

Abstract
The many-banded krait, Bungarus multicinctus, has been recorded as the animal resource of JinQianBaiHuaShe in the Chinese Pharmacopoeia. Characterization of its venoms classified chief phyla of modern animal neurotoxins. However, the evolutionary origin and diversification of its neurotoxins as well as biosynthesis of its active compounds remain largely unknown due to the lack of its high-quality genome. Here, we present the 1.58 Gbp genome of B. multicinctus assembled into 18 chromosomes with contig/scaffold N50 of 7.53 Mbp/149.8 Mbp. Major bungarotoxin-coding genes were clustered within genome by family and found to be associated with ancient local duplications. The truncation of glycosylphosphatidylinositol anchor in the 3′-terminal of a LY6E paralog released modern three-finger toxins (3FTxs) from membrane tethering before the Colubroidea divergence. Subsequent expansion and mutations diversified and recruited these 3FTxs. After the cobra/krait divergence, the modern unit-B of β-bungarotoxin emerged with an extra cysteine residue. A subsequent point substitution in unit-A enabled the β-bungarotoxin covalent linkage. The B. multicinctus gene expression, chromatin topological organization, and histone modification characteristics were featured by transcriptome, proteome, chromatin conformation capture sequencing, and ChIP-seq. The results highlighted that venom production was under a sophisticated regulation. Our findings provide new insights into snake neurotoxin research, meanwhile will facilitate antivenom development, toxin-driven drug discovery and the quality control of JinQianBaiHuaShe.
Key words: Medicinal snake, Neurotoxin origin, Antivenoms, Gene regulation, Chromatin
Graphical abstract
Post ancient duplication mutations shaped modern neurotoxins in the many-banded krait.
1. Introduction
Snake venom is an important source of drug development. At least eight drugs from snake venom toxins are proven effective for the treatment of human ailments, and others are used as a reference for drug design1, 2, 3. In addition, snakebite is one of the most substantial hidden public health threats and is estimated to cause 81,000–138,000 deaths and 400,000 permanent disabilities every year4, 5, 6. The development of safe and effective antivenoms is still ongoing5,7.
Snake toxins contain more than 20 well-established protein families, including phospholipases A2 (PLA2), zinc-dependent snake venom metalloproteinase, three-finger toxin (3FTx), and Kunitz-type inhibitors8. Toxins of different protein families are possibly recruited to venoms during the different stages of venomous snake evolution8,9. Current toxin evolutionary theories/phenomena include: (a) evolution of new functions by gene duplication10, 11, 12, (b) positive selection and accelerated evolution due to the higher number of observed mutations in exons than in introns13, 14, 15, (c) domain recombination/loss or gene-level exon shuffling12,16,17, (d) birth and death model18,19, (d) accelerated segment switch in exons to alter targeting20,21, (e) rapid accumulation of variations in exposed residues22, (f) arm race between predators and preys23, 24, 25, 26, 27, and (g) defense driving28.
Genome/transcriptome/proteome research has advanced and extended the evolutionary analysis of squamate, including snakes and their toxin development29, 30, 31, 32. Achievements referred to genome structure, adaption evolution33, 34, 35, axial patterning and limb development36,37, the originate and expansion of toxins19,36, toxin variation between species12,38, sex chromosome evolution39, 40, 41,tissue-specific expression42,43, gene expression regulation19,40,44. However, barring a few exceptions12,38,45,46, some fundamental questions regarding toxins’ evolutionary trajectories remain unaddressed. For instance, what are the genomic origin and ancestor of each toxin family? How many key events shaping the current toxins have occurred during evolution? How and when? What changes have prompted the specific expression of toxin genes in the venom gland9, 10, 11?
The many-banded krait, Bungarus multicinctus, has been recorded as the animal resource of JinQianBaiHuaShe in the Chinese Pharmacopoeia47,48. It is one of the most lethal snakes in the world because of its concurrent presynaptic toxicity and postsynaptic toxicity. Isolation and characterization of venoms from B. multicinctus in 1963 classified chief phyla of modern animal-target neurotoxins49. In the following decades, the constant applications of alpha-bungarotoxin (α-BgTx) have advanced our knowledge of nicotinic acetylcholine receptors and the corresponding cholinergic circuity50, 51, 52, 53, 54, 55. Beta-BgTx (β-BgTx), which is astoundingly lethal, is thus far the only known covalently-associated heterodimeric beta-neurotoxin10,56,57. Therefore, B. multicinctus is an ideal model for snake neurotoxin analysis. Here, we constructed a chromosome-level, highly contiguous B. multicinctus genome map to trace the origin and evolution of bungarotoxins in kraits. This work will facilitate innovations in antivenom, venom-driven drug development and the quality control of JinQianBaiHuaShe.
2. Materials and methods
2.1. Ethical approvals
This work was permitted by the Guangdong Wildlife Rescue Center (China). All snakes were fed and housed in Julong Artificial Breeding and Farming Center for Special Economic Animals. Before execution, the snakes were anesthetized using 100 mg of 2% phenobarbital. Details about the samples are listed in Supporting Information Table S1.
2.2. Genome sequencing, assembly, and annotation
High-molecular-weight DNA was extracted from the blood of a male B. multicinctus. PacBio SMRT 10 kbp library, Illumina (2 × 150 bp) shotgun libraries (insert 500 bp/800 bp), and Bionano optical maps (Nt.BspQI and Nb. BbvCI) were generated following the manufacturer's instructions and sequenced using PacBio Sequel, Illumina Hiseq X Ten, and Bionano Irys, respectively. Venom gland and muscle were obtained for Hi-C sequencing. MBoI was selected for Hi-C digestion after the chromatin was fixed in formaldehyde. Libraries for Hi-C sequencing were built using inserts around 350 bp and sequenced by Hiseq X Ten. A 50 kbp linked-read library was also constructed following 10 XGenomics' instruction and sequenced by Hiseq X Ten. The genome assembly data can be found in the Global Pharmacopoeia Genome Database (http://www.gpgenome.com/).
K-mer analysis was performed with jellyfish v2.3.0 (-m 23 -s 1000000000). Initial Pacbio assembly was conducted using Canu v1.71 (corOutCoverage = 80)58 and SMART denovo (wtpre -J 5000; wtzmo -z 10 -Z 16 -U -1 -A 1000 -m 0.1 -k16; wtclp -d 3 -k 300 -FT; wtlay -w 300 -s 200 -r 0.95 -c 1). The two drafts were merged by Quickmerge v0.2 (-hco 5.0 -c 1.5 -lm 5000 -l 300000). The merged contigs were polished using smrtlink v5.0 (--bestn 5 --minMatch 18 --nproc 4 --minSubreadLength 1000 --minAlnLength 500 --minPctSimilarity 70 --minPctAccuracy70 --hitPolicyrandombest --randomSeed 1) with PacBio long reads and Pilon v1.2.2 (default parameter) with Illumina short reads. After polishing, the assembly was orientated, scaffolded, and corrected using the TGH pipeline from Bionano Solve v3.1_08232017 (-d -U -N 6 -i 3 -f 0.1 -j 18) with Nt. BspQI optical map and Nb. BbvCI optical map59. Scaffolds were connected by Hi-C data using juicer v1.5, then manually curated with juicebox v1.11, and finally reassembled using 3D DNA v180419. The 10× data were assembled by Supernova v2.1.1.
The results from ab initio gene prediction (Augustus v3.3.1, GeneMark-ES v4.3.3 and SNAP v20131129) and homology gene prediction (blast v2.2.28 and Genewise v2.2.0) were incorporated using EVidenceModeler v1.1.160. Gene model generation and training were conducted using the full-length transcripts of PacBio Iso-seq pipeline and RNA-seq assemblies by Trinity v2.8.5. References for homology comparison included data from Ophiophagus hannah, D. acutus, P. molurus, A. carolinensis, and G. gallus. Assembly and gene prediction were evaluated by BUSCO v5.2.2 (vertebrata_odb10). Repetitive elements were annotated by RepeatMasker v4.0.7. De novo database construction was performed in RepeatModeler v1.0.861 with REPBASE as the comparison reference.
2.3. Genome evolution analysis
Gene family identification was first conducted in all versus all BLAST mode (v2.2.28, BLASTP, E-value cutoff 1e-5), followed by hcluster_sg v0.5.1 for clustering. Single-copy orthologous genes were aligned by MUSCLE v3.4 and then used for the construction of maximum likelihood (ML) phylogenetic tree by PhyML v3.0. Divergence time was inferred by MCMCtree from PAML package v4.9 (burn-in = 10,000, sample number = 100,000, sample frequency = 2, model = JC69). Calibration times were obtained from TimeTree (https:/timetree.org). Gene family expansion/contraction was calculated with CAFÉ v4.2.1. Synteny comparisons were analyzed using MCscan v0.9.662.
2.4. RNA sequencing and gene expression analysis
Total RNA was extracted from the heart, lung, muscle, kidney, liver, and venom gland using Qiagen RNeasy Kit (each sample was obtained in three biological replicates). A dataset of venom gland after venom depletion at different time (undepletion time, 3, 6 and 9 day, each with three replicates) was also used for gene expression analysis. RNA sequencing libraries were constructed using the Illumina mRNA-Seq Prep Kit and then sequenced by Illumina Hiseq X Ten. Venom replenishment data were referenced from our published work and retrieved at NCBI PRJNA608620. Raw reads were trimmed and filtered using Skewer v0.2263 (-m pe -q 30 -Q 20 -l 18 -t 32). Trimmed reads were mapped to B. multicinctus genome using HISAT v2.1.064. Transcript generation and counts were generated using StringTie v2.0.065. Differential gene expression was calculated using R package DEseq2 v1.26.066.
2.5. Proteomic analysis
The protein in venom samples was first quantified by Bradford method, from which 100 μg was alkylated with MTDTT and MIAA for 1 h respectively, and thereafter lysed with Trypsin at a ratio of 50:1 for 12 h. Before MS/MS analysis, venom proteins were separated into 16 fragments by an HPLC system based on Ultimate 3000, C18 column (3 mm, 0.10 mm × 20 mm) and C18 column (C18 1.9 mm, 0.15 mm × 120 mm), the flow speed was controlled at 600 nL/min. Moreover, MS/MS signals were generated and collected by Q-Exactive HF instrument (Thermo Scientific), proteins were qualitatively identified with MASCOT algorithm planted in Protein Discovery 2.0 and quantitatively identified with MaxQuant software.
2.6. Analysis of Hi-C data
Muscle and venom glands from the same sample were collected for Hi-C and ChIP-seq sequencing. Raw reads were trimmed by Skewer v0.22 (-m pe -q 30 -Q 20 -l 18 -t 32). The clean reads were mapped and processed using the Juicer pipeline to produce Hi-C maps. All contact matrices used for further analysis were KR-normalized in Juicer v1.5. Juicer Tools v1.14 was used for Hi-C maps analyses67. Contact matrix was extracted using dump, and data from different tissues were normalized as 1 × 109 input read pairs before comparison. Compartments were calculated using eigenvector with a bin size of 100 kbp, TADs were calculated using arrowhead with a bin size of 50 kbp, and loops were labeled using hiccups with resolutions of 5, 10, and 25 kbp68.
2.7. ChIP and data analysis
ChIP-seq was analyzed following the method of V.G. Tim et al.69. Muscle and venom gland fixed in formaldehyde (1%) were used for ChIP-seq, and each tissue was prepared in biological triplicates. Antibody ab47915 (abcam) for H3ac and #9733(CST) for H3K27me3 were selected for immunoprecipitation. Precipitated DNA was enriched, purified, and fragmented. Libraries were constructed using Illumina TrueSeq Sample Prep Kit. Libraries of raw DNA from each sample were used as the control. All libraries were sequenced using Hiseq X Ten. Raw reads were trimmed with Skewer v0.22 (-m pe -q 30 -Q 20 -l 18 -t 32). Reads were mapped using bwa v0.7.17 (mem -t 120 -k 18) and then filtered with samtools v1.9.0 (view -F 4 -q 30 -bS -@ 8). Peak calling was performed with MACS2 v2.2.7 (macs2 callpeak -c Input. bam -t H3Ac.bam -q 0.05 -f BAMPE -g 1.58e+9 -B -n H3Ac --outdir./Callpeak). Downstream annotation was conducted using R package Chipseeker v1.22.170, and motif finding was carried out with Homer package v3.2.171.
2.8. Testing enrichment of venom genes within clusters
Venom families with at least one member detected in the venom proteome or FPKM >50 were reserved for clustering analysis. In reference to Li et al.72, a cluster was defined as having at least two toxin-coding genes located within X bp. The effect of a range of X on the amount of clustering was then examined. For the global test: the number of genes per scaffold was retained, and a new starting position was randomly assigned for each gene. Empirical P values were computed as the number of duplicates where the number of genes in the clusters in the permuted set was larger than that in the clusters for the specified value of X. For the incremental test: all genes were initially set at a small nearest-neighborhood distance (1 kbp). The locations of the remaining nonclustered genes were then permuted within each scaffold, and the number of genes that would be clustered according to the second nearest-neighborhood distance (5 kbp) was computed and recorded. This process was repeated with the following distances (1, 5, 8, 10, 20, 50, 80, 100, 200, 500, and 800 kbp and 1, 5, 10, 50, and 100 Mbp). P value was set as the ratio of replicates where the number of clustered genes was higher than that in the given distance after 10,000 permutations.
2.9. Analysis of toxin-coding genes
Hmmer v3.0.0 and BLASTP program (with a cutoff E-value < 1e-5) were used for toxin gene identification. Reference genes were collected from NCBI based on published works. Manual curation was performed using Apollo v1.11.8. Signal peptides were predicted using SignalP v5.0 b with default parameters. Glycosylphosphatidylinositol (GPI) anchor sites were predicted by PredGPI predictor (https://gpcr2.biocomp.unibo.it/predgpi/pred.htm)73. Sequences were aligned using MAFFT v.760. Phylogenetic trees were built by PhyML v. 3.063 after Modeltest v3.7 prediction (best fit model for 3FTx: WAG + G; kunitz:VT + G; PLA2: WAG + G + I). The number of nucleotide substitutions per synonymous (dS) and nonsynonymous site (dN) for each pair of protein-coding genes was computed with the Nei–Gojobori method using MEGA X.
2.10. Protein interaction examined by pulldown assay
The interaction among proteins was verified by pulldown assay in vitro. Plasmids pGEX-4T1/A21(BM09280, the A chain of β-BgTx), pET28a/B22(BM19233, the B chain of β-BgTx), pET28a/B22-C55R (ΔBM19233 with a C55 -> R55), pET28a/Pilp3(BM19206, a Kunitz protein without C55), pET28a/γ-BgTx (BM02014) and pET28a/κ-BgTx (BM19133)were constructed for the expression of certain proteins with GST/His-tag. These proteins were expressed in E. coli and purified by Pierce™ Glutathione-Agarose or HisPur™ Ni-NTA Superflow Agarose (ThermoFisher Scientific). Afterward, 100 μg of each protein was used in each pulldown group. Glutathione-agarose bead volumes were scaled down to 50 μL and run on 4%–12% Bis-Tris gel (Invitrogen). GelCode™ Blue Stain Reagent (ThermoFisher Scientific) was used of visualization.
2.11. Protein interaction examined by surface plasmon resonance
The binding affinity of test proteinswas examined by surface plasmon resonance (SPR) assay in the Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences & Peking Union Medical College using a Biacore T200 instrument. The recombinant proteins with GST/His-tag were immobilized on CM5 sensor chips as ligands. N-Ethyl-N′-(3-dimethylaminopropyl) carbodiimide and N-hydroxysuccinimide were used in accordance with the standard primary amine coupling procedures. Then 0–1000 nmol/L recombinant proteins in HBS-EP (10 mmol/L HEPES, 150 mmol/L NaCl, 3 mmol/L EDTA, 0.005% (v/v) surfactant P20, pH 7.4) running buffer were injected at a flow rate of 30 μL/s for 180 s, and the channels were subsequently washed with the running buffer for 300 s. The association (ka) and dissociation (kd) rate constants and the equilibrium dissociation constant (KD = kd/ka) were determined by analyzing the sensorgram curves associated with the various concentrations using the Biacore Insight Evaluation Software.
2.12. Data availability statement
The raw data generated in this study were submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA682532 and PRJNA608620, to the GPGD under the link: http://www.gpgenome.com/species/148 and to the National Genomics Data Center (https://ngdc.cncb.ac.cn/) under the accession number: GWHBJIQ00000000, OMIX001664.
3. Results
3.1. Genomic feature of B. multicinctus
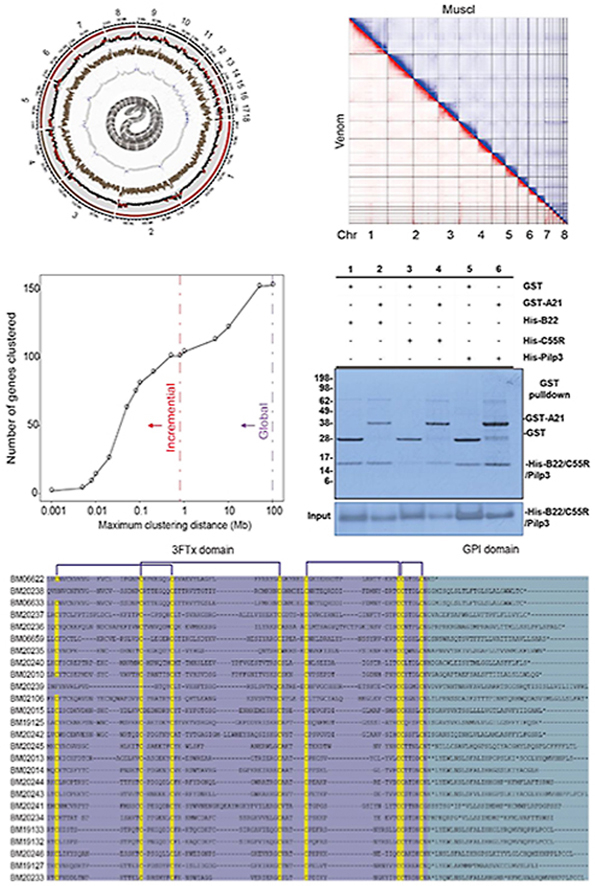
The 1.58 Gbp final assembly contained 11 macrochromosomes (>50 Mbp) and 7 microchromosomes (<50 Mbp) with a contig N50 of 7.53 Mbp and scaffold N50 of 149.80 Mbp (Fig. 1A, Supporting Information Fig. S1, Table S2). BUSCO assessment showed high integrity (C: 94.6% [S: 93.4%, D: 1.2%], F: 1.4%, M: 4.0%, n = 3354) (Supporting Information Table S3). A total of 20,246 protein-coding genes were annotated, including 270 toxin-coding genes from 34 families (Fig. 1B, Supporting Information Table S4). Approximately 790.81 Mbp sequences were identified as repetitive sequences, with 37.3% annotated as long interspersed elements (LINEs) (Supporting Information Table S5). CR1 was the most abundant long interspersed element co-occurring with simple sequence repeats and possibly contributed the most to B. multicinctus microsatellite expansion74. A phylogenetic tree was constructed on the basis of 387 single-copy orthologous genes conserved across 12 species (Fig. 1C and Supporting Information Fig. S2). As estimated, Archosauromorpha and Lepidosauromorpha diverged 280 million years ago (MYA), and B. multicinctus and O. hannah diverged around 20 (8.9–30.4) MYA. Large-scale genomic rearrangements, including the blocks containing toxin-coding genes, were visualized through synteny analysis (Fig. 1D and Supporting Information Fig. S3).
Figure 1.

The B. multicinctus genome indicated that toxin protein families originated independently from local tandem duplication. (A) The layers of the circus plot represent the chromosome length, repeat content, gene density and GC content (%) of the genome from the outside to the inside. Regions with repeat content greater than the third quantile are shown in brown and those with higher-than-average GC content are shown in blue. (B) Venom families identified in the genome. The identified proteins include members not expressed in the venom. Glycosyl hydrolase families (13,37,56) were presented as one. (C) Phylogenetic tree constructed using 387 single-copy orthologous genes conserved across 12 species speculates divergence times for each species. (D) Synteny maps of B. multicinctus aligned with H. sapiens and G. gallus. Light grey lines exhibit the synteny blocks. Dark grey lines exhibit the synteny blocks with venom genes. Red, yellow and green lines exhibit blocks with 3FTx, PLA2, and kunitz families respectively. (E) The incremental (red) and global (purple) tests show significant clustering of venom genes.
3.2. Toxin protein families originating independently from local tandem duplication
The highly contiguous genome enabled us to use the heuristic algorithm proposed by Li et al.72 to assess the spatial organization of venom genes on chromosomes. The results showed that more than 50% of venom genes were situated in clusters according to a cutoff of 100 kbp. Statistically significant clustering was observed at the cutoffs of 800 kbp and 10 Mbp in conservative incremental test and less conservative global test, respectively (Fig. 1E). The clustering genes increased the most (140% increase) when the cutoff was between 20 and 50 kbp, implying that most of the venom genes were located within relatively compact regions of the genome. Most clusters appeared to have originated from tandem duplication. At the 100 kbp cutoff, the only multivenom gene cluster consisted of one gene from the c-type lectin family and another from the cystatin family.
3.3. Tissue-specific expression and global epigenetic regulation of B. multicinctus genes
Six tissues (heart, kidney, lung, liver, muscle, and venom gland) were used for tissue-level expression analysis. A total of 13,499 expressed genes were detected (FPKM >1). Among these genes, 4273 exhibited a highly tissue-specific expression profile with Tau index ≥0.975, and 415 were exclusively expressed in venom glands (log2 fold change, lfc ≥1, false discovery rate, fdr ≤0.05) (Supporting Information Fig. S4A). Except for toxin-coding genes (toxin activity (GO:0090729)), all the venom gland up-regulated genes mainly enriched in several terms, such as Golgi membrane (GO:0000139), endoplasmic reticulum to Golgi vesicle-mediated transport (GO:0006888), protein N-linked glycosylation (GO:0006487), COPI-coated vesicle (GO:0030137), and protein folding (GO:0006457). These terms referred to several processes, such as peptides synthesis, protein sorting, disulfide bond formation, post-translation modification, vasodilatation, and muscle contraction. This result suggested the systematic specification of venom gland in the toxin synthesis and other physiological regulations. Among the 6071 venom gland-expressed genes, 576 were detected in venom proteome by mass spectrometry (Supporting Information Table S6).
Given that the venom gland and muscle vary hugely in gene expression profile, they were chosen for the following analysis. Chromatin interaction mapping showed distinctive topological features for these two tissues (Fig. 2A). About 10% of read-pairs were detected as Hi-C contacts in the dataset of muscle; meanwhile, 50.46% were detected for venom glands (Supporting Information Table S7). Contact decay curves implied the more condensed 3D chromatin organization in the B. multicinctus venom gland than in its muscle (Fig. 2C‒D, Supporting Information Table S8). Long-range interactions (>20 kbp) increased five times more than short-range interactions in the venom glands. For chromosomal interactions, the frequency for inter-MICs increased the most with an average ratio of 7.18, which was significantly larger than the number for junctions of inter/intra-MACs, intra-MICs, and junctions across MACs/MICs (Fig. 2B).
Figure 2.

3D chromatin organization and histone modification indicated the global epigenetic regulation of B. multicinctus genes. (A) Chromatin interaction in different tissues (red, venom gland; blue, muscle). (B) The increased ratio of chromosomal interactions in different chromosome groups when compare venom gland with muscle. (C, D) Whole-genome distance-contact frequency diagram with a 100-kb resolution in the venom gland (−0.94) and muscle (−0.88). Color: contact decay curve of different chromosomes; horizontal axis: distance between different sites on the chromosome; vertical axis: contact frequency. (E) Hi-C interaction map and colocalization of the genomic composition and various epigenetic marks on Chr7. The heatmap denotes the intrachromosomal Hi-C interaction frequencies among the pairwise 100-kb bins shown on the top. The PCA eigenvectors of the A and B compartments and genomic and epigenetic feature tracks are shown below in separate 100-kb bins, including abundances of genes and TEs, different histone modifications and mRNA expression levels (normalized by FPKM). (F) Bubble pattern of enriched pathways in the differential and conserved compartment domains (venom gland: A; muscle: B). The Y-axis represents the name of the pathway or function, and the X-axis represents the q values of each enriched gene family. The size of the bubble represents the number of genes in a signaling pathway or involved in a GO annotation. The color of the bubble indicates the ratio of the number of enriched genes.
In the venom glands, about 784.9 (account for 49.8% of the genome) regions were assigned to compartment A, which was considered as open chromatin (Fig. 2E, Supporting Information Figs. S4–S5). However, 9973 genes were assigned to the “closed chromatin” compartment B, implying the tissue-specific selection for chromatin regulation (Supporting Information Table S9)75,76. The 254 Mbp regions shifted from compartment B in muscle into compartment A in venom glands, and the genes in these regions were shared with the venom gland-specific expressed genes, including gene families such as toxin activity (GO:0090729), protein folding (GO:0006457), and Golgi membrane (GO:0000139) (Fig. 2F, Supporting Information Fig. S4B).
Hi-C matrix identified 1026 and 41 topologically associated domains (TADs) for venom gland and muscle, respectively (Supporting Information Table S10). All TADs in the muscle can be found in the venom glands, implying their conservation among tissues. By contrast, the remaining TADs in the venom glands were identified as specific. This observation indicated the nonrandom distribution of genome territories in venom glands77, 78, 79, 80. A total of 2006 loop peaks were found to be associated with 3827 distinct peak loci in the muscle and 7902 loops associated with 14,244 distinct peak loci in the venom glands (Supporting Information Table S11). Motifs enriched in loop peak loci included several regulatory elements such as RREB1, IRF1, EWSR1-FLT1 et al. Genes in the same loop tend to be coexpressed. An exemplary loop located in Chr1:10,400,000–10,700,000, containing 19 coexpressed genes (with an average Spearman correlation among all tissues = 0.7823), all of which belong to the MHC1 family (Supporting Information Fig. S6). Another typical loop is located at the terminal region of Chr2. Genes in this loop all belong to the PLA2 neurotoxins and are highly coexpressed (average Spearman correlation among all tissues = 0.9477, P-value < 0.001) (Fig. 6).
Figure 6.

The unit A of β-bungarotoxin lacked the Cys11‒Cys70 pair and were under global epigenetic regulation. (A) The ML phylogenetic tree and the amino acid sequence alignment of the GI-PLA2. (B, C) The expression characteristics of the many-banded krait GI-PLA2 in venom gland b and muscle c. Tracks from the top to bottom are H3ac counts (the red histogram), H3K27me3 counts (the navy histogram), the reads count of transcriptome (blue histogram) and gene structures (green blocks, where GI-PLA2 were indicated using red). Loops were displayed using arcs. Heatmaps implicated the contact frequency between chromosomal regions. The large square frames implicated the TADs and small square frame implicated peak loci.
We also detected histone modifications, including the active marker H3ac and the commonly suppressing marker H3K27me381. Totally 29,400/30,127 H3ac peaks and 9427/6812 H3K27me3 peaks were identified in venom gland/muscle ChIP-seq datasets. H3ac positively correlated with gene expressions (R = 0.56) and H3K27me3 negatively correlated with the gene expression (R = −0.45) (Supporting Information Fig. S7A‒C). Lengths of peaks were averagely around 2000 bp for H3ac and 500 bp for H3K27me3. For each marker, more than 10 percent peaks were located on the gene promoter regions (−1000 bp before transcriptional start sites, TSS, H3ac/H3K27me3: 17.58%/12.46% for muscle, 18.97%/13.48% for venom glands) (Fig. S7D). In the venom gland, H3ac was enriched in gene families, such as protein folding (GO:0006457), intracellular protein transport (GO:0006886), and chromatin silencing (GO:0006342); and H3K27me3 was enriched in mRNA splicing (GO:0000398), translation (GO:0006412), and rRNA processing (GO:0006364) following the chromatin organization features. H3ac peaks overlapped with serval regulatory elements in the promoter regions of venom glands, such as IRF1, EGR1, and ELF3; the corresponding transcriptional factors were highly expressed (Supporting Information Table S12).
3.4. The cytochrome P450 gene family and keratin gene family
Snake gall bladder is recorded as a materia medica in Chinese Pharmacopeia. Generally considering, bile acids are the main bioactive compounds of snake gall bladder. Bile acids are biosynthesized from cholesterol by a series decoration. Most enzymes engaged in this process are come from the cytochrome P450 (CYP) superfamily. In B. multicinctus genome, a total of 94 CYP coding genes were identified. These genes were distributed in 16 families. Compared to human, mouse, chick and naja, the CYP2, CYP3 and CYP4 gene families were under significant expansion or contraction (P < 0.05) (Supporting Information Table S13). Two CYP7 coding genes, three CYP8 coding genes and three CYP27 coding genes were identified. These genes may be responsible for the krait bile acids synthesis. Of them, 5 genes were specifically expressed in liver with a Tau index over 0.6 (Supporting Information Table S14). Gene structures of CYP7A1, CYP8B1 and CYP27A1 were conserved with those in human or mouse, indicating the relative conservation of bile acids metabolism in amniote (Supporting Information Fig. S8). However, the detail metabolites of each enzymes should be further verified.
Another traditional medication, the snake slough, comes from the epidermis of snake. It is mainly comprises of keratins. In the B. multicinctus genome, 23 alpha-keratins were annotated, including 13 type I and 10 type II, and separately located on Chr4, Chr1, and Chr3. Different from mammalian skin that only has alpha-keratins, snakes also possess beta-keratins impregnated in each stratum of its corneum. Eight beta-keratins were mostly located on Chr18, implicating their unique origin (Supporting Information Table S15). Analysis the composition of keratin coding genes will faciliatate the quality control of snake slough.
3.5. Ttruncation event in modern 3FTxs shaped by an ancient duplicated LY6 member
Thirty-three genes were identified belonging to the uPAR/LY6/CD59/snake toxin-receptor superfamily (PFAM clan:CL0117), including seven pseudogenes, (Supporting Information Table S4). Of these, 12 genes were detected in the RNA-Seq data with FPKM values > 100, and 10 were mapped to the venom proteomic data (Fig. 3B). The largest CL0117 cluster spanned 1.1 Mbp in the terminal region of Chr7, contained 16 genes, and showed high synteny with human Chr8 and G. gallus Chr2 (Figure 1, Figure 3A), which was consistent with ancient local gene duplication events. Phylogenetic analysis showed that the 3FTxs in elapids formed a large and well-supported monophyletic clade adjacent to human/gallus LY6E proteins (Supporting Information Fig. S9). Therefore, 3FTxs were created from the duplication and divergence of a LY6E-like protein.
Figure 3.

A truncation event in an ancient duplicated LY6 member shaped modern 3FTxs. (A) Genomic position for the 3FTx tandem arrays across vertebrates. Genes belong to the uPAR/LY6/CD59/snake toxin-receptor superfamily (Pfam: CL0117) and containing a glycosylphosphatidylinositol (GPI)-anchor tail were labeled using cyan, and those without GPI-anchor tail were labeled as red. Other genes were labeled as grey. (B) The statistics of 3FTxs. Gene labeled in purple indicate genes containing the glycosylphosphatidylinositol (GPI)-anchor tail; gene labeled in grey indicate genes losing the GPI-anchor tail. Numbers on labels denote the chromosome number. Following three columns show the gene length, transposable element (TE) number and the first intron size. For heatmaps, characteristic expression with amounts ≥100 are all marked as red.
A typical CL0117 superfamily gene comprises three parts: an N-terminal signal peptide, a well-conserved Ly-6 antigen/uPAE domain (with a characteristic three-finger structure formed by disulfide bridges), and a C-terminal GPI anchor tail for membrane tethering (Fig. 4A). However, the GPI anchor tail was not observed for all the venom proteomes expressing 3FTxs (Fig. 4B). Genome sequence analysis revealed that the conspicuous GPI anchor tail remained to flank with the stop codon of tail-truncated genes, suggesting the existence of nonsense mutations that truncated the 3FTxs (Fig. 4C). The GPI anchor remains were detectable either in the genomic loci or our full-length transcripts, indicating that the loss of GPI anchor coding sequences led to the loss of membrane-tethering function and ultimately allowed 3FTxs to be soluble and be secreted in venom.
Figure 4.

Sturcture features of 3FTxs in B. multicinctus. (A) Predicted tertiary structure of BM20242 (a LY6E ortholog in B. multicinctus) the GPI-anchor domain was indicated in black rectangle frame in dash, and the labled animo acid remain indicated the C-terminal of protein. (B) Predicted tertiary structure of BM20245 (the α-BgTx) unlike BM20242, its C-terminal GPI-anchor domain was missed. (C) Sequence alignment of the uPAR/LY6/CD59/snake toxin-receptor superfamily proteins in B. multicinctus. Purple shading shows the three-finger domain while the bluish grey shading shows the predicted GPI anchor. Yellow highlightings represent the cysteine pairs.
Expressed 3FTxs, such as γ-BgTx and κ-BgTx, share a conserved GPI anchor remain, which meant that all modern 3FTxs share a common ancestor. For example, the length, 3′ end, and GPI anchor remains of long-type 3FTx, α-BgTx, differed from those of other 3FTxs due to a fragment substitution occurring from the 4th to 23rd nucleotides behind the CCXXXXCN motif. This substitution changed the termination codon of α-BgTx. Sequences following this substitution were still conserved among paralogs, and their duplication sequence can be inferred from their structure difference. The gene + remain motif was detected in Elapidae, Colubridae, and Viperidae genomes/transcriptomes, implying that the soluble 3FTx ancestor emerged at least 45 MYA before the divergence of Colubroidea. With the phylogenetic tree as the basis, several rounds of duplication and functional diversification were presumed to occur after the truncation event because the same residues shared in the GPI anchor remained (Fig. 4C and Supporting Information Fig. S10). This finding verified B.G. Fry's hypothesis, which supposed that a certain type 3FTx (with lacking the 2nd and 3rd plesiotypic cysteines) might have originated from different duplication events ∼20 years ago18. In addition, our observation from genomic sequences was highly perceptible and conclusive.
The observed dN-to-dS ratios were greater than 1 for 3FTx gene pairs in the Nei-Gojobori model. In particular, the ratio was 8/0 for the κ-BgTx pair BM19132/BM19133, 11.53 for BM20233/BM19127, and 2.60 for γ-BgTxs BM02013/BM02014. These results indicated the strong divergence and functional diversification of 3FTx genes. Compared with that among exonic regions, greater sequence similarity was observed among the intronic regions of 3FTxs. These attributes suggested the accelerated evolution of the 3FTx family.
To examine the accelerated evolution fo 3FTx family, we reassembled and phased the Chr7:94,000,000‒96,000,000 into two homologous haploid contigs. Each of the haploid contigs contained 16 CL0117 superfamily coding genes (Fig. 5A). Among the 16 pair alleles, 5 pairs possessed variations with only one pair didn't have nonsynonymous mutations. All alleles with nonsynonymous mutations belong to the no GPI type (Fig. 5B). We further detected the SNP variation for CL0117 superfamily coding genes in whole genome wide (Supporting Information Table S16). The result showed that the average variation in genes without GPI anchor was significant higher than that in genes with GPI anchor (1.923 vs 0.154, P < 0.05), indicating strong selective pressure for 3FTxs (Fig. 5C).
Figure 5.

Genome phasing of B. multicinctus Chr7:94,000,000‒96,000,000. (A) Syntenies between refence and the haplotypes (P scaffold and H scaffold). (B) Heterozygous sites in different genes. The purple square represents Toxin_TOLIP domain, the orange square represents the UPAR_LY6 domain, the green dot represents nonsynonymous mutaitions, and the blue dot represents synonymous mutations.
3FTxs with GPI anchor domain loss were highly expressed with an average FPKM >30,000 (Supporting Information Table S17). Some LY6E-like genes that retained GPI anchors, such as BM02015, BM19125, and BM20242, were also expressed in the venom gland. According to the Ensembl released data, LY6E was expressed in the human salivary gland. These finding indicated that the LY6E ancestor genes were expressed in the venom gland, and certain genes, which were apparently recruited, showed elevated expression during 3FTx evolution. The expression of CL0117 genes was logarithmically negatively correlated with the gene length (R2 = 0.49), especially the length of the first intron (R2 = 0.74) (Fig. 3B and Supporting Information Fig. S11). Transposable element (TE) insertion was the major cause of the first intron length difference. Correspondingly, the signal peptides in the genes that retained GPI anchor domain showed more variation compared with those in 3FTxs. The 3 kbp sequences upstream of the promoters of CL0117 genes were extracted to construct a ML tree (Supporting Information Fig. S12). As expected, the sequences from soluble 3FTxs formed a specific clade. Motif screening revealed the binding sites of CREB3L4, FOXC1, PRDM1, ZBTB7B, ELF1, XBP1, NR1I3, SOX9, and IRF1. Consistently, the predicted transcription factors XBP1, FOXC1, and SOX9 were expressed with FPKM values > 20. More than 97% of the screened transcription factor binding sites were chimeric with TEs, highlighting the contribution of TE expansion to the shaping of the 3FTx regulatory network.
3.6. UnitA of β-BgTx is an adaption for the new type kunitz in krait
The PLA2 family of venoms is presumed to exert presynaptic or beta-neurotoxicity. In B. multicinctus, nine Asp49-type PLA2 encoding genes were identified, including four elapid group PLA2 (GI-PLA2) clustering on Chr2 and two viperid group PLA2s (GII-PLA2s) on Chr17 (Supporting Information Figs. S13–S14, Table S4). The coexistence of GI- and GII-PLA2s in the krait genome implied that the divergence of elapid and viperid PLA2 toxins may have originated from the different duplications of PLA2 ancestral genes.
Phylogenetics analysis indicated that BM19392 from GI-PLA2s was independent of the other three paralogs and clustered with the human pancreatic PLA2 (Fig. 6A). Another cluster contained only elapid genes, which lacked the pancreas loop-coding sequence and were annotated as GIA-PLA2s (Supporting Information Fig. S15). For the krait, only GIA-PLA2 genes were detected in the venom proteome datasets. These three genes belonged to the same Hi-C loop and were highly coexpressed (average Spearman correlation among all tissues = 0.95, P < 0.001), thereby indicating the importance of epigenetic regulation in PLA2 expression (Fig. 6B and C).
The average dN/dS ratio for all GI-PLA2s in B. multicinctus was approximately 3.38, which highlighted the dominant role of positive selection. The GIA-PLA2 lineage evolved much faster than the overall GI-PLA2 lineage with an average dN/dS of 5.18. BM19395 contained 14 cysteines, including Cys11 and Cys70 that formed a conserved disulfide bridge in snake GIA-PLA2s. BM19393 and BM09280 lacked this conserved Cys11‒Cys70 pair (because of a TGT- > TAT point mutation for Cys11- > Tyr and a G-T inversion TGT- > TTG for Cys70- > Leu). Instead, a new cysteine, Cys15, was formed from glycine by a GGC- > TGC substitution (Fig. 6A). This Cys15 was covalently linked to the Cys55 of unit B of β-BgTx.
The unit-B of β-BgTx belongs to the Kunitz gene family with a characteristic Cys55 in the C-terminus. A large tandem arrayed Kunitz family gene cluster was found on Chr5 and included four unit-B type genes. Most of the cluster members have corresponding mapping objects in the cobra genome, except for the unit-B type Kunitz genes (Supporting Information Figs. S16–S17)30. Unit-B genes were under strong positive selection with an observed dN/dS > 1. Cys55 is a typical result of a nonsynonymous mutation with a nucleotide transition from C to T and an AA transition from arginine to cysteine. Surface plasmon resonance experiment confirmed the necessary of this Cys55 for β-BgTx (Supporting Information Fig. S18).
Phylogenetic analysis showed that the unit-B type Kunitz did not solely appear in B. multicinctus and could also be found at least in B. candidus and B. fasciatus. This finding suggested that unit-B genes may have emerged after the divergence of cobras and kraits. A GIA-PLA2 in B. flaviceps (GenBank: GU190818.1)82 contains the Cys11–Cys70 pair and Cys15 but lacks the key alkaline residues in the reported binding-loop for unit-A56. This PLA2 seemed to be an intermediate form between nonenzymatic PLA2 and the mature unit-A of β-BgTx (Supporting Information Fig. S19‒S20). Hence, we postulated that A chain-type PLA2 matured during the divergence of Bungarus species at a later time than the unit-B emergence.
In addition, PLA2s have reported to potentiate cytotoxicity 3FTxs28. In co-immunoprecipitation/SPR test, the interactions between 3FTxs and PLA2s in venoms were examined (Supporting Information Fig. S21). Nevertheless, the evolutionary significance behind these interactions needs further exploration.
4. Discussion
Herbgenomics has faciliatated the research and development of several medicinal organisms47,83,84. We presented a highly continuous chromosome-level genome for B. multicinctus with a contig N50 larger than 7 Mbp. The high-quality assembly enabled us to quantitatively prove that toxin-coding gene families are generated from local gene duplication. We also successfully traced the evolutionary trajectory of toxin-coding genes. This tracing work will shed new lights for designing of artificial peptides for neuromodulation in a way of simulation of animal endogenous proteins from sequence to structure85. Moerover, deciphercing detail sequence of toxins can help for the antivenom research and help for the discover of new targets of toxins86.
Using the genome for B. multicinctus as the basis, we fully illustrated the scenario for 3FTx toxin evolution. For 3FTx formation, we theorized the following: (1) 3FTx toxins may arise from ancestors with the GPI region ended before the divergence of Colubroidea. If the expansion of LY6E was the onset of 3FTx's evolution, then the origin dated back to the divergence of amniotes; this finding was in accordance with the neuromodulation function of mammal and bird uPAR/LY6 genes87,88. (2) After truncation, several duplication/translocation events occurred in the 3FTx family in snake genome. (3) Duplication for truncated 3FTx may be random; for example, the number and composition of 3FTxs for N. naja and B. multicinctus were quite different from those in the same synteny locus (Supporting Information Fig. S22). (4) 3FTxs were under huge selection pressure, high dN/dS value can be observed even in ortholog 3FTxs in different species (Supporting Information Table S18). This theory explained the presence of many contemporary diverse 3FTx members in snakes. For the 3FTx recruitment scenario, we speculated the following: (1) ancestral 3FTx genes are expressed in the precursor tissues of venom gland; (2) during the formation of venom gland and the evolution of 3FTx family, the expression of certain 3FTx was refined; (3) TEs took part in the shaping portion of this process, either in the regulatory network construction or in the gene silence; and (4) epigenetic contribution should not be neglected during this process.
β-BgTx, a presynaptic neurotoxin isolated from the venom of B. multicinctus, consists of A chain (PLA2) and B chain (Kunitz proteins). The B chain of β-BgTx from B. multicinctus venom shows concentration- and time-dependent cytotoxicity against human neuroblastoma89. Our research showed that β-BgTx formed after the cobra/krait divergence with an extra cysteine residue emerging in its unit-B. A subsequent point substitution in unit-A enabled the β-BgTx covalent linkage. Therefore, unit-A matured later than unit-B during the evolution. We confirmed that PLA2s can interact a series proteins such as kunitz and 3FTxs. We suppose that PLA2s have the capacity to interact a lot of exogenous proteins. Notably, recent reports claimed that PLA2s exhibited strong virucidal activity against SARS-CoV-2 and HIV90,91.
Based on the evidence presented above, evolutions of bungarotoxins are under distinctly separate paths. They occurred in different geological times and involved into different forms such as the unique covalently-associated heterodimer of β-BgTx. However, some commonalities among which are unignorable to be stressed on. Firstly, neofunctionalization after tandem duplication is a key leading cause to toxins origination. This commonality helps us to remove the mystery of snake venom and hereby further focus on more specific problems in genome. Toxins derived from existed multiple gene families expanded before the differentiation of amniotic animals as synteny supported this idea. So genomic regions where toxin coding genes located are suspicious hotspots of tandem duplication. Parsing the composition of toxins located regions will help the exploration of toxin origination. This work requires not only genome comparison but also genetic operation. Secondly, except for functional genes, regulatory elements should be noticed. Variations of regulatory elements take a critical role in the evolution of snake. A well-known instance is the loss of snake limb. A slight long-range enhancer change leads to the significantly morphological transition37. Hi-C data and ChIP-seq data in this work are valuable resource for regulatory exploration. As expected, a series binding sites of transcriptional factors were identified. Validating theses binding sites is a huge and complex work. More importantly, till now, there is no authoritative model organism for venom transcription regulation examining. Repeats may involve in the shaping of regulatory elements. B. multicinctus genome contains astonishingly high-level simple sequence repeats (SSRs) (∼19.3 kb/Mb). LINEs may drive the expansion of SSRs through a process named “microsatellite seeding”74. Our results showed that CR1, a recently active element in squamate genomes, was the most abundant LINE type co-occurring with SSRs. Recently inserted repeats may bring new regulation pattern of functional genes. Thirdly, we have conducted genome phasing for the 3FTx region. To our surprise, the nonsynonymous rate between toxin-coding gene alleles is significantly higher than other genes. This finding indicated that toxin-coding genes were possibly still undergoing high-speed evolution. Moreover, there may contain more toxin-coding gene isotypes. Analysis of the trend of toxin-coding gene isotypes may contribute to finding the most efficient variation, and will benefit the modification of toxin-like peptide drugs. For the population analysis, cost and ethics issues should be considered. Notably, the paleogenome, which was awarded as this year's Nobel Prize, offered a series of DNA capture technologies92,93. These technologies will facilitate the reduction of both the tissues during DNA extraction and the data amount during sequencing. Last but not the least, we found comprehensive toxin–toxin interaction using Co-IP and SPR assay. The SPR assay provided a high-throughput way for interaction detection and afforded an unprecedented prospect of toxin–toxin interaction. Doley et al. indicated that some complexes exhibited much higher levels of pharmacological activities compared to individual components and played an significant role in pathophysiological effects during envenomation94. Another issue that should be considered is that if toxins can interact with each other, how about their orthologs in the snake's prey? Answering these questions will enhance the development of antivenom and facilitate the innovation of targeted drugs.
Venom production is complex and sophisticated at molecular, morphological, behavioral, and functional levels28. The expression of toxins is correlated with chromatin topological organization and histone modification. Except for toxin-coding genes, other genes involved in muscle contraction, protein folding, and proteolysis, were found to be expressed during venom replenishment. Analysis of the origin and regulation of snake toxins will facilitate the treatment of snakebite in animals and the development of endogenous antipsychotic, antitumor, and antiviral drugs.
Acknowledgments
The authors thank Prof. David T. Booth from the University of Queensland for the review and editing. This work was supported by the Fundamental Research Funds for the Central Public Welfare Research Institutes (ZZ13-YQ-047, ZXKT22006, China), quality standard system construction for the whole industry chain of Chinese medicine from Guangdong Provincial Drug Administration of China (002009/2019KT1261/2020ZDB25), the National Major Science and Technology Projects (2019ZX09201005, China), the Open Research Fund of Chengdu University of Traditional Chinese Medicino state Key Laboratory scluthwestern Chinese Medicine Resources (2022ZYXK2011006, China) and the National Key R&D Program of China (2019YFC1711100).
Author contributions
Design and supervision: Jiang Xu, Zhihai Huang, Shilin Chen. Experiment: Jiang Xu, Mingqian Li, He Su, Liang Le, Baosheng Liao, Xuejiao Liao, Haoyu Hu, Juan Lei, Lu Luo, Yiming Guo, Xiaohui Qiu, Dianyun Hou, Jin Pei, Yan Hua, Shiyu Chen. Data analysis and visualization: Jiang Xu, Shuai Guo, Xianmei Yin, Mingqian Li, Qiushi Li, Xuejiao Liao, Baosheng Liao, Yingjie Zhu, Jun Chen, Zhenzhan Chang, Nicholas Chieh Wu, Han Zhang. Writing: Jiang Xu, Lu Luo.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2022.11.015.
Contributor Information
Jiang Xu, Email: jxu@icmm.ac.cn.
Zhihai Huang, Email: zhhuang7308@163.com.
Shilin Chen, Email: slchen@icmm.ac.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Modahl C.M., Brahma R.K., Koh C.Y., Shioi N., Kini R.M. Omics technologies for profiling toxin diversity and evolution in snake venom: impacts on the discovery of therapeutic and siagnostic agents. Annu Rev Anim Biosci. 2020;8:91–116. doi: 10.1146/annurev-animal-021419-083626. [DOI] [PubMed] [Google Scholar]
- 2.Bordon K.C.F., Cologna C.T., Fornari-Baldo E.C., Pinheiro-Júnior E.L., Cerni F.A., Amorim F.G., et al. From animal poisons and venoms to medicines: achievements, challenges and perspectives in drug discovery. Front Pharmacol. 2020;11:1132. doi: 10.3389/fphar.2020.01132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bohlen C.J., Chesler A.T., Sharif-Naeini R., Medzihradszky K.F., Zhou S., King D., et al. A heteromeric Texas coral snake toxin targets acid-sensing ion channels to produce pain. Nature. 2011;479:410–414. doi: 10.1038/nature10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hunter C.J., Piechazek K.H., Nyarang'o P.M., Rennie T. Snakebite envenoming. Lancet (London, England) 2019;393:129–131. doi: 10.1016/S0140-6736(18)32762-4. [DOI] [PubMed] [Google Scholar]
- 5.The L. Snakebite-emerging from the shadows of neglect. Lancet (London, England) 2019;393:2175. doi: 10.1016/S0140-6736(19)31232-2. [DOI] [PubMed] [Google Scholar]
- 6.Murray K.A., Martin G., Iwamura T. Focus on snake ecology to fight snakebite. Lancet (London, England) 2020;395:e14. doi: 10.1016/S0140-6736(19)32510-3. [DOI] [PubMed] [Google Scholar]
- 7.Koh C.Y., Bendre R., Kini R.M. Repurposed drug to the rescue of snakebite victims. Sci Transl Med. 2020;12 doi: 10.1126/scitranslmed.abb6700. [DOI] [PubMed] [Google Scholar]
- 8.Fry B.G. From genome to "venome": molecular origin and evolution of the snake venom proteome inferred from phylogenetic analysis of toxin sequences and related body proteins. Genome Res. 2005;15:403–420. doi: 10.1101/gr.3228405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fry B.G., Vidal N., Norman J.A., Vonk F.J., Scheib H., Ramjan S.F., et al. Early evolution of the venom system in lizards and snakes. Nature. 2006;439:584–588. doi: 10.1038/nature04328. [DOI] [PubMed] [Google Scholar]
- 10.Kordis D., Gubensek F. Adaptive evolution of animal toxin multigene families. Gene. 2000;261:43–52. doi: 10.1016/s0378-1119(00)00490-x. [DOI] [PubMed] [Google Scholar]
- 11.Casewell N.R., Huttley G.A., Wüster W. Dynamic evolution of venom proteins in squamate reptiles. Nat Commun. 2012;3:1066. doi: 10.1038/ncomms2065. [DOI] [PubMed] [Google Scholar]
- 12.Giorgianni M.W., Dowell N.L., Griffin S., Kassner V.A., Selegue J.E., Carroll S.B. The origin and diversification of a novel protein family in venomous snakes. Proc Natl Acad Sci U S A. 2020;117:10911–10920. doi: 10.1073/pnas.1920011117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Juárez P., Comas I., González-Candelas F., Calvete J.J. Evolution of snake venom disintegrins by positive darwinian selection. Mol Biol Evol. 2008;25:2391–2407. doi: 10.1093/molbev/msn179. [DOI] [PubMed] [Google Scholar]
- 14.Nakashima K., Ogawa T., Oda N., Hattori M., Sakaki Y., Kihara H., et al. Accelerated evolution of Trimeresurus flavoviridis venom gland phospholipase A2 isozymes. Proc Natl Acad Sci U S A. 1993;90:5964–5968. doi: 10.1073/pnas.90.13.5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakashima K., Nobuhisa I., Deshimaru M., Nakai M., Ogawa T., Shimohigashi Y., et al. Accelerated evolution in the protein-coding regions is universal in crotalinae snake venom gland phospholipase A2 isozyme genes. Proc Natl Acad Sci U S A. 1995;92:5605–5609. doi: 10.1073/pnas.92.12.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Casewell N.R., Wagstaff S.C., Harrison R.A., Renjifo C., Wüster W. Domain loss facilitates accelerated evolution and neofunctionalization of duplicate snake venom metalloproteinase toxin genes. Mol Biol Evol. 2011;28:2637–2649. doi: 10.1093/molbev/msr091. [DOI] [PubMed] [Google Scholar]
- 17.Kini R.M. Accelerated evolution of toxin genes: exonization and intronization in snake venom disintegrin/metalloprotease genes. Toxicon. 2018;148:16–25. doi: 10.1016/j.toxicon.2018.04.005. [DOI] [PubMed] [Google Scholar]
- 18.Fry B.G., Wüster W., Kini R.M., Brusic V., Khan A., Venkataraman D., et al. Molecular evolution and phylogeny of elapid snake venom three-finger toxins. J Mol Evol. 2003;57:110–129. doi: 10.1007/s00239-003-2461-2. [DOI] [PubMed] [Google Scholar]
- 19.Vonk F.J., Casewell N.R., Henkel C.V., Heimberg A.M., Jansen H.J., McCleary R.J., et al. The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc Natl Acad Sci U S A. 2013;110:20651–20656. doi: 10.1073/pnas.1314702110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doley R., Pahari S., Mackessy S.P., Kini R.M. Accelerated exchange of exon segments in Viperid three-finger toxin genes (Sistrurus catenatus edwardsii; Desert Massasauga) BMC Evol Biol. 2008;8:196. doi: 10.1186/1471-2148-8-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doley R., Mackessy S.P., Kini R.M. Role of accelerated segment switch in exons to alter targeting (ASSET) in the molecular evolution of snake venom proteins. BMC Evol Biol. 2009;9:146. doi: 10.1186/1471-2148-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sunagar K., Jackson T.N., Undheim E.A., Ali S.A., Antunes A., Fry B.G. Three-fingered RAVERs: rapid accumulation of variations in exposed residues of snake venom toxins. Toxins. 2013;5:2172–2208. doi: 10.3390/toxins5112172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feldman C.R., Brodie E.D., Jr., Brodie E.D., 3rd, Pfrender M.E. The evolutionary origins of beneficial alleles during the repeated adaptation of garter snakes to deadly prey. Proc Natl Acad Sci U S A. 2009;106:13415–13420. doi: 10.1073/pnas.0901224106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feldman C.R., Brodie E.D., Jr., Brodie E.D., 3rd, Pfrender M.E. Constraint shapes convergence in tetrodotoxin-resistant sodium channels of snakes. Proc Natl Acad Sci U S A. 2012;109:4556–4561. doi: 10.1073/pnas.1113468109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGlothlin J.W., Chuckalovcak J.P., Janes D.E., Edwards S.V., Feldman C.R., Brodie E.D., Jr., et al. Parallel evolution of tetrodotoxin resistance in three voltage-gated sodium channel genes in the garter snake. Thamnophis sirtalis. Mol Biol Evol. 2014;31:2836–2846. doi: 10.1093/molbev/msu237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Healy K., Carbone C., Jackson A.L. Snake venom potency and yield are associated with prey-evolution, predator metabolism and habitat structure. Ecol Lett. 2019;22:527–537. doi: 10.1111/ele.13216. [DOI] [PubMed] [Google Scholar]
- 27.Gibbs H.L., Sanz L., Pérez A., Ochoa A., Hassinger A.T.B., Holding M.L., et al. The molecular basis of venom resistance in a rattlesnake-squirrel predator-prey system. Mol Ecol. 2020;29:2871–2888. doi: 10.1111/mec.15529. [DOI] [PubMed] [Google Scholar]
- 28.Kazandjian T.D., Petras D., Robinson S.D., van Thiel J., Greene H.W., Arbuckle K., et al. Convergent evolution of pain-inducing defensive venom components in spitting cobras. Science. 2021;371:386–390. doi: 10.1126/science.abb9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alföldi J., Di Palma F., Grabherr M., Williams C., Kong L., Mauceli E., et al. The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature. 2011;477:587–591. doi: 10.1038/nature10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suryamohan K., Krishnankutty S.P., Guillory J., Jevit M., Schröder M.S., Wu M., et al. The Indian cobra reference genome and transcriptome enables comprehensive identification of venom toxins. Nat Genet. 2020;52:106–117. doi: 10.1038/s41588-019-0559-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gemmell N.J., Rutherford K., Prost S., Tollis M., Winter D., Macey J.R., et al. The tuatara genome reveals ancient features of amniote evolution. Nature. 2020;584:403–409. doi: 10.1038/s41586-020-2561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ullate-Agote A., Burgelin I., Debry A., Langrez C., Montange F., Peraldi R., et al. Genome mapping of a LYST mutation in corn snakes indicates that vertebrate chromatophore vesicles are lysosome-related organelles. Proc Natl Acad Sci U S A. 2020;117:26307–26317. doi: 10.1073/pnas.2003724117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Campbell-Staton S.C., Cheviron Z.A., Rochette N., Catchen J., Losos J.B., Edwards S.V. Winter storms drive rapid phenotypic, regulatory, and genomic shifts in the green anole lizard. Science. 2017;357:495–498. doi: 10.1126/science.aam5512. [DOI] [PubMed] [Google Scholar]
- 34.Li J.T., Gao Y.D., Xie L., Deng C., Shi P., Guan M.L., et al. Comparative genomic investigation of high-elevation adaptation in ectothermic snakes. Proc Natl Acad Sci U S A. 2018;115:8406–8411. doi: 10.1073/pnas.1805348115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng C., Ren J.L., Deng C., Jiang D., Wang J., Qu J., et al. The Genome of shaw's sea snake (Hydrophis curtus) reveals secondary adaptation to its marine environment. Mol Biol Evol. 2020;37:1744–1760. doi: 10.1093/molbev/msaa043. [DOI] [PubMed] [Google Scholar]
- 36.Castoe T.A., de Koning A.P., Hall K.T., Card D.C., Schield D.R., Fujita M.K., et al. The Burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc Natl Acad Sci U S A. 2013;110:20645–20650. doi: 10.1073/pnas.1314475110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kvon E.Z., Kamneva O.K., Melo U.S., Barozzi I., Osterwalder M., Mannion B.J., et al. Progressive loss of function in a limb enhancer during snake evolution. Cell. 2016;167:633–642.e11. doi: 10.1016/j.cell.2016.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dowell N.L., Giorgianni M.W., Kassner V.A., Selegue J.E., Sanchez E.E., Carroll S.B. The deep origin and recent loss of venom toxin genes in rattlesnakes. Curr Biol. 2016;26:2434–2445. doi: 10.1016/j.cub.2016.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vicoso B., Emerson J.J., Zektser Y., Mahajan S., Bachtrog D. Comparative sex chromosome genomics in snakes: differentiation, evolutionary strata, and lack of global dosage compensation. PLoS Biol. 2013;11 doi: 10.1371/journal.pbio.1001643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin W., Wang Z.J., Li Q.Y., Lian J.M., Zhou Y., Lu B.Z., et al. Evolutionary trajectories of snake genes and genomes revealed by comparative analyses of five-pacer viper. Nat Commun. 2016;7 doi: 10.1038/ncomms13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gamble T., Castoe T.A., Nielsen S.V., Banks J.L., Card D.C., Schield D.R., et al. The discovery of XY sex chromosomes in a Boa and Python. Curr Biol. 2017;27:2148–2153.e4. doi: 10.1016/j.cub.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 42.Post Y., Puschhof J., Beumer J., Kerkkamp H.M., de Bakker M.A.G., Slagboom J., et al. Snake venom gland organoids. Cell. 2020;180:233–247.e21. doi: 10.1016/j.cell.2019.11.038. [DOI] [PubMed] [Google Scholar]
- 43.Reyes-Velasco J., Card D.C., Andrew A.L., Shaney K.J., Adams R.H., Schield D.R., et al. Expression of venom gene homologs in diverse python tissues suggests a new model for the evolution of snake venom. Mol Biol Evol. 2015;32:173–183. doi: 10.1093/molbev/msu294. [DOI] [PubMed] [Google Scholar]
- 44.Schield D.R., Card D.C., Hales N.R., Perry B.W., Pasquesi G.M., Blackmon H., et al. The origins and evolution of chromosomes, dosage compensation, and mechanisms underlying venom regulation in snakes. Genome Res. 2019;29:590–601. doi: 10.1101/gr.240952.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Whittington A.C., Mason A.J., Rokyta D.R. A single mutation unlocks cascading exaptations in the origin of a potent pitviper neurotoxin. Mol Biol Evol. 2018;35:887–898. doi: 10.1093/molbev/msx334. [DOI] [PubMed] [Google Scholar]
- 46.Margres M.J., Rautsaw R.M., Strickland J.L., Mason A.J., Schramer T.D., Hofmann E.P., et al. The tiger rattlesnake genome reveals a complex genotype underlying a simple venom phenotype. Natl Acad Sci U S A. 2021;118 doi: 10.1073/pnas.2014634118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su X., Yang L., Wang D., Shu Z., Yang Y., Chen S., et al. 1 K medicinal plant genome database: an integrated database combining genomes and metabolites of medicinal plants. Hortic Res. 2022;9 doi: 10.1093/hr/uhac075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liao B., Hu H., Xiao S., Zhou G., Sun W., Chu Y., et al. Global pharmacopoeia genome database is an integrated and mineable genomic database for traditional medicines derived from eight international pharmacopoeias. Sci China Life Sci. 2021;65:809–817. doi: 10.1007/s11427-021-1968-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang C.C., Lee C.Y. Isolation of neurotoxins from the venom of bungarus multicinctus and their modes of neuromuscular blocking action. Arch Int Pharmacodyn Ther. 1963;144:241–257. [PubMed] [Google Scholar]
- 50.Chang L., Lin S., Huang H., Hsiao M. Genetic organization of alpha-bungarotoxins from Bungarus multicinctus (Taiwan banded krait): evidence showing that the production of alpha-bungarotoxin isotoxins is not derived from edited mRNAs. Nucleic Acids Res. 1999;27:3970–3975. doi: 10.1093/nar/27.20.3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu L.F., Chang C.C., Liau M.Y., Kuo K.W. Genetic characterization of the mRNAs encoding alpha-bungarotoxin: isoforms and RNA editing in Bungarus multicinctus gland cells. Nucleic Acids Res. 1998;26:5624–5629. doi: 10.1093/nar/26.24.5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Denburg J.L., Eldefrawi M.E., O'Brien R.D. Macromolecules from lobster axon membranes that bind cholinergic ligands and local anesthetics (recpetors-procaine-acetylcholine-nicotine-Na+ and K+ gates) Proc Natl Acad Sci U S A. 1972;69:177–181. doi: 10.1073/pnas.69.1.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Greene L.A., Sytkowski A.J., Vogel Z., Nirenberg M.W. Bungarotoxin used as a probe for acetylcholine receptors of cultured neurones. Nature. 1973;243:163–166. doi: 10.1038/243163a0. [DOI] [PubMed] [Google Scholar]
- 54.Chang C.C., Huang M.C. Turnover of junctional and extrajunctional acetylcholine receptors of the rat diaphragm. Nature. 1975;253:643–644. doi: 10.1038/253643a0. [DOI] [PubMed] [Google Scholar]
- 55.Chang C.C., Su M.J. Does alpha-bungarotoxin inhibit motor endplate acetylcholinesterase?. Nature. 1974;247:480. doi: 10.1038/247480a0. [DOI] [PubMed] [Google Scholar]
- 56.Kwong P.D., McDonald N.Q., Sigler P.B., Hendrickson W.A. Structure of beta 2-bungarotoxin: potassium channel binding by Kunitz modules and targeted phospholipase action. Structure. 1995;3:1109–1119. doi: 10.1016/s0969-2126(01)00246-5. [DOI] [PubMed] [Google Scholar]
- 57.Šribar J., Oberčkal J., Križaj I. Understanding the molecular mechanism underlying the presynaptic toxicity of secreted phospholipases A(2): an update. Toxicon. 2014;89:9–16. doi: 10.1016/j.toxicon.2014.06.019. [DOI] [PubMed] [Google Scholar]
- 58.Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liao B., Shen X., Xiang L., Guo S., Chen S., Meng Y., et al. Allele-aware chromosome-level genome assembly of Artemisia annua reveals the correlation between ADS expansion and artemisinin yield. Mol Plant. 2022;15:1310–1328. doi: 10.1016/j.molp.2022.05.013. [DOI] [PubMed] [Google Scholar]
- 60.Haas B.J., Salzberg S.L., Zhu W., Pertea M., Allen J.E., Orvis J., et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008;9 doi: 10.1186/gb-2008-9-1-r7. R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zeng L., Kortschak R.D., Raison J.M., Bertozzi T., Adelson D.L. Superior ab initio identification, annotation and characterisation of TEs and segmental duplications from genome assemblies. PLoS One. 2018;13 doi: 10.1371/journal.pone.0193588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y., Tang H., Debarry J.D., Tan X., Li J., Wang X., et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49. doi: 10.1093/nar/gkr1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang H., Lei R., Ding S.W., Zhu S. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinf. 2014;15:182. doi: 10.1186/1471-2105-15-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., Salzberg S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Durand N.C., Shamim M.S., Machol I., Rao S.S., Huntley M.H., Lander E.S., et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016;3:95–98. doi: 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liao X., Guo S., Yin X., Liao B., Li M., Su H., et al. Hierarchical chromatin features reveal the toxin production in. Bungarus multicinctus. Chinese Med. 2021;16:90. doi: 10.1186/s13020-021-00502-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van Groningen T., Koster J., Valentijn L.J., Zwijnenburg D.A., Akogul N., Hasselt N.E., et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat Genet. 2017;49:1261–1266. doi: 10.1038/ng.3899. [DOI] [PubMed] [Google Scholar]
- 70.Yu G., Wang L.G., He Q.Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 71.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li Q., Ramasamy S., Singh P., Hagel J.M., Dunemann S.M., Chen X., et al. Gene clustering and copy number variation in alkaloid metabolic pathways of opium poppy. Nat Commun. 2020;11:1190. doi: 10.1038/s41467-020-15040-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pierleoni A., Martelli P.L., Casadio R. PredGPI: a GPI-anchor predictor. BMC Bioinf. 2008;9:392. doi: 10.1186/1471-2105-9-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pasquesi G.I.M., Adams R.H., Card D.C., Schield D.R., Corbin A.B., Perry B.W., et al. Squamate reptiles challenge paradigms of genomic repeat element evolution set by birds and mammals. Nat Commun. 2018;9:2774. doi: 10.1038/s41467-018-05279-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu P., Li T., Li R., Jia L., Zhu P., Liu Y., et al. 3D genome of multiple myeloma reveals spatial genome disorganization associated with copy number variations. Nat Commun. 2017;8:1937. doi: 10.1038/s41467-017-01793-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang M., Wang P., Lin M., Ye Z., Li G., Tu L., et al. Evolutionary dynamics of 3D genome architecture following polyploidization in cotton. Native Plants. 2018;4:90–97. doi: 10.1038/s41477-017-0096-3. [DOI] [PubMed] [Google Scholar]
- 78.Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Phillips-Cremins J.E., Sauria M.E., Sanyal A., Gerasimova T.I., Lajoie B.R., Bell J.S., et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–1295. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nora E.P., Lajoie B.R., Schulz E.G., Giorgetti L., Okamoto I., Servant N., et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao L., Xie L., Zhang Q., Ouyang W., Deng L., Guan P., et al. Integrative analysis of reference epigenomes in 20 rice varieties. Nat Commun. 2020;11:2658. doi: 10.1038/s41467-020-16457-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Siang A.S., Doley R., Vonk F.J., Kini R.M. Transcriptomic analysis of the venom gland of the red-headed krait (Bungarus flaviceps) using expressed sequence tags. BMC Mol Biol. 2010;11:24. doi: 10.1186/1471-2199-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu J., Liao B., Yuan L., Shen X., Liao X., Wang J., et al. 50th anniversary of artemisinin: from the discovery to allele-aware genome assembly of. Artemisia annua. Mol Plant. 2022;15:1243–1246. doi: 10.1016/j.molp.2022.07.011. [DOI] [PubMed] [Google Scholar]
- 84.Hu H., Shen X., Liao B., Luo L., Xu J., Chen S. Herbgenomics: a stepping stone for research into herbal medicine. Sci China Life Sci. 2019;62:1–8. doi: 10.1007/s11427-018-9472-y. [DOI] [PubMed] [Google Scholar]
- 85.Kryukova E.V., Egorova N.S., Kudryavtsev D.S., Lebedev D.S., Spirova E.N., Zhmak M.N., et al. From synthetic fragments of endogenous three-finger proteins to potential drugs. Front Pharmacol. 2019;10:748. doi: 10.3389/fphar.2019.00748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tsetlin V.I., Kasheverov I.E., Utkin Y.N. Three-finger proteins from snakes and humans acting on nicotinic receptors: old and new. J Neurochem. 2021;158:1223–1235. doi: 10.1111/jnc.15123. [DOI] [PubMed] [Google Scholar]
- 87.Falk E.N., Norman K.J., Garkun Y., Demars M.P., Im S., Taccheri G., et al. Nicotinic regulation of local and long-range input balance drives top-down attentional circuit maturation. Sci Adv. 2021;7 doi: 10.1126/sciadv.abe1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Artoni P., Piffer A., Vinci V., LeBlanc J., Nelson C.A., Hensch T.K., et al. Deep learning of spontaneous arousal fluctuations detects early cholinergic defects across neurodevelopmental mouse models and patients. Proc Natl Acad Sci U S A. 2020;117:23298–23303. doi: 10.1073/pnas.1820847116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cheng Y.C., Wang J.J., Chang L.S. B chain is a functional subunit of β-bungarotoxin for inducing apoptotic death of human neuroblastoma SK-N-SH cells. Toxicon. 2008;51:304–315. doi: 10.1016/j.toxicon.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 90.Siniavin A., Grinkina S., Osipov A., Starkov V., Tsetlin V., Utkin Y. Anti-HIV activity of snake venom phospholipase A2s: updates for new enzymes and different virus strains. Int J Mol Sci. 2022;23:1610. doi: 10.3390/ijms23031610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Siniavin A.E., Streltsova M.A., Nikiforova M.A., Kudryavtsev D.S., Grinkina S.D., Gushchin V.A., et al. Snake venom phospholipase A2s exhibit strong virucidal activity against SARS-CoV-2 and inhibit the viral spike glycoprotein interaction with ACE2. Cell Mol Life Sci. 2021;78:7777–7794. doi: 10.1007/s00018-021-03985-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hajdinjak M., Mafessoni F., Skov L., Vernot B., Hübner A., Fu Q., et al. Initial upper palaeolithic humans in Europe had recent neanderthal ancestry. Nature. 2021;592:253–257. doi: 10.1038/s41586-021-03335-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Massilani D., Skov L., Hajdinjak M., Gunchinsuren B., Tseveendorj D., Yi S., et al. Denisovan ancestry and population history of early East Asians. Science. 2020;370:579–583. doi: 10.1126/science.abc1166. [DOI] [PubMed] [Google Scholar]
- 94.Doley R., Kini R. Protein complexes in snake venom. Cell Mol Life Sci. 2009;66:2851–2871. doi: 10.1007/s00018-009-0050-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data generated in this study were submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA682532 and PRJNA608620, to the GPGD under the link: http://www.gpgenome.com/species/148 and to the National Genomics Data Center (https://ngdc.cncb.ac.cn/) under the accession number: GWHBJIQ00000000, OMIX001664.