

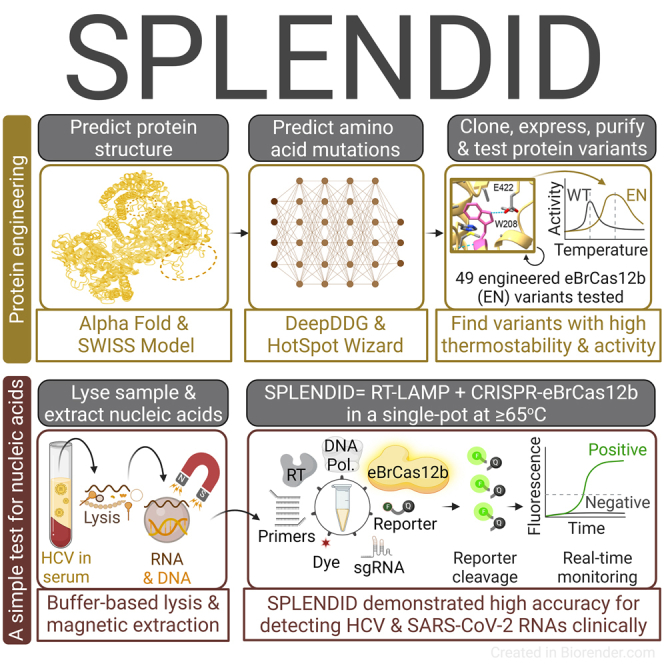
Summary
CRISPR-Cas-based diagnostics have the potential to elevate nucleic acid detection. CRISPR-Cas systems can be combined with a pre-amplification step in a one-pot reaction to simplify the workflow and reduce carryover contamination. Here, we report an engineered Cas12b with improved thermostability that falls within the optimal temperature range (60°C–65°C) of reverse transcription-loop-mediated isothermal amplification (RT-LAMP). Using de novo structural analyses, we introduce mutations to wild-type BrCas12b to tighten its hydrophobic cores, thereby enhancing thermostability. The one-pot detection assay utilizing the engineered BrCas12b, called SPLENDID (single-pot LAMP-mediated engineered BrCas12b for nucleic acid detection of infectious diseases), exhibits robust trans-cleavage activity up to 67°C in a one-pot setting. We validate SPLENDID clinically in 80 serum samples for hepatitis C virus (HCV) and 66 saliva samples for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) with high specificity and accuracy. We obtain results in as little as 20 min, and with the extraction process, the entire assay can be performed within an hour.
Keywords: CRISPR, Cas12b, protein engineering, thermal stability, diagnostics, hepatitis C, point-of-care diagnostics, COVID-19, RT-LAMP, one-pot detection
Graphical abstract
Highlights
-
•
Machine learning tools enable engineering of Cas to improve its thermostability
-
•
eBrCas12b allows RT-LAMP and CRISPR-Cas to be combined in one pot, creating SPLENDID
-
•
SPLENDID enables clinical detection of HCV RNA in serum with high accuracy
-
•
SPLENDID enables clinical detection of SARS-CoV-2 RNA in saliva with high accuracy
Nguyen et al. engineer the CRISPR-associated Cas protein to improve its thermostability. Engineered Cas allows combining target amplification and detection steps in a simple one-pot test that demonstrates high clinical accuracy for detecting HCV and SARS-CoV-2 RNAs and can be easily extended to other targets.
Introduction
Since the beginning of the coronavirus disease 2019 (COVID-19) pandemic, CRISPR-based diagnostic platforms have become a prominent detection technology that could identify the emergence of future pathogens via massive surveillance testing and potentially replace the traditional time-consuming quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) method.1,2,3,4,5 Recent advancements in CRISPR-based tests allow combination of a pre-amplification step, such as reverse transcription-loop-mediated isothermal amplification (RT-LAMP), and a CRISPR-Cas reaction into a convenient one-pot detection assay.6 RT-LAMP is a highly sensitive reaction that can result in detectable signals in as fast as 5–10 min.7 However, because the optimal temperature for the RT-LAMP reaction is around 60°C–65°C,7,8,9 coupling it with CRISPR-Cas systems has been less effective because of the lack of Cas thermostability in this range. The recently reported STOPCovid technology employs a thermostable Cas12b from Alicyclobacillus acidiphilus (AapCas12b), but one drawback of the system is that the Cas effector ceases to display enzymatic activity at temperatures higher than 60°C.6 In our previous study, we implemented a thermostable Cas12b derived from Brevibacillus sp. SYP-B805 (BrCas12b) that exhibits high enzymatic activity up to 63.4°C within a single-pot CRISPR-Cas diagnostic assay for detection of nucleic acids. Our one-pot assay successfully distinguished multiple severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants of concern (VOCs) with high sensitivity and specificity within 20 min.10 However, while BrCas12b allowed detection at a higher temperature than AapCas12b, the range of detection was still narrow. RT-LAMP primers are substantially dependent on temperature because of their loop-forming nature. Consequently, single-degree changes often result in significant improvement of target amplification, sensitivity, and specificity.7,11
To allow enzymatic activity at high temperatures so that complete coverage of the optimal range of RT-LAMP could be attained, we employed de novo structural analyses to engineer a more thermostable BrCas12b for better flexibility for use with the one-pot assay. We employed the capabilities of AlphaFold and SWISS-MODEL to obtain predicted structures of wild-type BrCas12b for visualization of amino acid interaction.12,13,14,15,16,17 These structures were entered into the stability prediction tools HotSpotWizard and DeepDDG, which helped us gain insight into potential mutations that encourage thermostability.18,19 These tools have been particularly useful for determination of hotspots and predicted ΔΔG to aid in protein engineering for enhanced stability, catalytic activity, and specificity. For instance, an engineered version of xylanase from Gloeophyllum trabeum was demonstrated by Wang et al.20 to have an improved temperature optimum up to 80°C and enhanced stability at low pH (2–7.5) compared with the wild-type enzyme, enabling applications in the feed and brewing industries. In another study, mutations D52N and S129A were introduced into pepsin based on ΔΔG calculations, and the half-life of these mutants was increased significantly.21 However, so far there have been no reports of improving the thermostability of Cas effectors for diagnostic purposes.
Here, we report engineered BrCas12b variants (eBrCas12b) that show up to a 13-fold increase in activity and melting temperature of 10°C higher than AapCas12b and 6°C higher than wild-type BrCas12b. When combined in a one-pot reaction with RT-LAMP, the engineered BrCas12b with the highest thermostability was observed to have robust detection capability up to 67°C. Because RT-LAMP is typically performed between 60°C and 65°C, the engineered BrCas12b variant enabled combination of the RT-LAMP amplification and CRISPR-mediated trans-cleavage assays to be carried out at a higher temperature range, allowing more versatility and ease of primer design. The one-pot assay, which we named SPLENDID (single-pot LAMP-mediated engineered BrCas12b for nucleic acid detection of infectious diseases), can be performed in low-resource areas and offers robust sensitivity and specificity in as little as 20 min.
Results
Identification of stabilizing mutants of BrCas12b
Type V and VI CRISPR-Cas systems, such as Cas12a, Cas12b, and Cas13a–Cas13d, have been harnessed for detection of nucleic acids.1,22,23,24,25 While Cas12a and Cas13a have been extensively deployed for on-site applications, Cas12b has recently emerged as a distinct class of enzyme, the majority of which are thermostable.26,27,28 Unlike Cas12a, Cas12b requires a CRISPR RNA (crRNA) and a trans-activating CRISPR RNA (tracrRNA) to carry out cleavage of double-stranded DNA (dsDNA) and subsequent collateral cleavage of single-stranded DNA (ssDNA), referred to as trans-cleavage activity (Figure 1A).26,27,28 Although CRISPR-Cas systems can detect nucleic acids, they suffer from a low sensitivity that lies within the picomolar range.23,29 Traditionally, CRISPR-based detection platforms are coupled with a target pre-amplification step to increase the sensitivity of detection.1,2,3,22 Amplification-free methods have also been developed and successfully executed.30,31 However, they require multiple guide RNAs to have detectable signal readouts, and as a result, it can be challenging for these assays to distinguish subvariants for genotyping purposes.
Figure 1.

Structural prediction of BrCas12b
(A) Schematics of BrCas12b trans-cleavage activity. BrCas12b forms a binary complex with its sgRNA and binds to target dsDNA, which activates its collateral ssDNA cleavage activity.
(B) Predicted structures of BrCas12b derived from AlphaFold and SWISS-MODEL were spatially aligned with its most homologous Cas12b family member, BthCas12b (PDB: 5WTI).32 The circled regions indicate misalignment between the two predicted models when aligned to the BthCas12b structure.
(C) The fitness landscape of BrCas12b was calculated by DeepDDG. Each tile represents a computationally predicted change in the free energy (ΔΔG) relative to the wild-type BrCas12b for the 20 amino acids.
(D) Probability of preservation of function of three residues (F208, L795, and T874) that are likely to increase the thermostability of the BrCas12b effector, calculated by Hotspot Wizard 3.0.
RT-LAMP has emerged as a powerful tool for molecular diagnostics because of its rapid isothermal amplification in as quickly as 5–10 min.7 Because four to six primers are needed to achieve such high sensitivity, false positives resulting from primer-dimer formation are a major limitation of this technology.33,34 Recent efforts have coupled RT-LAMP with a thermostable CRISPR-Cas complex in a one-pot reaction to overcome this drawback.6 The programmability of the CRISPR-Cas systems improves the specificity of detection. Upon amplification, correct base-pairing between the guide RNA and the amplified target serves as an additional checkpoint to provide accurate signal readouts. The one-pot reaction further eliminates the carryover contamination that is frequently observed with two-pot RT-LAMP-coupled platforms.10 Most RT-LAMP reactions work optimally between 60°C and 65°C;7,11 however, only a handful of Cas enzymes are functional at this temperature range.6,35 The discrepancies in reaction conditions and inhibitory effects between amplification and trans-cleavage have been major hurdles in developing such one-pot systems. Recently, we reported use of a thermostable Cas12b from Brevibacillus sp. SYP-B805 (BrCas12b) that outperformed previous platforms in a one-pot detection setting. Nevertheless, the thermostable nature of BrCas12b only allowed this system to work robustly up to 63.4°C.10
Previous studies have shown that engineering the crRNA for Cas12a enhances its trans-cleavage activity.29,36 However, the thermostability of Cas enzymes is mainly linked to their secondary and tertiary structures.37,38 Therefore, rather than focusing on modifying a single guide RNA (sgRNA) for BrCas12b, we aimed to engineer the enzyme itself not only to boost its activity but also its thermostability for better synergy with the RT-LAMP reaction. We employed structure-guided rational design to identify amino acid positions where mutations would likely be beneficial. To accomplish this, we predicted the structure of BrCas12b utilizing AlphaFold and SWISS-MODEL followed by sequence alignment of these models with the most homologous solved Cas12b ortholog, BthCas12b (PDB: 5WTI)12,13,14,15,17,32 to identify functional domains. Overall, the AlphaFold and SWISS-MODEL predictions were congruent with one another, with the exception of two minor regions close to the RuvC domains (Figure 1B). We next processed these predicted models via DeepDDG, a neural network-based algorithm to generate a fitness landscape of all amino acid positions.19 We observed that the majority of the mutations were destabilizing, except for hydrophobic substitutions in the REC1, RuvC, and UK-I regions (Figure 1C). Based on Gibbs free energy changes from the fitness landscape, we analyzed these mutations individually to determine predictors of thermostability in terms of hydrophobicity, compactness, hydrogen bonding, and salt bridges. In addition to these thermostability factors, we also applied the semi-rational protein design tool HotSpot Wizard 3.0 to select the best candidates for further validation.18 We pooled a few single-point mutations that were predicted to be destabilizing according to their ΔΔG values as controls. We identified the three most promising candidates (F208, L795, and T874) from consensus of both algorithms and their computed mutational landscape, which inferred the probability of preservation of function (Figure 1D).
Engineered BrCas12b showed robust activity at high temperature
We expressed and purified 35 single-point-mutated BrCas12b variants and characterized their thermostability and catalytic activity at different temperatures. Using differential scanning fluorimetry, we observed that 16 variants increased the melting temperatures (Tm) relative to the wild-type BrCas12b, whose apo form had a Tm of 61.4°C. Five variants (F208W, D567W, D868I, D868V, and D951F) displayed an increase of more than 2°C in their Tm compared with the wild type (Figures 2A and S1). To investigate whether a combination of these stabilizing mutations would display an additive effect on the overall stability of the protein, we stacked beneficial mutations on top of each other to create several multimutated variants. We observed that the majority of stacked mutations, such as F208W/D868V (FD), F208W/N524V/D868V (FND), F208W/N524V/T874H (FNT(H)), and F208W/N524V/L795I/D868V (FNLD) exhibited an overall additive increase in Tm; however, there were a few exceptions; for instance, a trimutant F208W/N524V/T874S (FNT(S)) decreased thermostability upon stacking and a hexamutant FNLD/T874S/A1015E (FNLDTA) showed no increase in Tm (Figures 2A–2C).
Figure 2.

Functional characterization of BrCas12b variants
(A) Change in Tm of BrCas12b variants compared with the wild-type effector (n = 4, two technical replicates examined over two independent experiments). Error bars represent mean ± SEM.
(B) Differential scanning fluorimetry plots in relative fluorescence units (RFUs) comparing the wild-type BrCas12b for selected variants screened from (A) (n = 4, two technical replicates examined over two independent experiments).
(C) Differential scanning fluorimetry plots in terms of derivative of RFU in (B). Global minima of the curve were determined to be the melting point of the protein (n = 4, two technical replicates examined over two independent experiments).
(D) The trans-cleavage activity of BrCas12b variants was determined at increasing temperatures. The heatmap represents the background-corrected RFU at t = 15 min (n = 2 biological replicates).
(E and F) Prediction accuracy in terms of thermostability for SWISS-MODEL and AlphaFold models (n = 4, two technical replicates examined over two independent experiments).
We performed an in vitro trans-cleavage assay on all variants to check for functional preservation at elevated temperatures. While the wild-type BrCas12b variants ceased to work above 65°C (possibly because of denaturation at high temperature), the engineered BrCas12b variants FN, FND, FNT(S), FNLD, R160E/FND (RFND), and FNLDTA showed robust activity up to 68°C (Figures 2D and S2). We also observed that some stabilizing mutants like D567W, T874H, and D951F were no longer active; by examining the predicted structure, we learned that these mutations were in the RuvC and UK-I regions and potentially affected Cas12b-mediated cleavage activity. We next sought to characterize the accuracy of the AlphaFold and SWISS-MODEL predictions by comparing the computationally predicted single-point mutants with the experimental observations. For SWISS-MODEL, only 13 of 25 mutants were experimentally consistent with the software calculations, yielding a prediction accuracy of 52%. On the other hand, the AlphaFold model resulted in 15 of 25 correct mutants, yielding a higher prediction accuracy of 60% (Figures 2E and 2F).
Engineered BrCas12b exhibited improved thermostability while maintaining specificity compared with its wild type
We next carried out time-dependent trans-cleavage assays to investigate the stability of BrCas12b variants compared with their wild type. Ribonucleoprotein (RNP) complexes were incubated between 10 and 60 min at different temperatures ranging from 60°C–75°C. The dsDNA target and a fluorescence-based reporter were then added to the RNP complex to initiate non-specific ssDNA cleavage. Based on kinetic measurements, we observed that the FND and RFND variants showed robust trans-cleavage activity at 68°C when the RNP complex was incubated for up to 20 min and 50 min, respectively. On the other hand, the wild-type BrCas12b failed to have detectable trans-cleavage activity under the same conditions (Figures 3A–3C). To investigate the specificity of these BrCas12b variants, we designed the dsDNA target with single-point or double-point mutations across its first 10 bases proximal to the protospacer adjacent motif (PAM) site. While the FND variants showed specificity comparable with the wild-type BrCas12b, RFND displayed a much lower tolerance to double-point mutations near the seed region than that observed in wild-type BrCas12b (Figures 3D–3F). Overall, the RFND variant exhibited the highest thermostability and specificity among all variants tested.
Figure 3.

Engineered BrCas12b variants exhibit exceptional stability compared with wild-type BrCas12b
(A–C) Time-dependent trans-cleavage activity at different temperatures for wild-type BrCas12b, the FND (F208W/N524V/D868V) variant, and the RFND (R160E/FND) variant, respectively. The heatmap represents background-corrected mean RFU at t = 15 min (n = 2 biological replicates).
(D–F) Specificity testing of wild-type BrCas12b, the FND variant, and the RFND variant against mutated activators (n = 2 biological replicates). Error bars represent mean ± SD.
(G) Structural insights into the mechanism of enhanced stability of BrCas12b variants at amino acid positions 208 and 868. The top panels display the wild-type BrCas12b with F208 (left) and D868 (right), while the bottom panels depict the engineered eBrCas12b with W208 (left) and V868 (right).
We hypothesized that the mutations F208W, N524V, and D868V contributed to the increase in Tm, while the R160E mutation enhanced the enzyme activity. By closely looking at the BrCas12b predicted structure, we observed that the tryptophan at position 208 further stabilized the interaction with His394, likely by hydrophobic π-π stacking and formation of an extra hydrogen bond with Glu422.39 Through biochemical assays, this mutation was observed to be the most contributing factor to the overall thermostability of the enzyme. Val868 allowed improved compactness in the hydrophobic core of the RuvC region through more interactions with nearby hydrophobic residues (Figure 3G). For the mutation R160E, we did not observe an increase in Tm via differential scanning fluorimetry; however, its trans-cleavage activity was higher than that of the wild-type BrCas12b. We hypothesized that conversion of R160E to a negatively charged residue may have subsequently led to enhancement in dsDNA cleavage.
Engineered BrCas12b variants showed robust activity up to 67°C in an RT-LAMP-mediated one-pot reaction
We aimed to investigate the thermostability of several engineered BrCas12b effectors in a one-pot setting. To accomplish this, we employed a LAMP primer set described in Broughton et al.2 that was used to detect the SARS-CoV-2 N gene and tested them at high temperatures ranging from 62°C–70°C with an increment of 1°C. Interestingly, these proof-of-principle primers worked robustly up to 68°C. We next coupled some of the most promising BrCas12b variants with the RT-LAMP reaction in a single pot at different temperatures ranging from 64°C–68°C. Notably, the FND, FNLD, RFND, F208W/N524V/P763C/D868V (FNPD), and FNLDTA variants showed a high detection signal at 64°C, with FND, FNLD, and RFND exhibiting an approximately 7-fold increase in activity compared with the wild-type BrCas12b (Figure 4A). At 65°C, the wild-type BrCas12b completely ceased activity because of denaturation at high temperatures (Figure 4B). The engineered BrCas12b variants worked robustly up to 66°C but performed poorly at 67°C, possibly because of reaching their thermostability limit (Figures 4C and 4D).
Figure 4.

RT-LAMP-coupled one-pot detection of BrCas12b variants
(A–D) One-pot detection of BrCas12b variants at 64°C, 65°C, 66°C, and 67°C, respectively (n = 3 biological replicates). FNT, F208W/N524V/T874S; FNLD, F208W/N524V/L795I/D868V; FNPD, F208W/N524V/P763C/D868V; FNLDTA, F208W/N524V/L795I/D868V/T874S/A1015E.
(E) Optimization of sucrose as an additive to the one-pot reaction at 67°C (n = 3 biological replicates).
(F) One-pot detection reaction of BrCas12b variants with 200 mM sucrose at 67°C (n = 3 biological replicates).
(G and H) PAM-dependent detection of the RFND variant with and without RT-LAMP reaction.
Recent studies have shown that additives can boost the activity of CRISPR-Cas reactions in a one-pot setting.6,40,41,42,43 For instance, Joung et al.6 used taurine in their STOPCovid reaction and observed enhanced detectable signals. Proline was also added to boost Cas12a activity in a two-pot setting.41 Therefore, we sought to leverage the one-pot reaction using BrCas12b variants by exploring several additives, such as taurine, mannitol, sucrose, trehalose, and betaine (Figures S3A–S3C). We selected the FNLDTA variant for testing because it showed the highest fluorescence signal at 67°C (Figure 4D). We discovered that adding sucrose to the one-pot reaction at a final concentration of 200 mM greatly enhanced eBrCas12b enzymatic activity up to approximately 2.5-fold (Figure 4E). We then applied 200 mM sucrose to the remaining variants and found that sucrose was able to restore their activity at 67°C (Figures 4F and S3A–S3C). Interestingly, we also observed that the activity of RFND was not enhanced at 64°C and 65°C but, rather, slightly reduced when 200 mM sucrose was incorporated into the one-pot reaction. We hypothesized that sucrose helps restore the function of RFND at the upper temperature limit and does not act as an enhancer but, rather, as a stabilizer against chemical denaturants and temperature by altering the protein solvation state and its interaction with the surroundings44,45,46 (Figures S3D–S3F). We called the one-pot assay utilizing the RFND variant and optimized sucrose condition SPLENDID.
CRISPR-Cas12b-mediated cleavage and detection of dsDNA are dependent on the presence of a short PAM upstream of the target sequence. It was empirically determined that the canonical PAM for BrCas12b was TTN.26,27,28,47 However, we speculated that, when targeting ssDNA, a PAM sequence may not be required because it shares similar enzymatic activity with Cas12a.23 Because the RT-LAMP reaction can result in ssDNA by-products because of its loop-forming nature at elevated temperatures,7 we hypothesized that PAM-less or near-PAM-less detection could be achieved by our RT-LAMP-coupled one-pot assay. This could potentially alleviate some challenges in primer and guide RNA designs. To investigate the PAM dependency of eBrCas12b, we designed a PAM library of dsDNA activators that were comprised of all possible 3-nt PAM sequences (NNN). We first sought to test the trans-cleavage of the RFND variant against these PAM combinations without the RT-LAMP step. We observed that the TTN PAM-containing activators displayed the highest activity with the engineered Cas12b, consistent with previous studies. We also noticed significant trans-cleavage activity with activators containing the ATA, ATT, and ATC PAM but not with ATG. Similarly, TAA-, TAT-, and TAC-containing activators had low levels of detection but not with TAG. Our results indicate that the presence of a single T at either the first or the second position is sufficient to initiate trans-cleavage of eBrCas12b provided that the third position contains A, T, or C. We next tested the detection of all the PAM-library activators in a one-pot setting (with RT-LAMP amplification). We observed that, while the TTH PAM-containing activators had the most robust detection, the majority of non-canonical PAM-containing sequences also showed detection, albeit at lower levels, with the only exceptions being CTC, CGC, and CCC PAMs, which showed no detection (Figures 4G and 4H). Our results demonstrate that it is possible to target a variety of non-canonical PAM sequences when amplified by RT-LAMP in a one-pot assay.
Validation of SPLENDID in hepatitis C- and SARS-CoV-2-infected samples
Screening for hepatitis C virus (HCV) is of high importance for early treatment because there are no vaccines available.48,49,50 Individuals infected with HCV can be cured within 8–12 weeks when the virus is detected early; otherwise, cirrhosis, hepatocellular carcinoma, and potential death can occur if left unnoticed.50,51 Traditional antibody assays offer up to 98% sensitivity and 99% specificity; however, these assays cannot distinguish between past and active infection. On the other hand, quantitative HCV RNA tests also exhibit a similar sensitivity, specificity, and low limit of detection (10–15 IU/mL),52,53,54 but these detection platforms are laborious, time consuming, and uneconomical. Therefore, as a proof of concept, we sought to develop a simple, rapid test for HCV using SPLENDID.
Prior to testing, a single-guide RNA was designed to target the 5′ untranslated region (5′ UTR) of the HCV RNA (+) genome (Figure 5A; Table S1). We then proceeded to clinically validate SPLENDID in 80 human serum samples (40 HCV-infected samples and 40 negative samples). Within the 40 HCV-infected samples, we had a genotypic distribution as follows: 34 samples of HCV genotype 1 (GT1), 4 samples of HCV genotype 2 (GT2), and 2 samples of HCV genotype 3 (GT3). We used LAMP primers as described in the study by Hongjaisee et al.55 but modified the forward inner primer (FIP) primer to make space for the guide-RNA binding. Crude samples were processed via a magnetic-based extraction to isolate viral RNA. These samples were randomized and blinded before undergoing SPLENDID testing (Figure 5B). Additionally, we performed an in-house qRT-PCR assay to determine the cycle threshold (Ct) value of each sample and compare the results with the clinically validated viral loads provided by HCV-TARGET and Boca Biolistics (Figure S4). All fluorescence-based measurements were compared with the qRT-PCR results to determine the sensitivity, specificity, and accuracy. Overall, we observed 7 false negatives and 1 false positive, achieving a specificity of 97.5%, an accuracy of 90.0%, and a sensitivity of 82.5% (Figures 5C and 5D). However, genotype-specific measurements indicated that we had a sensitivity of 91% (GT1), 50% (GT2), and 0% (GT3), respectively (Table S2). Our results show that, while the LAMP primers and guide RNA used in this study work robustly for HCV GT1, they perform poorly with other HCV genotypes. HCV GT2 contains multiple mutations within the LAMP primer binding region as well as the guide RNA binding region, which might explain the poor performance of our test with that genotype. The performance of SPLENDID for detection of diverse HCV genotypes can be improved simply by designing LAMP primers and guide RNA to be more specific to the target genotype. When analyzing the patient sample fluorescence results via a receiver operating characteristic (ROC) curve for the SPLENDID assay at time (t) = 25 min, we observed that the area under the curve (AUC) was 92.3%, indicating a high threshold for differentiation between positive and negative samples (Figure 5E).
Figure 5.

Clinical validation of SPLENDID in HCV-infected samples
(A) Schematic of the HCV RNA genome. Single-guide RNA was designed to target the 5′ untranslated region (5′ UTR) of the virus.
(B) Detailed steps of the SPLENDID assay from patient sample extraction via a magnetic-based method to the one-pot testing.
(C) Fluorescence measurements of 34 HCV-1, 4 HCV-2, and 2 HCV-3 as well as 40 healthy clinical samples from the SPLENDID assay. Different genotype signal readouts were taken at t = 25 min. The reaction was run at 65°C, and this temperature was determined by screening the RT-LAMP reaction alone in the absence of the CRISPR-eBrCas12b complex.
(D) Summary of clinical validation of SPLENDID in (D) in terms of sensitivity, specificity, and accuracy.
(E) Receiver operating characteristic (ROC) curve of the assay at t = 25 min.
Another clinically relevant disease that requires surveillance testing is COVID-19. SARS-CoV-2 quickly sparked a global pandemic and has become increasingly contagious. We therefore tailored the SPLENDID assay to detect SARS-CoV-2 in patient samples. Specifically, we targeted the N gene, which is less likely to mutate. We performed the one-pot assay on 66 SARS-CoV-2 saliva samples (29 positive and 37 negative) using SYTO 9 dye to track the RT-LAMP amplification and a fluorescence resonance energy transfer (FRET)-based hexachloro-fluorescein (HEX) reporter to monitor the trans-cleavage activity of the eBrCas12b RFND. We observed that the RT-LAMP reaction resulted in a high false-positive rate (8 of 37 negative samples were false positives). On the other hand, the SPLENDID assay produced no false positives (Figures S5A and S5B). Overall, the RT-LAMP reaction alone achieved 78.4% specificity, 86.4% accuracy, and 96.6% sensitivity; the SPLENDID assay attained 100% specificity, 97.0% accuracy, and 93.1% sensitivity (Figure S5). These results further solidified the importance of having the CRISPR-eBrCas12b in the rapid one-pot reaction to increase the specificity and accuracy of nucleic acid detection.
Discussion
CRISPR-based diagnostics offer many advantages over traditional testing methods for nucleic acid detection because of their high specificity, flexible programmability, and quick turnaround time. To increase sensitivity, the CRISPR-Cas reaction is often paired with a pre-amplification step such as reverse transcription-recombinase polymerase amplification (RT-RPA) or RT-LAMP. Here we report an eBrCas12b with substantial improvements in terms of thermostability compared with other CRISPR-based one-pot assays.
Of several mutations introduced to the wild-type BrCas12b to create engineered variants, we observed a substantial improvement in thermostability of the proteins at two residues, F208W and D868V, each with an around 2°C increase in Tm compared with the wild type. Structurally, these two residues are located at two distinct regions of the protein, with F208 at the intersection of the PAM-interacting domain and REC1 and with D868 being proximal to the catalytic pocket RuvCII domain. We hypothesized that F208W enabled enhanced π-π stacking with its neighbor H394, while D868V increased the compactness of its local region by increasing the hydrophobic surface interactions with its surroundings.
Our engineered detection assay using eBrCas12b, which we called SPLENDID, works robustly up to 67°C in a one-pot setting, encompassing the optimal temperature range of the RT-LAMP reaction. SPLENDID combines two promising rapid detection platforms, amplification-based RT-LAMP and CRISPR assays, into a single pot reaction with improved synergy for a robust two-step verification for the presence of target nucleic acids: (1) amplification of target and (2) collateral cleavage of BrCas12b. We applied SPLENDID to clinically validate 80 HCV and 66 SARS-CoV-2 patient samples. Early detection of HCV and SARS-CoV-2 is important for preventing acute complications and contagion. Current testing methods are costly, require specialized personnel and equipment, and may not distinguish between active and past infections. Our SPLENDID system utilizing the engineered thermostable BrCas12b (eBrCas12b) was able to successfully detect the presence of HCV in human serum samples with a specificity of 97.5%, an accuracy of 90.0% (overall) to 94.6% (GT1), and sensitivity of 82.5% (overall) to 91.1% (GT1). For SARS-CoV-2 testing, the assay achieved 97.0% accuracy, 100% specificity, and 93.1% sensitivity (Figure S5).
Compared with other CRISPR-based and non-CRISPR-based detection platforms, the SPLENDID assay demonstrated similar accuracy, specificity, and sensitivity while being a more rapid and convenient one-pot reaction with the potential to be deployed outside of the standard lab setting (Table S3). This detection platform can be applied toward many other pathogens because of the flexible programmability of the guide RNA design. We speculate that the sensitivity of our clinical validation testing heavily relied on the quality of the LAMP primer design. As a result, one way to improve the assay is to test many more LAMP primers to obtain the most optimal set.
Additionally, the SPLENDID assay can be performed in as little as 20 min, and the whole process, including sample extraction, takes up to an hour. When paired with a more rapid extraction method, the overall assay duration can be reduced. Additionally, one-pot reagents can potentially be lyophilized, as we discussed in our previous study, thus allowing administration in low-resource areas.10 We believe that the SPLENDID system has wide application for detection of emerging infectious diseases and can be quickly deployed to regions of interest at low cost.
Cas12b has been successfully repurposed for gene editing through protein engineering.26 We observed that several point mutations to the wild-type BrCas12b decreased its Tm while maintaining the dsDNA cleavage capability. Therefore, we believe that these mutations can be stacked to create lower melting point Cas12b variants capable of dsDNA cleavage at 37°C to be used for gene editing. Additionally, because of the similarity in structure and cleavage patterns between Cas12a and Cas12b, we imagine that Cas12b can be engineered to carry out precision base editing without dsDNA breaks.56,57 The design approaches of BrCas12b presented here demonstrate new engineering strategies for use of CRISPR-Cas systems in diagnostics and, potentially, therapeutic applications.
Limitations of the study
While SPLENDID offers many advantages as a rapid point-of-care diagnostic tool, we acknowledge that it has some limitations. SPLENDID still heavily relies on the robustness of the RT-LAMP reaction because of the low sensitivity of the trans-cleavage activity of CRISPR-Cas systems. Therefore, careful design of LAMP primers is crucial for the success of the assay. Additionally, although SPLENDID exhibited high sensitivity when validated in SARS-CoV-2 patient samples (93.1%), its sensitivity toward HCV patient samples was significantly lower (82.5% overall, 91% for GT1). For this reason, the assay needs further optimization to detect other genotypes and become practical for HCV on-field testing.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Rosetta™ 2(DE3)pLysS Singles Competent Cells |
Millipore Sigma | Cat# 71401 |
| NEB® 5-alpha Competent E. coli (High Efficiency) | New England Biolabs | Cat# C2987H |
| Biological samples | ||
| HCV Infected Patient Serum Samples | HCV-TARGET | N/A |
| Healthy Patient Serum Samples | Boca Biolistics | N/A |
| SARS-CoV-2 Infected Patient Saliva Samples | Clinical and Translational Biorepository; University of Florida | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| SYTO™ 9 Green Fluorescent Nucleic Acid Stain | Thermo Fisher Scientific | Cat# S34854 |
| IPTG | Gold Biotechnology | Cat# I2481C |
| NaCl | Fisher Scientific | Cat# BP358-212 |
| Tris-HCl | Gold Biotechnology | Cat# T-095-100 |
| Thermo Scientific™ PMSF Protease Inhibitor | Fisher Scientific | Cat# 36978 |
| TCEP-HCl | Gold Biotechnology | Cat# TCEP1 |
| Lysozyme | MP Biomedicals, LLC | Cat# 02100831-CF |
| Imidazole | AmBeed | Cat# A277098 |
| Deoxyribose Nuclease I (Bovine Pancreas) | Worthington Biochemical Corporation | Cat# LS002145 |
| Q5® polymerase | New England BioLabs | Cat# M0491S |
| TEV protease | Kapust et al.58 | Addgene #8827 |
| HEPES | Sigma Aldrich | Cat# H3375-500G |
| Critical commercial assays | ||
| Q5® Site-Directed Mutagenesis Kit |
New England BioLabs | Cat# E0554S |
| WarmStart® Multi-Purpose LAMP/RT-LAMP 2X Master Mix (with UDG) | New England BioLabs | Cat# M1708S |
| Luna® Probe One-Step RT-qPCR 4X Mix with UDG | New England BioLabs | Cat# M3019S |
| NEBuilder® HiFi DNA Assembly Cloning Kit | New England BioLabs | Cat# E5520S |
| Protein Thermal Shift™ Dye Kit | Thermo Fisher Scientific | Cat# 4461146 |
| Quick-DNA/RNA Viral MagBead Kit | Zymo Research | Cat# R2140 |
| Deposited data | ||
| 6xHis-MBP-BrCas12b-RFND | This Paper | Addgene# 195339 |
| 6xHis-MBP-BrCas12b-FNLDTA | This Paper | Addgene# 195340 |
| Oligonucleotides | ||
| RT-LAMP Primers SARS-CoV-2, see Table S1 | Broughton et al.2 | N/A |
| RT-LAMP Primers 5′ UTR of Hepatitis C, see Table S1 | Hongjaisee et al.55 | N/A |
| RT-qPCR HCV Primers and Probes, see Table S1 | Zauli et al.59 | N/A |
| RT-qPCR HCV Modified ZEN Probe, see Table S1 | Integrated DNA Technologies | N/A |
| Single-guide RNA Br_sgN_CoV2, see Table S1 | This Paper; Synthesized by Integrated DNA Technologies | N/A |
| Single-guide RNA Br_sgUTR_HCV, see Table S1 | This Paper; Synthesized by Integrated DNA Technologies | N/A |
| Reporter 1:/5HEX/TTTTTTTT/3IABkFQ/ | This Paper; Synthesized by Integrated DNA Technologies | N/A |
| Reporter 2:/5FAM/TTTTTTTTTT/3IABkFQ/ | This Paper; Synthesized by Integrated DNA Technologies | N/A |
| SARS-CoV-2 Activator PAM Library | Twist Biosciences | N/A |
| Recombinant DNA | ||
| 6xHis-MBP Destination Vector | Gift from Scott Gardia | Addgene# 29656 |
| pET28a+ Vector | Shilling et al.60 | Addgene# 154464 |
| Software and algorithms | ||
| Colab-Fold | Mirdita et al.61 | https://github.com/sokrypton/ColabFold |
| SWISS-MODEL | Waterhouse et al.13 | https://swissmodel.expasy.org/ |
| HotSpot Wizard 3.0 | Sumbalova et al.18 | https://loschmidt.chemi.muni.cz/hotspotwizard/ |
| DeepDDG | Cao et al.19 | http://protein.org.cn/ddg.html |
| NEB LAMP Primer Design Tool | New England Biolabs | https://lamp.neb.com/#!/ |
| PrimerExplorer v5 | Eiken Chemical Co. | https://primerexplorer.jp/e/ |
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com/scientificsoftware/prism/ |
| Other | ||
| Luria Broth | Fisher Scientific | Cat# BP9723-500 |
| Terrific Broth | Cold Spring Harbor Protocols | http://cshprotocols.cshlp.org/content/2015/9/pdb.rec085894.full?rss=1 |
| SOC Medium | New England Biolabs | B9020S |
| Antifoam 204 | Millipore Sigma | Cat# A8311-50ML |
| NEBuffer 2⋅1 | New England Biolabs | Cat# B9200S |
| 10X Nuclease-Free Duplex Buffer | Integrated DNA Technologies | Cat# 11-05-01-12 |
| Histrap 5 mL FF Column | Cytiva Life Sciences | Cat# 17524802 |
| HiTrap 1 mL SP HP Column | Cytiva Life Sciences | Cat# 29051324 |
| 0.22-Micron Filter | Millipore Sigma | Cat# GSWP14250 |
| SDS-PAGE 4–12% Gels | Genscript | Cat# M00653 |
| 50 kDa MWCO Amicon Ultra-15 Filter | Millipore Sigma | Cat# UFC9050 |
Resource availability
Lead contact
Further inquiries and information regarding resources and reagents should be directed to and will be fulfilled by the lead contact, Piyush Jain (jainp@ufl.edu).
Materials availability
Distribution of engineered BrCas12b mutant plasmids can be found on Addgene (plasmid # 195339 for 6xHis-MBP-BrCas12b-RFND and plasmid # 195340 for 6xHis-MBP-BrCas12b-FNLDTA).
Experimental model and subject details
Patient details
For this study, de-identified patient sample collection and processing were approved as a non-human study by the University of Florida Institutional Review Board (IRB202003085 and IRB202000781) and all ethical regulations were followed. For clinical validation, 80 patient samples consisting of human serum were obtained, 40 of which were HCV- positive and collected and de-identified by HCV-TARGET consortium; the other 40 derived from healthy patients were obtained from a commercial vendor, Boca Biolistics. All HCV-positive samples were collected by HCV-TARGET with obtaining a written informed consent from the patient. All SARS-CoV-2-positive saliva samples (n = 29) were obtained from the University of Florida CTSI Biorepository under the IRB202003085 protocol, which was approved as an exempt study with de-identified samples under confidentiality agreements signed by investigator (recipient investigator) and the code owner (collecting investigator). All healthy serum (n = 40) and saliva (n = 37) samples were procured by Boca Biolistics from their network of CAP/CLIA accredited partner laboratories across the United States as remnant (leftover) samples. These samples were delinked under the IIRB delinking protocol SOP 10–00114 Rev E.
Males and females greater than or equal to age 18 years who tested positive for HCV by an RNA test were included as positive. Males and females greater than or equal to age 18 years who tested negative for HCV by an RNA test were included as negative. For positive samples, fibrosis stage and prior treatment information is included in the supplementary information. Viral RNA was extracted from serum samples using Quick-DNA/RNA Viral MagBead Kit R2140 (Zymo Research. Extracted patient samples were chosen at random and blinded for one-pot testing. Viral RNA from saliva samples were extracted and quantified by following the CDC guidelines.
Cell lines
All BrCas12b variants were expressed in Rosetta 2(DE3)pLysS Singles Competent Cells (purchased from Millipore Sigma). Cells were kept frozen at −80°C until use. Cells were cultured as per supplier’s protocol with slight modifications. For transformation, frozen cells were thawed and then incubated with plasmids for 5 min on ice, heat-shocked for 30 s at 42°C in a water bath, placed back on ice for 2 min, and then cultured at 37°C in SOC medium (New England Biolabs) at 250 rpm for 1 h before spreading on antibiotic-containing agar plates. After spreading, the plates were incubated at 37°C for 12–48 h and then the colonies were picked, expanded in 10 mL Luria Broth (Fisher Scientific) containing the appropriate antibiotics, and sequenced by Sanger sequencing. Please refer to protein expression and purification section for more details.
Method details
Computational analyses
Protein structure predictions were performed on a local install of Colab-Fold with “AlphaFold2-ptm” parameters using A100 GPUs, where MSA search was done by MMseqs2 on Uniref. 100 database.61 For SWISS-MODEL predictions, protein sequences were input into the web-based tool via Expasy web server.13,15,16,17,62 Protein structures in pdb files were processed via both HotSpot Wizard 3.018 and DeepDDG19 web-based software.
Nucleic acid preparation
Double-stranded DNA targets, single-guide RNAs, and fluorescence-quencher reporters were synthesized by Integrated DNA Technologies (IDT). PAM Library targets for SARS-CoV-2 N gene was designed and synthesized by Twist Bioscience. For the trans-cleavage assay without RT-LAMP, the target-strand (TS) and non-target-strand (NTS) activators were ordered as 60-mer ssDNA oligos and annealed in a 5:1 (NTS:TS) ratio in 1X nuclease-free duplex buffer (IDT).
Site-directed mutagenesis
The wild-type BrCas12b gene was codon-optimized for E. coli expression, synthesized by Twist Bioscience, and cloned into pET28a+ vector. For a mutant with single-point or double-point mutations, mutagenesis was carried out using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs) following the manufacturer’s protocols. For simultaneous triple-point mutations, primers with intended mutations were designed and used to amplify each fragment of the BrCas12b using Q5 polymerase and assembled into a 6xHis-MBP destination vector (a gift from Scott Gardia, Addgene# 29656) using NEBuilder HiFi DNA Assembly Kit (New England Biolabs).
Protein expression and purification
Expression and purification of BrCas12b variants were performed according to a previous study with minor modifications.10 After transforming BrCas12b variants into Rosetta 2(DE3) pLysS Singles™ Competent Cells (Milipore Sigma), individual colonies were selected and propagated overnight (12–15 h) in 10 mL of Luria Broth (Fisher Scientific) containing the appropriate antibiotics. The bacterial cultures were then scaled up in 2 L of Terrific Broth containing the corresponding antibiotics and 5 drops of antifoam 204 (Sigma Aldrich). Once an OD600 of 0.5–0.8 was attained, the culture was cooled in an ice bath for 30 min, induced with 0.2 mM IPTG (isopropyl b-d-1-thiogalactopyranoside), and shaken at 18°C overnight (14–18 h). The bacterial cells were then pelleted (39,800 xg at 4°C), diluted in lysis buffer (0.5 M NaCl, 50 mM Tris-HCl, pH = 7.5, 1 mM PMSF, 0.5 mM TCEP, 0.25 mg/mL lysozyme, and 10 mg/mL deoxyribose nuclease I), and disrupted by sonication. The lysed cells were then spun down (39,800 xg at 4°C), and the supernatant was subjected to suction filtration through a 0.22-micron filter (Millipore Sigma). The filtered solution was purified using an FPLC Biologic Duoflow system (Bio-rad) through a Histrap 5 mL FF column (Cytiva), which was then washed (wash buffer: 0.5 M NaCl, 50 mM Tris-HCl, pH = 7.5, 20 mM Imidazole, 0.5 mM TCEP) before eluting (elution buffer: 0.5 M NaCl, 50 mM Tris-HCl pH = 7.5, 300 mM imidazole, 0.5 mM TCEP). For BrCas12b variants that were cloned into the 6xHis-MBP destination vector, an extra step of TEV site cleavage was performed to remove the MBP tag, and the protein was processed using Hitrap Heparin HP (Cytiva) purification. The elution was evaluated using SDS-PAGE; the fractions containing the protein were consolidated and concentrated (2000 xg at 4°C) in a 50 kDa MWCO Amicon Ultra-15 centrifugal filter unit (Millipore Sigma). The protein was stored in −20°C for further use, with back-up reserves of each protein kept at −80°C after dilution (buffer C: 150 mM NaCl, 50 mM HEPES, pH = 7, 0.5 mM TCEP).
Differential scanning fluorimetry
The melting temperature of each protein was mainly carried out in its apo form. BrCas12b was mixed in Protein Thermal Shift™ buffer (Thermofisher) in combination with 1X reaction buffer (100 nM NaCl, 50 nM Tris-HCl, pH = 7.5, 1mM DTT, and 10 mM MgCl2) to a final concentration of 500 nM. Protein Thermal Shift dye™ (Thermofisher) was then added to each mixture before being transferred to the qPCR StepOne Plus system (Thermofisher). The temperature profile was observed over a temperature range of 25°C–80°C at a ramp rate of 1%/s. Experiments were performed in duplicates and repeated twice.
Temperature- and time-dependent trans-cleavage assay
The thermostable BrCas12b variants and sgRNA were combined in 1X NEBuffer 2.1 (New England Biolabs) to a final concentration of 50 nM: 100 nM (sgRNA: BrCas12b) and then transferred to a pre-heated CFX96 Real-Time PCR system with C1000 Thermal Cycler module (Bio-rad) using a temperature gradient setting across each row, ranging from 60°C to 75°C (60°C, 61°C, 63°C, 65.9°C, 69.5°C, 72.5°C, 74.2°C, and 75°C). The ribonucleoprotein complexes were incubated for 10–60 min with an increment of 10 min. A fluorescence-based reporter (FQ) and dsDNA activator were then added to each mixture to a final concentration of 250 nM and 1 nM, respectively. The reactions were isothermally incubated at their corresponding complexation temperature for consistency. Fluorescent measurements were taken every 30 s for 120 cycles on the HEX channel (λex: 525/10 nm, λem: 570/10 nm). This experiment was repeated at incubation temperatures of 67°C and 68°C without the gradient setting to cover a better temperature distribution. All experiments were repeated twice.
One-pot RT-LAMP-coupled BrCas12b detection assay
LAMP primers were designed using NEB LAMP Primer Design Tool from New England Biolabs (https://lamp.neb.com/) and PrimerExplorer v5 from Eiken Chemical Co. (https://primerexplorer.jp/e/).
BrCas12b variants, single-guide RNA, fluorescence-quencher reporter, and 10X LAMP primers were combined in 1X Warmstart Multi-Purpose/RT-LAMP Master Mix with UDG (New England Biolabs) to a final concentration of 200 nM, 400 nM, 2000 nM, and 1X LAMP primers (200 nM F3/B3, 1600 nM FIP/BIP, and 800 nM LF/LB), respectively, yielding a volume of 22 mL. The activator (dsDNA/RNA) was added to the mixture (25 mL final volume), and the reaction was then transferred to a CFX96 Real-Time PCR system with a C1000 Thermal Cycler module (Bio-rad). Fluorescence measurements were taken every 30 s per cycle for 120 cycles.
PAM library
For trans-cleavage assays without the RT-LAMP step, BrCas12b and sgRNA activators were combined to a final concentration of 100 nM: 50 nM respectively in 1X NEBuffer 2.1 (New England Biolabs) and incubated at 65°C for 15 min. Fluorescence-based reporter (FQ) and 25 nM activators containing various PAM sequences were added to the reaction mixture containing the Cas12b: sgRNA complex to a final concentration of 250 nM and 1 nM, respectively. The entire reaction was then immediately transferred to a Bio-rad CFX96 Real-Time system with C1000 Thermal Cycler module. The reaction was isothermally incubated at 65°C, and fluorescence measurements were read every 30 s per cycle over 120 cycles.
In the experiment with RT-LAMP, the one-pot BrCas12b detection assay method was utilized. 3 μL of 10 p.m. activators containing various PAM sequences were added to the reaction. Both experiments were carried out in duplicates and repeated twice.
Patient samples extraction and processing
The Quick-DNA/RNA™ Viral MagBead extraction was conducted according to manufacturer protocol. 10 μL of Proteinase K (20 mg/mL) was combined with 200 μL of serum samples in 1.5 mL centrifuge tubes and incubated at room temperature (RT) for 15 min. DNA/RNA Shield™ (2X concentrate) was added to the serum sample containing Protease K in a 1:1 ratio. 800 μL Viral DNA/RNA Buffer was then incorporated into the combined mixture followed by the addition of 20 μL MagBinding Beads™ and vortexed for 10 min. The centrifuge tubes were placed in a magnetic stand until the beads were pelleted. The supernatant was aspirated and discarded. The beads were washed with 250 μL of MagBead DNA/RNA Wash 1, 250 μL MagBead DNA/RNA Wash 2, and two rounds of 250 μL of 100% ethanol. The tubes containing beads were air-dried for 10 min. DNA/RNA was eluted using 30 μL DNase/RNase-Free water and was subjected to a BrCas12b detection reaction.
RT-qPCR of HCV and SARS-CoV-2 clinical samples
HCV-specific primers and probe sequences were designed as described in Zauli et al.59 The probe was modified by adding an internal ZEN™ quencher from IDT to reduce the background signal. Ct values for 80 HCV serum and 66 SARS-CoV-2 saliva samples were obtained by using Luna Probe One-Step RT-qPCR 4X Mix with UDG (NEB# M3019S) and following manufacturer’s protocol. Briefly, Luna Probe One-Step RT-qPCR 4X Mix with UDG, forward and reverse primers, and probe were combined to a final concentration of 1X, 0.4 μM (each), and 0.2 μM, respectively. Once assembled, the master mix was added to a 96-well plate (Applied Biosystems). 3 μL of extracted patient sample were deposited in each well, to a total volume of 20 μL, to initiate the reaction. The 96-well plate was then inserted into the StepOnePlus Real-Time PCR system (Applied Biosystems), which set auto threshold to calculate the Ct values.
Quantification and statistical analysis
Patient samples containing positive HCV and SARS-CoV-2 pathogens were randomized and blinded. Data were analyzed and visualized using GraphPad Prism 8 (GraphPad Software, San Diego, CA) and results were expressed as mean ± SD.
Acknowledgments
We are grateful to the entire HCV-TARGET consortium, particularly the principal investigator, Dr. David Nelson, and the staff member, Lauren Morelli, at the University of Florida for providing us with their expert guidance and clinical samples for this study. We are thankful to the past and current members of the Jain Lab, Dr. Gary Wang’s Lab, and Dr. Fan Ren’s Lab at the University of Florida for sharing reagents and their insightful feedback. This work was financially supported by funds from the University of Florida (UF); the UF Herbert Wertheim College of Engineering; Dinesh O. Shah endowed professorship; NIH-NIAID R21AI156321; NIH-NIAID R21AI168795; and NIH-NIGMS R35GM147788. The funding sources did not have a role in the design of the study or the collection, analysis, or interpretation of data, nor in the writing of the manuscript.
Author contributions
P.K.J., L.T.N., S.R.R., L.G.Y., and N.C.M. designed the experiments. L.T.N., S.R.R., L.G.Y., and N.C.M. mainly performed experiments with support from other co-authors. L.C. and A.P. performed AlphaFold predictions of all BrCas12b variants. L.T.N., S.R.R., and L.G.Y. wrote the initial manuscript with support from the rest of the co-authors. The manuscript was proofread, edited, and approved by all authors.
Declaration of interests
L.T.N., S.R.R., and P.K.J. are listed as inventors on the patent applications related to the content of this work. P.K.J. is a co-founder of Genable Biosciences, LLC, Par Biosciences, LLC, and CRISPR, LLC.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community.
Published: May 8, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101037.
Supplemental information
Data and code availability
-
•
All data are presented in the main text as well as in the supplementary information. Predicted structures of all BrCas12b variants are included in the source files attached to this manuscript.
-
•
This paper does not contain original code.
-
•
Any additional information required to re-analyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Gootenberg J.S., Abudayyeh O.O., Lee J.W., Essletzbichler P., Dy A.J., Joung J., Verdine V., Donghia N., Daringer N.M., Freije C.A., et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017;356:438–442. doi: 10.1126/science.aam9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Broughton J.P., Deng X., Yu G., Fasching C.L., Servellita V., Singh J., Miao X., Streithorst J.A., Granados A., Sotomayor-Gonzalez A., et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020;38:870–874. doi: 10.1038/s41587-020-0513-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patchsung M., Jantarug K., Pattama A., Aphicho K., Suraritdechachai S., Meesawat P., Sappakhaw K., Leelahakorn N., Ruenkam T., Wongsatit T., et al. Clinical validation of a Cas13-based assay for the detection of SARS-CoV-2 RNA. Nat. Biomed. Eng. 2020;4:1140–1149. doi: 10.1038/s41551-020-00603-x. [DOI] [PubMed] [Google Scholar]
- 4.Hampton T. Virus surveillance and diagnosis with a CRISPR-based platform. Jama-J Am Med Assoc. 2020;324:430. doi: 10.1001/jama.2020.12669. [DOI] [PubMed] [Google Scholar]
- 5.Welch N.L., Zhu M., Hua C., Weller J., Mirhashemi M.E., Nguyen T.G., Mantena S., Bauer M.R., Shaw B.M., Ackerman C.M., et al. Multiplexed CRISPR-based microfluidic platform for clinical testing of respiratory viruses and identification of SARS-CoV-2 variants. Nat. Med. 2022;28:1083–1094. doi: 10.1038/s41591-022-01734-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joung J., Ladha A., Saito M., Kim N.G., Woolley A.E., Segel M., Barretto R.P.J., Ranu A., Macrae R.K., Faure G., et al. Detection of SARS-CoV-2 with SHERLOCK one-pot testing. N. Engl. J. Med. 2020;383:1492–1494. doi: 10.1056/NEJMc2026172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Notomi T., Okayama H., Masubuchi H., Yonekawa T., Watanabe K., Amino N., Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28:E63. doi: 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amaral C., Antunes W., Moe E., Duarte A.G., Lima L.M.P., Santos C., Gomes I.L., Afonso G.S., Vieira R., Teles H.S.S., et al. A molecular test based on RT-LAMP for rapid, sensitive and inexpensive colorimetric detection of SARS-CoV-2 in clinical samples. Sci. Rep. 2021;11:16430. doi: 10.1038/s41598-021-95799-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang X., Tang G., Ismail N., Wang X. Developing RT-LAMP assays for rapid diagnosis of SARS-CoV-2 in saliva. EBioMedicine. 2022;75:103736. doi: 10.1016/j.ebiom.2021.103736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen L.T., Macaluso N.C., Pizzano B.L.M., Cash M.N., Spacek J., Karasek J., Miller M.R., Lednicky J.A., Dinglasan R.R., Salemi M., Jain P.K. A thermostable Cas12b from Brevibacillus leverages one-pot discrimination of SARS-CoV-2 variants of concern. EBioMedicine. 2022;77:103926. doi: 10.1016/j.ebiom.2022.103926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong Y.P., Othman S., Lau Y.L., Radu S., Chee H.Y. Loop-mediated isothermal amplification (LAMP): a versatile technique for detection of micro-organisms. J. Appl. Microbiol. 2018;124:626–643. doi: 10.1111/jam.13647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L., et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kopp J., Schwede T. The SWISS-MODEL repository: new features and functionalities. Nucleic Acids Res. 2006;34:D315–D318. doi: 10.1093/nar/gkj056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guex N., Peitsch M.C., Schwede T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis. 2009;30:S162–S173. doi: 10.1002/elps.200900140. [DOI] [PubMed] [Google Scholar]
- 16.Studer G., Rempfer C., Waterhouse A.M., Gumienny R., Haas J., Schwede T. QMEANDisCo-distance constraints applied on model quality estimation (vol 36, pg 1765, 2020) Bioinformatics. 2020;36:2647. doi: 10.1093/bioinformatics/btaa058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertoni M., Kiefer F., Biasini M., Bordoli L., Schwede T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017;7:10480. doi: 10.1038/s41598-017-09654-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sumbalova L., Stourac J., Martinek T., Bednar D., Damborsky J. HotSpot Wizard 3.0: web server for automated design of mutations and smart libraries based on sequence input information. Nucleic Acids Res. 2018;46:W356–W362. doi: 10.1093/nar/gky417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao H., Wang J., He L., Qi Y., Zhang J.Z. DeepDDG: predicting the stability change of protein point mutations using neural networks. J. Chem. Inf. Model. 2019;59:1508–1514. doi: 10.1021/acs.jcim.8b00697. [DOI] [PubMed] [Google Scholar]
- 20.Wang X., Luo H., Yu W., Ma R., You S., Liu W., Hou L., Zheng F., Xie X., Yao B. A thermostable Gloeophyllum trabeum xylanase with potential for the brewing industry. Food Chem. 2016;199:516–523. doi: 10.1016/j.foodchem.2015.12.028. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Y., Miao Y., Zhi F., Pan Y., Zhang J., Yang X., Zhang J.Z.H., Zhang L. Rational design of pepsin for enhanced thermostability via exploiting the guide of structural weakness on stability. Front. Phys. 2021;9 doi: 10.3389/fphy.2021.755253. [DOI] [Google Scholar]
- 22.Gootenberg J.S., Abudayyeh O.O., Kellner M.J., Joung J., Collins J.J., Zhang F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science. 2018;360:439–444. doi: 10.1126/science.aaq0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J.S., Ma E., Harrington L.B., Da Costa M., Tian X., Palefsky J.M., Doudna J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 2018;360:436–439. doi: 10.1126/science.aar6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li L., Li S., Wu N., Wu J., Wang G., Zhao G., Wang J. HOLMESv2: a CRISPR-cas12b-assisted platform for nucleic acid detection and DNA methylation quantitation. ACS Synth. Biol. 2019;8:2228–2237. doi: 10.1021/acssynbio.9b00209. [DOI] [PubMed] [Google Scholar]
- 25.Qiao X., Gao Y., Li J., Wang Z., Qiao H., Qi H. Sensitive analysis of single nucleotide variation by Cas13d orthologs, EsCas13d and RspCas13d. Biotechnol. Bioeng. 2021;118:3037–3045. doi: 10.1002/bit.27813. [DOI] [PubMed] [Google Scholar]
- 26.Strecker J., Jones S., Koopal B., Schmid-Burgk J., Zetsche B., Gao L., Makarova K.S., Koonin E.V., Zhang F. Engineering of CRISPR-Cas12b for human genome editing. Nat. Commun. 2019;10:212. doi: 10.1038/s41467-018-08224-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tian Y., Liu R.R., Xian W.D., Xiong M., Xiao M., Li W.J. A novel thermal Cas12b from a hot spring bacterium with high target mismatch tolerance and robust DNA cleavage efficiency. Int. J. Biol. Macromol. 2020;147:376–384. doi: 10.1016/j.ijbiomac.2020.01.079. [DOI] [PubMed] [Google Scholar]
- 28.Teng F., Cui T., Feng G., Guo L., Xu K., Gao Q., Li T., Li J., Zhou Q., Li W. Repurposing CRISPR-Cas12b for mammalian genome engineering. Cell Discov. 2018;4:63. doi: 10.1038/s41421-018-0069-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen L.T., Smith B.M., Jain P.K. Enhancement of trans-cleavage activity of Cas12a with engineered crRNA enables amplified. Nat. Commun. 2020;11:6104. doi: 10.1038/s41467-020-20117-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fozouni P., Son S., Díaz de León Derby M., Knott G.J., Gray C.N., D'Ambrosio M.V., Zhao C., Switz N.A., Kumar G.R., Stephens S.I., et al. Amplification-free detection of SARS-CoV-2 with CRISPR-Cas13a and mobile phone microscopy. Cell. 2021;184:323–333.e9. doi: 10.1016/j.cell.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu T.Y., Knott G.J., Smock D.C.J., Desmarais J.J., Son S., Bhuiya A., Jakhanwal S., Prywes N., Agrawal S., Díaz de León Derby M., et al. Accelerated RNA detection using tandem CRISPR nucleases (vol 17, pg 982, 2021) Nat. Chem. Biol. 2021;17:1210. doi: 10.1038/s41589-021-00882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu D., Guan X., Zhu Y., Ren K., Huang Z. Structural basis of stringent PAM recognition by CRISPR-C2c1 in complex with sgRNA. Cell Res. 2017;27:705–708. doi: 10.1038/cr.2017.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Oliveira Coelho B., Sanchuki H.B.S., Zanette D.L., Nardin J.M., Morales H.M.P., Fornazari B., Aoki M.N., Blanes L. Essential properties and pitfalls of colorimetric Reverse Transcription Loop-mediated Isothermal Amplification as a point-of-care test for SARS-CoV-2 diagnosis. Mol. Med. 2021;27:30. doi: 10.1186/s10020-021-00289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hardinge P., Murray J.A.H. Reduced false positives and improved reporting of loop-mediated isothermal amplification using quenched fluorescent primers. Sci. Rep. 2019;9:7400. doi: 10.1038/s41598-019-43817-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ali Z., Aman R., Mahas A., Rao G.S., Tehseen M., Marsic T., Salunke R., Subudhi A.K., Hala S.M., Hamdan S.M., et al. iSCAN: an RT-LAMP-coupled CRISPR-Cas12 module for rapid, sensitive detection of SARS-CoV-2. Virus Res. 2020;288:198129. doi: 10.1016/j.virusres.2020.198129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim H., Lee W.J., Kim C.H., Oh Y., Gwon L.W., Lee H., Song W., Hur J.K., Lim K.S., Jeong K.J., et al. Highly specific chimeric DNA-RNA-guided genome editing with enhanced CRISPR-Cas12a system. Mol Ther-Nucl Acids. 2022;28:353–362. doi: 10.1016/j.omtn.2022.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hand T.H., Das A., Li H. Directed evolution studies of a thermophilic Type II-C Cas9. Method Enzymol. 2019;616:265–288. doi: 10.1016/bs.mie.2018.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar S., Tsai C.J., Nussinov R. Factors enhancing protein thermostability. Protein Eng. 2000;13:179–191. doi: 10.1093/protein/13.3.179. [DOI] [PubMed] [Google Scholar]
- 39.Fernández-Recio J., Vázquez A., Civera C., Sevilla P., Sancho J. The tryptophan/histidine interaction in alpha-helices. J. Mol. Biol. 1997;267:184–197. doi: 10.1006/jmbi.1996.0831. [DOI] [PubMed] [Google Scholar]
- 40.Louwrier A., van der Valk A. Can sucrose affect polymerase chain reaction product formation? Biotechnol. Lett. 2001;23:175–178. doi: 10.1023/A:1005656100993. [DOI] [Google Scholar]
- 41.Li Z., Zhao W., Ma S., Li Z., Yao Y., Fei T. A chemical-enhanced system for CRISPR-Based nucleic acid detection. Biosens Bioelectron. 192. 2021;192 doi: 10.1016/j.bios.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spiess A.N., Mueller N., Ivell R. Trehalose is a potent PCR enhancer: lowering of DNA melting temperature and thermal stabilization of Taq polymerase by the disaccharide trehalose. Clin. Chem. 2004;50:1256–1259. doi: 10.1373/clinchem.2004.031336. [DOI] [PubMed] [Google Scholar]
- 43.Jensen M.A., Fukushima M., Davis R.W. DMSO and betaine greatly improve amplification of GC-rich constructs in de novo synthesis. PLoS One. 2010;5:e11024. doi: 10.1371/journal.pone.0011024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee J.C., Timasheff S.N. The stabilization of proteins by sucrose. J. Biol. Chem. 1981;256:7193–7201. [PubMed] [Google Scholar]
- 45.Rozhkov S.P. Stabilization of protein by sucrose as revealed by the spin-label method. Biofizika. 1991;36:571–576. [PubMed] [Google Scholar]
- 46.Ruan K., Xu C., Li T., Li J., Lange R., Balny C. The thermodynamic analysis of protein stabilization by sucrose and glycerol against pressure-induced unfolding - the typical example of the 33-kDa protein from spinach photosystem II. Eur. J. Biochem. 2003;270:1654–1661. doi: 10.1046/j.1432-1033.2003.03485.x. [DOI] [PubMed] [Google Scholar]
- 47.Jain I., Minakhin L., Mekler V., Sitnik V., Rubanova N., Severinov K., Semenova E. Defining the seed sequence of the Cas12b CRISPR-Cas effector complex. RNA Biol. 2019;16:413–422. doi: 10.1080/15476286.2018.1495492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gupta E., Bajpai M., Choudhary A. Hepatitis C virus: screening, diagnosis, and interpretation of laboratory assays. Asian J. Transfus. Sci. 2014;8:19–25. doi: 10.4103/0973-6247.126683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Madhvi A., Hingane S., Srivastav R., Joshi N., Subramani C., Muthumohan R., Khasa R., Varshney S., Kalia M., Vrati S., et al. A screen for novel hepatitis C virus RdRp inhibitor identifies a broad-spectrum antiviral compound. Sci. Rep. 2017;7:5816. doi: 10.1038/s41598-017-04449-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spearman C.W., Dusheiko G.M., Hellard M., Sonderup M. Hepatitis C. Lancet. 2019;394:1451–1466. doi: 10.1016/S0140-6736(19)32320-7. [DOI] [PubMed] [Google Scholar]
- 51.Zeuzem S., Foster G.R., Wang S., Asatryan A., Gane E., Feld J.J., Asselah T., Bourlière M., Ruane P.J., Wedemeyer H., et al. Glecaprevir-pibrentasvir for 8 or 12 Weeks in HCV genotype 1 or 3 infection. N. Engl. J. Med. 2018;378:354–369. doi: 10.1056/NEJMoa1702417. [DOI] [PubMed] [Google Scholar]
- 52.Konerman M.A., Lok A.S. Diagnostic challenges of hepatitis C. JAMA. 2014;311:2536–2537. doi: 10.1001/jama.2014.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ishizaki A., Bouscaillou J., Luhmann N., Liu S., Chua R., Walsh N., Hess S., Ivanova E., Roberts T., Easterbrook P. Survey of programmatic experiences and challenges in delivery of hepatitis B and C testing in low- and middle-income countries. BMC Infect. Dis. 2017;17:696. doi: 10.1186/s12879-017-2767-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Terrault N.A. Hepatitis C elimination: challenges with under-diagnosis and under-treatment. F1000Res. 2019;8 doi: 10.12688/f1000research.15892.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hongjaisee S., Doungjinda N., Khamduang W., Carraway T.S., Wipasa J., Debes J.D., Supparatpinyo K. Rapid visual detection of hepatitis C virus using a reverse transcription loop-mediated isothermal ampli fi cation assay. Int. J. Infect. Dis. 2021;102:440–445. doi: 10.1016/j.ijid.2020.10.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li X., Wang Y., Liu Y., Yang B., Wang X., Wei J., Lu Z., Zhang Y., Wu J., Huang X., et al. Base editing with a Cpf1-cytidine deaminase fusion. Nat. Biotechnol. 2018;36:324–327. doi: 10.1038/nbt.4102. [DOI] [PubMed] [Google Scholar]
- 57.Wang X., Ding C., Yu W., Wang Y., He S., Yang B., Xiong Y.C., Wei J., Li J., Liang J., et al. Cas12a base editors induce efficient and specific editing with low DNA damage response. Cell Rep. 2020;31:107723. doi: 10.1016/j.celrep.2020.107723. [DOI] [PubMed] [Google Scholar]
- 58.Kapust R.B., Tözsér J., Fox J.D., Anderson D.E., Cherry S., Copeland T.D., Waugh D.S. Tobacco etch virus protease: Mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 59.Zauli D.A.G., Menezes C.L.P.d., Oliveira C.L.d., Mateo E.C.C., Ferreira A.C.d.S. In-house quantitative real-time PCR for the diagnosis of hepatitis B virus and hepatitis C virus infections. Braz. J. Microbiol. 2016;47:987–992. doi: 10.1016/j.bjm.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shilling P.J., Mirzadeh K., Cumming A.J., Widesheim M., Köck Z., Daley D.O. Improved designs for pET expression plasmids increase protein production yield in Escherichia coli. Commun Biol. 2020;3:214. doi: 10.1038/s42003-020-0939-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mirdita M., Schütze K., Moriwaki Y., Heo L., Ovchinnikov S., Steinegger M. ColabFold: making protein folding accessible to all. Nat. Methods. 2022;19:679–682. doi: 10.1038/s41592-022-01488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bienert S., Waterhouse A., de Beer T.A.P., Tauriello G., Studer G., Bordoli L., Schwede T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017;45:D313–D319. doi: 10.1093/nar/gkw1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data are presented in the main text as well as in the supplementary information. Predicted structures of all BrCas12b variants are included in the source files attached to this manuscript.
-
•
This paper does not contain original code.
-
•
Any additional information required to re-analyze the data reported in this work paper is available from the lead contact upon request.