) |
E. coli (51739) |
E. coli (25922) |
| 1 | PBS | 284 | n/a | n/a | 16 | 8 | 128 | 128 | 64 |
| 2 | PBS | 354 | 424 (0.004) | 4.67 | 16 | 8 | 64 | 64 | 32 |
| 3 | PBS | 354 | 417 (0.21) | 4.21 | 16 | 8 | 128 | 128 | 64 |
| 4 | PBS | 353 | 419 (0.31) | 4.44 | 32 | 16 | 128 | >128 | 64 |
λabs, λem, and Stokes shift are reported in nm, nm and 103 cm−1 respectively. Photophysical values reflect the average over three independent measurements.
MIC values are reported in γg/ml. ATCC designations for the respective bacterial strains are provided in parentheses.
To first establish the new derivatives as true functional puromycin surrogates, their antibacterial activity was evaluated against selected gram-positive and gram-negative bacteria. Minimal inhibitory concentration (MIC) values at which no metabolic activity was detected (MTT assay) were determined using the double dilution protocol (Table 1).[35] Puromycin is indiscriminate towards prokaryotic and eukaryotic cells, yet striking differences in bactericidal activity are evident between gram-positive and gram-negative bacteria (16 vs 128 μg/ml in S. aureus and S. marcescens, respectively). Overall, the puromycin analogues 2–4 displayed comparable antibiotic activity and selectivity trend, indicating that the thiopheno-pyrimidine heterocycle and the azetidine modification caused no major detrimental effect. Intriguingly, Thpuromycin 2 exhibited somewhat higher bactericidal activity in gram-negative bacteria compared to the native antibiotic. Slight decrease in potency was observed, however, upon introduction of the azetidine modification, which was further augmented by the difluoroazetidine modification in antibiotic 4.
To probe the potential utility of the new puromycin derivatives in eukaryotic systems, we examined the dose-dependent inhibition of GFP expression using a rabbit reticulocyte lysate translation system with a 5’-capped mRNA encoding the fluorescent protein.[36,37] The dose- and time-dependent inhibition of GFP translation by puromycin and its analogues monitoring the fluorescent protein production are shown in Figure 3. All derivatives terminate GFP expression at 8 μM, however, their IC50 values differed slightly, with puromycin being the most effective (< 1 μM), followed by antibiotic 3 (2 μM), antibiotic 2 (2.3 μM) and lastly 4 (2.8 μM) (Figures 3, S1). These results are in line with the MIC values measured for analogues 2–4, where 4 exhibited marginally lower activity.
Figure 3.

Dose-dependent inhibition of GFP expression with (top left) puromycin (1), (top right) thpuromycin (2), (bottom left) azetidine-modified Thpuromycin (3), and (bottom right) F2AzThpuromycin (4).
To explore the cellular uptake and potential for fluorescence imaging of the new derivatives, human embryonic kidney cells (HEK293T) were incubated with 10 μM of 1 and the fluorescent antibiotics for 0.5, 1 and 4 hr. The cells were visualized using fluorescence microscopy and total protein was assessed through western blotting (Figure 4a, Figure S2–S6[38]). As expected, puromycin exhibited no inherent fluorescence across all time points (Figure S3).[39] Similar observations were made for cells treated with 2 and azetidine-modified antibiotic 3 (Figure S4 and S5). Treatment with 4 yielded no discernable fluorescence after 30 min, but a clear signal was seen following 2 and 4 hr exposure (Figures 4a, S6), with diffused cytoplasmic fluorescence accompanied by intense punctate patterns (Figure 4a). We postulated these fluorescent regions were nucleoli, the progenitors of ribosome biogenesis, as has been previously proposed,[40] and recently observed in C. Elegans treated with click-ready O-propargyl puromycin (minimal colocalization with lysosomal LAMP1-GFP in transfected HEK293T cells excludes the lysosomes as the major punctate structures, see Figure S7, Table S3).[41] Such behavior may reflect trafficking or diffusion of puromycylated peptides into the nucleolus as observed,[41] as the patterns seen with treatment of F2AzThpuromcyin 4 are consistent with these previous observations.
Figure 4.

Live cell imaging of HEK293T cells (a) and primary rat hippocampal neurons (b) treated with 4 (10 μM). Brightfield channel (top), DAPI channel (middle, for compound visualization), and overlay (bottom).
Immunofluorescence was then employed to localize the puromycylated peptides in HEK293T cells. Following 0.5, 2 and 4h treatments of either puromycin or any of its analogues in the presence/absence of cycloheximide (CHX), the cells were fixed, permeabilized and treated with an anti-puromycin and anti-ribosomal (HPO-0100) antibodies,[42] along with the corresponding fluorescently tagged secondary antibodies (Goat Anti-Mouse AlexaFluor488 and Donkey Anti-Human Cy3, respectively). The anti-puromycin antibodies nicely bound peptides truncated by puromycin or compounds 2, 3, or 4, as revealed by the AlexaFluor-tagged secondary antibody (Figure 5a–d). No anti-puromycin immunofluorescence was detected in the blank (water treatment), indicating the incorporation of the antibiotic was necessary for visualization (Figure S8). Puromycin incorporated into peptides the fastest, with anti-puromycin immunofluorescence detected at 30 minutes and intensifying at 2 and 4 hours (Figure S9). Similar behavior was observed with the analogues 2–4, although their incorporation into cellular proteins appeared somewhat slower with little signal at 30 minutes (Figure S10–S12). Colocalization of anti-puromycin and anti-ribosome fluorescence (Figure 5a–d, Table S4, green and pink fluorescence, respectively) is evident after 2 hr of antibiotic treatment for all derivatives, although the anti-puromycin fluorescence appears more disperse after 4 hr (Figure S10–S12). Importantly, pretreatment with 50 μg/ml CHX (a translation inhibitor interfering with translocation[43]) resulted in decreased anti-puromycin fluorescence across all time points for every antibiotic tested without any effect on anti-ribosome immunostaining (Figure S9–S12).
Figure 5.

Immunofluorescence staining of HEK293T cells treated with (a) 10 μM puromycin (1), (b) 10 μM thpuromycin (2), (c) 10 μM AzThpuromycin (3), and (d) 10 μM F2AzThpuromycin (4) for 2 hours. Image channels top to bottom: DRAQ5 (nuclear stain), anti-puromycin (primary) and GAMAF488 (secondary), HPO-0100 (anti-ribosome, primary) and DAH Cy3 (secondary), overlay. (e) Fixed primary rat hippocampal neuron images stained with DRAQ5 (top), compound 4 (second row), anti-puromycin (third row), and anti-ribosome (fourth row). Images are overlayed (bottom).
Studies of neuronal cell protein synthesis have utilized puromycin to tag nascent peptides formed as a response to extracellular signals.[13] Neuronal cultures were therefore incubated for 4 hr with 4, the most emissive analogue. Cells were then fixed, permeabilized, and subjected to anti-puromycin/anti-ribosome primary antibodies and DRAQ5 (a nuclear stain). Anti-puromycin antibody immunofluorescence indicated the presence of puromycylated peptides throughout the neuronal cell body, axons, and dendrites (Figure 5e, Table S5). Anti-ribosomal antibody immunofluorescence was observed in the main cell body, although weaker fluorescence was present in the thicker axons. Strikingly, peptides labelled with antibiotic 4 exhibited an inherent intense fluorescence within neurons that was not apparent in HEK293T cells. This result corroborates prior observations regarding rates of diffusion of puromycylated peptides, where higher rates were observed in mammalian cells compared to neuronal compartments.[2,6] Additionally, fluorescence from antibiotic 4 colocalized nicely with the anti-puromycin fluorescence and was primarily visible in the soma (Figure 5e). Fluorescence was also visible in certain axon branches (Figure 5e). Anti-puromycin immunofluorescence also visualized the antibiotic-terminated peptides in the smaller neuronal structures such as axons and dendrites. Lastly, live neurons were treated with F2AzThpuromycin 4 to ascertain its ability to fluorescently tag newly synthesized proteins in a non-cytotoxic manner (Figure 4b). Images following a 4 hr treatment with 4 suggest the puromycylated peptides are primarily visible in the soma, as observed with the fixed neurons, although fluorescence in other neuronal structures is also visible, highlighting the ability of compound 4 to tag newly synthesized peptides and be visualized in real-time in live neurons (Figure 4b).
In summary, the newly synthesized azetidino-modified thiopheno-based puromycin analogues function similarly to their native counterpart yet exhibit unique and useful photophysical features that facilitate live- and fixed-cell protein synthesis imaging experiments. In particular, antibiotic 4 produced intense fluorescence in HEK293T cells and neurons, paralleling the mechanism of action of native puromycin, as suggested by fluorescence colocalization of antibody staining and CHX inhibition.
Inherently emissive puromycin analogs are unique among reagents currently used for puromycin-directed experiments, as they do not require a follow up chemistry or antibody staining for visualization. As such, they lend themselves to experiments in live cells, where secondary conjugation reactions might be problematic, and fixation followed by antibody staining is not an option. While some commercial puromycin-based reagents, such as O-propargyl puromycin,[41] can also be used for live-cell labelling, we stress that the current landscape of puromycin-based fluorescent probes either suffer from significant bulk due to the conjugation of a large fluorophore to the puromycin skeleton (e.g. dC-puromycin), which impacts cellular uptake and activity, or require additional treatments for visualization, which limits their utility for real-time experiments. Inherently emissive puromycin analogs with similar structure and activity to the native antibiotics have the potential to excel in this space on their own, or in combination with existing reagents.
Understandably, as small fluorophores with a purine-like footprint, such analogs display limited brightness. Yet, analogs such as 4 were sufficiently bright to be imaged in cells and neurons. The data provided in the manuscript establish that the new synthetic analogs display very similar activity to that of the natural product, both in eukaryotic- and prokaryotic-based assays. We thus surmise that their cytotoxicity and optimal working concentrations will vary by cell type and outcome desired, as established for the natural antibiotic.[44–46] Beyond revealing new avenues for tolerable structural modifications of the puromycin nucleobase core, our observations indicate that live-cell fluorescence tagging of newly synthesized peptides can be performed in real-time in eukaryotic and neuronal cells without the need for extraneous treatments.
Supplementary Material
Acknowledgements
We thank the National Institutes of Health (via grant number GM139407), the UCSD Mass Spectrometry Facility, Anthony Mrse (UCSD NMR Facility), and Milan Gembicky (UCSD Crystallography Facility)
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Rattan SIS, in Principles of Medical Biology (Eds.: Bittar EE, Bittar N), Elsevier, 1995, pp. 247–263. [Google Scholar]
- [2].Hershey JWB, Sonenberg N, Mathews MB, Cold Spring Harb Perspect Biol 2012, 4, a011528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jackson RJ, Hellen CUT, Pestova TV, Nat Rev Mol Cell Biol 2010, 11, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dermit M, Dodel M, Mardakheh FK, Mol. Biosyst 2017, 13, 2477–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dieterich DC, Link AJ, Graumann J, Tirrell DA, Schuman EM, Proc. Nat. Acad. Sci. USA 2006, 103, 9482–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Heiman M, Kulicke R, Fenster RJ, Greengard P, Heintz N, Nat Protoc 2014, 9, 1282–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Thomas A, Lee P-J, Dalton JE, Nomie KJ, Stoica L, Costa-Mattioli M, Chang P, Nuzhdin S, Arbeitman MN, Dierick HA, PLOS ONE 2012, 7, e40276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sankaran L, Pogell BM, Antimicrob. Agents Chemother. 1975, 8, 721–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Porter J, Hewitt R, Hesseltine CW, Krupka G, Lowery JA, Wallace WS, Bohonos N, Williams JHH, Antibiot. Chemother. 1952, 2, 409–410. [PubMed] [Google Scholar]
- [10].Yarmolinsky MB, Haba GLDL, Proc. Natl. Acad. Sci. USA 1959, 45, 1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nathans D, Neidle A, Nature 1963, 197, 1076. [DOI] [PubMed] [Google Scholar]
- [12].Miyamoto-Sato E, Nemoto N, Kobayashi K, Yanagawa H, Nucleic Acids Res. 2000, 28, 1176–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Aviner R, Comput Struct Biotechnol J 2020, 18, 1074–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kim E, Jung H, BMB Rep 2015, 48, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ge J, Zhang C-W, Ng XW, Peng B, Pan S, Du S, Wang D, Li L, Lim K-L, Wohland T, Yao SQ, Angew Chem Int Ed Engl 2016, 55, 4933–4937. [DOI] [PubMed] [Google Scholar]
- [16].Albinsson B, J. Am. Chem. Soc. 1997, 119, 6369–6375. [Google Scholar]
- [17].Ballini J-P, Daniels M, Vigny P, Eur. Biophys. J. 1988, 16, 131–142. [DOI] [PubMed] [Google Scholar]
- [18].Shin D, Sinkeldam RW, Tor Y, J. Am. Chem. Soc. 2011, 133, 14912–14915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rovira AR, Fin A, Tor Y, J. Am. Chem. Soc. 2015, 137, 14602–14605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rovira AR, Fin A, Tor Y, Chem. Sci. 2017, 8, 2983–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kuchlyan J, Martinez-Fernandez L, Mori M, Gavvala K, Ciaco S, Boudier C, Richert L, Didier P, Tor Y, Improta R, Mély Y, J. Am. Chem. Soc. 2020, 142, 16999–17014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sanches de Araújo AV, Valverde D, Canuto S, Borin AC, J. Phys. Chem. A 2020, 124, 6834–6844. [DOI] [PubMed] [Google Scholar]
- [23].Ludford PT, Li Y, Yang S, Tor Y, Org. Biomol. Chem. 2021, 19, 6237–6243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bucardo MS, Wu Y, Ludford PT, Li Y, Fin A, Tor Y, ACS Chem. Biol. 2021, 16, 1208–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cong D, Li Y, Ludford III PT, Tor Y, Chem. Eur. J. 2022, 28, e202200994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kilin V, Gavvala K, Barthes NPF, Michel BY, Shin D, Boudier C, Mauffret O, Yashchuk V, Mousli M, Ruff M, Granger F, Eiler S, Bronner C, Tor Y, Burger A, Mély Y, J. Am. Chem. Soc. 2017, 139, 2520–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Grytsyk N, Richert L, Didier P, Dziuba D, Ciaco S, Mazzoleni V, Lequeu T, Mori M, Tor Y, Martinez-Fernandez L, Improta R, Mély Y, Int. J. Biol. Macromol. 2022, 213, 210–225. [DOI] [PubMed] [Google Scholar]
- [28].Steinbuch KB, Tor Y, Handbook of Chemical Biology of Nucleic Acids, Springer Nature, Singapore, 2022, pp. 1–24. [Google Scholar]
- [29].Hadidi K, Tor Y, Chem. Eur. J. 2022, 28, e202200765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30]. CCDC Deposition Number 2140402.
- [31]. See supporting information for details.
- [32]. Reproducible photophysical measurements of compounds 2–4 in aqueous media could only be achieved in buffered solutions.
- [33]. We speculate that compounds 2–4 may exhibit higher emission in more apolar media based on the photophysical characteristics of parent nucleobases. 27
- [34]. Native Puromycin is virtually non-emissive. Attempts to measure puromycin’s emission spectra were unsuccessful.
- [35].Wiegand I, Hilpert K, Hancock REW, Nat. Protoc. 2008, 3, 163–175. [DOI] [PubMed] [Google Scholar]
- [36].“In Vitro Translation Using Rabbit Reticulocyte Lysate | Springer Nature Experiments,” can be found under https://experiments.springernature.com/articles/10.1385/0-89603-288-4:215, n.d.
- [37].“Rabbit Reticulocyte Lysate, Nuclease-Treated,” can be found under https://www.promega.com/products/protein-expression/cell-free-protein-expression/rabbit-reticulocyte-lysate-system_-nuclease-treated/, n.d.
- [38]. Compounds were treated with 10 μM puromycin. Treatments with low concentrations of puromycin leads only to addition at the C-terminus and minimal cytotoxicity. 12, 44–46
- [39]. Given puromycin is non-emissive, the control was also used to confirm non-detectable cellular autofluorescence under the imaging conditions used.
- [40].David A, Dolan BP, Hickman HD, Knowlton JJ, Clavarino G, Pierre P, Bennink JR, Yewdell JW, J. Cell. Biol. 2012, 197, 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Enam SU, Zinshteyn B, Goldman DH, Cassani M, Livingston NM, Seydoux G, Green R, eLife 2020, 9, e60303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].“Human Antibody Against Ribosomal P Antigen,” can be found under http://www.immunovision.com/hpo-0100/, n.d.
- [43].Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Liu JO, Nat. Chem. Biol. 2010, 6, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chang B, Xu Q, Guo H, Zhong M, Shen R, Zhao L, Zhao J, Ma T, Chu Y, Zhang J, Fang J, J. Med. Chem. 2023, 66, 3250–3261. [DOI] [PubMed] [Google Scholar]
- [45].Buhr F, Kohl-Landgraf J, tom Dieck S, Hanus C, Chatterjee D, Hegelein A, Schuman EM, Wachtveitl J, Schwalbe H, Angew Chem Int Ed Engl 2015, 127, 3788–3792. [DOI] [PubMed] [Google Scholar]
- [46].Abbas M, Elshahawi SI, Wang X, Ponomareva LV, Sajid I, Shaaban KA, Thorson JS, J. Nat. Prod. 2018, 81, 2560–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.